

Abstract
Cell death is a key feature of neurological diseases, including stroke and neurodegenerative disorders. Studies in a variety of ischemic/hypoxic mouse models demonstrate that poly(ADP-ribose) polymerase 1 (PARP-1)-dependent cell death, also named PARthanatos, plays a pivotal role in ischemic neuronal cell death and disease progress. PARthanatos has its unique triggers, processors, and executors that convey a highly orchestrated and programmed signaling cascade. In addition to its role in gene transcription, DNA damage repair, and energy homeostasis through PARylation of its various targets, PARP-1 activation in neuron and glia attributes to brain damage following ischemia/reperfusion. Pharmacological inhibition or genetic deletion of PARP-1 reduces infarct volume, eliminates inflammation, and improves recovery of neurological functions in stroke. Here, we reviewed the role of PARP-1 and PARthanatos in stroke and their therapeutic potential.
Keywords: NAD+, oxidative stress, PARP-1, PARthanatos, stroke
1 |. INTRODUCTION
Stroke is an acute and lethal cerebrovascular disorder. As a leading cause of death worldwide, new attacks and recurrent stroke affect approximately 13.7 million people globally each year (Hasan et al., 2018; Virani et al., 2020). Ischemic stroke accounts for more than 80% of stroke cases. It is characterized by rapid disruption of cerebral arterial blood flow and lack of oxygen to the affected area leading to neuronal cell death. On the other hand, stroke has also been ranked the third leading cause of disability worldwide. It may cause some long-term severe complications or sequelae including paralysis, speech problems, dementia as well as loss of advanced brain functions like learning and memory (Powers, 2020). Therefore, stroke interferes with patients’ daily life and sometimes even causes severe life threatening, which has brought a heavy economic burden to the society. So far, the most effective stroke therapeutic strategy is recanalization or reperfusion by either intravenous thrombolysis or endovascular thrombectomy. However, endovascular thrombectomy is usually restricted within a very narrow time window—6 hr from the first symptoms onset (Campbell et al., 2019)—, which has been recently extended to 24 hr (https://www.medpagetoday.com/meetingcoverage/isc/70735). Neuronal cell death is a key feature following ischemic stroke and contributes to the significant loss of brain functions.
Different types of cell death including apoptosis, necrosis, necroptosis, ferroptosis, pyroptosis, and autophagy have been implicated in neuron loss following stroke based on various studies from in vitro cell cultures as well as in vivo animal models (Li et al., 2020; Naito et al., 2020; Wang et al., 2018; Wang et al. 2016; Wang et al., 2009; Wang et al., 2011). However, the precise cell death mechanism in stroke remains obscure. Poly(ADP-ribose) polymerase 1 (PARP-1)-dependent cell death (PARthanatos) is a unique type of cell death program, which is different from apoptosis, necrosis and other types of cell death as we reviewed previously [Table 1 (Wang et al., 2009)]. Emerging evidence indicates that PARP-1 and PARthanatos play a pivotal role in ischemic stroke. Here, we reviewed the recent progress of PARP-1 and PARthanatos in stroke and their effects on neuronal cell death, inflammation and metabolic regulation.
TABLE 1.
Neuroprotective effects by PARP inhibitors in ischemic brain
| Application timing | ||||||||
|---|---|---|---|---|---|---|---|---|
| Inhibitor | Target | Animal model | Species | Before ischemia | After ischemia | Effect on infarction | Effect on neurological functions | References |
| DPQ | PARP–1 | MCAO | Rat | 2 hr | 2 hr | Reduction | Not applicable | Takahashi et al. (1997) |
| MCAO | Rat | – | 30 min | Reduction | Not applicable | Takahashi et al. (1999) | ||
| MCAO | Rat | – | 0 hr | Reduction | Not applicable | Lo et al. (1998) | ||
| MCAO | Rat | 15 min | – | Reduction | Not applicable | Takahashi & Greenberg, 1999) | ||
| 3AB | PARP | MCAO | Rat | 30 min | – | Reduction | Not applicable | Tokime et al. (1998) |
| MCAO | Rat | 15 min | – | Reduction | Improve motor function | Couturier et al. (2003) | ||
| MCAO | Rat | 30 min | Recanalization | Reduction | Improve motor function | Ding et al. (2001) | ||
| FR247304 | PARP–1 | MCAO | Rat | 10 min | Recanalization | Reduction | Not applicable | Iwashita et al. (2004) |
| MCAO | Mouse | 2 hr | 6 hr | Reduction | Not applicable | Abdelkarim et al. (2001) | ||
| PJ34 | PARP–1/2 | MCAO | Mouse | – | 0 and 3 hr | Reduction | Improve sensory motor function | El Amki et al. (2018) |
| tCCAO | Rat | – | 8 hr | Reduction | Not applicable | Hamby et al. (2007) | ||
| DR2313 | PARP–1/2 | MCAO | Rat | 6 hr | or 2 hr | Reduction | Not applicable | Nakajima et al. (2005) |
| JPI–289 | PARP–1 | MCAO | Rat | – | 2 hr | Reduction | Improve sensory motor function | Kim et al. (2018b) |
| HYDAMTIQ | PARP–1/2 | MCAO | Rat | – | 0.5h or 4h | Reduction | Improve sensory motor function | Moroni et al. (2012) |
| MP–124 | PARP–1 | MCAO | Rhesus monkey | – | 0, 3 or 6 hr | Reduction | Improve overall neurological function | Matsuura et al. (2011) |
| Nicotinamide | SIRT1/PARP–1 | MCAO | Mouse | – | 1 hr | Reduction | Not applicable | Liu et al. (2009) |
| NU1025 | Non-selective | MCAO | Rat | 1 hr | – | Reduction | Improve overall neurological function | Kaundal et al. (2006) |
| Olaparib | PARP–1/2 | MCAO | Mouse | – | 0 hr | Reduction | Improve overall neurological function | Teng et al. (2016) |
| TES–448 | PARP–1 | pCCAO | Rat | – | 10 min, 3 hr, 6 hr | Reduction | Not applicable | Klofers et al. (2017) |
| ISO | PARP–1/iNOS | MCAO | rat | – | 1 hr | Reduction | Improve overall neurological function | Singh et al. (2014) |
Abbreviations: 3AB, 3-amino-benzamide; DPQ, 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone; DR2313, 2-methyl-3,5,7,8-tetrahydrothiopyrano[4,3-d] pyrimidine-4-one; FR247304, 5-chloro-2-[3-(4-phenyl-3,6-dihydro-1(2H)-pyridinyl) propyl]-4(3H)-quinazolinone; HYDAMTIQ,2-((dimethylamino)methyl)-9-hydroxythieno[2,3-c] isoquinolin-5(4H)-one; ISO, 1,5-Isoquinolinediol; MCAO, middle cerebral artery occlusion; pCCAO, permanent common carotid artery occlusion; PJ34, N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N, N-dimethylacetamide; tCCAO, transient common carotid artery occlusion.
2 |. PARP-1-DEPENDENT CELL DEATH
PARP-1 is a well-characterized nuclear enzyme that belongs to the PARP superfamily containing 17 members (Gupte et al., 2017; Kim et al., 2020). It functions as a DNA damage sensor and accounts for about 90% of poly(ADP-ribose) (PAR) production in response to DNA damage or oxidative stress. Besides its role in DNA damage repair, PARP-1 is involved in multiple other biological processes including DNA replication, gene transcription, centromere and spindle assembly/disassembly, cell differentiation, inflammation, and chromatin structure regulation (Gupte et al., 2017; Kim et al., 2020; Wang et al., 2009, 2019). However, PARP-1 hyperactivation causes a unique type of cell death termed PARthanatos, which was named after PAR that is a product of PARP-1 activation and Thanatos who is the Greek personification of death and mortality (Wang et al. 2016; Wang et al., 2009, 2011; Yu et al., 2002; Fatokun et al., 2014; Galluzzi et al., 2018). PARthanatos is a type of programmed necrotic cell death but distinct from other forms of cell death, including apoptosis, necrosis, necroptosis, and autophagy. It has several key features: (1) PARP-1 is a central player in the process as its hyperactivation initiates this unique cell death pathway. Pharmacological inhibition or genetic deletion of PARP-1 blocks PARthanatos (Eliasson et al., 1997; Kam et al., 2018; Zhang et al., 1994); (2) nuclear shrinkage and large DNA fragmentation (>10 kb) are observed in the cell undergoing PARthanatos (Wang et al. 2016; Wang et al., 2009; Yu et al., 2002); and (3) caspase activation is dispensable for PARthanatos. PARthanatos cannot be blocked by pan-caspase inhibitors, such as z-VAD-fmk or boc-aspartyl-fmk (BAF) (Yu et al., 2002). Increasing evidences show that PARthanatos is involved in numerous neurological diseases including Alzheimer’s disease (AD) (Abeti et al., 2011; Love et al., 1999), Parkinson’s disease (PD) (Kam et al., 2018), Huntington’s disease (HD) (Chidambaram et al., 2017; Paldino et al., 2020), amyotrophic lateral sclerosis (ALS) (Rulten et al., 2014), traumatic brain injury (d’Avila et al., 2012) and stroke (Eliasson et al., 1997; Meng et al., 2018; Zhang et al., 1994).
2.1 |. PARP-1 and its activation in ischemic stroke
PARP-1 is the most extensively studied nuclear enzyme in the PARP superfamily (Gupte et al., 2017; Kim et al., 2020). It contains three major functional domains: (1) an N-terminal DNA-binding domain, containing three zinc-finger motifs and a nuclear localization sequence (NLS), which recognizes both DNA double- and single-strand breaks; (2) a central BRCA1 C terminus (BRCT) auto-modification domain that is a target of self-poly-(ADP-ribosyl)ation; and (3) a C-terminal catalytic domain containing a tryptophan–glycine–arginine-rich (WGR) motif and a PARP signature motif that is the nicotinamide adenine dinucleotide (NAD) binding site essential for PAR synthesis (Figure 1) (Clark et al., 2012; Kameshita et al., 1984; Kraus & Lis, 2003; Luo & Kraus, 2012; Pinnola et al., 2007; Thomas et al., 2014). PARP-1 is directly activated by DNA damage including DNA alkylation and strand nicks and breaks (Lautier et al., 1993; Wang et al., 2019). Oxidative stress is another trigger that induces excessive DNA damage, PARP-1 activation, and PARthanatos (Figure 2) (Park et al., 2020; Wang et al., 2009, 2019). In addition, a variety of environmental stimuli, including free radicals, hydrogen peroxide, hydroxyl radical, peroxynitrite, ionizing radiation, and alkylating chemotherapy drugs, also trigger DNA damage and PARP-1 activation (Wang et al., 2009, 2019).
FIGURE 1.
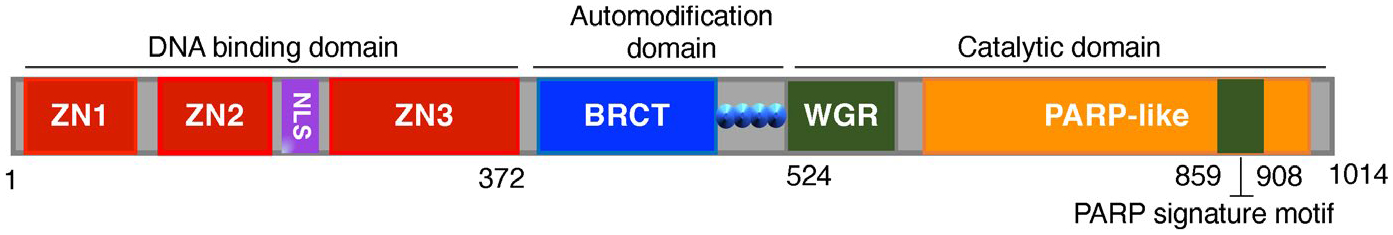
The functional domains of human PARP-1. Human PARP-1 contains a DNA-binding domain consisting of three zinc-binding motifs (Zn1, Zn2, and Zn3) and a nuclear localization signal (NLS) at its N-terminus, an auto-modification domain with BRCA1 C terminus (BRCT) motif in the center, and a catalytic domain with a WGR (tryptophan–glycine–arginine-rich) motif and a PARP signature motif at its C-terminus
FIGURE 2.
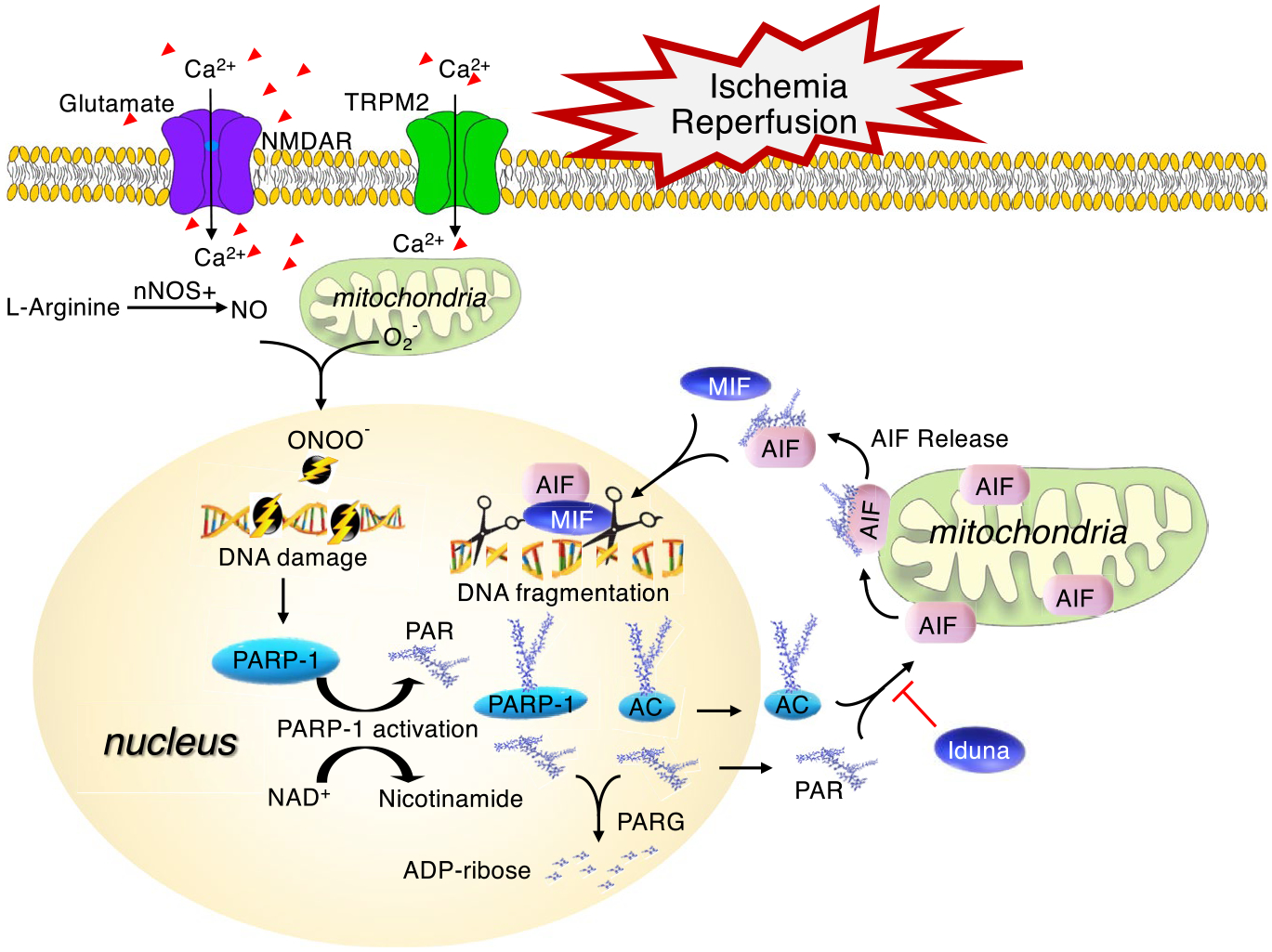
PARthanatos signaling pathway following ischemia and reperfusion. Following ischemia and reperfusion injury, the excess release of glutamate activates NMDA receptor (NMDAR) and causes extracellular calcium influx, which leads to nitric oxide (NO) production and reactive oxygen species (ROS, e.g., superoxide, hydrogen peroxide, and peroxynitrite (ONOO−)) generation. Peroxynitrite can directly damage DNA and causes PAPR-1 hyperactivation. PARP-1 uses NAD+ as the substrate to generate poly-ADP-ribose (PAR) and catalyzes the addition of PAR to different accept proteins (AC) including PARP-1 itself, which might lead to energy depletion. Then, free PAR and/or PARylated accept proteins are translocated from nucleus to mitochondria and trigger AIF release from mitochondria. AIF recruits MIF to the nucleus where MIF functions as a nuclease and cuts DNA into a large fragmentation leading to chromatinolysis and subsequent cell death. TRPM2 receptor (TRPM2R) is regulated by free intracellular PAR and may amplify PARthanatos signaling by increasing calcium influx. In contrast, poly(ADP-ribose) glycohydrolase (PARG) dynamically cleaves PAR into mono ADP-ribose, which suppresses PAR death signal. In addition, Iduna is a PAR-dependent E3 ligase and interferes with PARthanatos by blocking PAR-AIF cross talk
Following ischemic stroke, massive excitatory neurotransmitter glutamate is released. Subsequently, N-methyl-d-aspartate (NMDA) receptor is activated leading to calcium influx, nitric oxide (NO) production, and reactive oxygen species (ROS) generation (Figure 2) (Wang et al., 2009). Previous studies showed that NO levels in the cortex are strikingly increased within minutes following middle cerebral artery occlusion (MCAO) and reperfusion (Malinski et al., 1993). NO then rapidly reacts with superoxide to generate excessive unstable oxidant peroxynitrite (ONOO−), which induces chromosomal DNA nicks and breaks to trigger PARP-1 activation (Endres et al., 1998). In line with this, formation of PAR is detected as early as 5 min after 2-hr MCAO, peaks at 1 hr after reperfusion, then decreases rapidly at 6 hr and back to the basal level at 24 hr after reperfusion in ischemic cortex (Endres et al., 1997, 1998). Moreover, PARP-1 activation and PAR production have been found mainly in human neurons within the ischemic infarct core area predominantly during initial 18–24 hrs, and a few PAR is detected in glia and infiltrated macrophages in the adjacent area after 24 hr (Love et al., 2000). These studies provide the direct evidence of PARP-1 activation in ischemic stroke. However, studies on quantification of PARP-1 activation and its functional differences in the core and peri-core areas are still lacking.
2.2 |. PARP-1 activation-impaired NAD+ metabolism in ischemic stroke
NAD+ is a cofactor and substrate of hundreds of enzymes that are involved in various fundamental metabolic and biological processes to sustain cell survival. It exists in different subcellular compartments including mitochondria, cytosol, and nucleus as either the oxidized forms NAD+ and NADP+ or the reduced forms NADH and NADPH. Interestingly, NAD+ pools in these subcellular compartments have non-redundant functions (Cambronne & Kraus, 2020). In the cytosol, NAD+ is essential for glycolysis and the production of lactate from pyruvate, whereas NAD+ in the mitochondrial matrix is required for redox homeostasis, fatty acid catabolism, tricarboxylic acid (TCA) cycle, and oxidative phosphorylation. NAD+ can be consumed by sirtuins in mitochondria, cytosol, and nucleus, which have an impact on gene expression, genome stability, and metabolism (Covarrubias et al., 2021).
PARP-1 is another major consumer of cellular NAD+ in the nucleus. Upon PARP-1 activation, it uses NAD+ as the substrate and transforms NAD+ into the different length of PAR, which are further transferred to a variety of nuclear proteins (PARylation), including histones, DNA polymerases, topoisomerases, DNA ligase-2, transcription factors, and PARP-1 itself (Lautier et al., 1993). Although the basal PAR levels are very low, hyperactivation of PARP-1 leads to 10–500-fold increase in PAR formation, which varies in lengths, branching frequencies, and complexity (Aberle et al., 2020). PARP-1-dependent PARylation plays the crucial roles in DNA damage sensing, repair and genome stability maintenance, as well as gene transcription (Izhar et al., 2015; Lanz et al., 2019; Wang et al., 2019). On the other hand, PARylation is energetically challenging and may result in cellular NAD+ depletion and energetic collapse (Figure 3). Multiple previous studies have shown that the ipsilateral cellular levels of NAD+ are significantly reduced as compared to its contralateral levels at 2–24 hrs following ischemic stroke (Endres et al., 1997; Hu et al., 2017). It is no doubt that energy depletion plays an important role in ischemic cell death, although previous studies in primary cell cultures indicate that energy depletion may not be a primary factor in PARP-1-mediated cell death (Fossati et al., 2007). Recently, it was shown that PARP-1 activation is associated with mitochondrial dysfunction and causes energy depletion (Figure 3) (Andrabi et al., 2014). More specifically, PARP-dependent energy depletion occurs through inhibition of glycolysis but not NAD+ depletion as NAD+ depletion by a nicotinamide phosphoribosyltransferse inhibitor FK866 does not alter glycolysis or mitochondrial function (Andrabi et al., 2014). These studies indicate that energy depletion alone might not be sufficient to mediate PARthanatos, but it does not exclude that PARP-1-dependent energy depletion is a part of ischemic cell death mechanisms. Further studies are required to investigate whether PARP-1-regulated alteration of NAD+ metabolism contributes to PARthanatos in stroke.
FIGURE 3.
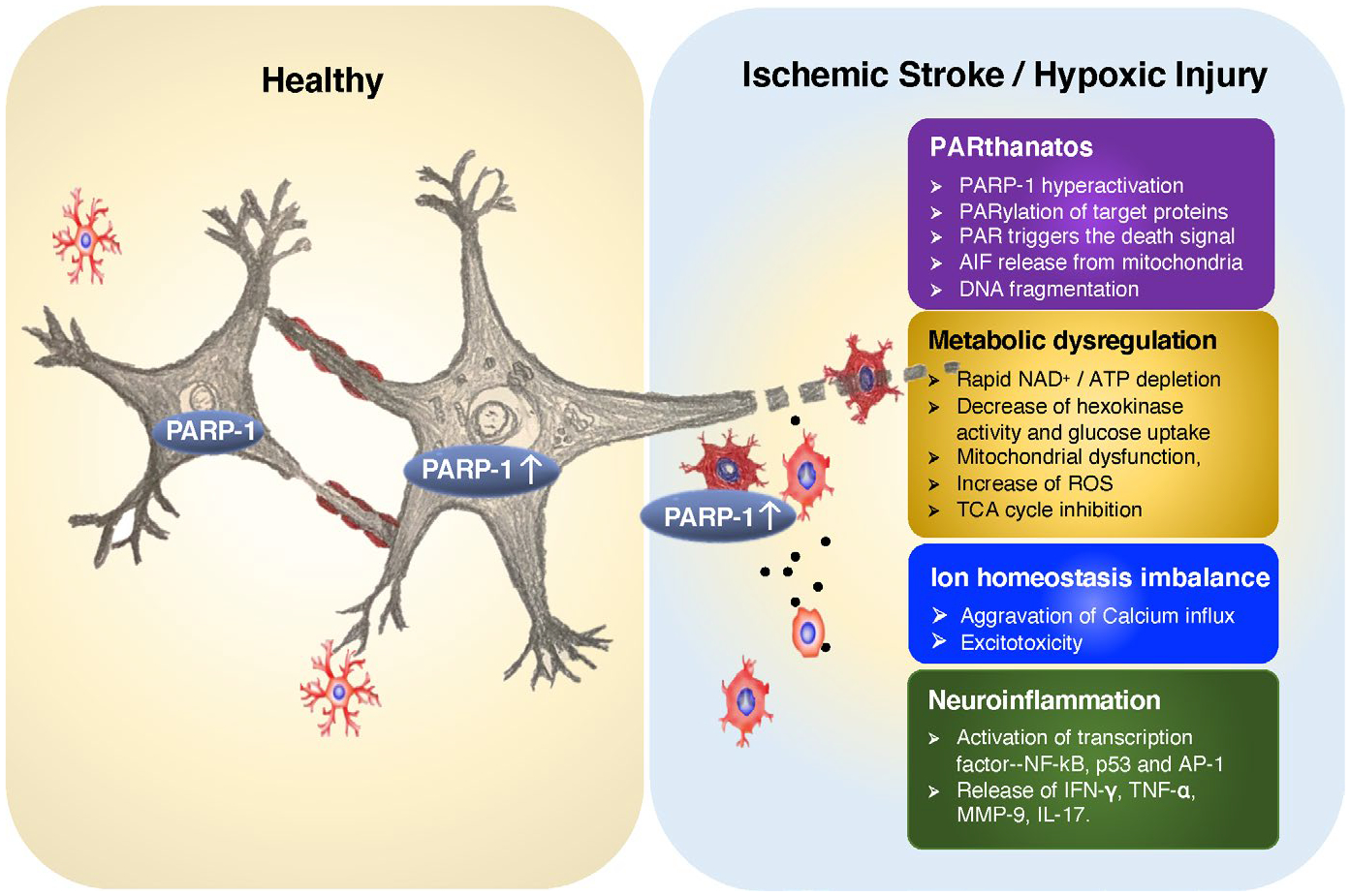
Multifaceted effects of PARP-1 activation on neuron and microglia following ischemia/hypoxia. PARP-1 has multifaceted effects on neuron and glial cells and causes neuronal cell death following ischemic/hypoxic injury. First, PARP-1 hyperactivation leads to PAR accumulation, which enables nuclear-mitochondria cross talk and triggers AIF release and subsequent PARthanatos. Second, PARP-1 hyperactivation causes NAD+ depletion and regulates metabolic reprogramming, including inhibition of intracellular glucose uptake and hexokinase activity, ROS increase, and TCA cycle inhibition. Third, PARP-1 activation participates in regulation of ion homeostasis during oxidative stress by generation of PAR, which aggravates calcium influx through TRPM2 and AMPA receptors leading to a vicious cycle of more calcium influx and more excitotoxicity. Fourth, PARP-1 activation plays a role in microglial activation and neuroinflammation by activating transcription factors (such as NF-κB, p53, and AP-1) and their downstream gene expression
2.3 |. Apoptosis-inducing factor (AIF)—a key mediator of PARthanatos in ischemic stroke
AIF is a mitochondrial flavoprotein with a vital function in bioenergetics and a lethal function when it moves to the nucleus (Wang et al., 2009, 2011). It mainly locates in the mitochondrial intermembrane space with its N-terminus attached to the inner membrane and another small portion of AIF is associated with the outer membrane (Yu et al., 2009). AIF is important for mitochondrial complex I assembly and contributes to the oxidative phosphorylation process under physiological conditions. AIF has also been implicated to function as a ROS scavenger (Polster, 2013; Wang et al., 2009). Apart from its vital function, AIF has been recognized as a key cell death mediator in PARthanatos (Figure 2). Microinjection of AIF antibody protects against PARthanatos (Yu et al., 2002).
During the process of PARthanatos, PARP-1 hyperactivation leads to the excessive accumulation of PAR. PAR itself functions as a death signal (Andrabi et al., 2006), and translocates from the nucleus to mitochondria, where it interacts with AIF on the conserved PAR binding motif and triggers AIF release from mitochondria (Wang et al., 2011; Yu et al., 2006). Mutation of PAR-binding domain on the C-terminus of AIF almost completely abolishes AIF nuclear translocation and PARthanatos (Wang et al., 2011). Therefore, both AIF and PAR are required for nucleus–mitochondria cross talk to mediate PARthanatos.
The translocation of AIF from mitochondria to nucleus is considered a key step of PARthanatos. AIF is initially synthesized in the cytosol as a precursor of 67 kDa and processed into its mature form (62 kDa) after transporting into mitochondria. Calpain I has been shown to cleave AIF at L101/G102 into a soluble truncated AIF (tAIF) of 57 kDa following oxygen-glucose deprivation or transient global ischemia (Cao et al., 2007). Then, tAIF is disassociated from the inner membrane and translocated into the nucleus to mediate ischemic cell death. Over-expression of a calpain inhibitor calpastatin or knockdown of calpain I reduces tAIF nuclear translocation in CA1 neurons after global ischemia and suppresses ischemic cell death (Cao et al., 2007). Recently, another study also showed that calpain I-mediated AIF truncation and AIF nuclear translocation cause DNA fragmentation and myocyte cell death (Chelko et al., 2021). Therefore, calpain I may play a role in mitochondrial AIF cleavage and release in ischemic stroke. Future study is needed to determine whether calpain I-mediated release of AIF is a parallel pathway independent of PAR, or whether calpain I plays a role in PAR-mediated AIF release during PARthanatos.
2.4 |. Macrophage migration inhibitory factor (MIF)-executor of PARthanatos in ischemic stroke
AIF nuclear translocation is often associated with nuclear shrinkage, chromatin condensation, and large DNA fragmentation in PARthanatos. However, AIF itself does not have the obvious nuclease activity. Studies from C. elegans models showed that AIF homolog wah-1 cooperates with mitochondrial endonuclease CPS-6/endonuclease G (EndoG) to promote DNA degradation and cell death (Wang et al., 2002). However, EndoG does not seem to be a primary contributor for large DNA fragmentation in PARthanatos in mammals since knockout of EndoG fails to block PARP-1 hyperactivation-induced large DNA fragmentation and cell death (Wang et al. 2016). Through two sequential high-throughput screenings for AIF-interacting proteins critical for PARthanatos, we recently identified MIF as a PARP-1 activity-associated nuclease (PAAN) that requires Mg2+ or Ca2+ for its 3’ nuclease activity (Wang et al. 2016). MIF was previously known as a secreted protein involved in inflammation, immune response, and cancers and its expression is up-regulated following hypoxia and ischemic stroke (Bloom & Bennett, 1966; Bloom et al., 2016; Oda et al., 2008). Following PARP-1 hyperactivation, AIF is released from mitochondria and recruits MIF to the nucleus, where MIF cleaves DNA into 10-to 50-kb large DNA fragments (Wang et al. 2016). Moreover, depletion of MIF, disruption of AIF and MIF interaction, or suppression of MIF’s nuclease activity protects neurons from NMDA-induced cytotoxicity or ischemic cell death. It also has long-lasting histological and behavioral rescue in the transient focal ischemic model of stroke (Wang et al. 2016). Thus, MIF functions as the executive nuclease in PARthanatos to chop genomic DNA into large fragmentation, and subsequently causes chromatinolysis and cell death (Figure 2).
3 |. MITOCHONDRIAL OXIDATIVE STRESS AND METABOLIC ALTERATION: CAUSE OR CONSEQUENCE OF PARP-1 ACTIVATION IN ISCHEMIC STROKE?
Brain is highly dependent on aerobic respiration because of its high demand for energy and vulnerability to oxidative stress. Mitochondria produce ATP and maintain the homeostasis of ROS including superoxide and hydrogen peroxide under physiological conditions. Unlike the basal level of ROS that contributes to physiological regulation and modulation of cell signaling and can be cleared by intracellular scavengers, excessive ROS is accumulated as a result of insufficient oxygen in stroke and other neurological diseases and irreversibly oxidizes many critical cellular building blocks, including nucleic acids, lipids, and proteins, thereby altering their functions and cell viability (Crack & Taylor, 2005; Hernansanz-Agustín et al., 2014, 2017; Khoshnam et al., 2017). Oxidative stress, no matter acute or chronic, remains to be the key causal factor in many neurological disorders including stroke and degenerative disorders, since oxidative molecules are endogenous inducer of DNA damage and PARP-1 activation leading to PARthanatos.
On the other hand, the notable disturbances in cerebral glucose metabolism in ischemic patients were uncovered a decade ago by tracing glucose uptake using 18F-FDG-PET (Bunevicius et al., 2013). Glucose utilization is dramatically decreased in the ischemic core, but elevated in the peri-ischemic area to support peri-ischemic cells for survival (Bunevicius et al., 2013). PARP-1 is activated excessively in the core area and inhibits hexokinase activity and glycolysis following ischemic injury (Figure 3). Hexokinase is a rate-limiting enzyme to initiate the first step of glycolysis by phosphorylating glucose to glucose-6-phosphate. It is attached to the outer mitochondrial membrane and contains a putative PAR-binding motif. Upon PARP-1 activation during ischemic stroke, PAR interacts with hexokinase and triggers the release of hexokinase from mitochondria, and subsequently, reduces hexokinase activity (Fouquerel et al., 2014). In addition, oxidative phosphorylation is impaired under hypoxia, leading to reduced ATP production but ROS generation. As such, the reduced glycolytic metabolic flux (basal glycolysis, glycolytic capacity, glycolytic reserve, and lactate production) in the ischemic core regions could be the downstream metabolic impact of PARP-1 activation. In addition, PARP-1 also directly modulates mitochondrial bioenergetics as determined by mitochondrial oxygen consumption rate and reserved respiratory capacity (Andrabi et al., 2014) and regulates the TCA cycle through affecting NAD+/NADH levels (Dölle et al., 2013). PAR also has a direct effect on mitochondrial membrane potential collapse (Cipriani et al., 2005). Thus, PARP-1 activation may inhibit glycolysis and cause energy depletion, leading to altered cellular metabolism.
Together, PARP-1 activation in the nucleus has a strong connection with the status of mitochondrial oxidative stress and cellular metabolic reprogramming. The crosstalk between nucleus and mitochondria seems to be critical for the cell fate following stroke. Future studies are required to understand the deep layer of connections of PARP-1 activation with mitochondrial oxidative stress and cellular metabolic reprogramming in ischemic cell death.
4 |. PARP-1 ACTIVATION IN NEUROINFLAMMATION AND ISCHEMIC STROKE
The inflammatory response in brain following stroke involves glial activation and migration, which contribute to cell death (Skaper, 2003). PARP-1 participates in the progression of the inflammatory response in brain (Figure 3). PARP-1 is activated by TNF-α treatment and, in turn, controls TNF-α-induced inflammatory responses in glial cells following ischemic stroke (Skaper, 2003). Genetic or pharmacological inhibition of PARP-1 blocks TNF-α-induced inflammatory response in microglia (Chang & Alvarez-Gonzalez, 2001; Chiarugi & Moskowitz, 2003; Kauppinen & Swanson, 2005; Madinier et al., 2009; Ullrich et al., 2001), indicating that PARP-1 is required for microglial activation. The underlying molecular signaling transduction involves TNF-α receptor 1, calcium entry, activation of phosphatidylcholine-specific phospholipase C, and activation of the MEK1/ERK2 protein kinase cascade (Kauppinen et al., 2013; Vuong et al., 2015). In addition, PARP-1 interacts with transcription factors including NF-κB, p53, and AP-1 and functions as a transcriptional co-activator to control transcription of genes involved in inflammatory response (Nakajima et al., 2004). PARP-1 PARylates NF-κB at its PAR-binding motif in vitro, although the significance of NF-κB PARylation remains unclear (Kameoka et al., 2000; Pleschke et al., 2000). Various animal studies showed potential neuroprotective effects of PARP-1 inhibitor 3-aminobenzamide (3-AB) on inhibition of neuroinflammation and neuronal cell death following ischemic stroke (Koh et al., 2004) and also traumatic brain injury (Lescot et al., 2010). Moreover, treatment of PARP-1 inhibitor JPI-289 decreases pro-inflammatory cytokines (IFN-γ, TNF-α, and IL-17) in stroke patients (Noh et al., 2018). Collectively, these studies support that targeting PARP-1 is protective under conditions of stroke.
The neurovascular unit, which consists of neurons, astrocytes, microglial cells, oligodendrocytes, endothelial cells, smooth muscular cells, pericytes, and basal membranes of the key structure of blood–brain barrier (BBB), plays an important role in inflammation following ischemic brain injury (Rom et al., 2016; Wang et al., 2020). Activation and migration of glial cells as well as infiltration of neutrophils and macrophages significantly contribute to the release of inflammatory factors and breakdown of basal lamina of the capillaries. Post-stroke inflammatory response prompts a vicious circle, which ultimately causes collapse of neurovascular unit functions and cell death. Thus, inhibition of inflammatory response and preservation of functions of neurovascular unit are critical for stroke recovery. Targeting PARP-1 as an approach to inhibit neuroinflammation and preserve the integrity of BBB has been explored recently (Rom et al., 2016; Wang et al., 2020). PARP-1 deletion substantially eliminates TNF-α-induced inflammatory responses in brain microvasculature and reduces BBB permeability through suppressing protein levels of adhesion molecules and activity of GTPases (Rom et al., 2016; Wang et al., 2020). These studies indicate that PARP-1 may also play an important role in neurovascular unit and neuroinflammation in ischemic stroke.
5 |. PARP-1 REGULATES ION INFLUX IN ISCHEMIC/HYPOXIC BRAIN INJURY
TRPM2 is a non-selective cation channel highly expressed in brain and involved in influx of extracellular Ca2+. It has been reported that PARP-1 activation is required for TRPM2 channel opening in response to oxidative stress (Fonfria et al., 2004). Poly(ADP-ribose) glycohydrolase (PARG) is a known enzyme responsible for the degradation of PAR into free ADP-ribose and also regulates TRPM2-mediated Ca2+ flux, leading to cell death (Blenn et al., 2011). These studies indicate that ADP-ribose as the main metabolite of PARP-1/PARG system might be the key driver regulating TRPM2 and Ca2+ influx. In addition, another study showed that PARP-1 hyperactivation induced by an alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) increases the expression of Ca2+-permeable AMPA receptors and causes pyramidal cell death in the hippocampal CA1 region (Gerace et al., 2014). These studies indicate that PARP-1 and ADP-ribose may play a role in regulation of ion influx during oxidative stress (Figure 3).
6 |. PARP-1 AND HYPOXIA-INDUCIBLE FACTOR (HIF) IN ISCHEMIC/HYPOXIC BRAIN INJURY
HIF is a master regulator of hypoxia response (Luo & Wang, 2018). It consists of an inducible α-subunit and a constitutively expressed β-subunit (Luo & Wang, 2018). Expression of HIF-1α is increased in the penumbra tissues and ischemic core regions following stroke (Bergeron et al., 1999; Demougeot et al., 2004). HIF induces hundreds of genes whose proteins are involved in angiogenesis, epigenetics, and metabolism (Luo & Wang, 2018) and has also been implicated to play a role in ischemic stroke, although its precise role still remains debating (Shi, 2009). Both detrimental and beneficial effects of HIF-1 were previously reported in ischemic stroke, indicating that HIF may contribute to cell death after a severe and prolonged ischemia but promote cell survival following mild ischemic injury (Shi, 2009). Similar to HIF’s highly context-dependent functions in ischemic stroke, PARP-1 has been known to promote DNA repair and cell survival in response to mild DNA damage and triggers cell death following severe DNA damage and brain injury (Wang et al. 2016; Wang et al., 2009, 2019). Despite the importance and similarity of PARP-1 and HIF in ischemic stroke, little is known whether PARP-1 and HIF cooperate to regulate neuronal cell fate decision-making following the brain injury, except that PARP-1 has been shown to regulate HIF-1 expression following hypoxia in mouse brain (Martinez-Romero et al., 2009). In addition, previous studies showed that PARP-1 interacts and forms a complex with HIF-1α, thereby regulating HIF-1 transcriptional activation in myelogenous leukemia cells as well as B cells upon hypoxic stress (Elser et al., 2008; Hulse et al., 2018). It would be interesting to study how PARP-1 and HIF directly cooperate and impact on ischemic stroke outcome in the future.
7 |. POTENTIAL THERAPIES BY TARGETING PARP-1 AND PARTHANATOS
Given the importance of PARP-1 in PARthanatos, metabolism and neuroinflammation in ischemic stroke (Figure 3), PARP-1 becomes a potential therapeutic target to prevent ischemic brain injury. So far, four PARP-1 inhibitors including olaparib, veliparib, niraparib, and rucaparib have been approved by FDA to treat BRCA1/2-mutant ovarian cancer and metastatic breast and prostate cancers by suppressing PARP-1 functions in DNA damage repair and increasing synthetic lethality (Ledermann et al., 2012; Mirza et al., 2016; Swisher et al., 2017). Although the application of PARP inhibitors in the clinical trials for stroke treatment is far behind as compared to their application in cancer therapy, increasing number of preclinical studies showed great enthusiasm to use PARP inhibitors to treat acute ischemic brain injury and chronic neurological and systematic disorders (Table 1). For example, Dawson laboratory recently showed that PARP inhibitors including veliparib, rucaparib, and talazoparib suppress pathologic α-synuclein aggregation and increase cell viability in PD models (Kam et al., 2018). Moreover, PARP inhibitor olaparib has been shown to attenuate TDP-43-induced motor neuron cell death in models of ALS (Duan et al., 2019). Currently, multiple clinical trials of PARP-1 inhibitors are underway to treat acute ischemic stroke (JPI-289, phase2, NCT03062397) (Kim et al., 2018a), ischemic acute kidney injury (Jang et al., 2020), myocardial ischemia, diabetes, diabetes-associated cardiovascular dysfunction, shock, and traumatic central nervous system injury.
One possible limitation of PARP-1 inhibitors on treating stroke might be sexual dimorphism, although it is still under debating. Inhibition of PARP-1 hyperactivation has been proven to reduce ischemia-induced PAR formation and AIF nuclear translocation in both sexes (Yuan et al., 2009). However, the detrimental effect of PARP-1 activation on ischemic cell death in both sexes remains different. Several studies reported that PARP-1 inhibitors reduce stroke-induced lesion volume and improve behavior mainly in male but not female animals (Charriaut-Marlangue et al., 2018; Liu et al., 2011), which may be partially caused by impaired PARthanatos in females (Sharma et al. 2011) or different levels of estrogen and androgen in female and male mice (Dang et al., 2011; Vagnerova et al., 2010). 17β-estradiol and progesterone treatment have been shown to be protective and improve behavioral function in both males and ovariectomized females (Dang et al., 2011). It has also been reported that estrogen does not directly inhibit the enzymatic activity of PARP (Mabley et al., 2005). However, androgen–androgen receptor signaling is required for PARthanatos in male MCAO mouse models as the reduction in infarction caused by PJ-34 in wild-type mice is lost after removal of testicular androgens, which could be reversed by androgen treatment (Vagnerova et al., 2010). Meanwhile, XX and XY neurons exhibit differential vulnerability independent of sex hormone effects, in response to various cytotoxic agents (Du et al., 2004). Another study showed that delayed treatment of PARP-1 inhibitor PJ34 reduces microglial activation and neuroinflammation to similar levels in both male and female mice, but inhibition of inflammatory cytokines (iNOS, IL-1β, and MMP-9) by PARP-1 inhibitor is more profound in male MCAO mice, and PARP-1 inhibitor-caused improvement of neurological performance is also more prominent in males (Chen et al., 2020). Future studies are required to further investigate the role of PARP-1 and its inhibitors in ischemic brain injury in both sexes and underlying deep mechanisms.
Besides PARP-1 itself, its downstream signaling factors in PARthanatos are also attractive therapeutic targets. Iduna is a PARylation-dependent E3 ligase (Kang et al., 2011). It ubiquitinates and degrades PARylated substrates and protects against NMDA-induced excitotoxicity and ischemic stroke-induced neuronal cell death in mice (Andrabi et al., 2011). Thus, Iduna could be one of the potential target to reduce ischemic brain injury (Figure 2). Harlequin mice with about 80% of AIF reduction display resistance to NMDA-induced neurotoxicity (Wang et al., 2011). We have shown that preventing AIF nuclear translocation, interfering AIF-MIF interaction, or inhibiting MIF nuclease activity may potentially block or reduce PARthanatos in the model of ischemic stroke (Wang et al. 2016).
Melatonin (N-acetyl-5-methoxytrptamine), a natural hormone with antioxidative and anti-inflammatory properties, has a neuroprotective effect in various models of injury including stroke, traumatic brain injury, and spinal cord injury (Andrabi et al., 2015). Melatonin may suppress PARP-1 activation and inhibit PARthanatos following ischemic stroke as it inhibits the upstream factors of PARthanatos including Ca2+ elavation and mitochondrial oxidative damage (Andrabi et al., 2015). Similarly, propofol (2, 6-diisopropylphenol), a widely used intravenous anesthetic agent, inhibits PARthanatos in vitro and in vivo via suppressing ROS production, Ca2+ releasing, and mitochondrial depolarization. Melatonin and propofol may offer alternative therapeutic approaches to prevent ischemic cell death (Zhong et al., 2018).
Ischemic pre-conditioning via exposure to a non-lethal ischemic stress renders cells less susceptible to severe insults, which could be another strategy protecting neurons from ischemic stroke (Kitagawa et al., 1997; Wang et al., 2015). The mechanisms of ischemic pre-conditioning are complicated, involving changes in gene expression, activation of protein kinase C, post-translational modification, and metabolic regulation (Stenzel-Poore et al., 2003; Wang et al., 2015; Zhang et al., 2011). Mild activation of PARP-1 also contributes to the neuroprotective effects of ischemic pre-conditioning. Ischemic pre-conditioning increases the enzymatic activity of PARP-1 as well as its product PAR. Application of PARP inhibitor before ischemic pre-conditioning abolishes its protective effects (Gerace et al., 2012). The beneficial effect of mild PARP-1 activation is likely related to its DNA repair functions during ischemic preconditioning; however, clear evidence remains limited and insufficient and requires further investigation.
8 |. CONCLUSION
PARthanatos is a unique cell death program distinct from many other known cell deaths like apoptosis, necrosis, and necroptosis, and attributes to ischemic stroke and degenerative disorders (Figure 2). Under conditions of stroke, PARP-1 is hyperactivated upon oxidative insults. PAR then functions as a death signal and triggers AIF release from mitochondria to the nucleus. Subsequently, AIF recruits MIF to the nucleus where MIF cleaves genomic DNA into large fragments and causes neuronal cell death. Genetic deletion or pharmacological inhibition of PARP-1, AIF, or MIF reduces NMDA-induced cytotoxicity and ischemic neuronal cell death. Further studies are required to fully understand PARthanatos, which may help develop inhibitors to specifically block PARthanatos in stroke as well as other neurological diseases.
ACKNOWLEDGMENTS
Works from the authors’ laboratories were supported by grants from the National Institutes of Health (NIH) NS078049, R35GM124693, and R01AG066166, Darrell K Royal Research Fund, Welch Foundation (I-1939-20170325), TIBIR Grant, the University of Texas (UT) Southwestern Medical Center Startup funds and UT Rising Stars to Y.W., NIH (R01CA222393), the CPRIT (RP190358), American Cancer Society-Lisa Dean Mosely Foundation (RSG-19-229-01-DMC), the Mary Kay Foundation (08-19), and the Welch Foundation (I-1903-20190330) to W.L.. W.L. is a CPRIT Scholar in Cancer Research.
Funding information
National Institute on Aging, Grant/Award Number: R01AG066166; Welch Foundation, Grant/Award Number: I-1903-20190330 and I-1939-20170325; University of Texas Rising Stars; American Cancer Society, Grant/Award Number: RSG-19-229-01-DMC; Mary Kay Foundation, Grant/Award Number: 08-19; Cancer Prevention and Research Institute of Texas, Grant/Award Number: RP190358; National Institute of Neurological Disorders and Stroke, Grant/Award Number: NS078049; National Institute of General Medical Sciences, Grant/Award Number: R35GM124693; Texas Institute for Brain Injury and Repair, University of Texas Southwestern Medical Center; Darrell K Royal Research Fund; National Cancer Institute, Grant/Award Number: R01CA222393
Abbreviations:
- AIF
apoptosis-inducing factor
- ATP
adenosine triphosphate
- BBB
blood–brain barrier
- EndoG
endonuclease G
- HIF
hypoxia-inducible factor
- MCAO
middle cerebral artery occlusion
- Melatonin
N-acetyl-5-methoxytrptamine
- MIF
macrophage migration inhibitory factor
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- NAD
nicotinamide adenine dinucleotide
- NLS
nuclear localization signal
- NMDA
N-methyl-D-aspartate
- NO
Nitric oxide
- PAR
Poly(ADP-ribose)
- PARG
Poly(ADP-ribose) glycohydrolase
- PARP-1
Poly(ADP-ribose) polymerase 1
- PBM
PAR binding motif
- PD
Parkinson’s disease
- Propofol
2, 6-diisopropylphenol
- ROS
reactive oxygen species
- tAIF
truncated AIF
- TCA
tricarboxylic acid
- WGR
tryptophan–glycine–arginine-rich motif
Footnotes
CONFLICT OF INTERESTS
The authors declare that they have no conflict of interests.
DATA AVAILABILITY STATEMENT
Data sharing not applicable - no new data generated.
REFERENCES
- Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, & Endres M (2001). Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. International Journal of Molecular Medicine, 7, 255–260. 10.3892/ijmm.7.3.255 [DOI] [PubMed] [Google Scholar]
- Aberle L, Krüger A, Reber JM, Lippmann M, Hufnagel M, Schmalz M, Trussina IREA, Schlesiger S, Zubel T, Schütz K, Marx A, Hartwig A, Ferrando-May E, Bürkle A, & Mangerich A (2020). PARP1 catalytic variants reveal branching and chain length-specific functions of poly(ADP-ribose) in cellular physiology and stress response. Nucleic Acids Research, 48, 10015–10033. 10.1093/nar/gkaa590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeti R, Abramov AY, & Duchen MR (2011). β-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain, 134, 1658–1672. 10.1093/brain/awr104 [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kang HC, Haince J-F, Lee Y-I, Zhang J, Chi Z, West AB, Koehler RC, Poirier GG, Dawson TM, & Dawson VL (2011). Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nature Medicine, 17, 692–699. 10.1038/nm.2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu S-W, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, & Dawson TM (2006). Poly(ADP-ribose) (PAR) polymer is a death signal. Proceedings of the National Academy of Sciences, 103, 18308–18313. 10.1073/pnas.0606526103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SS, Parvez S, & Tabassum H (2015). Melatonin and ischemic stroke: Mechanistic roles and action. Advances in Pharmacological Sciences, 2015, 384750. 10.1155/2015/384750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagné JP, Poirier GG, Dawson VL, & Dawson TM (2014). Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proceedings of the National Academy of Sciences, 111, 10209–10214. 10.1073/pnas.1405158111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron M, Yu AY, Solway KE, Semenza GL, & Sharp FR (1999). Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. European Journal of Neuroscience, 11, 4159–4170. 10.1046/j.1460-9568.1999.00845.x [DOI] [PubMed] [Google Scholar]
- Blenn C, Wyrsch P, Bader J, Bollhalder M, & Althaus FR (2011). Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cellular and Molecular Life Sciences, 68, 1455–1466. 10.1007/s00018-010-0533-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom BR, & Bennett B (1966). Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science, 153, 80–82. 10.1126/science.153.3731.80 [DOI] [PubMed] [Google Scholar]
- Bloom J, Sun S, & Al-Abed Y (2016). MIF, a controversial cytokine: A review of structural features, challenges, and opportunities for drug development. Expert Opinion on Therapeutic Targets, 20, 1463–1475. 10.1080/14728222.2016.1251582 [DOI] [PubMed] [Google Scholar]
- Bunevicius A, Yuan H, & Lin W (2013). The potential roles of 18F-FDG-PET in management of acute stroke patients. BioMed Research International, 2013, 634598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne XA, & Kraus WL (2020). Location, location, location: Compartmentalization of NAD(+) synthesis and functions in mammalian cells. Trends in Biochemical Sciences, 45, 858–873. 10.1016/j.tibs.2020.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, & Donnan GA (2019). Ischaemic stroke. Nature Reviews Disease Primers, 5, 70. 10.1038/s41572-019-0118-8 [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, & Chen J (2007). Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. Journal of Neuroscience, 27, 9278–9293. 10.1523/JNEUROSCI.2826-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W-J, & Alvarez-Gonzalez R (2001). The sequence-specific DNA binding of NF-κB is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. Journal of Biological Chemistry, 276, 47664–47670. 10.1074/jbc.M104666200 [DOI] [PubMed] [Google Scholar]
- Charriaut-Marlangue C, Leconte C, Csaba Z, Chafa L, Pansiot J, Talatizi M, Simon K, Moretti R, Marchand-Leroux C, Baud O, & Besson VC (2018). Sex differences in the effects of PARP inhibition on microglial phenotypes following neonatal stroke. Brain, Behavior, and Immunity, 73, 375–389. 10.1016/j.bbi.2018.05.022 [DOI] [PubMed] [Google Scholar]
- Chelko SP, Keceli G, Carpi A et al. (2021). Exercise triggers CAPN1-mediated AIF truncation, inducing myocyte cell death in arrhythmogenic cardiomyopathy. Science Translational Medicine, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li X, Xu S, Zhang M, Wu Z, Zhang X, Xu Y, & Chen Y (2020). Delayed PARP-1 inhibition alleviates post-stroke inflammation in male versus female mice: Differences and similarities. Frontiers in Cellular Neuroscience, 14, 77. 10.3389/fncel.2020.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, & Moskowitz MA (2003). Poly(ADP-ribose) polymerase-1 activity promotes NF-κB-driven transcription and microglial activation: Implication for neurodegenerative disorders. Journal of Neurochemistry, 85, 306–317. 10.1046/j.1471-4159.2003.01684.x [DOI] [PubMed] [Google Scholar]
- Chidambaram SB, Vijayan R, Sekar S, Mani S, Rajamani B, & Ganapathy R (2017). Simultaneous blockade of NMDA receptors and PARP-1 activity synergistically alleviate immunoexcitotoxicity and bioenergetics in 3-nitropropionic acid intoxicated mice: Evidences from memantine and 3-aminobenzamide interventions. European Journal of Pharmacology, 803, 148–158. 10.1016/j.ejphar.2017.03.023 [DOI] [PubMed] [Google Scholar]
- Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, & Chiarugi A (2005). Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. Journal of Biological Chemistry, 280, 17227–17234. 10.1074/jbc.M414526200 [DOI] [PubMed] [Google Scholar]
- Clark NJ, Kramer M, Muthurajan UM, & Luger K (2012). Alternative modes of binding of poly(ADP-ribose) polymerase 1 to free DNA and nucleosomes. Journal of Biological Chemistry, 287, 32430–32439. 10.1074/jbc.M112.397067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier JY, Ding-Zhou L, Croci N, Plotkine M, & Margaill I (2003). 3-Aminobenzamide reduces brain infarction and neutrophil infiltration after transient focal cerebral ischemia in mice. Experimental Neurology, 184, 973–980. 10.1016/S0014-4886(03)00367-4 [DOI] [PubMed] [Google Scholar]
- Covarrubias AJ, Perrone R, Grozio A, & Verdin E (2021). NAD(+) metabolism and its roles in cellular processes during ageing. Nature Reviews Molecular Cell Biology, 22, 119–141. 10.1038/s41580-020-00313-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crack PJ, & Taylor JM (2005). Reactive oxygen species and the modulation of stroke. Free Radical Biology and Medicine, 38, 1433–1444. 10.1016/j.freeradbiomed.2005.01.019 [DOI] [PubMed] [Google Scholar]
- Dang J, Mitkari B, Kipp M, & Beyer C (2011). Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain, Behavior, and Immunity, 25, 715–726. 10.1016/j.bbi.2011.01.013 [DOI] [PubMed] [Google Scholar]
- d’Avila JC, Lam TI, Bingham D, Shi J, Won SJ, Kauppinen TM, Massa S, Liu J, & Swanson RA (2012). Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. Journal of Neuroinflammation, 9, 31. 10.1186/1742-2094-9-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demougeot C, Van Hoecke M, Bertrand N, Prigent-Tessier A, Mossiat C, Beley A, & Marie C (2004). Cytoprotective efficacy and mechanisms of the liposoluble iron chelator 2,2’-dipyridyl in the rat photothrombotic ischemic stroke model. Journal of Pharmacology and Experimental Therapeutics, 311, 1080–1087. 10.1124/jpet.104.072744 [DOI] [PubMed] [Google Scholar]
- Ding Y, Zhou Y, Lai Q, Li J, Gordon V, & Diaz FG (2001). Long-term neuroprotective effect of inhibiting poly(ADP-ribose) polymerase in rats with middle cerebral artery occlusion using a behavioral assessment. Brain Research, 915, 210–217. 10.1016/S0006-8993(01)02852-9 [DOI] [PubMed] [Google Scholar]
- Dölle C, Rack JG, & Ziegler M (2013). NAD and ADP-ribose metabolism in mitochondria. FEBS Journal, 280, 3530–3541. 10.1111/febs.12304 [DOI] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, & Clark RS (2004). Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. The Journal of Biological Chemistry, 279, 38563–38570. 10.1074/jbc.M405461200 [DOI] [PubMed] [Google Scholar]
- Duan Y, Du A, Gu J, Duan G, Wang C, Gui X, Ma Z, Qian B, Deng X, Zhang K, Sun LE, Tian K, Zhang Y, Jiang H, Liu C, & Fang Y (2019). PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Research, 29, 233–247. 10.1038/s41422-019-0141-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Amki M, Lerouet D, Garraud M, Teng F, Beray-Berthat V, Coqueran B, Barsacq B, Abbou C, Palmier B, Marchand-Leroux C, & Margaill I (2018). Improved reperfusion and vasculoprotection by the Poly(ADP-Ribose)polymerase inhibitor PJ34 after stroke and thrombolysis in mice. Molecular Neurobiology, 55, 9156–9168. 10.1007/s12035-018-1063-3 [DOI] [PubMed] [Google Scholar]
- Eliasson MJL, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang Z-Q, Dawson TM, Snyder SH, & Dawson VL (1997). Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nature Medicine, 3, 1089–1095. 10.1038/nm1097-1089 [DOI] [PubMed] [Google Scholar]
- Elser M, Borsig L, Hassa PO, Erener S, Messner S, Valovka T, Keller S, Gassmann M, & Hottiger MO (2008). Poly(ADP-ribose) polymerase 1 promotes tumor cell survival by coactivating hypoxia-inducible factor-1-dependent gene expression. Molecular Cancer Research, 6, 282–290. 10.1158/1541-7786.MCR-07-0377 [DOI] [PubMed] [Google Scholar]
- Endres M, Scott G, Namura S, Salzman AL, Huang PL, Moskowitz MA, & Szabó C (1998). Role of peroxynitrite and neuronal nitric oxide synthase in the activation of poly(ADP-ribose) synthetase in a murine model of cerebral ischemia-reperfusion. Neuroscience Letters, 248, 41–44. 10.1016/S0304-3940(98)00224-9 [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, & Moskowitz MA (1997). Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. Journal of Cerebral Blood Flow and Metabolism, 17, 1143–1151. 10.1097/00004647-199711000-00002 [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, & Dawson TM (2014). Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. British Journal of Pharmacology, 171, 2000–2016. 10.1111/bph.12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, & McNulty S (2004). TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. British Journal of Pharmacology, 143, 186–192. 10.1038/sj.bjp.0705914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossati S, Cipriani G, Moroni F, & Chiarugi A (2007). Neither energy collapse nor transcription underlie in vitro neurotoxicity of poly(ADP-ribose) polymerase hyper-activation. Neurochemistry International, 50, 203–210. 10.1016/j.neuint.2006.08.009 [DOI] [PubMed] [Google Scholar]
- Fouquerel E, Goellner EM, Yu Z, Gagné J-P, Barbi de Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, Migaud M, Van Houten B, Poirier GG, & Sobol RW (2014). ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Reports, 8, 1819–1831. 10.1016/j.celrep.2014.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, … Kroemer G (2018). Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death and Differentiation, 25, 486–541. 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerace E, Masi A, Resta F, Felici R, Landucci E, Mello T, Pellegrini-Giampietro DE, Mannaioni G, & Moroni F (2014). PARP-1 activation causes neuronal death in the hippocampal CA1 region by increasing the expression of Ca(2+)-permeable AMPA receptors. Neurobiology of Diseases, 70, 43–52. 10.1016/j.nbd.2014.05.023 [DOI] [PubMed] [Google Scholar]
- Gerace E, Scartabelli T, Formentini L, Landucci E, Moroni F, Chiarugi A, & Pellegrini-Giampietro DE (2012). Mild activation of poly(ADP-ribose) polymerase (PARP) is neuroprotective in rat hippocampal slice models of ischemic tolerance. European Journal of Neuroscience, 36, 1993–2005. 10.1111/j.1460-9568.2012.08116.x [DOI] [PubMed] [Google Scholar]
- Gupte R, Liu Z, & Kraus WL (2017). PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes & Development, 31, 101–126. 10.1101/gad.291518.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby AM, Suh SW, Kauppinen TM, & Swanson RA (2007). Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke, 38, 632–636. 10.1161/01.STR.0000250742.61241.79 [DOI] [PubMed] [Google Scholar]
- Hasan TF, Rabinstein AA, Middlebrooks EH, Haranhalli N, Silliman SL, Meschia JF, & Tawk RG (2018). Diagnosis and management of acute ischemic stroke. Mayo Clinic Proceedings, 93, 523–538. 10.1016/j.mayocp.2018.02.013 [DOI] [PubMed] [Google Scholar]
- Hernansanz-Agustín P, Izquierdo-Álvarez A, Sánchez-Gómez FJ, Ramos E, Villa-Piña T, Lamas S, Bogdanova A, & Martínez-Ruiz A (2014). Acute hypoxia produces a superoxide burst in cells. Free Radical Biology and Medicine, 71, 146–156. 10.1016/j.freeradbiomed.2014.03.011 [DOI] [PubMed] [Google Scholar]
- Hernansanz-Agustín P, Ramos E, Navarro E, Parada E, Sánchez-López N, Peláez-Aguado L, Cabrera-García JD, Tello D, Buendia I, Marina A, Egea J, López MG, Bogdanova A, & Martínez-Ruiz A (2017). Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biology, 12, 1040–1051. 10.1016/j.redox.2017.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Manaenko A, Bian H, Guo Z, Huang JL, Guo ZN, Yang P, Tang J, & Zhang JH (2017). Hyperbaric oxygen reduces infarction volume and hemorrhagic transformation through ATP/NAD(+)/Sirt1 pathway in hyperglycemic middle cerebral artery occlusion rats. Stroke, 48, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse M, Caruso LB, Madzo J, Tan Y, Johnson S, & Tempera I (2018). Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by Epstein-Barr virus latent membrane protein 1. PLoS Pathogens, 14, e1007394. 10.1371/journal.ppat.1007394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashita A, Tojo N, Matsuura S et al. (2004). A novel and potent poly(ADP-ribose) polymerase-1 inhibitor, FR247304 (5-chloro-2-[3-(4-phenyl-3,6-dihydro-1(2H)-pyridinyl)propyl]-4(3H)-quinazolinone), attenuates neuronal damage in in vitro and in vivo models of cerebral ischemia. Journal of Pharmacology and Experimental Therapeutics, 310, 425–436. [DOI] [PubMed] [Google Scholar]
- Izhar L, Adamson B, Ciccia A, Lewis J, Pontano-Vaites L, Leng Y, Liang AC, Westbrook TF, Harper JW, & Elledge SJ (2015). A Systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Reports, 11, 1486–1500. 10.1016/j.celrep.2015.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HR, Lee K, Jeon J, Kim J-R, Lee JE, Kwon GY, Kim Y-G, Kim DJ, Ko J-W, & Huh W (2020). Poly (ADP-Ribose) polymerase inhibitor treatment as a novel therapy attenuating renal ischemia-reperfusion injury. Frontiers in Immunology, 11, 564288. 10.3389/fimmu.2020.564288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam TI, Mao X, Park H et al. (2018). Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science, 362, eaat8407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameoka M, Ota K, Tetsuka T, Tanaka Y, Itaya A, Okamoto T, & Yoshihara K (2000). Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. The Biochemical Journal, 346(Pt 3), 641–649. [PMC free article] [PubMed] [Google Scholar]
- Kameshita I, Matsuda Z, Taniguchi T, & Shizuta Y (1984). Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. Journal of Biological Chemistry, 259, 4770–4776. 10.1016/S0021-9258(17)42913-9 [DOI] [PubMed] [Google Scholar]
- Kang HC, Lee Y-I, Shin J-H, Andrabi SA, Chi Z, Gagne J-P, Lee Y, Ko HS, Lee BD, Poirier GG, Dawson VL, & Dawson TM (2011). Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proceedings of the National Academy of Sciences, 108, 14103–14108. 10.1073/pnas.1108799108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaundal RK, Shah KK, & Sharma SS (2006). Neuroprotective effects of NU1025, a PARP inhibitor in cerebral ischemia are mediated through reduction in NAD depletion and DNA fragmentation. Life Sciences, 79, 2293–2302. 10.1016/j.lfs.2006.07.034 [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Gan L, & Swanson RA (2013). Poly(ADP-ribose) polymerase-1-induced NAD(+) depletion promotes nuclear factor-κB transcriptional activity by preventing p65 de-acetylation. Biochimica Et Biophysica Acta, 1833, 1985–1991. 10.1016/j.bbamcr.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, & Swanson RA (2005). Poly(ADP-Ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. The Journal of Immunology, 174, 2288–2296. 10.4049/jimmunol.174.4.2288 [DOI] [PubMed] [Google Scholar]
- Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, & Moghaddam HF (2017). Pathogenic mechanisms following ischemic stroke. Neurological Sciences, 38, 1167–1186. 10.1007/s10072-017-2938-1 [DOI] [PubMed] [Google Scholar]
- Kim DS, Challa S, Jones A, & Kraus WL (2020). PARPs and ADP-ribosylation in RNA biology: From RNA expression and processing to protein translation and proteostasis. Genes & Development, 34, 302–320. 10.1101/gad.334433.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kim YS, Kim HY, Noh M-Y, Kim JY, Lee Y-J, Kim J, Park J, & Kim SH (2018a). Early treatment with Poly(ADP-Ribose) polymerase-1 inhibitor (JPI-289) reduces infarct volume and improves long-term behavior in an animal model of ischemic. Stroke, 55, 7153–7163. 10.1007/s12035-018-0910-6 [DOI] [PubMed] [Google Scholar]
- Kim Y, Kim YS, Kim HY, Noh MY, Kim JY, Lee YJ, Kim J, Park J, & Kim SH (2018b). Early treatment with poly(ADP-Ribose) polymerase-1 inhibitor (JPI-289) reduces infarct volume and improves long-term behavior in an animal model of ischemic stroke. Molecular Neurobiology, 55, 7153–7163. 10.1007/s12035-018-0910-6 [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Mandai K, Matsushita K, Hori M, & Yanagihara T (1997). Ischemic tolerance in hippocampal CA1 neurons studied using contralateral controls. Neuroscience, 81, 989–998. 10.1016/S0306-4522(97)00229-7 [DOI] [PubMed] [Google Scholar]
- Klofers M, Kohaut J, Bendix I, Herz J, Boos V, Felderhoff-Muser U, & Dzietko M (2017). Effects of poly(ADP-Ribose) polymerase-1 inhibition in a neonatal rodent model of hypoxic-ischemic injury. BioMed Research International, 2017, 2924848. 10.1155/2017/2924848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S-H, Park Y, Song CW, Kim JG, Kim K, Kim J, Kim M-H, Lee SR, Kim DW, Yu H-J, Chang D-I, Hwang SJ, & Kim SH (2004). The effect of PARP inhibitor on ischaemic cell death, its related inflammation and survival signals. European Journal of Neuroscience, 20, 1461–1472. 10.1111/j.1460-9568.2004.03632.x [DOI] [PubMed] [Google Scholar]
- Kraus WL, & Lis JT (2003). PARP goes transcription. Cell, 113, 677–683. 10.1016/S0092-8674(03)00433-1 [DOI] [PubMed] [Google Scholar]
- Lanz MC, Dibitetto D, & Smolka MB (2019). DNA damage kinase signaling: Checkpoint and repair at 30 years. The EMBO Journal, 38, e101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautier D, Lagueux J, Thibodeau J, Menard L, & Poirier GG (1993). Molecular and biochemical features of poly (ADP-ribose) metabolism. Molecular and Cellular Biochemistry, 122, 171–193. 10.1007/BF01076101 [DOI] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, & Matulonis U (2012). Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. New England Journal of Medicine, 366, 1382–1392. 10.1056/NEJMoa1105535 [DOI] [PubMed] [Google Scholar]
- Lescot T, Fulla-Oller L, Palmier B, Po C, Beziaud T, Puybasset L, Plotkine M, Gillet B, Meric P, & Marchand-Leroux C (2010). Effect of acute poly(ADP-ribose) polymerase inhibition by 3-AB on blood-brain barrier permeability and edema formation after focal traumatic brain injury in rats. Journal of Neurotrauma, 27, 1069–1079. 10.1089/neu.2009.1188 [DOI] [PubMed] [Google Scholar]
- Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, & Wang G (2020). Ferroptosis: Past, present and future. Cell Death & Disease, 11, 88. 10.1038/s41419-020-2298-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Gharavi R, Pitta M, Gleichmann M, & Mattson MP (2009). Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. NeuroMolecular Medicine, 11, 28–42. 10.1007/s12017-009-8058-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lang J, Li J, Benashski SE, Siegel M, Xu Y, & McCullough LD (2011). Sex differences in the response to poly(ADP-ribose) polymerase-1 deletion and caspase inhibition after stroke. Stroke, 42, 1090–1096. 10.1161/STROKEAHA.110.594861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Bosque-Hamilton P, & Meng W (1998). Inhibition of poly(ADP-ribose) polymerase: Reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke, 29, 830–836. 10.1161/01.STR.29.4.830 [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, & Wilcock GK (1999). Increased poly(ADP-ribosyl) ation of nuclear proteins in Alzheimer’s disease. Brain, 122, 247–253. 10.1093/brain/122.2.247 [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, & Wilcock G (2000). Neuronal death in brain infarcts in man. Neuropathology and Applied Neurobiology, 26, 55–66. 10.1046/j.1365-2990.2000.00218.x [DOI] [PubMed] [Google Scholar]
- Luo W, & Wang Y (2018). Epigenetic regulators: Multifunctional proteins modulating hypoxia-inducible factor-alpha protein stability and activity. Cellular and Molecular Life Sciences, 75, 1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, & Kraus WL (2012). On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes & Development, 26, 417–432. 10.1101/gad.183509.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabley JG, Horváth EM, Murthy KGK, Zsengellér Z, Vaslin A, Benkő R, Kollai M, & Szabó C (2005). Gender differences in the endotoxin-induced inflammatory and vascular responses: Potential role of poly(ADP-ribose) polymerase activation. Journal of Pharmacology and Experimental Therapeutics, 315, 812. 10.1124/jpet.105.090480 [DOI] [PubMed] [Google Scholar]
- Madinier A, Bertrand N, Mossiat C, Prigent-Tessier A, Beley A, Marie C, & Garnier P (2009). Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS One, 4,e8101. 10.1371/journal.pone.0008101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinski T, Bailey F, Zhang Z, & Chopp M (1993). Nitric oxide measured by a porphyrinic micro sensor in rat brain after transient middle cerebral artery occlusion. Journal of Cerebral Blood Flow & Metabolism, 13, 355–358. 10.1038/jcbfm.1993.48 [DOI] [PubMed] [Google Scholar]
- Martinez-Romero R, Canuelo A, Martinez-Lara E, Javier Oliver F, Cardenas S, & Siles E (2009). Poly(ADP-ribose) polymerase-1 modulation of in vivo response of brain hypoxia-inducible factor-1 to hypoxia/reoxygenation is mediated by nitric oxide and factor inhibiting HIF. Journal of Neurochemistry, 111, 150–159. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Egi Y, Yuki S, Horikawa T, Satoh H, & Akira T (2011). MP-124, a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, ameliorates ischemic brain damage in a non-human primate model. Brain Research, 1410, 122–131. 10.1016/j.brainres.2011.05.069 [DOI] [PubMed] [Google Scholar]
- Meng D, He W, Huang P, Liu D, Zhong L, Yu R, & Li J (2018). Polymorphism of PARP-1 indicates an increased risk and a worse initial severity of ischemic stroke. Personalized Medicine, 15,355–360. [DOI] [PubMed] [Google Scholar]
- Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Mądry R, Christensen RD, Berek JS, Dørum A, Tinker AV, du Bois A, González-Martín A, … Matulonis UA (2016). Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New England Journal of Medicine, 375, 2154–2164. 10.1056/NEJMoa1611310 [DOI] [PubMed] [Google Scholar]
- Moroni F, Cozzi A, Chiarugi A, Formentini L, Camaioni E, Pellegrini-Giampietro DE, Chen Y, Liang S, Zaleska MM, Gonzales C, Wood A, & Pellicciari R (2012). Long-lasting neuroprotection and neurological improvement in stroke models with new, potent and brain permeable inhibitors of poly(ADP-ribose) polymerase. British Journal of Pharmacology, 165, 1487–1500. 10.1111/j.1476-5381.2011.01666.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito MG, Xu D, Amin P, Lee J, Wang H, Li W, Kelliher M, Pasparakis M, & Yuan J (2020). Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proceedings of the National Academy of Sciences, 117, 4959–4970. 10.1073/pnas.1916427117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Kakui N, Ohkuma K, Ishikawa M, & Hasegawa T (2005). A newly synthesized poly(ADP-ribose) polymerase inhibitor, DR2313 [2-methyl-3,5,7,8-tetrahydrothiopyrano[4,3-d]-pyrimidine-4-one]: Pharmacological profiles, neuroprotective effects, and therapeutic time window in cerebral ischemia in rats. Journal of Pharmacology and Experimental Therapeutics, 312, 472–481. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Nagaso H, Kakui N, Ishikawa M, Hiranuma T, & Hoshiko S (2004). Critical role of the automodification of poly(ADP-ribose) polymerase-1 in nuclear factor-kappaB-dependent gene expression in primary cultured mouse glial cells. Journal of Biological Chemistry, 279, 42774–42786. [DOI] [PubMed] [Google Scholar]
- Noh MY, Lee WM, Lee SJ, Kim HY, Kim SH, & Kim YS (2018). Regulatory T cells increase after treatment with poly (ADP-ribose) polymerase-1 inhibitor in ischemic stroke patients. International Immunopharmacology, 60, 104–110. 10.1016/j.intimp.2018.04.043 [DOI] [PubMed] [Google Scholar]
- Oda S, Oda T, Nishi K, Takabuchi S, Wakamatsu T, Tanaka T, Adachi T, Fukuda K, Semenza GL, & Hirota K (2008). Macrophage migration inhibitory factor activates hypoxia-inducible factor in a p53-dependent manner. PLoS One, 3, e2215. 10.1371/journal.pone.0002215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paldino E, D’Angelo V, Laurenti D, Angeloni C, Sancesario G, & Fusco FR (2020). Modulation of inflammasome and pyroptosis by olaparib, a PARP-1 inhibitor, in the R6/2 mouse model of huntington’s disease. Cells, 9, 2286. 10.3390/cells9102286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Kam TI, Dawson TM, & Dawson VL (2020). Poly (ADP-ribose) (PAR)-dependent cell death in neurodegenerative diseases. International Review of Cell and Molecular Biology, 353, 1–29. [DOI] [PubMed] [Google Scholar]
- Pinnola A, Naumova N, Shah M, & Tulin AV (2007). Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. Journal of Biological Chemistry, 282, 32511–32519. 10.1074/jbc.M705989200 [DOI] [PubMed] [Google Scholar]
- Pleschke JM, Kleczkowska HE, Strohm M, & Althaus FR (2000). Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. Journal of Biological Chemistry, 279, 40974–40980. 10.1074/jbc.M006520200 [DOI] [PubMed] [Google Scholar]
- Polster BM (2013). AIF, reactive oxygen species, and neurodegeneration: A “complex” problem. Neurochemistry International, 62, 695–702. 10.1016/j.neuint.2012.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers WJ (2020). Acute ischemic stroke. New England Journal of Medicine, 383, 252–260. 10.1056/NEJMcp1917030 [DOI] [PubMed] [Google Scholar]
- Rom S, Zuluaga-Ramirez V, Reichenbach NL, Dykstra H, Gajghate S, Pacher P, & Persidsky Y (2016). PARP inhibition in leukocytes diminishes inflammation via effects on integrins/cytoskeleton and protects the blood-brain barrier. Journal of Neuroinflammation, 13,254. 10.1186/s12974-016-0729-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gómez-Herreros F, Hafezparast M, & Caldecott KW (2014). PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Research, 42, 307–314. 10.1093/nar/gkt835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma J, Nelluru G, Wilson MA, Johnston MV, & Hossain MA (2011). Sex-specific activation of cell death signalling pathways in cerebellar granule neurons exposed to oxygen glucose deprivation followed by reoxygenation. ASN Neuro, 3(2), AN20100032. 10.1042/AN20100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H (2009). Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Current Medicinal Chemistry, 16, 4593–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Sharma G, Singh N, & Hanif K (2014). A comparative study of neuroprotective effect of single and combined blockade of AT1 receptor and PARP-1 in focal cerebral ischaemia in rat. International Journal of Stroke, 9, 560–568. 10.1111/j.1747-4949.2012.00916.x [DOI] [PubMed] [Google Scholar]
- Skaper SD (2003). Poly(ADP-Ribose) polymerase-1 in acute neuronal death and inflammation: A strategy for neuroprotection. Annals of the New York Academy of Sciences, 993, 217–228. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, & Simon RP (2003). Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: Similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet, 362, 1028–1037. 10.1016/S0140-6736(03)14412-1 [DOI] [PubMed] [Google Scholar]
- Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM, Kristeleit RS, Ma L, Bell-McGuinn KM, Brenton JD, Cragun JM, Oaknin A, Ray-Coquard I, Harrell MI, Mann E, … McNeish IA (2017). Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. The Lancet Oncology, 18, 75–87. 10.1016/S1470-2045(16)30559-9 [DOI] [PubMed] [Google Scholar]
- Takahashi K, & Greenberg JH (1999). The effect of reperfusion on neuroprotection using an inhibitor of poly(ADP-ribose) polymerase. NeuroReport, 10, 2017–2022. 10.1097/00001756-199907130-00005 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Greenberg JH, Jackson P, Maclin K, & Zhang J (1997). Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. Journal of Cerebral Blood Flow and Metabolism, 17, 1137–1142. 10.1097/00004647-199711000-00001 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, & Greenberg JH (1999). Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Research, 829, 46–54. 10.1016/S0006-8993(99)01335-9 [DOI] [PubMed] [Google Scholar]
- Teng F, Zhu L, Su J, Zhang X, Li N, Nie Z, & Jin L (2016). Neuroprotective effects of Poly(ADP-ribose)polymerase Inhibitor Olaparib in transient cerebral ischemia. Neurochemical Research, 41,1516–1526. 10.1007/s11064-016-1864-6 [DOI] [PubMed] [Google Scholar]
- Thomas CJ, Kotova E, Andrake M, Adolf-Bryfogle J, Glaser R, Regnard C, & Tulin AV (2014). Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via poly(ADP-ribosyl)ation. Molecular Cell, 53, 831–842. 10.1016/j.molcel.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, & Ueda K (1998). Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. Journal of Cerebral Blood Flow and Metabolism, 18, 991–997. 10.1097/00004647-199809000-00008 [DOI] [PubMed] [Google Scholar]
- Ullrich O, Diestel A, Eyüpoglu IY, & Nitsch R (2001). Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nature Cell Biology, 3, 1035–1042. 10.1038/ncb1201-1035 [DOI] [PubMed] [Google Scholar]
- Vagnerova K, Liu K, Ardeshiri A, Cheng J, Murphy SJ, Hurn PD, & Herson PS (2010). Poly (ADP-ribose) polymerase-1 initiated neuronal cell death pathway–do androgens matter? Neuroscience, 166, 476–481. 10.1016/j.neuroscience.2009.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virani SS, Alonso A, Benjamin EJ et al. (2020). Heart disease and stroke statistics—2020 update: A report from the american heart association. Circulation, 141, e139–e596. [DOI] [PubMed] [Google Scholar]
- Vuong B, Hogan-Cann AD, Alano CC, Stevenson M, Chan WY, Anderson CM, Swanson RA, & Kauppinen TM (2015). NF-κB transcriptional activation by TNFα requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. Journal of Neuroinflammation, 12, 229. 10.1186/s12974-015-0448-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma C, Zhu J, Rao G, & Li H (2020). Effect of 3-aminobenzamide on the ultrastructure of astrocytes and microvessels after focal cerebral ischemia in rats. Dose-Response, 18(1), 155932581990124. 10.1177/1559325819901242. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang P, Shao BZ, Deng Z, Chen S, Yue Z, & Miao CY (2018). Autophagy in ischemic stroke. Progress in Neurobiology, 163–164,98–117. 10.1016/j.pneurobio.2018.01.001 [DOI] [PubMed] [Google Scholar]
- Wang X, Yang C, Chai J, Shi Y, & Xue D (2002). Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science, 298, 1587–1592. 10.1126/science.1076194 [DOI] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science, 354(6308), aad6872. 10.1126/science.aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dawson VL, & Dawson TM (2009). Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Experimental Neurology, 218, 193–202. 10.1016/j.expneurol.2009.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, & Dawson TM (2011). Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Science Signalling, 4, ra20. 10.1126/scisignal.2000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Luo W, & Wang Y (2019). PARP-1 and its associated nucleases in DNA damage response. DNA Repair, 81, 102651. 10.1016/j.dnarep.2019.102651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Reis C, Applegate R 2nd, Stier G, Martin R, & Zhang JH (2015). Ischemic conditioning-induced endogenous brain protection: Applications pre-, per- or post-stroke. Experimental Neurology, 272, 26–40. 10.1016/j.expneurol.2015.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, & Dawson VL (2006). Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proceedings of the National Academy of Sciences, 103, 18314–18319. 10.1073/pnas.0606528103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, & Dawson VL (2002). Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science, 297, 259–263. 10.1126/science.1072221 [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang Y, Frydenlund DS, Ottersen OP, Dawson VL, & Dawson TM (2009). Outer mitochondrial membrane localization of apoptosis-inducing factor: Mechanistic implications for release. ASN Neuro, 1, AN20090046. 10.1042/AN20090046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Siegel C, Zeng Z, Li J, Liu F, & McCullough LD (2009). Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Experimental Neurology, 217, 210–218. 10.1016/j.expneurol.2009.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dawson V, Dawson T, & Snyder S (1994). Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science, 263, 687–689. 10.1126/science.8080500 [DOI] [PubMed] [Google Scholar]
- Zhang N, Yin Y, Han S, Jiang J, Yang W, Bu X, & Li J (2011). Hypoxic preconditioning induced neuroprotection against cerebral ischemic injuries and its cPKCgamma-mediated molecular mechanism. Neurochemistry International, 58, 684–692. [DOI] [PubMed] [Google Scholar]
- Zhong H, Song R, Pang Q, Liu Y, Zhuang J, Chen Y, Hu J, Hu J, Liu Y, Liu Z, & Tang J (2018). Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell Death & Disease, 9, 932. 10.1038/s41419-018-0996-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable - no new data generated.