

Abstract
Brain atrophy occurs in aging even in the absence of dementia, but it is unclear to what extent this is due to undetected preclinical Alzheimer disease. Here we examine a cross-sectional cohort (ages 18-88) free from confounding influence of preclinical Alzheimer disease, as determined by amyloid PET scans and three years of clinical evaluation post-imaging. We determine the regional strength of age-related atrophy using linear modeling of brain volumes and cortical thicknesses with age. Age-related atrophy was seen in nearly all regions, with greatest effects in the temporal lobe and subcortical regions. When modeling age with the estimated derivative of smoothed aging curves, we found that the temporal lobe declined linearly with age, subcortical regions declined faster at later ages, and frontal regions declined slower at later ages than during midlife. This age-derivative pattern was distinct from the linear measure of age-related atrophy and significantly associated with a measure of myelin. Atrophy did not detectably differ from a preclinical Alzheimer disease cohort when age ranges were matched.
Keywords: Normal Aging, Volumetrics, Preclinical Alzheimer disease, Magnetic Resonance Imaging (MRI)
1. Introduction
Older adults constitute an increasingly large fraction of our society, making research on brain aging important for public health. Cerebral atrophy associated with aging is in particular a concern due to its association with cognitive decline, independent of known neurodegenerative diseases (Armstrong et al., 2020; Fletcher, Gavett, et al., 2018). Previous studies have shown regional variability and non-linear changes in this atrophy occurring with age. In general, these studies show the strongest atrophy in frontal and temporal regions, and a pattern of accelerated atrophy in temporal regions (Irwin et al., 2018; Lockhart & DeCarli, 2014). It has been hypothesized that these non-linear regional patterns may in part be due to mid-life increases in cerebral myelination causing the appearance of reduced gray matter density (Irwin et al., 2018). However, myelin may also be acting as a proxy for other regional properties of the brain such as intracortical circuit complexity and aerobic glycolysis levels (Glasser et al., 2014).
Measures of age-related atrophy are complicated by abundant confounding factors inherent within studies of aging. One major factor is cardiovascular disease, with atrophy correlating with white matter hyperintensities (Coutu et al., 2017; Habes et al., 2021), high blood pressure (Armstrong, An, et al., 2019; Lockhart & DeCarli, 2014), and diabetes (Hamed, 2017; Suzuki et al., 2019). Some studies indicate sex or gender differences in age-related atrophy, with greater atrophy in men for select regions (Armstrong, Huang, et al., 2019; Chételat et al., 2010; Jack et al., 2015; Lockhart & DeCarli, 2014; Wang et al., 2019). Additionally, apolipoprotein E ε4 (APOE4) - the greatest genetic risk factor for sporadic Alzheimer disease (AD) - has also been associated with greater rates of atrophy even in the unimpaired (Armstrong, An, et al., 2019; Erten-Lyons et al., 2013; Irwin et al., 2018; Kelly et al., 2018; Mishra et al., 2018; Raz et al., 2010; Smith et al., 2012). A previous study has shown that this effect of APOE4 is linked to increasing amyloid levels, indicative of the preclinical stage of AD (Mishra et al., 2018).
Preclinical AD is characterized by the absence of cognitive symptoms and the presence of parenchymal deposits of amyloid-β peptide, one of the hallmarks of AD. Despite its association with atrophy (Becker et al., 2011; Chételat et al., 2012; Dickerson et al., 2009; Fagan et al., 2009; Fjell et al., 2010; Fletcher et al., 2016; Fletcher, Filshtein, et al., 2018; Oh et al., 2014; Pettigrew et al., 2017; Schott et al., 2010; Storandt et al., 2009; Xie et al., 2020), preclinical AD can only be detected on an individual basis using measures of amyloid. As such, it often goes undetected in studies of aging populations and may be contaminating results. For example, screening out participants with preclinical AD has been shown to reduce variability and age-related decline in neuropsychological tests (Hassenstab et al., 2016) and resting-state functional connectivity measures (Brier et al., 2014). However, it is unclear if this confound extends to measures of atrophy.
Prior studies have assessed the impact of undetected Alzheimer pathology (Armstrong, Huang, et al., 2019; Fjell, McEvoy, et al., 2013; Fjell, Westlye, et al., 2014; Fjell, McEvoy, et al., 2014; Knopman et al., 2013), using either measures of amyloid pathology or longitudinal tracking to ensure no cognitive impairment develops. However, sample sizes were small in these studies and screening used longitudinal tracking or amyloid measures separately. In this study we use cognitively normal participants from longitudinal AD studies, allowing us to screen a large cohort for preclinical AD using both amyloid PET and longitudinal tracking of cognition in the same individuals. Using this screened cohort, we measure age-related volumetric changes across the brain that occur independent of preclinical AD.
2. Methods
2.1. Participants
The n = 383 participants in the Normal Aging Cohort came from 2 open source databases: Open Access Series of Imaging Studies (OASIS) (LaMontagne et al., 2019) and the Dominantly Inherited Alzheimer Network (DIAN). The n = 115 participants in the Preclinical AD Cohort were all from OASIS. All procedures in this retrospective study were HIPAA compliant and approved by the Washington University Institutional Review Board; informed consent was gained for all participants.
Both the Normal Aging and the Preclinical AD Cohorts only included participants who were evaluated as “Cognitively normal” or “No dementia” in their clinical assessment and who had a global Clinical Dementia Rating (CDR™) (Morris, 1993) of 0 within 1 year of Magnetic Resonance Imaging (MRI). The Normal Aging Cohort, which has been previously described (Koenig et al., 2020), only included participants who remained CDR 0 for a minimum of 3 years after MRI. Participants over age 45 were only included if they additionally had a negative amyloid PET scan (defined in section 2.4) within 1 year of their MRI. The longitudinal CDR and negative amyloid PET scan limited the possibility that the participants in the Normal Aging Cohort were in the preclinical stage of Alzheimer disease. The Preclinical AD Cohort differed from the Normal Aging Cohort in that it required a positive amyloid PET scan and did not require longitudinal CDR assessment.
While the Normal Aging Cohort included participants from DIAN, a study on autosomal dominant Alzheimer disease caused by rare mutations, only non-mutation carriers (control group) were included. DIAN was used due to its similarity to studies in the OASIS database and because DIAN has amyloid PET data available in the 45-60 age range. When compared to OASIS participants in the overlapping age range (age 42-59), there were no differences in volumetric data after multiple comparisons (section 2.6 and Supplemental Table S1). Both OASIS and DIAN include self-reported race and gender. We use the term gender and not sex to match the terminology of the questionnaire used, but participants were offered only “Male” and “Female” as options and sex was not assessed separately.
2.2. Clinical assessment
Experienced clinicians, blinded to amyloid status, evaluated each participant for the possibility of a clinical diagnosis of dementia, and only those considered to be cognitively normal were included in this study. Their assessment, outlined previously (Morris et al., 2006), integrated results from a semi-structured interview conducted with the participant and a knowledgeable collateral source, a thorough neurological examination, and bedside measures of cognitive function (including Mini Mental State Exam (MMSE) (Folstein et al., 1975) among others).
2.3. MR imaging
The MR imaging parameters for OASIS are approximate due to the variety of studies included. Scanner strength was primarily 3T (n = 19 were 1.5T) within OASIS, while DIAN was 3T. OASIS T1-weighted images (MPRAGE) primarily had 2 set of parameters. The first used TR = 2.3 s, TE = 3.16 ms, TI = 1 s, a flip angle of 8 degrees, and a spatial resolution of 1 × 1 × 1. The second used TR = 2.3 s, TE = 2.95 ms, TI = 0.9 s, a flip angle of 9 degrees, and a spatial resolution of 1 × 1 × 1 or 1 × 1 × 1.2 mm3 . DIAN T1 scans had TR = 2.3 s, TE = 2.95 ms, TI = 0.9 s, a flip Angle of 9 degrees, and a spatial resolution of 1 × 1 × 1.2 mm3 .
Volumetric T1-weighted images underwent regional tissue segmentation with FreeSurfer (version 5.0 or 5.1 for 1.5T scans and version 5.3 for 3T scans) (Fischl, 2012). Regional volumes (cortical and subcortical) were adjusted for head size with a regression approach using intracranial volume (Buckner et al., 2004). Left and right hemispheric data were combined by summing volumes and averaging cortical thicknesses.
2.4. PET imaging
[11C]-Pittsburgh compound B was used as the amyloid tracer in DIAN participants, with dosage ~15 mCi, and data collected 40-70 minutes post-injection. Within OASIS, 287 participants were imaging using [11C]-Pittsburgh compound B, with dosage ~13 mCi and data collected 30-60 minutes post-injection. The remaining 75 participants were imaged using Florbetapir ([18F]-AV45), with dosage ~10 mCi and data collected 50-70 minutes post-injection.
PET images were processed with an in-house pipeline using FreeSurfer-derived regions (Su, 2014/2021) and a cerebellar cortex reference region. Signal spillover was addressed with partial volume correction, specifically with a regional spread function (geometric transfer matrix) technique based on the scanner point spread function and the relative distance between regions (Su et al., 2013, 2015). The mean cortical Standard Uptake Value Ratio with Regional Spread Function applied (SUVR rsf) was defined as the average SUVR rsf from the precuneus, prefrontal cortex, gyrus rectus, and lateral temporal regions (Su et al., 2019).
A negative amyloid PET scan was defined as having a mean cortical SUVR rsf<1.42 (Centiloid<16.4) for [11C]-Pittsburgh compound B PET or SUVR rsf<1.19 (Centiloid<20.6) for Florbetapir PET. The Centiloid conversion process, used to more easily compare the two amyloid tracers, is documented in detail in the initial Centiloid paper (Klunk et al., 2015), with specific equations in follow-up papers (Su et al., 2018, 2019). Harmonization procedures such as this are imperfect, and so to remain as accurate as possible we used cutoffs determined individually for each tracer and then converted into Centiloid, as opposed to a unified Centiloid cutoff.
2.5. T1w/T2w myelin maps
This study uses a spatial map of the ratio of T1w/T2w image intensities in a cohort of 1071 healthy young adults (ages 22-37, mean 29) from the Human Connectome Project (Glasser et al., 2014, 2016a,b; Glasser and Van Essen, 2011). The original map was averaged within each region of the Desikan-Killiany atlas used by FreeSurfer to allow comparison. Prior work has shown that this ratio correlates with cerebral cortical myelin content due to differences in lipids, free and myelin-bound water, and iron content (Glasser & Van Essen, 2011).
2.6. Statistics
We first examined if gender, MMSE, APOE4, race, and education influenced linear models of each regional volume (after normalization for intracranial volume) and each cortical thickness in the Normal Aging Cohort. A separate linear model was run for every factor and regional volumeor thickness pairing, with a Bonferroni-Holm corrected p < 0.05 considered significant. Bonferroni-Holm, which progressively adapts the significance threshold, was done separately for each of the five factors, and across the 101 examined brain regions. Race in this study was self-reported and binarized to Caucasian and non-Caucasian due to the relatively small percentage of non-Caucasian participants. As few significant correlations were observed for any of these factors, we did not include these as covariates in the remaining analyses.
We next modeled each regional volume and thickness by age. We used the resulting standardized coefficients (β weights) to compare the strength and directionality of age-related atrophy across regions. We then addressed non-linear changes that occur with age using the estimated derivative of normal aging curves. Normal aging curves were determined by smoothing the Normal Aging Cohort’s data for each FreeSurfer region with a locally weighted scatter-plot smoother regression, resulting in a nonlinear estimate of age-related atrophy. By correlating age with the estimated derivative at each age, we estimate the pattern of age-related atrophy across the lifespan. We display examples of these normal aging curves and their estimated derivatives in Fig. 1. As these are cross-sectional data, the estimated derivative is the change in the region’s smoothed average by age, not an individual participant’s trajectory over time. As with the previous analysis, we again corrected each set of p-values for multiple comparisons across the 101 regions using Bonferroni-Holm.
Fig. 1.
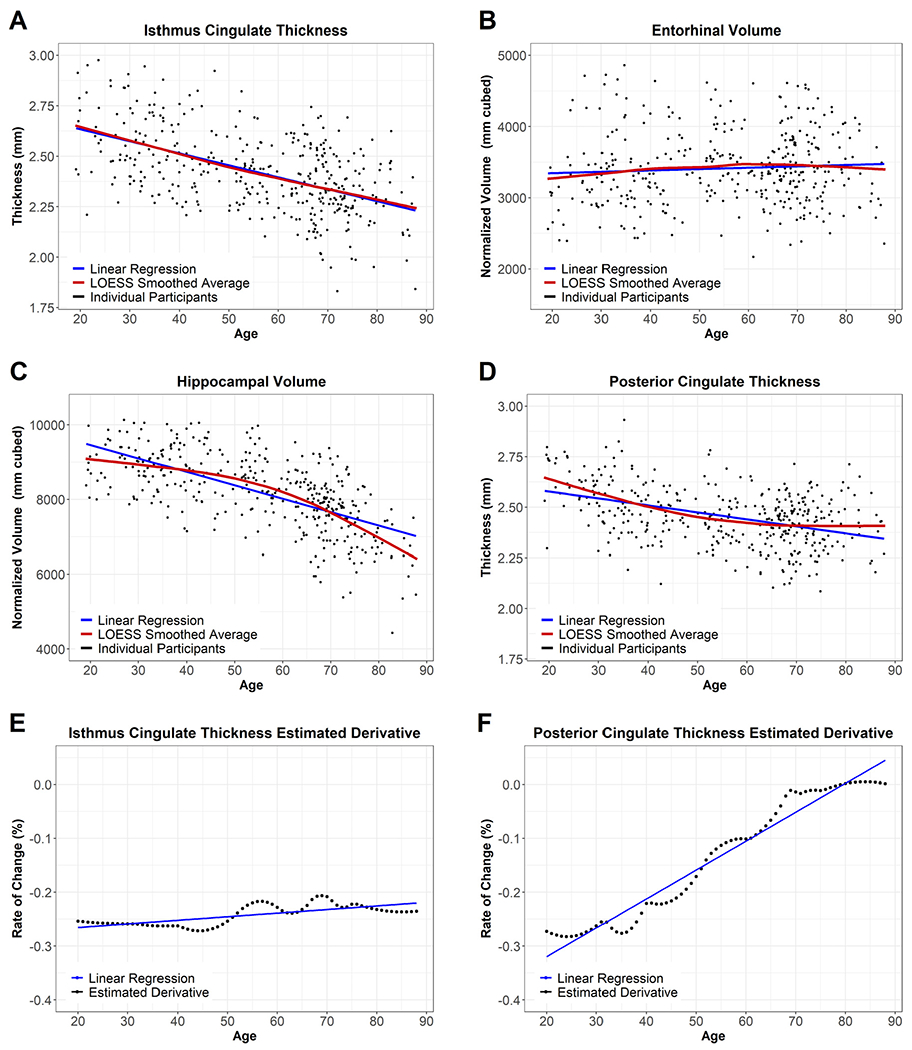
Example Regions in the Normal Aging Cohort. Fig. 2 displays regional maps of the standardized β weights from the linear models used to assess age-related atrophy. Fig. 2A displays the overall age-effect, taken from a direct comparison of participants’ ages and regional volume/thickness (blue line in Fig. 1A-D). A darker purple indicates more atrophy with age, while yellow indicates a lack of atrophy. Fig. 2B displays the pattern of atrophy with age, taken from the association of the age-derivative with age (blue line in Fig. 1E-F). Blue in Fig. 2B indicates regions whose rate of atrophy becomes less severe as age increases, while red indicates regions whose atrophy accelerates at later ages. To maintain the color schemes, the lateral ventricles are displayed with a reversed sign.
The 34 beta values for each of the four resulting cortical maps (from linear models of cortical volumes or thicknesses; as predicted by age or the age-derivative) were correlated with the myelin map described above. As this is a spatial correlation, Spearman’s rank correlation was used. To maintain consistency, these p-values were also corrected for multiple comparisons across the 4 pairings using Bonferroni-Holm.
Finally, we used linear models to assess the impact of amyloid on regional volumes and thicknesses. The Preclinical AD Cohort (amyloid positive) and the participants above age 60 in the Normal Aging Cohort were combined, and a linear model was run for each region using age, amyloid positivity, and their interaction. This process was also repeated by replacing amyloid positivity with a continuous measure of amyloid (Centiloid). Each set of p-values was corrected for multiple comparisons across the 101 regions using Bonferroni-Holm.
3. Results
3.1. Demographics
Demographics for both cohorts and the subset of the Normal Aging Cohort above age 60 are listed in Table 1. As expected, the Normal Aging Cohort had a lower frequency of APOE4 alleles and lower amyloid levels than the Preclinical AD Cohort. No regions in the Normal Aging Cohort showed significant associations with APOE4 status, MMSE, or years of education, and few regions showed significant associations with gender or race after correction for multiple comparisons (Supplemental Table S1). As such, the later analyses did not adjust for these factors.
Table 1.
Demographics.
| Normal Aging Cohort | Normal Aging Cohort (Age>60) | Preclinical AD Cohort | |
|---|---|---|---|
| n | 383 | 192 | 115 |
| n by Data Source | |||
| DIAN | 134 | 0 | 0 |
| OASIS | 249 | 192 | 115 |
| Age (median) | 18-88 (60) | 60-88 (70) | 61-89 (74) |
| Gender (% M) | 35.8 | 33.9 | 47.8 |
| MMSE (median) | 24-30 (30)a | 26-30 (30) | 23-30 (29) |
| APOE4 (% with an e4 allele) | 24.8 | 22.4 | 55.6 |
| Race (% Caucasian) | 89.8a | 89.1 | 90.4 |
| Education (years) (median) | 9-22 (16) | 10-20 (16) | 8-20 (16) |
| Amyloid Mean Cortical SUVR rsf – Centiloid (median) | −9.34-19.0 (−0.880)a | −9.34-19.0 (−0.453) | 16.4-141 (63.4) |
indicates missing data: 2 MMSEs, 6 Races, and 124 Amyloid (all from those under age 45) from the Normal Aging Cohort
Significant differences by gender were observed in intracranial volume ( β = 0.601, corrected p < 0.001), fusiform volume (β = 0.200, corrected p = 0.008), frontal pole volume (β = 0.182, corrected p = 0.03), lateral occipital volume (β = 0.178, p = 0.04), amygdala volume (β = 0.230, corrected p < 0.001), and lateral ventricle volume (β = −0.197, p = 0.01). Significant differences by race were in cuneus volume (β = 0.186, p = 0.03), inferior temporal volume (β = 0.182, p = 0.04), lateral occipital volume (β = 0.198, p = 0.01), middle temporal volume (β = 0.220, p = 0.001), and optic chiasm volume (β = −0.195, p = 0.01). In these models, a positive β weight indicates larger volumesor thicknesses in men or Caucasians, respectively.
3.2. Regional variation in strength of age-related atrophy
Almost all regions showed a significant association between atrophy and age in the Normal Aging Cohort (Supplemental Table S2). The only non-significant regional measures were caudal anterior cingulate thickness, entorhinal volume, temporal pole volume, corpus callosum posterior volume, intracranial volume, total subcortical gray matter volume, and fifth ventricle (cavum septum pallucidum) volume. While volumetric measures of the remaining regions were significantly associated with age, the strength of that relationship varied. The strongest age effects were seen in the temporal lobe and subcortical regions (Fig. 2A). Of the regions and composites not pictured in Fig. 2A, summary measures such as total cortex volume and total gray matter volume also showed some of the strongest age effects (Supplemental Table S2).
Fig. 2.
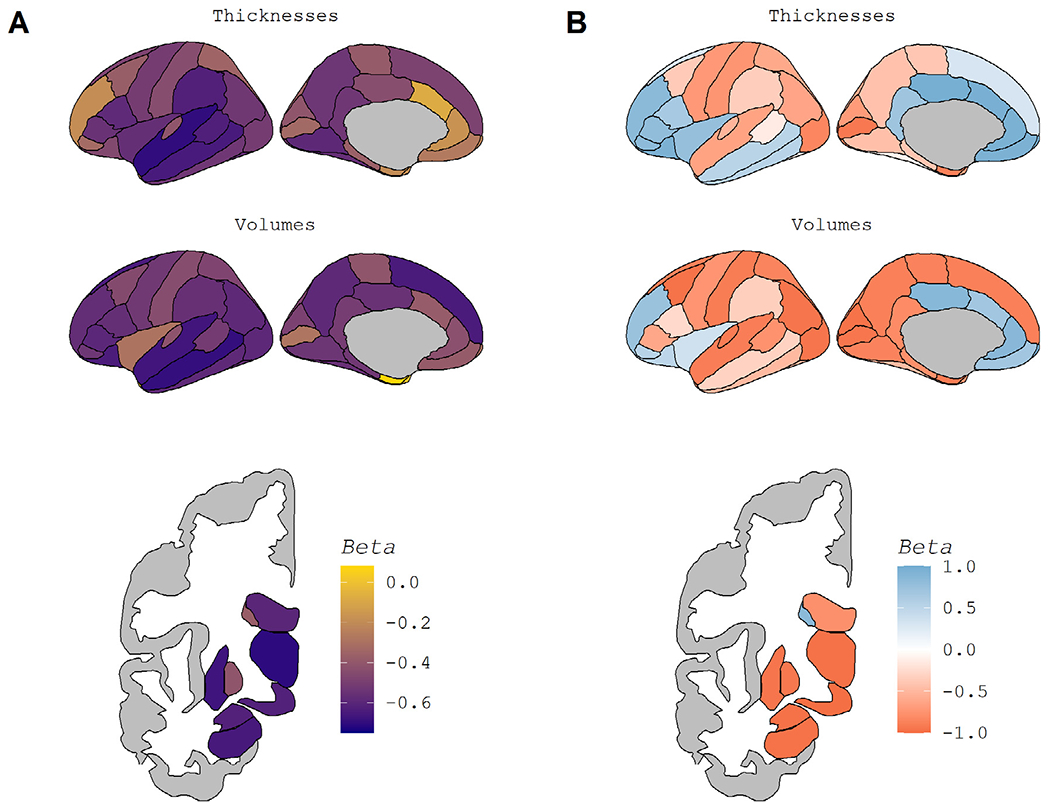
Regional Maps of Age-Related Atrophy. Fig. 2 displays regional maps of the standardized β weights from the linear models used to assess age-related atrophy. Fig. 2A displays the overall age-effect, taken from a direct comparison of participants’ ages and regional volume and/or thickness (blue line in Fig. 1A-D). A darker purple indicates more atrophy with age, while yellow indicates a lack of atrophy. Fig. 2B displays the pattern of atrophy with age, taken from the association of the age-derivative with age (blue line in Fig. 1E-F). Blue in Fig. 2B indicates regions whose rate of atrophy becomes less severe as age increases, while red indicates regions whose atrophy accelerates at later ages. To maintain the color schemes, the lateral ventricles are displayed with a reversed sign. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.3. Regional variation in non-linear patterns of age-related atrophy
The previous section used standardized β weights from linear models to compare the strength of the relationship between age and regional volumetrics. Select regions declined in a linear fashion. Many regions showed non-linear patterns, with atrophy appearing to accelerate or decelerate at older ages. We assessed the non-linear pattern of each region by smoothing our data to create normal aging curves and then estimating the derivative of that curve at each age. Fig. 1 displays examples of these normal aging curves and the corresponding estimated derivatives, and the normal aging curves for all examined regions can be seen at https://github.com/benzinger-icl/SARA or viewed interactively at https://lnkoenig.shinyapps.io/NormalAgingVolumetrics_ShinyApp/.
Almost all regions’ age-derivative showed a significant association with age (Supplemental Table S2). The regions showing non-significant correlations of age were banks of the superior temporal sulcus thickness, fusiform thickness, and pars opercularis volume. Non-significance in this case indicates no relationship, i.e. rate of atrophy did not change linearly across the age range suggesting linear decline or no atrophy with age. The strength of the association between age and the age-derivative, again represented using β weights, is displayed spatially in Fig. 2B and appears distinct from the age-association pattern in Fig. 2A. Of those regions that showed the most age-related atrophy, the temporal cortex showed an overall linear decline with age, while atrophy in subcortical regions appears to accelerate with age. In contrast, frontal regions appear to show higher rates of atrophy at midlife as opposed to late life. Of the regions not pictured in Fig. 2B, the corpus callosum stood out as a region stable at younger ages that atrophies rapidly in old age.
3.4. Relationship of T1w/T2w myelin content and slope of age-related atrophy
To quantify if the spatial patterns we observed in Fig. 2 related to myelin levels, we correlated each set of 34 cortical beta weights in Fig. 2 to an average T1w/T2w myelin map. This myelin map was generated on a separate cohort of healthy young adults (ages 22-37, mean 29) and was and is displayed in Fig. 3. The regional pattern of the strength of age-related atrophy was not significantly associated with the regional map of myelin (rho = −0.060, corrected p = 0.74 for cortical volumes; rho = −0.348, corrected p = 0.09 for cortical thicknesses). However, the regional pattern of the estimated derivative βs was significantly associated with the regional map of myelin (rho = −0.640, corrected p < 0.001 for volume; rho = −0.546, corrected p = 0.003 for thickness). The directionality of the correlation is such that regions with higher myelin content are more likely to follow the pattern shown in Fig. 1C, with atrophy that accelerates in late life. Conversely, lower myelin regions were more likely to show the pattern in Fig. 1D: atrophy greatest in midlife and tapered at older ages. While this result emphasizes the distinctness of the two patterns, the moderate correlation suggests other factors are also at play.
Fig. 3.
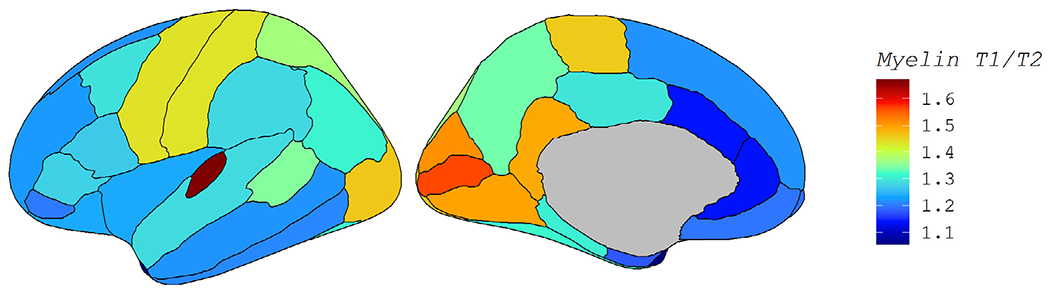
T1w/T2w Myelin Map. Fig. 3 displays the cerebral cortical myelin map that was correlated with each of the four regional maps in Fig. 2. Myelin content was measured by the ratio of T1w/T2w image intensities in a separate cohort of healthy adults.
3.5. Atrophy in preclinical AD vs normal aging
The impact of amyloid was assessed using those over age 60 in the Normal Aging Cohort (amyloid negative) and the Preclinical AD Cohort (amyloid positive). Linear models used age, amyloid, and age*amyloid to predict regional volumesor thicknesses. No significant effects of amyloid or amyloid*age were found after accounting for age and correcting for multiple comparisons (Supplemental Table S3, with examples in Fig. 4).
Fig. 4.
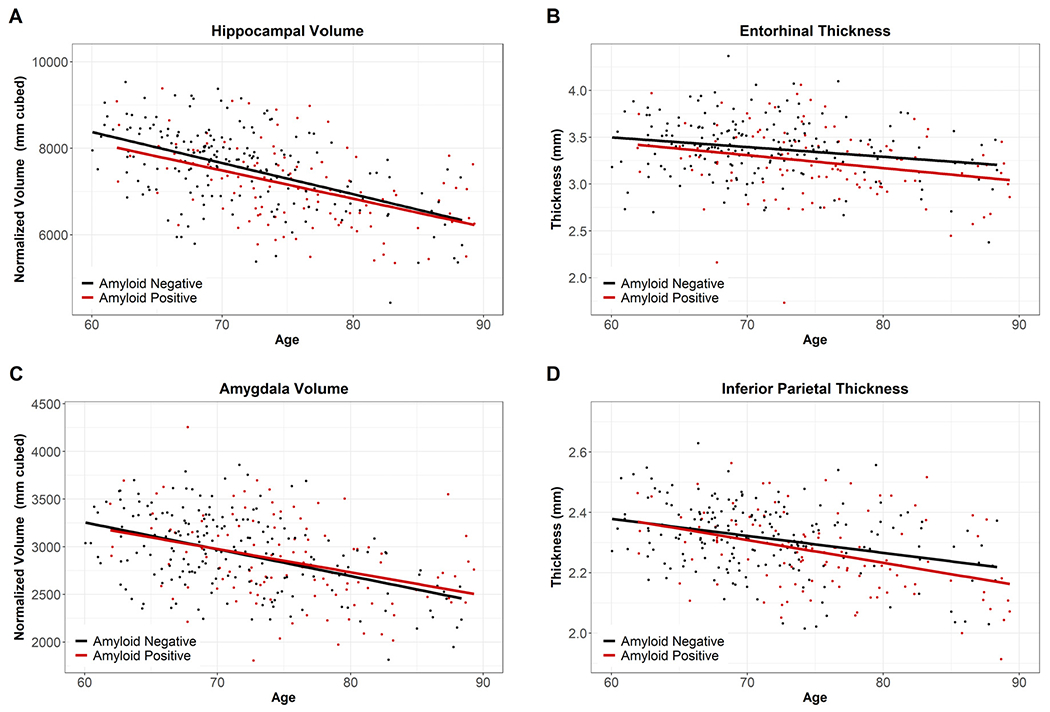
Example Regions for Normal Aging Cohort vs. Preclinical AD Cohort. Fig. 4 displays the overlap of the Normal Aging Cohort (Amyloid Negative, black) and the Preclinical Cohort (Amyloid Positive, red), indicating our non-significant findings for amyloid and age* amyloid. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
In this paper we report regional variation in age-related atrophy, with different spatial patterns for the effect size of age-related atrophy and in the non-linear pattern observed across the lifespan. Temporal regions showed the greatest association with age, while frontal and cingulate areas showed a deceleration of atrophy with age (i.e. higher atrophy in mid-life than late-life). This reduced rate of atrophy in late life contrasted to most regions which showed accelerating atrophy in late life. This pattern of non-linearity was spatially related to myelin levels determined by T1w/T2w intensity ratio. As this ratio was determined in a separate cohort of healthy adults, this suggests that the observed pattern is the end result of a fundamental organizational property of the brain. The lack of correlation between myelin and the direct association with age further supports that the two observed patterns are unique. The direction of the myelin and age-derivative correlation suggests that regions that characteristically have higher myelin content in midlife are more vulnerable to accelerated atrophy in later life. While causality is not clear, this could in part be due to the greater vulnerability of myelinating cells to oxidative stress (Nasrabady et al., 2018). No differences were detected between our Normal Aging Cohort and our Preclinical AD Cohort, though a larger sample may reveal subtle differences.
Limitations of this study include its cross-sectional design, the lack of diversity in our participants, and our inability to control for vascular influences on structural brain measures in these analyses. Group averages in aging volumetrics have been shown to be commensurate across cross-sectional and longitudinal designs (Fjell, Westlye, et al., 2013; Fotenos et al., 2005). However, by looking only at a single time-point per participant, we were unable to assess possible subtypes of patterns of aging in individuals. Our Normal Aging Cohort, collated from several studies of aging and Alzheimer disease, is predominantly highly educated and Caucasian. A more representative cohort may show greater age-related atrophy due to the association of social inequities with chronic health conditions and other social determinants of health. As such, our study may be closer to a measure of ‘healthy aging’ than the ‘normal aging’ an average individual in our society experiences.
While our cohort may not be representative of the broader population, it does reduce the probability that some unmeasured factors are confounding our measures of aging. Vascular disease is one such unmeasured factor that is common within the population represented in this study and likely impacts our results. Differences in blood pressure, even in non-hypertensive individuals, have correlated with volumetric differences (Lockhart & DeCarli, 2014). Additionally, regional volumetrics may be influenced by other non-AD neurodegenerative pathologic processes that are less common and more difficult to detect (e.g. argyrophilic grain disease, primary aging-related tauopathy, hippocampal sclerosis of aging, limbic-predominant aging-related TDP-43 encephalopathy neuropathologic change, aging related tau astrogliopathy, frontotemporal lobar degeneration, Lewy body disease). For our detected pathology, amyloid, we are limited in that we did not follow our Preclinical AD participants longitudinally. We would expect some but not all of these participants to develop AD in the near future, and these two subgroups would likely have different rates and patterns of atrophy. One final limitation is that the FreeSurfer regions used in this study were relatively coarse regions defined based on gyral and sulcal landmarks that contain significant structural and functional heterogeneity. This limits the neurobiological interpretability of regional effects as compared to cortical areas based on multiple modalities (Glasser, Coalson, et al., 2016) or more homogeneous functional regions (Gordon et al., 2016).
Despite these limitations, our results indicate that age-related atrophy is a regionally heterogeneous process, with severity of atrophy and lifespan pattern of atrophy varying independently across regions. We also showed that age-related atrophy is not significantly associated with amyloid positivity in the absence of cognitive symptoms. This suggests that volumetric studies in older adults do not need to include amyloid PET scans to screen for pre-clinical AD or track their participants longitudinally for dementia if they instead use the same rigorous dementia screening we used at baseline (integrating a comprehensive history with a trusted collateral source and neurological examination). Previous similar studies had smaller sample sizes and were unable to screen by both longitudinal cognition and amyloid levels, giving a new weight to our negative findings. Future studies should further investigate the association we saw between myelin levels and lifespan pattern of atrophy, as well as the potential influence of non-AD neurodegenerative pathologies.
5. Conclusions
The amount of atrophy that occurs with age and the pattern of decline over the lifespan exhibit two unique spatial patterns, with only the second pattern associating with regional myelination. Broadly, these patterns indicated greatest atrophy in the temporal lobe and subcortical regions, with linear patterns of decline in temporal lobe regions, accelerating decline in subcortical regions, and decelerating declines in frontal regions. Despite measures of amyloid PET and longitudinal CDR, our aging cohort and preclinical Alzheimer disease cohort did not show measurable differences in atrophy.
Supplementary Material
Abbreviations:
- AD
Alzheimer disease
- APOE4
Apolipoprotein E ε4
- CDR™
Clinical Dementia Rating™
- CV
Cortical Volume
- DIAN
Dominantly Inherited Alzheimer Network
- MMSE
Mini Mental State Exam
- MRI
Magnetic Resonance Imaging
- NA
Not Applicable
- OASIS
Open Access Series of Neuroimaging Studies
- PET
Positron Emission Tomography
- SUVR RSF
Standard Uptake Value Ratio (Regional Spread Function applied)
- SV
Subcortical Volume measure
- T
Cortical Thickness measure
Footnotes
Author credit
The revision system has this CRediT Author Statement as a required document to upload, but the instructions indicate that it is encouraged but not required. We are happy to provide a statement if necessary, but do not have one prepared at this time.
Disclosure statement
Authors declare that data contained in the manuscript being submitted have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neurobiolaging.2021.09.010.
References
- Armstrong NM, An Y, Beason-Held L, Doshi J, Erus G, Ferrucci L, Davatzikos C, Resnick SM, 2019. Predictors of neurodegeneration differ between cognitively normal and subsequently impaired older adults. Neurobiol. Aging 75, 178–186. doi: 10.1016/j.neurobiolaging.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong NM, An Y, Shin JJ, Williams OA, Doshi J, Erus G, Davatzikos C, Ferrucci L, Beason-Held LL, Resnick SM, 2020. Associations between cognitive and brain volume changes in cognitively normal older adults. Neuroimage 223, 117289. doi: 10.1016/j.neuroimage.2020.117289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong NM, Huang C-W, Williams OA, Bilgel M, An Y, Doshi J, Erus G, Davatzikos C, Wong DF, Ferrucci L, Resnick SM, 2019. Sex differences in the association between amyloid and longitudinal brain volume change in cognitively normal older adults. NeuroImage: Clinical 22, 101769. doi: 10.1016/j.nicI.2019.101769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JA, Hedden T, Carmasin J, Maye J, Rentz DM, Putcha D, Fischl B, Greve DN, Marshall GA, Salloway S, Marks D, Buckner RL, Sperling RA, Johnson KA, 2011. Amyloid-β Associated Cortical Thinning in Clinically Normal Elderly. Ann. Neurol 69 (6), 1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Thomas JB, Snyder AZ, Wang L, Fagan AM, Benzinger T, Morris JC, Ances BM, 2014. Unrecognized preclinical Alzheimer disease confounds rsfcMRI studies of normal aging. Neurology 83 (18), 1613–1619. doi: 10.1212/WNL.0000000000000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Head D, Parker J, Fotenos AF, Marcus DS, Morris JC, Snyder AZ, 2004. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: Reliability and validation against manual measurement of total intracranial volume. Neuroimage 23 (2), 724–738. doi: 10.1016/j.neuroimage.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Chételat G, Villemagne VL, Pike KE, Baron J-C, Bourgeat P, Jones G, Faux NG, Ellis KA, Salvado O, Szoeke C, Martins RN, Ames D, Masters CL, Rowe CC, 2010. Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain 133 (11), 3349–3358. doi: 10.1093/brain/awq187. [DOI] [PubMed] [Google Scholar]
- Chételat G, Villemagne VL, Villain N, Jones G, Ellis KA, Ames D, Martins RN, Masters CL, Rowe CC, 2012. Accelerated cortical atrophy in cognitively normal elderly with high β-amyloid deposition. Neurology 78 (7), 477–484. doi: 10.1212/WNL.0b013e318246d67a. [DOI] [PubMed] [Google Scholar]
- Coutu J-P, Lindemer ER, Konukoglu E, Salat DH, 2017. Two distinct classes of degenerative change are independently linked to clinical progression in Mild Cognitive Impairment. Neurobiol. Aging 54, 1–9. doi: 10.1016/j.neurobiolaging.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL, 2009. The Cortical Signature of Alzheimer’s Disease: Regionally Specific Cortical Thinning Relates to Symptom Severity in Very Mild to Mild AD Dementia and is Detectable in Asymptomatic Amyloid-Positive Individuals. Cereb. Cortex 19 (3), 497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erten-Lyons D, Dodge HH, Woltjer R, Silbert LC, Howieson DB, Kramer P, Kaye JA, 2013. Neuropathologic Basis of Age-Associated Brain Atrophy. JAMA Neurol. 70 (5), 616. doi: 10.1001/jamaneurol.2013.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM, 2009. Decreased cerebrospinal fluid Aβ42 correlates with brain atrophy in cognitively normal elderly. Ann. Neurol 65 (2), 176–183. doi: 10.1002/ana.21559 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, 2012. FreeSurfer. Neuroimage 62 (2), 774–781. doi: 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB, 2014. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog. Neurobiol 117, 20–40. doi: 10.1016/j.pneurobio.2014.02.004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB, 2013. Brain Changes in Older Adults at Very Low Risk for Alzheimer’s Disease. J. Neurosci 33 (19), 8237–8242. doi: 10.1523/JNEUROSCI.5506-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, Blennow K, Brewer JB, Dale AM, 2010. Brain Atrophy in Healthy Aging Is Related to CSF Levels of Aβ1-42. Cerebral Cortex (New York, NY) 20 (9), 2069–2079. doi: 10.1093/cercor/bhp279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Grydeland H, Amlien I, Espeseth T, Reinvang I, Raz N, Dale AM, Walhovd KB, 2014. Accelerating Cortical Thinning: Unique to Dementia or Universal in Aging? Cereb. Cortex 24 (4), 919–934. doi: 10.1093/cercor/bhs379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Grydeland H, Amlien I, Espeseth T, Reinvang I, Raz N, Holland D, Dale AM, Walhovd KB, 2013. Critical ages in the life course of the adult brain: Nonlinear subcortical aging. Neurobiol. Aging 34 (10), 2239–2247. doi: 10.1016/j.neurobiolaging.2013.04.006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher E, Filshtein TJ, Harvey D, Renaud A, Mungas D, DeCarli C, 2018. Staging of amyloid β, t-tau, regional atrophy rates, and cognitive change in a nondemented cohort: Results of serial mediation analyses. Alzheimer’s & Dementia : Diagnosis, Assessment & Disease Monitoring 10, 382–393. doi: 10.1016/j.dadm.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher E, Gavett B, Harvey D, Farias ST, Olichney J, Beckett L, DeCarli C, Mungas D, 2018. Brain volume change and cognitive trajectories in aging. Neuropsychology 32 (4), 436–449. doi: 10.1037/neu0000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher E, Villeneuve S, Maillard P, Harvey D, Reed B, Jagust W, DeCarli C, 2016. β-amyloid, hippocampal atrophy and their relation to longitudinal brain change in cognitively normal individuals. Neurobiol. Aging 40, 173–180. doi: 10.1016/j.neurobiolaging.2016.01.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR, 1975. Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res 12 (3), 189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fotenos AF, Snyder AZ, Girton LE, Morris JC, Buckner RL, 2005. Normative estimates of cross-sectional and longitudinal brain volume decline in aging and AD. Neurology 64 (6), 1032–1039. doi: 10.1212/01.WNL.0000154530.72969.11. [DOI] [PubMed] [Google Scholar]
- Glasser MF, Coalson TS, Robinson EC, Hacker CD, Harwell J, Yacoub E, Ugurbil K, Andersson J, Beckmann CF, Jenkinson M, Smith SM, Van Essen DC, 2016a. A multi-modal parcellation of human cerebral cortex. Nature 536 (7615), 171–178. doi: 10.1038/nature18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser MF, Goyal MS, Preuss TM, Raichle ME, Van Essen DC, 2014. Trends and properties of human cerebral cortex: Correlations with cortical myelin content. Neuroimage 93 (Pt 2), 165–175. doi: 10.1016/j.neuroimage.2013.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser MF, Smith SM, Marcus DS, Andersson JLR, Auerbach EJ, Behrens TEJ, Coalson TS, Harms MP, Jenkinson M, Moeller S, Robinson EC, Sotiropoulos SN, Xu J, Yacoub E, Ugurbil K, Van Essen DC, 2016b. The Human Connectome Project’s neuroimaging approach. In: Nature Neuroscience, 19. Nature Publishing Group, pp. 1175–1187. doi: 10.1038/nn.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser MF, Van Essen DC, 2011. Mapping human cortical areas in vivo based on myelin content as revealed by T1- and T2-weighted MRI. J. Neurosci 31 (32), 11597–11616. doi: 10.1523/JNEUROSCI.2180-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon EM, Laumann TO, Adeyemo B, Huckins JF, Kelley WM, Petersen SE, 2016. Generation and Evaluation of a Cortical Area Parcellation from Resting-State Correlations. Cereb. Cortex 26 (1), 288–303. doi: 10.1093/cercor/bhu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habes M, Pomponio R, Shou H, Doshi J, Mamourian E, Erus G, Nasrallah I, Launer LJ, Rashid T, Bilgel M, Fan Y, Toledo JB, Yaffe K, Sotiras A, Srinivasan D, Espeland M, Masters C, Maruff P, Fripp J, … Davatzikos C, 2021. The Brain Chart of Aging: Machine-learning analytics reveals links between brain aging, white matter disease, amyloid burden, and cognition in the iSTAGING consortium of 10,216 harmonized MR scans. Alzheimer’s & Dementia 17 (1), 89–102. doi: 10.1002/alz.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamed SA, 2017. Brain injury with diabetes mellitus: Evidence, mechanisms and treatment implications. Expert Review of Clinical Pharmacology 10 (4), 409–428. doi: 10.1080/17512433.2017.1293521. [DOI] [PubMed] [Google Scholar]
- Hassenstab J, Chasse R, Grabow P, Benzinger TL, Fagan AM, Xiong C, Jasielec MS, Grant EA, Morris JC, 2016. Certified normal: Alzheimer’s disease biomarkers and normative estimates of cognitive functioning. Neurobiol. Aging 43, 23–33. doi: 10.1016/j.neurobiolaging.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin K, Sexton C, Daniel T, Lawlor B, Naci L, 2018. Healthy Aging and Dementia: Two Roads Diverging in Midlife? Frontiers in Aging Neuroscience 10 (275). doi: 10.3389/fnagi.2018.00275 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Knopman DS, Vemuri P, Mielke MM, Lowe V, Senjem ML, Gunter JL, Machulda MM, Gregg BE, Pankratz VS, Rocca WA, Petersen RC, 2015. Age, Sex, and APOE ε4 Effects on Memory, Brain Structure, and β-Amyloid Across the Adult Life Span. JAMA Neurol. 72 (5), 511–519. doi: 10.1001/jamaneurol.2014.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DA, Seidenberg M, Reiter K, Nielson KA, Woodard JL, Smith JC, Durgerian S, Rao SM, 2018. Differential 5-year brain atrophy rates in cognitively declining and stable APOε4 elders. Neuropsychology 32 (6), 647–653. doi: 10.1037/neu0000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD, Jagust WJ, Johnson KA, Mathis CA, Minhas D, Pontecorvo MJ, Rowe CC, Skovronsky DM, Mintun MA, 2015. The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s & Dementia 11 (1), 1–15. doi: 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC, 2013. Selective Worsening of Brain Injury Biomarker Abnormalities in Cognitively Normal Elderly Persons With β-Amyloidosis. JAMA Neurol. 70 (8), 1030. doi: 10.1001/jamaneurol.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig LN, Day GS, Salter A, Keefe S, Marple LM, Long J, LaMontagne P, Massoumazada P, Snider BJ, Kanthamneni M, Raji CA, Ghoshal N, Gordon BA, Miller-Thomas M, Morris JC, Shimony JS, Benzinger TLS, 2020. Select Atrophied Regions in Alzheimer disease (SARA): An improved volumetric model for identifying Alzheimer disease dementia. NeuroImage: Clinical 26, 102248. doi: 10.1016/j.nicl.2020.102248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMontagne PJ, Benzinger TL, Morris JC, Keefe S, Hornbeck R, Xiong C, Grant E, Hassenstab J, Moulder K, Vlassenko AG, Raichle ME, Cruchaga C, Marcus D, 2019. OASIS-3: Longitudinal Neuroimaging, Clinical, and Cognitive Dataset for Normal Aging and Alzheimer Disease. MedRxiv doi: 10.1101/2019.12.13.19014902. [DOI] [Google Scholar]
- Lockhart SN, DeCarli C, 2014. Structural Imaging Measures of Brain Aging. Neuropsychol. Rev 24 (3), 271–289. doi: 10.1007/s11065-014-9268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Blazey TM, Holtzman DM, Cruchaga C, Su Y, Morris JC, Benzinger TLS, Gordon BA, 2018. Longitudinal brain imaging in preclinical Alzheimer disease: Impact of APOE ε4 genotype. Brain 141 (6), 1828–1839. doi: 10.1093/brain/awy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, 1993. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43 (11), 2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA, 2006. The Uniform Data Set (UDS): Clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis. Assoc. Disord 20 (4), 210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- Nasrabady SE, Rizvi B, Goldman JE, Brickman AM, 2018. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathologica Communications 6 (1), 22. doi: 10.1186/s40478-018-0515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Madison C, Villeneuve S, Markley C, Jagust WJ, 2014. Association of Gray Matter Atrophy with Age, β-Amyloid, and Cognition in Aging. Cerebral Cortex (New York, NY) 24 (6), 1609–1618. doi: 10.1093/cercor/bht017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew C, Soldan A, Sloane K, Cai Q, Wang J, Wang M-C, Moghekar A, Miller MI, Albert M, Research Team BIOCARD, 2017. Progressive medial temporal lobe atrophy during preclinical Alzheimer’s disease. NeuroImage. Clinical 16, 439–446. doi: 10.1016/j.nicl.2017.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Ghisletta P, Rodrigue KM, Kennedy KM, Lindenberger U, 2010. Trajectories of brain aging in middle-aged and older adults: Regional and individual differences. Neuroimage 51 (2), 501–511. doi: 10.1016/j.neuroimage.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JM, Bartlett JW, Fox NC, Barnes J, 2010. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Aβ1-42. Ann. Neurol 68 (6), 825–834. doi: 10.1002/ana.22315. [DOI] [PubMed] [Google Scholar]
- Smith CD, Andersen AH, Gold BT, 2012. Structural Brain Alterations before Mild Cognitive Impairment in ADNI: Validation of Volume Loss in a Predefined Antero-Temporal Region. Journal of Alzheimer’s Disease 31 (s3), S49–S58. doi: 10.3233/JAD-2012-120157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M, Mintun MA, Head D, Morris JC, 2009. Cognitive Decline and Brain Volume Loss as Signatures of Cerebral Amyloid-β Peptide Deposition Identified With Pittsburgh Compound B: Cognitive Decline Associated With Aβ Deposition. Arch. Neurol 66 (12), 1476–1481. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. 2021. Ysu001/PUP [C]. Original work published 2014. https://github.com/ysu001/PUP. [Google Scholar]
- Su Y, Blazey TM, Snyder AZ, Raichle ME, Marcus DS, Ances BM, Bateman RJ, Cairns NJ, Aldea P, Cash L, Christensen JJ, Friedrichsen KA, Hornbeck RC, Farrar AM, Owen CJ, Mayeux R, Brickman AM, Klunk WE, Price JC, … Benzinger TL, 2015. Partial volume correction in quantitative amyloid imaging. Neuroimage 107 (3–5), 55–64. doi: 10.1016/j.neuroimage.2014.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, Mintun MA, Benzinger TL, 2013. Quantitative Analysis of PiB-PET with FreeSurfer ROIs. PLoS One 8 (11), e73377. doi: 10.1371/journal.pone.0073377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Flores S, Hornbeck RC, Speidel B, Vlassenko AG, Gordon BA, Koeppe RA, Klunk WE, Xiong C, Morris JC, Benzinger TLS, 2018. Utilizing the Centiloid scale in cross-sectional and longitudinal PiB PET studies. NeuroImage: Clinical 19, 406–416. doi: 10.1016/j.nicl.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Flores S, Wang G, Hornbeck RC, Speidel B, Joseph-Mathurin N, Vlassenko AG, Gordon BA, Koeppe RA, Klunk WE, Jack CR, Farlow MR, Salloway S, Snider BJ, Berman SB, Roberson ED, Brosch J, Jimenez-Velazques I, van Dyck CH, … Benzinger TL, 2019. Comparison of Pittsburgh compound B and florbetapir in cross-sectional and longitudinal studies. Alzheimer’s & Dementia: Diagnosis. Assessment & Disease Monitoring 11, 180–190. doi: 10.1016/j.dadm.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Venkataraman AV, Bai W, Guitton F, Guo Y, Dehghan A, Matthews PM, 2019. Associations of Regional Brain Structural Differences With Aging, Modifiable Risk Factors for Dementia, and Cognitive Performance. JAMA Network Open 2 (12), e1917257. doi: 10.1001/jamanetworkopen.2019.17257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu Q, Luo J, Hu M, Zuo C, 2019. Effects of Age and Sex on Subcortical Volumes. Frontiers in Aging Neuroscience 11 (259). doi: 10.3389/fnagi.2019.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Wisse LEM, Das SR, Vergnet N, Dong M, Ittyerah R, Flores R.de, Yushkevich PA , Wolk DA , 2020. Longitudinal atrophy in early Braak regions in preclinical Alzheimer’s disease. Hum. Brain Mapp 41 (16), 4704–4717. doi: 10.1002/hbm.25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.