

Abstract
(4,4'-Bisfluorophenyl)methoxymethyl (BFPM) group of uridine ureido nitrogen shows good relative stability in a variety of chemical transformation reactions for uridine. The BFPM group can be cleaved via 2% of TFA in CH2Cl2 without affecting the Boc group.
Keywords: Uridine, Ureido nitrogen, Diarylmethoxymethyl group, (Bis(4-fluorophenyl)methoxy)methyl group, Chemical transformations, muraymycin synthesis
Graphical Abstract
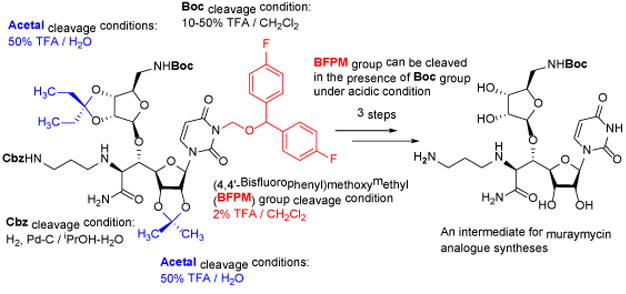
Uridine is a building block of a large number of biologically important natural products.1 Uridine-containing antibiotics, muraymycins and capuramycin, have been of our interest in developing novel antibacterial and anticancer drug leads.2a-k Muraymycin analogues, APPB and APPU, display unique antiproliferative activity with strong anti-metastatic activity against certain solid cancers (e.g., pancreatic and cervical cancers) by targeting N-acetylglucosaminephosphotransferase1 (DPAGT1).2a,b Protection of the uridine ureido nitrogen is essential to achieve the synthesis of uridine-containing complex natural products. Benzyloxymethyl (BOM) group has widely been utilized as a convenient protecting group for the uridine ureido nitrogen.3 However, BOM deprotection of uridine derivatives under hydrogenation conditions often resulted in unpredictable results with over-reduction followed by cleavage of the N-glycoside bond.4 Alternatively, strong Lewis acids are applied, however, acid-sensitive glyosyl bonds are often cleaved during the BOM-deproection steps.4a Thus, we determined that the BOM protecting group is not suitable for systematic synthesis of DPAGT1 inhibitors.2b To this end, we have introduced (2,6-dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)methoxymethyl chloride [1, monomethoxydiphenylmethoxymethyl chlroide (MPPM-Cl) and (2,6-dichloro-4-methoxyphenyl) (2,4-dichlorophenyl) methoxymethyl chloride [2, monomethoxytetrachlorodiphenylmethoxymethyl (MTPM-Cl) for the protection of the uridine ureido nitrogen for synthesis of complex uridine-containing natural products (A in Figure 1).5 MPPM and MTPM groups exhibited excellent stability against a variety of Brønsted and Lewis acids such as 20% TFA, 10% TMSOTf, 10% HCl, 30% HF, 90% AcOH, 30% TsOH, La(OTf)3, BF3•OEt2, and TiCl4 at room temperature, however, these groups could be deprotected with 30% TFA in CH2Cl2.5a The precursors of MPPM and MTPM groups require synthesizing in two chemical steps including Friedel–Crafts and carbonyl reduction reactions. Thus, we have sought a convenient diphenyl methoxymethyl group that can 1) readily be synthesized with commercially available building blocks, and 2) tolerate to a wide range of chemical transformations applied for modifications of uridine under neutral and basic conditions. The other consideration is that deprotections of MPPM and MTPM groups require 30% or higher concentrations of TFA in CH2Cl2. Under these conditions, Boc, silyl, and ketal groups are removed. To synthesize the versatile intermediate E (Figure 1b) for systematic generation of DPAGT1 inhibitors, it is required that the uridine ureido nitrogen protecting group is stable against a series of Lewis and Brønsted acids (e.g., BF3•OEt2, CF3CO2H/H2O, AcOH, Ti(OiPr)4), and can be deprotected without the cleavage of the Boc group. To fulfil these criteria, we rescreened 4,4’-disubstituted diphenylmethoxymethyl groups. Relative stability of 3-((bisphenyl)methoxy)methyl)-1-(6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione derivatives (1-4) against BF3•OEt2 in CH2Cl2, TFA in CH2Cl2, 4N HCl in dioxane, 80% AcOH at 50 °C, and a hydrogenation condition (H2, 10% Pd-C in iPrOH-H2O) is summarized in Table 1.
Figure 1. Acid-cleavable diphenylmethoxylmethyl groups for uridine ureido nitrogen.


a: MPPM and MTPM groups introduced for the syntheses of muraymycin D1 and its analogues (previous works)
b: Reinvestigation of a convenient diphenylmethoxymethyl group to synthesize the intermediate E (this work)
Table 1.
Relative reactivity of differently substituted diphenylmethymethyl protecting groups.

| ||||
|---|---|---|---|---|
| reactivityb╲compound | 1: X, Y = H | 2: X = OMe, Y = F | 3: X = F, Y = F | 4: X = Cl, Y = Cl |
| BF3•OEt2 (10 equiv.) / CH2Cl2, RT | H | H | L | L |
| 1% TFA / CH2Cl2 | H | H | M | L |
| 2% TFA / CH2Cl2 | H | H | H | L |
| 5% TFA / CH2Cl2 | H | H | H | H |
| 10% TFA / CH2Cl2 | H | H | H | H |
| 4N HCl / dioxane | Lc | Hc | Lc | Lc |
| 80% AcOH / H2O 50 °C | M c | M c | Lc | Lc |
| H2, Pd-C / iPrOH-H2O | L | L | L | L |
H (High) indicates the protecting group is readily cleaved.; M (Marginal) indicates that the protecting group may be stable or may be cleaved slowly.; L (Low) indicates that the protecting group is stable.
Recation time: 3 h.
The acetonide group was cleaved.
The (bisphenyl)methoxymethyl and (fluorophenyl)(methoxyphenyl)methoxymethyl groups of uridine acetonide (1 and 2) were readily cleaved under BF3•OEt2 (10 equiv.) in CH2Cl2 and showed acid instability against 1-10%
TFA in CH2Cl2. On the other hand, the (4,4'-bisfluorophenyl)methoxymethyl (BFPM) and (4,4'-bischlorophenyl)methoxymethyl (BCPM) groups were intact under BF3•OEt2 (10 equiv.) in CH2Cl2. These preliminary studies may indicate that BFPM is stable under the chemical transformations (steps 1~7 and 9) and can be deprotected selectively without affecting the Boc group (step 8) in Figure 1b. In contrast, the BCPM group is difficult to remove selectively in the presence of the Boc group(s).
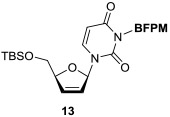
In order to demonstrate utility of the BFPM group as a protecting group for the uridine ureido nitrogen, the BFPM-protected uridine 5 was transformed to a wide range of advanced building blocks that are utilized for the syntheses of bioactive uridine derivatives. Selected examples are summarized in Table 2. Ketal formations of 5 under TsOH•H2O in acetone provided the corresponding 2’,3’-protected derivative 3 in 89% yield (entry 1). The primary TBS group of 6 could be deprotected selectively with 10% HF in CH3CN to furnish 7 without deprotection of the BFPM group (entry 2). The trityl group of 8 was selectively removed by using BF3•OEt2 in the presence of TolSH without deprotection of the BFPM group (entry 3). The secondary alcohol 6 could be oxidized under CrO3•Pyridine•Ac2O in CH2Cl2 to afford 9 in >81% yield. Reduction of the C3’-ketone of 9 with LiBH4 in THF furnished a 1 : 1 mixture of 10α and 10β (6) in 90% yield; in this reduction condition, no carbonyl reductions of the uridine base and cleavage of the BFPM group were observed (entry 5). Selective hydrogenolytic deprotection of the benzyl group of 11 was carried out via 10% Pd-C in iPrOH-water to furnish 3 without over-reduction of the uracil double bond (entry 6). Olefination of the carbonodithioate 12 was achieved by using nBu3SnH and AIBN in toluene at 100 °C to provide 2',3'-didehydro-2',3'-dideoxy derivative 13 in 85% yield, during which cleavage of the BFPM group was not observed (entry 7). Hydrogenation of the 2’,3’-double bond of 13 was also achieved under a kinetically-controlled hydrogenation condition with 10% Pd-C in MeOH to provide the BFPM-protected uridine-2',3'-dideoxy derivative 14 in 93% yield (entry 8). Thus, it was experimentally proved that the BFPM group is a durable protecting group for the uridine ureido nitrogen to synthesize a wide variety of uridine derivatives.
Table 2.
Functionalizations of BFPM-protected uridines.
| Entry | Starting material | Conditions | Product | Yield (%)a |
|---|---|---|---|---|
| 1 |

|
Acetone/TsOH•H2O, rt, 3h |

|
89 |
| 2 |

|
10% HF / CH3CN, rt, 10 min. |
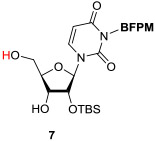
|
83 |
| 3 |

|
BF3•OEt2 (0.5 equiv), TolSH / CH2Cl2, rt, 1h |

|
84 |
| 4 |

|
CrO3•Pyridine•Ac2O / CH2Cl2, rt, 0.5h |
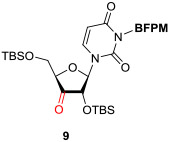
|
>81 |
| 5 |

|
LiBH4 / THF, 0 °c, 1h |

|
90 |
| 6 |

|
H2 (1 atm), 10% Pd-C / iPrOH-watert, 2h |
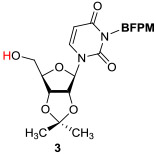
|
88 |
| 7 |

|
nBuSnH, AIBN / toluene, 100 °C, 3h |

|
85 |
| 8 |
|
H2 (1 atm), 10% Pd-C MeOH, 2h |

|
93b |
Product was isolated by SiO2 chromatography.
no over-reduction was observed.
We applied the BFPM group for the synthesis of the muraymycin analogue intermediate 16 (E in Figure 1b). The advanced intermediate 15 was synthesized according to the synthetic scheme established previously (Figure 1b); the synthetic procedures and physical data for the new molecules are summarized in Supporting Information.2c The acetals of 15 were deported with 50% TFA in H2O, under which the Boc and BFPM groups were intact. The BFPM group could be deprotected with 2% TFA in CH2Cl2 within 2h at room temperature without deprotection of the Boc group. Finally, hydrogenation with 10% Pd-C in iPrOH-H2O furnished 16 with 76% overall yield for 3 chemical steps (Scheme 1).
Scheme 1.
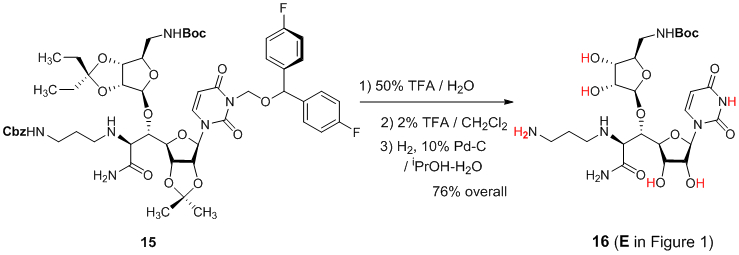
Deprotections of the protecting groups of 15, leading to the advanced intermediate 16.
In conclusion, we have reinvestigated diphenyl methoxymethyl protecting groups for the uridine ureido nitrogen. The (4,4'-bisfluorophenyl)methoxymethyl (BFPM) group displays excellent relative stability in general chemical transformations for advancing uridine. Uniquely, the BFPM group is tolerated in aqueous acidic and hydrogenation conditions, but can be cleaved with 2% TFA in CH2Cl2. Thus, the BFPM group can be distinguished from the Boc group. BFPM-Cl can readily be synthesized from commercially available bis(4-fluorophenyl)methanol in two chemical steps with high yield using inexpensive chemicals. Application of the BFPM group is not be limited to uridine; all reported synthetic intermediates possessing the benzyloxymethyl (BOM) or its related groups can be replaced with BFPM group.6 In this regard, the BFPM group has been applied to primary and secondary alcohol protections in our laboratory.
General experimental details
All chemicals were purchased from commercial sources and used without further purification unless otherwise noted. THF, CH2Cl2, and DMF were purified via Innovative Technology's Pure-Solve System. All reactions were performed under an Argon atmosphere. All stirring was performed with an internal magnetic stirrer. Reactions were monitored by TLC using 0.25 mm coated commercial silica gel plates (EMD, Silica Gel 60F254). TLC spots were visualized by UV light at 254 nm, or developed with ceric ammonium molybdate or anisaldehyde or copper sulfate or ninhydrin solutions by heating on a hot plate. Reactions were also monitored by using SHIMADZU LCMS-2020 with solvents: A: 0.1% formic acid in water, B: acetonitrile. Flash chromatography was performed with SiliCycle silica gel (Purasil 60 Å, 230-400 Mesh). Proton magnetic resonance (1H-NMR) spectral data were recorded on 400, and 500 MHz instruments. Carbon magnetic resonance (13C-NMR) spectral data were recorded on 100 and 125 MHz instruments. For all NMR spectra, chemical shifts (δH, δC) were quoted in parts per million (ppm), and J values were quoted in Hz. 1H and 13C NMR spectra were calibrated with residual undeuterated solvent (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm; CD3CN: δH = 1.94 ppm, δC = 1.32ppm; CD3OD: δH =3.31 ppm, δC =49.00 ppm; DMSO-d6: δH = 2.50 ppm, δC = 39.52 ppm; D2O: δH = 4.79 ppm) as an internal reference. The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, dd = double doublets, t = triplet, q = quartet, quin = quintet, hept = heptet, m = multiplet, br = broad. Infrared (IR) spectra were recorded on a Perkin-Elmer FT1600 spectrometer. HPLC analyses were performed with a Shimadzu LC-20AD HPLC system. HR-MS data were obtained from a Waters Synapt G2-Si (ion mobility mass spectrometer with nanoelectrospray ionization).
Procedure for synthesis of 4,4'-((chloromethoxy)methylene)bis(fluorobenzene) (BFPM-Cl)
To a stirred suspension of NaH (1.4 g, 60% in oil, 35 mmol) in THF (17 mL), bis(4-fluorophenyl)methanol (3.8 g, 17 mmol) was added at 0 °C. After being stirred for 0.5 h, chloromethyl methyl sulfide (2.9 mL, 39 mmol) was added. After 3 h at 0 °C, the reaction mixture was quenched with sat. aq. NH4Cl, and extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 99/1) to afford (bis(4-fluorophenyl))methoxymethyl methyl sulfide as pale yellow oil (4.0 g, 82%). IR (thin film) νmax = 2922, 1603, 1507, 1433, 1297, 1223, 1181, 1155, 1098, 1048, 1014, 964, 877, 860, 832, 793, 781, 731, 684 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.29 (dd, J = 8.5, 5.5 Hz, 4H), 7.02 (t, J = 8.6 Hz, 4H), 5.85 (s, 1H), 4.61 (s, 2H), 2.19 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 163.44, 160.99, 136.86, 136.83, 129.06 (2C), 128.98 (2C), 115.49 (2C), 115.28 (2C), 72.53, 13.95; HRMS (ESI+) m/z calcd for C15H14F2NaOS [M + Na] 303.0631, found: 303.0637.
To a stirred solution of (bis(4-fluorophenyl))methoxymethyl methyl sulfide (4.0 g, 14 mmol) in CH2Cl2 (28 mL) was added sulfuryl chloride (3.5 mL, 43 mmol) at 0 °C. After being stirred for 3 h at 0 °C, the reaction mixture was concentrated in vacuo. The crude mixture (BFPM-Cl) was used for next reaction without purification. IR (thin film) νmax = 2930, 1602, 1507, 1409, 1316, 1225, 1157, 1139, 1101, 1057, 1015, 966, 928, 859, 836, 767 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.29 (dd, J = 8.5, 5.4 Hz, 4H), 7.05 (t, J = 8.6 Hz, 4H), 5.88 (s, 1H), 5.44 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 133.66, 133.63, 129.51, 129.42, 129.27, 129.19, 115.68, 115.63, 115.46, 115.42, 79.56, 62.71; HRMS (ESI+) m/z calcd for C14H11ClF2NaO [M + Na] 291.0364, found: 291.0370.
3-((Bis(4-fluorophenyl)methoxy)methyl)-1-((3R,4S)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (5)
To a stirred solution of BFPM-Cl (14 mmol) in DMF (14 mL), uridine (3.5 g, 14 mmol) and DBU (4.3 mL, 29 mmol) were added. After being stirred for 8 h at r.t., the reaction mixture was quenched with 1.0 M HCl, extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 2/1 - CHCl3/MeOH = 88/12) to afford 5 as yellow oil (5.2 g, 77% for 2 steps). [a] + 0.027 (c = 0.31, CHCl3); IR (thin film) νmax = 3453 (br), 2959, 2929, 2874, 2861, 1723, 1664, 1603, 1508, 1460, 1379, 1274, 1224, 1155, 1122, 1074, 1015, 834, 744 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.55 (d, J = 8.2 Hz, 1H), 7.29 (dd, J = 8.6, 5.5 Hz, 4H), 6.98 (t, J = 8.7 Hz, 4H), 5.70 (s, 1H), 5.68 (d, J = 8.2 Hz, 1H), 5.60 (d, J = 4.2 Hz, 1H), 5.51 (s, 2H), 4.34 – 4.28 (m, 2H), 4.23 (q, J = 2.8 Hz, 1H), 3.96 (dd, J = 11.9, 2.6 Hz, 1H), 3.82 (dd, J = 11.8, 2.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 163.43, 162.34, 160.98, 151.83, 139.23, 137.41, 137.38, 128.64, 128.57, 128.56, 128.49, 115.38 (2C), 115.17 (2C), 101.72, 93.52, 86.00, 82.05, 75.40, 71.14, 69.80, 62.07; HRMS (ESI+) m/z calcd for C23H22F2N2NaO7 [M + Na] 499.1293, found: 499.1302.
3-((Bis(4-fluorophenyl)methoxy)methyl)-1-((3aR,4R,6aR)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (3)
To a stirred solution of 5 (5.2 g, 11 mmol) and 2,2-dimethoxypropane (6.8 mL, 55 mmol) in acetone (11 mL), TsOH•H2O (0.21 g, 1.1 mmol) was added. After being stirred for 3 h at r.t., the reaction mixture was quenched with sat. aq. NaHCO3, extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 1/1 to 1/2) to afford 3 as yellow foam (5.1 g, 89%). [α] - 0.129 (c = 0.61, CHCl3); IR (thin film) νmax = 3474 (br), 2935, 1716, 1667, 1604, 1508, 1457, 1275, 1221, 1156, 1078, 835 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.29 (ddd, J = 8.4, 5.4, 2.3 Hz, 4H), 7.24 (d, J = 8.1 Hz, 1H), 6.98 (td, J = 8.7, 2.0 Hz, 4H), 5.70 (s, 1H), 5.66 (d, J = 8.1 Hz, 1H), 5.51 (s, 3H), 4.96 (dd, J = 6.5, 3.3 Hz, 1H), 4.91 (dd, J = 6.5, 2.9 Hz, 1H), 4.29 (q, J = 3.0 Hz, 1H), 3.93 (dd, J = 12.0, 2.5 Hz, 1H), 3.81 (dd, J = 12.0, 3.3 Hz, 1H), 1.58 (s, 3H), 1.37 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 163.38, 162.26, 160.93, 150.88, 141.15, 137.48, 128.55, 128.54, 128.47, 128.46, 115.35, 115.33, 115.14, 115.12, 114.39, 102.09, 96.51, 86.72, 83.59, 82.06, 80.17, 69.87, 62.70, 27.24, 25.23; HRMS (ESI+) m/z calcd for C26H26F2N2NaO7 [M + Na] 539.1606, found: 539.1620.
Selective desilylation of 3-((Bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3R,4R)-3-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-4-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (6)
To a stirred solution of 6 (13 mg, 0.018 mmol) in CH3CN (0.16 mL), 50% HF in H2O (0.04 mL) was added. After being stirred for 10 min at r.t., the reaction mixture was diluted with H2O, extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 1/1 - 1/2) to afford 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3R,4R)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione as colorless oil (7, 9.0 mg, 83%). - 0.058 (c = 0.29, CHCl3); IR (thin film) νmax = 3480 (br), 2954, 2930, 2858, 1714, 1665, 1604, 1508, 1459, 1273, 1259, 1224, 1155, 1114, 1076, 1014, 862, 836, 812, 782 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.49 (d, J = 8.1 Hz, 1H), 7.29 (ddd, J = 8.5, 5.4, 2.6 Hz, 4H), 6.99 (t, J = 8.6 Hz, 4H), 5.73 (d, J = 8.1 Hz, 1H), 5.69 (s, 1H), 5.55 (d, J = 5.0 Hz, 1H), 5.46 (s, 2H), 4.55 (t, J = 5.0 Hz, 1H), 4.20 – 4.16 (m, 1H), 4.15 – 4.12 (m, 1H), 3.98 (dd, J = 12.2, 2.1 Hz, 1H), 3.82 (dd, J = 12.1, 2.1 Hz, 1H), 0.87 (s, 9H), 0.05 (s, 3H), 0.02 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 163.40, 162.32, 160.96, 150.98, 140.76, 137.42, 137.38, 128.71, 128.64, 128.63, 128.56, 115.34, 115.32, 115.13, 115.10, 102.22, 93.42, 85.22, 81.50, 74.05, 70.70, 69.27, 62.18, 25.59, 17.91, −4.86, −5.21; HRMS (ESI+) m/z calcd for C29H36F2N2NaO7Si [M + Na] 613.2158, found: 613.2162.
Detritylation of 3-((Bis(4-fluorophenyl)methoxy)methyl)-1-((3aR,4R,6aR)-2,2-dimethyl-6-((trityloxy)methyl)tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)pyrimidine-2,4(1H,3H)-dione (8)
To a stirred solution of 8 (65 mg, 0.086 mmol) and TolSH (16 mg, 0.13 mmol) in CH2Cl2 (0.43 mL), BF3•OEt2 (5.3 μL, 0.043 mmol) was added at r.t. After being stirred for 1 h, the reaction mixture was quenched with sat. aq. NaHCO3, extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 1/1 - 1/2) to afford 3 as yellow foam (45 mg, 84%).
Oxidation of 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3R,4R)-3-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-4-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (6)
To a stirred suspension of CrO3 (35 mg, 0.35 mmol) in CH2Cl2 (0.30 mL), Ac2O (99 mL, 1.04 mmol) and pyridine (0.17 mL, 2.09 mmol) were added at 0 °C. After being stirred for 0.5 h at 0 °C, 0.5 h at r.t., 6 (61 mg, 0.087 mmol) was added. After 0.5 h, the reaction mixture was diluted with EtOAc, filtered through celite and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 9/1 to 8/2) to afford 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3S)-3-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-4-oxotetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione as colorless oil (9, 49 mg, 81%). [α] + 0.525 (c = 0.46, CHCl3); IR (thin film) νmax = 2955, 2930, 2858, 1786, 1723, 1672, 1604, 1508, 1454, 1272, 1255, 1222, 1070, 834, 780 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.77 (d, J = 8.2 Hz, 1H), 7.30 (dd, J = 8.4, 5.4 Hz, 4H), 6.98 (t, J = 8.4 Hz, 4H), 6.27 (d, J = 7.9 Hz, 1H), 5.79 (d, J = 8.1 Hz, 1H), 5.69 (s, 1H), 5.49 (d, J = 9.8 Hz, 1H), 5.44 (d, J = 9.8 Hz, 1H), 4.24 (s, 1H), 4.12 (d, J = 7.9 Hz, 1H), 3.93 (d, J = 11.7 Hz, 1H), 3.89 (d, J = 11.3 Hz, 1H), 0.88 (s, 9H), 0.79 (s, 9H), 0.09 (s, 3H), 0.05 (s, 3H), 0.02 (s, 3H), −0.09 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 208.35, 163.42, 162.17, 160.97, 151.03, 137.75, 137.44, 137.42, 137.39, 128.69, 128.66, 128.61, 128.58, 115.33 (2C), 115.11 (2C), 103.18, 85.51, 81.85, 81.40, 69.36, 62.84, 25.72 (3C), 25.26 (3C), 18.19, 18.04, −4.88, −5.51, −5.64, −5.79; HRMS (ESI+) m/z calcd for C35H48F2N2NaO7Si2 [M + Na] 725.2866, found: 725.2889.
Reduction of 9
To a stirred solution of 9 (12 mg, 0.018 mmol) in THF (0.35 mL), LiBH4 (4.0M in THF, 8.8 μL, 0.035 mmol) was added at 0 °C. After being stirred for 1 h, the reaction mixture was quenched with sat. aq. NaHCO3, extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 85/15 to 80/20) to afford mixture of 10α (6) and 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3R,4S)-3-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-4-hydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (10β, 11 mg, 90%) as colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 7.81 (d, J = 8.2 Hz, 1H), 7.30 (ddd, J = 7.9, 5.3, 2.3 Hz, 4H), 7.02 – 6.94 (m, 4H), 5.72 (s, 1H), 5.69 (s, 1H), 5.58 (d, J = 8.2 Hz, 1H), 5.49 (d, J = 9.8 Hz, 1H), 5.46 (d, J = 9.8 Hz, 1H), 4.40 (s, 1H), 4.30 (dd, J = 12.3, 3.4 Hz, 1H), 4.22 – 4.18 (m, 2H), 4.15 (s, 1H), 4.10 (s, 1H), 0.91 (s, 9H), 0.90 (s, 9H), 0.19 (s, 3H), 0.15 (s, 3H), 0.14 (s, 3H), 0.13 (s, 3H).
Debenzylation of 1-((3aR,4R,6aR)-6-((benzyloxy)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-3-((bis(4-fluorophenyl)methoxy)methyl)pyrimidine-2,4(1H,3H)-dione (11)
To a stirred solution of 11 (28 mg, 0.046 mmol) in a 10/1 mixture of iPrOH/H2O (0.5 mL) was added 10% Pd/C (2.8 mg). H2 gas was introduced and the reaction mixture was stirred under H2 atmosphere. After 2 h, the solution was filtered through Celite and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc = 1/1 to 1/2) to afford 3 as yellow foam (21 mg, 88%).
Deoxygenations of 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3R,4S)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione
To a stirred suspension of NaH (11 mg, 60% in oil, 0.28 mmol) in THF (0.46 mL), 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R,3R,4S)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (55 mg, 0.092 mmol) was added at 0 °C. After being stirred for 0.5 h, CS2 (17 mL, 0.28 mmol) was added. After 2 h, MeI (17 mL, 0.28 mmol) was added. After 4 h, the reaction mixture was quenched with sat. aq. NaHCO3, and extracted with EtOAc. The combined extract was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexanes/EtOAc = 90/10 to 85/15) to afford O,O'-((2R,3R,4R)-2-(3-((bis(4-fluorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-3,4-diyl) S,S'-dimethyl bis(carbonodithioate) as pale yellow oil (12, 70 mg, 99%). [α] – 0.250 (c = 1.42, CHCl3); IR (thin film) νmax = 2955, 2929, 2858, 1721, 1675, 1604, 1508, 1456, 1260, 1222, 1198, 1095, 1075, 833, 810, 780 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.78 (d, J = 8.2 Hz, 1H), 7.31 – 7.27 (m, 4H), 6.98 (t, J = 8.5 Hz, 4H), 6.56 (d, J = 7.2 Hz, 1H), 6.22 (d, J = 5.5 Hz, 1H), 6.03 (dd, J = 7.0, 5.7 Hz, 1H), 5.71 – 5.67 (m, 2H), 5.50 (d, J = 9.9 Hz, 1H), 5.47 (d, J = 9.8 Hz, 1H), 4.45 (s, 1H), 4.02 (dd, J = 11.4, 1.5 Hz, 1H), 3.92 (dd, J = 11.1, 1.5 Hz, 1H), 2.61 (s, 3H), 2.53 (s, 3H), 0.94 (s, 9H), 0.17 (s, 3H), 0.15 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 214.74, 214.41, 163.41, 163.38, 162.18, 160.96, 160.93, 151.06, 138.18, 137.48, 137.45, 137.39, 137.36, 128.74, 128.66 (2C), 128.58, 115.30 (2C), 115.09 (2C), 102.86, 85.95, 84.31, 81.48, 79.91, 78.85, 69.63, 63.25, 25.93 (3C), 18.33, −5.36, −5.65; HRMS (ESI+) m/z calcd for C33H40F2N2NaO7S4Si [M + Na] 793.1353, found: 793.1361.
To a stirred solution of 12 (46 mg, 0.059 mmol) in toluene (0.60 mL) were added nBu3SnH (80 mL, 0.30 mmol) and AIBN (29 mg, 0.18 mmol). After being stirred for 3 h at 100 °C, the reaction mixture was concentrated in vacuo. The crude product was purified by silica gel column chromatography (10% w/w anhydrous K2CO3-SiO2, hexanes/EtOAc = 85/15 to 80/20) to afford 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R)-5-(((tert-butyldimethylsilyl)oxy)methyl)-2,5-dihydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione as colorless oil (13, 28 mg, 85%). [α] – 0.177 (c = 0.70, CHCl3); IR (thin film) νmax = 2956, 2929, 2858, 1718, 1669, 1604, 1508, 1458, 1258, 1223, 1139, 1075, 836, 808, 781 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 7.74 (d, J = 8.2 Hz, 1H), 7.31 (dt, J = 8.9, 5.3 Hz, 4H), 7.00 (d, J = 2.9 Hz, 1H), 6.97 (dd, J = 8.5, 2.7 Hz, 4H), 6.24 (d, J = 5.9 Hz, 1H), 5.76 (d, J = 5.6 Hz, 1H), 5.72 (s, 1H), 5.61 (d, J = 8.1 Hz, 1H), 5.51 (s, 2H), 4.88 (s, 1H), 3.91 (dd, J = 11.7, 2.8 Hz, 1H), 3.83 (dd, J = 11.7, 2.6 Hz, 1H), 0.88 (s, 9H), 0.06 (d, J = 1.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 163.36, 162.73, 160.91, 151.27, 139.71, 137.64, 137.61, 134.30, 128.65, 128.63, 128.57, 128.54, 126.44, 115.28 (2C), 115.07 (2C), 101.80, 90.25, 87.28, 81.66, 69.66, 64.10, 25.86 (3C), 18.48, −5.42, −5.60; HRMS (ESI+) m/z calcd for C29H34F2N2NaO5Si [M + Na] 579.2103, found: 579.2119.
Hydrogenation of 13
To a stirred solution of 13 (14 mg, 0.026 mmol) in MeOH (5.0 mL) was added 10% Pd/C (2.8 mg). H2 gas was introduced and the reaction mixture was stirred under H2 atmosphere. After 2 h, the solution was filtered through Celite and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes/EtOAc = 85/15 to 80/20) to afford 3-((bis(4-fluorophenyl)methoxy)methyl)-1-((2R)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione as colorless oil (14, 13 mg, 93%). [α] + 0.171 (c = 1.13, CHCl3); IR (thin film) νmax = 2955, 2929, 2858, 1713, 1663, 1604, 1507, 1459, 1276, 1260, 1222, 1127, 1073, 1014, 998, 863, 836, 807, 779 cm−1; 1H NMR (400 MHz, Chloroform-d) δ 8.01 (d, J = 8.1 Hz, 1H), 7.30 (ddd, J = 8.2, 5.4, 2.3 Hz, 4H), 6.97 (td, J = 8.7, 3.1 Hz, 4H), 5.99 (dd, J = 6.5, 2.6 Hz, 1H), 5.72 (s, 1H), 5.59 (d, J = 8.2 Hz, 1H), 5.52 (d, J = 9.9 Hz, 1H), 5.49 (d, J = 9.8 Hz, 1H), 4.14 (td, J = 8.0, 1.8 Hz, 1H), 4.06 (dd, J = 11.6, 2.2 Hz, 1H), 3.69 (dd, J = 11.6, 2.2 Hz, 1H), 2.41 – 2.31 (m, 1H), 2.02 – 1.83 (m, 3H), 0.92 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 163.34, 162.89, 160.90, 150.78, 139.29, 137.72, 137.69, 137.65, 137.61, 128.65, 128.57 (2C), 128.48, 115.26 (2C), 115.04 (2C), 100.72, 86.84, 82.23, 81.72, 69.58, 63.33, 33.52, 25.86 (3C), 23.99, 18.41, −5.52, −5.61; HRMS (ESI+) m/z calcd for C29H36F2N2NaO5Si [M + Na] 581.2259, found: 581.2267.
Deprotections of tert-butyl (((2R,3S,4R,5S)-5-((1S,2S)-3-amino-2-((3-aminopropyl)amino)-1-((2S,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-3-oxopropoxy)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)carbamate (15)
To a stirred solution of 15 (17 mg, 0.016 mmol) in H2O (0.5 mL) was added TFA (0.5 mL). After being stirred for 2 h at r.t., all volatile were evaporated in vacuo. To a stirred solution of the crude mixture in CH2Cl2 (1.0 mL) was added TFA (0.02 mL). After 2 h, all volatile were evaporated in vacuo. To a stirred solution of the crude mixture in iPrOH-H2O (10:1, 1.0 mL) was added 10% Pd/C (10 wt % 6.8 mg). H2 gas was introduced and the reaction mixture was stirred for 6 h under H2. The solution was filtered through Celite and concentrated in vacuo. The crude mixture was purified by C18 reverse-phase HPLC [column: Luna® (100 Å, 10 μm, 250 x 10 mm), solvents: 25:75 MeOH:0.05 M NH4HCO3 in H2O, flow rate: 3.0 mL/min, UV: 254 nm] to afford tert-butyl (((3aR,4R,6S,6aR)-6-((1S,2S)-3-amino-2-((3-(((benzyloxy)carbonyl)amino)propyl)amino)-1-((4R,6R)-6-(3-((bis(4-fluorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-3-oxopropoxy)-2,2-diethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)carbamate as colorless oil (16, 7.2 mg, 76% for 3 steps, retention time: 13.0 min): [α] + 0.386 (c = 0.35, MeOH); IR (thin film) νmax = 3329 (br), 3312 (br), 2979, 2929, 1678, 1572, 1508, 1459, 1392, 1367, 1279, 1131, 1115, 1077, 1057, 1018 cm−1; 1H NMR (400 MHz, Deuterium Oxide) δ 7.76 (d, J = 7.8 Hz, 1H), 5.79 (s, 1H), 5.76 (d, J = 7.8 Hz, 1H), 5.05 (s, 1H), 4.25 – 4.13 (m, 3H), 4.03 (dd, J = 10.5, 6.8 Hz, 3H), 3.52 (d, J = 6.4 Hz, 1H), 3.36 – 3.31 (m, 2H), 3.29 (s, 1H), 2.97 (t, J = 7.3 Hz, 2H), 2.65 (t, J = 7.1 Hz, 2H), 1.82 – 1.73 (m, 2H), 1.34 (s, 9H); 13C NMR (101 MHz, D2O) δ 176.10, 161.92, 161.73, 160.96, 146.67, 140.44, 109.60, 102.04, 90.59, 90.39, 82.37, 81.77, 80.86, 79.92, 75.26, 74.88, 41.07, 38.32, 38.19, 27.69 (3C); HRMS (ESI+) m/z calcd for C24H40N6NaO12 [M + Na] 627.2602, found: 627.2613.
Supplementary Material
Acknowledgment
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM114611. M.K. thanks UTRF (University of Tennessee Health Science Center) for generous financial support (Innovation award R079700292). NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant..
Funding Information
NIH/GM114611
Footnotes
Supporting Information
Syntheses of new materials and NMR spectra of the compounds in the Experimental Section
References
- (1).(a) Arbour CA; Imperiali B Bioorg. Med. Chem 2020, 28, 115661. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wiegmann D; Koppermann S; Wirth M; Niro G; Leyerer K; Ducho C Beilstein J. Org. Chem 2016, 12, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shelton J; Lu X; Hollenbaugh JA; Cho JH; Amblard F; Schinazi RF Chem. Rev 2016, 116, 14379. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Serpi M; Ferrari V; Pertusati F J. Med. Chem 2016, 59, 10343. [DOI] [PubMed] [Google Scholar]; (e) Yamamoto T; Koyama H; Kurajoh M; Shoji T; Tsutsumi Z; Moriwaki Y Clinica. Chimica. Acta 2011, 412, 1712. [DOI] [PubMed] [Google Scholar]; (f) Winn M; Goss RJM; Kimura K; Bugg TDH Nat. Prod. Rep 2010, 27, 279. [DOI] [PubMed] [Google Scholar]
- (2).(a) Reports from our group: Mitachi K; Kansal R; Hevener K; Gillman C; Yun H-G; Husain M; Miranda-Carboni G; Glazer E; Clemons WM Kurosu M J. Med. Chem 2020, 63, 10855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kurosu M Future Med. Chem 2019, 11, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mitachi K; Kurosu SM; Gillman CD; Yun H-G; Clemons WM; Kurosu M MethodsX 2019, 6, 2305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mitachi K; Kurosu S; Eslamimehr S; Lemieux M; Ishizaki Y; Clemons W; Kurosu M Org. Lett 2019, 21, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mitachi K; Yun H-G; Kurosu SM; Eslamimehr S; Lemieux MR; Klaić L; Clemons WM; Kurosu M ACS Omega 2018, 3, 1726. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kurosu M J. Mol. Pharm. Org. Process Res 2018, 6, 141. [PMC free article] [PubMed] [Google Scholar]; (g) Mitachi K; Aleiwi BA; Schneider CM; Siricilla S; Kurosu M J.Am. Chem. Soc 2016, 138, 12975. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Siricilla S; Mitachi K; Wan B; Franzblau SG; Kurosu M J. Antibiot 2015, 68, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wang Y; Siricilla S; Aleiwi BA; Kurosu M Chem. Eur. J 2013, 19, 13847. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kurosu M; Li K Heterocycles 2009, 77, 217. [Google Scholar]; (k) Kurosu M; Li K; Crick DC Org. Lett 2009, 11, 2393. Reports from other groups: [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Biecker AL; Liu X; Thorson JS; Yang Z; Van Lanen SG Molecules 2019, 24, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Ichikawa S; Yamaguchi M; Matsuda A Curr. Med. Chem 2015, 22, 3951. [DOI] [PubMed] [Google Scholar]; (n) Knapp S; Nandan SR J. Org. Chem 1994, 59, 281. [Google Scholar]; (o) Patel B; Kerr RV; Malde AK; Zunk M; Bugg TDH; Grant G; Rudrawar S Chem. Med. Chem 2020, 15, 1429. [DOI] [PubMed] [Google Scholar]; (p) Heib A; Niro G; Weck SC; Koppermann S; Ducho C Molecules 2020, 25, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Leyerer K; Koppermann S; Ducho C Eur. J. Org. Chem 2019, 45, 7420. [Google Scholar]; (r) Wiegmann D; Koppermann S; Ducho C Molecules 2018, 23, 3085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Katsuyama A; Ichikawa S Chem. Pharm. Bull 2018, 66, 123. [DOI] [PubMed] [Google Scholar]; (t) Yamashita A; Norton E; Peterson PJ; Rasmussen BA; Singh G; Yang Y; Mansour TS; Ho DM Bioorg. Med. Chem. Lett 2003, 13, 3345. [DOI] [PubMed] [Google Scholar]
- (3).(a) Jarowickia K; Kocienski PJ Chem. Soc. Perkin Trans. 1 1999, 1589. [Google Scholar]; (b) Kunz H; Waldman H Protecting Groups In Comprehensive Organic Synthesis; Trost BM, Fleming I, Eds.; Pergamon Press: Oxford, United Kingdom, 1991; Vol. 6, pp 631. Chapter 3.1. [Google Scholar]
- (4).(a) Aleiwi BA; Kurosu M Tetrahedron Lett. 2012, 53. 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Connor DS; Klein GW; Taylor GN; Boeckman RK; Medwid JB Org. Syn 1972, 52, C10. [Google Scholar]; (c) Hanessian S; Kloss J; Sugawara T J. Am. Chem. Soc 1986, 108, 2758. [Google Scholar]; (d) Goff DA; Harris RN; Bottaro JC; Bedford CD J. Org. Chem 1986, 51, 4711. [Google Scholar]; (e) Prakash TP; Manoharan M; Kawasaki AM; Fraser AS; Lesnik EA; Sioufi N; Leeds JM; Teplova M; Egli M Biochemistry 2002, 41, 11642. [DOI] [PubMed] [Google Scholar]
- (5).(a) Wang Y; Kurosu M Tetrahedron 2012, 68, 4797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kurosu M; Li K Synthesis 2009, 21, 3633. [Google Scholar]; (c) Kurosu M; Li K Org. Lett 2009, 11, 911. [DOI] [PubMed] [Google Scholar]; (d) Kurosu M; Biswas K; Crick DC Org. Lett 2007, 9, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Takahashi D, Yano T, Fukui T Org Lett. 2012, 14, 4514. [DOI] [PubMed] [Google Scholar]; (b) Saudi M; van Aerschot A Molecules 2013, 18, 8524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mezaache R; Harkat H; Obszynski J; Benkouider A; Blanc A; Weibel JM; Pale P Tetrahedron Lett. 2014, 55, 7167. [Google Scholar]; (d) Li J; Zhang X; Shen H; Liu Q; Pan J; Hu W; Chen C Advanced Syn. Catal 2015, 357, 3115. [Google Scholar]; (e) Howard KT; Duffy BC; Linaburg MR; Chisholm JD Org. Biom. Chem 2016, 14, 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tran VH; La MT; Kim HK Org. Biomol. Chem 2019, 17, 6221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.