

ABSTRACT
Osteopontin (OPN) is produced by tumor cells as well as by myeloid cells and is enriched in the tumor microenvironment (TME) of many cancers. Given the roles of OPN in tumor progression and immune suppression, we hypothesized that targeting OPN with aptamers that have high affinity and specificity could be a promising therapeutic strategy. Bi-specific aptamers targeting ligands for cellular internalization were conjugated to siRNAs to suppress OPN were created, and therapeutic leads were selected based on target engagement and in vivo activity. Aptamers as carriers for siRNA approaches were created including a cancer targeting nucleolin aptamer Ncl-OPN siRNA and a myeloid targeting CpG oligodeoxynucleotide (ODN)-OPN siRNA conjugate. These aptamers were selected as therapeutic leads based on 70–90% OPN inhibition in cancer (GL261, 344SQ, 4T1B2b) and myeloid (DC2.4) cells relative to scramble controls. In established immune competent 344SQ lung cancer and 4T1B2b breast cancer models, these aptamers, including in combination, demonstrate therapeutic activity by inhibiting tumor growth. The Ncl-OPN siRNA aptamer demonstrated efficacy in an immune competent orthotopic glioma model administered systemically secondary to the ability of the aptamer to access the glioma TME. Therapeutic activity was demonstrated using both aptamers in a breast cancer brain metastasis model. Targeted inhibition of OPN in tumor cells and myeloid cells using bifunctional aptamers that are internalized by specific cell types and suppress OPN expression once internalized may have clinical potential in cancer treatment.
KEYWORDS: Osteopontin, nucleolin, aptamers, CpG ODN, siRNA, brain cancer, breast cancer, lung cancer
Introduction
Glioblastoma (GBM) is the most common and aggressive cancer in the brain, and about 15,000 people develop this disease per year in the United States. Despite great advances in treatment of many forms of malignancy, GBM remains a vexing clinical problem, with a median survival time of 12–15 months and with less than 3–5% of patients surviving beyond 5 years. Over the last several decades, immune therapies such as vaccines, immune checkpoint blocking antibodies, and chimeric antigen receptor T cells have achieved promising efficacy in multiple clinical trials of cancer patients.1,2 However, these immune therapeutic strategies have not resulted in “cures” in GBM patients because of tumor resistance and recurrence including constraints to drug and immune access, tumor heterogeneity, low mutational burden, and severe tumor-imposed T cell and other immune dysfunction. Two major cell components in the tumor microenvironment (TME) responsible for tumor escape from immune surveillance include glioblastoma stem cells (GSCs) and glioblastoma-infiltrating microphages/microglia (GIMs).
Elevated osteopontin (OPN) expression has been reported in multiple tumor types and is considered a key contributor to tumor malignancy and progression.3 The gene encoding OPN is secreted phosphoprotein 1 (SPP1). OPN is expressed in both tumor and myeloid cells that invade tumor. We have previously shown that tumor- and myeloid cell-derived OPN are critical for glioblastoma (GBM) development, and OPN deficiency in either GBM cells or non-tumor cells results in a marked reduction in intratumoral immune suppressive M2 macrophages while increasing T cell effector activity.4 Blockade of secreted OPN by its neutralizing antibody exerts minimal in vivo anti-glioblastoma activity in contrast to the diminished in vivo tumorigenicity of OPN gene SPP1 knockdown in glioma cells.4,5 This implies that intracellular OPN is an important contributor to tumor malignancy. Within the tumor microenvironment (TME), secreted OPN displays complex activities. For instance, it functions as a chemokine that attracts myeloid cells and is an alternative immune checkpoint ligand.6,7 Both malignant cells and myeloid cells have receptors for OPN. Whether tumor-derived OPN differs structurally or functionally from myeloid-derived OPN remains to be clarified. The existence of intracellular OPN also contributes to the pleiotropic activities of OPN via immune modulation.8 Inhibiting OPN expression prior to translation of corresponding message should, in principle, decrease intracellular and secreted OPN. siRNA-mediated RNA interference strategies could accomplish this, such as has been demonstrated by decreasing expression of transporter associated with antigen processing (TAP) and the signal transducer and activator of transcription 3 (STAT3).9 TAP loads antigens onto major histocompatibility complex and its inhibition allows for alternative antigens that are not typically presented to be loaded onto the cell surface for antigen presentation and immune activation. Targeting TAP can elicit protective cytotoxic T cell immunity against TAP-deficient tumors.10,11 STAT3 siRNA has been previously selected as a therapeutic strategy secondary to the dual role of this pathway essential in tumor progression and myeloid-mediated immune suppression.12,13
Nucleolin is a non-ribosomal phosphoprotein that influences a wide range of cellular activities such as cell adhesion, cell division and migration, regulation of rRNA transcription, modification, and processing of nascent pre-rRNA, telomerase maintenance, and DNA repair.14 Nucleolin is highly expressed in a broad range of human and murine cancers,15 and its cellular distribution is often altered in tumors.16 Because of its high-level expression across cancers, nucleolin has been suggested as a potential target for the delivery of therapy to tumors.17 The nucleolin-targeting aptamer, AS1411, was developed and has been used for this purpose, and is reactive with human and murine target proteins. This 26-mer DNA aptamer with G-quadruplex structure, renders the aptamer heat stable and resistant to DNase/RNase degradation.18
CpG oligodeoxynucleotides (ODNs) are commonly used as vaccine adjuvants. Their endocytic receptor, toll-like receptor 9 (TLR9), is expressed in glioma tissues and on myeloid cells including antigen presenting cells (APCs). GBM patients with tumors that have high expression of TLR9 have a poor prognosis.19,20 ODNs trigger down regulation of their endocytic receptor TLR9, which is followed by apoptosis of GL261 murine GBM cells in vitro and in vivo. The effects of CpG treatments are also known to enhance the antigen-presenting capacity of immune cells, and to shift immune response toward CD8 + T cells while decreasing the number of CD4+ CD25+ regulatory T cells (Tregs).21 However, a phase II clinical trial demonstrated that intraoperative injection of CpG ODN into surgical resection cavities of GBM patients failed to improve patient survival relative to standard of care treatment without administration of CpG ODN.22 This outcome is likely because innate immune response induced by the CpG adjuvant are insufficient for tumor clearance in human subjects, and additional anti-tumor specific T cell immunity is required. Nonetheless, CpG ODNs can be used as targeting moieties for both cancer and myeloid cells.
RNA inference (RNAi) holds great potential for therapeutic application, and an increasing number of FDA-approved RNAi therapeutics, especially small interfering RNAs (siRNAs) have been used to treat cancer patients.23 Short single-stranded RNA aptamers are effective as delivery molecules targeting specific cell types, thereby reducing off-target effects or other unwanted side effects.24 As such, we developed a novel RNA therapeutic strategy consisting of aptamer-siRNA chimeras: nucleolin aptamer-OPN siRNA targeting cancer cells and CpG ODN-OPN siRNA targeting myeloid cells. As such, we hypothesized that we could exploit nucleolin and TLR9 as targeting moieties to modulate OPN to achieve an antineoplastic therapeutic effect. Because of its high specificity to GIMs and GSCs in the glioblastoma TME, the bispecific nucleolin aptamer and the CpG ODN-OPN siRNA are promising therapeutics that are potentially less toxic than traditional cytotoxic therapies for primary brain tumors. Furthermore, this work may lead to highly innovative nucleic acid–based and TME-specific immune therapeutics for GBM that may also have implications for other cancers that also have high levels of OPN expression. Finally, this analysis contributes to a broader understanding of gene silencing and consequent immune modulation, specific to the TME, via a novel therapeutic approach using modified and aptamer-linked nucleic acid therapeutics.
Materials and Methods
Study Approval
All in vivo mouse experiments were approved, in accordance with Laboratory Animal Resources Commission standards, by the institutional Animal Care and Use Committee and conducted according to the approved protocol 08–06-11831.
Cell lines
GL261 was maintained in Dulbecco’s Modified Eagle Media with 10% fetal bovine serum (FBS), glutamine and essential amino acids. 4T1. B2b cells (murine breast cancer with brain metastatic capacity, from Dr. Debeb Bisrat’s laboratory at MD Anderson Cancer Center), 344SQ (murine lung cancer with brain metastatic capacity from Dr. Jonathan Kurie at MD Anderson Cancer Center), the glioblastoma cell lines D54, U87, and U251, and the dendritic cell-line DC2.4 (EMD Millipore, Burlington, MA) were cultured in 10% FBS RPMI supplemented with glutamine and essential amino acids. Derivation and culture of glioblastoma stem cells (GSCs) have been previously described.25 Briefly, the tissues were enzymatically digested with Papain dissociation system (Worthington Biomedical). After a single-cell suspension was prepared, erythrocytes were lysed using 1× RBC lysis buffer (eBioscience). Trypan blue staining confirmed >80% cell viability. Dissociated tumor cells were cultured in DMEM/F-12 containing 20 ng/mL epidermal growth factor, basic fibroblast growth factor (Sigma), and B27 (1:50; Invitrogen) as a neural stem cell–permissive medium (neurosphere medium) at a density of 3 × 106 per 60 mm dish to form spheres. After primary sphere formation was noted, sphere cells were dissociated for characterization of their properties as glioblastoma multiforme cancer-initiating cells such as immune phenotyping, cell self-renewal, differentiation, and tumorigenesis. All human cell lines were authenticated by the Cytogenetics and Cell Authentication Core at MD Anderson Cancer Center on 12/2021.
Aptamers, siRNA, and conjugate synthesis
Nucleic acid aptamers, ODNs, and siRNAs were purchased from Integrated DNA Technologies, Inc., (Coralville, IA) and TriLink (San Diego, CA). The sequences of these oligonucleotides including CpG1668 ODN, CpG19 ODN, nucleolin aptamer AS1411, sense and anti-sense strands of mouse SPP1, mouse TAP2 and human SPP1 siRNAs are listed in Supplementary Table 1. RNA was expressed as “r”; DNA as standard A, C, G, and T; 2’ O-methyl RNA bases as “m”; and internal C9 spacer as “iSp9”. “*” in CpG ODN sequences represented the linkage of phosphorothioate. To assemble the aptamers, 100 ~ 500 nmol/l of CpG-ODN or AS1411 with equal molar sense and anti-sense siRNA of OPN or TAP2 was mixed in 1x dPBS. The conjugate mixture was heated to 95°C for 4 minutes and then was cooled down gradually to 22°C within 45 minutes (Chu et al., Nucleic Acid Res 2006). Properly annealed aptamers were confirmed by 2% Tris-borate-EDTA agarose gel. Once single band of the annealed oligos at the correct size on the gel is confirmed for nearly 100% purity, the product was directly used for downstream application.
Transfection
Transfection was conducted following a previous protocol with modification.26 1 × 105 cells of GL261 were seeded in 24-well plates and the transfection was carried out for 24 hours. 100–500 nmol/l of CpG-siRNA or Ncl-siRNA aptamers were added to each well with antibiotic-free culture media. Cells were harvested to extract total RNA or protein 48 hours upon transfection. When lipofectamine 2000 was used for siRNA transfection of cultured cells, the incubation time was limited to 8 hours and was replaced with fresh medium. For 344SQ, 4T1B2b, DC2.1 and U87, cells were seeded at amounts of 1.5x105/well in 24-well plates, transfection was carried out after cell seeding overnight. For OPN aptamer and siRNA in vivo transfection, 20 μg of the RNA oligonucleotides or scramble control in 10 μL of PBS mixed with the vehicle (80 μL PBS + 10 μL Lipofectamine 2000; Invitrogen, Carlsbad, CA) or the vehicle control (90 μL PBS + 10 μL Lipofectamine 2000) per dose was used for intravenous infusion.
Real time qPCR
Total RNA was isolated from each well following the manufacture real time PCR protocol. Briefly, cells were dissolved in 200 µl of TRIzol (Invitrogen). After incubation for 5 min, the samples were mixed with 100 µl chloroform. They were centrifuged at 15,000 rpm for 10 minutes. The upper-phase liquid was collected, and total RNA was precipitated with isopropanol. The samples were washed with 70% ethanol and the pelleted RNA was resuspended in 20 µl of DEPC water. Total RNA was reversely transcribed to cDNA in a 20 µl reaction containing synthesis buffer, 0.2 mM dNTPs, reverse transcriptase (ThermoFisher, Waltham, MA) and 1 µM primers. Reactions were performed at 42°C for 30 minutes followed by 94°C for 2 minutes. Real-time PCR (10 µl) contained QuantaBio SYBR Buffer and Taq DNA polymerase (VWR, Radnor, PA), 1 nmol/l forward and reverse primers and 15–20 ng cDNA reaction. The reactions were conducted on an ABI 7500 real-time PCR machine (Applied Biosystems, Foster City, CA) using the cycling parameters: 95°C for 2 min, followed by 45 cycles of 95°C for 15 seconds, 60°C for 1 minute. The amounts of OPN mRNA in transfected and control cells were normalized with the GAPDH control mRNA. OPN qPCR primers specific to the conserved regions among the SPP1 isoforms are as follows: mouse SPP1 forward primer – 5ʹAGAGCGGTGAGTCTAAGGAGT 3’; mouse SPP1 reverse primer – 5’ TGCCCTTTCCGTTGTTGTCC 3’; human SPP1 forward primer – 5’ GGAGTTGAATGGTGCATACAAGG 3’; human SPP1 reverse primer – 5’ CCACGGCTGTCCCAATCAG 3’. All data represent the average of three independent experimental replicates.
Immunofluorescence and immunohistochemistry (IHC) staining
Paraffin-fixed slides of de-identified human GBM specimens were deparaffinized in xylene and rehydrated through a graded ethanol series (100%, 95%, and 70%). Antigen retrieval was carried out using DAKO solution (pH 6.0) for 30 minutes in a steam bath. Nonspecific protein binding was blocked on the slides with 10% normal goat serum for 60 minutes and then stained with either a rabbit TLR9 Ab at 1:100 dilution (LSbio, Seattle, WA) or a rabbit anti-nucleolin Ab (1:100, Sigma). The secondary antibody was an Alexa Fluor® 488 goat anti-rabbit antibody at 1:1000 dilution or secondary anti-rabbit Ab-HRP, respectively. To determine the correlation between TLR9 in macrophages and the cellular localization, the TLR9 stained tissue slides were co-stained with Alexa Fluor® 647-anti-Iba1 Ab (Sigma-Aldrich, St. Louis, MO, 1:1000) and Alexa Fluor® 555 anti-Na+/K+ ATPase Ab (Sigma, 1:100). Counterstaining was performed with DAPI (1:5000, in PBS) for 5 mins followed by standard PBS washing steps and mounting.
Western Blot
Total cell lysates were isolated using RIPA Buffer (ThermoFisher) supplemented with protease inhibitors (Roche, Indianapolis, IN) and the phosphatase inhibitor phenylmethylsulfonyl fluoride (Sigma). The concentration of the isolated proteins was determined using the BCA Protein Assay Reagent (Pierce, Rockford, IL). The protein (30 µg) was separated on a 3–20% Tris glycine acrylamide gel (Invitrogen) and transferred under electrophoresis to PVDF membranes (Millipore, Billerica, MA). Membranes were then incubated with the primary antibodies against the mouse OPN (Invitrogen PA5-34579), human OPN (Abcam, Cambridge, MA), or β-actin (Sigma), and the secondary antibodies. Detected protein signals were visualized using ECL (BioRad, Hercules, CA) and normalized with the corresponding β-actin signals. The protein bands on the membranes were quantified with ImageJ software (https://imagej.nih.gov/ij/).
IsoPlexis single cell-based cytokine array
CD11b+ myeloid cells were purified from healthy donor buffy coats using MACS CD11b positive selection (Miltenyi Biotech). 1 × 105 CD11b+ cells per well were seeded in 24-well plates in the presence of 500 nmol/l of CpG19 ODN-control siRNA or CpG19 ODN-human OPN siRNA at 37°C, 5% CO2 for 36 hours. The treated cells were harvested, rinsed, and stained with Alexa Fluor 647 conjugated viability dye according to the manufacture human monocyte protocol (IsoPlexis, Branford, CT). Approximately 30 μL of cell suspension was loaded into the IsoCode Chip and incubated at 37°C, 5% CO2 for an additional 16 hours. Protein secretion from approximately 1,000 single cells were captured by the 32-plex antibody barcoded chip and analyzed by fluorescence ELISA-based assay.
Animal Models
Wild-type (WT) C57BL/6 J and Balb/c mice were purchased from Jackson Laboratories (Bar Harbor, ME). 1 × 106 344SQ cells in a volume of 100 μL of PBS were subcutaneously implanted in the posterior flank of 129S6 syngeneic male mice purchased from Charles River Laboratories. When the tumor became palpable on day 7, mice were intraperitoneally (i.p.) treated with a 0.5 nmol aptamer-siRNA chimera for total 6 doses (n = 5/group and one dose every 3 days). Similarly, 4T1. B2b cells were injected into the right hind flanks of Balb/c female mice at a dose of 5 × 104 cells in 100 µl PBS. When palpable tumors formed on day 6, the mice (n = 5/group) were treated by i.p. injection of 1 nmol/dose aptamer-siRNA chimeras for three doses (one dose every 3 days). The number of treatments is based on the tumor growth kinetics of the different models using the principles of maximum numbers of doses possible prior to reaching unacceptable animal tumor burden. Tumors were measured every 3 days. Tumor volume was calculated with slide calipers using the following formula: V = (L × W × H)/2, where V is volume (mm3), L is the long diameter, W is the short diameter, and H is the height. Animals were monitored regularly and euthanized when they exhibited signs of morbidity or when the size of the subcutaneous tumor required sacrifice (typically at 5–6 weeks).
To induce intracerebral tumors in C57BL/6 J mice, GL261 cells were collected in logarithmic growth phase, washed twice with PBS, mixed with an equal volume of 10% methyl cellulose in Improved modified Eagle’s Zinc Option medium, and loaded into a 250-μl syringe (Hamilton, Reno, NV) with an attached 25-gauge needle. The needle was positioned 2 mm to the right of bregma and 4 mm below the surface of the skull at the coronal suture using a stereotactic frame (Kopf Instruments, Tujunga, CA). The intracerebral tumorigenic dose for GL261 cells was 2 × 104 in a total volume of 5 μl. Mice were then randomly assigned to control and treatment groups (n = 5/group). Aptamer-siRNA chimera systemic delivery by i.p. injection started on day 6 after GL261 cell implantation: 0.5 nmol/dose, 5 doses 3 or 4 days apart. The mice were observed twice per week for survival recording, and when they showed signs of neurological deficit (lethargy, failure to ambulate, lack of feeding, or loss of >20% body weight), they were compassionately killed.
Alexa 647 labeled aptamer enrichment and imaging in intracranial brain tumors
The CpG-OPN and the Ncl-OPN aptamers were fluorescently tagged with Alexa Fluor 647 using anti-link oligonucleotide annealing at the 3’ ends of the aptamers. To determine whether the aptamers were deposited in brain tumors, C57BL/6 mice with intracerebral GL261 tumors established for 20 days that were neurologically symptomatic were injected intravenously with 500 pmol of Alexa Fluor 647–conjugated aptamer duplexes. After 2 hours, the mice were euthanized and perfused with 25 ml PBS per mouse by cardiac infusion, and thereafter their brains were harvested and imaged using an IVIS 200 fluorescence imager.
Statistics
The distribution of each continuous variable was summarized by its mean, SD, and range. The distribution of each categorical variable was summarized in terms of its frequencies and percentages. Four groups significance was assessed with One-Way ANOVA and Bonferroni’s multiple comparison test, and two group comparison was employed with unpaired two tailed t test. Kaplan–Meier curves were used to estimate unadjusted time to event variables. Log-rank tests were used to compare each time-to-event variable between groups. P values of less than 0.05 (two-sided) were considered statistically significant. Statistical analysis was carried out using GraphPad Prism version 8.0 software (Graphpad Software, Inc).
Results
OPN is a frequently expressed in a variety of cancers
Using the Human Protein Atlas (https://www.proteinatlas.org) and GlioVis (https://www.gliovis.bioinfo.cnio.es), cancer (n = 17) profiling demonstrated that OPN was highest in GBM, lung, and renal cancers (Figure 1a). Among other types of gliomas, elevated OPN mRNA was more likely to be associated with astrocytic gliomas than oligodendrogliomas and normal brain (i.e., non-tumor) (Figure 1b). To evaluate if there would be a therapeutic benefit to targeting OPN in some of these cancer indications, a siRNA to inhibit OPN was used in combination with an osteopontin aptamer (OPN R3) in an immunocompetent glioma model. C57BL/6 mice bearing GL261 intracranial tumors were treated seven times over 20 days with either scrambled control, the OPN aptamer R3, OPN siRNA, or OPN siRNA in combination with the OPN aptamer R3. Multiple treatments were well tolerated as indicated by the absence of adverse events, behavior changes, or signs of neurological toxicity in animal subjects. Median survival (MS) was greater for mice treated with both the OPN siRNA and OPN aptamer R3 (MS duration 29 days) relative to those treated with scrambled control (MS duration 23.5 days, P < .01), OPN aptamer R3 (MS duration, 23 days; P < .01), or OPN siRNA (MS duration, 21.5 days; P < .01) (Figure 1c).
Figure 1.

A) SPP1 expression among 17 major cancer types. The data are compiled from the Human Protein Atlas (https://www.proteinatlas.org) and GlioVis (https://www.gliovis.bioinfo.cnio.es). FPKM: Fragments Per Kilobase of exon per Million reads. B) OPN mRNA expression levels in non-tumor normal brain, oligodendrogliomas, oligoastrocytoma, astrocytoma, and glioblastomas. *P < .0001. Non-tumor vs Oligodendroglioma P < .0001; Oligodendroglioma vs Oligoastrocytoma P < .0001; Oligoastrocytoma vs Astrocytoma P < .0001; Astrocytoma vs Glioblastoma P < .0001. C) Treatment schema (top panel) and Kaplan-Meier survival analysis (bottom panel) of mice with intracerebral (i.c.) GL261 treated intraperitoneally with scramble control, OPN R3, OPN siRNA, and the combination starting on day 6 after tumor implantation. The median survival durations in the treatment groups are as follows: scramble control (n = 8), 23.5 days; OPN aptamer R3 (n = 8), 23 days; OPN siRNA (n = 8), 21.5 days; and the combination (n = 8), 29 days. Statistics: combination versus scramble control, P < .01; combination versus OPN aptamer R3, P < .01, combination versus OPN siRNA, P < .01.
Tumor cell nucleolin expression and myeloid cell TLR9 for therapeutic targeting
In cancer cells, the nucleolin protein is reportedly distributed not only in the nucleolus and cytoplasm but also on the tumor cell surface,27 consistent with our IHC staining of patient GBM tissue (Figure 2a). AS1411 is a nucleolin-specific DNA molecule having a G-rich quadruplex structure that is resistant to nuclease degradation (Figure 2b). We treated murine GL261 cells as well as human D54, U87, and U251 GBM cells with Cy3 fluorophore labeled AS1411 and analyzed treated cells by flow cytometry. The results from this experiment showed nucleolin expression on the surface of 58.9% GL261 cells (Figure 2c), and on 24%, 33.1%, and 31.6% of D54, U87, and U251 cells, respectively (Figure 2d). This same approach was applied to the analysis of TLR9 expression on myeloid cells, CpG ODN as a probe, The results show TLR9 as being highly expressed in the cytoplasm and on the cell surface of Iba-1+ myeloid cells extracted from patient GBMs (Figure 2e). Flow cytometry analysis revealed TLR9 expression on murine GL261 cells (figure 2f) as well as on human glioma stem cells GSCs (Figure 2g), indicating that TLR9 targeted agents would interact with both cell types.
Figure 2.

Characterization of cell targeting moieties. A) Representative immunohistochemistry staining of nucleolin expression in human glioblastoma specimen. B) Structure of AS1411, a nucleolin specific DNA molecule having a G-rich quadruplex structure resistant to nuclease degradation. C) Cy3 fluorophore labeled AS1411 was co-incubated with murine GL261 cells and D) human D54, U87, and U251 glioma cells. E) Multiplex fluorescent imaging of glioblastoma demonstrating TLR9 was found to be highly expressed in the cytoplasm and on the cell surface of Iba-1+ myeloid cells. DAPI nuclear staining: blue; TLR9: green; Na-K ATPase membrane: red; Iba1: white. F) Flow cytometry analysis demonstrated TLR9 expression on murine GL261 cells. G) TLR9 expression on human glioblastoma stem cells (GSCs). Isotype control is white curve, marker expression is gray curve.
Development of OPN targeting aptamers
As motivated by the results above, we synthesized OPN-targeting aptamers by connecting a CpG ODN with a C9 spacer to an OPN siRNA duplex coupled with linker segments, which are henceforth designated as the CpG-OPN aptamer (Figure 3a). The same strategy was used for the creation of the Ncl-OPN aptamer except the AS1411 region replaced the CpG ODN region (Figure 3b). Gel electrophoresis confirmed nearly 100% annealing efficacy by showing a single band of the conjugated products with the correct lengths of 70 and 90 base pairs, respectively. To determine if the aptamer constructs were functional in vitro, control aptamer with scrambled sequence, CpG-OPN, or the Ncl-OPN were co-cultured with either mouse glioma GL261 cells or myeloid DC 2.4 cells. Quantitative PCR demonstrated that both aptamers decreased murine OPN mRNA levels relative to GAPDH in each cell line. For DC2.4 cells, treatment with CpG-OPN aptamer was more effective at reducing the OPN transcript relative to Ncl-OPN (Figure 3d). Aptamer treatments also reduced OPN protein levels in vitro, as indicated by results from western blot analysis which showed OPN protein reductions of 93% and 61% in DC2.4 cells treated with CpG-OPN and Ncl-OPN aptamers, respectively. Treatments with Ncl-OPN aptamer but not the CpG-OPN aptamer, caused modest reductions in human OPN mRNAs: a result suggesting species-dependent responses to aptamer treatments (Figure 3e). Furthermore, using murine-specific OPN siRNA aptamer duplexes, we determined intracellular OPN expression level of GL261, 344SQ and 4T1.B2b by Western Blot and found all of them had high OPN expression (figure 3f).
Figure 3.

A) Schema demonstrating the construction of the CpG-OPN aptamer. B) Schema demonstrating the construction of the Ncl-OPN aptamer. C) Agarose gel analysis showing the annealed final products at the correct molecular weight. D) Quantification of OPN and GAPDH mRNA after co-culture of either GL261 gliomas cells (Blue) or DC2.4 myeloid cells (Orange) with either controls (Ctrl) or the CpG-OPN or Ncl-OPN aptamers. E) Western blot analysis of OPN and actin protein in either DC2.4 myeloid cells or human U87 glioblastoma cells after co-incubation with scramble controls (Ctrl-Ctrl) or the CpG-OPN or Ncl-OPN aptamers at 200 nM. F) Western blot analysis of OPN and actin protein in mouse GL261, 344SQ and 4T1.B2b incubating with 200 nM Ctrl-Ctrl, CpG-OPN or Ncl-OPN aptamers. The ratio is the calculated amount of OPN expression relative to the control set at 1.
Benchmarking of the OPN aptamers relative to other devised strategies
Because there was only modest efficacy in the glioma preclinical model, we evaluated alternative oncology indications. OPN, TAP and STAT3 are all highly expressed and important in lung cancer progression, metastasis, and immune resistance.3,26,28–31 Given prior work involving the use of aptamers to suppress the expression of immune response modifiers, we synthesized additional siRNA aptamers (scramble control; Ncl-TAP2; CpG-TAP2; and OPN-STAT3) for use in comparing treatment effects against the CpG-OPN and Ncl-OPN aptamers (Supplementary Figure 1). The murine lung cancer line 344SQ with brain metastatic propensity, that was derived from a spontaneous and subcutaneous lesion from p53R172HΔg/+K-rasLA1/+ mice32 was used to test aptamers in vivo, with tumor cells engrafted in immune competent syngeneic 129 v host mice. Tumors were established in the flank of mice and animals were subsequently treated with siRNA aptamers by i.p. injection with treatments every other day over the course of two weeks. Tumor sizes were measured by caliper, with volumes plotted against time for comparing effects of each aptamer on tumor growth (Supplementary Figure 2). The results show that OPN siRNA aptamers were most effective (P = .002 for CpG-OPN vs aptamer control, P = .0223 for CpG-TAP2 vs aptamer control).
CpG ODN targeting OPN modulates the immune response
Next, we treated bone marrow derived myeloid cells with the CpG aptamer construct and observed cellular upregulation of CD40, indicating that the CpG aptamer conjugation does not impede the CpG immune modulatory effects (Figure 4a). An evaluation of the cytokine secretome from treated cells revealed similar profiles although there were lower levels of GM-CSF, IL-18, and MIF from the CpG-OPN aptamer treatments relative to CpG-NT (P < .001) attributed to cellular response heterogeneity (Figure 4b). SPP1 knocked-down efficacy in CpG-OPN siRNA treated myeloid cells was validated with qPCR (Figure 4c).
Figure 4.
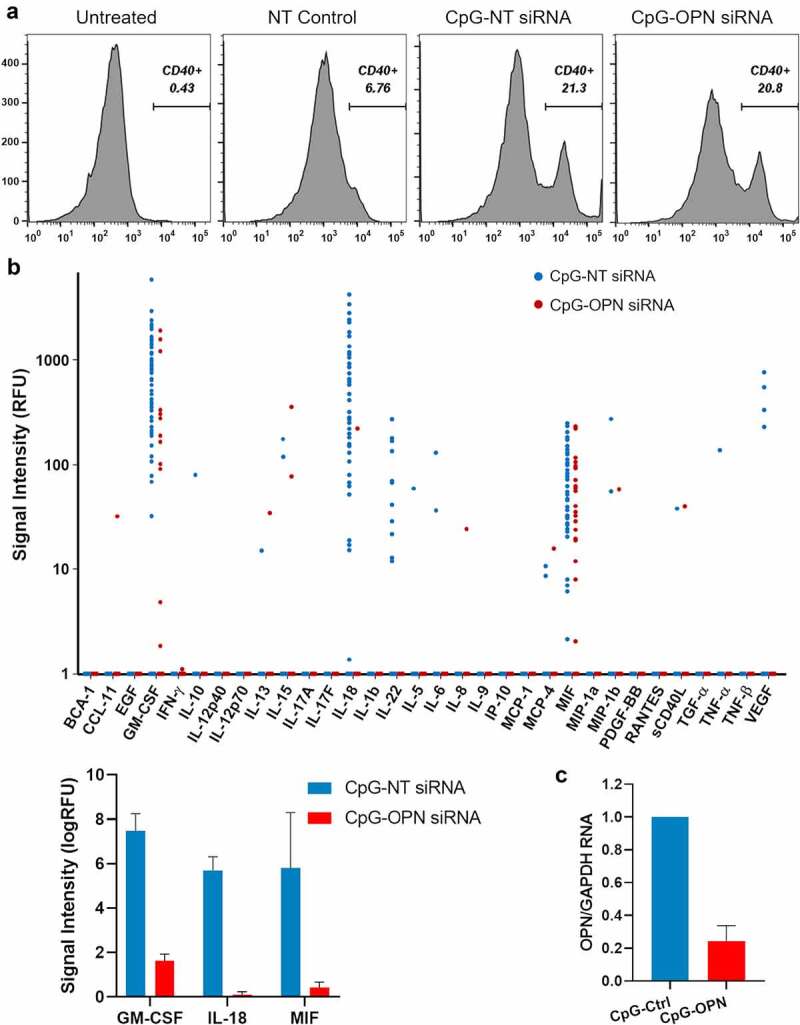
A) Flow cytometry data of CD40 expression on mouse bone marrow derived myeloid cells treated with a non-targeting (NT) control siRNA, CpG- NT siRNA, or CpG-OPN siRNA. B) IsoPlexis analysis of the cytokine secretome from human PBMC CD11b+ myeloid cells (n = 2) treated with CpG-NT siRNA or CpG-OPN siRNA. The most deregulated cytokines, GM-CSF, IL-18, and MIF, were plotted as a bar graph (p < .0001 for all). RFU: Relative fluorescence unit. C) Quantification of OPN and GAPDH mRNA in CpG-NT or CpG-OPN treated CD11b+ cells prior to the IsoPlexis analysis.
OPN targeting aptamers exert a therapeutic effect in vivo, using models of lung and breast cancer
Results from in vivo testing with the murine 344SQ lung cancer immunocompetent model showed that both the Ncl-OPN and the CpG-OPN aptamers inhibited tumor growth relative to scramble control (P = .0006 and P = .1852, respectively). To test if there would be additional therapeutic activity by targeting cancer and myeloid cells, these aptamers were evaluated in vivo and were found to suppress tumor growth but no synergistic effect (P = .1243) (Figure 5a). Ncl-TAP and CpG-TAP were also evaluated in the 344SQ lung cancer model, with results showing that suppression of TAP insignificantly delayed tumor growth relative to the scramble control using Ncl-TAP (P = .6881) and CpG-TAP (P = .0858). Combined TAP aptamer treatment was ineffective in this model relative to scrambled control (P = .4927) (Figure 5b). These aptamers were also not therapeutically efficacious when used in treating mice with intracranial 344SQ tumors (Supplementary Figure 3).
Figure 5.
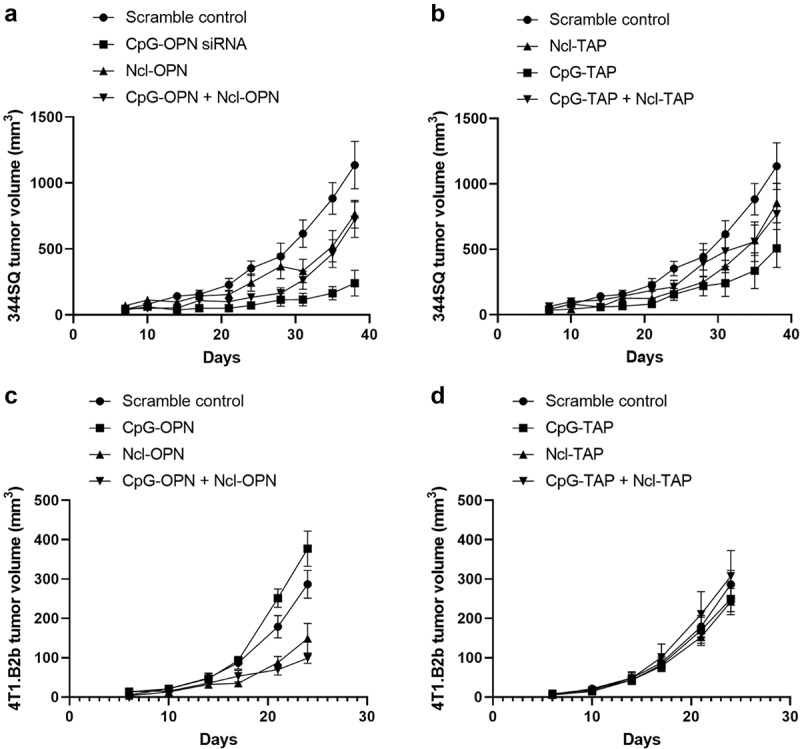
Lung and breast cancer tumor growth is suppressed in vivo when treated with CpG-OPN and Ncl-OPN aptamers. A) 344SQ murine lung cancer cells (originally derived from a spontaneous subcutaneous metastatic lesion in p53R172HΔg/+K-rasLA1/+ mice) were subcutaneously implanted into the posterior flank of syngeneic 129S mice. The aptamers were administered intraperitoneally every 3 days for 5 doses starting on day 7 (0.5 nmol/dose). There were 3 treatment groups, 5 mice/ group. The volumes of subcutaneous 344SQ tumor are shown from day 7 to 38. Error Bars represent standard error of the mean (SEM). P = .0006 for CpG-OPN relative to scramble control; P = .1852 for Ncl-OPN relative to scramble control; P = .1243 for their combination compared to scramble control. B) Same model as A. Ncl-TAP, CpG-TAP, and the combination relative to scramble control. C) 4T1.B2b murine breast cancer cells (originally derived from a brain metastatic lesion in 4T1 tumor bearing mice) were subcutaneously implanted into the posterior flank of syngeneic Balb/c mouse. The aptamer-siRNAs were administered intraperitoneally every 4 days for 3 doses starting on day 7 (1 nmol/dose). There were 3 treatment groups, 5 mice/ group. The volumes of subcutaneous 4T1 tumor were shown from day 6 to 24. P = .0302 and P = .0018 for Ncl-OPN and the combination of CpG-OPN and Ncl-OPN relative to scramble control. D) Same model as C but there was no statistically significant difference between experimental cohorts.
We tested an additional brain metastasis model for response to systemic aptamer administration to address whether the lack of efficacy observed in the prior experiment was tumor model specific. For this, we utilized a murine 4T1 breast cancer line derived from a brain metastasis in an immune competent syngeneic host (Supplementary Figure 4). Monotherapy treatment with the Ncl-OPN aptamer reduced tumor growth relative to the scramble control (P = .0302), showing added benefit when including CpG-OPN aptamer as part of a combination treatment (P = .0018) (Figure 5c). For this tumor model no growth suppression was noted upon treatment with the CpG-TAP and/or Ncl-TAP (Figure 5d). Nonetheless, we did observe a correlation of anti-tumor activity of the OPN siRNA aptamers to their variable silencing efficacy on intracellular OPN. For example, Ncl-mOPN siRNA exerted anti-GL261 glioma activity but this was not case for CpG-mOPN siRNA – consistent with Ncl duplex capable of inhibiting OPN, whereas CpG OPN duplex did not. CpG-mOPN siRNA was more effective than Ncl-mOPN siRNA in treating 344SQ lung cancer because OPN was suppressed more in CpG-mOPN siRNA treated 344SQ cells than Ncl-mOPN siRNA. In contrast, Ncl-mOPN siRNA was superior to CpG-mOPN siRNA on delaying 4T1.B2b tumor progression due to higher potency of Ncl-mOPN siRNA inhibiting OPN expression.
OPN aptamer distribution in the TME and therapeutic effect in mice with brain tumors
OPN in tumor and myeloid cells promotes the malignant biology of GBM, as indicated by the effects of OPN knockout in tumor cells.4 As such, we wanted to ascertain the therapeutic activity of these aptamers in the indication of glioma. These aptamers were fluorescently tagged (Figure 6a) and verified to bind to GL261 (Supplementary Figure 5). There was no significant difference in tagged aptamer binding to the 4T1.B2b, 344SQ and GL261 cell lines that would correlate with differences in therapeutic efficacy (Supplementary Figure 5A). To ascertain if the aptamers have access to the intracranial TME following systemic administration, fluorescently tagged aptamers were intravenously administered into glioma-bearing mice. Two hours later, fluorescent imaging was performed of the tumor-bearing mouse brains ex vivo (Figure 6b). While the tagged scrambled aptamer demonstrated minimal access to the TME, substantial access was indicated for CpG-OPN and Ncl-OPN aptamers (Figure 6c). To test the therapeutic effect of the aptamers in the GL261-C57BL6 model, aptamers were intraperitoneally administered 5 times, once every three days, beginning on day 6 or 7 after tumor implantation. 40% of mice treated with Ncl-OPN aptamer survived long term (Figure 6c). In contrast, the other tested aptamer siRNA complexes such as CpG-OPN, Ncl-TAP, and CpG-TAP exerted little anti-GL261 glioma activity (Supplementary Figure 5B). To ascertain the aptamer activity against breast cancer brain metastasis, mice with 4T1.B2b intracranial tumor were systemically treated with the aptamers via tail vein delivery. 4T1.B2b was derived from 4T1 after two continuous rounds of selection from peripheral circulation to the brain microenvironment in syngeneic Balb/c mouse recipients. Although monotherapy with either OPN aptamer failed to indicate anti-tumor activity, the combination of both CpG-OPN and Ncl-OPN increased survival to 49 days relative to control of 39 days (P = .0063) (Figure 6e), suggesting that dual targeting of tumor cells and myeloid cells will be important for achieving benefit from treatment, at least for certain types of cancer.
Figure 6.
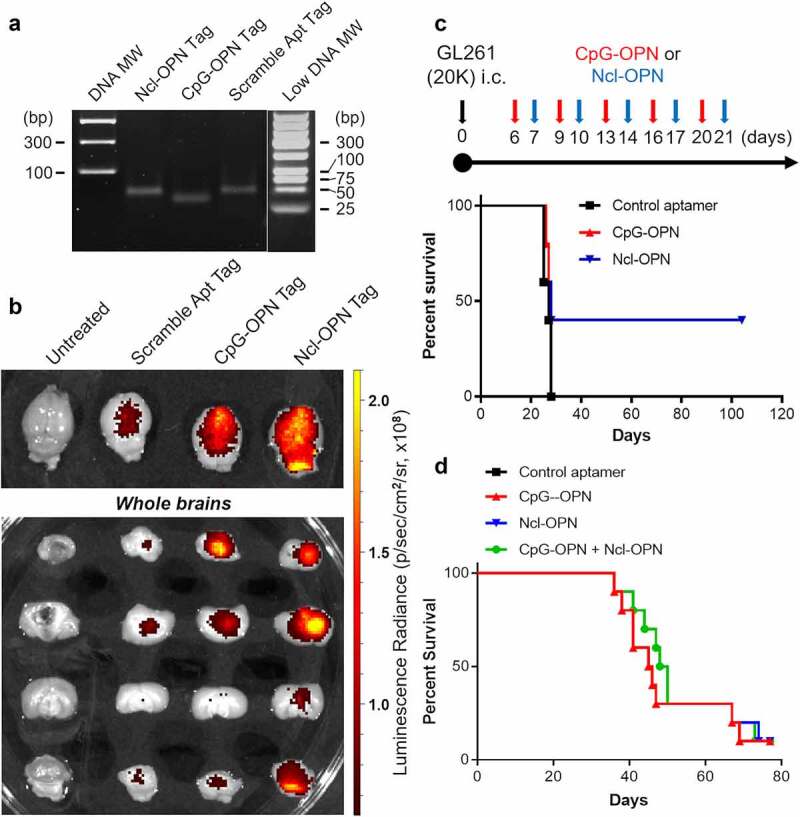
A) Agarose gel demonstrating florescent-tagged aptamers. B) Fluorescently tagged aptamers were intravenously administered in glioma-bearing mice. Two hours later, fluorescent imaging was performed of the extracted brains from untreated mice and those treated with a scrambled, CpG-OPN and Ncl-OPN aptamers. C) Treatment schema (top panel) and Kaplan-Meier survival analysis (bottom panel) of mice with intracerebral (i.c.) GL261 treated intraperitoneal with scramble control, CpG-OPN and the Ncl-OPN aptamer starting on day 6 or 7 after tumor implantation. There was no difference in median survival but there were 40% long term-survivors. D) Kaplan-Meier survival analysis of mice with immune competent breast cancer brain metastasis, 4T1.B2b (10,000 cells per mouse) intracranial implanted tumors treated i.v. with the scramble control, CpG-OPN, Ncl-OPN and combinatorial aptamers starting on day 3 for six doses (1 nmol per dose, a dose every 2 days). The median survival durations in the treatment groups are as follows: scramble control (n = 10), 39 days; CpG-OPN R3 (n = 10), 45.5 days; Ncl-OPN (n = 10), 41 days; and the combination of aptamers (n = 10), 49 days. Statistics: combination versus scramble control, P = .0063; combination versus CpG-OPN, P = .57, combination versus Ncl-OPN, P = .21.
In Fig.6d. the black line representing control aptamer treated group is hidden. Please fix.
Discussion
Aptamer-siRNA chimeras have appeared as one of the most efficient strategies for target delivery modules because of their high specificity and binding affinity, and fully automated synthesis. Nucleotide-based therapeutics have broad appeal for multiple cancer types and given their small size could potentially overcome the limitations of blood-brain-barrier (BBB) permeability33 thereby addressing the clinical unmet need for therapeutics that reside in the central nervous system. We have previously demonstrated that OPN mediates crosstalk between glioma cells and myeloid cells and is a critical chemokine attracting immune cells into the glioblastoma microenvironment both in vitro and in vivo, and that OPN ablation delays glioma progression and prolongs survival.4 Consequently, OPN is a promising therapeutic target. However, our prior data indicated that OPN neutralizing antibodies have no anti-glioma activity against GL261 mouse glioma, although they exerted anti-tumor activity in other cancer types.34,35 This discrepancy may be due to differences in cancer dependencies on the degree of secreted OPN and/or the lack of sufficient BBB penetration of the antibody. It has been reported that different cellular localizations of OPN are associated with distinct activities.3,36 Because the antagonistic OPN antibodies only target the extracellular secreted OPN, the intracellular OPN production remains. As such, we devised an aptamer strategy using OPN siRNA to suppress OPN mRNA, the source of OPN within the cell, and its subsequent secreted protein product. To target cytoplasmic OPN production, the siRNA was conjugated to a nucleolin or TLR9 targeting moiety such as AS1411 or CpG ODN, respectively. Given that the OPN aptamers demonstrated modest efficacy in delaying tumor growth in two cancer lineages, alternative targets will be considered.
This study provides a rational to select nucleolin and CpG ODN aptamers for respective targeting of tumor cells and stromal myeloid cells. In glioblastoma, the nucleolin protein is distributed not only in the nucleolus and cytoplasm but also on the tumor cell surface. Cy3 fluorophore labeled AS1411 binds to the surface of murine GL261 glioma cell line and human glioma cell lines indicating that glioma cell targeting by the AS1411 aptamer via nucleolin is a feasible strategy. Short CpG containing ODNs are a potent immune adjuvant, and their cognate receptor TLR9 is enriched in myeloid-derived APCs.37 Our study demonstrates that TLR9 is highly expressed on the Iba-1+ macrophages in the glioblastoma indicating CpG ODN is an ideal targeting ligand for macrophages. Moreover, the GL261 murine glioma also expresses TLR9 on the cell surface, as do human GSCs. Dual TLR9 expression on myeloid cells and GSCs makes the CpG-OPN aptamer capable of simultaneous targeting of these two major OPN-producing cell types in the TME. The therapeutic activity of the OPN aptamers is partially determined by their OPN knock-down activity within tumor cells in the TME. If there was sufficient signal of therapeutic response to warrant further translational efforts, these biomarkers could have been an area of future investigation. Lead therapeutic candidates can also be evaluated in the future in combination with other standards-of-care including immune checkpoint inhibitors. We observed a fraction of long-term survivors of GL261 glioma mice treated with Ncl-OPN siRNA but not CpG-OPN siRNA. This may be due to varied OPN inhibitory capacity on the glioma cells between these two aptamer complexes. Intrinsic factors may play a role in the survivors such as nucleotide therapeutic uptake to the tumor microenvironment or clearance rate. Further investigation is warranted to identify the associated factors and mechanisms. Efficacy of the combination of Ncl-OPN siRNA and CpG-OPN siRNA was observed in the 4T1.B2b breast cancer brain metastatic mouse model. A marked unmet need is for therapeutics that can treat breast cancer brain metastasis,38 and as such dual aptamer conjugates may be a promising strategy.
A major obstacle that needs to be overcome for delivery of siRNAs into the cytoplasm of the targeted tumor cells and/or immune cells is the negative charge of nucleotides and endosomal degradation. A strategy that could overcome this limitation is to embed the cell-specific aptamers into siRNA encapsulating nanoparticles, which could improve the delivery efficacy of naked siRNAs passing through the cellular barriers. OPN mRNA in breast cancer cells was knocked-down by 40% with a OPN siRNA delivered with polymer-based nanoparticles that led to significant reduction of in vivo tumor growth.39 Increasingly sophisticated nanoparticle systems, also relying on targeting moieties for BBB penetration and/or improved target cell transfection efficacy, may provide an avenue toward clinical application that demonstrate more robust signals of therapeutic response.
Our proposed dual-targeting therapeutics involving direct conjugation of a siRNA molecule to a nucleic acid aptamer which reduces the complexity of manufacturing of these reagents. Systemic administration requires greater therapeutic doses (leading to higher treatment costs) and carries a greater risk for harmful side effects owing to greater exposure of non-targeted tissues. Improvements that would minimize the necessary dose of the complex would reduce both the cost of treatment and the risk for harmful side effects. To circumvent degradation by serum nuclease, a portion of the nucleotides in the siRNAs and aptamers evaluated in this study has been modified with a nuclease-resistant 2-O methyl moiety and a phosphorothioate linkage, which enhances the in vivo stability.40 Because some of the tested aptamers are species specific, human-specific aptamers will need to be synthesized and validated prior to initiating clinical trials. Future studies should also evaluate the potential of OPN targeting, using the aptamer approach, to enhance the effects of temozolomide or radiation therapy in GBM. Moreover, the overall approach may have important implications in other tumors as well, as OPN engages signaling pathways41 important for neoplastic cell survival and growth and this remains to be established in future studies.
Supplementary Material
Funding Statement
This study was funded by the Ben and Catherine Ivy Foundation and Provost Retention Funds (ABH), and the MDACC Institutional Research Grant (JW).
Novelty statement
This manuscript demonstrates that nucleolin and CpG aptamers can be exploited to target intracellular therapeutic targets in both cancer and myeloid cells, respectively. Using fluorescent tags, conjugated aptamers were shown to pass through the blood-brain barrier and can be used as therapeutics for both gliomas and brain metastasis in preclinical models.
Disclosure statement
The authors have declared that no conflict of interest exists related to the subject matter of this manuscript.
Data Availability Statement
The data used to support the findings of this study are available within this article.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med. 2018;378(5):449–13. published Online First: 2018/02/01. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urbani F, Ferraresi V, Capone I, Macchia I, Palermo B, Nuzzo C, Torsello A, Pezzotti P, Giannarelli D, Pozzi AF, et al. Clinical and Immunological Outcomes in High-Risk Resected Melanoma Patients Receiving Peptide-Based Vaccination and Interferon Alpha, With or Without Dacarbazine Preconditioning: a Phase II Study. Front Oncol. 2020;10. doi: 10.3389/fonc.2020.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sangaletti S, Tripodo C, Sandri S, Torselli I, Vitali C, Ratti C, Botti L, Burocchi A, Porcasi R, Tomirotti A, et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014;74(17):4706–4719. published Online First: 2014/07/19. doi: 10.1158/0008-5472.Can-13-3334. [DOI] [PubMed] [Google Scholar]
- 4.Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, Grami Z, Kong L-Y, Ling X, Caruso H, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. 2019;129(1):137–149. published Online First: 2018/10/12. doi: 10.1172/jci121266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamour V, Le Mercier M, Lefranc F, Hagedorn M, Javerzat S, Bikfalvi A, Kiss R, Castronovo V, Bellahcène A.. Selective osteopontin knockdown exerts anti-tumoral activity in a human glioblastoma model. Int Journal of Cancer. 2010;126(8):1797–1805. doi: 10.1002/ijc.24751. [DOI] [PubMed] [Google Scholar]
- 6.Butti R, Kumar TVS, Nimma R, Banerjee, P, Kundu, IG, Kundu, GC, et al. Osteopontin Signaling in Shaping Tumor Microenvironment Conducive to Malignant Progression. Adv Exp Med Biol. 2021;1329:419–441. published Online First: 2021/10/20. doi: 10.1007/978-3-030-73119-9_20. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Liu H, Zhao Y, Yue D, Chen C, Li C, Zhang Z, Wang C. Tumor-associated macrophages (TAMs)-derived osteopontin (OPN) upregulates PD-L1 expression and predicts poor prognosis in non-small cell lung cancer (NSCLC). Thorac Cancer. 2021;12(20):2698–2709. doi: 10.1111/1759-7714.14108. published Online First: 2021/08/24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue M, Shinohara ML. Intracellular osteopontin (iOPN) and immunity. Immunol Res. 2011;49(1–3):160–172. doi: 10.1007/s12026-010-8179-5. published Online First: 2010/12/08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hossain DM, Dos Santos C, Zhang Q, Kozlowska A, Liu H, Gao C, Moreira D, Swiderski P, Jozwiak A, Kline J, et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood. 2014;123(1):15–25. published Online First: 2013/10/31. doi: 10.1182/blood-2013-07-517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers B, Grufman P, Fredriksson V, Andersson K, Roseboom M, Laban S, Camps M, Wolpert EZ, Wiertz EJHJ, Offringa R, et al. Induction of protective CTL immunity against peptide transporter TAP-deficient tumors through dendritic cell vaccination. Cancer Res. 2007;67(18):8450–8455. doi: 10.1158/0008-5472.CAN-07-1092. [DOI] [PubMed] [Google Scholar]
- 11.Doorduijn EM, Sluijter M, Querido BJ, Oliveira CC, Achour A, Ossendorp F, van der Burg SH, van Hall T. TAP-independent self-peptides enhance T cell recognition of immune-escaped tumors. J Clin Invest. 2016;126(2):784–794. doi: 10.1172/jci83671. published Online First: 2016/01/20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei J, Wang F, Kong LY, Xu S, Doucette T, Ferguson SD, Yang Y, McEnery K, Jethwa K, Gjyshi O, et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 2013;73(13):3913–3926. published Online First: 2013/05/03. doi: 10.1158/0008-5472.Can-12-4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9(1):67–78. published Online First: 2010/01/08. doi: 10.1158/1535-7163.Mct-09-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia W, Yao Z, Zhao J, Guan Q, Gao L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 2017;186:1–10. published Online First: 2017/07/29. doi: 10.1016/j.lfs.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Mosafer J, Mokhtarzadeh A. Cell Surface Nucleolin as a Promising Receptor for Effective AS1411 Aptamer-Mediated Targeted Drug Delivery into Cancer Cells. Curr Drug Deliv. 2018;15(9):1323–1329. doi: 10.2174/1567201815666180724104451. published Online First: 2018/07/25 [DOI] [PubMed] [Google Scholar]
- 16.Ugrinova I, Petrova M, Chalabi-Dchar M, Bouvert, P, et al. Multifaceted Nucleolin Protein and Its Molecular Partners in Oncogenesis. Adv Protein Chem Struct Biol. 2018;111:133–164. published Online First: 2018/02/21. doi: 10.1016/bs.apcsb.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Wei T, Zhao J, Huang Y, Deng H, Kumar A, Wang C, Liang Z, Ma X, Liang X-J, et al. Multifunctional aptamer-based nanoparticles for targeted drug delivery to circumvent cancer resistance. Biomaterials. 2016;91:44–56. published Online First: 2016/03/21. doi: 10.1016/j.biomaterials.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Bates PJ, Laber DA, Miller DM, Thomas SD, Trent JO. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp Mol Pathol. 2009;86(3):151–164. doi: 10.1016/j.yexmp.2009.01.004. published Online First: 2009/05/21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leng L, Jiang T, Zhang Y. TLR9 expression is associated with prognosis in patients with glioblastoma multiforme. J Clinical Neuroscience. 2012;19(1):75–80. doi: 10.1016/j.jocn.2011.03.037. [DOI] [PubMed] [Google Scholar]
- 20.Miyar A, Habibi I, Ebrahimi A, Mansourpour D, Mokarizadeh A, Rajabi A, Farshgar R, Eshaghzadeh M, Zamani-Ahmadmahmudi M, Nodushan SMHT, et al. Predictive and prognostic value of TLR9 and NFKBIA gene expression as potential biomarkers for human glioma diagnosis. J Neurol Sci. 2016;368:314–317. doi: 10.1016/j.jns.2016.07.046. [DOI] [PubMed] [Google Scholar]
- 21.Andaloussi AE, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme1. Neuro-Oncology. 2006;8(3):234–243. doi: 10.1215/15228517-2006-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ursu R, Carpentier A, Metellus P, Lubrano V, Laigle-Donadey F, Capelle L, Guyotat J, Langlois O, Bauchet L, Desseaux K, et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma—a phase II multicentric, randomised study. Eur J Cancer. 2017;73:30–37. doi: 10.1016/j.ejca.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Bajan S, Hutvagner G. RNA-Based Therapeutics: from Antisense Oligonucleotides to miRNAs. Cells. 2020;9(1):137. doi: 10.3390/cells9010137. published Online First: 2020/01/16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adachi T, Nakamura Y. Aptamers: a Review of Their Chemical Properties and Modifications for Therapeutic Application. Molecules. 2019;24:4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113–1125. doi: 10.1093/neuonc/noq082. published Online First: 2010/07/30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrido G, Schrand B, Rabasa A, Levay, A, Eramo, F, Berezhnoy, A, Modi, S, Gefen, T, Marijt, K, Doorduijn, E, Dudeja, V, van Hall, T, Gilboa, E, et al. Tumor-targeted silencing of the peptide transporter TAP induces potent antitumor immunity. Nat Commun. 2019;10(1):3773. doi: 10.1038/s41467-019-11728-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrara B, Belbekhouche S, Habert D, Houppe C, Vallée B, Bourgoin-Voillard S, Cohen JL, Cascone I, Courty J. Cell surface nucleolin as active bait for nanomedicine in cancer therapy: a promising option. Nanotechnology. 2021;32(32). doi: 10.1088/1361-6528/abfb30. published Online First: 2021/04/24 [DOI] [PubMed] [Google Scholar]
- 28.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, Zhang C, Liang W, Scuto A, Weng S, et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71(21):6601–6610. published Online First: 2011/09/17. doi: 10.1158/0008-5472.Can-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Liu H, Zhao Y, Yue D, Chen C, Li C, Zhang Z, Wang C, et al. Tumor-associated macrophages (TAMs)-derived osteopontin (OPN) upregulates PD-L1 expression and predicts poor prognosis in non-small cell lung cancer (NSCLC). Thoracic Cancer. 2021;20(12):2698–2709. doi: 10.1111/1759-7714.14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao C, Cui Y, Chang S, Huang J, Birkin E, Hu M, Zhi X, Li W, Zhang L, Cheng S, Jiang WG, et al. OPN promotes the aggressiveness of non-small-cell lung cancer cells through the activation of the RON tyrosine kinase. Sci Rep. 2019;9(1):18101. published Online First: 2019/12/04. doi: 10.1038/s41598-019-54843-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamort AS, Giopanou I, Psallidas I, Stathopoulos GT. Osteopontin as a Link between Inflammation and Cancer: the Thorax in the Spotlight. Cells. 2019;8(8):815. doi: 10.3390/cells8080815. published Online First: 2019/08/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26(48):6896–6904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
- 33.Wei J, Gilboa E, Calin GA, Heimberger AB. Immune Modulatory Short Noncoding RNAs Targeting the Glioblastoma Microenvironment. Front Oncol. 2021;11:682129. published Online First: 2021/09/18. doi: 10.3389/fonc.2021.682129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim E-K, Jeon I, Seo H, Park Y-J, Song B, Lee K-A, Jang Y, Chung Y, Kang C-Y. Tumor-Derived Osteopontin Suppresses Antitumor Immunity by Promoting Extramedullary Myelopoiesis. Cancer Res. 2014;74(22):6705–6716. doi: 10.1158/0008-5472.Can-14-1482. [DOI] [PubMed] [Google Scholar]
- 35.Mi Z, Guo H, Russell MB, Liu Y, Sullenger BA, Kuo PC. RNA aptamer blockade of osteopontin inhibits growth and metastasis of MDA-MB231 breast cancer cells. Molecular Therapy. 2009;17(1):153–161. doi: 10.1038/mt.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leavenworth JW, Verbinnen B, Wang Q, Shen E, Cantor H. Intracellular osteopontin regulates homeostasis and function of natural killer cells. Proceedings of the National Academy of Sciences. 2015;112(2):494–499. doi: 10.1073/pnas.1423011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adamus T, Kortylewski M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp Oncol (Pozn). 2018;22(1a):56–60. doi: 10.5114/wo.2018.73887. published Online First: 2018/04/10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hosonaga M, Saya H, Arima Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer Metastasis Rev. 2020;39(3):711–720. doi: 10.1007/s10555-020-09881-y. published Online First: 2020/05/14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-David-Naim M, Dagan A, Grad E, Aizik G, Nordling-David M, Morss Clyne A, Granot Z, Golomb G. Targeted siRNA Nanoparticles for Mammary Carcinoma Therapy. Cancers (Basel). 2019;11(4):442. doi: 10.3390/cancers11040442. published Online First: 2019/04/03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16(3):181–202. doi: 10.1038/nrd.2016.199. published Online First: 2016/11/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao H, Chen Q, Alam A, Cui J, Suen KC, Soo AP, Eguchi S, Gu J, Ma D. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. 2018;9(3):356. doi: 10.1038/s41419-018-0391-6. published Online First: 2018/03/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to support the findings of this study are available within this article.