

ABSTRACT
Group B streptococcus (Streptococcus agalactiae, GBS) is an important cause of life-threatening disease in newborns. Pregnant women colonized with GBS can transmit the bacteria to the developing fetus, as well as to their neonates during or after delivery where infection can lead to sepsis, meningitis, pneumonia, or/and death. While intrapartum antibiotic prophylaxis (IAP) is the standard of care for prevention of invasive GBS disease in some countries, even in such settings a substantial residual burden of disease remains. A GBS vaccine administered during pregnancy could potentially address this important unmet medical need and provide an adjunct or alternative to IAP for the prevention of invasive GBS disease in neonates. A hurdle for vaccine development has been relatively low disease rates making efficacy studies difficult. Given the well-accepted inverse relationship between anti-GBS capsular polysaccharide antibody titers at birth and risk of disease, licensure using serological criteria as a surrogate biomarker represents a promising approach to accelerate the availability of a GBS vaccine.
KEYWORDS: GBS;invasive disease;infants;IAP;capsular polysaccharide;conjugate vaccine;maternal immunization;serocorrelate
Introduction
Group B streptococcus (Streptococcus agalactiae, GBS) is an encapsulated opportunistic Gram positive pathogen that usually harmlessly colonizes the lower intestinal and rectovaginal area of many adults. Colonization is dynamic, in that GBS carriage can be lost and reacquired.1 When protection by the immune system is impaired, such as due to immunological immaturity in young infants and immunosenescence in older adults, invasive disease can manifest as sepsis, meningitis or pneumonia. Most invasive neonatal GBS disease is diagnosed within the first week of life and is defined as early onset disease (EOD). Invasive GBS disease occurring between the first week and third month of life is considered late onset disease (LOD).2 A meta-analysis of epidemiological studies reported between 2000 and 2017 determined an overall (EOD + LOD) global incidence rate of invasive neonatal GBS disease of 0.49/1,000 live births and an associated 8.4% mortality rate.3 Even when not fatal, invasive GBS disease in infants causes significant morbidity with serious long-term sequelae. In surveillance conducted from 2003 to 2007 in the United States (U.S.), GBS was found to be responsible for more than 86% of bacterial meningitis episodes in children <2 months of age.4 In a separate study, 11% of infants with GBS meningitis died, and of those surviving, ~19% had neurologic sequelae including cognitive delay, cerebral palsy, blindness, or hearing loss.5 The recent Full Value of Vaccine assessment from the World Health Organization (WHO) estimated an annual burden of ~392,000 invasive GBS cases in infants with ~91,000 deaths and ~40,000 with neurodevelopmental impairment. Additionally, GBS was associated with ~46,000 stillbirths and 518,000 preterm births.6 These sobering figures reinforce the urgent need for development of an efficacious vaccine to address this substantial unmet medical need.
A study of GBS isolates from colonized mothers and their infants, who were either colonized or had GBS EOD, demonstrated that bacteria are likely transferred from the mother to the baby in utero or during passage through the birth canal.7 The maternal rectovaginal colonization rate in the U.S. has been estimated at 25%, with a global rate of 18%.8 Although premature labor, intrapartum fever and prolonged ruptured membranes increase the chance of EOD in the newborn, the main risk factor is the colonization status of the mother within a few weeks prior or during labor.9 A landmark study by Boyer and Gotoff10 reported an incidence rate of EOD at 1.9/1,000 (61 cases/32,384 live births) among babies born from colonized mothers between 1973 and 1981.
In addition to impact of colonization on the infant, GBS can cause ascending infections in pregnant women that may directly affect the developing fetus. In pregnant women, GBS may be associated with chorioamnionitis which may result in stillbirth or preterm delivery, and puerperal sepsis which may be fatal.11 GBS infection inutero occurring proximal to the time of delivery may also potentially account for some instances of GBS EOD where infection is documented on the first day of life. As support for this notion, a recent report documented the presence of GBS genomic DNA in approximately 5% of placental samples collected prior to onset of labor.12 Furthermore, a separate study found that 2.6% of infants born by elective cesarian section to GBS-colonized mothers were already colonized with GBS at delivery.13 In a single-center historical cohort study of a total of 60,029 births, performed between 2003 and 2015 in the U.S., 0.1% of the maternal population had invasive GBS disease.14 Data from meta-analyses conducted in high-income countries suggest that the incidence of invasive maternal GBS disease was 0.38/1,000 pregnancies,15 which is slightly higher than the current incidence of EOD in infants in the U.S. (0.25/1,000 live births).16 The multifactorial causes of stillbirth and preterm delivery makes the role of maternal GBS colonization hard to quantify. Conservatively, 1–4% of stillbirths have been attributed to GBS,17 and maternal GBS colonization has been estimated to increase the risk for preterm delivery by 21–85%.18
Partial protection against invasive neonatal GBS disease following introduction of universal GBS screening and intrapartum antibiotic prophylaxis
Given the high rates of invasive infant disease in the U.S., prevention of invasive GBS using prophylactic antibiotics was evaluated in clinical trials. Intrapartum antibiotic prophylaxis (IAP) provided to GBS-colonized pregnant women during labor was shown to reduce the incidence of EOD and colonization of the neonate.19 As a result, IAP was introduced in the U.S. in the late 1990s. Guidelines were expanded in 2002 to include universal screening for rectovaginal GBS carriage during the 35-37th week of pregnancy.
Introduction of IAP has reduced the incidence of EOD in the U.S. from ~1.7/1,000 live births in 1990 to ~0.26/1,000 live births in 2010.20 While adoption of universal screening and administration of IAP has been highly effective in preventing EOD, the initial reduction largely achieved in the early 2000s subsequently plateaued.20 The remaining, constant incidence rate illustrates that the IAP intervention following screening is not 100% effective for a variety of reasons including missed screenings in women with limited access and false-negative culture results.21 In one study, 61% of women with term infants that developed GBS EOD had negative screening results, suggesting that acquisition occurred between the screening test and delivery, or the screening result was falsely negative.21 Additionally, universal screening and IAP have not meaningfully affected the incidence of LOD.22,23 Because the majority of cases of infant meningitis in the U.S. are associated with GBS LOD, the incidence of GBS meningitis has similarly not decreased.
Finally, the impact of long-term widespread use of prophylactic antibiotics on drug resistance is a concern. Combined with prophylaxis to prevent infections following cesarian deliveries, antibiotics are administered in approximately 40% of all labor and delivery procedures in the U.S., leading to a growing apprehension about development of antimicrobial resistance among human bacterial pathogens associated with maternal and neonatal disease.24,25 The concern was highlighted at the time that universal screening and use of IAP occurred in the U.S. was recommended with inclusion of the explicit caveat that antibiotic resistance would need to be monitored until a vaccine became available.26 Resistance to first-line beta-lactam antibiotics has not yet been seen; however, substantial resistance has been documented to second-line antibiotics such as erythromycin and clindamycin given to individuals for whom beta-lactam antibiotics such as penicillin are contraindicated,27 who constitute up to 10% of the U.S. population who report having an allergic reaction in their past.28 In other high-income countries (HICs [e.g., UK]), a risk-based approach to IAP has been implemented out of concerns for overuse of antibiotics, medicalization of labor and delivery, the transient nature of colonization, and resource concerns. The risk-based approach focuses on screening women with a history of GBS or a history of an GBS-infected child, in addition to other factors such as premature labor, intrapartum fever and/or amnionitis, and prolonged rupture of membranes.29,30 This approach has significant limitations since a considerable proportion of infant GBS disease cases may not be associated with identified risk factors.20 However, neither approach has fully eliminated GBS disease in infants.
In low- and middle-income countries (LMICs), where healthcare access and standards of prenatal care may vary or the resources or infrastructure for significant preventative interventions are not available, the rates of GBS disease are significantly higher. For instance, one surveillance study in South Africa estimated the rate of GBS EOD to be 1.41 cases per 1,000 live births and LOD to be 1.18 cases per 1,000 births.31 Moreover, the limited medical infrastructure in these regions makes capture of all cases difficult so the true burden may be under-represented. This is illustrated by the example of a multi-site study that evaluated reported GBS cases for which inexplicably no cases were recorded at a study site in Bangladesh. By comparison, this study recorded GBS EOD/LOD cases in infants from Panama, Dominican Republic, and Hong Kong with estimates of 0.77/0.58, 2.35/0.17, 0.76/0.38 cases per 1,000 live births, respectively.32
A maternal GBS vaccine as an alternative to IAP
Maternal GBS immunization offers an alternative approach to screening and antibiotic prophylaxis for GBS disease prevention in infants. There is a long history of success in protecting infants and adults against diseases caused by encapsulated bacteria using vaccines that target the capsular polysaccharide (CPS) and generate functional antibodies.33 Experience with investigational GBS conjugate vaccines in the literature suggests that this is a promising approach.34,35 However, active immunization of infants would not be effective in the case of GBS disease, since the time of highest risk is from birth through the first 3 months of life. In addition, the risk timeframe also includes the intrapartum period with the majority of EOD occurring within the first 24 h. The preponderance of disease during peripartum and early life thus precludes having sufficient time to develop an immune response in vaccinated neonates to provide coverage against GBS during the period of risk. Therefore, maternal immunization could serve as a mechanism to ensure that sufficient protective antibodies, including anti-CPS IgG, are actively transported across the placenta from the mother to the baby via the neonatal Fc receptor (FcRN) with selective subclass preference for IgG1, followed by IgG2, IgG3, and IgG4 (Figure 1(a)).36,37 Thus, this would lead to sufficient antibody levels circulating in the infant at the time of birth and the immediate postpartum period persisting throughout the period of risk (Figure 1(b)). These antibodies, if present at the appropriate levels, could protect the baby from GBS disease. Administering a vaccine to pregnant women to induce target-specific antibodies meant to cross the placenta and prevent disease in the infant enhances the pathway of natural immunity and is a well-established strategy for prevention of neonatal disease. Vaccination of pregnant women has been successfully used as a public health tool globally in the prevention of neonatal tetanus and more recently for prevention of pertussis in young infants, and to protect women and their infants against influenza.38 In addition, recent guidance from the U.S. Centers for Disease Control and Prevention provides recommendations for the use of COVID-19 vaccines in pregnant women.39 Polysaccharide conjugate vaccines are ideally suited for this purpose, as adults have a mature immune system and generally demonstrate robust, relatively rapid immunological responses with functional activity after a single vaccine dose. Immunization with a GBS conjugate vaccine may also reduce maternal GBS carriage, potentially providing additional benefit for prevention of transmission and consequent reduction of infant disease.40 Finally, there are estimates that a GBS vaccine with moderate (>70%) efficacy implemented in an LMIC setting could prove cost-effective by averting over one-third of GBS cases and deaths.41
Figure 1.
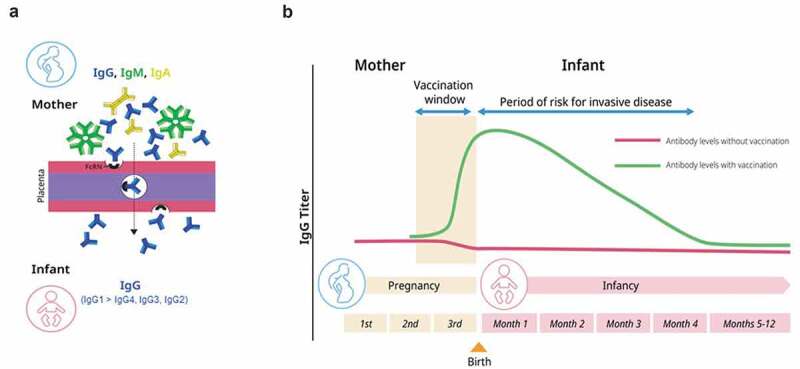
Schematic representation of the presence of immunoglobulins in infants due to maternal vaccination. (a) Maternal IgG is selectively transported across the placenta by the neonatal Fc receptor (FcRN). (b) Maternal vaccines augment or induce maternal antibody levels to protect the infant from infectious disease in the first few months of life.
GBS capsular polysaccharides are important virulence factors and have been linked to protective responses
GBS express the CPS as a mechanism of immune evasion. The CPS contains terminal sialic acid moieties that interfere with binding of the complement component C3 and thus shield the pathogen from the alternative complement pathway of the immune system.42 There are 10 capsular serotypes of GBS (Ia, Ib-IX), which are composed of five sugar components, glucose, galactose, N-acetyl glucosamine and N-acetyl neuraminic acid (sialic acid), and, in the case of type VIII, rhamnose.43 Serotype specificity is conferred by the specific arrangement of monosaccharides within each repeated polysaccharide unit. All GBS serotypes have been found to cause disease, but their prevalence is variable.44,45 Globally, serotypes Ia and III are responsible for the majority of EOD and LOD disease accounting in one study for 66%–80% of cases by region (Figure 2)3,46 with the remaining disease attributed primarily to serotypes Ib, II, IV, and V.
Figure 2.
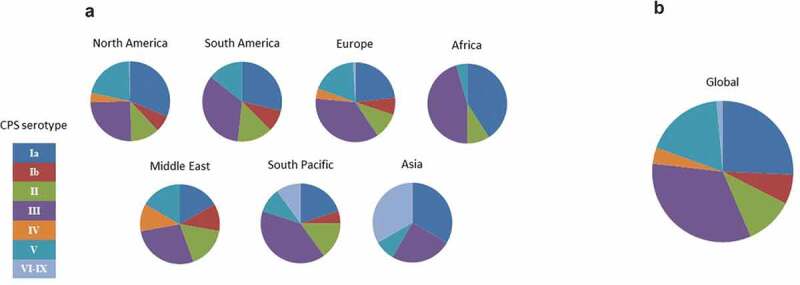
Global prevalence of GBS serotypes causing neonatal disease (2004–2013). (a) Distribution of serotypes by region. (b) Overall global distribution. Adapted from Buurman et al.46
In the late 1970s, a study among women colonized with GBS serotype III described an association between low maternal serotype III-specific IgG concentrations and infant susceptibility to EOD caused by serotype III strains.47 This observation was later also made for serotypes Ia and V.48,49 Preclinical research efforts further corroborated the protective capacity of anti-CPS antibodies.46,50,51 These seminal findings established the rationale for induction of anti-CPS IgG antibodies as a mechanism to protect against invasive GBS disease and paved the way for development of GBS polysaccharide vaccines.
GBS glycoconjugate vaccines in clinical development
Given the public health need for prophylactic interventions to prevent invasive GBS disease among neonates and infants in HIC and LMIC settings, development of a GBS vaccine has been an active area of research since the 1980s. Pioneering work from Carol Baker, Dennis Kasper and colleagues tested the first CPS vaccines and later established that monovalent GBS CPS glycoconjugates for serotypes Ia, Ib, II, III and V were immunogenic and could facilitate the transfer of antibodies induced from vaccinating the mother to their fetus.52–56 Subsequently, GlaxoSmithKline (GSK) advanced a trivalent investigational vaccine (serotypes Ia, Ib, III) through Phase 2 clinical development, demonstrating safety and good tolerability as well as immunogenicity in both non-pregnant and pregnant women, with GBS-specific antibody transfer from mothers to infants.57–60 At the time of this writing, there are no active or imminent trials announced for this vaccine.
Pfizer is developing a hexavalent GBS CPS conjugate vaccine (GBS6) that represents the highest valency vaccine formulation to date and offers the most extensive coverage against invasive GBS disease. GBS6 includes CPS from serotypes Ia, Ib, II, III, IV, and V individually conjugated to CRM197 and offers coverage against up to 97% or more of strains causing infant disease globally.46 As most women of childbearing age will have been colonized with GBS at various times in their lives before becoming pregnant,1 their immune response should have been primed to recognize GBS CPS, and thus, a single administration with GBS6 may be sufficient to induce an anamnestic protective response.34 To demonstrate preclinical efficacy for GBS6, a murine maternal transfer challenge model was utilized where dams were immunized with GBS6 or placebo and pups were subsequently challenged with GBS strains expressing the CPS serotypes covered by the vaccine after birth. Whereas >80% mortality was found upon challenge of pups born to dams administered placebo, GBS6 vaccination conferred significant protection against challenge for pups born to immunized dams with 88–100% of pups surviving.46 Additionally, passive administration of pregnant dams with monoclonal IgG antibodies specific for the CPS serotypes in GBS6 also protected their pups against challenge. Taken together, these findings indicated that immunization with GBS6 elicited protective antibodies that were transferred to pups and protected against GBS challenge. Importantly, these results provided supportive preclinical evidence for advancement of GBS6 to clinical studies.
Pfizer began clinical development of GBS6 in 2017. The program began with the conduct of a first-in-human study in the U.S. that was a Phase 1/2 dose escalation study evaluating three dose levels and two formulations of GBS6 in healthy adult men and women 18–49 years of age.34 This study demonstrated that GBS6 was well tolerated in healthy adults. From a safety perspective, administration of GBS6 was primarily characterized by mild pain at the injection site seen more frequently in participants dosed with the adjuvanted formulation of the vaccine. Few differences in systemic reactions or other adverse events were observed in vaccine recipients as compared to placebo. Importantly, the post-vaccination fever rate was low. GBS6 elicited robust immune responses for all dose levels and formulations. IgG geometric mean concentrations peaked as early as 2 weeks after vaccination, were sustained through at least 1 month, and then declined gradually. The immune response, however, persisted for at least 6 months after vaccination, as demonstrated by preserved fold rises in IgG concentrations from pre-vaccination levels (Figure 3). These results supported further evaluation of GBS6 in pregnant women.
Figure 3.
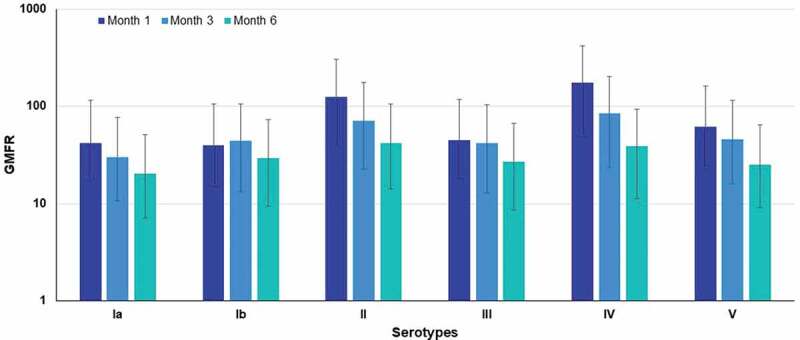
Serotype-specific IgG geometric mean fold rise (GMFR) from baseline at 1, 3 and 6 months following GBS6 vaccination for 120 μg (20 μg CPS/serotype/dose) dose level formulated without aluminum phosphate.
A Phase 1/2 randomized controlled trial in pregnant women in South Africa and the UK was subsequently initiated in 2018 and is ongoing at the time of writing this review (NCT03765073). This study aims to determine the safety and immunogenicity of GBS6 in pregnant women, as well as establish whether sufficient antibodies are transferred to their newborns to reduce the risk of GBS infection.
Anti-Capsular GBS IgG concentrations are linked to protection of infants
Based on the established inverse relationship between anti-CPS IgG titer at birth and risk of invasive GBS disease, elucidation of the protective titers associated with anti-CPS antibody could be linked to induced antibody levels for the development of GBS vaccines. To determine the serotype-specific protective level of maternally transferred anti-CPS IgG, multiple seroepidemiology studies conducted over the previous decades have set out to determine protective antibody levels based on naturally induced anti-CPS IgG (Table 1). Practical considerations for capture of sufficient number of cases have generally been limited to the most prevalent serotypes, Ia and III. Moreover, these studies were not harmonized and different protocols, immunogenicity assays and reference standards were used, as well as different statistical methods (Table 2). Most recent studies have used the standardized human reference sera established by Dr. Carol Baker, which are composed of immune sera from subjects with strong immune responses following immunization with monovalent tetanus toxoid conjugates.61 Despite this improvement, immunological data still could not be compared, as they were generated using different assays and reagents that were not standardized. In addition, the use of maternal or infant sera to derive the estimated protective level is of importance. Given that antibody levels in the infant may be influenced by placental transfer rates and selectivity for IgG isotypes, thresholds based on maternal sera may not adequately define a protective level in infants. Indeed, several studies have indicated that the protective threshold for transferred anti-GBS CPS IgG is lower than the maternal level.57,62,63 The protective threshold derived from transferred IgG in the infant is thus likely more relevant for defining a risk of acquiring EOD and LOD, as this better represents available antibodies at the site of infection.
Table 1.
Summary of prior seroepidemiological studies for GBS
| Serotype |
Region |
EOD/LOD |
Healthy controls |
Ref |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ia |
III |
V |
|
|
|
|
||||
| Proposed protective threshold (serum source) |
Cases | Proposed protective threshold (serum source) |
Cases | Proposed protective threshold (serum source) |
Cases | |||||
| 1 | 5 μg/mLa (maternal) 4 μg/mLa (cord) |
50 | 10 μg/mLa (maternal) 7 μg/mLa (cord) |
26 | – | – | USA | EOD | Colonized infants | 49, 66 |
| 2 | 0.5 μg/mLa (maternal) | 17 | 0.5 μg/mLa (maternal) | 9 | 0.5 μg/mLa (maternal) | 7 | USA | EOD | Infants from colonized mothers | 48 |
| 3 | 6 μg/mLb (maternal) | 27 | 3 μg/mLb (maternal) | 29 | – | – | South Africa | EOD + LOD | Infants from colonized mothers | 62 |
| 4 | 1 μg/mLb (maternal) | 8 | 1 μg/mLb (maternal) | 23 | – | – | Europe | EOD | Infants from colonized mothers | 64 |
| 5 | 2.31 μg/mLb (maternal) 0.93 μg/mLb (cord/infant) |
19 | 3.41 μg/mLb (maternal) 1.08 μg/mLb (cord/infant) |
39 | – | – | South Africa | EOD + LOD | Infants from colonized mothers | 67 |
aInferred by conditional logistic regression analysis.
bCalculated by Bayesian analysis.
Abbreviations: EOD, early onset disease; LOD, late onset disease; Ref, reference.
Table 2.
Assay formats and reference standards that have been used to quantify anti-GBS capsule polysaccharide IgG antibodies in published seroepidemiological studies
| Assay format | Immobilized antigen | Sera | Ref | |
|---|---|---|---|---|
| 1 | Monoplex direct binding ELISA | In-house preparation of type Ia CPS | Derived from an individual immunized with a 4-valent CPS vaccine (Serum 20, NABI Inc.) | 49 |
| 2 | Monoplex direct binding ELISA | In-house preparation of type III CPS adsorbed with methylated HSA | Derived from an individual immunized with 4-valent CPS vaccine that includes III (Serum 19, NABI Inc.) | 66 |
| 3 | Monoplex direct binding ELISA | In-house CPS preparation conjugated to HSA | Sera from individuals immunized with five monovalent CPS-TT conjugates (Ia, Ib, II, III, V). Calibrated by immunoprecipitation, RABA and correction with IgM/IgA concentrations by ELISA | 48 |
| 4 | Multiplex Luminex immunoassay | Novartis (GSK)-produced CPS, chemically coupled to microspheres | Purified pooled human gammaglobulin (South Africa) calibrated to Baker et al. reference standards | 67, 71 |
| 5 | Monoplex direct binding ELISA | GSK-produced CPS conjugated to HSA | Baker et al. reference standards | 64 |
Abbreviations: CPS, capsular polysaccharide; CPS-TT, capsular polysaccharide tetanus toxoid conjugate vaccine; ELISA, enzyme-linked immunosorbent assay; GSK, GlaxoSmithKline; HSA, human serum albumin; RABA, radio-antigen binding assay; Ref, reference; TT, tetanus toxoid.
As one example, Baker et al. determined that CPS serotypes Ia- and III-specific IgG concentrations of >0.5 μg/mL in maternal sera corresponded to >90% risk reduction for EOD in infants (compared to CPS-specific IgG concentrations of <0.1 μg/mL in maternal sera) when analyzed by logistic regression methodology.48 Similar analyses for serotype V found that serotype-specific IgG concentration of 0.5 μg/mL corresponded to approximately 70% reduction in risk of EOD.48 Importantly, when a different statistical approach was used (Bayesian method), a risk reduction of 70% was found for antibodies to these three serotypes when maternal serotype-specific IgG titers were >1 µg/mL.48 The similarity of the results using different statistical approaches provides strong support for anti-CPS IgG antibody levels correlating to a reduced risk of GBS infection. Results reported by Fabbrini et al. for serotypes Ia and III in European infants were generally consistent with these findings, predicting risk reductions for EOD by Bayesian methodology of ~75–80% for maternal serum anti-GBS CPS serotype IgG concentrations greater than 1 μg/mL.64 Interestingly, protective thresholds for the different serotypes within each study were largely similar, a pattern that portends the possibility that a serotype-nonspecific protective titer may be possible. Indeed, if confirmed, this would be similar to what has been found for the protective threshold in infants for anti-capsular antibodies directed against Streptococcus pneumoniae.65 In two prior studies conducted in the U.S., Lin et al. correlated maternal serum anti-CPS IgG titers of ≥5 μg/mL and ≥10 μg/mL for serotypes Ia and III, respectively, with 80–90% reduced risk of EOD using logistic regression analysis.49,66 Separate studies have also been conducted by researchers at the University of the Witwatersrand in South Africa that have attempted to define a protective threshold for invasive GBS disease through the first 3 months of life. An initial study assessed both EOD and LOD in paired maternal and infant sera taken at the time of recorded infection. Calculated IgG anti-CPS thresholds for serotypes Ia and III in this study were determined to be ≥ 3–6 μg/mL based on maternal sera, levels that were markedly higher than the threshold of ≥0.5 μg/mL obtained from cognate infant sera.62 A subsequent study from this group evaluated protective thresholds through the first 90 days of life in maternal sera and infant sera for which both cord blood samples as well as sera taken at the time of infection were included.67 This study estimated that serum titers of ≥0.93 µg/mL and ≥1.08 µg/mL of serotypes Ia- and III-specific IgG, respectively, were associated with >90% risk reduction against invasive GBS disease with the cognate serotype strains when analyzed with the combination cord and time-of-infection serum titers. Furthermore, in agreement with the prior finding, the protective thresholds estimated from maternal sera exceeded those obtained from sera reflective of titers in the infant. Despite these similarities, it is not possible to ascertain what protective antibody level would really need to be achieved by vaccination, unless the same assay was used for these studies. An approach to how this can be accomplished will be described in the following sections.
Development of a standardized IgG assay to assess anti-CPS IgG antibody levels
Measurements of immune responses to either vaccination or natural infection are often useful in predicting clinical outcomes, even in the absence of a direct causal relationship. Protection against invasive GBS disease in newborn babies has been shown to correlate with serotype-specific IgG concentrations at the time of birth.48,49,62,64,66 As introduced above, the major limitation of these prior seroepidemiology studies has been the lack of a standardized assay to allow direct comparison of protective threshold IgG concentration estimates. The use of non-standardized assays makes it difficult to compare the magnitude and/or quality of immune responses across studies. A brief overview of the different assay formats that has been used to support the prior studies is provided in Table 2.
To support the clinical development of GBS6, Pfizer developed and validated a Luminex-based six-plex IgG assay to simultaneously measure serum antibodies to GBS6 vaccine serotypes Ia, Ib, II, III, IV and V.34 This assay was adopted by the GBS assay standardization consortium, funded by the Bill and Melinda Gates Foundation (BMGF, grant# OPP1153630). At the time of writing this review, an interlaboratory study is underway with participating laboratories to evaluate precision and repeatability of the assay across different laboratories. A primary goal of this study is to develop a panel of quality control serum samples with consensus IgG antibody concentration estimates for each serotype. An international reference standard serum for the six CPS serotypes will also be needed and development of this important reagent is an additional priority. This new standardized IgG assay would allow for a better understanding of the relationship between GBS CPS-specific serum IgG antibody concentrations and the relative risk of disease in infants. Importantly, it will permit comparability across seroepidemiological studies and relieve a limitation that has hampered elaboration of an internationally recognized protective titer to date. An international reference standard also would permit potential comparatiblity across studies that use different immunoassays but the same standard.
Statistical methods to establish the risk–titer relationship
A range of statistical techniques are available to estimate the antibody thresholds of protection utilizing data obtained from natural history studies. Studies described previously individually employed one or more methods to derive potential protective levels. These methods rely on transformation of titers for cases and non-cases into a defined risk–titer curve. The key quantity to be estimated is the probability of invasive GBS disease (caused by a given serotype), as a function of the infantʻs IgG value at birth for that serotype. Consistent with previous seroepidemiology studies, this risk function can be estimated by both Bayesian and frequentist approaches. Carey et al.68 introduced a Bayesian approach in which a parametric form of the distributions of antibody levels among cases and controls was assumed. A Gibbs sampling procedure was then used to combine these distribution functions and an assumed unconditional population risk to derive the probability of disease conditional on the titer exceeding a given value. The weighted logistic regression approach of Rose and van der Laan69 has also been applied to estimate the risk function, using weighting that accounts for the case-control design. From this method, the serotype-specific antibody titers which result in reductions in risk of disease across a range of risk levels can be derived. Once the risk function has been established, it can further be possible to predict the level of vaccine efficacy that may be achieved by an investigational GBS vaccine. This can be done by applying the titer distributions to the estimated risk function for the vaccine and placebo groups.
Approaches to licensure of a maternal GBS vaccine
There are four conceptual approaches to licensing a vaccine, three are illustrated in Figure 4. The fourth, known as “the animal rule”, is not considered for this review. This approach only comes into play if there is no feasible way to conduct clinical studies that can be used to demonstrate efficacy. Though an efficacy study with a disease endpoint may be difficult to conduct due to the size required and time it would take to conduct such a study, a serological approach for licensure may be possible, negating the need to conduct licensure based on the animal rule. Variations of the three licensure pathways outlined here are also accepted by national regulatory authorities in other regions, and thus the U.S. regulatory pathways will be given as representative examples.
Figure 4.
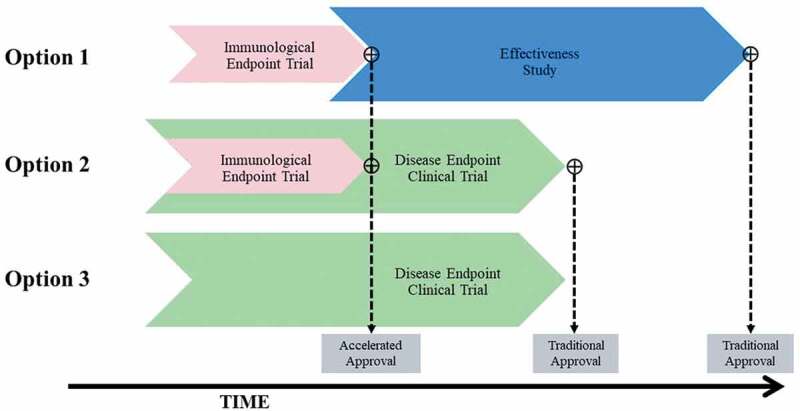
Overview of potential approaches to licensure of a maternal GBS6 vaccine. Effectiveness study refers to a clinical endpoint trial that is conducted under real-world settings after vaccine licensure. Disease endpoint clinical trial refers to an efficacy trial with GBS disease as the primary study endpoint. Accelerated approval is not applicable to Option 3.
The accelerated approval pathway of the U.S. Food and Drugs Administration (FDA) permits licensure based on a surrogate endpoint, that can include immunological endpoints such as antibody levels that reasonably predict clinical benefit. The protective risk-titer curve determined by the strategy detailed above, that makes use of a serological biomarker as an immunological surrogate, could constitute such an endpoint and could potentially be used as the basis for accelerated approval with post-approval commitments to conduct an efficacy or effectiveness study.
The licensure pathway for a GBS vaccine was discussed during a Vaccine and Related Biological Products Advisory Committee (VRBPAC) held at the request of the FDA in May 2018. The committee agreed that immunologic endpoints could be used to demonstrate effectiveness and support the U.S. licensure of a GBS vaccine, particularly if an immunological correlate or surrogate could be validated in the ongoing serologic studies using a standardized assay.70 This approach would need to be confirmed in post-licensure studies. A description of the main approaches is detailed below.
For both Options 1 and 2, a rigorous approach as detailed above would be required to provide evidence that the antibody titers achieved in newborn infants are sufficient to protect them from invasive GBS disease through a period of significant risk. While licensure using an immunological endpoint may provide advantages in terms of shortening the time to availability of a GBS vaccine, it should be noted that development of a universally agreed upon protective titer (whether a surrogate or correlate) requires agreement from regulators, vaccine manufacturers, and experts in the field. It is possible that licensure through this pathway may be complex. Alternatively, licensure of a maternal GBS vaccine could be achieved through the conduct of an efficacy trial; however, given the low incidence of invasive GBS disease in neonates in certain regions, this could require enrollment of up to 80,000 participants. Even in regions with the highest incidence, an efficacy trial would require enrollment of tens of thousands of pregnant women. The details and merits of each option are described below.
Licensure Option 1: pre-licensure immunogenicity study and confirmatory post-licensure effectiveness studies
Option 1 would use an immunological endpoint to support accelerated approval followed by traditional approval with a confirmatory post-licensure effectiveness trial. There are several options for the conduct of a confirmatory trial that include observational studies and the use of real-world evidence (such as vaccine surveillance data) to establish vaccine efficacy. Randomization strategies such as cluster-based randomization may be less complex than traditional pre-licensure subject level randomized clinical trials. Such a strategy would enable licensure much sooner given the smaller size of a pivotal study compared to a conventional efficacy trial approach pre-licensure. This approach was supported for GBS vaccine development during the May 2018 VRBPAC.70 This option would have to consider the timing of the effectiveness study in relation to availability of licensed vaccine and the FDAʻs general requirement to have post-licensure confirmatory trials underway at the time the accelerated approval is granted. Timing may require regulatory negotiation or conduct of an effectiveness trial in regions where a vaccine would need to be supplied.
In Option 1, effectiveness studies would need to be conducted in regions where the vaccine has been introduced and is available, particularly if use of surveillance data would be needed. The least costly and most time-efficient studies would need to be conducted in regions of high disease incidence, and could link to obstetric and pregnancy surveillance systems to monitor incidence as a measure of effectiveness. A caveat of this approach is that deployment to regions that require post-approval effectiveness studies may be delayed; however, pre-qualification by the WHO may ease the path for a vaccine that is not licensed by a traditional route. While an accelerated approval pathway enables early licensure, it does not guarantee its use, and slow uptake may delay implementation of an effectiveness study. Furthermore, in LMICs where efficacy or clinical disease endpoint data are traditionally required prior to approval and recommendations for use, conduct of an effectiveness study may require the vaccine to be provided prior to licensure. Effectiveness studies could take advantage of alternative study designs to facilitate evaluation of the vaccine. For example, the use of large observational studies or effectiveness studies using cluster randomization in lieu of individual randomization schemes would make study conduct much less complex. In addition, effectiveness studies often use fewer and less stringent study measures for evaluation which is advantageous from an operational perspective. Thus, while effectiveness studies would still require a large number of pregnant women to be enrolled, they may be less costly than a pre-licensure Phase 3 clinical trial.
Licensure Option 2: combined immunogenicity and efficacy study
Option 2 describes an efficacy trial with early licensure after an embedded immunogenicity analysis with a serological endpoint by an accelerated approval pathway. The surrogate of protection may be a measure of antibody titers in cord blood or sera from infants born to vaccinated mothers. While the overall trial size would be based on a clinical disease endpoint, the immunogenicity assessments would rely on achieving an agreed upon level of immune response related to a consensus serocorrelate in a minimum number of exposed subjects. This would utilize a surrogate of protection that represents a measure reasonably likely to predict vaccine efficacy and be determined by the strategy detailed above. As with Option 1, one of the most important benefits of this approach, however, is that a GBS vaccine would be licensed and available for use to pregnant women and their at risk infants much sooner than if a traditional approval pathway was sought from the onset. While the efficacy portion would still require a large sample size, completion of the clinical disease endpoint trial would serve as the confirmatory data needed to convert the accelerated approval to traditional approval. The advantages of this approach include making the vaccine available earlier as well as confirming the protective antibody level in a single disease endpoint clinical trial. This combination hybrid approach would enable operational efficiencies and minimize the overall timeline to traditional approval. For example, after the required number of subjects have been enrolled and completed visits for the immunogenicity assessment, analysis and regulatory submissions can occur concurrently while the efficacy portion of the study continues. The most salient disadvantage of this approach is the cost and time required to enroll enough subjects to meet the clinical efficacy endpoint.
Licensure Option 3: pre-licensure efficacy study
In the event that an immunological threshold of protection cannot be established or agreed upon, licensure of a GBS vaccine could be achieved through the conduct of a conventional disease endpoint Phase 3 placebo-controlled randomized clinical trial (Option 3). Efficacy could be established by comparing the effect of the vaccine or placebo in reducing incidence of invasive GBS disease in infants. This study would need to be conducted in countries with high incidence of GBS disease. The highest reported rates of invasive disease are in South Africa with 1–3 cases/1,000 infants per year.71,72 Assuming a vaccine efficacy rate of 75%, a Phase 3 efficacy trial would require a minimum of ~28,000 enrolled participants in the most optimistic scenario (assume lower 95% confidence interval [LCI] of 0% and an incidence of 3/1000) and upwards of 80,000 pregnant women (assuming an LCI of 20% and incidence rate of 1/1000) in a pessimistic scenario. Conduct of such a study, particularly as infants and mothers will need follow-up for at least 612 months, carries a high degree of operational challenge and risk. A minimum of at least 200 sites would be required to enroll the smallest number of subjects over an 8-12–month enrollment period. Such a study would likely take several years to complete and would require significant investment while causing delay to availability of a potentially life-saving vaccine.
Summary and future perspectives
Invasive GBS disease remains an important cause of infant morbidity and mortality, for which the development of an efficacious vaccine remains a global health imperative. After several decades of research into the protective correlates and immunobiology of GBS CPS, the prospect of the first GBS conjugate vaccine becoming available is now on the horizon with the development of GBS6. Licensure of GBS6 by a pathway requiring conduct of an efficacy study would be complex and likely require many years to complete. Regulatory approval based on an immune threshold of protection derived from natural history studies is feasible and offers advantages for an expedited path to licensure. Such an approach has some precedent drawing from the established pathways for approval of new vaccines for Streptococcus pneumoniae, Neisseria meningitidis and Haemophilus influenzae b (Hib), where licensure based on immune thresholds derived from the efficacy studies of the foundational vaccines is well established. For Hib, a threshold of protection of 1 µg/mL anti-capsule antibody was initially proposed based on immunization studies with the isolated polyribosyl ribitol phospate (PRP) capsule as immunogen. The protective threshold for antibodies induced by PRP conjugates was subsequently found to be lower (~0.15 µg/mL), an effect attributed in part to the affinity maturation and consequent higher quality antibody associated with glycoconjugate immunization.73 For GBS anti-CPS antibodies, if natural immunity is similar to that induced by an unconjugated polysaccharide, it is conceivable that antibody thresholds derived from natural exposure may overestimate the protective level provided by immunization with a CPS conjugate that could induce anti-CPS antibodies with increased avidity with help from T cells directed against the carrier protein.
In HICs where IAP is the established standard of care, a GBS vaccine may be used as an adjunct public health tool to reduce residual cases of EOD not addressed by IAP, and to prevent LOD where IAP has not had an impact. Given concerns around antibiotic stewardship and the rise of antimicrobial resistance, an efficacious vaccine may also ultimately replace IAP. Deployment of a maternal GBS vaccine in LMICs where IAP is not available or readily implementable could provide substantial benefit for reduction of GBS disease in early life, and maternal vaccination could be integrated as part of existing private market and national immunization programs. Agreement on acceptable pathways to licensure and registration will need to be addressed on an individual basis with national and regional regulatory bodies; however, where acceptable, the use of an immune correlate offers the possibility for accelerated regulatory approval to make available a vaccine to prevent a devastating disease affecting young infants worldwide.
Acknowledgements
The authors would like to thank the following Pfizer Inc. colleagues: Robert Donald, Natalie Silmon de Monerri, Jessica Atwell, and Yasmeen Agosti for critical review of the manuscript and Katheryn Liby for assistance in the graphics preparation. The authors also thank Nataliya Kushnir, Pfizer Inc. for scientific writing assistance.
Funding Statement
The development of this manuscript was supported by Pfizer Inc.
Disclosure statement
The authors are employees of Pfizer Inc. at time of writing and may own Pfizer stock.
References
- 1.Foxman B, Gillespie BW, Manning SD, Marrs CF.. Risk factors for group B streptococcal colonization: potential for different transmission systems by capsular type. Ann Epidemiol. 2007;17(11):854–12. doi: 10.1016/j.annepidem.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker CJ, Barrett FF, Gordon RC, Yow MD. Suppurative meningitis due to streptococci of Lancefield group B: a study of 33 infants. J Pediatr. 1973;82(4):724–29. doi: 10.1016/S0022-3476(73)80606-7. [DOI] [PubMed] [Google Scholar]
- 3.Madrid L, Seale AC, Kohli-Lynch M, Edmond KM, Lawn JE, Heath PT, Madhi SA, Baker CJ, Bartlett L, Cutland C, et al. Infant Group B streptococcal disease incidence and serotypes worldwide: systematic review and meta-analyses. Clin Infect Dis. 2017;65(suppl_2):S160–S72. doi: 10.1093/cid/cix656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thigpen MC, Whitney CG, Messonnier NE, Zell ER, Lynfield R, Hadler JL, Harrison LH, Farley MM, Reingold A, Bennett NM, et al. Bacterial meningitis in the United States, 1998–2007. N Engl J Med. 2011;364(21):2016–25. doi: 10.1056/NEJMoa1005384. [DOI] [PubMed] [Google Scholar]
- 5.Libster R, Edwards KM, Levent F, Edwards MS, Rench MA, Castagnini LA, Cooper T, Sparks RC, Baker CJ, Shah PE. Long-Term outcomes of group B streptococcal meningitis. Pediatrics. 2012;130(1):e8–e15. doi: 10.1542/peds.2011-3453. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization . Group B streptococcus vaccine: full value of vaccine assessment. 2 Nov 2021. https://www.who.int/publications/i/item/9789240037526.
- 7.Baker CJ, Barrett FF. Transmission of group B streptococci among parturient women and their neonates. J Pediatr. 1973;83(6):919–25. doi: 10.1016/S0022-3476(73)80524-4. [DOI] [PubMed] [Google Scholar]
- 8.Russell NJ, Seale AC, Oʻ-Sullivan C, Le Doare K, Heath PT, Lawn JE, Bartlett L, Cutland C, Gravett M, Ip M, et al. Risk of early-onset neonatal group B streptococcal disease with maternal colonization worldwide: systematic review and meta-analyses. Clin Infect Dis. 2017;65(suppl_2):S152–S9. doi: 10.1093/cid/cix655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puopolo KM, Lynfield R, Cummings JJ.Committee On Fetus Newborn, Committee On Infectious Diseases. Management of Infants at risk for group B streptococcal Disease. Pediatrics. 2019;144:e20191881. [DOI] [PubMed] [Google Scholar]
- 10.Boyer KM, Gotoff SP. Strategies for chemoprophylaxis of GBS early-onset infections. Antibiot Chemother. 1985;35:267–80. [DOI] [PubMed] [Google Scholar]
- 11.Baker CJ. The spectrum of perinatal group B streptococcal disease. Vaccine. 2013;Suppl 31:D3–6. doi: 10.1016/j.vaccine.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 12.de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, Parkhill J, Charnock-Jones DS, Smith GCS. Human placenta has no microbiome but can contain potential pathogens. Nature. 2019;572(7769):329–34. doi: 10.1038/s41586-019-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin FY, Weisman LE, Azimi P, Young AE, Chang K, Cielo M, Moyer P, Troendle JF, Schneerson R, Robbins JB. Assessment of intrapartum antibiotic prophylaxis for the prevention of early-onset group B Streptococcal disease. Pediatr Infect Dis J. 2011;30(9):759–63. doi: 10.1097/INF.0b013e31821dc76f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards JM, Watson N, Focht C, Wynn C, Todd CA, Walter EB, Heine RP, Swamy GK. Group B Streptococcus (GBS) colonization and disease among pregnant women: a historical cohort study. Infect Dis Obstet Gynecol. 2019;2019:5430493. doi: 10.1155/2019/5430493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall J, Adams NH, Bartlett L, Seale AC, Lamagni T, Bianchi-Jassir F, Lawn JE, Baker CJ, Cutland C, PT Heath, et al. Maternal disease with group B Streptococcus and serotype distribution worldwide: systematic review and meta-analyses. Clin Infect Dis. 2017;65(suppl_2):S112–S24. doi: 10.1093/cid/cix660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Centers for Disease Control and Prevention . Active bacterial core surveillance report, emerging infections program network, group B Streptococcus, 2018. 19 May 2020. https://www.cdc.gov/abcs/reports-findings/survreports/gbs18.html.
- 17.Seale AC, Bianchi-Jassir F, Russell NJ, Kohli-Lynch M, Tann CJ, Hall J, Madrid L, Blencowe H, Cousens S, Baker CJ, et al. Estimates of the burden of group B Streptococcal disease worldwide for pregnant women, stillbirths, and children. Clin Infect Dis. 2017;65(suppl_2):S200–S19. doi: 10.1093/cid/cix664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi-Jassir F, Seale AC, Kohli-Lynch M, Lawn JE, Baker CJ, Bartlett L, Cutland C, Gravett MG, Heath PT, Ip M, et al. Preterm birth associated with group B Streptococcus maternal colonization worldwide: systematic review and meta-analyses. Clin Infect Dis. 2017;65(suppl_2):S133–S42. doi: 10.1093/cid/cix661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyer KM, Gotoff SP. Prevention of early-onset neonatal group B streptococcal disease with selective intrapartum chemoprophylaxis. N Engl J Med. 1986;314(26):1665–69. doi: 10.1056/NEJM198606263142603. [DOI] [PubMed] [Google Scholar]
- 20.Verani JR, McGee L, Schrag SJ. Prevention of perinatal group B streptococcal disease-revised guidelines from CDC. 2010. MMWR Recomm Rep. 2010;59:1–36. [PubMed] [Google Scholar]
- 21.Van Dyke MK, Phares CR, Lynfield R, Thomas AR, Arnold KE, Craig AS, Mohle-Boetani J, Gershman K, Schaffner W, Petit S, et al. Evaluation of universal antenatal screening for group B streptococcus. N Engl J Med. 2009;360(25):2626–36. doi: 10.1056/NEJMoa0806820. [DOI] [PubMed] [Google Scholar]
- 22.Schrag SJ, Verani JR. Intrapartum antibiotic prophylaxis for the prevention of perinatal group B streptococcal disease: experience in the United States and implications for a potential group B streptococcal vaccine. Vaccine. 2013;Suppl 31:D20–6. doi: 10.1016/j.vaccine.2012.11.056. [DOI] [PubMed] [Google Scholar]
- 23.Jordan HT, Farley MM, Craig A, Mohle-Boetani J, Harrison LH, Petit S, Lynfield R, Thomas A, Zansky S, Gershman K, et al. Revisiting the need for vaccine prevention of late-onset neonatal group B streptococcal disease: a multistate, population-based analysis. Pediatr Infect Dis J. 2008;27(12):1057–64. doi: 10.1097/INF.0b013e318180b3b9. [DOI] [PubMed] [Google Scholar]
- 24.Ledger WJ, Blaser MJ. Are we using too many antibiotics during pregnancy? BJOG. 2013;120(12):1450–52. doi: 10.1111/1471-0528.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoll BJ, Puopolo KM, Hansen NI, Sanchez PJ, Bell EF, Carlo WA, Cotten CM, DʻAngio CT, Kazzi SN, Poindexter BB, et al. Early-Onset neonatal sepsis 2015 to 2017, the rise of escherichia coli , and the need for novel prevention strategies. JAMA Pediatr. 2020;174(7):e200593. doi: 10.1001/jamapediatrics.2020.0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrag S, Gorwitz R, Fultz-Butts K, Schuchat A. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. MMWR Recomm Rep. 2002;51:1–22. [PubMed] [Google Scholar]
- 27.Hayes K, Oʻ-Halloran F, Cotter L. A review of antibiotic resistance in Group B Streptococcus: the story so far. Crit Rev Microbiol. 2020;46(3):253–69. doi: 10.1080/1040841X.2020.1758626. [DOI] [PubMed] [Google Scholar]
- 28.Centers for Disease Control and Prevention . Evaluation and diagnosis of penicillin allergy for healthcare professionals. 31 Oct 2017. https://www.cdc.gov/antibiotic-use/community/pdfs/penicillin-factsheet.pdf.
- 29.Royal College of Obstetricians & Gynecologists . Group B Streptococcal disease, Early-onset (Green-top guideline no. 36). 13 Sept 2017. https://www.rcog.org.uk/en/guidelines-research-services/guidelines/gtg36/2017.
- 30.UK National Screening Committee . Group B Streptococcus. Antenatal screening programme. March 2017. https://view-health-screening-recommendations.service.gov.uk/group-b-streptococcus/2017.
- 31.Dangor Z, Cutland CL, Izu A, Kwatra G, Trenor S, Lala SG, Madhi SA. Temporal changes in invasive group B Streptococcus serotypes: implications for vaccine development. PLoS One. 2016;11(12):e0169101. doi: 10.1371/journal.pone.0169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera L, Saez-Llorens X, Feris-Iglesias J, Ip M, Saha S, Adrian PV, Madhi SA, Boudville IC, Cunnington MC, Casellas JM, et al. Incidence and serotype distribution of invasive group B streptococcal disease in young infants: a multi-country observational study. BMC Pediatr. 2015;15(1):143. doi: 10.1186/s12887-015-0460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein–polysaccharide conjugate vaccines. Nat Rev Immunol. 2009;9(3):213–20. doi: 10.1038/nri2494. [DOI] [PubMed] [Google Scholar]
- 34.Absalon J, Segall N, Block SL, Center KJ, Scully IL, Giardina PC, Peterson J, Watson WJ, Gruber WC, Jansen KU, et al. Safety and immunogenicity of a novel hexavalent group B streptococcus conjugate vaccine in healthy, non-pregnant adults: a phase 1/2, randomised, placebo-controlled, observer-blinded, dose-escalation trial. Lancet Infect Dis. 2021;21(2):263–74. doi: 10.1016/S1473-3099(20)30478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sobanjo-Ter Meulen A, Abramson J, Mason E, Rees H, Schwalbe N, Bergquist S, Klugman KP., et al. Path to impact: A report from the Bill and Melinda Gates Foundation convening on maternal immunization in resource-limited settings; Berlin – January 29–30, 2015. Vaccine. 2015;33(47):6388–95. doi: 10.1016/j.vaccine.2015.08.047. [DOI] [PubMed] [Google Scholar]
- 36.Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, and Carneiro-Sampaio M. IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol. 2012;2012:985646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilcox CR, Holder B, Jones CE. Factors affecting the FcRn-mediated transplacental transfer of antibodies and implications for vaccination in pregnancy. Front Immunol. 2017;8:1294. doi: 10.3389/fimmu.2017.01294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abu Raya B, Edwards KM, Scheifele DW, Halperin SA. Pertussis and influenza immunisation during pregnancy: a landscape review. Lancet Infect Dis. 2017;17(7):e209–e22. doi: 10.1016/S1473-3099(17)30190-1. [DOI] [PubMed] [Google Scholar]
- 39.Centers for Disease Control and Prevention . COVID-19 vaccines while pregnant or breastfeeding. 7 Oct 2021. https://www.cdc.gov/coronavirus/2019-ncov/vaccines/recommendations/pregnancy.html.
- 40.Hillier SL, Ferrieri P, Edwards MS, Ewell M, Ferris D, Fine P, Carey V, Meyn L, Hoagland D, Kasper DL, et al. A phase 2, randomized, control trial of Group B Streptococcus (GBS) Type III capsular polysaccharide-tetanus toxoid (GBS III-TT) vaccine to prevent vaginal colonization with GBS III. Clin Infect Dis. 2019;68(12):2079–86. doi: 10.1093/cid/ciy838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russell LB, Kim S-Y, Cosgriff B, Pentakota SR, Schrag SJ, Sobanjo-Ter Meulen A, Verani JR, Sinha A. Cost-Effectiveness of maternal GBS immunization in low-income sub-Saharan Africa. Vaccine. 2017;35(49):6905–14. doi: 10.1016/j.vaccine.2017.07.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marques MB, Kasper DL, Pangburn MK, Wessels MR. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect Immun. 1992;60(10):3986–93. doi: 10.1128/iai.60.10.3986-3993.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berti F, Adamo R. Recent mechanistic insights on glycoconjugate vaccines and future perspectives. ACS Chem Biol. 2013;8(8):1653–63. doi: 10.1021/cb400423g. [DOI] [PubMed] [Google Scholar]
- 44.Madzivhandila M, Adrian PV, Cutland CL, Kuwanda L, Schrag SJ, Madhi SA. Serotype distribution and invasive potential of group B streptococcus isolates causing disease in infants and colonizing maternal-newborn dyads. PLoS One. 2011;6(3):e17861. doi: 10.1371/journal.pone.0017861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melin P, Efstratiou A. Group B streptococcal epidemiology and vaccine needs in developed countries. Vaccine. 2013;Suppl 31:D31–42. doi: 10.1016/j.vaccine.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 46.Buurman ET, Timofeyeva Y, Gu J, Kim JH, Kodali S, Liu Y, Mininni T, Moghazeh S, Pavliakova D, Singer C, et al. A novel hexavalent capsular polysaccharide conjugate vaccine (GBS6) for the prevention of neonatal group B streptococcal infections by maternal immunization. J Infect Dis. 2019;220(1):105–15. doi: 10.1093/infdis/jiz062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker CJ, Kasper DL. Correlation of maternal antibody deficiency with susceptibility to neonatal group B streptococcal infection. N Engl J Med. 1976;294(14):753–56. doi: 10.1056/NEJM197604012941404. [DOI] [PubMed] [Google Scholar]
- 48.Baker CJ, Carey VJ, Rench MA, Edwards MS, Hillier SL, Kasper DL, Platt R. Maternal antibody at delivery protects neonates from early onset group B streptococcal disease. J Infect Dis. 2014;209(5):781–88. doi: 10.1093/infdis/jit549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin FY, Philips JB 3rd, Azimi PH, Weisman LE, Clark P, Rhoads GG, Regan J, Concepcion NF, Frasch CE, Troendle J, et al. Level of maternal antibody required to protect neonates against early-onset disease caused by group B Streptococcus type Ia: a multicenter, seroepidemiology study. J Infect Dis. 2001;184(8):1022–28. doi: 10.1086/323350. [DOI] [PubMed] [Google Scholar]
- 50.Boyer KM, Kendall LS, Papierniak CK, Klegerman ME, Gotoff SP. Protective levels of human immunoglobulin G antibody to group B streptococcus type Ib. Infect Immun. 1984;45(3):618–24. doi: 10.1128/iai.45.3.618-624.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wessels MR, Paoletti LC, Guttormsen HK, Michon F, Dʻ-Ambra AJ, Kasper DL. Structural properties of group B streptococcal type III polysaccharide conjugate vaccines that influence immunogenicity and efficacy. Infect Immun. 1998;66(5):2186–92. doi: 10.1128/IAI.66.5.2186-2192.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker CJ, Paoletti LC, Rench MA, Guttormsen H-K, Carey VJ, Hickman ME, Kasper D. Use of capsular polysaccharide–tetanus toxoid conjugate vaccine for type II group B streptococcus in healthy women. J Infect Dis. 2000;182(4):1129–38. doi: 10.1086/315839. [DOI] [PubMed] [Google Scholar]
- 53.Baker CJ, Paoletti LC, Rench MA, Guttormsen HK, Edwards MS, Kasper DL. Immune response of healthy women to 2 different group B streptococcal type V capsular polysaccharide–protein conjugate vaccines. J Infect Dis. 2004;189(6):1103–12. doi: 10.1086/382193. [DOI] [PubMed] [Google Scholar]
- 54.Baker CJ, Paoletti LC, Wessels MR, Guttormsen H-K, Rench MA, Hickman ME, Kasper D. Safety and immunogenicity of capsular polysaccharide–tetanus toxoid conjugate vaccines for group B Streptococcal types Ia and Ib. J Infect Dis. 1999;179(1):142–50. doi: 10.1086/314574. [DOI] [PubMed] [Google Scholar]
- 55.Baker CJ, Rench MA, McInnes P. Immunization of pregnant women with group B streptococcal type III capsular polysaccharide-tetanus toxoid conjugate vaccine. Vaccine. 2003;21(24):3468–72. doi: 10.1016/S0264-410X(03)00353-0. [DOI] [PubMed] [Google Scholar]
- 56.Kasper DL, Paoletti LC, Wessels MR, Guttormsen HK, Carey VJ, Jennings HJ, Baker CJ. Immune response to type III group B streptococcal polysaccharide-tetanus toxoid conjugate vaccine. J Clin Invest. 1996;98:2308–14. doi: 10.1172/JCI119042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donders GG, Halperin SA, Devlieger R, Baker S, Forte P, Wittke F, Slobod KS, Dull PM. Maternal immunization with an investigational trivalent group B streptococcal vaccine: a randomized controlled trial. Obstet Gynecol. 2016;127(2):213–21. doi: 10.1097/AOG.0000000000001190. [DOI] [PubMed] [Google Scholar]
- 58.Leroux-Roels G, Bebia Z, Maes C, Aerssens A, De Boever F, Grassano L, Buffi G, Margarit I, Karsten A, Cho S, et al. Safety and immunogenicity of a second dose of an investigational maternal trivalent group B streptococcus vaccine in nonpregnant women 4–6 years after a first dose: results from a phase 2 trial. Clin Infect Dis. 2020;70(12):2570–79. doi: 10.1093/cid/ciz737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Madhi SA, Cutland CL, Jose L, Koen A, Govender N, Wittke F, Olugbosi M, Sobanjo-ter Meulen A, Baker S, Dull PM, et al. Safety and immunogenicity of an investigational maternal trivalent group B streptococcus vaccine in healthy women and their infants: a randomised phase 1b/2 trial. Lancet Infect Dis. 2016;16(8):923–34. doi: 10.1016/S1473-3099(16)00152-3. [DOI] [PubMed] [Google Scholar]
- 60.Madhi SA, Koen A, Cutland CL, Jose L, Govender N, Wittke F, Olugbosi M, Sobanjo-ter Meulen A, Baker S, Dull PM, et al. Antibody kinetics and response to routine vaccinations in infants born to women who received an investigational trivalent group B streptococcus polysaccharide CRM197-conjugate vaccine during pregnancy. Clin Infect Dis. 2017;65(11):1897–904. doi: 10.1093/cid/cix666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pannaraj PS, Edwards MS, Ewing KT, Lewis AL, Rench MA, Baker CJ. Group B streptococcal conjugate vaccines elicit functional antibodies independent of strain O-acetylation. Vaccine. 2009;27(33):4452–56. doi: 10.1016/j.vaccine.2009.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dangor Z, Kwatra G, Izu A, Adrian P, Cutland CL, Velaphi S, Ballot D, Reubenson G, Zell ER, Lala SG, et al. Correlates of protection of serotype-specific capsular antibody and invasive Group B Streptococcus disease in South African infants. Vaccine. 2015;33(48):6793–99. doi: 10.1016/j.vaccine.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 63.Swamy GK, Metz TD, Edwards KM, Soper DE, Beigi RH, Campbell JD, Grassano L, Buffi G, Dreisbach A, Margarit I, et al. Safety and immunogenicity of an investigational maternal trivalent group B streptococcus vaccine in pregnant women and their infants: results from a randomized placebo-controlled phase II trial. Vaccine. 2020;38(44):6930–40. doi: 10.1016/j.vaccine.2020.08.056. [DOI] [PubMed] [Google Scholar]
- 64.Fabbrini M, Rigat F, Rinaudo CD, Passalaqua I, Khacheh S, Creti R, Baldassarri L, Carboni F, Anderloni G, Rosini R, et al. The protective value of maternal group B streptococcus antibodies: quantitative and functional analysis of naturally acquired responses to capsular polysaccharides and pilus proteins in European maternal sera. Clin Infect Dis. 2016;63(6):746–53. doi: 10.1093/cid/ciw377. [DOI] [PubMed] [Google Scholar]
- 65.Jodar L, Butler J, Carlone G, Dagan R, Goldblatt D, Kayhty H, Klugman K, Plikaytis B, Siber G, Kohberger R, et al. Serological criteria for evaluation and licensure of new pneumococcal conjugate vaccine formulations for use in infants. Vaccine. 2003;21(23):3265–72. doi: 10.1016/S0264-410X(03)00230-5. [DOI] [PubMed] [Google Scholar]
- 66.Lin FY, Weisman LE, Azimi PH, Philips JB 3rd, Clark P, Regan J, Rhoads GG, Frasch CE, Gray BM, Troendle J, et al. Level of maternal IgG anti–Group B streptococcus type III antibody correlated with protection of neonates against early-onset disease caused by this pathogen. J Infect Dis. 2004;190(5):928–34. doi: 10.1086/422756. [DOI] [PubMed] [Google Scholar]
- 67.Madhi SA, Izu A, Kwatra G, Jones S, Dangor Z, Wadula J, Moultrie A, Adam Y, Pu W, Henry O, et al. Association of group B streptococcus (GBS) serum serotype-specific anticapsular immunoglobulin G concentration and risk reduction for invasive GBS disease in South African infants: an observational birth-cohort, matched case-control study. Clin Infect Dis. 2021;73(5):e1170–e80. doi: 10.1093/cid/ciaa1873. [DOI] [PubMed] [Google Scholar]
- 68.Carey VJ, Baker CJ, Platt R. Bayesian inference on protective antibody levels using case-control data. Biometrics. 2001;57(1):135–42. doi: 10.1111/j.0006-341X.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- 69.Rose S, van der Laan MJ. Simple optimal weighting of cases and controls in case-control studies. Int J Biostat. 2008;4(1):Article 19. doi: 10.2202/1557-4679.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Food and Drug Administration (FDA). Center for Biologics Evaluation and Research (CBER) . Approaches for demonstrating effectiveness of group B streptococcus (GBS) vaccines intended for use in pregnant women to protect the newborn infant. 152nd meeting of the vaccines and related biological products advisory committee (VRBPAC). 17 May 2018. https://www.fda.gov/media/113958/download.
- 71.Dangor Z, Lala SG, Cutland CL, Koen A, Jose L, Nakwa F, Ramdin T, Fredericks J, Wadula J, Madhi SA. Burden of invasive group B Streptococcus disease and early neurological sequelae in South African infants. PLoS One. 2015;10(4):e0123014. doi: 10.1371/journal.pone.0123014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quan V, Verani JR, Cohen C, von Gottberg A, Meiring S, Cutland CL, Schrag SJ, Madhi SA. Invasive group B streptococcal disease in South Africa: importance of surveillance methodology. PLoS One. 2016;11(4):e0152524. doi: 10.1371/journal.pone.0152524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. 2010;17(7):1055–65. doi: 10.1128/CVI.00131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]