

Abstract
A major challenge in the analysis of tissue imaging data is cell segmentation, the task of identifying the precise boundary of every cell in an image. To address this problem we constructed TissueNet, a dataset for training segmentation models that contains more than 1 million manually labeled cells, an order of magnitude more than all previously published segmentation training datasets. We used TissueNet to train Mesmer, a deep learning-enabled segmentation algorithm. We demonstrated that Mesmer is more accurate than previous methods, generalizes to the full diversity of tissue types and imaging platforms in TissueNet, and achieves human-level performance. Mesmer enabled the automated extraction of key cellular features, such as subcellular localization of protein signal, which was challenging with previous approaches. We then adapted Mesmer to harness cell lineage information in highly multiplexed datasets and used this enhanced version to quantify cell morphology changes during human gestation. All code, data, and models are released as a community resource.
Introduction
Understanding the structural and functional relationships present in tissues is a challenge at the forefront of basic and translational research. Recent advances in multiplexed imaging have expanded the number of transcripts and proteins that can be quantified simultaneously1–12, opening new avenues for large-scale analysis of human tissue samples. Ambitious collaborative efforts such as the Human Tumor Atlas Network13, the Human BioMolecular Atlas Program14, and the Human Cell Atlas15 are using these methods to comprehensively characterize the location, function, and phenotype of the cells in the human body. However, the tools needed for analysis and interpretation of these datasets at scale do not yet exist. The clearest example is the lack of a generalized algorithm for locating single cells in images. Unlike flow cytometry or single-cell RNA sequencing, tissue imaging is performed with intact specimens. Thus, to extract single-cell data, each pixel must be assigned to a cell, a process known as cell segmentation. Since the features extracted through this process are the basis for downstream analyses16, inaccuracies at this stage can have far-reaching consequences for interpreting image data. The difficulty of achieving accurate, automated cell segmentation is due in large part to the differences in cell shape, size, and density across tissue types17, 18. Machine-learning approaches developed to meet this challenge19–24 have fallen short for tissue imaging data. A common pitfall is the need to perform manual, image-specific adjustments to produce useful segmentations. This lack of full automation poses a prohibitive barrier given the increasing scale of tissue imaging experiments.
Deep learning algorithms for computer vision are increasingly being used for a variety of tasks in biological image analysis, including nuclear and cell segmentation25–31. These algorithms are capable of achieving high accuracy, but require substantial amounts of annotated training data. Generating ground-truth data for cell segmentation is time intensive, and as a result, existing datasets are of modest size (104-105 annotations). Moreover, most public datasets26,27,32–38 annotate the location of cell nuclei rather than that of whole cells, meaning that models trained on these datasets are only capable of performing nuclear segmentation, not cell segmentation. Thus, the lack of available data, combined with the difficulties of deploying pre-trained models to the life science community39–42, has hampered progress in whole-cell segmentation.
Here, we sought to close these gaps by creating an automated, simple, and scalable algorithm for nuclear and whole-cell segmentation that performs accurately across a diverse range of tissue types and imaging platforms. Developing this algorithm required two innovations: (1) a scalable approach for generating large volumes of pixel-level training data and (2) an integrated deep learning pipeline that uses these data to achieve human-level performance. To address the first challenge, we developed a crowdsourced, human-in-the-loop approach for segmenting cells where humans and algorithms work in tandem to produce accurate annotations (Figure 1a). We used this pipeline to create TissueNet, a comprehensive segmentation dataset of >1 million paired whole-cell and nuclear annotations. TissueNet contains twice as many nuclear and 16 times as many whole-cell labels as all previously published datasets combined. To address the second challenge, we developed Mesmer, a deep learning-enabled pipeline for scalable, user-friendly segmentation of tissue imaging data. To enable broad use by the scientific community, we harnessed DeepCell, an open-source collection of software libraries, to create a web interface for using Mesmer, as well as plugins for ImageJ and QuPath. We have made all code, data, and trained models available under a permissive license as a community resource, setting the stage for application of these modern, data-driven methods to a broad range of research challenges.
Figure 1: A human-in-the-loop approach enables scalable, pixel-level annotation of large image collections.
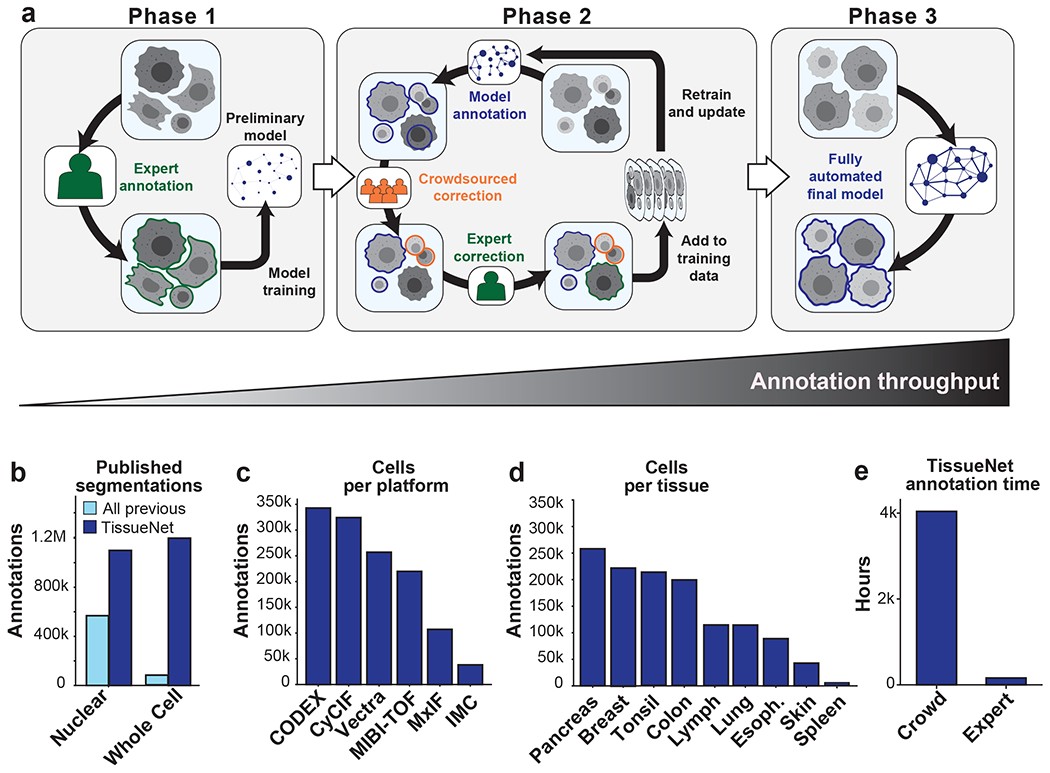
a, This approach has three phases. During phase 1, annotations are created from scratch to train a model. During phase 2, new data are fed through a preliminary model to generate predictions. These predictions are used as a starting point for correction by annotators. As more images are corrected, the model improves, which decreases the number of errors, increasing the speed with which new data can be annotated. During phase 3, an accurate model is run without human correction. b, TissueNet has more nuclear and whole-cell annotations than all previously published datasets. c, The number of cell annotations per imaging platform in TissueNet. d, The number of cell annotations per tissue type in TissueNet. e, The number of hours of annotation time required to create TissueNet.
A human-in-the-loop approach for constructing TissueNet
Existing annotated datasets for cell segmentation are limited in scope and scale (Figure 1b)26,27,32–38. This limitation is largely due to the linear, time-intensive approach used to construct them, which requires the border of every cell in an image to be manually demarcated. We therefore implemented a three-phase approach to create TissueNet. In the first phase, expert human annotators outlined the border of each cell in 80 images. These labeled images were used to train a preliminary model (Figure 1a, left; Methods). The process then moved to the second phase (Figure 1a, middle), where images were first passed through the model to generate predicted annotations and then sent to crowdsourced annotators to correct errors. The corrected annotations underwent final inspection by an expert prior to being added to the training dataset. When enough new data were compiled, a new model was trained and phase two was repeated. Each iteration yielded more training data, which led to improved model accuracy, fewer errors that needed to be manually corrected, and a lower marginal cost of annotation. This virtuous cycle continued until the model achieved human-level performance. At this point, we transitioned to the third phase (Figure 1a, right), where the model was run without human assistance to produce high-quality predictions.
Human-in-the-loop pipelines require specialized software that is optimized for the task at hand. Although prior work has used the human-in-the-loop approach to create segmentation datasets22,43–45, existing tools were not optimized for crowdsourcing or for correcting large quantities of tissue image data. We therefore developed DeepCell Label46, a browser-based graphical user interface optimized for editing existing cell annotations in tissue images (Extended Data Figure 1a; Methods). DeepCell Label is supported by a scalable cloud backend that dynamically adjusts the number of servers according to demand (Extended Data Figure 1b). Using DeepCell Label, we trained annotators from multiple crowdsourcing platforms to identify whole-cell and nuclear boundaries. To further simplify our annotation workflow, we integrated DeepCell Label into a pipeline that allowed us to prepare and submit images for annotation, have users annotate those images, and download the results. The images and resulting labels were used to train and update our model, completing the loop (Extended Data Figure 1c; Methods).
As a result of the scalability of our human-in-the-loop approach to data labeling, TissueNet is larger than the sum total of all previously published datasets26,27,32–38 (Figure 1b), with 1.3 million whole-cell annotations and 1.2 million nuclear annotations. TissueNet contains 2D data from six imaging platforms (Figure 1c), nine organs (Figure 1d), and includes both histologically normal and diseased tissue (e.g., tumor resections). TissueNet also encompasses three species, with images from human, mouse, and macaque. Constructing TissueNet required >4,000 person hours, the equivalent of nearly 2 person-years of full-time effort (Figure 1e).
Mesmer is a deep learning algorithm cell segmentation
To address the requirements for both accuracy and speed in cell segmentation, we created Mesmer, a deep learning-based algorithm for nuclear and whole-cell segmentation of tissue data. Mesmer’s model consists of a ResNet50 backbone coupled to a Feature Pyramid Network with four prediction heads (two for nuclear segmentation and two for whole-cell segmentation) that are attached to the top of the pyramid (Extended Data Figure 2a; Methods)47–49. The input to Mesmer is a nuclear image (e.g., DAPI) to define the nucleus of each cell and a membrane or cytoplasm image (e.g., CD45 or E-cadherin) to define the shape of each cell (Figure 2a). These inputs are normalized50 (to improve robustness), tiled into patches of fixed size (to allow processing of images with arbitrary dimensions), and fed to the deep learning model. The model outputs are then untiled51 to produce predictions for the centroid and boundary of every nucleus and cell in the image. The centroid and boundary predictions are used as inputs to a watershed algorithm52 to create the final instance segmentation mask for each nucleus and each cell in the image (Methods).
Figure 2: Mesmer delivers accurate nuclear and whole-cell segmentation in multiplexed images of tissues.
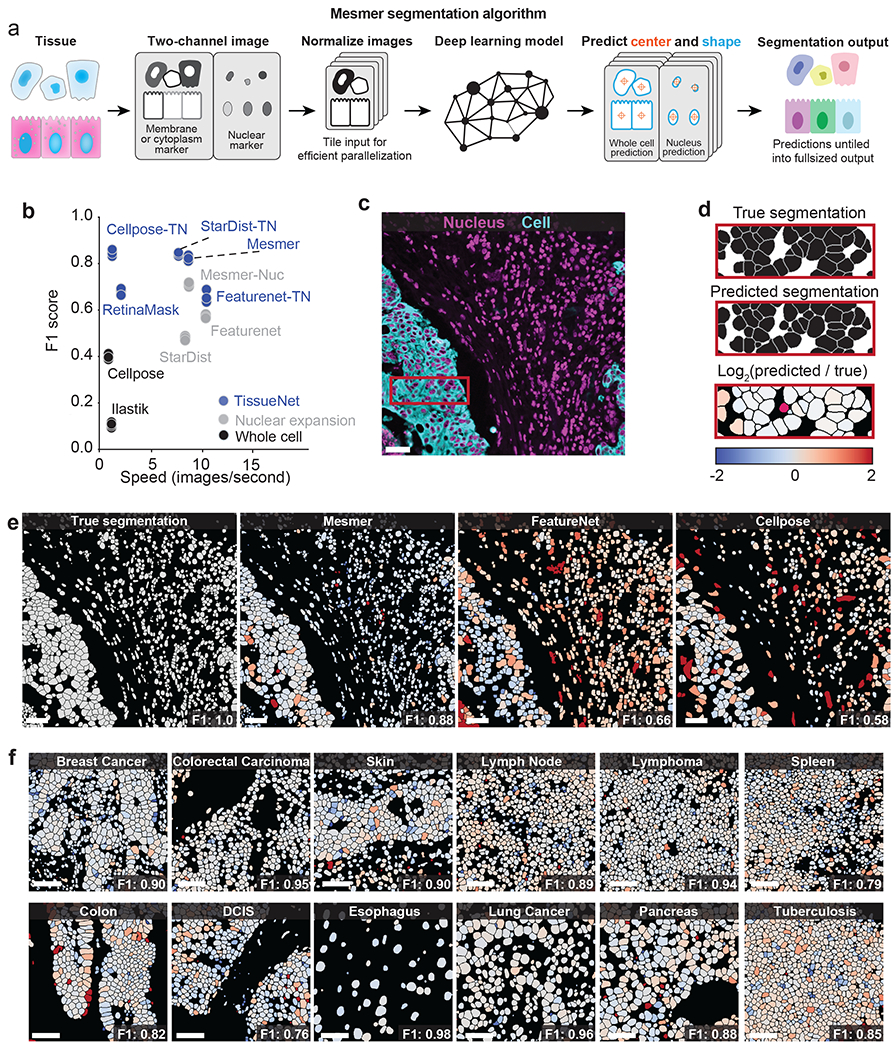
a, Diagram illustrating the key steps in the Mesmer segmentation pipeline. b, Speed versus accuracy comparison of Mesmer and previously published models, as well as architectures we retrained on TissueNet. Accuracy is measured by the F1 score (Methods) between the predicted segmentations and the ground-truth labels in the test set of TissueNet, where 0 indicates no agreement and 1 indicates perfect agreement. c, Color overlay of representative image of colorectal carcinoma. d, Inset showing the ground truth (top) and predicted (middle) labels from a small region in c, along with a visual representation of segmentation accuracy (bottom). Predicted segmentations for each cell are colored by the log2 of the ratio between the predicted area and ground-truth area. Predicted cells that are too large are red, while predicted cells that are too small are blue. e, Ground-truth segmentation labels for the image in c, along with the predicted labels from Mesmer and previously published models, each colored by the log2 as in d. As seen visually, Mesmer offers substantially better performance than previous methods. f, Mesmer generalizes across tissue types, imaging platforms, and disease states. The F1 score is given for each image. In all panels, scale bars are 50 μm.
To evaluate Mesmer’s accuracy, we performed comprehensive benchmarking against previously published, pre-trained algorithms as well as deep learning models that were retrained on TissueNet. These comparisons allowed us to understand the relative contributions of deep learning methodology and training data to overall accuracy. We first compared Mesmer’s performance against two pre-trained algorithms: FeatureNet26, which we previously used16 to perform nuclear segmentation followed by expansion to analyze a cohort of MIBI-TOF breast cancer samples, and Cellpose28, a recently published algorithm for whole-cell segmentation of microscopy data. Overall, we observed higher accuracy for Mesmer (F1=0.82) than both FeatureNet (F1=0.63) and Cellpose (F1=0.41) (Figure 2b, Extended Data Figure 2c–f).
We then compared Mesmer’s performance against a range of supervised segmentation methods22,26,53,54 that were trained on TissueNet. FeatureNet, RetinaMask, and Ilastik did not achieve equivalent performance to Mesmer, even when trained on TissueNet (Figure 2b, Extended Data Figure 2c–f). In contrast, Cellpose and StarDist obtained equivalent performance to Mesmer when trained on TissueNet (Figure 2b). Last, we compared Mesmer with a model trained to perform nuclear segmentation followed by a pixel expansion (a common method16,55–57 to approximate the entire cell for existing nuclear segmentation algorithms), and found that Mesmer achieved superior performance (Extended Data Figure 2g). These comparisons were not affected by our choice of metrics, as we observed similar trends for recall, precision, and Jaccard index (Extended Data Figure 2c–f).
In addition to differences in accuracy, the algorithms that we benchmarked differed substantially in their speed. Mesmer was only 13% slower than FeatureNet, despite a significant increase in model capacity, and was 20 times faster than Cellpose (Figure 2b). RetinaMask and Illastik also suffered from slow processing times (Figure 2b). These differences in speed are primarily due to differences in post-processing between the various methods, which accounts for most of the computational time (Extended Data Figure 2b).
To visualize the performance differences between Mesmer and the published, pre-trained versions of FeatureNet and Cellpose, we used all three algorithms to segment an image of colorectal carcinoma (Figure 2c). We compared segmentation predictions to the ground-truth data, coloring each cell by the ratio of the predicted area to the ground-truth area (Figure 2d). Overall, Mesmer more effectively captured the true size of each cell in the image (Figure 2e). In comparison, FeatureNet poorly captured the true size of each cell, which is expected given that this model approximates cell shape by performing nuclear segmentation followed by expansion. In line with its lower recall score (Extended Data Figure 2c), Cellpose failed to identify a large fraction of the cells in the image (Figure 2e), likely due to the relative scarcity of tissue images in the data used to train Cellpose.
Next, we examined Mesmer’s segmentation predictions across a range of tissue types (Figure 2f). Mesmer’s errors were unbiased, with an equal number of cells that were too large or too small. Further, Mesmer’s errors were not correlated with the true size of the cell (Extended Data Figure 2g). In contrast, methods that rely on nuclear segmentation and expansion tend to overestimate the size of most small cells and underestimate the size of most large cells (Extended Data Figure 2g). Taken together, this benchmarking demonstrates that Mesmer is a significant advance over prior segmentation methods.
Mesmer achieves human-level performance for segmentation
Our results thus far (Figure 2f, Figure 3a) suggested that Mesmer performed well across all of TissueNet without manual adjustment. However, given that cell morphology and image characteristics can vary depending on organ site, disease state, and imaging platform17,18, training a specialist model on data from a single platform or single tissue type could lead to superior performance when compared to a model trained on all of TissueNet. To evaluate Mesmer’s generalizability, we benchmarked performance against models that were trained using a subset of TissueNet that was either tissue- or platform-specific. We observed comparable performance between Mesmer and the specialist models (Figure 3b, c). We next sought to evaluate how specialist models performed when evaluated on data not seen during training. In general, specialist models had poor performance when evaluated on data types not seen during training (Extended Data Figure 3b). Dataset size likewise played an important role, as models trained on small subsets of TissueNet did not perform as well as those trained on the entire dataset (Extended Data Figure 3d–h).
Figure 3: Mesmer performs whole-cell segmentation across tissue types and imaging platforms with human-level accuracy.
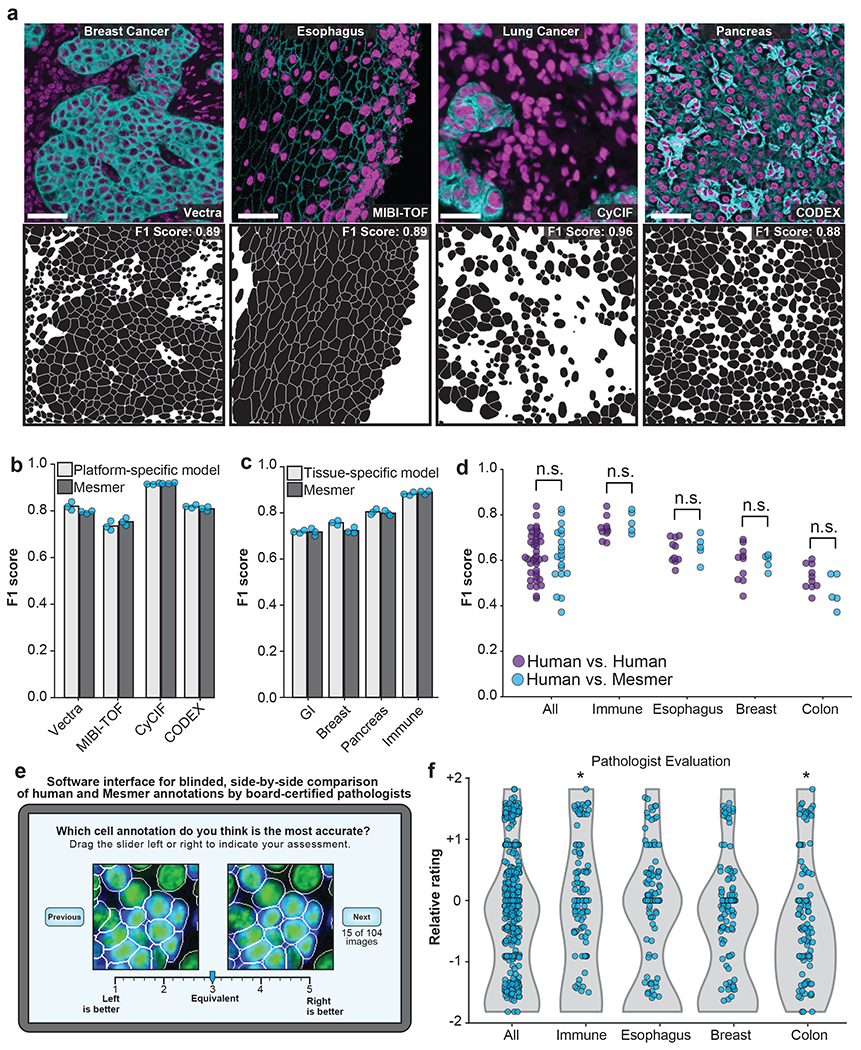
a, Sample images, predicted segmentations, and F1 scores for distinct tissues and imaging platforms visually demonstrate that Mesmer delivers accurate cell segmentation for all available imaging platforms. b, Mesmer has accuracy equivalent to specialist models trained only on data from a specific imaging platform (Methods), with all models evaluated on data from the platform used for training. c, Mesmer has accuracy equivalent to specialist models trained only on data from a specific tissue type (Methods), with all models evaluated on data from the tissue type used for training. GI, gastrointestinal. d, F1 scores evaluating the agreement between segmentation predictions for the same set of images. The predictions from five independent expert annotators were compared against each other (human vs. human) or against Mesmer (human vs. Mesmer). No statistically significant differences between these two comparisons were found, demonstrating that Mesmer achieves human-level performance. e, Workflow for pathologists to rate the segmentation accuracy of Mesmer compared with expert human annotators. f, Pathologist scores from the blinded comparison. A positive score indicates a preference for Mesmer while a negative score indicates a preference for human annotations. Pathologists displayed no significant preference for human labels or Mesmer’s outputs overall. When broken down by tissue type, pathologists displayed a slight preference for Mesmer in immune tissue (p=0.02), and a slight preference for humans in colon tissue (p=0.01), again demonstrating that Mesmer has achieved human-level performance. n.s., not significant; *p<0.05, two-sample t-test for d, one-sample t-test for f. All scale bars are 50 μm.
The metrics for model accuracy used here treated human-annotated data as ground truth, but even expert annotators can disagree with one another. We therefore compared Mesmer’s segmentation predictions with predictions from five independent expert human annotators. We evaluated all pairs of human annotators against one another, using one annotator as the “ground truth” and the other as the prediction. We then evaluated Mesmer’s predictions against predictions from each of these five annotators. We detected no significant differences between human-to-human and human-to-Mesmer F1 scores (p=0.93) (Figure 3d), suggesting that Mesmer performed on par with human annotators.
To further evaluate Mesmer’s performance relative to humans, we enlisted four pathologists to perform a blinded evaluation of segmentations from the human annotators and Mesmer. Each pathologist was shown paired images containing a prediction from Mesmer and an annotation from a human (Figure 3e). When evaluated in aggregate, the pathologists rated Mesmer’s predictions and the expert annotator’s predictions equivalently (Figure 3f). Breaking down the evaluation by tissue type, we observed only modest differences in pathologist evaluation, with Mesmer performing slightly better than the annotators for some tissues and the annotators performing slightly better in others. Taken together, the preceding analyses demonstrate that Mesmer performs whole-cell segmentation with human-level performance. This outcome is also likely for Cellpose and StarDist, given that they achieve performance equivalent to Mesmer when trained on TissueNet (Figure 2b). To our knowledge, no previous cell segmentation algorithm has achieved parity with human performance for tissue data.
To finish our performance analysis, we sought to understand Mesmer’s limitations by identifying images for which Mesmer produced low-quality cell segmentations. Inaccurately segmented images were characterized by low signal-to-noise ratio, heterogeneous staining, and focus issues (Extended Data Figure 3i). To characterize the impact of each of these factors on model performance, we evaluated model accuracy after blurring, resizing, or adding image noise. While Mesmer was robust to moderate image distortion, performance suffered as the distortions increased in magnitude (Extended Data Figure 3j–l)—as expected, since these manipulations remove information from the images.
Mesmer enables accurate analysis of tissue imaging data
Cell segmentation is the first step for quantitative analysis of tissue imaging data and serves as the foundation for subsequent single-cell analysis. Thus, Mesmer’s ability to generate both whole-cell and nuclear segmentation predictions should enable analyses that were difficult to perform with previous segmentation algorithms. One example is predicting the subcellular localization of proteins within cells, which can be used to quantify the nuclear translocation of transcription factors58,59 or degree of membrane staining of HER2 for the assessment of breast cancer60. To explore the accuracy of subcellular signal prediction, we stained breast cancer samples with a panel of phenotyping markers and imaged them with MIBI-TOF61 (Figure 4a; Methods). We created an integrated multiplexed image analysis pipeline, ark-analysis62, that integrates Mesmer’s segmentation predictions with downstream analysis. We extracted the compartment-specific expression of each marker using both the predicted and ground truth segmentation masks (Methods). Subcellular localization predictions from Mesmer agreed with those from the ground-truth data (Figure 4b). We observed predominantly nuclear expression for known nuclear markers (e.g., Ki67 and HH3) and non-nuclear expression for membrane markers (e.g., E-cadherin and HER2; Figure 4b).
Figure 4: Mesmer enables accurate analysis of multiplex imaging data.
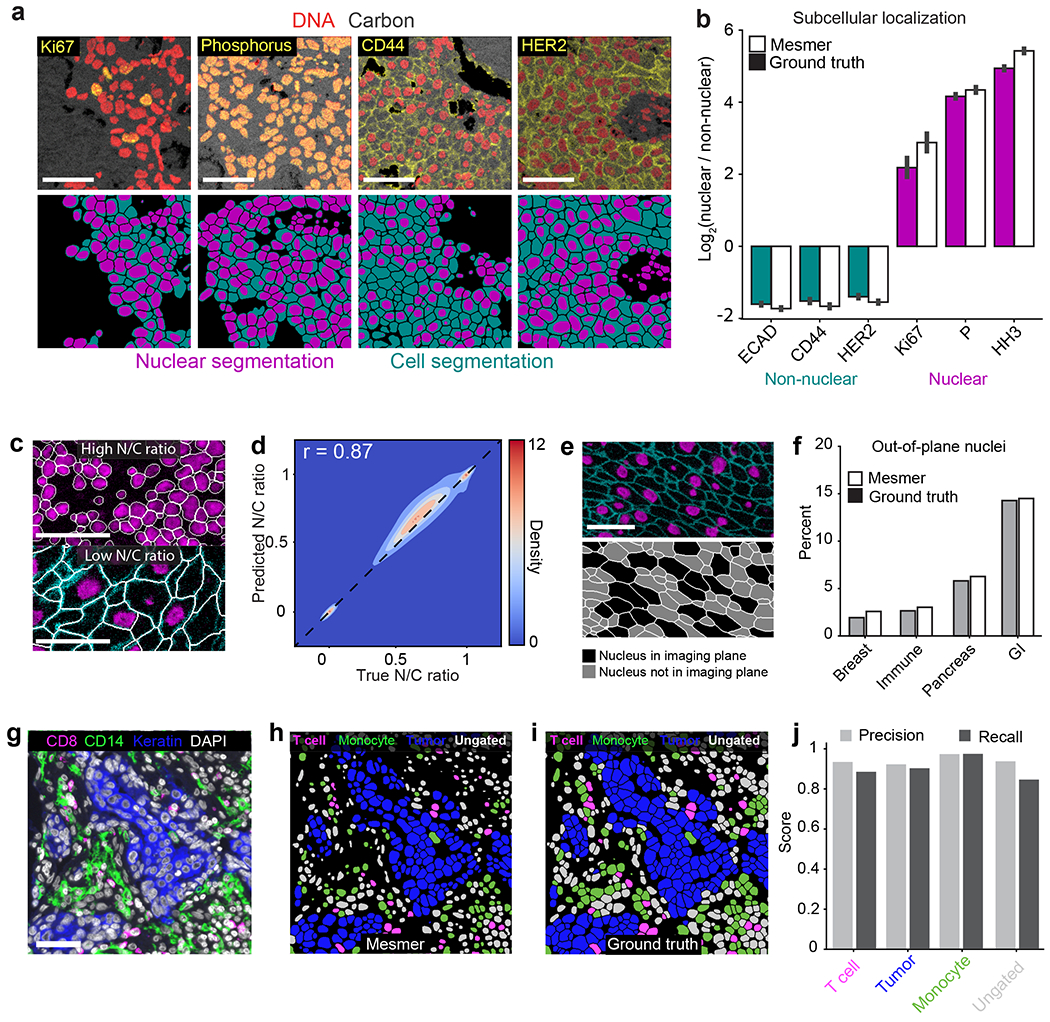
a, Color overlays showing staining patterns for nuclear and non-nuclear proteins (top), with associated nuclear and whole-cell segmentation predictions (bottom). b, Quantification of subcellular localization of the proteins in a for predicted and ground-truth segmentations. The agreement between localization for prediction and ground-truth segmentations indicates that Mesmer accurately quantifies protein localization patterns at the single-cell level. n=1069 cells. Data are presented as mean +/− 95% confidence interval. c, Example image of a tissue with a high N/C ratio (top) and a low N/C ratio (bottom). The N/C ratio is one of several metrics used for quantifying cell morphology (Methods). d, A Pearson’s correlation contour plot of the accuracy of N/C ratio predictions across the entire test split of TissueNet demonstrates that Mesmer accurately quantifies cell morphology. e, Representative image of a tissue with many nuclei outside the imaging plane (top), along with corresponding segmentations colored by whether the nucleus is or is not in the imaging plane. f, Quantification of the number of cells with an out-of-plane nucleus in the predicted and ground-truth segmentations. These cells are detected by Mesmer but would be missed by nuclear segmentation-based methods. GI, gastrointestinal. g, Representative image of the expression of multiple informative proteins in a breast cancer sample. h, Predicted segmentation colored by cell lineage. i, Ground-truth segmentation colored by cell lineage. j, Quantification of precision and recall of each cell type in the ground-truth and predicted segmentations demonstrates that Mesmer produces accurate cell-type counts. All scale bars are 50 μm.
As Mesmer also provides automated analysis of the relationship between each individual nucleus and cell, it should enable automatic scoring of the nuclear/cytoplasmic ratio, which is widely used by pathologists to evaluate malignancies63. We used Mesmer to generate nuclear and whole-cell segmentations for every cell in the test set of TissueNet. We then computed the nuclear/whole-cell (N/C) ratio, which is conceptually similar to the nuclear/cytoplasm ratio but has higher numerical stability for cells with little cytoplasm (e.g., immune cells; Methods). Mesmer accurately captured cells with low and high N/C ratios (Figure 4c), and there was a strong correlation (Pearson’s r = 0.87) between the predicted and ground-truth N/C ratios across all cells in TissueNet (Figure 4d).
This analysis identified a subpopulation of cells with an N/C ratio of 0 (Figure 4e), indicating that no nucleus was observed in that cell. These cells arise when the imaging plane used to acquire the data captures the cytoplasm, but not the nucleus. We quantified the proportion of cells with an out-of-plane nucleus across the tissue types in TissueNet for both the predicted and ground-truth segmentation labels and found good agreement between predicted and true rates of out-of-plane nuclei (Figure 4f). The highest proportion of out-of-plane nuclei occurred in gastrointestinal tissue (Figure 4f), presumably due to the elongated nature of the columnar epithelium. Cells with out-of-plane-nuclei are missed by nucleus-based segmentation approaches but are captured by Mesmer.
Cell classification is a common task following segmentation. Inaccuracies in segmentation can lead to substantial bias in the identification and enumeration of the cells present in an image. To benchmark how Mesmer’s predictions impact this process, we analyzed a cohort of breast cancer samples generated with the Vectra platform. Each image was stained with a panel of lineage-defining markers (Figure 4g), which we used to classify each cell as either a T cell, monocyte, tumor cell, or ungated. We selected two distinct regions from three patients and generated both predicted and ground-truth segmentations for all the cells in the image. We classified all cells from the predicted (Figure 4h) and ground-truth (Figure 4i) segmentations into these categories using the same gating scheme (Methods). We then computed the precision and recall for each cell type across the patients. We observed strong agreement between the two annotations (Figure 4j), showing that Mesmer’s segmentation predictions enable accurate classification of the diversity of cells present in these images.
Lineage-aware segmentation quantifies morphological changes
We have demonstrated that models trained on TissueNet can harness the two channels present in this data to accurately segment cells across a diversity of tissue types. However, some tissue types have complex morphologies that cannot be accurately captured with only two channels of imaging data. For example, the decidua, the mucosal membrane of the uterus, shows substantial variation in cell size and cell shape arising from the interaction between maternal and fetal cells. This complexity is compounded by the tight juxtaposition of these cells with one another and the non-convex geometries that they can assume64, with small round cells nestled within concavities of much larger cells.
This complex morphology makes segmentation challenging when using a single membrane channel, even for an expert annotator (Figure 5a, top). However, information about the location and shape of each cell can be attained by including additional markers that are cell-type specific (Figure 5a, bottom). These additional markers provide crucial information about cell morphology during model training that is lost when they are combined into a single channel. We used MIBI-TOF to generate a multiplexed imaging dataset from the human decidua with six lineage specific markers (Greenbaum, Averbukh, Soon et al., manuscript in preparation) and then used DeepCell Label to generate lineage-aware ground-truth segmentations from a subset of the images. We modified our deep learning architecture to accept these six channels of input data and trained a model using this dataset (Methods). The resulting lineage-aware segmentation pipeline accurately performed whole-cell segmentation, despite the complex cell morphologies in these images (Figure 5b).
Figure 5: Lineage-aware segmentation yields morphological profiling of cells in the decidua during human pregnancy.
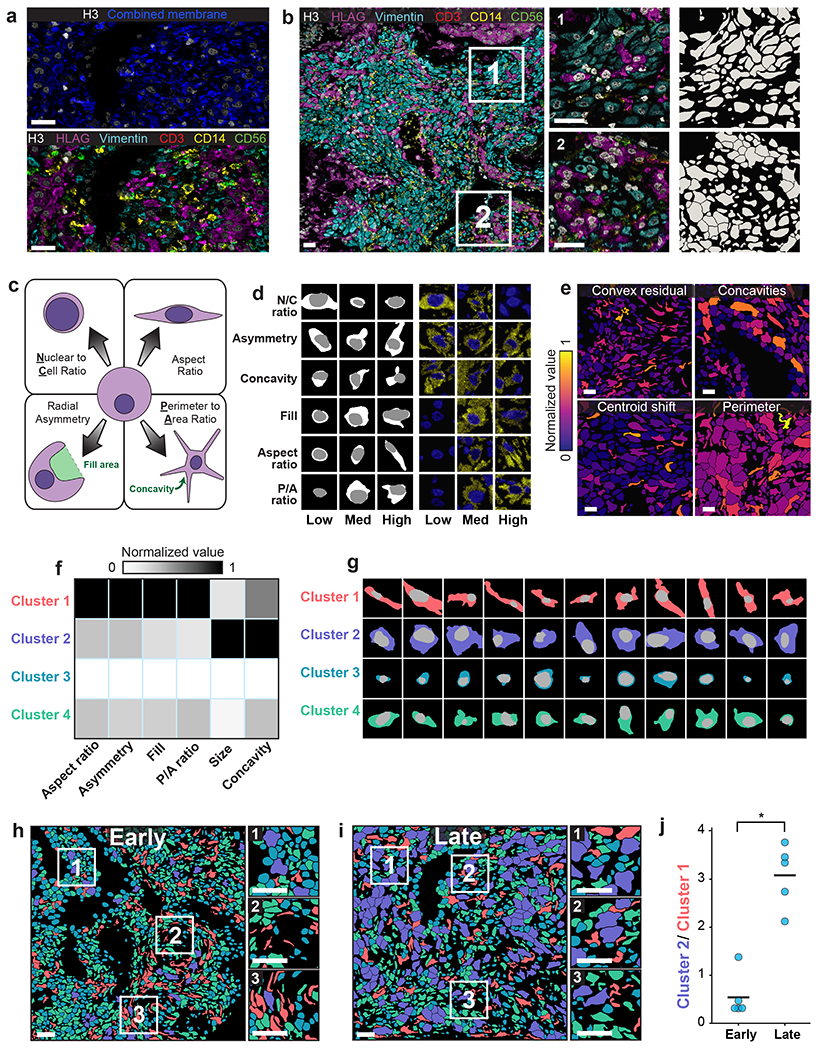
a, Color overlay showcasing the challenge of distinguishing cells with only a single combined membrane channel (top), paired with a version of the same image containing all six channels used for lineage-aware segmentation (bottom). b, Representative image of the diverse morphology of cell types in the human decidua (left), along with insets (right) with corresponding segmentation predictions. c, Diagram illustrating the morphology metrics that we defined to enable automated extraction of cell-shape parameters (Methods). d, Predicted segmentations (left) placing cells on a spectrum from low to high for each morphology metric, along with the corresponding imaging data for those cells (right). e, Cell segmentations in four representative images colored by morphology metrics demonstrate the accurate quantification of diverse features. f, Heatmap of inputs to k-means clustering used to identify distinct cell populations based on cell morphology. g, Example cells belonging to each cluster illustrate the morphological differences between cells belonging to each cluster. h,i Representative images of maternal decidua in early (h) and late (i) gestation, with segmentations colored by cluster. j, Quantification of the ratio between cluster 2 and cluster 1 cells in early pregnancy versus late pregnancy. Cluster 2 cells become more prominent in the later time point while cluster 1 cells become rarer. p=0.0003, two-sample t-test. All scale bars are 50 μm.
We used this lineage-aware segmentation pipeline to quantify morphological changes of cells in the decidua over time. We first defined a series of morphological metrics to capture the diversity of cell shapes in this dataset (Figure 5c; Methods). Manual inspection demonstrated accurate assignment of cells within each category (Figure 5d). We then created an automated pipeline that computed these metrics for every cell in an image62. We applied our pipeline to this dataset and found that these metrics captured key morphological features of the cell shapes that we observed (Figure 5e). We then performed k-means clustering on the cell morphology profiles (Methods) and identified four distinct clusters (Figure 5f, g). To determine how these cellular morphologies changed over time in the human decidua, we divided the samples into two groups based on age: early (6-8 weeks) and late (16-18 weeks) gestational age. Coloring each cell by its cluster highlighted the difference in cell morphology between the two gestational age groups (Figure 5h, i). We observed an abundance of cluster 1 cells (elongated) in the early time point and an abundance of cluster 2 cells (large and globular) at the late timepoint (Figure 5j). This shift likely reflects the morphological transformation undergone by maternal stromal cells during decidualization65. Our analysis demonstrates that whole-cell segmentation can make cell morphology a quantitative observable, bridging the historical knowledge of pathologists and modern multiplexed imaging methods.
DeepCell supports community-wide deployment of Mesmer
To facilitate the deployment of deep learning models, our group previously created DeepCell37,39,46,66, a collection of linked, open-source software libraries for cellular image analysis. Here we used DeepCell to make Mesmer accessible to the broader biological imaging community, with two distinct deployment solutions based on the volume of data that must be processed (Figure 6). The first solution is geared toward moderate amounts of data (<103 1-megapixel images) and centers around our web portal https://deepcell.org, which hosts the full Mesmer pipeline. Users can access Mesmer through this web portal directly or submit images through plugins that we have made for ImageJ21 and QuPath67, two popular image-analysis tools. This web portal is served by a scalable backend (created by DeepCell Kiosk39), which dynamically adjusts the server’s computational resources to match the volume of data being submitted. This strategy increases computational resources to support large volumes of data during times of high demand, while reducing these resources during times of low demand to reduce costs.
Figure 6: Cloud-native and on-premise software facilitates deployment of Mesmer.
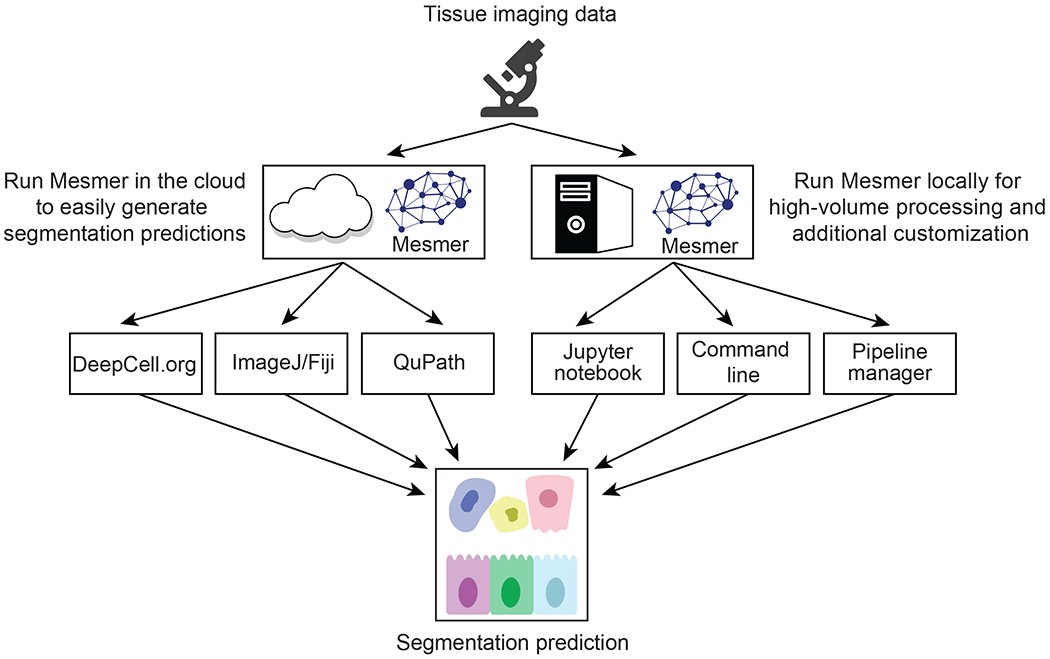
A centralized web server, https://deepcell.org, hosts a version of the Mesmer pipeline. Users with moderate amounts of data (<103 1-megapixel images) to process can access this pipeline through a web portal. Alternatively, users can use ImageJ and QuPath plugins that submit data to the https://deepcell.org web server and retrieve the results. We have also created a containerized version of Mesmer that is compatible with existing workflow managers, so that users with larger amounts of data (>103 1-megapixel images) to process can benefit from our work.
The second deployment solution is targeted toward users with larger volumes of data (>103 1-megapixel images) and who need more control over the execution of the described algorithm. For these users, we provide a Docker image that contains the full Mesmer pipeline. The image generates a Docker container locally on a user’s computational infrastructure and can be installed with a one-line command. This container can be used to launch an interactive Jupyter Notebook that processes data with Mesmer. The container can also be configured as an executable, making it possible to integrate Mesmer within existing image-analysis workflows. In addition, we have developed a software package specifically for analyzing multiplexed imaging data, ark-analysis62, that integrates cloud-based segmentation predictions with downstream analysis and visualization.
Discussion
Cell segmentation has been a major bottleneck for the tissue imaging community, as prior methods19,22–24 required extensive manual curation and parameter tuning to produce usable results. Our experience has shown that these shortcomings can lead to months-long delays in analysis. Mesmer provides a single unified solution to cell segmentation for the most widely used fluorescence and mass spectrometry imaging platforms in a user-friendly format. Mesmer achieves human-level accuracy across a variety of tissues and imaging modalities while requiring no manual parameter tuning from the end user. To make Mesmer widely available, we created cloud-based and local software solutions that position users of all backgrounds to generate accurate predictions for their data. Mesmer’s speed and scalability should facilitate the analysis of the large volumes of multiplexed imaging data currently being generated by consortia around the world. Mesmer was trained on tissue imaging data from both fluorescence- and mass spectrometry-based platforms. Analyses of hematoxylin and eosin images and images of cells in cell culture have been achieved by prior work26,28,38,68,69; making these functionalities available through DeepCell will be the focus of future work.
Along with Mesmer, here we present the initial release of TissueNet, a comprehensive cell segmentation dataset for tissue images. TissueNet contains paired nuclear and whole-cell annotations for >1 million cells from nine organs and six imaging platforms. Prior tissue datasets16,28 were not large enough to train accurate whole-cell segmentation models. As a result, previous efforts to generate accurate tissue-segmentation models have focused on nuclear segmentation27,29,38, meaning that nearly all previous benchmarking has been limited to the evaluation of nuclear-segmentation models27,29,53. TissueNet will enable these valuable efforts to move from nuclear segmentation to whole-cell segmentation, facilitating the development and benchmarking of a new class of tissue-segmentation algorithms. To expand upon this initial release of TissueNet, we are currently constructing a seamless mechanism for investigators to add their own annotated data. Future releases of TissueNet will likely include higher-dimensional data from a wider diversity of tissue types, imaging techniques, and species.
Constructing TissueNet required a new process for generating annotations. Rather than using manual annotation by experts, we used a human-in-the-loop approach. Because annotators in such approaches correct model mistakes rather than create annotations from scratch, annotation time is linked to model performance. As model performance improves, the marginal cost of annotation is reduced, delivering the scalability required for annotating large collections of biological images. Here, we built on prior work22,43–45 by integrating the human-in-the-loop annotation framework with crowdsourcing. This integration increases the speed at which annotation can be performed by distributing work across a crowdsourced labor pool and decreases the annotation burden that deep learning methods place on experts. Thus, crowdsourcing can help meet the data-annotation needs of the biological imaging community. We also demonstrated accurate segmentations for a tissue with complex cell morphologies, the human decidua, using lineage-aware segmentation and a custom six-channel model. While this lineage-aware model was limited to our specific dataset, we believe that it indicates the potential of general-purpose, lineage-aware segmentation models.
Our experience developing TissueNet and Mesmer raises a natural question: how much data is enough? We observed diminishing returns to training data at ~104-105 labels (Extended Data Figure 3c). We believe that the effort required to go beyond this scale is warranted when accuracy is a paramount concern, for example for models applied across projects or in clinical contexts, which is the case for Mesmer. This effort is also worthwhile for generating gold-standard datasets to benchmark model performance. For other use cases, smaller datasets and bespoke models would likely suffice.
Future challenges include the need for a standardized, cross-tissue antibody panel for cell segmentation. Development of such a panel would be a significant advance and would synergize with the work presented here. Whole-cell segmentation in 3D70 is another challenge that will become more prominent as imaging throughput increases to allow routine collection of such datasets. Existing deep learning approaches for 3D instance segmentation are promising71, but a 3D equivalent of TissueNet to power future models currently does not exist. This work can serve as a starting point for these efforts, as it yields accurate prediction in 2D slices of tissues (Extended Data Figure 4). Now that accurate cell segmentation is available to the community, many scientific insights can be expected from the diversity of data currently being generated.
Methods
Creating TissueNet
Human-in-the-loop annotation in the crowd with DeepCell Label
Labeling multiplexed imaging data presents a unique software engineering challenge. Labeling software must allow users to view multiple channels at once, so that they can use all available information to identify cell and nuclear boundaries. This software also needs specialized labeling operations to enable efficient labeling of densely packed fields of cells. Further operations are needed for creating labels from scratch and for editing existing labels; the latter is a key requirement for our human-in-the-loop framework. The final constraint is that this software needs to be browser based, which is essential for crowdsourcing. While existing software packages address specific aspects of these challenges, to date none have met all of the necessary requirements for human-in-the-loop data annotation of multiplexed image data.
To meet this challenge, we previously developed DeepCell Label46, a software package by which humans and algorithms collaboratively create and correct annotations for biological images. DeepCell Label consists of a frontend, which enables users to visualize and interact with images and labels (Extended Data Figure 1a), and a backend, which serves images and labels (stored in cloud buckets) to the frontend (Extended Data Figure 1b). This backend is built on Elastic Beanstalk, a scalable web framework that allows our application to scale as the number of users increases. This scalability enables multiple users to work on the same collection of data at the same time while maintaining responsiveness. A database keeps track of user access and stores key metadata involved in the annotation process. The DeepCell Label software is available at https://github.com/vanvalenlab/deepcell-label.
Because DeepCell Label is cloud-native, it is compatible with any crowdsourcing platform that supports HTML iframes. We have successfully used two crowdsourcing platforms to perform crowdsourced labeling of multiplexed imaging data with DeepCell Label: Appen (https://appen.com) and Anolytics (https://anolytics.ai). DeepCell Label enables our human-in-the-loop framework to blend expert and novice human annotators to increase the scale of creating dense, pixel-level labels for biological images (Figure 1). An example of the instructions provided for the crowd annotators can be found here: https://github.com/vanvalenlab/publication-figures/blob/mesmer_update/2021-Greenwald_Miller_et_al-Mesmer/Example_annotation_instructions.docx
Cropping and stitching of labeled images
We found that supplying smaller image crops led to significantly better crowd annotation of dense images (data not shown). Large images can be overwhelming for annotators to examine and are difficult to navigate at the high zoom level necessary for accurate pixel-level annotation. These two issues significantly increase the time required to complete each job. To alleviate these issues, we created a pipeline to crop and stitch images as part of the annotation process (Extended Data Figure 1c). Input images are cropped to a predetermined size, generally 270x270 pixels, with an overlap between adjacent crops. We keep track of the necessary metadata for each crop to facilitate stitching the image back together. Each crop is independently annotated, with crops from the same image being randomly assigned to annotators. Following annotation, these crops are stitched back together. Cells at the boundary between crops are merged based on maximum overlap. Once each image has been stitched back together, it is quality-controlled by an internal expert to correct stitching artifacts and remaining errors from the annotator output. The finalized annotations are stored with the corresponding image data in .npz files to facilitate easy loading and manipulation in Python.
Combining labeled data for model training
To construct the dataset used for model training, individual .npz files containing annotated images from a single experiment were combined. During this process, the data were randomly split 80%/10%/10% into training, validation, and testing fractions. We applied automated quality control to each image, such as removing cells with area <15 pixels and removing images with <20 cells. Finally, we cropped each image to 256x256 pixels, the input size that the model expects.
TissueNet construction
Our goal in creating TissueNet was to use it to power general-purpose tissue segmentation models. To ensure that models trained on TissueNet would serve as much of the imaging community as possible, we made two key choices. First, all data in TissueNet contains two channels, a nuclear channel (such as DAPI) and a membrane or cytoplasm channel (such as E-cadherin or Pan-Keratin). Although some highly multiplexed platforms are capable of imaging dozens of markers at once1,2,4,6, restricting TissueNet to include only the minimum number of channels necessary for whole-cell segmentation maximizes the number of imaging platforms where the resulting models can be used. Second, the data in TissueNet are derived from a wide variety of tissue types, disease states, and imaging platforms. This diversity of data allows models trained on TissueNet to handle data from many different experimental setups and biological contexts. The images included in TissueNet were acquired from the published and unpublished works of labs who routinely perform tissue imaging55,61,72–77.
Each dataset was manually inspected to identify images suitable for model training. To be included, images from each dataset needed to have robust nuclear staining of all cells, as well as membranous/cytoplasmic staining of a substantial subset of the cells. For datasets with multiple potential nuclear and membrane markers, the best marker (high signal-to-noise ratio, ubiquitous expression) was chosen for each cell type. For multiplexed datasets containing more than one high-quality nuclear or membrane marker, these channels were added together (after rescaling) if doing so increased the coverage of relevant cell compartments across the cells in the image. Selected images were fed through the human-in-the-loop data pipeline to create the final labeled dataset.
Mesmer algorithm design
Deep learning model architecture
The deep learning models used for segmentation are based on feature pyramid networks. Briefly, these networks consist of a ResNet5049 backbone that is connected to a feature pyramid. Prior to entering the backbone model, images are concatenated with a coordinate map. We use backbone layers C3-C5 and pyramid layers P3-P7; for the pyramid layers we use depthwise convolutions for computational efficiency. We attach two semantic segmentation heads to the top of the feature pyramid that perform upsampling to produce predictions the same size as the input image.
Label image transforms
We employed a deep learning approach to segmentation that is inspired by previous work30,54. For each image we use a deep learning model to predict two distinct transforms. The first transform is a prediction of whether each pixel belongs to the cell interior, cell boundary, or background. We call this transform a “pixel-wise transform”. The second transform captures the distance of each pixel inside a cell to that cell’s centroid. If the distance of a cell’s pixel to that cell’s centroid is r, then we compute , where and β is a hyper-parameter that we take to be 1. We call this the “inner distance transform”. One key difference between our formula and the work where this strategy was first proposed30 is the introduction of α, which makes this transform relatively indifferent to differences in cell size. We use the softmax loss for the semantic head assigned to the pixel-wise transform and the mean squared error for the semantic head assigned to the inner distance transform. Similar to prior work, we scale the softmax loss by 0.01 to stabilize training.
Mesmer preprocessing
To handle the variation in staining intensity and signal-to-noise ratio across tissue types and imaging platforms, we normalize each image prior to running it through our deep learning model. We first perform 99.9% scaling to reduce the influence of extremely bright, isolated pixels. We then utilize Contrast Limited Adaptive Histogram Equalization (CLAHE)50 to normalize each image to have the same dynamic range.
Mesmer post-processing
The output of the deep learning model is two sets of predictions, one for the interior/border/background transform and a second for the inner distance transform. We use marker-based watershed52 as a post-processing step to convert these continuous predictions into discrete label images where the pixels belonging to each cell are assigned a unique integer id. To perform this post-processing step, a peak-finding algorithm78 is first applied to the prediction image for the inner distance transform to locate the centroid of each cell in the image. These predictions are thresholded at a value of 0.1. The interior class of the prediction image for the pixel-wise transform is thresholded at a value of 0.3. The cell centroid locations and interior pixel prediction image are used as inputs to the marker-based watershed algorithm to produce the final label image. We smooth the transforms with a gaussian filter to eliminate minor variations and perform additional processing that removes holes and all objects with an area <15 pixels from the final prediction.
Model training
All models were trained using the Adam optimizer79 with a learning rate of 10−4, clipnorm of 0.001, and batch size of 8 images. During training, each image is augmented by performing random flips, rotations, crops, and scaling to increase the diversity of the training dataset. We use 80% of the data for training, 10% for validation, and 10% for testing. We evaluate the loss on the validation dataset after each epoch, and only save the model weights if the loss decreases from the previous value. The test set is only used to evaluate the final trained model. The deep learning model architecture code is available at: https://github.com/vanvalenlab/deepcell-tf
Cell segmentation benchmarking
Classifying error types
We previously described a methodology for classifying segmentation error types37, which we used here. We first construct a cost matrix between all cells in the ground-truth and predicted images, where the cost for each pair of cells is defined as (1 - the intersection over union) between cells. We use this value to determine which predicted cells have a direct, 1-to-1 mapping with ground-truth cells; these cells are classified as accurately segmented. For all other cells, we generate a graph in which the nodes are cells and the edges are connections between cells with nonzero intersection over union. Predicted cells with no edges are labeled as false positives, since they do not have a corresponding match in the ground-truth data. True cells with no edges are labeled as false negatives, since they do not have a corresponding match in the predicted data. If a single predicted cell has edges to multiple ground-truth cells, then this cell is labeled as a merge. If a single ground-truth cell has edges to multiple predicted cells, then this cell is labeled as a split. Finally, if none of the above criteria are satisfied, and there are edges between multiple ground truth and predicted cells, then we categorize such cells as “other.”
Benchmarking model performance
To evaluate model accuracy, we created three random splits of TissueNet, each with a different training, validation, and testing set. These three separate versions of TissueNet were used to train models in triplicate for all model comparisons in this and subsequent sections. Each model was trained using the training and validation splits and evaluated using the corresponding test split, which was never seen during training. We used our error classification framework to calculate the types of errors present in each image in the test split and reported the average and standard deviation across the replicates. All deep learning models were trained for 100 epochs with a fixed number of steps per epoch to control for differences in dataset size.
Comparison with alternate models and architectures
To evaluate the relative performance of distinct deep learning architectures, we compared our approach with several alternative methods: Cellpose28 (whole-cell), StarDist53 (nuclear), RetinaMask54 (trained on TissueNet), FeatureNet16 (nuclear), FeatureNet26 (trained on TissueNet), and Ilastik22. For all models other than Ilastik, we trained on the entire train split of the TissueNet dataset, using the default settings as supplied in the original, respective papers. For Ilastik, we manually annotated two images from each tissue type using the Ilastik interface, rather than using the entire train split, to more accurately mirror how this software is used in practice. All models were evaluated on the same test splits of TissueNet.
Mesmer performance analysis
Nuclear expansion comparison
To compare Mesmer with the current nuclear-based segmentation approaches listed above, we generated whole-cell labels using Mesmer, as well expanded nuclear labels, for all of the images in the test set. Nuclear expansion predictions were generated from nuclear predictions by applying a morphological dilation with a disk of radius 3 pixels as the structuring element. To characterize the error modes of each approach, we selected predictions that mapped directly to a single ground-truth cell using our error-type classification approach (see Classifying error types). Following identification of the corresponding ground-truth cell, we computed the ratio of predicted cell size to true cell size for each prediction.
Specialist model evaluation
To evaluate how a specialist model trained on only a subset of the data compared to a generalist model trained on the entire dataset, we identified the four most common tissue types and four most common imaging platforms in TissueNet. Each of these four tissue types had images from multiple imaging platforms, and each of the four imaging platforms had images from multiple tissue types. For each of the eight specialist models, we identified the images in the training and validation split that belonged to that class and used that subset for model training. We then evaluated the trained specialist model on the data in the test split that belonged to that class and compared this performance to the generalist model evaluated on the same portion of the test split.
Dataset size evaluation
To evaluate how training dataset size impacts model accuracy, we divided TissueNet into bins of increasing size. Each bin of increasing size contained all data from the previous bins, with new data added. This strategy ensured that each bin is a superset of the previous bin, rather than each bin being a random draw from the whole dataset. Bins of increasing size were generated for the training and validation splits while holding the test split constant. We trained models on the progressively larger bins and evaluated all models on the same complete test set.
Inter-annotator agreement
To determine the degree to which annotators agreed with one another, we recruited five expert annotators (lab members or PhD students) to annotate the same set of images. For each of the four images, all five annotators generated segmentations from scratch, without using model predictions. We also generated model predictions for these same four images, which were not included in the training data. We then computed the F1 score between all pairs of annotators, as well as between each annotator and the model.
Pathologist evaluation
To evaluate the relative accuracy of Mesmer and human annotators, we enlisted four board-certified pathologists to evaluate segmentation accuracy. Each pathologist was shown pairs of images; one image contained the segmentation predictions from Mesmer and the other contained the segmentation prediction from one of our expert annotators. We selected 13 random crops from each of the four images. Each crop was displayed to each pathologist twice, with the same Mesmer prediction each time, but matched to a different expert annotator prediction, for a total of 104 comparisons.
Image distortion quantification
To determine how image quality impacts model performance, we systematically degraded images in the test set and assessed the corresponding decrease in F1 score. To simulate out-of-focus images, we performed a gaussian blur with increasing sigma. The blurred images were then passed through the model to generate predictions. To determine how image resolution impacts model performance, we downsampled each image to represent low-resolution data. We then upsampled back to the original size and ran the upsampled images through the model. To simulate low signal-to-noise ratio and high background staining, we added uniform random noise of increasing magnitude to each pixel. The noise-corrupted images were passed through the model to generate predictions.
Analyzing multiplexed imaging datasets
Generating subcellular segmentation predictions
We created a custom analysis pipeline that integrates nuclear and whole-cell segmentation predictions. This pipeline takes as inputs the predictions from Mesmer of each cell and each nucleus in the image. We first link each cell mask with its corresponding nuclear mask using maximum overlap, splitting nuclei that are larger than their corresponding cell. We use these linked masks to extract the counts per compartment for all channels of imaging data. The counts for each marker within each compartment are summed and normalized by cell area. Our multiplex image analysis pipeline is available at: https://github.com/angelolab/ark-analysis
We used this pipeline to compute the subcellular localization of a panel of phenotypic markers with known profiles. We stained a tissue microarray of ductal carcinoma in-situ samples61, imaged them with MIBI-TOF, and ran the above pipeline. For each channel, we selected fields of view in which the marker showed clear expression and computed the localization within each cell, after removing the bottom 20% lowly expressing cells within each marker. We performed the same procedure using the ground-truth labels generated by the human annotators and used the computed localization from the true labels to assess the accuracy of our predictions.
Computing nucleus to whole-cell ratio (N/C)
Traditionally, the nuclear to cytoplasm ratio assessed by pathologists is the ratio between the area of the nucleus and the area of the cytoplasm63. However, as a quantitative measure, this formulation runs into issues with division by zero for immune or stromal cells that have no detectable cytoplasm. To alleviate this issue, we instead use the nucleus to whole-cell ratio (N/C), which uses the whole-cell area rather than the cytoplasm’s area. The nuclear and whole-cell areas are always greater than zero, thus avoiding division by zero and leading to more stable estimates while maintaining the same qualitative interpretation. Cells with high N/C ratios have larger nuclei relative to their overall cell size, and cells with low N/C ratios have smaller nuclei relative to their overall cell size.
Evaluating accuracy of N/C ratio predictions
To determine the accuracy of our N/C ratio predictions, we ran Mesmer on the entire test split of TissueNet. We computed the nuclear and cell predictions for each cell in the image. For each cell, we computed the N/C ratio by first matching each nucleus to its corresponding cell, and then calculated the ratios of their respective areas. We reported the Pearson correlation between the true N/C and predicted N/C for all predicted cells with a direct match in the ground-truth data.
Assessing frequency of out-of-plane nuclei
Multiplexed imaging platforms analyze tissue slices that represent a 2D cut through a 3D structure. As a result, sometimes the nucleus of a given cell is not captured in the image plane/tissue section, whereas the rest of the cell is. Given that Mesmer is trained to separately identify nuclei and cells, cells whose nucleus is out of the imaging plane can still be identified and segmented. To determine the frequency of this occurrence, and to validate that these predictions were not merely segmentation artifacts, we compared the incidence of cells with an out-of-plane nucleus in the ground-truth data and Mesmer predictions from the TissueNet test set. For each ground-truth or predicted cell, we identified the corresponding nucleus. Cells without a matching nucleus were classified as out of plane.
Quantifying accuracy of cell type predictions
To determine how the accuracy of Mesmer’s segmentation predictions influenced downstream quantification of cell type, we analyzed a cohort of breast cancer samples acquired on the Vectra platform. We selected two fields of view each from three patients. Each patient’s sample was stained with DAPI, CD8, CD14, and Pan-Keratin to identify major cell subpopulations. We generated segmentation predictions for all images with Mesmer, as well as ground-truth labels with our human-in-the-loop pipeline. We then extracted the counts of each marker in each cell and used hierarchical gating to define cell populations. Thresholds for gating were determined by manual inspection of the histogram for size-normalized counts of each marker. We used the same thresholds for the ground-truth and predicted segmentations. We matched each ground-truth cell with the predicted cell with maximal IOU. We then determined whether these matching cells were of the same, or different, assigned cell type based on our gating scheme. Matching cells with the same assigned cell type were labeled as true positives. Matching cells which did not have the same assigned cell type and unmatched predicted cells were labeled as false positive. Unmatched ground-truth cells were labeled as false negative.
Decidual cell morphology
Ethical approval
Approval for this study was obtained from the Institutional Review Boards of Stanford University and University of California San Francisco. All participants provided informed consent.
Training a six-channel Mesmer model
To establish the potential of a model that takes in multiple lineage markers, we replaced Mesmer’s model with a lineage-aware variant. We stained samples of human decidua with a panel of markers to define the cell lineages present, then generated images using the MIBI-TOF platform (Greenbaum et al., manuscript in prep). We manually generated whole-cell segmentation labels for 15 of these images using HH3 to define the nucleus and CD3, CD14, CD56, HLAG, and vimentin to define the shape of the cells in the image. We modified the model architecture to accept six channels of input data and trained it using the settings described above.
Generating cell morphology information
To quantify the range of cell shapes and morphologies present in the image data, we created an automated pipeline that extracts key features from each cell segmentation in an image. We extract morphological information using the regionprops function in the scikit-image78 library. We use the following default features from regionprops: area, perimeter, centroid, convex area, equivalent diameter, convex image, and major axis length. These features are transformed as described below to create the selected morphology metrics. Our analysis pipeline is available at: https://github.com/angelolab/ark-analysis
Many of the metrics relate to the difference between the cell shape and the corresponding convex hull. A convex hull for a given segmentation is defined as the smallest possible convex shape that completely contains the cell shape. For shapes that are already convex and do not have any concave angles, the convex hull and the cell are equivalent. For shapes that do have concavities, the convex hull fills in these areas.
In addition to the N/C ratio, we computed the following five morphology metrics:
Asymmetry: the distance between the centroid of the convex hull and the centroid of the cell, normalized by the square root of the area of the cell. The centroid of the convex hull is far from the centroid of the cell when extra mass is added to the convex hull in an imbalanced fashion, indicating that the original cell was not symmetrical.
Concavities: the number of concavities present in each cell. We only include concavities that have an area of at least 10 pixels2 and a perimeter-to-area ratio < 60 to avoid counting very small deviations from convexity. This approach summarizes how many unique indentations and divots are present in each cell.
Fill: the difference in area between the convex hull and the cell, normalized by the area of the convex hull. This ratio is effectively the proportion of the convex hull that was newly added and quantifies the fraction of the cell that is composed of divots and indentations.
Aspect ratio: the ratio between the major axis length and the equivalent diameter. Major axis length is the length of the major axis of an ellipse with the same moments as the original cell and serves as a proxy for the length of the longest diagonal of the cell. Equivalent diameter is the diameter of a circle with the same area as the cell. The ratio of these two quantities gives an estimate of cell elongation.
Perimeter to area ratio: The ratio between the perimeter squared of the cell and the area of the cell. We use perimeter squared rather than perimeter itself for better consistency across cell sizes.
Identifying morphological clusters
We classified the cells based only on the above five morphology metrics, which we computed for the images in the decidua cohort. We first normalized each metric independently, and then performed k-means clustering with k=4. We plotted the mean value of metric in each cluster to identify the features that separated them from one another and performed hierarchical clustering on the resulting output.
Model deployment
DeepCell Kiosk: A scalable, cloud-based solution for hosting deep learning models
We previously described the construction of DeepCell Kiosk, our cloud-based deployment system39. This software dynamically adjusts the amount of compute resources needed at any one time to match demand; since the load on the server is quite low most of the time, this strategy delivers economical hosting of the web portal for community use. When demand increases, compute resources are automatically increased. The Kiosk is available at: https://github.com/vanvalenlab/kiosk-console.
Generating predictions from Mesmer using cloud deployments
To facilitate quick and easy access to Mesmer, we used the Kiosk to generate several easy ways to predict cell segmentation. We created a web portal that allows anyone to upload their data and instantly receive results. This web-based interface facilitates point-and-click upload and download of results, with no installation required. We have also created plugins for ImageJ21 and QuPath67 that automatically send data to the Kiosk and return predictions to the user. These predictions can then be used within ImageJ or QuPath for downstream analyses of interest. Detailed tutorials and documentation can be found at: https://github.com/vanvalenlab/intro-to-deepcell.
Generating predictions from Mesmer using local deployments
Although cloud deployment offers a fast, intuitive way for users with little computational experience to generate predictions, it offers less fine-grained control over the input and output parameters. Further, web portals are not ideal for integration with existing image-processing workflows. For users with more computational expertise, we have created local deployments of Mesmer to facilitate future model development and integration with existing workflows. To facilitate training and model development, we provide example Jupyter and Colab notebooks. For integration with existing computational workflows, we provide a command line interface and docker container. Finally, we have also made our open-source multiplex image analysis pipeline available for users who want an end-to-end solution for segmenting, quantifying, and analyzing image data. A guide showing users how to use these resources is available at: https://github.com/vanvalenlab/intro-to-deepcell.
Statistics and Reproducibility
The image shown in Figure 2c is a crop from a single patient sample; this experiment was not repeated. The images shown in Figures 2f and 3a are crops from individual patient samples with the highest F1 scores; this experiment was repeated across all images in the test split of TissueNet, with results reported in Figure 2b and 3b–c. The images shown in Figure 4c are crops from individual patient samples; this experiment was repeated across all samples in the test split of TissueNet, with the results reported in Figure 4d. The images shown in Figure 4e are crops from a single patient sample; this experiment was repeated across all samples in the test split of TissueNet, with the results reported in Figure 4f. The images shown in Figure 4g–i are crops from a single patient sample; this experiment was repeated twice in two distinct crops for the three patients in this dataset, with the results reported in Figure 4j. The images shown in Figures 5a–b, 5e, and 5h–i are crops from individual patient samples; this experiment was repeated across all 10 patients in this cohort. The images in Supplementary Figure 3i are crops from individual patient samples; this experiment was repeated across all samples in the test split of TissueNet.
Software
This project would not have been possible without numerous open-source Python packages including jupyter80, keras81, matplotlib82, numpy83, pandas84, scikit-image78, scikit-learn85, seaborn86, tensorflow87, and xarray88. Specific versions for each package can be found at: https://github.com/vanvalenlab/deepcell-tf/blob/master/requirements.txt
Extended Data
Extended Data Figure 1.
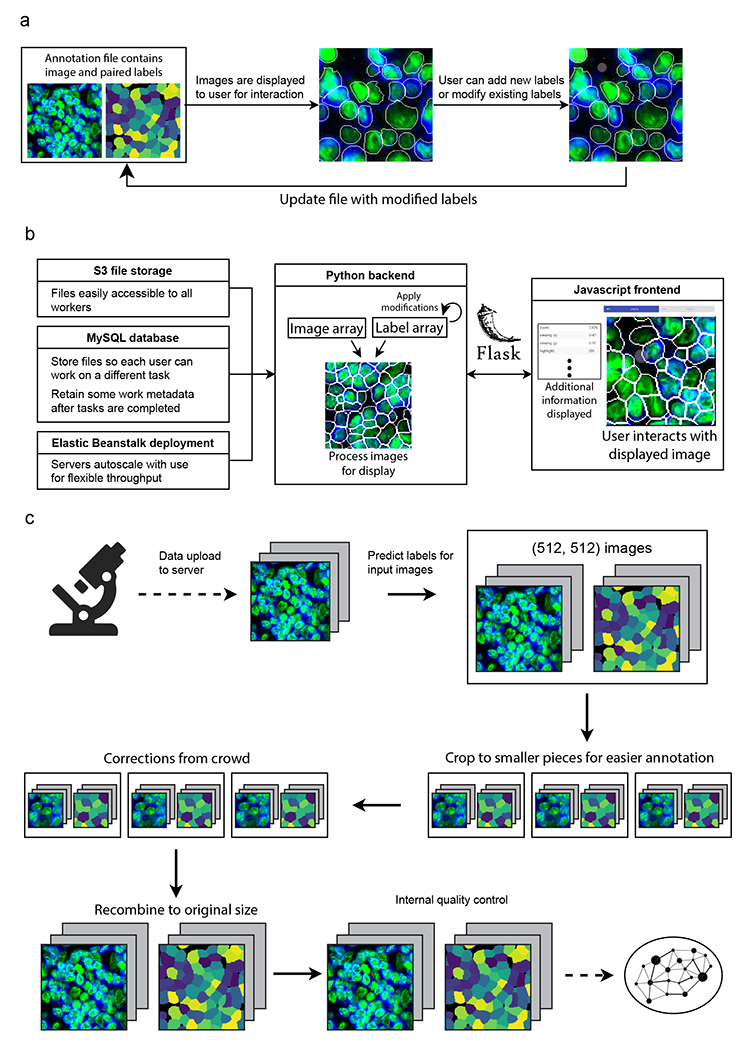
a, How multichannel images are represented and edited in DeepCell Label. b, Scalable backend for DeepCell Label that dynamically adjusts required resources based on usage, allowing concurrent annotators to work in parallel. c, Human-in-the-loop workflow diagram. Images are uploaded to the server, run through Mesmer to make predictions, and cropped to facilitate error correction. These crops are sent to the crowd to be corrected, stitched back together, run through quality control to ensure accuracy, and used to train an updated model.
Extended Data Figure 2.
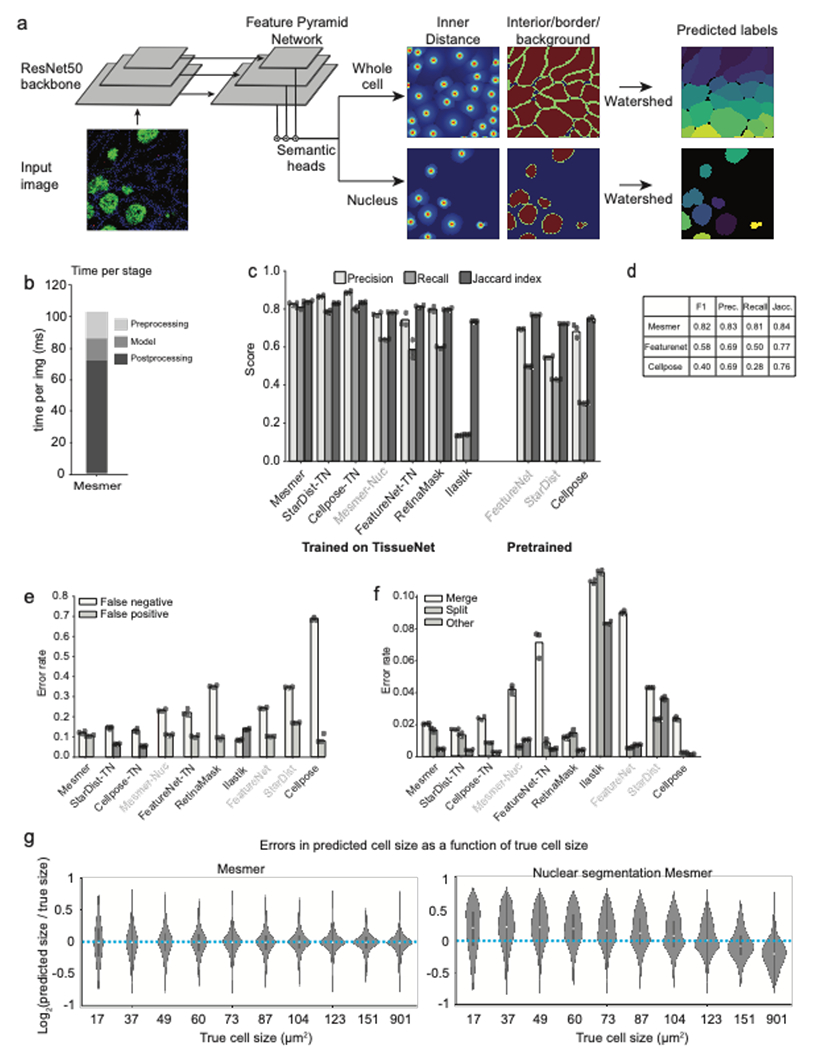
a, PanopticNet architecture. Images are fed into a ResNet50 backbone coupled to a feature pyramid network. Two semantic heads produce pixel-level predictions. The first head predicts whether each pixel belongs to the interior, border, or background of a cell, while the second head predicts the center of each cell. b, Relative proportion of preprocessing, inference, and post-processing time in PanopticNet architecture. c, Evaluation of precision, recall, and Jaccard index for Mesmer and previously published models (right) and models trained on TissueNet (left). d, Summary of TissueNet accuracy for Mesmer and selected models to facilitate future benchmarking efforts e,f Breakdown of most prevalent error types (e) and less prevalent error types (f) for Mesmer and previously published models illustrates Mesmer’s advantages over previous approaches. g, Comparison of the size distribution of prediction errors for Mesmer (left) with nuclear segmentation followed by expansion (right) shows that Mesmer’s predictions are unbiased.
Extended Data Figure 3.
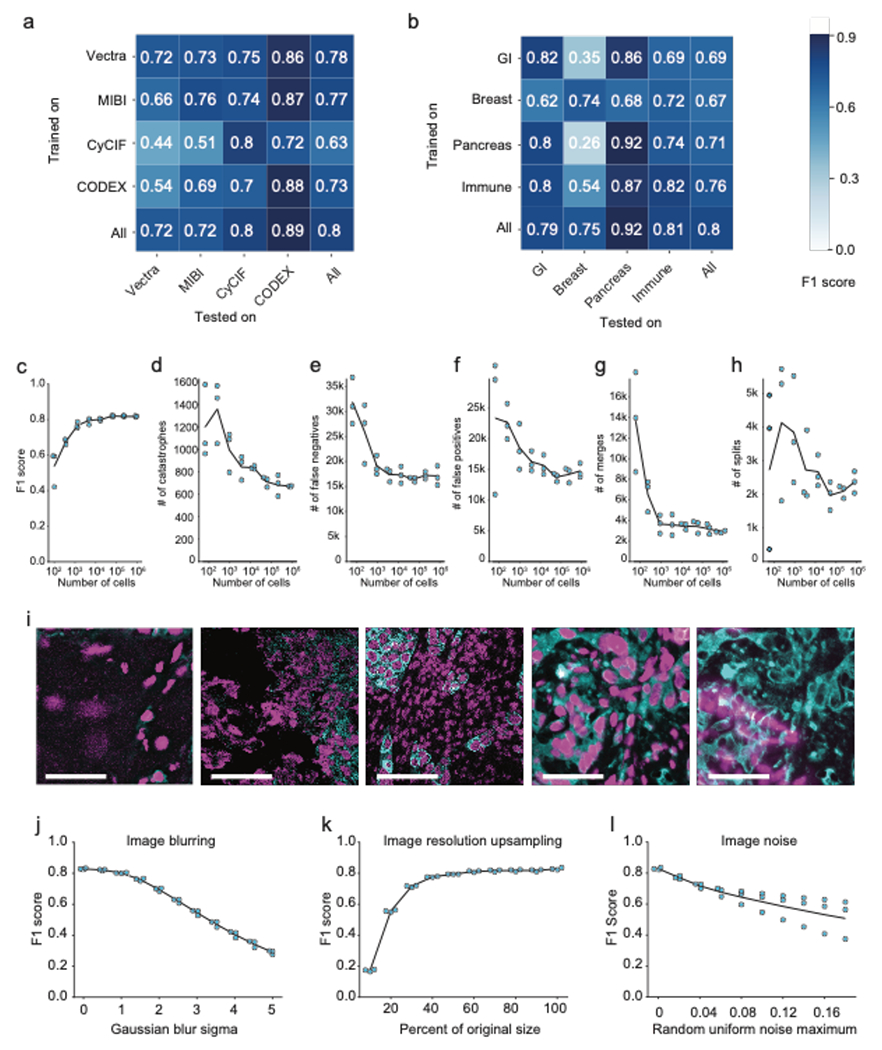
a, Accuracy of specialist models trained on each platform type (rows) and evaluated on data from other platform types (columns) indicates good agreement within immunofluorescence and mass spectrometry-based methods, but not across distinct methods. b, Accuracy of specialist models trained on each tissue type (rows) and evaluated on data from other tissue types (columns) demonstrates that models trained on only a single tissue type do not generalize as well to other tissue types. c, Quantification of F1 score as a function of the size of the dataset used for training. d-h, Quantification of individual error types as a function of the size of the dataset used for training. i, Representative images where Mesmer accuracy was poor, as determined by the image specific F1 score. j, Impact of image blurring on model accuracy. k, Impact of image downsampling and then upsampling on model accuracy. l, Impact of adding random noise to image on model accuracy. All scale bars are 50 μM.
Extended Data Figure 4.
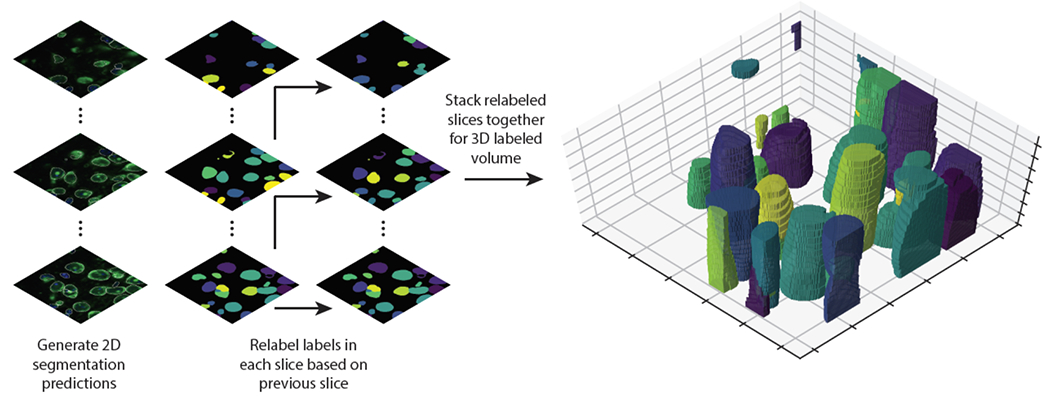
Proof of principle for using Mesmer’s segmentation predictions to generate 3D segmentations. A z-stack of 3D data is fed to Mesmer, which generates separate 2D predictions for each slice. We computationally link the segmentations predictions from each slice to form 3D objects. This approach can form the basis for human-in-the-loop construction of training data for 3D models.
Supplementary Material
{kind=link}
Acknowledgments
We thank Katy Borner, Long Cai, Markus Covert, Andrej Karpathy, Steve Quake, and Matt Thomson for interesting discussions; David Glass and Erin McCaffrey for feedback on the manuscript; Tiffany Vora for copy editing; Roshan Angoshtari, Graham Barlow, Bernd Bodenmiller, Christopher Carey, Robert Coffey, Alea Delmastro, Colt Egelston, Michal Hoppe, Hartland Jackson, Anand Jeyasekharan, Sizun Jiang, Youn Kim, Erin McCaffrey, Eliot McKinley, Michael Nelson, Siok-Bian Ng, Gary Nolan, Sanjay Patel, Yanfen Peng, Darci Philips, Rumana Rashid, Scott Rodig, Sandro Santagata, Christian Schuerch, Daniel Schulz, Diana Simons, Peter Sorger, Jason Weirather, and Yuan Yuan for providing imaging data for TissueNet; the crowd annotators who powered our human-in-the-loop pipeline; and all patients who donated samples for this study.
Funding
This work was supported by grants from the Shurl and Kay Curci Foundation, the Rita Allen Foundation, the Susan E. Riley Foundation, the Pew Heritage Trust, the Alexander and Margaret Stewart Trust, the Heritage Medical Research Institute, the Paul Allen Family Foundation through the Allen Discovery Centers at Stanford and Caltech, the Rosen Center for Bioengineering at Caltech, and the Center for Environmental and Microbial Interactions at Caltech (D.V.V.). This work was also supported by 5U54CA20997105, 5DP5OD01982205, 1R01CA24063801A1, 5R01AG06827902, 5UH3CA24663303, 5R01CA22952904, 1U24CA22430901, 5R01AG05791504, and 5R01AG05628705 from NIH, W81XWH2110143 from DOD, and other funding from the Bill and Malinda Gates Foundation, Cancer Research Institute, the Parker Center for Cancer Immunotherapy, and the Breast Cancer Research Foundation (M.A.). N.F.G. was supported by NCI CA246880-01 and the Stanford Graduate Fellowship. B.J.M. was supported by the Stanford Graduate Fellowship and Stanford Interdisciplinary Graduate Fellowship. T.D. was supported by the Schmidt Academy for Software Engineering at Caltech.
Footnotes
Code Availability
All software for dataset construction, model training, deployment, and analysis is available on our github page https://github.com/vanvalenlab/intro-to-deepcell. All code to generate the figures in this paper is available at: https://github.com/vanvalenlab/publication-figures/tree/master/2021-Greenwald_Miller_et_al-Mesmer
Competing interests
M.A. is an inventor on patent US20150287578A1. M.A. is a board member and shareholder in IonPath Inc. T.R. has previously consulted for IonPath Inc. D.V.V and E.M. have filed a provisional patent for this work. The remaining authors declare no competing interests.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article
Data availability
The TissueNet dataset is available at datasets.deepcell.org for non-commercial use.
References
- 1.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Keren L et al. MIBI-TOF: A multiplexed imaging platform relates cellular phenotypes and tissue structure. Sci Adv 5, eaax5851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang W, Hennrick K & Drew S A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum Pathol 44, 29–38 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Lin J-R et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 7, e31657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerdes MJ et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc National Acad Sci 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goltsev Y et al. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moffitt JR et al. Molecular, spatial and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nat Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rozenblatt-Rosen O et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181, 236–249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder MP et al. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regev A et al. The Human Cell Atlas White Paper. Arxiv (2018). [Google Scholar]
- 16.Keren L et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174, 1373–1387.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milo R & Phillips R Cell Biology by the Numbers. (Garland Sciences, 2015). [Google Scholar]
- 18.Mescher A Junqueira’s Basic Histology: Text and Atlas. (McGraw Hill, 2013). [Google Scholar]
- 19.McQuin C et al. CellProfiler 3.0: Next-generation image processing for biology. Plos Biol 16, e2005970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nat Methods 16, 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Chaumont F. de et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods 9, 690–696 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Belevich I, Joensuu M, Kumar D, Vihinen H & Jokitalo E Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. Plos Biol 14, e1002340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronneberger O, Fischer P & Brox T U-Net: Convolutional Networks for Biomedical Image Segmentation. Arxiv (2015). [Google Scholar]
- 26.Valen DAV et al. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. Plos Comput Biol 12, e1005177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caicedo JC et al. Nucleus segmentation across imaging experiments: the 2018 Data Science Bowl. Nat Methods 16, 1247–1253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Hollandi R et al. nucleAIzer: A Parameter-free Deep Learning Framework for Nucleus Segmentation Using Image Style Transfer. Cell Syst 10, 453–458.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koyuncu CF, Gunesli GN, Cetin-Atalay R & Gunduz-Demir C DeepDistance: A Multi-task Deep Regression Model for Cell Detection in Inverted Microscopy Images. Arxiv (2019). [DOI] [PubMed] [Google Scholar]
- 31.Yang L et al. NuSeT: A deep learning tool for reliably separating and analyzing crowded cells. Plos Comput Biol 16, e1008193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu W et al. CCDB:6843, mus musculus, Neuroblastoma. CIL. doi: 10.7295/W9CCDB6843. [DOI] [Google Scholar]
- 33.Koyuncu CF, Cetin□Atalay R & Gunduz□Demir C Object□Oriented Segmentation of Cell Nuclei in Fluorescence Microscopy Images. Cytom Part A 93, 1019–1028 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Ljosa V, Sokolnicki KL & Carpenter AE Annotated high-throughput microscopy image sets for validation. Nat Methods 9, 637–637 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar N et al. A Multi-Organ Nucleus Segmentation Challenge. Ieee T Med Imaging 39, 1380–1391 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verma R et al. Multi-organ Nuclei Segmentation and Classification Challenge 2020. (2020) doi: 10.13140/rg.2.2.12290.02244/1. [DOI] [Google Scholar]
- 37.Moen E et al. Accurate cell tracking and lineage construction in live-cell imaging experiments with deep learning. bioRxiv 803205 (2019) doi: 10.1101/803205. [DOI] [Google Scholar]
- 38.Gamper J et al. PanNuke Dataset Extension, Insights and Baselines. Arxiv (2020). [Google Scholar]
- 39.Bannon D et al. DeepCell Kiosk: scaling deep learning–enabled cellular image analysis with Kubernetes. Nat Methods 18, 43–45 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haberl MG et al. CDeep3M—Plug-and-Play cloud-based deep learning for image segmentation. Nat Methods 15, 677–680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouyang W, Mueller F, Hjelmare M, Lundberg E & Zimmer C ImJoy: an open-source computational platform for the deep learning era. Nat Methods 16, 1199–1200 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Chamier L. von et al. ZeroCostDL4Mic: an open platform to use Deep-Learning in Microscopy. Biorxiv 2020.03.20.000133 (2020) doi: 10.1101/2020.03.20.000133. [DOI] [Google Scholar]
- 43.Hughes AJ et al. Quanti.us: a tool for rapid, flexible, crowd-based annotation of images. Nat Methods 15, 587–590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ouyang W, Le T, Xu H & Lundberg E Interactive biomedical segmentation tool powered by deep learning and ImJoy. F1000research 10, 142 (2021). [Google Scholar]
- 45.Wolny A et al. Accurate and versatile 3D segmentation of plant tissues at cellular resolution. Elife 9, e57613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeepCell Label github. https://github.com/vanvalenlab/deepcell-label.
- 47.Lin T-Y et al. Feature Pyramid Networks for Object Detection. Arxiv (2016). [Google Scholar]
- 48.Tan M, Pang R & Le QV EfficientDet: Scalable and Efficient Object Detection. Arxiv (2019). [Google Scholar]
- 49.He K, Zhang X, Ren S & Sun J Deep Residual Learning for Image Recognition. Arxiv (2015). [Google Scholar]
- 50.Zuiderveld K Graphics Gems. Viii Image Process 474–485 (1994) doi: 10.1016/b978-0-12-336156-1.50061-6. [DOI] [Google Scholar]
- 51.Chevalier G Smoothly Blend Image Patches. https://github.com/Vooban/Smoothly-Blend-Image-Patches. [Google Scholar]
- 52.Meyer F & Beucher S Morphological segmentation. J Vis Commun Image R 1, 21–46 (1990). [Google Scholar]
- 53.Weigert M, Schmidt U, Haase R, Sugawara K & Myers G Star-convex Polyhedra for 3D Object Detection and Segmentation in Microscopy. 2020 Ieee Winter Conf Appl Comput Vis Wacv 00, 3655–3662 (2020). [Google Scholar]
- 54.Fu C-Y, Shvets M & Berg AC RetinaMask: Learning to predict masks improves state-of-the-art single-shot detection for free. Arxiv (2019). [Google Scholar]
- 55.Schürch CM et al. Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 182, 1341–1359.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali HR et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat Cancer 1, 163–175 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Gaglia G et al. HSF1 phase transition mediates stress adaptation and cell fate decisions. Nat Cell Biol 22, 151–158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson DE et al. Oscillations in NF-κB Signaling Control the Dynamics of Gene Expression. Science 306, 704–708 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Kumar KP, McBride KM, Weaver BK, Dingwall C & Reich NC Regulated Nuclear-Cytoplasmic Localization of Interferon Regulatory Factor 3, a Subunit of Double-Stranded RNA-Activated Factor 1. Mol Cell Biol 20, 4159–4168 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolff AC et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J Clin Oncol 31, 3997–4013 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Risom T et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. bioRxiv 2021.01.05.425362 (2021) doi: 10.1101/2021.01.05.425362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.ark analysis github. https://github.com/angelolab/ark-analysis. [Google Scholar]
- 63.Diagnostic Cytology and Its Histopathologic Bases. (J.B. Lippincott Company, 1979). [Google Scholar]
- 64.Erlebacher A Immunology of the Maternal-Fetal Interface. Annual Review of Immunology 31, 387–411 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Garrido-Gomez T et al. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc National Acad Sci 114, E8468–E8477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.deepcell-tf github. https://github.com/vanvalenlab/deepcell-tf (n.d.).
- 67.Bankhead P et al. QuPath: Open source software for digital pathology image analysis. Sci Rep-uk 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Graham S et al. Hover-Net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med Image Anal 58, 101563 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Tsai H-F, Gajda J, Sloan TFW, Rares A & Shen AQ Usiigaci: Instance-aware cell tracking in stain-free phase contrast microscopy enabled by machine learning. Softwarex 9, 230–237 (2019). [Google Scholar]
- 70.Kiemen A et al. In situ characterization of the 3D microanatomy of the pancreas and pancreatic cancer at single cell resolution. bioRxiv 2020.12.08.416909 (2020) doi: 10.1101/2020.12.08.416909. [DOI] [Google Scholar]
- 71.Cao J et al. Establishment of a morphological atlas of the Caenorhabditis elegans embryo using deep-learning-based 4D segmentation. Nat Commun 11, 6254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schulz D et al. Simultaneous Multiplexed Imaging of mRNA and Proteins with Subcellular Resolution in Breast Cancer Tissue Samples by Mass Cytometry. Cell Syst 6, 531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKinley ET et al. Optimized multiplex immunofluorescence single-cell analysis reveals tuft cell heterogeneity. Jci Insight 2, e93487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patel SS et al. The microenvironmental niche in classic Hodgkin lymphoma is enriched for CTLA-4- positive T-cells that are PD-1-negative. Blood 134, 2059–2069 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jackson HW et al. The single-cell pathology landscape of breast cancer. Nature 578, 615–620 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Rashid R et al. Highly multiplexed immunofluorescence images and single-cell data of immune markers in tonsil and lung cancer. Sci Data 6, 323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCaffrey EF et al. Multiplexed imaging of human tuberculosis granulomas uncovers immunoregulatory features conserved across tissue and blood. bioRxiv 2020.06.08.140426 (2020) doi: 10.1101/2020.06.08.140426. [DOI] [Google Scholar]
- 78.Walt S van der et al. scikit-image: image processing in Python. Peerj 2, e453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kingma DP & Ba J Adam: A Method for Stochastic Optimization. Arxiv (2014). [Google Scholar]
- 80.Kluyver T et al. Jupyter Notebooks -- a publishing format for reproducible computational workflows. in Positioning and Power in Academic Publishing: Players, Agents and Agendas (ed. Schmidt FL and B.) 87–90. [Google Scholar]
- 81.Keras. Francis Chollet and others. 2015. https://keras.io. [Google Scholar]
- 82.Hunter JD Matplotlib: A 2D Graphics Environment. Comput Sci Eng 9, 90–95 (2007). [Google Scholar]
- 83.Harris CR et al. Array programming with NumPy. Nature 585, 357–362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.The pandas development team.pandas-dev/pandas: Pandas. February 2020. 10.5281/zenodo.3509134. (n.d.). [DOI] [Google Scholar]
- 85.Pedregosa F et al. Scikit-learn: Machine Learning in Python. Arxiv (2012). [Google Scholar]
- 86.Michael Waskom and the seaborn development team. mwaskom/seaborn. September, 2020. 10.5281/zenodo.592845. (n.d.). [DOI] [Google Scholar]
- 87.Abadi M et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. Arxiv (2016). [Google Scholar]
- 88.Hoyer S & Hamman J xarray: N-D labeled Arrays and Datasets in Python. Journal of Open Research Software 1, 10 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TissueNet dataset is available at datasets.deepcell.org for non-commercial use.