

Abstract
It is a challenge to effectively reactivate preexisting tumor-infiltrating lymphocytes (TILs) without causing severe toxicity. IL-12 can potently activate lymphocytes, but its clinical use is limited by its short half-life and dose-related toxicity. In this study, we developed a tumor-conditional IL-12 (pro-IL-12), which masked IL-12 with selective extracellular receptor binding domains of the IL-12 receptor, while preferentially and persistently activating TILs after being unmasking by matrix metalloproteases expressed by tumors. Systemic delivery of pro-IL-12 demonstrated reduced toxicity but better control of established tumors compared to IL-12-Fc. Mechanistically, antitumor responses induced by pro-IL-12 were dependent on TILs and IFNγ. Furthermore, direct binding of IL-12 to IL12R on CD8+, not CD4+, T cells was essential for maximal effectiveness. Pro-IL-12 improved the efficacy of both immune checkpoint blockade and targeted therapy when used in combination. Therefore, our study demonstrated that pro-IL-12 could rejuvenate TILs, which then combined with current treatment modalities while limiting adverse effects for treating established tumors.
One Sentence Summary:
A tumor-specific pro-IL-12 rejuvenated preexisting TILs to induce tumor growth delay without inducing systemic toxicity.
Introduction
IL-12 plays a critical role in modulating the tumor immune environment, including activating and expanding TILs. IL-12 is a heterodimeric cytokine (75 kDa) composed of two different polypeptide chains, an α-chain (p35 subunit) and a β-chain (p40 subunit), which are covalently linked by a disulfide bond. A functional IL-12 receptor complex is composed of two β-type cytokine receptor subunits (1). Mouse IL-12 receptor β1 is the subunit primarily responsible for high affinity binding to IL-12, and the expression of IL-12 receptor β2 alone is insufficient to mediate IL-12 responsiveness (2). The systemic administration of recombinant mouse IL-12 elicits potent antitumor effects in numerous mouse models (3). IL-12 is a critical cytokine for helper T cell differentiation and antibody production (4). Additionally, IL-12 stimulates the effector functions of activated T cells and natural killer (NK) cells via induction of cytotoxic enzymes and cytokines, mainly interferon-gamma (IFNγ) (3, 5). Local expression of IL-12 in the tumor can result in faster tumor rejection and the generation of antitumor memory immune responses (6–11). Unfortunately, the systemic administration of IL-12 at therapeutic doses is limited due to its short half-life in vivo and lethal on-target, off-tumor side effects (12, 13). Therefore, significant efforts have been invested in developing an IL-12 that can be preferentially delivered to the tumor microenvironment (TME), such as immunocytokines that couple IL-12 to antibodies against tumor-associated antigens (14–16). However, in these designs, either extremely high or specific expression of tumor antigens is required. Additionally, some cytokines are always unintentionally bound and consumed before reaching the TME due to interactions with high-affinity receptors on various immune cells (17). Moreover, targets that are not neoantigens can be expressed on non-tumor cells, which consume the cytokines (11). Therefore, systemic administration of IL-12 to treat well-established tumors without causing severe toxicity remains a challenge.
There is a clear need to develop a next generation of cytokines that are engineered to preferentially activate within the tumor, allowing them to have both increased efficacy and reduced toxicity. Matrix metalloproteases (MMPs) are zinc-dependent endopeptidases that degrade extracellular matrix (ECM) proteins to break barriers for cancer invasion and metastasis (18–21). Clinical studies and murine models have demonstrated that MMPs are highly expressed in both primary and metastatic tumors, as well as tumor-associated myeloid cells (22–25). Some groups utilized tumor-specific MMP expression by designing protease-activated antibodies (pro-antibodies) that connect masking peptides to the antibodies via an MMP susceptible linker (26). However, due to the difficulties of short peptides to completely block antibodies and their strong immunogenicity, it is challenging to evaluate the competency and durability of pro-antibodies. We proposed that the use of high-affinity subdomains within endogenous receptors could overcome both limitations. In this study, we designed a new pro-IL-12 consisting of IL-12 masked by IL-12 extracellular receptor binding domains that could be released using an MMP cleavable linker. It appeared that this pro-IL-12 could be preferentially activated inside the TME, with limited systemic sequelae. Thus, pro-IL-12 could be more effective than existing IL-12 designs in targeting and treating tumors while limiting side effects.
Results
IL-12-Fc fusion proteins potently control tumor growth
To explore if IL-12 levels in the TME had a differential effect on cancer patient prognosis, we used data from The Cancer Genome Atlas (TCGA) to compare overall survival for high vs. low IL-12B expression in several tumors, including colon cancer, breast cancer, and melanoma (Supplementary Fig. 1A–C). In all three cases, high IL12B expression correlated with superior survival and a hazard ratio <0.7 compared to low IL12B expression, indicating that high levels of IL-12 within the TME could lead to improved patient outcomes. Therefore, we decided to engineer an IL-12 that could accumulate within the TME and then assessed the potentially improved tumor control in mouse tumor models.
Our first improvement of IL-12 was a fusion with the human immunoglobulin Fc domain to extend in vivo half-life as well as simplify the purification process. To that end, we constructed and compared homodimer and heterodimer versions of IL-12-Fc-fusion proteins (homo-IL-12-Fc and hetero-IL-12-Fc). These designs consisted of two subunits of mouse IL-12 linked to the N-terminus of Fc in parallel or series, respectively (Fig. 1A). We then produced the fusion proteins by using the 293F cell line, purified them with a protein A-Sepharose column, and analyzed them by reducing and non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After confirming that both hetero-IL-12-Fc and homo-IL-12-Fc were expressed as the expected stable fusion proteins (Supplementary Fig. 2A), we set out to determine how their activity compared to that of recombinant mouse IL-12 (rmIL-12). Using Hek-Blue™ IL-12 cells, which could quantify the bioactivity of human and murine IL-12, we observed that both hetero-IL-12-Fc and homo-IL-12-Fc induced STAT-4 signaling in a dose-dependent manner, similar to rmIL-12 (Fig. 1B). These data demonstrated that the addition of Fc did not reduce the in vitro activity of IL-12, and in fact, might increase potency in vivo by prolonging its half-life. Then, we analyzed the antitumor activity of recombinant IL-12 (rIL-12) and IL-12-Fc using MC38, an immunogenic murine tumor model. Compared to equimolar doses of conventional rIL-12, hetero-IL-12-Fc presented more effective tumor elimination and prolonged mouse survival (Supplementary Fig. 2B and 2C).
Fig. 1. IL-12-Fc fusion proteins potently control tumor growth.

(A) Schematic structure of hetero-IL-12-Fc and homo-IL-12-Fc.
(B) Activity of fusion proteins as detected by the Hek-Blue™ IL-12 reporter cell line. Reporter cells were cultured with serial dilutions of each protein for 24 hours. In the cell culture supernatant, secreted embryonic alkaline phosphatase (SEAP) was detected by reading the OD value at 630nm with a microplate reader.
(C-D) Tumor growth curves of C57BL/6N mice inoculated with 5×105 MC38 cells and then teated with indicated doses of homo-IL-12-Fc (n=5/group): 66.7pmol, 13.3pmol, 6.67pmol. PBS (n=5) (C). Hetero-IL-12-Fc: 6.67pmol (n=14), 1.33pmol (n=13), 0.27pmol (n=10). PBS (n=14) (D) by i.p. injection three times (days 13, 16, and 19). Tumor size was measured twice per week.
(E) Tumor growth curves of C57BL/6N mice (n=9/group) inoculated with 5×105 MC38 cells and then treated with PBS, 6.67pmol homo-IL-12-Fc or hetero-IL-12-Fc fusion protein by i.p. injection on days 13, 16, and 19.
(F) Tumor growth curves of C57BL/6N mice (n=9/group) inoculated with 3×105 B16-F10 cells and then treated with PBS, 33.3pmol hetero-IL-12-Fc or homo-IL-12-Fc fusion protein by i.p. injection on days 10, 13, and 16.
Data indicate mean ± SEM and are repeated two or three independent experiments. Statistical analysis for E and F was performed using A two-way analysis of variance (ANOVA).*P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We next tested the in vivo antitumor activity of homo-IL-12-Fc and hetero-IL-12-Fc using a series of doses to determine the minimum effective schedule. We hypothesized that homo-IL-12-Fc with its increased avidity would be more potent than hetero-IL-12-Fc. Mice bearing established MC38 tumors were treated with various doses of each fusion protein intraperitoneally (i.p.). Both hetero-IL-12-Fc and homo-IL-12-Fc could eradicate the tumor in a dose-dependent manner. But unexpectedly, hetero-IL12-Fc required a much lower dose for similar results (Fig. 1C, 1D). To confirm these findings, we directly compared the two fusion proteins within the same experiment. Hetero-IL12-Fc demonstrated greater potency than homo-IL-12-Fc in controlling MC38 tumor growth at equimolar dose (Fig. 1E). To determine if these fusion proteins could also control poorly immunogenic tumors, we performed a similar experiment using B16-F10. Once again, we observed that both proteins could control tumor growth and that hetero-IL-12-Fc was more potent than homo-IL-12-Fc at equimolar dose (Fig. 1F). These findings could be explained by differences in clearance kinetics in vivo. Consistent with that hypothesis, hetero-IL-12-Fc exhibited higher levels of stability and bioactivity over time when compared to homo-IL-12-Fc, as determined by serum concentration and in vitro activity after the incubation with serum (Supplementary Fig. 3A and 3B). The additional disulfide-bond of p35-p40 might allow hetero-IL-12-Fc to have greater in vivo stability, leading to superior antitumor potency. Therefore, hetero-IL-12-Fc was the better candidate for further design iterations. Despite their effective antitumor activity, both hetero-IL-12-Fc and homo-IL-12-Fc caused cytokine storms in mice (Supplementary Fig. 3C and 3D). The toxicity was most likely because their active site was exposed during the circulation, allowing for on-target, off-tumor activation of immune cells.
Pro-IL-12 shields IL-12 activity in vitro and minimizes systemic toxicity in vivo
One approach that has been used to minimize immune related adverse events during immunotherapy is the development of pro-antibodies that remain inactive in healthy tissue until a tumor-associated protease cleavage event unmasks them (27). Taking inspiration from theses pro-antibodies, we asked whether it was possible to design a next-generation pro-IL-12 that was inactive in circulation and preferentially activated inside tumor tissues. Pro-IL-12 would consist of a long peptide-masking domain to block IL-12 activity in circulation, a protease cleavage domain to release the masking domain within the TME, IL-12, and an Fc domain to increase the half-life. For our masking domain, we decided to use portions of the endogenous IL-12 receptor to utilize naturally generated high affinity interactions without introducing potentially immunogenic sites. For our protease cleavage domain, we decided on MMPs as our protease of choice based on reports that extraordinarily high levels of active MMP within tumors can cleave shielding peptides to expose fluorescent substrates (28–30). In particular, MMP14 is a membrane-bound MMP expressed on tumor cells and inflammatory cells that is greatly enriched inside the TME, with minimal leakage out of the TME (31, 32). MMP-14 was significantly enriched in most human solid tumors but not adjacent normal tissues (Supplementary Fig. 4A). The amino acid sequence substrate of SGRSENIRTA recognized by MMP14 was chosen as the cleavable substrate linker. For the MMP hydrolysis efficiency of this peptide sequence, MMP14 is 79%, while MMP2 is 4%, and MMP9 is 9% (33). Thus, the SGRSENIRTA substrate was highly MMP14-specific. We hypothesized that pro-IL-12 would be mostly inactive due to the specific binding of the receptor domains before cleavage, and then switched to an active state in the TME after cleavage by MMP14.
For our final design, we selected the binding motifs of the cytokine-binding homology region (CHR) in the extracellular domains (ECD) of IL-12 receptor β1 and linked them to the N-terminus of hetero-IL-12-Fc via a cleavable MMP14 substrate sequence (Supplementary Fig. 4B). We purified pro-IL-12 and analyzed the product by reducing and non-reducing SDS-PAGE (Supplementary Fig. 4C). Our data showed that pro-IL-12 produced by 293F cells was precisely the expected size and isolated with high purity, and pro-IL-12 serum half-life was similar to that of hetero-IL-12-Fc (Supplementary Fig. 4D). Because cytokine stability was the prerequisite for broader and more reliable translational application, we further tested the stability and activity of pro-IL-12 after long-term storage and observed that pro-IL-12 activity was well maintained at 4 °C for at least 6 months (Supplementary Fig. 4E).
To determine whether the chosen ECD of IL-12Rβ1 could sufficiently block IL-12 activity, we performed a Hek-Blue™ IL-12 reporter assay with purified pro-IL-12, which showed a 10-fold decrease of IL-12 activity compared to hetero-IL-12-Fc (Supplementary Fig. 5A). Sterically linking the receptor via a protein linker allowed IL-12Rβ1 to bind to IL-12 more effectively by holding it in place, but even then, there was a limitation to the blocking efficiency (Supplementary Fig. 5A). To strengthen self-blockade, we also constructed a pro-IL-12 containing both Rβ1 and Rβ2 domains (pro-R1R2). The data (Supplementary Fig. 5B) showed that pro-R1R2 could further block IL-12 bioactivity up to 100-fold. However, pro-R1R2 also had reduced antitumor efficacy in vivo (Supplementary Fig. 5C). To achieve a balance of efficacy and toxicity, we chose Rβ1 for pro-IL-12 empirically. To determine the restored pro-IL-12 activity after cleavage and separation from the ECD, pro-IL-12 was incubated with rhMMP14 at 37°C overnight. The subsequent byproduct showed almost 100% cleavage by Western Blot and greatly increased IL-12 activity as measured by Hek-Blue™ IL-12 reporter assay (Fig. 2A, 2B). Moreover, we compared the antitumor effect of pro-IL-12 containing a cleavable MMP linker, SGRSENIRTA, with that containing a non-cleavable G4S linker. Cleavable pro-IL-12 significantly inhibited tumor growth, while non-cleavable (Nsub) pro-IL-12 showed reduced antitumor efficacy in vivo (Supplementary Fig. 5D). This result demonstrated that the cleavable MMP-sensitive sequence enhanced pro-IL-12’s bioactivity in vivo.
Fig. 2. Pro-IL-12 shields IL-12 activity in vitro and reduces systemic toxicity in vivo.
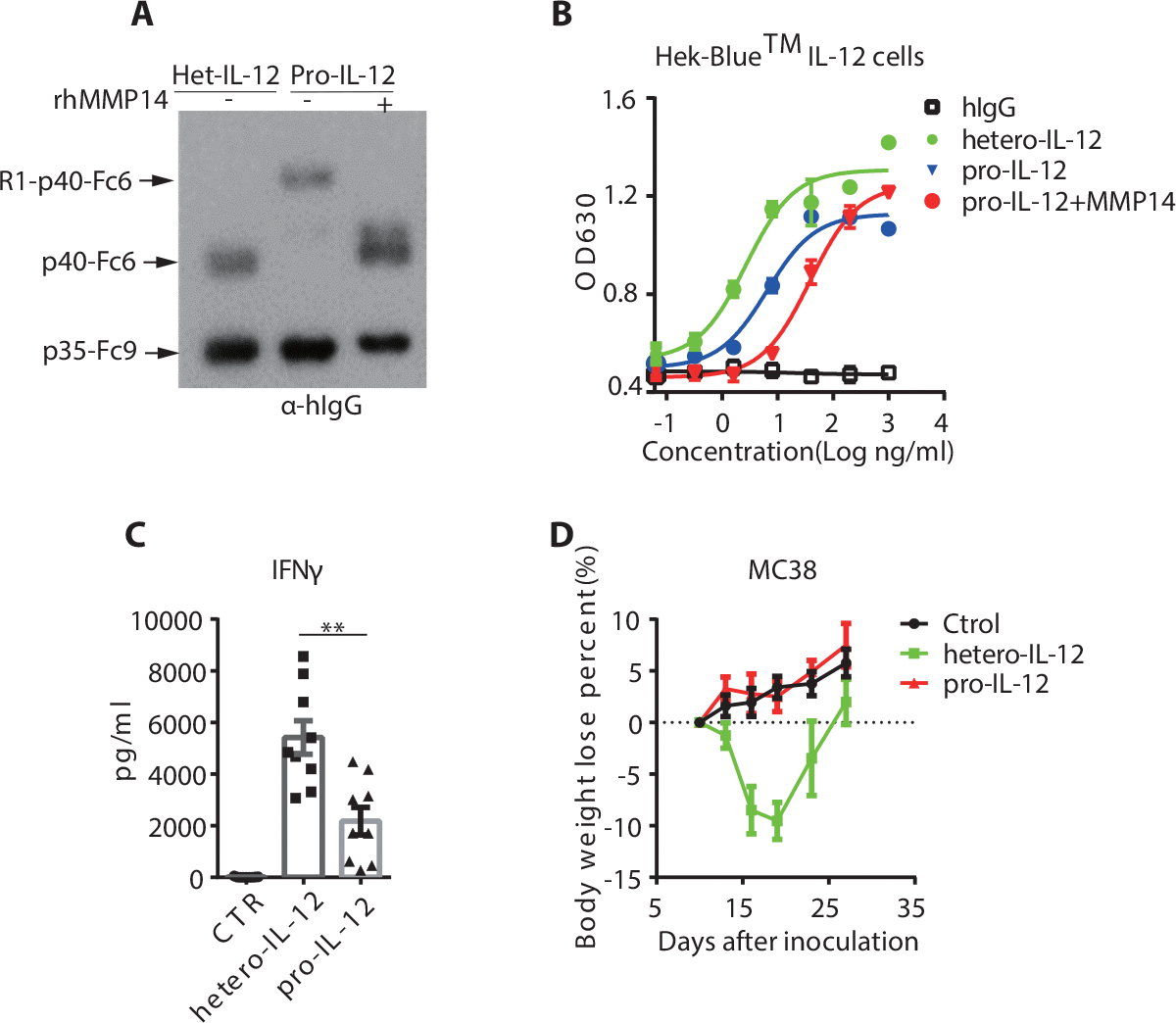
(A-B) Successful cleavage of pro-IL-12 by MMP14. 5μg of each pro-IL-12 fusion protein was co-cultured with or without 200ng activated MMP14 at 37°C overnight. The MMP14-digested pro-IL-12 under different conditions was analyzed by Western Blot (A) and Hek-Blue™ IL-12 reporter cell line (B).
(C) Serum IFNγ levels of C57BL/6N mice (n=9/group) bearing MC38 tumors after treatment with PBS, 33.3pmol of hetero-IL-12-Fc or pro-IL-12. Serum was collected six hours after the third injection and measured by Cytometric Bead Array (CBA).
(D) Body weight loss of C57BL/6N mice (n=9/group) inoculated with 5×105 MC38 cells and then treated with PBS, 33.3pmol of hetero-IL-12 or pro-IL-12 by i.p. injection on days 10, 13, and 16. Mouse body weight was measured every two days, starting from day 0 of the first injection.
Data indicate mean ± SEM and are repeated two or three independent experiments. Statistical analysis for C was performed using unpaired two-tailed t-tests.*P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We next investigated whether pro-IL-12’s reduced activity in vitro translated to reduced toxicity in vivo. Mice bearing large, established MC38 tumors were treated with hetero-IL-12-Fc, pro-IL-12, or PBS control, at a high dose via i.p. injection. High levels of serum inflammatory cytokines, including IFNγ, TNFα, IL-6, and IL10, were observed in the hetero-IL-12-Fc treated group six hours after the final treatment. In contrast, mice treated with pro-IL-12 showed significantly reduced levels of IFNγ (Fig. 2C), while other serum inflammatory cytokines also showed a decreasing trend (Supplementary Fig. 5E). We detected the IL-12 associated inflammation in mice organs, such as the liver. Hetero-IL-12-Fc induced significant immune cell infiltration in liver tissue. In contrast, pro-IL-12 resulted in no liver inflammation comparable to untreated control mice (supplementary Fig. 5F). We also compared body weight changes and observed that mice from the hetero-IL-12-Fc group started to lose weight quickly after the first treatment. However, the body weight of mice treated with pro-IL-12 remained mostly unchanged throughout the treatment (Fig. 2D). These data suggested that masking hetero-IL-12-Fc with ECD of IL-12 receptor β1 reduced toxicity when administered systemically.
Pro-IL-12 still effectively controls both primary and metastatic tumors
After demonstrating that pro-IL-12 was less toxic than hetero-IL-12-Fc, we wanted to know if it was at least as good if not better at controlling tumors due to preferential activation within the TME. To assess how well pro-IL-12 accumulates in the TME, MC38 tumor-bearing mice were injected i.p. with pro-IL-12, and the fusion protein level in various tissues was examined by ELISA. As anticipated, pro-IL-12 had a significantly greater concentration in the tumor as compared to a few non-tumor tissues (Fig. 3A). Additional bioluminescence data showed that pro-IL-12 intensified within tumor tissue after tail vein injection, further supporting our hypothesis (Supplementary Fig. 6A). In melanoma patients treated with rhIL-12, a robust and prolonged increase of IFNγ is associated with favorable clinical responses (34). To examine if pro-IL-12 not only accumulated within the TME but also was bioactive, we quantified the level of IL-12 related cytokines within tumor tissue after the treatment. MC38 tumor-bearing mice treated with pro-IL-12 exhibited sustained level of intratumoral IFNγ for at least 4 days following the treatment (Supplementary Fig. 6B). Furthermore, pro-IL-12 increased other pro-inflammatory cytokines in the tumor, such as TNF (Supplementary Fig. 6C).
Fig. 3. Pro-IL-12 could effectively control both primary and metastatic tumors.
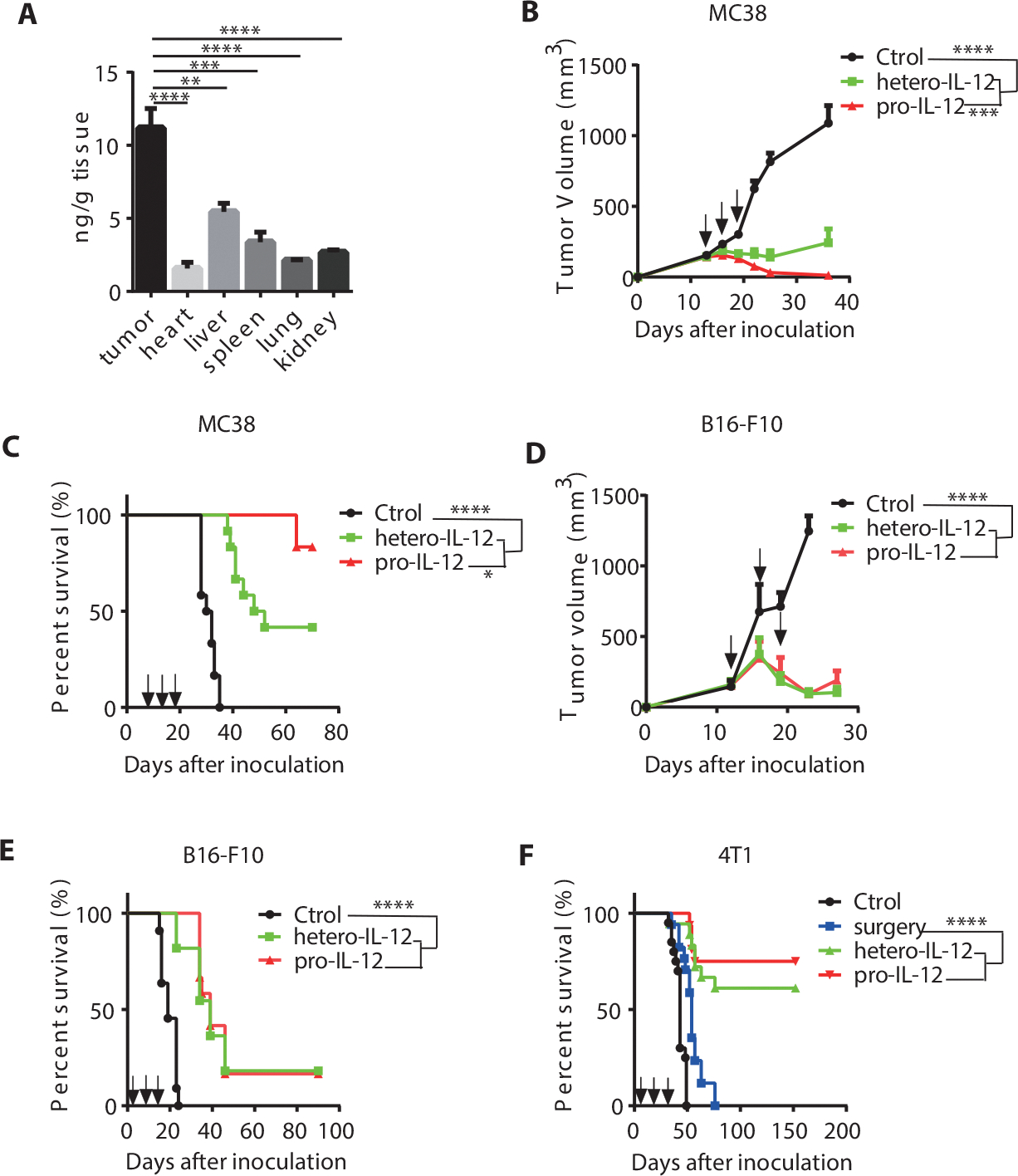
(A) Biodistribution of pro-IL-12 in various tissues after 2.4 μg pro-IL-12 was injected i.p. into MC38 tumor-bearing mice (n=6/group). HIgG ELISA was performed to quantify the amount pro-IL-12 in each homogenate, normalized by total tissue weight.
(B-C) Tumor growth and survival curves of C57BL/6N mice (n=12/group) inoculated with 5×105 MC38 cells and then treated with PBS, 3.33pmol of hetero-IL-12-Fc, or pro-IL-12 by i.p. injection on days 13, 16, and 19. Tumor size was measured twice per week (B), and the mouse survival curve was shown as (C).
(D-E) Tumor growth and survival curves of C57BL/6N mice inoculated with 3×105 B16-F10 cells and then treated with PBS (n=8), 33.3pmol of hetero-IL-12-Fc (n=11), or pro-IL-12 (n=11) by i.p. injection on days 13, 16, and 19. Tumor size was measured twice per week (D), and the mouse survival curve was shown in (E).
(F) Survival curves of BALB/C mice inoculated with 1.5×105 4T-1 cells and then treated with PBS(n=20), 3.33pmol of hetero-IL-12-Fc (n=18), or pro-IL-12 (n=16) by i.p. injection on days 9, 12, and 15 or only surgery (n=17). All primary tumors were resected on day 16, except for the no-surgery group, as a control.
Data indicate mean ± SEM and are repeated two or three independent experiments. Statistical analysis for A and D was performed using unpaired two-tailed t-tests. For B, a two-way analysis of variance (ANOVA) was applied. Statistical analysis for the survival curve data was performed using log-rank tests.*P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Upon confirming that pro-IL-12 preferentially resided in the TME and generated a sustained cytokine response, we explored whether those characteristics translated to differences in antitumor efficacy in vivo. Mice bearing MC38 tumors were treated with pro-IL-12, hetero-IL-12-Fc, or PBS control by i.p. injection. At low doses, pro-IL-12 and hetero-IL-12-Fc were both capable of initially suppressing primary tumor growth and extending mouse survival (Fig. 3B and 3C). Pro-IL-12 reduced toxicity and had greater antitumor efficacy than hetero-IL-12.
The potency of pro-IL-12 in vivo led us to characterize it in various refractory tumor models further. For a poorly immunogenic and cold tumor model, we treated B16-F10 tumor-bearing mice with hetero-IL-12-Fc, pro-IL-12, and PBS control by i.p. injection. In this case, we observed effective tumor control by both hetero-IL-12-Fc and pro-IL-12, and prolonged survival of mice over the control group (Fig. 3D and 3E). For a metastatic tumor model, we used 4T1 mammary carcinoma, which can spontaneously metastasize from the primary tumor 10 days after local implantation (35). We treated the mice bearing metastatic 4T1 with pro-IL-12, hetero-IL-12-Fc, or PBS control prior to surgical removal of the local tumor 16 days post inoculation as a neoadjuvant strategy. The mice could not survive with surgery alone. But we found that both preoperative hetero-IL12 and pro-IL-12 treatment significantly extended mouse survival, with 82% of pro-IL-12 treated mice remaining tumor-free 130 days post treatment (Fig. 3F). These data suggested that pro-IL-12 could effectively control tumor growth.
Pro-IL-12 efficacy is mediated by preexisting intratumor T cells
Among its various functions, IL-12 provides essential signals for the activation, functional activity, and survival of NK cells and T cells (36). To identify crucial cellular responders to pro-IL-12, we first assessed the relative contribution of innate immunity, including NK cells, and adaptive immunity, including T cells, to the pro-IL-12 induced antitumor response. MC38-bearing Rag1 KO mice lacking adaptive immune cells and WT C57BL/6N mice were treated with pro-IL-12 or PBS control. Unlike in WT mice, where pro-IL-12 could eliminate tumors, the therapeutic effect of pro-IL-12 was abolished in Rag1 KO mice (Fig. 4A and 4B). This result suggested that innate immune cells were not sufficient, whereas adaptive immune cells were completely indispensable for pro-IL-12 antitumor effects. To account for the possibility that Rag1 KO mice had inherent defects in their innate immune cells, we examined individual innate immune cells by a series of cell depletion in MC38 tumor-bearing WT mice treated with pro-IL-12. We observed that neither the depletion of NK cells (Supplementary Fig. 7A) nor neutrophils affected the therapeutic effects of pro-IL-12 (supplementary Fig. 7B). IL-12 reprograms inhibitory cells such as tumor-associated macrophages (TAMs) via switching immunosuppressive M2-like macrophages to a pro-immunogenic M1-like phenotype (37, 38). To confirm if IL-12-driven macrophages were necessary to control tumors, we depleted them using Clophosome. After macrophage depletion, we unexpectedly observed that the therapeutic function of pro-IL-12 still persisted (supplementary Fig. 7C), suggesting that macrophages were not required for pro-IL-12 tumor control. As a whole, these results revealed that innate immunity was not sufficient for pro-IL-12 therapy.
Fig. 4. Intratumoral CD8+ T cells and IFNγ are necessary for pro-IL-12 induced antitumor effects.
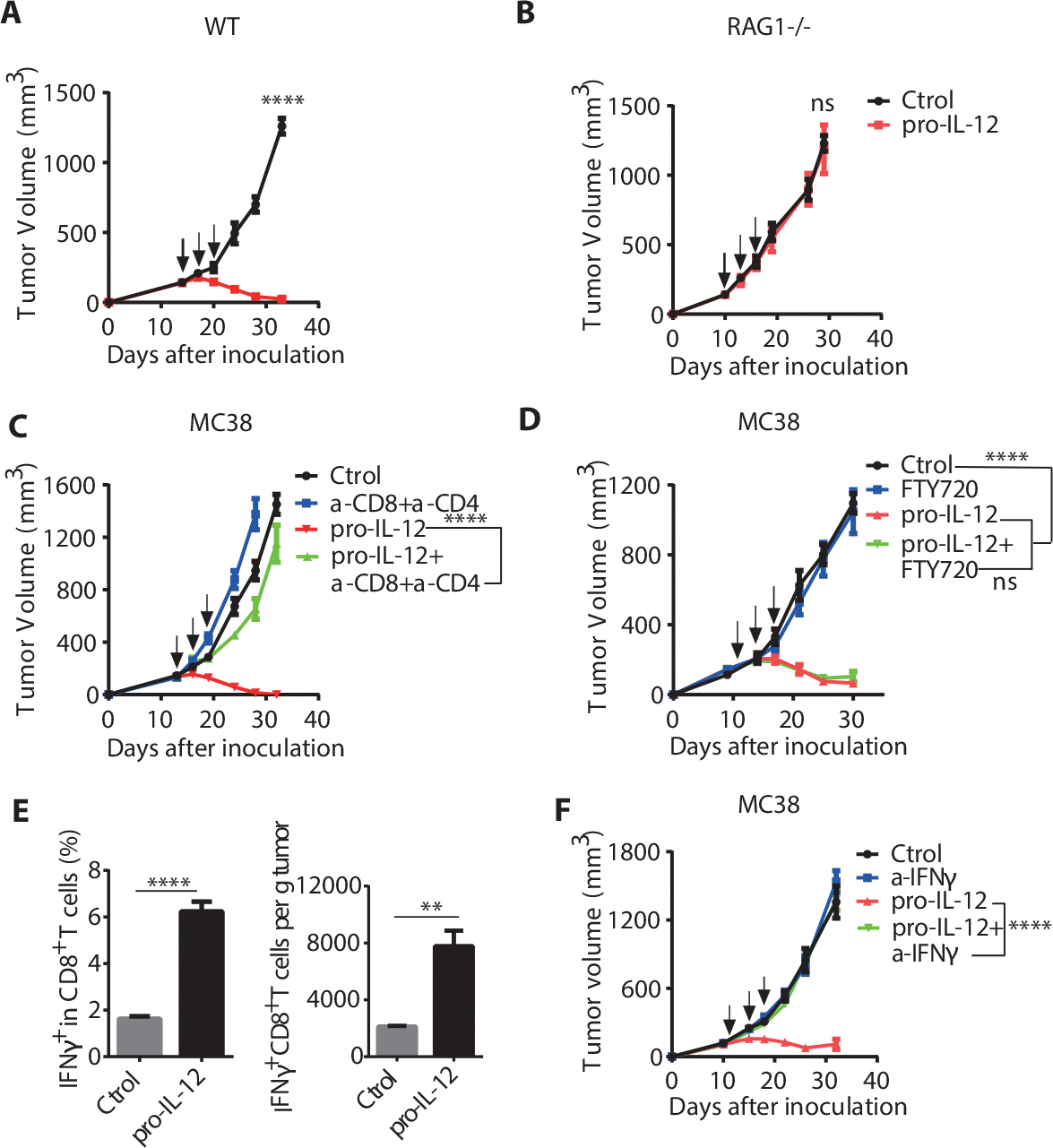
(A-B) Tumor growth curves of C57BL/6N (A) (n=8/group) or Rag1−/− (B) (n=6/group) mice bearing MC38 tumors that were treated with PBS or 3.33pmol of pro-IL-12 by i.p. injection when the tumor volume was 130–160mm3.
(C) Tumor growth curves of C57BL/6N mice (n=8/group) bearing MC38 tumors that were depleted of T cells and then treated with PBS or 3.33pmol of pro-IL-12 by i.p. injection on days 13, 16, and 19. For T cell depletion, mice were i.p. injected with 200 μg of anti-CD8 antibody or anti-CD4 antibody prior to treatment with pro-IL-12 or PBS. Tumor growth was measured twice per week.
(D) Tumor growth curves of MC38 tumor-bearing mice (n=11/group) given FTY720 and then treated with 3.33pmol pro-IL-12 by i.p. injection on days 11, 14, and 17. FTY720 was administered 25μg by i.p. injection every other day starting 10 days after tumor cell inoculation. Tumor growth was measured twice per week.
(E) Percent and the absolute number of IFNγ+ CD8+ T cells within the tumors of C57BL/6N mice (n=8/group) inoculated with 8×105 MC38-OVA cells and then treated with PBS or 33.3pmol of pro-IL-12 by i.p. injection. After four days, mice were i.p. injected with 500μl 0.5mg/ml Brefeldin A solution. Six hours later, tumor tissues were collected, and FACS analysis was performed
(F) Tumor growth curves of C57BL/6N mice bearing MC38 tumors that were IFNγ blocked and then treated with PBS (n=10), α-IFNγ (n=10), 3.33pmol of pro-IL-12 (n=10), 3.33pmol of pro-IL-12+α-IFNγ (n=11) by i.p. injection on days 12, 15, and 18. To block IFNγ, mice were i.p. injected with 500 μg of anti-IFNγ one day before pro-IL-12 treatment and then once every three days for three times. Tumor growth was measured twice per week.
Data indicate mean ± SEM and are repeated two or three independent experiments. Statistical analysis for C, E and F was performed using unpaired two-tailed t-tests. For A, B and D, a two-way analysis of variance (ANOVA) was applied. *P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Most studies show that IL-12 induced antitumor immune responses can be attributed to Th1 cells when considering adaptive immune cells (39–41). It is widely accepted that IL-12 can activate CD4+ T cells for their antitumor activity, thus driving strong Th1 immunity that is required for strong CTLs. When we used both anti-CD8 and anti-CD4 depletion antibodies together during pro-IL-12 treatment, the efficacy of pro-IL-12 was entirely diminished (Fig. 4C). To determine which of these T cell subsets was also important in the context of pro-IL-12, we depleted either CD4+ or CD8+ T cells alone in MC38-bearing mice before treating them with pro-IL-12. The therapeutic effects of pro-IL-12 treatment persisted after CD4+ T cell depletion (Supplementary Fig. 7D). But when we depleted the CD8+ T cells, the therapeutic effects were largely abolished (Supplementary Fig. 7E). These data revealed that the therapeutic effect of pro-IL-12 was dependent on T cells, with CD8+ T cells playing a more significant role.
These T cells can potentially originate from two primary sources: preexisting T cells inside tumor tissues or newly activated T cells migrating from peripheral lymphoid tissues into the tumor. FTY720, a sphingosine 1-phosphate (S1P) receptor agonist, can potently inhibit naïve and effector lymphocytes’ egression from the LN into the circulation and peripheral tissues, limiting migration of primed T cells into tumor tissues (42). To determine whether preexisting intratumoral T cells were sufficient for pro-IL-12 therapy, we used FTY720 to inhibit migratory T cells from entering the TME. MC38-bearing mice were given FTY720 one day before pro-IL-12 treatment and every other day during the therapy, and few T cells were detected among PBMCs after treatment. Antitumor effects of pro-IL-12 were still well maintained under FTY720 treatment (Fig. 4D), indicating that preexisting T cells are sufficient for pro-IL-12 therapy.
IFNγ is necessary for pro-IL-12 induced antitumor effects.
For preexisting intratumoral T cells to mediate tumor rejection, it was most likely the case that pro-IL-12 could either directly or indirectly activate tumor-specific CTLs. To monitor the effect of pro-IL-12 on tumor-specific T cells, MC38-OVA tumor-bearing mice were treated with pro-IL-12 or PBS control by intratumoral (i.t.) injection. Cells were then isolated from tumor draining lymph nodes (DLN) and co-cultured with OTI or non-related control peptides. An IFNγ ELISPOT assay showed significantly more IFNγ producing cells in the DLN of pro-IL-12 treated mice than in the control group (Supplementary Fig. 7F). The OVA-tetramer analysis also showed an increase in tumor-specific CD8+ T cells within the tumor (Supplementary Fig. 7G), suggesting that pro-IL-12 could yield a more robust tumor-specific CD8+ T cell response. PD-1+Tim-3+ T cells are highly dysfunctional compared to PD-1– Tim-3– antigen-specific CD8+ T cells in tumors. Worse prognosis is associated with high-level expressions of PD-1 and Tim-3 in patients’ tumors (43–45). In tumor-bearing mice after pro-IL-12 treatment, we observed that the frequency of PD-1+ Tim-3+ OVA-specific CD8+ T cells in tumors was reduced, and the frequency and absolute number of PD-1− TIM3− OVA-specific CD8+ T cells were significantly enhanced (Supplementary Fig. 8A, 8B and 8C). Moreover, the absolute number of PD-1− Tim-3− Ki-67+ OVA-specific CD8+ T cells was increased after pro-IL-12 treatment, which was not observed in PD-1+ Tim-3+ Ki-67+ CD8+ T cells (Supplementary Fig. 8D and 8E). We also quantified Tregs by FACS analysis after pro-IL-12 therapy. Pro-IL-12 treatment significantly reduced the Tregs in the TME (Supplementary Fig. 8F and 8G). Together, these data suggested that pro-IL-12 expanded functional CTLs and restricted the development of exhausted T cells in the TME.
Mechanistically, the binding of IL-12 to T cells caused a cascade of phenotypic changes, including pro-inflammatory cytokine production. Among the cytokines induced by IL-12, IFNγ was most highly expressed (Supplementary Fig. 3D) and best associated with favorable clinical outcomes (34). To observe how pro-IL-12 affected IFNγ production by CTLs, MC38-OVA-bearing mice were treated with pro-IL-12 or PBS control by i.p. injection, and tumor tissue was examined via flow cytometry. Indeed, the ratio and the absolute number of CD8+ IFNγ+ T cells inside the tumor were both significantly increased on day 4 in the pro-IL-12 treatment group compared to the PBS group (Fig. 4E and Supplementary Fig. 7G). To investigate whether IFNγ release plays a crucial role in tumor control, we treated MC38 tumor-bearing mice with an IFNγ-specific neutralizing antibody along with pro-IL-12 therapy. Following the IFNγ neutralization, the antitumor activity of pro-IL-12 was abolished entirely, and the tumor became resistant to pro-IL-12 treatment (Fig. 4F). These results implied that IFNγ was essential for pro-IL-12 induced antitumor therapeutic effects.
IL-12R on CD8+ T cells but not CD4+ T cells is required for tumor rejection
After determining the essential role of IFNγ for pro-IL-12 therapy, we wanted to ascertain the major cellular source of the cytokine. IL-12 receptors are primarily expressed on T and NK cells, and both cell types produce IFNγ in response to IL-12 (46–48). To test the relative contribution of T and NK cell subsets to extracellular IFNγ levels after the stimulation with IL-12, we cultured splenocytes from C57BL/6 mice ex vivo after excluding CD3+, NK1.1+, CD4+, or CD8+ cells by FACS. We then added PBS or hetero-IL12-Fc and incubated the cells for 48h before measuring IFNγ in the media. As expected, the hetero-IL12-Fc treatment caused a dramatic increase in IFNγ levels when administered to bulk splenocytes compared to PBS control (Fig. 5A). This effect was completely abrogated by excluding all T cells (-CD3), suggesting a limited role of NK cells in producing IFNγ. Consistent with that finding, the exclusion of NK cells did not have a significant effect on IFNγ. In contrast, exclusion of CD4+ T cells (-CD4) reduced IFNγ levels by about 30% and exclusion of CD8+ T cells (-CD8) reduced IFNγ levels by almost 80% (Fig. 5A). These results were consistent with our previous depletion data, which showed an essential role of CD8+ T cells for pro-IL-12 efficacy in tumor control (Supplementary Fig. 7E).
Fig. 5. IL-12R on CD8+ T cells but not CD4+ T cells is required for tumor immunity.
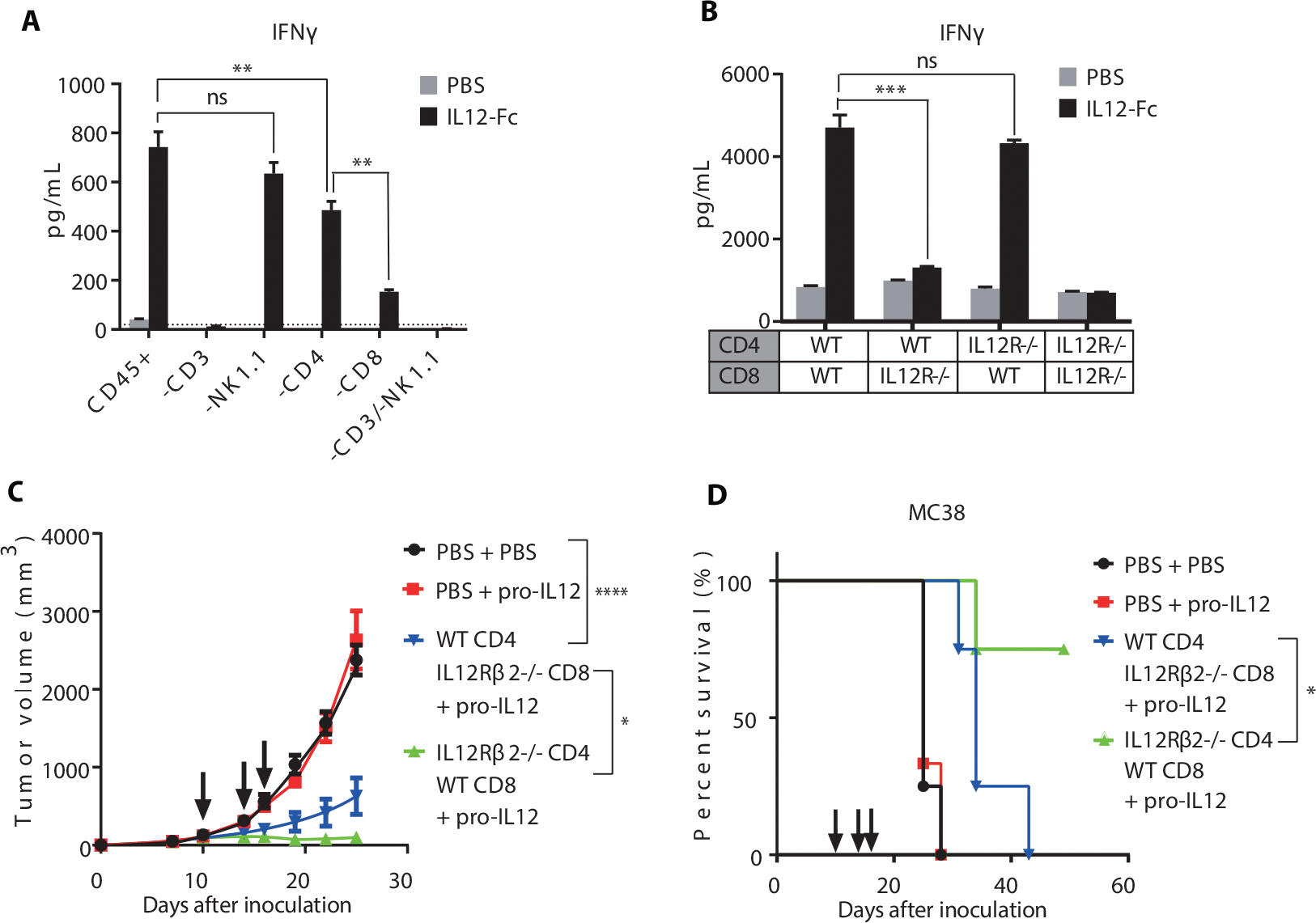
(A) IFNγ levels in the media of splenocytes from C57BL/6 mice sorted by FACS to exclude various cell populations and cultured ex vivo in the presence of PBS or het-IL-12-Fc. After 48h, media was collected and measured for IFNγ by CBA.
(B) IFNγ levels in the media of CD4+ and CD8+ T cells from WT or IL12RB2−/− C57BL/6 mice purified by MACS and cultured ex vivo in the presence of PBS or het-IL-12-Fc. After 48h, media was collected and measured for IFNγ by CBA.
(C-D) Tumor growth and survival curves of RAG1−/−mice (n=4/group) reconstituted with T cells from WT or IL12Rβ2−/− C57BL/6 mice, inoculated with MC38 tumors, and treated with PBS or 0.25ug pro-IL-12 by i.p. injection on days 10, 14, and 16. CD4+ and CD8+ T cells were purified by MACS from the spleens of WT or IL12Rβ2−/− C57BL/6 mice and adoptively transferred to RAG1 −/− mice 6 days before inoculation with 1×106 MC38 cells. Tumor size was measured twice per week (C), and the mouse survival curve was shown as (D).
Data indicate mean ± SEM and are repeated two or three independent experiments. Statistical analysis for A and B was performed using one way analysis of variance (ANOVA) with Tukey’s multiple comparison. For C, a two-way ANOVA was applied. For D, log-rank tests were used. *P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
However, in this system, it was still unclear if IL-12 acts directly on CD8+ T cells or acts on another cell type such as Th1 helper cells to stimulate CD8+ T cells in a cascade of effects through a second messenger. To more directly determine the contribution of IL-12 binding to IL-12R on CD4+ and CD8+ T cells, we purified both cell types from the spleens, either WT or IL12Rβ2−/− C57BL/6 mice, and mixed them ex vivo in all four combinations. IL12Rβ2 contains the signaling domain of the IL12R heterodimeric complex, so these IL12Rβ2−/− cells are completely deficient in IL-12 signaling (49). Once again, the cells were incubated with either PBS or IL12-Fc and we measured media IFNγ 48 hours later (Fig. 5B). Strikingly, IL12Rβ2−/− CD8+ cells mixed with WT CD4+ cells produced a minimal amount of IFNγ after treatment with IL12-Fc compared to PBS treated control. In contrast, WT CD8+ cells mixed with IL12Rβ2−/− CD4+ cells generated almost as much IFNγ as the full WT control. These results suggested that the vast majority of IFNγ produced following IL-12 treatment came from direct binding to CD8+ T cells, with only minor contributions from CD4+cells.
To confirm that this phenotype was physiologically relevant to the antitumor effect of pro-IL-12, we adoptively transferred either WT CD4+ and IL12Rβ2−/− CD8+ cells or IL12Rβ2−/− CD8+ and WT CD4+ cells to RAG1 KO mice before inoculating with MC38 and treating with pro-IL-12. Mice lacking IL12R on CD8+ cells showed partial tumor control before ultimately succumbing to the tumor challenge (Fig. 5C and 5D). However, mice lacking IL12R on CD4+ cells showed significant tumor regression, with half of the mice achieving complete tumor-free survival for more than 50 days. As a whole, these data showed that IL12R expression on CD8+ T cells was not only necessary for maximal IFNγ production but it was also sufficient for in vivo tumor control.
Pro-IL-12 combined with TKI therapy to induce robust tumor regression
TUBO is a HER2/neu-dependent mammary carcinoma derived from BALB/c mice transgenic with the rat neu oncogene. The TUBO model, representative of cold tumors, presents similarly to clinical EGFR/HER2-driven tumors with poor response to immunotherapy due to insufficient TILs (50–53). Unlike the B16-F10 and MC38 tumor models, pro-IL-12 failed to control even partially established TUBO tumors (Fig. 6A). EGFR TKIs function by directly blocking oncogenic signals to induce tumor cell death (53), enhancing tumor immunogenicity or innate sensing, and resulting in increased TILs. Second and third-generation EGFR-TKI therapy can rapidly reduce tumor burden in patients with EGFR- or Her2-dependent cancers, but a high relapse rate becomes the major clinical problem. In mice, as in human, TKI therapy partially delayed established TUBO tumor growth, but all the treated tumors relapsed later (Supplementary Fig. 9A). We measured and compared the frequency and the absolute number of T cells inside the TUBO tumor six days after TKI treatment, and we found that TILs were significantly increased in TKI treated mice (Fig. 6B). We proposed that additional pro-IL-12 treatment could work together with enhanced TIL responses from TKI therapy to treat cold tumors in which monotherapy was insufficient. We compared pro-IL-12 or TKI alone to TKI combined with pro-IL-12 by administering TKI every four days by gavage and pro-IL-12 every two days by i.p injection. We observed that the combination treatment could effectively control tumor growth, prevent tumor relapse, and prolong the survival of mice, compared to both TKI and pro-IL-12 alone (Fig. 6C and 6D). The effect of this combined treatment was abolished by T cell depletion (Fig. 6C and 6D). Oncogene-driven tumors often resist immunotherapy. Our data suggested that pro-IL-12 could overcome resistance to TKI targeted therapy in patients with advanced EGFR/HER2-dependent tumors in a T-cell dependent manner.
Fig. 6. Pro-IL-12 in combination with TKI and anti-PD-L1 anti-tumor therapies.
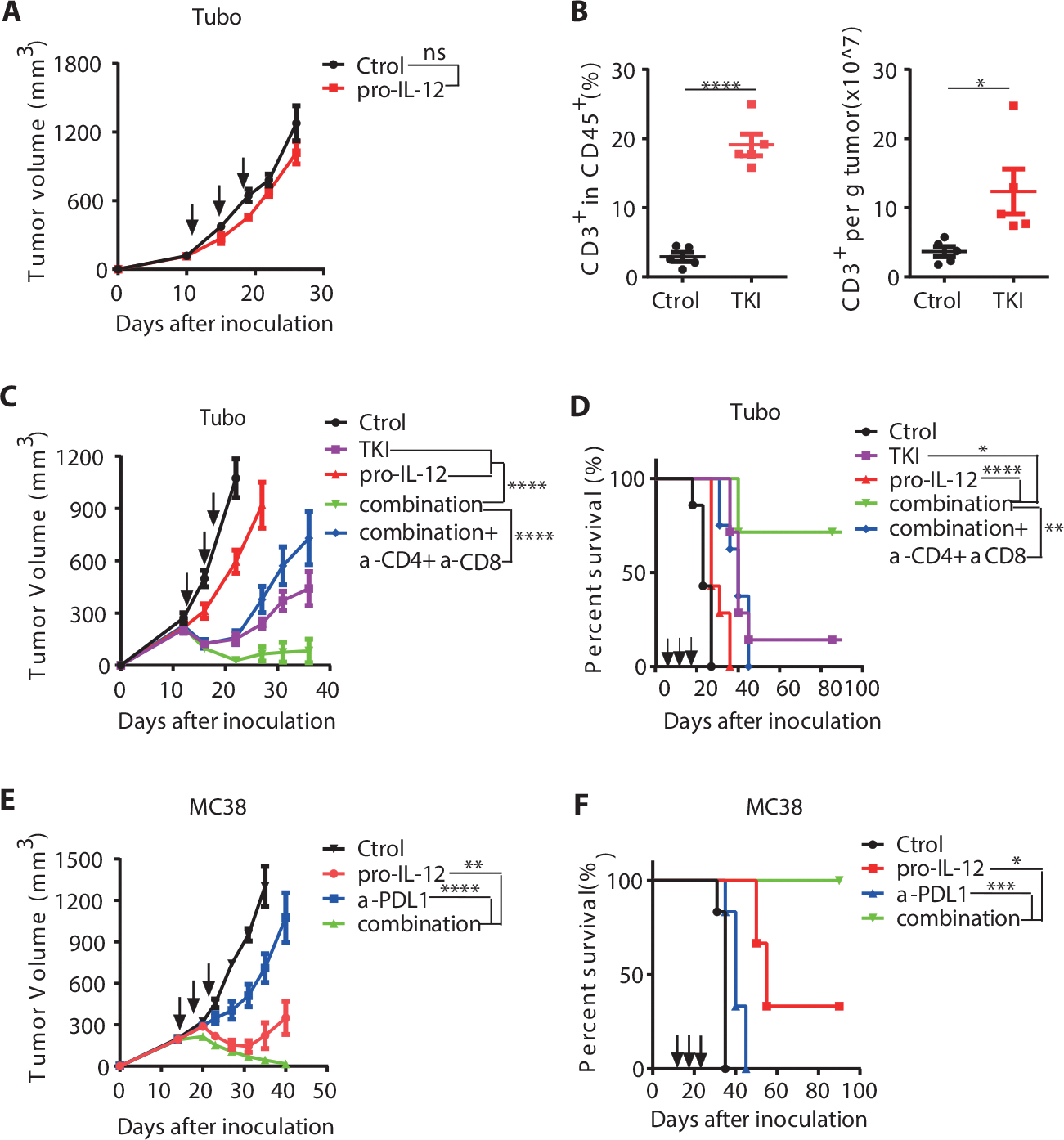
(A) Tumor growth curves of BALB/C mice (n=7/group) that were inoculated with 5×105 TUBO cells and then treated with PBS or 3.33pmol pro-IL-12 by i.p. injection on days 12, 15, 18. Tumor growth was measured twice per week.
(B) Absolute number and percent of CD3+ T cells within tumors of BABL/C mice inoculated with 5×105 TUBO cells and then treated with 1mg of TKI through oral gavage on days 13 and 18. Total T cell ratio and number were detected by flow cytometry six days after the first TKI treatment (n=5/group). Tumor growth was measured at the indicated time points for TKI treatment in (Supplementary Fig. 8A).
(C-D) Tumor growth curves of BABL/C mice (n=7/group) inoculated with 5×105 TUBO cells and then treated with PBS, TKI, pro-IL-12, or both. 1mg of TKI was given through oral gavage on days 13 and 18, whereas 6.67 pmol of pro-IL-12 was injected i.p. on days 13, 16, 19. An additional combination treatment group (n=8) was given 200 μg of depleting anti-CD8 and anti-CD4 antibody by i.p. injection alongside pro-IL-12 treatment every 3 days for a total of three doses. Tumor size was measured twice per week (C), and the mouse survival curve was shown as (D).
(E-F) Tumor growth curves of C57BL/6N mice (n=6/group) inoculated with 5×105 MC38 cells and then treated with PBS, pro-IL-12, anti-PDL1, or both. 3.33pmol of Pro-IL-12 was injected i.p. on days 15, 18, and 21, whereas 200μg of anti-PDL1 was injected i.p. on days 15, 19, and 23. Tumor size was measured twice per week (E), and the mouse survival curve was shown as (F).
Data indicate mean ± SEM and are repeated two or three independent experiments. Statistical analysis for B was performed using unpaired two-tailed t-tests. For A, C and E, a two-way analysis of variance (ANOVA) was applied. Statistical analysis for the survival curve data was performed using log-rank tests.*P <0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Pro-IL-12 combined with anti-PD-L1 to induce robust tumor regression
Immune checkpoint blockades (ICBs) such as anti-PD-1/PD-L1 are well recognized for their efficacy against a broad range of tumors, but many patients develop the resistance even after initial responses. There is an urgent need to overcome both primary and secondary resistance. Like many clinical responders, mice bearing advanced MC38 tumors partially respond to but ultimately develop resistance to ICB therapy. One possible explanation for why ICB alone is insufficient to control tumor growth is that simply removing suppression may not restore sufficient immunity against the tumor, and additional T cell cytokines are needed to sustain antitumor immunity. Given the impressive ability of pro-IL-12 to enhance tumor-specific T cell responses, we evaluated whether pro-IL-12 therapy could improve the function of ICB. IL-12 could induce IFN that increases PD-L1 on tumor tissues to regulate T cell function negatively. Therefore, we hypothesized that anti-PD-L1 could be improved with pro-IL-12.
To detect whether PD-L1 expression was increased in tumor tissue after pro-IL-12 treatment, we isolated and analyzed tumor cells (CD45−), DCs (CD11C+ MHCII+), MDSCs (CD45+ CD11b+ Gr-1+), and macrophages (CD45+ CD11b+ F4/80+) within the TME. We observed the upregulation of PD-L1 in all of these cells in the pro-IL-12 treated mice compared to the control group (Supplementary Fig. 9B and 9C). This result indicated that pro-IL-12 promoted tumor-specific T cell responses, accompanied by increased PD-L1 expression in the TME. To determine whether pro-IL-12 could prevent tumor relapse after anti-PD-L1 treatment, we inoculated mice with MC38 and treated them with pro-IL-12 every two days, anti-PD-L1 every three days, or both by i.p. injection. We observed that the combination of pro-IL-12 with anti-PD-L1 resulted in complete tumor rejection and prolonged survival of mice, which was superior to either treatment alone (Fig. 6E and 6F). These data thus demonstrated that pro-IL-12 could overcome resistance to ICB and that the combination of PD-L1 blockade therapy with pro-IL-12 led to maximal antitumor activity. Overall, these data revealed the therapeutic potential of pro-IL-12 for advanced tumors in combination with anti-PD-L1 treatment in the clinic.
Human pro-IL-12 exhibits the same characteristics as mouse pro-IL-12
Mouse IL-12 is structurally similar to human IL-12, but the two molecules are not identical. To determine whether our pro-IL-12 design is translatable to human use, we first created a human hetero-IL-12-Fc (hu-het-IL-12) using human IL-12 subunits. When administered to human peripheral blood mononuclear cells (PBMCs), hu-het-IL-12 induced high levels of IFNγ production, similar to mouse het-IL-12-Fc (Supplementary Fig. 10A). Importantly, CD8+ PBMCs alone produced much higher levels of IFNγ than CD4+ PBMCs alone (Supplementary Fig. 10A), mimicking our data with mouse splenocytes. We then created an analogous pro-IL-12 using human IL12Rβ1 (hu-pro-IL-12) and tested its activity in vitro using Hek-Blue™ IL-12 reporter cells (Supplementary Fig. 10B). Hu-pro-IL-12 had approximately 4-fold less activity than both recombinant human IL-12 and hu-het-IL-12, and its activity was fully restored after incubation with recombinant MMP14. Concerned that the blocking efficiency was lower than that of murine pro-IL-12, we also generated a human pro-IL-12 using ECD from both human IL12Rβ1 and IL12Rβ2 (R1/R2-hu-pro-IL12). R1/R2-hu-pro-IL-12 had a 32-fold lower activity than both recombinant human IL-12 and hu-het-IL12, and its activity was still rescued after incubation with recombinant MMP14 (Supplementary Fig. 10C). Using a humanized mouse model, we looked to see if human pro-IL-12 could induce human immune cells to reject human tumors. We inoculated immunodeficient NSG mice with the immunogenic HCT116-cGAS human colon cancer cell line, reconstituted them i.v. with human cord blood mononuclear cells, and treated them with three equimolar doses of hu-IL12-Fc, R1/R2-hu-pro-IL-12, or PBS control. Compared to the untreated control, the IL12-Fc and prodrug treated groups showed a significant reduction in tumor size, suggesting antitumor immune activation (Supplementary Fig. 10D). Although both IL-12-treated groups displayed treatment-related body weight loss, the prodrug-treated group recovered within 10 days of the final dose, whereas the IL12-Fc group remained low body weight during the entire experiment. Altogether, human pro-IL-12 showed similar blocking/restoration characteristics as murine pro-IL-12, induced IFNγ from the same cell types, and successfully mediated tumor rejection, giving further support to the clinical viability of our design.
Discussion
IL-12 is a potent inducer of cell-mediated immunity that can stimulate the effector functions of activated T cells and NK cells to reject solid tumors. The release of cytotoxic enzymes and cytokines, mainly IFNγ, from T and NK cells mediates a favorable immune phenotype in the TME and is reportedly required for maximal antitumor efficacy (3–5). However, clinical administration of IL-12 has been limited due to its short half-life, low efficacy, and dose-limiting systemic toxicity (12, 13). Our study developed a tumor-conditional pro-IL-12 to overcome the limitations of current IL-12 based therapies and provided a framework for future pro-cytokine designs. We first generated a recombinant IL-12-Fc fusion protein to extend the in vivo half-life of IL-12. From that structure, we further engineered a pro-IL-12 whose active site was blocked by IL-12’s natural extracellular receptor binding domains. The bioactivity of pro-IL-12 could be recovered when an MMP cleavable peptide linker was cut by tumor-enriched MMP14. We observed that pro-IL-12 significantly reduced serum inflammatory cytokines, did not incur body weight loss, and generated a sustained proinflammatory environment within the tumor. Systemic treatment with pro-IL-12 resulted in effective tumor control and prolonged mouse survival in three separate tumor models. Mechanistically, this next-generation IL-12 acted directly on preexisting, intratumoral, tumor-specific CD8+ T cells causing them to release IFNγ within the TME. Pro-IL-12 could improve outcomes when combined with both TKI targeted therapy and ICB therapy, suggesting that it might be used in tandem with existing therapies to improve response rates in challenging patients.
Until now, the most pressing barrier preventing IL-12 from reaching clinical viability is its severe toxicity. The frequent systemic and subcutaneous administration of recombinant IL-12 has shown dose-dependent severe adverse events resulting from binding to peripheral receptors on circulating immune cells (46). Fc-fused IL-12 extended half-life in vivo and induced high levels of serum inflammatory factors. This outcome is consistent with previously reported clinical toxicity caused by rapidly increased proinflammatory cytokines, such as IFNγ, TNFα, and IL-6 (54). Local delivery of pleiotropic cytokines is one way to avoid the overwhelming “cytokine storm” while still preserving antitumor effector mechanisms. In fact, intratumor injection of IL-12 can be remarkably more effective for tumor rejection and generation of antitumor memory immune responses (6–11). Currently, most attempts at generating a tumor-targeting IL-12 are immune-cytokines that couple IL-12 to antibodies against tumor-associated antigens (14–16). However, these immune-cytokines are limited by the specificity of their antibody targets and could also bind to the same antigen expressed on non-tumor cells, causing off-target toxicity. Some immune-cytokines have been found in all tissues in mice 2–3 days after i.v. administration (11). Additionally, immune-cytokines can be consumed in the periphery before reaching the TME due to competing interactions with high affinity receptors of cytokines on various circulating immune cells (17). To overcome these weaknesses, we constructed a next-generation pro-IL-12, whose binding to receptor-expressing cells could be spatially manipulated to prevent peripheral inflammation while preserving potent antitumor effects.
Our pro-IL-12 was preferentially cleaved and activated by MMP14 enriched in tumors, allowing precise IL-12 activity inside tumors with limited systemic toxicity. Unlike similar studies with pro-antibodies that utilize synthetic peptides to block antibody bioactivity (26, 55–57), pro-IL-12 used its natural receptor binding domain to cover IL-12 reversibly and avoided host immunogenicity. After incubation with MMP14 in vitro, pro-IL-12 was almost completely cleaved, and IL-12 activity was significantly recovered. In vivo, pro-IL-12 had significantly reduced serum inflammatory cytokines and negligible weight loss compared to het-IL-12-Fc when administered to mice. Finally, pro-IL-12 maintained potent antitumor effects with increased quantity and activity of tumor-specific T cells within the TME, leading to the tumor clearance.
Locally administered IL-12 has been previously reported to reverse the tumor immunosuppressive microenvironment. This reversion can be through converting suppressive TAMs to cytotoxic macrophages (58) or altering the suppressive activities of tumor-associated MDSC (59). By depleting different subtypes of immune cells in the MC38 mouse colorectal cancer model, we found that the therapeutic effect of pro-IL-12 also did not require NK cells or CD4+ T cells but depended on CD8+ T cells. Mechanistically, IL-12 is thought to indirectly act on CTLs through Th1 polarization of CD4+ T cells. While the role IL-12 plays in Th1 polarization has been relatively well studied (47), the relative importance of IL-12 direct signaling in CD8+ T cells is still not fully understood. Surprisingly, we found that IL-12 binding to CD4+ T cells only had a minor contribution to IFNγ levels and was not necessary for complete tumor control. In contrast, direct binding of pro-IL-12 to CD8+ T cells was essential for maximal IFNγ production and antitumor efficacy. Therefore, IL-12 signaling on CD8+ T cells was both necessary and sufficient for tumor clearance after treatment with pro-IL-12.
CD8+ T cell infiltration and activation within the tumor are strongly correlated with better prognosis in the clinic and preclinical tumor models (50). Some oncogene-driven cancers with poor T cell infiltration do not respond to immunotherapy but are sensitive to targeted therapy (50, 51, 60). ICB has become the major clinically utilized immunotherapy, but most patients fail to achieve complete tumor-free survival (61–63). We observed that IL-12 could rejuvenate TILs to and work in tandem with ICB therapy.
Various strategies have been attempted to improve IL-12 antitumor efficacy with reduced toxicity. For example, in current clinical trials, intra-tumor delivery of IL-12 via plasmid or viral vectors has demonstrated that locally delivered IL-12 can generate systemic adaptive immune responses, activate or reactivate tumor-infiltrating CD8+ T cells, and lead to primary tumor control and inhibition of metastases (6–11). The advantage of our approach was that we could utilize systemic delivery methods while still achieving a tumor-specific local response, allowing for the treatment of poorly accessible tumors.
In conclusion, we developed a next-generation IL-12 pro-cytokine with a superior half-life, limited peripheral toxicity, tumor-targeted activation, and potent antitumor efficacy compared to recombinant IL-12. With its ability to increase and activate tumor-specific CTLs within the tumor, pro-IL-12 could treat tumors in various settings. Moreover, its design was highly modular and easily usable as a framework for other cytokines and engineered fusion proteins. Overall, pro-IL-12 therapy improved upon nearly all limitations of existing IL-12 designs, holding high translational promise for antitumor immunotherapy.
Materials and Methods
Study Design:
This study aimed to explore whether and how optimizing next-generation IL-12 could treat tumors while limiting toxic side effects. Mouse tumor models were used to assess the anti-tumor efficacy of IL-12-Fc and pro-IL-12 by tumor growth and survival, and toxicity by body weight loss, inflammatory cytokine production, and histology. We produced IL-12-Fc and pro-IL-12 as a comparison using the 293F expression system. The tumor-bearing mice were assigned to different groups by tumor size (the average size is similar among different groups). The sample size is specified in each figure legend, and samples were not blinded or randomized during experiments or analysis.
Mice
Female (6–10-weeks-old) BALB/c and C57BL/6N mice were purchased from Vital River Laboratories (Beijing, China) or The Jackson Laboratory (Bar Harbor, Maine). C57BL/6N Rag1 KO mice were purchased from the model animal research center of Nanjing University or The Jackson Laboratory. C57BL/6N IL12Rβ2 KO and NSG mice were purchased from The Jackson Laboratory. All mice were maintained under specific pathogen-free conditions in the animal facilities of the Institute of Biophysics and the University of Texas Southwestern Medical Center. Animal care and experiments were carried out under institutional protocol and guidelines. All studies were approved by the Animal Care and Use Committee of the Institute of Biophysics and the University of Texas Southwestern Medical Center.
Tumor inoculation and treatments
5×105–1×106 of MC38, 7–8×105 of MC38-OVA, 2.5–5×105 of B16-F10, 5×105 of TUBO cells, or 4×106 of HCT116-cGAS were injected subcutaneously into the right flank of 6–10-week-old mice. 1.5×105 of 4T1 cells were injected into the mammary fat pad. Tumor volumes were measured twice a week and calculated by length× width× height/ 2. After the tumor was established, mice were treated with three intraperitoneal injections PBS, 3.33pmol of hetero-IL-12-Fc, homo-IL-12-Fc, and pro-IL-12, respectively, every two days for three times unless otherwise indicated. To block lymphocyte trafficking, mice were injected intravenously with 25 μg of FTY720 one day before pro-IL-12 treatment. For the 4T1 tumor model, the primary tumor was surgically removed on days 16, 3.33pmol pro-IL-12 was given post-surgery. For depletion of CD8+ T cells and CD4+ T cells, 200 μg of anti-CD8 Ab (TIB210) or anti-CD4 Ab (GK1.5) was injected i.p. beginning with pro-IL-12 treatment every 3 days for a total of three doses. Anti-NK1.1 Ab (PK136) was injected i.p. at a dose of 400 μg, beginning with pro-IL-12 treatment every 3 days for a total of three doses. Anti-Ly-6G Ab (1A8) was injected i.t. at a dose of 50μg one day before pro-IL-12 treatment every 3 days for three doses in total. Clophosome (100μl/mouse) was injected one day before pro-IL-12 treatment every 3 days for a total of two doses. Anti-IFNγ (XMG1.2) was i.p. injected 500 μg/mouse one day before pro-IL-12 treatment. For the IL12Rβ2 KO adoptive transfer experiments, spleens were extracted from donor mice, homogenized, treated with ACK RBC lysis buffer, and sorted using EasySep™ Mouse CD8+ (19853, STEMCELL) or CD4+ (19852, STEMCELL) T Cell Isolation Kit according to manufacturer’s protocol. ~7×106 CD4+ and ~3.5×106 CD8+ cells were then transferred via i.v. tail vein injection to each recipient mouse 6 days before MC38 inoculation. For humanized mouse experiments, ~8×106 human cord blood mononuclear cells were transferred via i.v. retro-orbital injection 7 days after HCT116-cGAS inoculation.
Production and characterization of IL-12 prodrug fusion proteins
For both human and mouse IL-12, the p35 and p40 were fused to the N-terminus of hIgG-Fc via a (G4S)3 linker, respectively, to generate the hetero-IL-12-Fc. Three repeats of G4S were used to establish conformational flexibility (64) for heterodimer p35-p40 assembly. The heterodimerization of p35 and p40 was facilitated by the knobs-into-holes design of Fc, which technique was reported previously (65). P40 was fused to the “Knobs” (T366W) Fc (CH2CH3), and p35 was linked to the “Holes” (T366S, L368A, and Y407V) Fc. For the expression of hetero-IL-12-Fc, two plasmids, one encoding p35-(G4S)3 linker-hole Fc and the other encoding p40-(G4S)3 linker-knob Fc, were transiently co-transfected in HEK293F cells. For the expression of homo-IL-12-Fc, one plasmid in the format of p40-(G4S)3 linker-p35-(G4S)3 linker-WT human IgG Fc was transfected into HEK293F cells. For the generation of pro-IL-12, two plasmids, one encoding p35-(G4S)3 linker or R2-ECD-linker-p35-(G4S)3) linker-hole Fc and the other encoding R1 ECD-linker-p40-(G4S)3 liner-knob Fc were inserted into pEE12.4 vector (Lonza). Both plasmids were transiently co-transfected in HEK293F cells to form either pro-IL12 with R1 or pro-IL12 with R1 and R2. The linker between ECD and p35 or p40 subunits is a 10-amino acid protease-cleavable substrate (SGRENIRTA (33)), which can be cleaved by MMP14 and is flanked by flexible G4S peptides on both sides. Pro-IL-12-Nsub is identical to other designs of pro-IL-12, except SGRENIRTA is replaced with G4S. Supernatants were collected on day 7 after transfection. The fusion protein was purified using a Protein A-Sepharose column according to the manual (Repligen Corporation) and analyzed by reducing and non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Quantitative biodistribution studies
Biodistribution of pro-IL-12 in various tissues after 2.4 μg pro-IL-12 was i.p. injected into MC38 tumor-bearing mice. ELISA was performed to quantify the amount pro-IL-12 in each homogenate, normalized by total tissue weight. Human Fc was used as a biomarker in the ELISA, which would not detect endogenous mouse IL-12.
Fluorescence imaging
Fluorescent dyes with maleimide active groups can be coupled to biological molecules with sulfhydryl groups such as polypeptides, proteins or small molecules. Pro-IL-12 was labeled with Cy5.5 and purified by washing away the unbound Cy5.5. 10 μg fluorescently-labeled pro-IL-12 was then injected via tail vein into C57BL/6 mice bearing subcutaneous MC38 tumors. The accumulation of Cy5.5-labeled protein within tumors was measured by the in vivo imaging system (IVIS) Spectrum (PerkinElmer). The fluorescence imaging data were analyzed by Living Image software (PerkinElmer).
Cell lines and reagents
Invitrogen™ FreeStyle™ 293-F Cells (R79007) were grown in SMM 293-TI medium (M293TI, Sino Biological) or EX-CELL® 293 Serum-Free Medium (14571C, Sigma Aldrich). B16-F10, MC38, and 4T1 cells were purchased from ATCC. MC38-OVA was selected from single-cell clones after being transduced by lentivirus-expressing the OVA. TUBO was cloned from a spontaneous mammary tumor in a BALB/c Neu-transgenic mouse (66). HCT116-cGAS was generated as previously described. TUBO, MC38, B16-F10 were cultured in 5% CO2 and maintained in vitro in Dulbecco’s modified Eagle’s medium, supplemented with 10% heat-inactivated fetal bovine serum, 2mmol/l L-glutamine, 0.1mmol/l Minimum Essential Medium nonessential amino acids, 100U/ml penicillin, and 100mg/ml streptomycin. 4T1 cells, purified mouse splenocytes, and human PBMCs (70025, STEMCELL) were maintained in vitro in complete RPMI 1640 medium, supplemented with 10% heat-inactivated fetal bovine serum. Hek-Blue™ IL-12 cells require two kinds of cell culture medium. The growth medium is the complete DMEM with 100 mg/ml Normocin™, and the test medium is the growth medium with extra HEK-Blue™ Selection. All cell lines were routinely tested using a mycoplasma contamination kit (R&D). Anti-CD8 Ab (TIB210), FcγRII/III blocking Ab (2.4G2), anti-CD4 Ab (GK1.5), and anti-NK1.1 Ab (PK136), anti-IFNγ Ab (XMG1.2), anti-Ly-6G Ab (1A8), and Anti-PD-L1 Ab (10F.9G2) were purchased from Bio X Cell (USA). The TKI-Afatinib was purchased from Shanghai Bojing Chemical Co., Ltd FTY720 was purchased from Sigma.
Flow cytometric analysis
Tumor tissues were collected, cut into small pieces, and re-suspended in digestion buffer (RPMI-1640 medium with 1 mg/ml type IV collagenase and 100 μg/ml DNase I). Tumors were digested for 45 min at 37 °C, then passed through a 70μm cell strainer to make single-cell suspensions. Cells suspended in FACS buffer (1% bovine serum albumin and 0.05% NaN3) were blocked with anti-CD16/32 Ab (anti-FcγIII/ II receptor, clone 2.4G2) for 30 min (Supplementary Fig. 11F) and then stained with specific antibodies for 30 min on ice. For intracellular IFNγ, KI67, Foxp3 staining, samples were fixed, permeabilized, and stained with anti-mouse IFNγ, anti-mouse KI67 or anti-mouse Foxp3. DAPI or LIVE/DEAD™ fixable yellow dye (ThermoFisher) was used to exclude dead cells. Samples were analyzed on a FACSCalibur or Fortessa flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (TreeStar).
Measurement of IFNγ-secreting T cells by ELISPOT assay
Draining lymph nodes (DLNs) from tumor-bearing mice were isolated, and single-cell suspensions were prepared. Cells isolated from draining lymph nodes were co-cultured with 5μg/ml OTI or negative control peptide for 48 hours. The IFNγ production was determined with an IFNγ ELISPOT assay kit according to the manufacturer’s protocol (BD Biosciences). The cytokine spots were enumerated with the Immuno-Spot Analyzer (CTL).
In vitro digestion conditions for pro-IL-12
Recombinant hMMP-14 (R&D Systems) was activated by rhFurin in an Activation Buffer (50 mM Tris, 1 mM CaCl2, 0.5% (w/v) Brij-35, pH 9.0) at 37 °C for 2 hours. Pro-IL-12 was co-cultured with activated rhMMP14 in an Assay Buffer (50 mM Tris, 3 mM CaCl2, 1 μM ZnCl2, pH 7.5) at 37 °C overnight. Protein cleavage was confirmed by Western-Blot analysis. Pro-IL-12 was only pre-treated with protease for in vitro cleavage tests and not at any other point.
Hek-Blue™ IL-12 reporter assay
Hek-Blue™ IL-12 reporter cell line was purchased from InvivoGen. These cells were generated by stably introducing the human genes of the IL-12 receptor and IL-12 signaling pathway into HEK293 cells. For detecting IL-12 biological activity, Hek-Blue™ IL-12 reporter cells were incubated overnight at 37°C with different IL-12 fusion protein samples (hetero-IL-12-Fc, homo-IL-12-Fc, or pro-IL-12) serial-diluted 5-fold from 13.3μM. The levels of secreted embryonic alkaline phosphatase (SEAP) in cell culture supernatants were determined using a spectrophotometer at 630–650 nm.
Measurement of IFNγ by ex vivo/in vitro stimulation
Spleens were extracted from mice, homogenized, and treated with ACK RBC lysis buffer to collect mouse splenocytes. Cells were then sorted by FACS using a FACSMelody™ (BD) with antibodies against CD45 (30-F11), CD3Ɛ (145–2C11), CD4 (GK1.5), CD8α (53–6.7), and NK1.1 (PK136) or by MACS using EasySep™ Mouse CD8+ or CD4+ T Cell Isolation Kits (STEMCELL). 1–2×104 sorted cells were then incubated on 96-well u-bottom plates pre-coated with anti-CDƐ (145–2C11) antibody and cultured in complete RPMI containing 0.01–0.1μg IL-12-Fc or an equivalent volume of PBS. After 48 hours incubation, IFNγ was measured by CBA using Mouse IFNγ Flex Set (558296, BD) according to the manufacturer’s protocol. For human PBMCs, cells were sorted by MACS using EasySep™ Human CD8+ (17953, STEMCELL) or CD4+ (17952, STEMCELL) T Cell Isolation Kits according to manufacturer’s protocol. 1×104 sorted cells were then incubated on 96-well u-bottom plates in complete RPMI with ImmunoCult™ Human CD3/CD28 T Cell Activator (10971, STEMCELL), adding either 0.1μg of hu-IL-12-Fc or an equivalent volume of PBS. After 48 hours incubation, IFNγ was measured by R&D Human IFN-gamma DuoSet™ ELISA Kit (DY285B, Fisher) according to the manufacturer’s protocol.
Patient data analysis
An online database Kaplan–Meier plotter (http://bioinfo.henu.edu.cn/DatabaseList.jsp) was used to generate the survival plot (67–69).
Statistical analysis
All analyses were performed using GraphPad Prism statistical software (GraphPad Software Inc., San Diego, CA). A two-way analysis of variance (ANOVA) was used to analyze tumor growth, with post hoc Tukey’s multiple comparison to compare main column effect or Sidak’s multiple comparisons test to compare each column effect if significant. Survival analysis was done using Log-rank test. All the other data were analyzed using unpaired two-tailed t-tests or one-way ANOVA with Tukey’s multiple comparisons. A value of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Kaiting Yang, Eric Hsu, Zhichen Sun, Yong Liang, Hairong Xu, Jiao Shen, and Huan Xia for providing experiment materials and helpful discussions. We thank the faculties in the animal facility of the Institute of Biophysics, Chinese Academy of Science, and UT Southwestern Medical Center.
Funding:
National Key R&D Program of China grant 2018ZX10301–404 National Institutes of Health, Integrative Immunology Training Grant 2T32AI005285–41A1. National Cancer Institute of the National Institutes of Health, National Research Service Award (NRSA) 1F30CA254023–01A1. This work was also supported by Cancer Prevention and Research Institute of Texas (CPRIT) grant RP180725 given to Yang-Xin Fu.
Footnotes
Conflicts of interest Statement
D.X, H.P, and Y-X.F. are inventors on patent application CN 110396133A related to pro-IL12 that is assigned to Immune Targeting, Inc. Y.W. is an employee of Immune Targeting, Inc., which provided human pro-IL12 protein. The other authors declare that they have no competing interests.
Data and materials availability
All data associated with this study are present in the paper or the Supplementary Materials. The materials that support the findings of this study are available from the corresponding authors on reasonable request. Requests for the mouse or human versions of pro-IL-12 should be directed to Dr. Hau Peng as the primary contact.
Reference and notes
- 1.Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu C-Y, Gately MK, Gubler U, A functional interleukin 12 receptor complex is composed of two β-type cytokine receptor subunits. Proceedings of the National Academy of Sciences 93, 14002–14007 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu C.-y., Ferrante J, Gately MK, Magram J, Characterization of IL-12 receptor beta1 chain (IL-12Rbeta1)-deficient mice: IL-12Rbeta1 is an essential component of the functional mouse IL-12 receptor. The Journal of Immunology 159, 1658–1665 (1997). [PubMed] [Google Scholar]
- 3.Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, Kulig P, Becher B, New insights into IL-12-mediated tumor suppression. Cell Death Differ 22, 237–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trinchieri G, Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3, 133–146 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Vignali DAA, Kuchroo VK, IL-12 family cytokines: immunological playmakers. Nature Immunology 13, 722–728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson-Abelson MR, Purohit VS, Pang WM, Iyer V, Odunsi K, Demmy TL, Yokota SJ, Loyall JL, Kelleher RJ Jr, Balu-Iyer S, IL-12 delivered intratumorally by multilamellar liposomes reactivates memory T cells in human tumor microenvironments. Clinical Immunology 132, 71–82 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahvi D, Henry M, Albertini M, Weber S, Meredith K, Schalch H, Rakhmilevich A, Hank J, Sondel P, Intratumoral injection of IL-12 plasmid DNA–results of a phase I/IB clinical trial. Cancer gene therapy 14, 717–723 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Núñez N, Stowe CL, Friebel E, Nannini F, Rindlisbacher L, Roberts TA, Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nature Communications 12, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bramson JL, Hitt M, Addison CL, Muller WJ, Gauldie J, Graham FL, Direct intratumoral injection of an adenovirus expressing interleukin-12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum Gene Ther 7, 1995–2002 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB, In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer research 60, 3832–3837 (2000). [PubMed] [Google Scholar]
- 11.Sharifi J, Khawli LA, Hu P, King S, Epstein AL, Characterization of a phage display-derived human monoclonal antibody (NHS76) counterpart to chimeric TNT-1 directed against necrotic regions of solid tumors. Hybridoma and hybridomics 20, 305–312 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Cohen J, Clinical-Trials - Il-12 Deaths - Explanation and a Puzzle. Science 270, 908–908 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Ryan JL, Effects of single-dose interleukin-12 exposure on interleukin-12 associated toxicity and interferon-γ production. Blood 90, 2541–2548 (1997). [PubMed] [Google Scholar]
- 14.Pasche N, Wulhfard S, Pretto F, Carugati E, Neri D, The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clinical cancer research : an official journal of the American Association for Cancer Research 18, 4092–4103 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, Lan Y, Sabzevari H, Schlom J, Greiner JW, The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, Williford J-M, Alpar AT, Raczy MM, Gray LT, Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nature biomedical engineering 4, 531–543 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD, Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proceedings of the National Academy of Sciences 112, 3320–3325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Y, Liu X, Chen Z, Zhu J, Xiong Y, Li K, Dong J, Li X, Interaction between Cancer Cells and Stromal Fibroblasts Is Required for Activation of the uPAR-uPA-MMP-2 Cascade in Pancreatic Cancer Metastasis. 13, 3115–3124 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Hojilla CV, Mohammed FF, Khokha R, Matrix metalloproteinases and their tissue inhibitors direct cell fate during cancer development. British Journal of Cancer 89, 1817–1821 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deryugina EI, Quigley JP, Matrix metalloproteinases and tumor metastasis. Cancer metastasis reviews 25, 9–34 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Kai K, Plaks V, Werb Z, Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 141, 0–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu LS, Chen L, Ding WX, Li K, Wu JJ, Elevated expression of both MDR1 and MMP-2 genes in metastasized lymph node of invasive ductal breast cancer. Eur Rev Med Pharmacol Sci 16, 2037–2043 (2012). [PubMed] [Google Scholar]
- 23.Hofmann UB, Westphal JR, van Muijen GN, Ruiter DJ, Matrix metalloproteinases in human melanoma. Journal of Investigative Dermatology 115, 337–344 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Gurgel DC, Valenca-Junior JT, Dornelas CA, Vieira RB, Maia-Filho JT, Lima-Junior RC, Ribeiro RA, Almeida PR, Immunoexpression of metalloproteinases 2 and 14 and TIMP-2 inhibitor in main types of primary gastric carcinomas and lymph node metastasis. Pathology oncology research : POR 21, 73–81 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Garcia MF, Gonzalez-Reyes S, Gonzalez LO, Junquera S, Berdize N, Del Casar JM, Medina M, Vizoso FJ, Comparative study of the expression of metalloproteases and their inhibitors in different localizations within primary tumours and in metastatic lymph nodes of breast cancer. Int J Exp Pathol 91, 324–334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, Hall SS, Ruoslahti E, Daugherty PS, Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. Journal of controlled release : official journal of the Controlled Release Society 161, 804–812 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Autio KA, Boni V, Humphrey RW, Naing A, Probody therapeutics: an emerging class of therapies designed to enhance on-target effects with reduced off-tumor toxicity for use in immuno-oncology. Clinical Cancer Research 26, 984–989 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bremer C, Tung CH, Weissleder R, In vivo molecular target assessment of matrix metalloproteinase inhibition. Nature Medicine 7, 743–748 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Matrisian LM, Near-Infrared Optical Proteolytic Beacons for In Vivo Imaging of Matrix Metalloproteinase Activity. Methods in Molecular Biology 622, 279–304 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie BW, Mol IM, Keereweer S, van Beek ER, Que I, Snoeks TJ, Chan A, Kaijzel EL, Lowik CW, Dual-wavelength imaging of tumor progression by activatable and targeting near-infrared fluorescent probes in a bioluminescent breast cancer model. PloS one 7, e31875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turunen SP, Tatti-Bugaeva O, Lehti K, Membrane-type matrix metalloproteases as diverse effectors of cancer progression. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1864, 1974–1988 (2017). [DOI] [PubMed] [Google Scholar]
- 32.McGowan P, Duffy M, Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Annals of oncology 19, 1566–1572 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Kridel SJ, Sawai H, Ratnikov BI, Chen EI, Li W, Godzik A, Strongin AY, Smith JW, A Unique Substrate Binding Mode Discriminates Membrane Type-1 Matrix Metalloproteinase from Other Matrix Metalloproteinases. J Biol Chem 277, 23788 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, Atkins MB, Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-γ induction is associated with clinical response. Clinical Cancer Research 6, 1678–1692 (2000). [PubMed] [Google Scholar]
- 35.Pulaski BA, Ostrand-Rosenberg S, Mouse 4T1 breast tumor model. Current protocols in immunology 39, 20.22. 21–20.22. 16 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Smyth MJ, M., Taniguchi SEA Street, The Anti-Tumor Activity of IL-12: Mechanisms of Innate Immunity That Are Model and Dose Dependent. J Immunol 165, 2665–2670 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Ursa LT, Luisa L, Maja C, Urska K, Michele FV, Gregor S, Emanuela S, Gene Electrotransfer of Plasmid-Encoding IL-12 Recruits the M1 Macrophages and Antigen-Presenting Cells Inducing the Eradication of Aggressive B16F10 Murine Melanoma. Mediators of Inflammation 2017, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magdalena JB, Natalia K, Sybilla M, Tomasz C, Jolanta P. a.-P. a., Justyna C, Ryszard S, Daria S, Klaudia K, Stanis?aw S, M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PloS one 13, e0191012- (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zilocchi C, Stoppacciaro A, Chiodoni C, Parenza M, Terrazzini N, Colombo MP, Interferon gamma-independent rejection of interleukin 12-transduced carcinoma cells requires CD4+ T cells and Granulocyte/Macrophage colony-stimulating factor. J Exp Med 188, 133–143 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsung K, Meko JB, Peplinski GR, Tsung YL, Norton JA, IL-12 induces T helper 1-directed antitumor response. The Journal of Immunology 158, 3359–3365 (1997). [PubMed] [Google Scholar]
- 41.Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately M, Antitumor and antimetastatic activity of interleukin 12 against murine tumors. The Journal of experimental medicine 178, 1223–1230 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiba K, FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Therapeut 108, 308–319 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Nakano M, Ito M, Tanaka R, Yamaguchi K, Ariyama H, Mitsugi K, Yoshihiro T, Ohmura H, Tsuruta N, Hanamura F, PD-1+ TIM-3+ T cells in malignant ascites predict prognosis of gastrointestinal cancer. Cancer science 109, 2986–2992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuai W, Xu X, Yan J, Zhao W, Li Y, Wang B, Yuan N, Li Z, Jia Y, Prognostic impact of PD-1 and Tim-3 expression in tumor tissue in stage I-III colorectal cancer. BioMed Research International 2020, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Granier C, Dariane C, Combe P, Verkarre V, Urien S, Badoual C, Roussel H, Mandavit M, Ravel P, Sibony M, Tim-3 expression on tumor-infiltrating PD-1+ CD8+ T cells correlates with poor clinical outcome in renal cell carcinoma. Cancer research 77, 1075–1082 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Trinchieri G, Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 13, 155–168 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A, Interleukin-12: biological properties and clinical application. Clinical Cancer Research 13, 4677–4685 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH, The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annual review of immunology 16, 495–521 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Wu C, Wang X, Gadina M, O’Shea JJ, Presky DH, Magram J, IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J Immunol 165, 6221–6228 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, Xu L, Liang Y, Guo J, Bian Y, A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nature communications 10, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ali H, Provenzano E, Dawson S-J, Blows F, Liu B, Shah M, Earl H, Poole C, Hiller L, Dunn J, Association between CD8+ T-cell infiltration and breast cancer survival in 12 439 patients. Annals of oncology 25, 1536–1543 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer cell 18, 160–170 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z, Han C, Dong C, Shen A, Hsu E, Ren Z, Lu C, Liu L, Zhang A, Timmerman C, Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity. Science immunology 4, (2019). [DOI] [PubMed] [Google Scholar]
- 54.Rubinstein MP, Cloud CA, Garrett TE, Moore CJ, Schwartz KM, Johnson CB, Craig DH, Salem ML, Paulos CM, Cole DJ, Ex vivo interleukin-12-priming during CD8+ T cell activation dramatically improves adoptive T cell transfer antitumor efficacy in a lymphodepleted host. Journal of the American College of Surgeons 214, 700–707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Science translational medicine 5, 207ra144–207ra144 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Pai C-CS, Simons DM, Lu X, Evans M, Wei J, Wang Y.-h., Chen M, Huang J, Park C, Chang A, Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. The Journal of clinical investigation 129, 349–363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puskas J, Skrombolas D, Sedlacek A, Lord E, Sullivan M, Frelinger J, Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology 133, 206–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watkins SK, Egilmez NK, Suttles J, Stout RD, IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. The Journal of Immunology 178, 1357–1362 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Steding CE, Wu S. t., Zhang Y, Jeng MH, Elzey BD, Kao C, The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology 133, 221–238 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong Z-Y, Zhang J-T, Liu S-Y, Su J, Zhang C, Xie Z, Zhou Q, Tu H-Y, Xu C-R, Yan L-X, EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology 6, e1356145 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zou W, Wolchok JD, Chen L, PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jenkins RW, Barbie DA, Flaherty KT, Mechanisms of resistance to immune checkpoint inhibitors. British journal of cancer 118, 9–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Argos P, An Investigation of Oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. Journal of Molecular Biology 211, 943–958 (1990). [DOI] [PubMed] [Google Scholar]
- 65.R. J. B.B., P. L. G., Paul C, ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Engineering, 7 (1996). [DOI] [PubMed] [Google Scholar]
- 66.Rovero S, Amici A, Di Carlo E, Bei R, Nanni P, Quaglino E, Porcedda P, Boggio K, Smorlesi A, Lollini PL, Landuzzi L, Colombo MP, Giovarelli M, Musiani P, Forni G, DNA vaccination against rat Her-2/neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol 165, 5133–5142 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416. e411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan Z, Wang Q, Sun X, Ban B, Lu Z, Dang Y, Xie L, Zhang L, Li Y, Zhu W, OSbrca: a web server for breast cancer prognostic biomarker investigation with massive data from tens of cohorts. Frontiers in oncology 9, 1349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang L, Wang Q, Wang L, Xie L, An Y, Zhang G, Zhu W, Li Y, Liu Z, Zhang X, OSskcm: an online survival analysis webserver for skin cutaneous melanoma based on 1085 transcriptomic profiles. Cancer Cell International 20, 1–8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The materials that support the findings of this study are available from the corresponding authors on reasonable request. Requests for the mouse or human versions of pro-IL-12 should be directed to Dr. Hau Peng as the primary contact.