

Abstract
Ion channels are macromolecular complexes whose function is exquisitely tuned by interacting proteins. Fluorescence Resonance Energy Transfer (FRET) is a powerful methodology that is adept at quantifying ion channel protein-protein interactions in living cells. For FRET experiments, the interacting partners are tagged with appropriate donor and acceptor fluorescent proteins. If the fluorescently-labeled molecules are in close proximity, then photoexcitation of the donor results in non-radiative energy transfer to the acceptor, and subsequent fluorescence emission of the acceptor. The stoichiometry of ion channel interactions and their relative binding affinities can be deduced by quantifying both the FRET efficiency and the total number of donors and acceptors in a given cell. In this chapter, we discuss general considerations for FRET analysis of biological interactions, various strategies for estimating FRET efficiencies, and detailed protocols for construction of binding curves and determination of stoichiometry. We focus on implementation of FRET assays using a flow cytometer given its amenability for high-throughput data acquisition, enhanced accessibility, and robust analysis. This versatile methodology permits mechanistic dissection of dynamic changes in ion channel interactions.
Keywords: Fluorescence Resonance Energy Transfer, FRET 2-hybrid assay, Flow Cytometry, Stoichiometry, Live-cell binding
1. INTRODUCTION
Ion channels are macromolecular complexes whose functions are exquisitely tuned by a rich repertoire of regulatory proteins (Handbook of Ion Channels, 2015; Isacoff, Jan, & Minor, 2013). This process enables ion flux to be orchestrated with subcellular precision to support diverse physiological roles. Disruption of ion channel regulation has emerged as a pivotal factor for many human ailments including ion channelopathies (Cannon, 2007; Schwartz, Ackerman, & Wilde, 2017). As such, elucidating ion channel assembly and dynamic interactions are vital for identifying and understanding in-depth physiological and pathophysiological mechanisms.
An impressive collection of cell biological and biophysical methods has been applied in recent years for studying ion channel assemblies as described in other chapters. These methods have distinct spatial and temporal resolutions and possess unique advantages in probing various facets of ion channel macromolecular complexes (Algar, Hildebrandt, Vogel, & Medintz, 2019). Techniques such as X-ray crystallography and single-particle cryo-electron microscopy offer sub-nanometer or atomic-scale resolution. Optical approaches have also been broadly adapted to study ion channel complexes in situ (Sigal, Zhou, & Zhuang, 2018; Zhang, Carver, Choveau, & Shapiro, 2016). Super-resolution microscopy including stochastic optical reconstruction microscopy (STORM), photoactivated localization microscopy (PALM) (Betzig et al., 2006), stimulated emission depletion microscopy (STED) (Hein, Willig, & Hell, 2008), and stochastic optical fluctuation imaging (SOFI) (Dertinger, Colyer, Iyer, Weiss, & Enderlein, 2009) have 20-100 nm resolution, and permit analysis of subcellular organization of individual molecules and assemblies in their physiological environment (Godin, Lounis, & Cognet, 2014; Sigal et al., 2018). In comparison, Fluorescence Resonance Energy Transfer (FRET) is advantageous for monitoring biological interactions in living cells, given its exquisite ability to identify co-localization of molecules within 1-10 nm (Sekar & Periasamy, 2003; Vogel, van der Meer, & Blank, 2014). Furthermore, FRET is rapid and well-suited for tracking dynamic changes in protein-protein interactions. In addition, FRET measurements are facile with minimal sample preparation requirements and can be obtained from a variety of instrumentation with some platforms amenable to high throughput analysis (Algar et al., 2019).
Conceptually, FRET is a spectroscopic phenomenon (Forster, 1948; Lackowicz, 1983; Selvin, 1995). A FRET system at its core is composed of two fluorophores: (1) a donor and (2) an acceptor. When the donor fluorophore is excited, it typically de-excites via direct emission of a photon with its characteristic wavelength. However, when the acceptor fluorophore is in close-proximity and in the right orientation, the donor may instead transfer energy to the acceptor via long-range dipole-dipole coupling (Clegg, 1992; Selvin, 1995). Subsequently, the acceptor may de-excite by emitting a photon with a longer wavelength. FRET refers to this process of non-radiative energy transfer (Forster, 1948). The efficiency of energy transfer from the donor to the acceptor fluorophore is known as FRET efficiency. This parameter can be estimated via a number of approaches by considering the donor and acceptor fluorescence with different excitation strategies. The conditions for FRET are quite stringent. First, the emission spectrum of the donor fluorophore must overlap with the excitation spectrum of the acceptor. Second, FRET is highly sensitive to the relative spatial arrangement of the two fluorophores including both the distance between the fluorophores and the relative orientation of the two dipole moments. In fact, the rate of FRET is inversely proportional to the sixth-power of the distance between the two fluorophores much like London dispersion forces (Clegg, 1992). Each donor-acceptor FRET pair has a characteristic distance—known as the Förster distance (R0)—where the likelihood of energy transfer between the two fluorophores is 50%. FRET efficiency measurements exhibit its highest sensitivity as the distance of separation between the donor and acceptor approaches R0. Importantly, R0 depends on: (1) the quantum yield of the donor (QD), i.e., the efficiency of photon emission; (2) the molar extinction coefficient (εA) of the acceptor; (3) the spectral overlap between the two fluorophores (J(λ)); and (4) the relative orientation of two molecules, typically indicated by the parameter κ2 (Clegg, 1992; Selvin, 1995). The exquisite sensitivity of FRET for molecular distances makes it a highly desirable method to detect biomolecular interactions and protein conformational dynamics (Algar et al., 2019).
Furthermore, over the past three decades, the expanding palette of genetically encoded fluorophores (Bajar, Wang, Zhang, Lin, & Chu, 2016; Shaner, Steinbach, & Tsien, 2005) and protein labeling strategies (England, Luo, & Cai, 2015; Hoffmann et al., 2010) have enabled a wide range of biological applications for FRET. In particular, FRET or related processes have been applied as an atomic-scale ruler to track conformational changes of ion channel domains such as the voltage-sensing domain (Glauner, Mannuzzu, Gandhi, & Isacoff, 1999), G-protein-coupled conformational changes of GIRK channels (Raveh, Riven, & Reuveny, 2009), BK channel gating ring (Miranda et al., 2013), and others (Wang et al., 2019). Moreover, numerous FRET-based biosensors have been engineered to monitor dynamic changes in cellular signaling and various physiologically relevant parameters (Tsien, 2009), such as cellular concentrations of second messenger molecules like Ca2+ (Miyawaki et al., 1997) and cAMP (Zaccolo et al., 2000), physical properties like membrane potential (Zou et al., 2014) and cellular forces (Grashoff et al., 2010), and the biological activity of signaling molecules such as kinases, phosphatases, and proteases (Hochreiter, Garcia, & Schmid, 2015; Zhang, Ma, Taylor, & Tsien, 2001). In addition, FRET has also emerged as a valuable tool for the detection of biomolecular interactions in intact cells (Chen, Puhl, & Ikeda, 2007; Erickson, Alseikhan, Peterson, & Yue, 2001; Hoppe, Christensen, & Swanson, 2002; Wouters, Verveer, & Bastiaens, 2001; You et al., 2006). In this chapter, we discuss the usage of FRET to quantify stoichiometry and relative affinities for ion channel interactions, including specific protocols for implementing FRET 2-hybrid assays using a flow cytometer (Hochreiter, Kunze, Moser, & Schmid, 2019; Lee, Sang, & Yue, 2016; Ujlaky-Nagy, Nagy, Szollosi, & Vereb, 2018).
2. OBTAINING FRET EFFICIENCY FROM LIVE CELLS
2.1. FRET Efficiency.
FRET efficiency (E) is a commonly reported measure of FRET that corresponds to the probability that following photoexcitation, a donor fluorophore de-excites via energy transfer as opposed to direct photon emission or other non-radiative pathways. Mathematically, the efficiency of the energy transfer for a given donor-acceptor pair is related to the rate of energy transfer (kT), the rate of fluorescence decay (kD), and the rate of all non-radiative de-excitation pathways (kd,nr) as follows (Algar et al., 2019; Clegg, 1992; Selvin, 1995):
| (1) |
Physically, FRET efficiency is inversely related to the sixth power of the distance between the donor and acceptor (R). Direct estimation of the molecular distances from FRET efficiencies, however, is often impractical as estimation of the Förster distance (R0) typically assumes rapid rotation of the fluorophores resulting in random dipole orientations (Vogel et al., 2014). Although this assumption is true for small organic dyes that rotate on a picosecond timescale, large fluorescent proteins rotate slowly (20ns) and thus may result in a heterogenous dipole distribution resulting in biased distance estimates (Partikian, Olveczky, Swaminathan, Li, & Verkman, 1998; Sarkar et al., 2017). Practically, FRET efficiency can be experimentally determined by measuring either fluorescence lifetimes or intensities (Algar et al., 2019). Specifically, when an acceptor is nearby, the fluorescence decay of the donor (τDA) is accelerated resulting in shorter lifetimes compared to that in the absence of the acceptor (τD). Similarly, FRET efficiency also relates to the donor fluorescence intensity, as the presence of the acceptor quenches the fluorescence of the donor. Thus, E can be estimated from the ratio of fluorescence intensities of the donor-acceptor complex (IDA) to that of the donor in the absence of the acceptor (ID).
2.2. Approaches to measure FRET efficiency.
Several experimental strategies for obtaining FRET efficiencies from biological samples have been reviewed extensively (Algar et al., 2019; Vogel, Thaler, & Koushik, 2006). Approaches for quantification utilize measurements of fluorescence intensity, fluorescence lifetime, and anisotropy. Although these methods utilize different instrumentation, they follow two general strategies (Butz et al., 2016) – (1) measuring quenching of the donor fluorescence, and (2) detecting enhancement in acceptor fluorescence due to sensitized emission. Here, we briefly describe a few of these approaches before delving further into three-cube FRET methodology which is versatile and can be implemented on multiple platforms using either a conventional epifluorescence microscope or a flow cytometer (Butz et al., 2016; Ujlaky-Nagy et al., 2018).
(1). Acceptor photobleaching / FRET-FLIM —
A direct approach to measure FRET efficiency involves determining changes in donor fluorescence both in the presence and the absence of the acceptor. The acceptor photobleaching approach measures the donor fluorescence intensity both before and after photobleaching of the acceptor (Wallrabe & Periasamy, 2005). If FRET existed between the donor and acceptor, then the donor fluorescence is enhanced following photobleaching of the acceptor. The fractional increase in donor fluorescence can be used to directly obtain FRET efficiency. A second possibility is to use Fluorescence Lifetime Imaging (FLIM) (Clegg, Holub, & Gohlke, 2003). Here, the donor’s fluorescence lifetime is measured by quantifying the fluorescence decay following a brief excitation light pulse both before and after photobleaching of the acceptor. If FRET exists between the donor and acceptor, then following acceptor photobleaching, the fluorescence lifetime of the donor is prolonged. The FRET efficiency can then be determined by computing the ratio of the donor lifetimes before and after photobleaching. The advantage of these approaches is that they offer a direct measurement of FRET; however, a weakness is that they require photo-destruction of the acceptor and, therefore, are not amenable for dynamic FRET measurements (Algar et al., 2019).
(2). Spectral imaging / Linear Unmixing —
In principle, FRET efficiency may be deduced non-destructively by considering the acceptor fluorescence emission following the excitation of the donor (Neher & Neher, 2004; Zimmermann, 2005). However, as fluorescent proteins have broad excitation and emission spectra, there is significant overlap between the donor and acceptor spectra (Neher & Neher, 2004). Spectral imaging and linear unmixing obtains FRET efficiency by deconstructing the donor, the acceptor, and the sensitized emission signals from the aggregate spectrum that is obtained using spectral imaging microscopy (Thaler, Koushik, Blank, & Vogel, 2005; Zheng, 2006). Specifically, the aggregate spectrum of the donor-acceptor undergoing FRET is obtained at two different excitation wavelengths, typically corresponding to the donor excitation and the acceptor excitation (Zheng, 2006). In addition, the individual spectra of the donor and acceptor are determined from cells expressing the donor or the acceptor alone. These measurements combined with a fitting procedure to estimate individual fluorescence components which are then used to estimate FRET efficiency (Zheng, 2006). The advantage of this approach is that measurements are robust as fits are performed using complete donor and acceptor spectra. The disadvantage, however, is that specialized equipment is required to obtain fluorescence spectra.
(3). Three-cube FRET —
Conceptually similar to spectral imaging / linear unmixing, three-cube FRET simplifies the spectral unmixing problem by obtaining only three different fluorescence signals from each individual cell: (1) the donor signal, (2) the acceptor signal, and (3) the FRET signal to correct for crosstalk of the donor-acceptor fluorophores (Chen et al., 2007; Erickson, Liang, Mori, & Yue, 2003; Hoppe et al., 2002; Wlodarczyk et al., 2008). Subsequently, the corrected fluorescence emissions are used to obtain sensitized emission of the acceptor due to FRET, and ultimately, FRET efficiency as described in depth in subsequent sections. This approach has gained popularity as it is non-destructive, rapid, and relatively straightforward to implement using commonly available equipment. The weakness, however, is Three-cube FRET calculations require careful instrumentation-specific calibration (Butz et al., 2016).
(4). Anisotropy measurements –
As the excitation and emission spectra of many protein fluorophores are often broad, considerable overlap may exist between the excitation and emission spectra of a given fluorophore. As such, FRET may occur between identical fluorophores that are spatially clustered, a process known as homo-FRET (Thaler, Koushik, Puhl, Blank, & Vogel, 2009). This approach is particularly useful for determining clustering of a fluorescent-tagged protein or subunit stoichiometry. As identical fluorophores are involved in homo-FRET, this process does not affect the emission spectrum of fluorescence lifetime. As such afore-mentioned approaches are not suitable. Instead, steady-state or time-resolved fluorescence anisotropy may be used for quantifying homo-FRET (Devauges et al., 2014; Thaler et al., 2009). The rate of anisotropy decay typically corresponds to rotational mobility of a fluorophore. However, homo-FRET causes increased randomization of fluorophores and reduces anisotropy.
3. GENERAL OVERVIEW OF THREE-CUBE FRET MEASUREMENTS
Several formalisms of the Three-cube FRET methodology have been developed to date and have been applied to many biological problems (Algar et al., 2019). The focus of this chapter is to elucidate methods for deducing relative binding affinities and stoichiometry of macromolecular interactions in live cells.
The three-cube FRET method can be readily adapted to various data acquisition platforms including epifluorescence and confocal microscopes (Butz et al., 2016; Chen et al., 2007; Erickson et al., 2003; Hoppe et al., 2002), commercially available flow cytometers (Lee et al., 2016), and plate readers (Liao, Song, & Liu, 2015). A variety of fluorescent protein FRET pairs with varying spectral properties and brightness are also appropriate for monitoring protein-protein interactions (Bajar et al., 2016). For epifluorescence microscopy-based setup, the Donor, Acceptor, and FRET fluorescent cubes should be optimized for a given FRET pair (Butz et al., 2016). The advantages of microscopy-based setups are: (1) depending on the optics of the microscope and the quantum efficiency of the imaging device, highly reliable fluorescence measurements are possible even if protein expression is low; (2) the ability to discern sub-cellular differences in FRET, and (3) the feasibility of obtaining kinetic changes in FRET from the same cell. The disadvantage of this approach is that the throughput is often limited.
Commercially available flow cytometers are equipped with multiple lasers and several emission channels well-suited for probing a wide-range of fluorescent proteins and permitting reliable FRET measurements (Lee et al., 2016; Ujlaky-Nagy et al., 2018). The availability of multiple channels also enables simultaneous measurement of fluorescent signals from additional fluorophores that may not be involved in FRET but serve as expression markers. The advantages for the flow cytometry approach are (1) vastly improved throughput as FRET efficiency can be quantified from thousands of individual cells in seconds, (2) standardized instrumentation and analysis procedure enhances accessibility with minimal training requirements, and (3) reduced selection bias as large populations of cells can be assessed. As previous protocols have elaborated on the implementation of three-cube FRET assays using a microscopy-based setup (Butz et al., 2016), here we focus on the flow cytometric approach.
The typical workflow for Three-cube FRET experiments is as follows: (1) determine spectral crosstalk parameters using cells expressing either the donor or acceptor alone, (2) obtain instrument-specific calibration constants using a series of donor-acceptor dimers, (3) obtain raw Donor, Acceptor, and FRET signals from samples to be studied; (4) calculate fundamental fluorescence signals by unmixing the raw signals using instrument-specific spectral cross-talk parameters; (5) use the fundamental fluorescence signals and instrument-specific calibration constants to calculate FRET efficiency using acceptor-centric or donor-centric approaches to estimate donor and acceptor concentrations; (6) correlate FRET efficiencies with donor and acceptor concentrations to obtain relative binding affinities and stoichiometry.
4. DEDUCING STOICHIOMETRY AND RELATIVE BINDING AFFINITIES FROM FRET 2-HYBRID ASSAYS
The process of FRET changes two features of the fluorescence emission spectrum of the bound donor-acceptor complex: (1) the donor emission is quenched as a result of FRET, and (2) the acceptor emission is enhanced. These spectral changes imply that FRET efficiency can be determined using two distinct metrics: (1) a donor-centric measure (EA) that reports the fractional reduction in the donor’s fluorescence intensity as a result of FRET (Fig. 1A) and (2) an acceptor-centric measure (ED) that quantifies the fractional enhancement in the acceptor’s fluorescence intensity due to FRET (Fig. 1A). If the donor and the acceptor molecules associate with a 1:1 stoichiometry, then the maximal donor-centric ED obtained at saturating concentrations of the acceptor is equivalent to the maximal acceptor-centric EA determined at saturating concentrations of the donor (Fig. 1B) (Ben-Johny, Yue, & Yue, 2016; Hoppe et al., 2002). Both metrics report the true time-independent transition probability of energy transfer between the donor-acceptor pair. However, if the complex contained multiple donors and acceptors, then the donor-centric ED metric reports the expected number of energy transfer even per donor molecule in the complex while the acceptor-centric EA metric reports the expected number of energy transfer even per acceptor molecule in the complex (Ben-Johny et al., 2016). Mathematically, for a complex containing nD donors and nA acceptors,
| (2) |
where Eij represent the time-independent transition probability of energy transfer (or pairwise FRET efficiency) between ith donor and jth acceptor (Fig. 1C). The stoichiometry of the complex, i.e. the ratio of the number of donors to the number of acceptors (nD / nA), is given by the ratio EA,max / ED,max (Ben-Johny et al., 2016).
Figure 1.
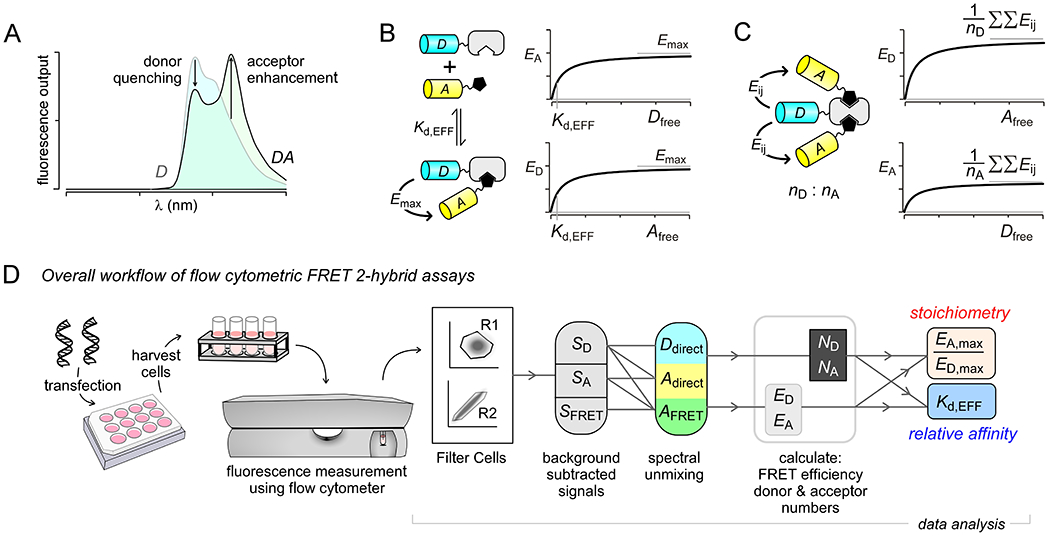
Conceptual framework of FRET 2-hybrid assay for probing the stoichiometry and binding of macromolecular complexes in live cells. (A) Changes in donor and acceptor emission spectra due to FRET. The donor emission is quenched as a result of FRET while the acceptor emission is enhanced. (B) Schematic shows 1:1 interaction of binding partners labeled with donor and acceptor fluorophores. Stochastic expression of the FRET pairs results in variable binding. When the donor and acceptor fluorophores are bound, they undergo FRET. FRET efficiencies can be quantified as either donor-centric ED (fractional quenching) or acceptor-centric EA (sensitized emission) methods. Correlating EA versus the free concentration of the acceptor (Afree) or ED versus the free concentration of the donor (Dfree) yields FRET-binding curves. With 1:1 binding, the saturating maximal FRET efficiency of both curves should equal while the concentration, and relative binding affinity can be measured as Dfree or Afree concentration where FRET efficiency is half maximal. (C) For binding interactions with stoichiometry of nD:nA, saturating EA and ED values are different. The ratio EA,max / ED,max yields the stoichiometry nD/nA. (D) Schematic shows overall workflow for FRET 2-hybrid experiments. Cells are transfected with FP-tagged proteins and analyzed using a flow cytometer. FRET efficiency and relative binding affinities are obtained following filtering for single cells, background subtraction, and spectral unmixing.
To obtain relative binding affinities, the relative donor and acceptor concentrations in each cell are estimated based on the intensity of the donor corrected for FRET and the intensity of the acceptor (Butz et al., 2016; Chen et al., 2007; Erickson et al., 2003; Hochreiter et al., 2019; Hoppe et al., 2002; Wlodarczyk et al., 2008). Subsequently, a binding model is imposed based on the estimated stoichiometry of the interaction. Both EA and ED FRET efficiencies from each cell are iteratively fit to obtain the free parameter Kd,EFF.
| (3a and 3b) |
Importantly, the free donor (Dfree) and acceptor (Afree) concentrations are be obtained as follows:
| (4) |
| (5) |
Interactions with non 1:1 stoichiometry are more complex as they involve multiple binding affinities and FRET efficiencies corresponding to sub-states resulting in an increase in the number of free parameters. If so, robust fits may not be always be feasible. In some scenarios, previous studies have utilized simplifying assumptions such as independent and identical binding to estimate aggregate affinities (Ben-Johny et al., 2016). The overall workflow for FRET 2-hybrid analysis is provided in Fig. 1D.
5. PROTOCOL
5.1. Materials, Equipment, & Reagents
Nunc cell-culture treated 12 Well plate
Polyethylenimine 25kD (Polysciences 23966-1)
5 mL round bottom tubes
LSR II flow cytometer
Ca2+-free Phosphate Buffer Saline (PBS) (Corning MT21040CV)
Cycloheximide (Sigma)
Cell Culture media
Dulbecco’s Modified Eagle’s Medium (Corning 15017CV)
FBS (Cytiva SH30070.03)
L-glutamine (Corning MT25005CI)
Penicillin/Streptomycin (Gibco 15140122)
Gentamicin 50 mg/mL (Quality Biological 120098661)
Plasmids
Cerulean
Venus
Cerulean – 5AA – Venus
Cerulean – 32AA – Venus
Cerulean – 40AA – Venus
Cerulean – Traf – Venus
Cerulean and venus-tagged binding partners.
Equipment
BD LSR II flow cytometer
Software
MATLAB analysis software (http://github.com/manubenjohny/FACS_FRET/)
5.2. Cell Culture and Transfection of HEK293 cells.
Here, we describe the protocol for FRET 2-hybrid analysis of ion channel interactions with using Cerulean and Venus FRET pairs. Custom Matlab software is provided for data analysis (Github).
General Considerations for constructing FP tagged proteins
As ion channels are large macromolecules, the location of the fluorescent tag may be critical to obtain high FRET signals. Typically, FPs are inserted at distinct locations of the two binding partners to identify optimal FRET pairs. Electrophysiological analysis is used to ensure that FP insertion does not disrupt ion channel function. If one of the FRET pairs exhibit weaker expression, it is advantageous to attach this partner with venus, given the higher brightness of this fluorophore when compared to Cerulean.
Cell Culture
-
1
Human Embryonic Kidney (HEK293) cells from ATCC (CRL-1573) are grown and maintained in DMEM supplemented with 10% FBS, 100 units/mL of Penicillin-Streptomycin, and 2 mM L-glutamine. Add 10 μg/mL of Gentamicin. Cells were maintained at 37°C at 5% CO2 concentration.
-
2
Seed HEK293 cells at ~ 1x105 density per well into a 12 well plate 1 – 2 days prior to transfection.
Transfection
-
3
Prepare 1 μg/ μL PEI stock solution by dissolving in endotoxin-free ddH2O. To dissolve PEI, the solution is heated to ~80°C. After cooling to room temperature, neutralize to pH 7.0, sterilize using 0.22μm filter, and store at −20°C.
-
4For each FRET 2-hybrid experiment, several controls are necessary for the calculation of various parameters:
- Spectral crosstalk parameters:
- ‘Blank’ – or sham transfection of pBluescript to estimate background fluorescence.
- ‘Donor’ – Cerulean-N3 plasmid containing Cerulean alone.
- ‘Acceptor’ – Venus-N3 plasmid containing Venus alone.
- Instrument specific calibration and positive control
- ‘Cer-5-Ven’ – Cerulean tethered to Venus with a 5 amino acid linker.
- ‘Cer-32-Ven’ – Cerulean tethered to Venus with a 32 amino acid linker.
- ‘Cer-40-Ven’ – Cerulean tethered to Venus with a 40 amino acid linker.
- ‘Cer-T-Ven’ – Cerulean and Venus bookending Traf protein.
- Spurious FRET
- Cerulean-N3 and Venus-N3 co-transfected to estimate collisional or spurious FRET.
-
5
For transfections of HEK293 cells, we utilize Polyethyleneimine (PEI) 25 kDA linear polymer.
-
6
For each control and test condition, place 100 μL of serum-free DMEM media into a sterile 1.5 mL tube.
-
7
For control conditions, add 0.5 – 1 μg of each plasmid from step 4 into each tube.
-
8
For test conditions, mix cerulean-tagged and venus-tagged binding partners at various ratios. Typically, multiple DNA ratios are tested to optimized expression. To boost expression, 0.5 μg of T-antigen may also be co-transfected.
-
9
Add PEI at a 2:1 or 3:1 ratio of PEI (μg) to total DNA (μg). Vigorously mix PEI stock solution before addition to the serum-free DMEM solution.
-
10
Incubate DNA-PEI mixture for 10 min before addition to cells.
-
11
Flow cytometry experiments are typically performed 1 – 3 days post transfection.
Alternative approach – Ca2+ phosphate transfection.
For holo-channel FRET experiments, high expression of FP-tagged ion channels is desired to attain binding saturation and to reduce variability. In some cases, PEI transfection may not yield appropriately high expression. If so, the Ca2+-phosphate transfection approach may utilized. This approach may also be advantageous if multiple ion channel subunits are to be co-expressed.
-
12
Prepare the following reagents: (1) ‘2X HBS’ solution containing 280 mM NaCl; 50 mM HEPES; 1.5 mM Na2HPO4 with pH adjusted to 7.10 using NaOH; (2) 2 mM CaCl2 solution; and (3) sterile ddH2O.
-
13
For each transfection, prepare two 1.5 mL sterile tubes:
Into Tube 1, place 100 μL of 2x HBS solution.
Into Tube 2, mix appropriate concentrations of plasmids encoding FRET-binding pairs along with 6.8 μL of 2 mM CaCl2 solution and dilute to a total volume of 50 μL.
-
14
Dropwise mix Tube 2 into Tube 1 and incubate for 5-15 min to allow formation of Ca2+-phosphate crystals. Longer incubation can enhance maximal expression.
-
15
Dropwise add the Ca2+-phosphate DNA mixtures on to cells.
-
16
Following 2-5 hrs of incubation, rinse cells twice with 1 mL of Ca2+-free Phosphate Buffer Saline (PBS).
-
17
Replace PBS with fresh DMEM media with appropriate supplements.
5.3. Harvesting HEK293 cells for flow cytometric analysis
-
18
Approximately 2 hours prior to experimentation, add 100 μM cycloheximide (final concentration) to arrest protein translation and to allow fluorophores to fully mature.
-
19
Prior to experimentation, remove DMEM media and rinse once with Ca2+-free PBS to remove phenol red.
-
20
Incubate cells in Ca2+-free PBS for 2–3 mins and gently titurate cells using 1000 μL of PBS to dislodge the cells.
-
21
Collect cells in a 1.5 mL tube and spin down at 1800g for 3 mins to pellet the cells.
-
22
Aspirate media and resuspend in 200 μL of PBS (Ca2+ free). Tyrode solution may also be used for resuspending cells. Of note, extracellular Ca2+ can cause clumping of cells and is therefore avoided for typical experiments. Nonetheless, for experiments that require intracellular Ca2+ elevation, we have used Tyrode solution containing up to 10 mM Ca2+ in conjunction with 10 μM ionomycin.
-
23
Collect cells into 5mL round bottom tubes.
5.4. Flow Cytometry Data Acquisition
Any commonly available flow cytometers with appropriate excitation and emission channels can be used for FRET analysis (Lee et al., 2016). The following steps detail data acquisition using BD LSR II.
-
24
Turn on the instrument and follow start up procedures as required.
-
25
Set up data acquisition on FACSDiva software by specifying various acquisition channels. For Cerulean – Venus FRET pair on LSR II, we acquire data from ‘BV421’, ‘FITC’, and ‘BV510’ channels corresponding to ‘Donor’, ‘Acceptor’, and ‘FRET’ channels. In addition, FSC (forward scatter) and SSC (side scatter) signals are collected to gate for cell size and single cells including both height and area. For convenience, the data acquisition sheet displays several plots: (i) SSC-H (side scatter height) is plotted against FSC-H (forward scatter height), (ii) FSC-H plotted against FSC-A (forward scatter area), (iii) Donor signal (BV421-A) plotted against the Acceptor signal (FITC-A), (iv) FRET signal (BV510-A) plotted against the Donor (BV421-A) signal, and (v) the FRET signal plotted against the Acceptor signal.
-
26
For set up, acquire preview data from ‘Blank’ cells at low sample flow rate and adjust both ‘FSC’ and ‘SSC’ voltages to isolate a population of HEK293 cells. Here, the low sample flow rate is utilized to prevent depleting the sample volume for final data acquisition Typically, we use voltages of 300 and 350 respectively.
-
27
The gains for ‘BV421’, ‘FITC’, and ‘BV510’ channels can be adjusted and corrected for using fluorescent beads. In practice, we utilize a standard gain across experiments and over multiple days for simplicity of data analysis. The variation in expression of cerulean and venus fluorophores are sufficiently broad that with typical transfections a complete binding curve can be constructed without adjusting the gain. For LSR II, the gain is set to 300 for all three channels. The cytometer settings may be saved and recalled for future experiments.
-
28
Obtain fluorescence data from various controls in step 4 and all experimental conditions. The sample flow rate is adjusted to medium speed (1000-2000 events/sec). Flow rate may be adjusted to either high or low speed depending on cell density. All compensations are performed post-hoc and there is no need to set up compensation on the instrument. For each sample, we typically gate for cells that express both Cerulean and Venus and collect a total of 50000 – 100000 cells.
-
29
Export all data files in FCS format for data analysis and follow instrument shut down procedures.
5.5. FRET Data Analysis
The companion MATLAB analysis software FACS-FRET(Github) is able to read flow cytometry standard (FCS) files, with analysis prebuilt for BD LSR II, BD Fortessa, and Attune flow cytometers. Data analysis software can be customized by specifying ‘Donor’, ‘Acceptor’, and ‘FRET’ channels for a given FRET pair and for a specific instrument. The data analysis process involves multiple stages as elaborated below.
Gating for single cells
In the first stage, the population of single HEK293 cells is selected through sequential gating of FSC and SSC signals using the MATLAB program: FilterFACSData.m (Fig. 2A–2C).
Figure 2.
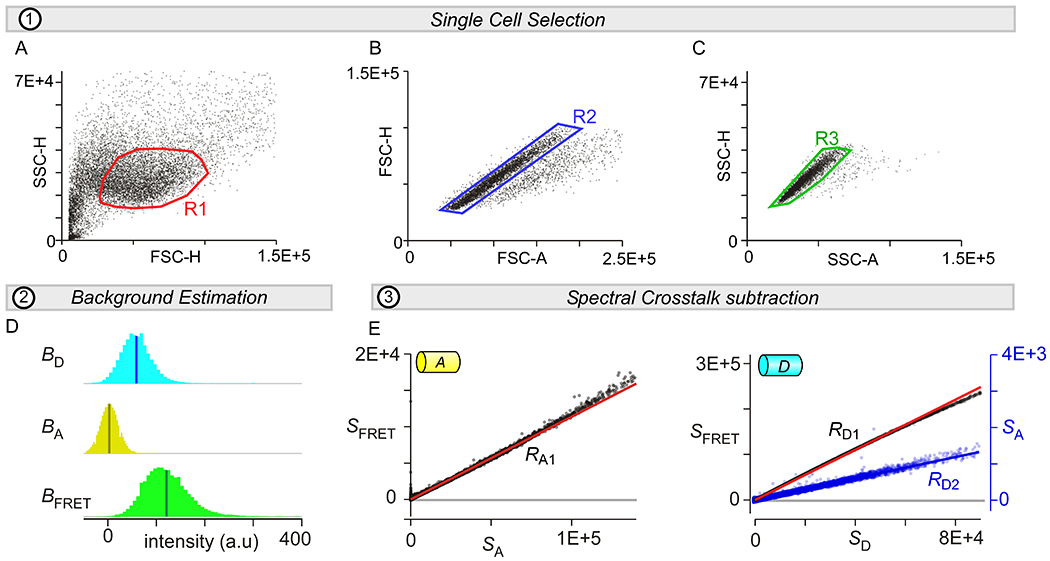
Filtering single cells and subtracting spectral crosstalk. (A-C) Three-step sequential gating of single cells by forward-scatter (FSC) and side-scatter (SSC) signals. Briefly, SSC-H is plotted again FSC-H and a region of interest R1 is selected (panel A). Second, FSC-H and FSC-A signals from cells within R1 is used to select region of interest R2 (panel B). Finally, SSC-H and SSC-A signals for cells within R2 are utilized to select single cells (R3) that will be subject to further analysis (panel C). (D) Background signals from untransfected or sham-transfected cells are used to estimate autofluorescence in the donor, acceptor, and FRET channels. (E) Left, the spectral crosstalk of the acceptor into the FRET channel. The slope provides estimate for RA1 parameter. Right, the spectral crosstalk of the donor into the FRET (left axis) and acceptor (right axis) channels. The slopes provide estimates for RD1 and RD2 parameters, respectively.
-
30
Plot SSC-H versus FSC-H. Draw a region of interest (R1) to filter the single cell population.
-
31
For cells within the R1 region, plot FSC-H versus FSC-A. Draw another region of interest R2 to isolate single-cells.
-
32
Plot SSC-H versus SSC-A for cells in the R2 region. Draw region R3 to specify the single-cell population.
-
33
Filter all data sets using Region of Interests R1-R3. This filtered data is used for all subsequent analysis.
Background and spectral crosstalk subtraction.
Following single cell selection and filtering, FRET 2-hybrid analysis is performed using the MATLAB program: FACS_FRET.m.
-
34
To estimate background fluorescence, the average Donor, Acceptor, and FRET signals are computed from single un-transfected or sham-transfected cells (‘blank’) filtered based on the R1-R3 criteria. For each cell, the background fluorescence for each channel is subtracted from each fluorescence emission channel. In the FACS-FRET software, this is accomplished by the ‘Background’ button (Fig. 2D).
-
35Spectral crosstalk parameters are subsequently determined as follows (Fig. 2E):
- To obtain acceptor crosstalk, we probe cells expressing the acceptor alone and determine the slope of the linear regression (RA1) of the FRET emission (SFRET) with the acceptor emission (SA). In the FACS-FRET software, this computation is performed by the ‘Setup RA’ button.
- To obtain donor bleed-through, we analyze cells expressing cerulean alone and measure emission in the FRET channel and in the acceptor channel. RD1 is estimated as the slope of a linear regression of FRET emission (SFRET) to the donor emission (SD). RD2 is determined as the slope of the linear regression of acceptor emission (SA) to the donor emission (SD). In the FACS-FRET software, this computation is performed by the ‘Setup RD’ button.
- If cells contain additional fluorophores beyond the FRET pair, then crosstalk from these fluorophores should also be subtracted. For instance, mCherry (Z) may be utilized as an expression marker. Although mCherry does not have any appreciable crosstalk with the Donor, Acceptor, or FRET channels, there is significant bleed-through of cerulean and venus into the mCherry channel. The amount of crosstalk can be determined by computing the slope (RD3) of the linear regression between the red fluorescence emission (SR) and SD in cells expressing cerulean alone and the slope (RA2) of the linear regression between SR and SA.
-
36For each cell, the spectral crosstalk is subtracted by the following matrix operation.
(6)
Obtaining instrument specific calibration coefficients.
Fluorescence measurements from a series of Cerulean–Venus dimers with linkers of varying lengths are used to estimate instrument-specific calibration coefficients gA/gD corresponding the relative absorption of the acceptor to that of the donor, and fA/fD corresponding to the ratio of the relative emission of the acceptor to the donor (Lee et al., 2016). Typically, 3–4 different dimers suffice to obtain robust estimate of the two ratios.
-
37
Use the matrix operation in Eq. 6 to unmix SD, SA, SFRET signals to obtain Ddirect, Adirect, and AFRET for cells collected for all dimers.
-
38
Plot AFRET / Ddirect versus Adirect / Ddirect (Fig. 3A) and fit a linear regression between these entities to estimate both the slope and the intercept. The slope corresponds to gD/gA and the intercept to fA/fD. In some cases, such as when the number of data points per dimers vary, the median value of the ratios may be used to obtain robust fit parameters. In the FACS-FRET software, this computation is performed by the ‘Setup Dimers’ button.
- 39
-
40The total concentration of the acceptor is given by (Ben-Johny et al., 2016):
Although this process provides the ratios gA/gD and fA/fD, it does not yield the absolute brightness of the donor and the acceptor fluorophores necessary for obtaining ND and NA values and absolute concentrations. One approach is to empirically measure the fluorescence emission of FITC beads of known brightness to estimate venus brightness. Subsequently, cerulean brightness is approximated from the venus brightness with the ratio (gAfA)/(gDfD). An alternative approach is to assume a gD · fD = 1. Thus, Eq. 9-10 can be reduced to:(9-10) (11) -
41
To validate the calibration constants, we ensure that the ratio ND/NA for all dimers is ~1 (Fig. 3D).
Figure 3.
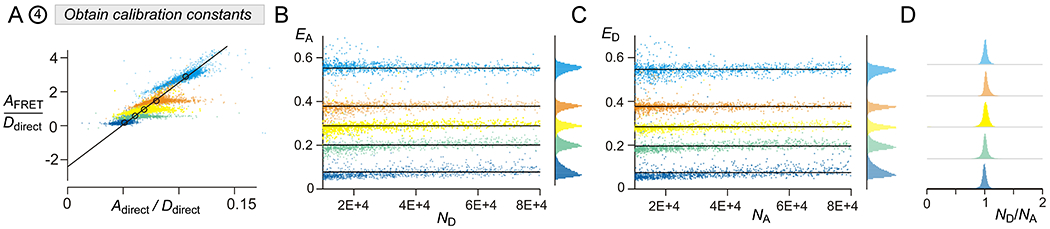
Obtaining instrument specific calibration coefficients and measuring FRET efficiencies for various cerulean – venus dimers (A) Calculation of donor-centric (ED) and acceptor-centric (ED) FRET efficiencies require calculation of two instrument specific parameter gA / gD and fA / fD corresponding to the ratio of absorption and emissions of the donor and acceptor via FRET cube. Three cube FRET measurements are obtained from cells expressing cerulean-venus dimers and spectral unmixing is used to estimate Ddirect, Adirect, and AFRET. The ratio AFRET / Ddirect is plotted against Adirect/Ddirect. The intercept yields fA / fD while the slope corresponds to gD/gA. (B) Acceptor-centric FRET efficiencies (EA) obtained from cells expressing Cer-5AA-Ven (light blue), Cer-32AA-Ven (orange), Cer-40AA-Ven (yellow), Cer-50AA-Ven (green), and Cer-traf-Ven (deep blue). (C) Donor-centric FRET efficiencies (ED) for the same dimers. Format as in panel B. (D) Histogram of single-cell estimates for ND/NA for various cerulean-venus dimers. ND / NA ~ 1 for all dimers, as expected.
Subtracting ‘spurious’ or collisional FRET.
Strong co-expression of cerulean and venus fluorophores can lead to concentration-dependent collisional FRET that results from the donor and acceptor fluorophore being within FRET distance by random chance (Butz et al., 2016). Collisional FRET is estimated from cells expressing only cerulean and venus fluorophores alone.
-
42
Perform spectral unmixing procedure on raw fluorescence intensities from cells co-expressing Cerulean N3 with Venus N3.
-
43
Determine EA, and ED using Eq. 7–8, as well as NA, and ND using Eq. 10–11.
-
44
Plot EA versus ND and ED versus NA and estimate slopes using linear regression. In both cases, the intercepts are set to 0.
-
45
The slopes are used for subtraction of spurious FRET from EA and ED for various experimental conditions. In the FACS-FRET software, this step is accomplished by the ‘Spurious FRET’ subroutine.
Obtain FRET 2-hybrid binding curves for experimental conditions.
-
3
Perform spectral unmixing procedure on raw fluorescence intensities for all data points from the experimental conditions, and determine EA, and ED using Eq. 7–8 as well as NA and ND using Eq. 10–11.
-
4
Iteratively fit an appropriate binding model. The stoichiometry is estimated by comparing the saturating values of EA and ED. For 1:1 interactions, EA,max = ED,max and Eqs. 3–5 may be used to iteratively estimate the binding affinity by least squares minimization. If the saturating values are different, the ratio EA,max / ED,max yields the stoichiometry, and simplifying assumptions such as independent and identical binding must be made to estimate Kd,eff.
6. APPLICATIONS
We highlight a few applications of FRET 2-hybrid assays using epifluorescence microscopy and flow cytometry.
CaM interaction with the myosin Va and CaV1.2
The ubiquitous Ca2+-binding protein calmodulin elicits vital Ca2+-dependent regulation of various cellular proteins. Here we highlight two applications of FRET 2-hybrid assay to study CaM interaction with: (1) Myosin Va and (2) CaV1.2 carboxy-tail (Ben-Johny et al., 2016). The neck domain of the myosin Va contains 6 tandem IQ domains, each of which could interact with a CaM. Epifluorescence microscopy-based FRET has been used to study the stoichiometry of CaM interaction with various truncations of myosin Va (Fig. 4A–D) (Ben-Johny et al., 2016). In this experiment, CaM is tagged with cerulean and Myosin Va segments are attached to venus. Analysis of EA,max and ED,max confirms that CaM stoichiometry correlates with the number of IQ domains in Myosin Va. Similarly, FRET 2-hybrid assay has been used to probe both binding and stoichiometry of CaM to CaV1.2. Fig. 4E shows flow cytometry-based FRET 2-hybrid analysis of cerulean-CaM with the carboxy-tail of CaV1.2. As in previous studies, the saturating value of EA and ED are identical confirming 1:1 binding at low Ca2+ levels (Ben-Johny et al., 2016).
Figure 4.
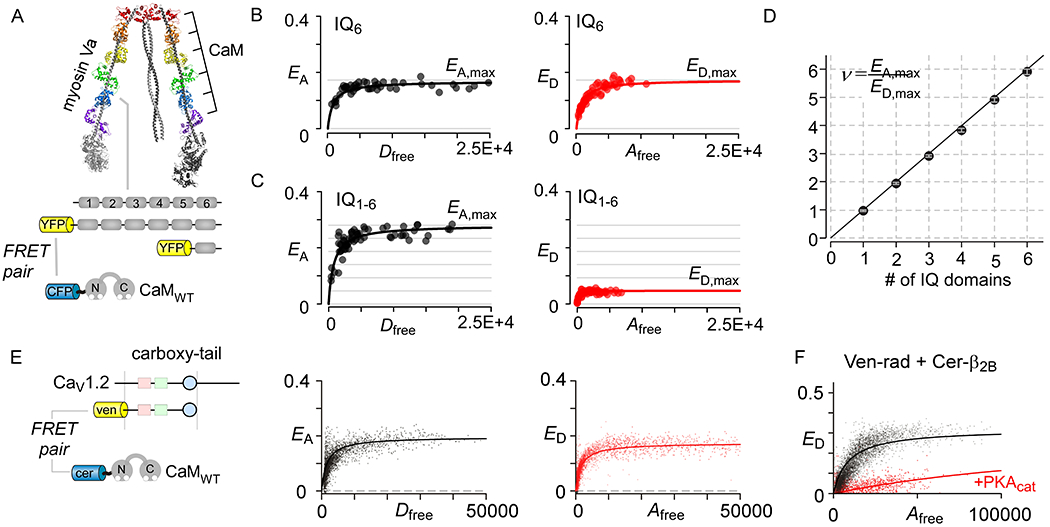
Application of FRET 2-hybrid analysis to deduce stoichiometry and binding. (A) Cryo-electron microscopy of Myosin Va shows the interaction of 6 CaM molecules with the neck domain that contains 6 tandem IQ domains (PDB code, 2DFS) (J. Liu, Taylor, Krementsova, Trybus, & Taylor, 2006). (B) FRET 2-hybrid analysis of a single IQ domain shows identical saturating values for acceptor-centric (EA,max) and donor-centric (ED,max) FRET efficiencies suggesting 1:1 binding. (C) Analysis of the entire neck region containing IQ1–6 shows that saturating value for acceptor-centric (EA,max) is ~ 6-fold higher than the donor-centric (ED,max) FRET efficiencies. This suggest that there are 6 donor molecules (i.e. CaM) per acceptor in the complex. (D) Correlation of estimated stoichiometries for various truncations of myosin Va neck region using FRET with the expected stoichiometry given the number of IQ domains in each truncation. (E) Flow-cytometric FRET 2-hybrid assay confirms robust interaction of Cer-tagged CaM with ven-tagged carboxy-tail segment of CaV1.2. As EA,max = ED,max, the stoichiometry is 1:1. (F) Flow-cytometric FRET 2-hybrid assay shows PKA-dependent change in binding of the Cerulean tagged CaVβ2B subunit with Venus tagged Rad. These findings illustrate the ability of FRET to resolve changes in binding due to signaling events. Panels B-D were reproduced with permission from Ben-Johny et al, 2016.
PKA regulation of CaVβ subunit binding to RGK proteins
The mechanism of β-adrenergic upregulation of CaV1.2 currents in the heart was recently shown to be initiated by PKA phosphorylation of the RGK protein Rad (G. Liu et al., 2020). At baseline, Rad binds to the CaVβ and inhibit channel openings. Phosphorylation of Rad dislodges it from the CaVβ subunit and relieves CaV1.2 inhibition. Flow-cytometry based FRET 2-hybrid assay is well-suited to monitoring PKA dependent conformational changes (G. Liu et al., 2020; Papa et al., 2020). Fig. 4F shows baseline binding of venus-tagged Rad with cerulean-tagged CaVβ2b subunit. Co-expression of PKA catalytic subunit diminishes FRET suggesting that phosphorylation of Rad disrupts CaVβ2b interaction.
7. CHALLENGES AND TROUBLESHOOTING
Here, we review commonly-encountered challenges with live-cell FRET 2-hybrid assays and potential strategies for troubleshooting.
Mismatch in expression of FRET pairs.
One commonly-encountered source of variability in FRET efficiency measurements from live-cells is the uneven expression of FRET pairs. In particular, construction of FRET binding curves relies on having a broad range of donor and acceptor concentrations in cells (Butz et al., 2016). However, a complexity is that a large mismatch in the donor versus acceptor concentration could lead to errors in estimation of unmixed fluorescence signals. In turn, this error could result in variability in the FRET efficiencies computed. One approach is to filter cells based on the ratio of the number of donors to the number of acceptors in the cell. Typically, only cells where 0.1 < ND / NA < 10 are considered for construction of binding curves. Narrowing this range can reduce variability in the FRET curves. Too narrow a range, however, can result in too few cells to obtain a binding curve. Cells that have low donor and acceptor expression are also excluded from analysis. In the FACS FRET software, the ‘Filtering’ window allows for setting custom limits for gating cells based on overall expression.
Nonlinearities in spectral crosstalk.
For spectral decomposition of raw fluorescence signals, the matrix operation in Eq. 6 assumes linearity of the spectral crosstalk parameters. For some instruments, however, there are nonlinearities in the fluorescence detectors that may violate this assumption particularly near saturation of the detector. In this case, data analysis is restricted to cells within the linear range (Lee et al., 2016).
Effect of immature fluorophores
Incomplete maturation of fluorophores is also an important source of variability for FRET efficiency measurements (B. Liu et al., 2018). If mature donors were closely-juxtaposed to immature acceptors, then no energy transfer would occur between these pairs. In this case, the apparent donor-centric ED measurement would be reduced, but the accepter-centric EA measurement would be unaffected. Similarly, if immature donors are near mature acceptors, no energy transfer occurs. In this case, however, only the apparent acceptor-centric EA measurement is diminished. From the perspective of deducing binding parameters, partial maturation of fluorophores may impact estimates of both stoichiometry and binding affinity. Based on previous studies, the effect of fluorophore maturation on stoichiometry (ν) can be estimated as follows:
| (12) |
Where fm,D and fm,A are the fraction of mature donors and acceptors respectively (Ben-Johny et al., 2016; Wlodarczyk et al., 2008). That is, the measured stoichiometry will deviate from the actual stoichiometry by the ratio fm,D / fm,A. The effect of immature fluorophores on binding is more complex and challenging to quantify. For 1:1 binding interactions, the ED-Afree binding curve should yield an identical Kd,EFF as EA-Dfree curve. With partial maturation, the estimated Kd,EFF for the two relations may differ. In practice, the effects of immature fluorescent proteins can be minimized by blocking protein translation using cycloheximide and allowing already-produced fluorophores to mature completely (Lee et al., 2016). Of note, an empirical estimate for fm,D / fm,A for any given FRET pair can be estimated by computing ND/NA for donor-acceptor dimers.
Effect of endogenous proteins
A key advantage of FRET 2-hybrid assay is the ability to deduce relative binding affinities within living cells. However, as FRET requires fluorescently-labeled proteins, the relative binding affinities obtained may be biased by the presence of unlabeled endogenous proteins. In particular, if the free concentration of the unlabeled endogenous variant far exceeds the true affinity of the binding pair, then the estimated relative binding affinity would approximate the concentration of the endogenous protein rather than the true affinity. Theoretical modeling as showcased in previous studies can provide some estimates for error due to the presence of endogenous binding proteins (Yang, Johny, & Yue, 2014). Experimentally, shRNA knockdown or CRISPR/Cas-based approaches can be used to reduce levels of endogenous binding partners. Importantly, stoichiometry estimates are obtained from saturating EA and ED value that are only attained only when the labeled binding partner is over-expressed well beyond that of the unlabeled endogenous variant. As such, the presence of endogenous proteins minimally affects stoichiometry estimates (Ben-Johny et al., 2016).
8. SUMMARY
FRET has emerged as a powerful and versatile methodology for studying biological molecules in living cells. This chapter provides an overview of FRET methodology for deducing relative binding and stoichiometry of ion channel macromolecular complexes using a flow cytometer and provides strategies to overcome commonly-encountered experimental challenges. Usage of flow cytometry for FRET data acquisition brings forth key advantages: (1) enhanced throughput as fluorescence from thousands of cells can be measured in a few seconds and samples can be collected from 96-well format using autosamplers, (2) reduced sampling bias as FRET curves are constructed from 10000-50000 cells using standardized procedures, and (4) increased availability of commercial flow cytometers at core facilities improves accessibility of FRET experiments.
This overall platform sets the stage for future advancements for FRET applications. A bevy of FRET biosensors have been developed to quantify key biological parameters such as, second messenger molecules and the activity of cell signaling molecules (Tsien, 2009). Beyond FRET 2-hybrid assay, flow-cytometry based analysis of FRET biosensors may facilitate single-cell analysis of these physiologically relevant parameters and lend new insights into population level variability in signaling (Lee et al., 2016). Furthermore, typical flow cytometers are equipped with many fluorescent channels. Thus, it is possible to simultaneously deduce multiple interactions within the same cell or to quantify changes in binding due to cell signaling. Finally, FRET-activated cell-sorting methods and emerging CRISPR-screening strategies may be combined to establish forward screening approaches for the discovery of novel regulators of key biological interactions (Yu & Yusa, 2019).
ACKNOWLEDGEMENTS
This work was funded by NIH NINDS R01 NS110672.
REFERENCES
- Algar WR, Hildebrandt N, Vogel SS, & Medintz IL (2019). FRET as a biomolecular research tool - understanding its potential while avoiding pitfalls. Nat Methods, 16(9), 815–829. doi: 10.1038/s41592-019-0530-8 [DOI] [PubMed] [Google Scholar]
- Bajar BT, Wang ES, Zhang S, Lin MZ, & Chu J (2016). A Guide to Fluorescent Protein FRET Pairs. Sensors (Basel), 16(9). doi: 10.3390/s16091488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M, Yue DN, & Yue DT (2016). Detecting stoichiometry of macromolecular complexes in live cells using FRET. Nat Commun, 7, 13709. doi: 10.1038/ncomms13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, … Hess HF (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science, 313(5793), 1642–1645. doi: 10.1126/science.1127344 [DOI] [PubMed] [Google Scholar]
- Butz ES, Ben-Johny M, Shen M, Yang PS, Sang L, Biel M, … Wahl-Schott C (2016). Quantifying macromolecular interactions in living cells using FRET two-hybrid assays. Nat Protoc, 11(12), 2470–2498. doi: 10.1038/nprot.2016.128 [DOI] [PubMed] [Google Scholar]
- Cannon SC (2007). Physiologic principles underlying ion channelopathies. Neurotherapeutics, 4(2), 174–183. doi: 10.1016/j.nurt.2007.01.015 [DOI] [PubMed] [Google Scholar]
- Chen H, Puhl HL 3rd, & Ikeda SR (2007). Estimating protein-protein interaction affinity in living cells using quantitative Forster resonance energy transfer measurements. J Biomed Opt, 12(5), 054011. doi: 10.1117/1.2799171 [DOI] [PubMed] [Google Scholar]
- Clegg RM (1992). Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol, 211, 353–388. [DOI] [PubMed] [Google Scholar]
- Clegg RM, Holub O, & Gohlke C (2003). Fluorescence lifetime-resolved imaging: measuring lifetimes in an image. Methods Enzymol, 360, 509–542. doi: 10.1016/s0076-6879(03)60126-6 [DOI] [PubMed] [Google Scholar]
- Dertinger T, Colyer R, Iyer G, Weiss S, & Enderlein J (2009). Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc Natl Acad Sci U S A, 106(52), 22287–22292. doi: 10.1073/pnas.0907866106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devauges V, Matthews DR, Aluko J, Nedbal J, Levitt JA, Poland SP, … Ameer-Beg SM (2014). Steady-state acceptor fluorescence anisotropy imaging under evanescent excitation for visualisation of FRET at the plasma membrane. PLoS One, 9(10), e110695. doi: 10.1371/journal.pone.0110695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- England CG, Luo H, & Cai W (2015). HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem, 26(6), 975–986. doi: 10.1021/acs.bioconjchem.5b00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, & Yue DT (2001). Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron, 31(6), 973–985. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, & Yue DT (2003). FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron, 39(1), 97–107. [DOI] [PubMed] [Google Scholar]
- Forster T (1948). Zwischenmolekulare Energiewanderung and Fluoresczenz. Ann. Phys, 2, 57–75. [Google Scholar]
- Glauner KS, Mannuzzu LM, Gandhi CS, & Isacoff EY (1999). Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature, 402(6763), 813–817. doi: 10.1038/45561 [DOI] [PubMed] [Google Scholar]
- Godin AG, Lounis B, & Cognet L (2014). Super-resolution microscopy approaches for live cell imaging. Biophys J, 107(8), 1777–1784. doi: 10.1016/j.bpj.2014.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, … Schwartz MA (2010). Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature, 466(7303), 263–266. doi: 10.1038/nature09198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handbook of Ion Channels. (2015). (Zheng J & Trudeau M Eds. 1 ed.). Boca Raton, FL.: CRC Press. [Google Scholar]
- Hein B, Willig KI, & Hell SW (2008). Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc Natl Acad Sci U S A, 105(38), 14271–14276. doi: 10.1073/pnas.0807705105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter B, Garcia AP, & Schmid JA (2015). Fluorescent proteins as genetically encoded FRET biosensors in life sciences. Sensors (Basel), 15(10), 26281–26314. doi: 10.3390/s151026281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter B, Kunze M, Moser B, & Schmid JA (2019). Advanced FRET normalization allows quantitative analysis of protein interactions including stoichiometries and relative affinities in living cells. Sci Rep, 9(1), 8233. doi: 10.1038/s41598-019-44650-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Gaietta G, Zurn A, Adams SR, Terrillon S, Ellisman MH, … Lohse MJ (2010). Fluorescent labeling of tetracysteine-tagged proteins in intact cells. Nat Protoc, 5(10), 1666–1677. doi: 10.1038/nprot.2010.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe A, Christensen K, & Swanson JA (2002). Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys J, 83(6), 3652–3664. doi: 10.1016/S0006-3495(02)75365-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isacoff EY, Jan LY, & Minor DL Jr. (2013). Conduits of life’s spark: a perspective on ion channel research since the birth of neuron. Neuron, 80(3), 658–674. doi: 10.1016/j.neuron.2013.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackowicz JR (1983). Energy Transfer. In Principles of Fluorescence Spectroscopy (pp. 443–472). New York, NY: Springer. [Google Scholar]
- Lee SR, Sang L, & Yue DT (2016). Uncovering Aberrant Mutant PKA Function with Flow Cytometric FRET. Cell Rep, 14(12), 3019–3029. doi: 10.1016/j.celrep.2016.02.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JY, Song Y, & Liu Y (2015). A new trend to determine biochemical parameters by quantitative FRET assays. Acta Pharmacol Sin, 36(12), 1408–1415. doi: 10.1038/aps.2015.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Mavrova SN, van den Berg J, Kristensen SK, Mantovanelli L, Veenhoff LM, … Boersma AJ. (2018). Influence of Fluorescent Protein Maturation on FRET Measurements in Living Cells. ACS Sens, 3(9), 1735–1742. doi: 10.1021/acssensors.8b00473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, … Marx SO (2020). Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature, 577(7792), 695–700. doi: 10.1038/s41586-020-1947-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Taylor DW, Krementsova EB, Trybus KM, & Taylor KA (2006). Three-dimensional structure of the myosin V inhibited state by cryoelectron tomography. Nature, 442(7099), 208–211. doi: 10.1038/nature04719 [DOI] [PubMed] [Google Scholar]
- Miranda P, Contreras JE, Plested AJ, Sigworth FJ, Holmgren M, & Giraldez T (2013). State-dependent FRET reports calcium- and voltage-dependent gating-ring motions in BK channels. Proc Natl Acad Sci U S A, 110(13), 5217–5222. doi: 10.1073/pnas.1219611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, & Tsien RY (1997). Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature, 388(6645), 882–887. [DOI] [PubMed] [Google Scholar]
- Neher RA, & Neher E (2004). Applying spectral fingerprinting to the analysis of FRET images. Microsc Res Tech, 64(2), 185–195. doi: 10.1002/jemt.20078 [DOI] [PubMed] [Google Scholar]
- Papa A, Kushner JS, Hennessey JA, Katchman AN, Zakharov SI, Chen BX, … Marx SO (2020). Adrenergic CaV1.2 Activation via Rad Phosphorylation Converges at alpha1C I-II Loop. Circ Res. doi: 10.1161/CIRCRESAHA.120.317839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partikian A, Olveczky B, Swaminathan R, Li Y, & Verkman AS (1998). Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J Cell Biol, 140(4), 821–829. doi: 10.1083/jcb.140.4.821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh A, Riven I, & Reuveny E (2009). Elucidation of the gating of the GIRK channel using a spectroscopic approach. J Physiol, 587(Pt 22), 5331–5335. doi: 10.1113/jphysiol.2009.180158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P, Davis KA, Puhl HL 3rd, Veetil JV, Nguyen TA, & Vogel SS (2017). Deciphering CaMKII Multimerization Using Fluorescence Correlation Spectroscopy and Homo-FRET Analysis. Biophys J, 112(6), 1270–1281. doi: 10.1016/j.bpj.2017.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Ackerman MJ, & Wilde AAM (2017). Channelopathies as Causes of Sudden Cardiac Death. Card Electrophysiol Clin, 9(4), 537–549. doi: 10.1016/j.ccep.2017.07.005 [DOI] [PubMed] [Google Scholar]
- Sekar RB, & Periasamy A (2003). Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol, 160(5), 629–633. doi: 10.1083/jcb.200210140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvin PR (1995). Fluorescence resonance energy transfer. Methods Enzymol, 246, 300–334. doi: 10.1016/0076-6879(95)46015-2 [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, & Tsien RY (2005). A guide to choosing fluorescent proteins. Nat Methods, 2(12), 905–909. doi: 10.1038/nmeth819 [DOI] [PubMed] [Google Scholar]
- Sigal YM, Zhou R, & Zhuang X (2018). Visualizing and discovering cellular structures with super-resolution microscopy. Science, 361(6405), 880–887. doi: 10.1126/science.aau1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler C, Koushik SV, Blank PS, & Vogel SS (2005). Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys J, 89(4), 2736–2749. doi: 10.1529/biophysj.105.061853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler C, Koushik SV, Puhl HL 3rd, Blank PS, & Vogel SS (2009). Structural rearrangement of CaMKIIalpha catalytic domains encodes activation. Proc Natl Acad Sci U S A, 106(15), 6369–6374. doi: 10.1073/pnas.0901913106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY (2009). Indicators based on fluorescence resonance energy transfer (FRET). Cold Spring Harb Protoc, 2009(7), pdb top57. doi: 10.1101/pdb.top57 [DOI] [PubMed] [Google Scholar]
- Ujlaky-Nagy L, Nagy P, Szollosi J, & Vereb G (2018). Flow Cytometric FRET Analysis of Protein Interactions. Methods Mol Biol, 1678, 393–419. doi: 10.1007/978-1-4939-7346-0_17 [DOI] [PubMed] [Google Scholar]
- Vogel SS, Thaler C, & Koushik SV (2006). Fanciful FRET. Sci STKE, 2006(331), re2. doi: 10.1126/stke.3312006re2 [DOI] [PubMed] [Google Scholar]
- Vogel SS, van der Meer BW, & Blank PS (2014). Estimating the distance separating fluorescent protein FRET pairs. Methods, 66(2), 131–138. doi: 10.1016/j.ymeth.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrabe H, & Periasamy A (2005). Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol, 16(1), 19–27. doi: 10.1016/j.copbio.2004.12.002 [DOI] [PubMed] [Google Scholar]
- Wang S, Lee SJ, Maksaev G, Fang X, Zuo C, & Nichols CG (2019). Potassium channel selectivity filter dynamics revealed by single-molecule FRET. Nat Chem Biol, 15(4), 377–383. doi: 10.1038/s41589-019-0240-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk J, Woehler A, Kobe F, Ponimaskin E, Zeug A, & Neher E (2008). Analysis of FRET signals in the presence of free donors and acceptors. Biophys J, 94(3), 986–1000. doi: 10.1529/biophysj.107.111773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters FS, Verveer PJ, & Bastiaens PI (2001). Imaging biochemistry inside cells. Trends Cell Biol, 11(5), 203–211. [DOI] [PubMed] [Google Scholar]
- Yang PS, Johny MB, & Yue DT (2014). Allostery in Ca(2)(+) channel modulation by calcium-binding proteins. Nat Chem Biol, 10(3), 231–238. doi: 10.1038/nchembio.1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X, Nguyen AW, Jabaiah A, Sheff MA, Thorn KS, & Daugherty PS (2006). Intracellular protein interaction mapping with FRET hybrids. Proc Natl Acad Sci U S A, 103(49), 18458–18463. doi: 10.1073/pnas.0605422103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JSL, & Yusa K (2019). Genome-wide CRISPR-Cas9 screening in mammalian cells. Methods, 164-165, 29–35. doi: 10.1016/j.ymeth.2019.04.015 [DOI] [PubMed] [Google Scholar]
- Zaccolo M, De Giorgi F, Cho CY, Feng L, Knapp T, Negulescu PA, … Pozzan T (2000). A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat Cell Biol, 2(1), 25–29. doi: 10.1038/71345 [DOI] [PubMed] [Google Scholar]
- Zhang J, Carver CM, Choveau FS, & Shapiro MS (2016). Clustering and Functional Coupling of Diverse Ion Channels and Signaling Proteins Revealed by Super-resolution STORM Microscopy in Neurons. Neuron, 92(2), 461–478. doi: 10.1016/j.neuron.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma Y, Taylor SS, & Tsien RY (2001). Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A, 98(26), 14997–15002. doi: 10.1073/pnas.211566798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J (2006). Spectroscopy-based quantitative fluorescence resonance energy transfer analysis. Methods Mol Biol, 337, 65–77. doi: 10.1385/1-59745-095-2:65 [DOI] [PubMed] [Google Scholar]
- Zimmermann T (2005). Spectral imaging and linear unmixing in light microscopy. Adv Biochem Eng Biotechnol, 95, 245–265. doi: 10.1007/b102216 [DOI] [PubMed] [Google Scholar]
- Zou P, Zhao Y, Douglass AD, Hochbaum DR, Brinks D, Werley CA, … Cohen AE (2014). Bright and fast multicoloured voltage reporters via electrochromic FRET. Nat Commun, 5, 4625. doi: 10.1038/ncomms5625 [DOI] [PMC free article] [PubMed] [Google Scholar]