

Abstract
Macrophages’ response to stimulation is a dynamic process, which coordinates the orderly adoption and resolution of various immune functions. Accumulating work over the past decade has demonstrated that during the immune response macrophage metabolism is substantially rewired to support important cellular processes, including the production of bioactive molecules, intercellular communication, and regulation of intracellular signaling and transcriptional programming. Here we particularly discuss an important concept emerging from recent studies – metabolic rewiring during the immune response is temporally structured. We review the regulatory mechanisms that drive the dynamic remodeling of metabolism and examine the implications of these metabolic dynamics in coordination of macrophages’ functional transitions.
Keywords: immunometabolism, dynamics, metabolic regulation, inflammatory response and resolution, macrophage
Macrophages’ diverse functional states are coupled to distinct metabolic programs
Macrophages are highly plastic innate immune cells. Upon stimulation, macrophages can quickly polarize into diverse functional states which play roles in tissue homeostasis, host defense against pathogens, and repair of damaged tissue [1]. An increasing body of work has shown that macrophage activation is coupled to metabolic reprogramming and has characterized the common and unique metabolic features associated with specific polarization states, as has been nicely reviewed [2-4]. Such specific metabolic rewiring during polarization is functionally important, as it not only provides energy but can also support production of bioactive molecules, influence intracellular signaling, and modulate transcriptional and epigenetic programming [2-6].
A concept which has gained more attention recently is that macrophage metabolism varies, not only with diverse stimuli, but also across time in response to the same signal [7-9]. A key example is the response to pathogen associated molecular patterns (PAMPs). When exposed to PAMPs, macrophages swiftly adopt a pro-inflammatory state. However, after some time, there is a shift to a less inflammatory and more pro-resolution phenotype. These dynamics allow for a rapid response while also mitigating harm that can occur from prolonged inflammation [10]. Recent work has revealed that metabolism is dynamically rewired alongside and in support of these functional transitions [7-9,11]. Though the investigation of the temporal structure of metabolic rewiring is just beginning, it has the potential to lead to a more holistic understanding of the physiological immune response. Importantly, it can also provide insight into pathological conditions in which the proper timing of metabolic and functional transitions is disrupted, such as chronic inflammation or immune paralysis.
Here, largely focusing on the example of macrophages stimulated with the prototypical PAMP, lipopolysaccharide (LPS), we review the functional roles of metabolic rewiring and explore recent work showing the temporal structure of this rewiring. We further discuss the molecular mechanisms controlling the dynamic metabolic changes and the connection of these dynamics to functional transitions across time. Lastly, we discuss the important insights which can be gleaned from considering time when studying macrophage metabolism and the vital questions left to be answered.
Macrophage metabolism supports and regulates immune functions via multiple mechanisms
Metabolism supports the production of bioactive compounds and extracellular signals
Many bactericidal or signaling compounds produced by macrophages are derived from metabolism, linking the rewiring of metabolic pathways directly to function (Figure 1). In response to LPS, macrophages dramatically increase the production of reactive nitrogen species (RNS) and reactive oxygen species (ROS), both of which, through their capacity to cause oxidative damage, have direct anti-microbial activity [12,13]. RNS are derived from nitric oxide (NO), which is generated from arginine by inducible NO synthase (iNOS). Up regulation of iNOS and the rewiring of arginine metabolism is one of the most classic examples of metabolic rewiring coupled to pro-inflammatory activation [13,14]. Activated macrophages also release lipid-derived molecules which can signal in autocrine or paracrine fashions. Most well-known are eicosanoids, a diverse family of oxidized fatty acids. Eicosanoid production is partially regulated by the remodeling of lipid metabolism to expand the availability of the eicosanoid precursor, arachidonic acid (AA) [15]. Indeed, recent work showed that increased triglyceride (TG) synthesis upon LPS stimulation supports production of the eicosanoid, prostaglandin E2, by promoting expansion of lipid droplets and AA containing TGs within them [16]. Extracellular nucleotides and nucleosides, through activation of purinergic receptors, also act as immune regulatory signals [17]. In response to PAMP exposure, macrophages release ATP which acts as an inflammatory signal. However, plasma membrane proteins can quickly hydrolyze ATP to adenosine, which then signals to dampen pro-inflammatory functions and promote resolution [17]. More recent work has shown that some TCA cycle-derived metabolites, such as succinate and itaconate, are released by macrophages and play a functional role extracellularly [18]. Extracellular itaconate is bactericidal via its ability to inhibit the bacterial isocitrate lyase [19]. Extracellular succinate can signal via the succinate receptor 1 which is present on numerous cell types including macrophages, but the broader impact of this signaling is still not fully understood [20]. Macrophages can influence the microenvironment not just through release but also via depletion of certain compounds. For example, activated macrophages increase tryptophan uptake and degradation, which not only results in an extracellular accumulation of the immunomodulatory tryptophan metabolite kynurenine, but also a depletion of extracellular tryptophan. Both changes promote an immunosuppressive microenvironment [21]. In these ways macrophage metabolism can greatly influence the extracellular milieu during the response to PAMPs.
Figure 1. Macrophage metabolic rewiring supports the production and release of bioactive compounds.

In response to PAMP stimulation, macrophages reprogram their metabolism to support the production of bioactive and bactericidal compounds that are released into the extracellular space. Reprogramming of the TCA cycle leads to extracellular accumulation of itaconate. A shift in arginine metabolism flux to nitric oxide production leads to an increase of reactive nitrogen species (RNS). Reactive oxygen species (ROS) production also increases. Itaconate, RNS and ROS all play a direct bactericidal role either by inhibiting key metabolic enzymes in bacteria (itaconate, RNS, ROS) or due to broad oxidative damage of pathogen cell components (ROS, RNS). Other metabolically derived compounds are released and signal in an autocrine or paracrine fashion to shape the immune response. Lipid mediators, such as eicosanoids, are produced due to a reprogramming of lipid metabolism. Succinate can be released and accumulate in the extracellular space due to inhibition of the TCA cycle enzyme succinate dehydrogenase (SDH). Both eicosanoids and succinate can promote both inflammatory and anti-inflammatory functions depending on the context and target cell type. Activated macrophages increase tryptophan degradation leading to increased release of the tryptophan metabolite kynurenine and depletion of extracellular tryptophan, both of which promote an anti-inflammatory microenvironment. Additionally, macrophages and dying cells can release ATP, which acts to promote inflammatory functions by signaling to purinergic receptors. The extracellular ATP can be converted by the macrophage to its metabolite, adenosine, which generally promotes anti-inflammatory functions and contributes to resolution of the inflammatory response.
Macrophage metabolism modulates key intracellular regulatory events
A key outcome of macrophage stimulation is the activation of the inflammasome and its caspase-1 activity, which processes the inflammatory cytokines IL1β and IL18, and the pore forming protein gasdermin D, to their mature forms [22]. Though the mechanisms are not yet fully understood, inflammasome activation can be induced by diverse signals, many of which are derived from or influenced by metabolic rewiring [23] (Figure 2). Release of mitochondrial DNA (mtDNA) or its oxidized form (ox-mtDNA) to the cytosol can activate the NLRP3 (NOD- LRR- and pyrin containing protein 3) and AIM2 (absent in melanoma 2) inflammasomes [22]. Recent work showed that the release of ox-mtDNA is dependent on the catalytic activity of the mitochondrial enzyme CMPK2, which can convert dCMP to dCDP. CMPK2 is upregulated upon LPS stimulation to promote synthesis of mtDNA [24]. The release of ox-mtDNA or mtDNA is also sensitive to changes in mitochondrial membrane lipid composition. In response to stimulation, increased choline uptake supports the production of phosphatidylcholine (PC), which is required for the inhibition of excessive mitophagy and proper release of ox-mtDNA [25]. Cholesterol synthesis, on the other hand, is actively suppressed upon LPS stimulation due to accumulation of its negative regulator, 25-hydroxycholesterol. This avoids overaccumulation of cholesterol in the mitochondrial membrane, preventing excessive mtDNA release and overactivation of the AIM2 inflammasome [26]. Recent work has also suggested a possible role for itaconate in suppressing the NLRP3 inflammasome, via its ability to post-translationally modify cysteine residues [27,28]. Hooftman et al. showed that the itaconate derivative, 4-OI, can modify NLRP3 protein directly, and proposed this leads to impaired interaction between NLRP3 and the inflammasome component NEK7 (NIMA related kinase 7) [27]. Bamboushkova et al. proposed that modification of gasdermin-D by itaconate suppresses inflammasome activation, by impairing the interplay between gasdermin-D and caspase-1 activation [28]. The inflammasome is also responsive to oxidative stress, and thus both ROS and RNS have been implicated in control of the inflammasome (Box 1). These demonstrate some, among many, ways in which metabolism can influence inflammasome activation, as discussed in excellent recent reviews [23,29,30].
Figure 2. Macrophage metabolism influences inflammasome activation.

Activation of the inflammasome allows for processing of the inflammatory cytokines, IL1β and IL18, and gasdermin D (GSDMD) to their mature and active forms. Inflammasome activation is influenced by changes in multiple metabolic pathways, many of which impinge on mitochondria. Release of mitochondrial DNA (mtDNA) or its oxidized form (ox-mtDNA) from mitochondria serves as an inflammasome activating signal and is regulated by multiple metabolic changes. In response to stimulation, upregulation of the mitochondrial metabolic enzyme, CMPK2, which participates in the synthesis of mitochondrial dCTP and dUTP, supports the production of mtDNA, while alteration in cholesterol and choline production influence mitochondrial membrane composition and mtDNA release. Reactive oxygen species (ROS) from both mitochondrial and cytosolic sources has also been implicated in the activation of the inflammasome. Mitochondrial ROS (mtROS) has been shown to accumulate in response to stimulation and is reported to originate either from complex III of the electron transport chain or from complex I via a phenomenon termed reverse electron transport (RET). In response to activation, there is also an accumulation of reactive nitrogen species (RNS) and the TCA cycle metabolite, itaconate, both of which have been shown to inhibit inflammasome activation.
Box 1: ROS and RNS production and function in macrophages.
A key feature of macrophages’ response to PAMPs is the production of ROS and RNS. RNS are mainly derived from nitric oxide which is produced by iNOS. ROS have been shown to be produced at multiple sites including NADPH oxidase (NOX), xanthine oxidase and the mitochondrial electron transport chain (ETC) [12,32,63]. Studies have proposed multiple mechanisms controlling mitochondrial ROS (mtROS) production in macrophages, including reverse electron transport (RET) during which increased succinate oxidation through complex II, along with an increase in the mitochondrial membrane potential leads to backward electron flow in the ETC and ultimately to formation of ROS by complex I [32,84].
Not only are ROS and RNS important for their direct bactericidal effects in the phagosome and extracellular space, but they also play important signaling roles intracellularly. These reactive species have been shown to target numerous signaling pathways including NFκB, HIF-1α and Jak/Stat signaling [12,85]. ROS have also been implicated in the assembly and activation of the inflammasome, though the exact source of ROS and mechanisms controlling this connection remain not fully understood [12,79,85]. RNS, conversely, has been shown to inhibit the assembly and activation of the inflammasome, at least partially due to nitrosylation of inflammasome components (for more comprehensive discussion of the targets of ROS/RNS see [12,79,85])
Though RNS and ROS are critical for immune function, excessive accumulation of these species can cause unwanted damage, requiring maintenance of a tight redox balance. The negative effects of ROS and RNS are mitigated by the thioredoxin (TRX) and glutaredoxin (GRX) antioxidant systems and by activation of the transcription factor NRF2 [79]. Interestingly, the same molecule required to produce ROS and RNS via NOX and iNOS, NADPH, is also utilized by the GRX and TRX systems to control ROS/RNS induced damage.
Both ROS and RNS influence macrophage bioenergetics by impairing oxidative metabolism [9,66-71]. Activation of the immune response by macrophages requires engagement of energetically costly processes such as protein synthesis and membrane remodeling. However, accumulation of ROS and RNS inhibits multiple TCA cycle and ETC proteins, shutting down ATP production from this source. This results in macrophages which are heavily reliant on glycolytically derived ATP. This adoption of a highly glycolytic metabolism even in the presence of oxygen is a common feature of PAMP stimulated macrophages [69].
Metabolism also has a vital role in controlling the transcriptional landscape during the response to PAMP stimulation. In part, this is mediated by the control of transcription factors. An example is the metabolic control of hypoxia inducible-1α (HIF-1α), a regulator of the inflammatory response [31]. HIF-1α is largely regulated by its degradation, which is promoted by α-ketoglutarate (α-KG) dependent prolyl-hydroxylases (PHDs). PHDs are inhibited by ROS, itaconate, and succinate, all which accumulate in activated macrophages, leading to stabilization of HIF-1α [7,32,33]. Work has also shown that the glycolytic enzyme pyruvate kinase M2 (PKM2) is a regulator of HIF-1α. LPS stimulation leads to an upregulation of the enzymatically inactive form of PKM2, which can bind to and promote HIF-1α transcriptional activity [34]. Another key transcription factor in macrophages is NRF2 (nuclear factor-erythroid factor 2-related factor 2), which acts to upregulate antioxidant and suppress inflammatory genes [35]. NRF2 is known to be activated by ROS/RNS-mediated oxidative stress and recently a role for itaconate has also been proposed. Derivatives of itaconate were shown to induce NRF2, through modification of the NRF2 inhibitor KEAP1 (Kelch-like ECH-associated protein 1), or via its direct conjugation to the antioxidant glutathione, leading to increased oxidative stress [36,37].
Metabolites can additionally influence transcription by acting as substrates, inhibitors, or activators of epigenetic modifications of histones or DNA. Alterations in histone acetylation and methylation, are key to regulating gene expression in response to macrophage stimulation [38]. Recent work has shown that the production of cytosolic acetyl-CoA, the substrate for histone acetylation, supports de novo histone acetylation at the promoters of IL6 and IL1β and their increased transcription [9]. Another investigation showed that in response to LPS, pentose phosphate pathway (PPP) and one-carbon metabolism contribute to the production of methylation substrate, SAM (S-adenosyl methionine) which drives an increase in H3K36trimethylation required for IL1β transcription [39]. Histone methylation in LPS stimulated macrophages is additionally regulated by a high succinate:α-ketoglutarate (α-KG) ratio which inhibits α-KG-dependent demethylases [7,40]. These examples illustrate that metabolism, via control of transcription factors and the epigenetic landscape, can have important influence over gene expression in macrophages.
Via the regulation of the inflammasome, transcription factors, epigenetic modifications, and more, metabolism has great influence over the functional state of macrophages. Interestingly, several metabolites, such as itaconate or ROS/RNS, have been shown to regulate immune function through multiple mechanisms ([41], Box 1). An important determinant of the exact functional role of a metabolite is its subcellular localization. For instance, itaconate, which is mainly produced in mitochondria but can be exported, can inhibit the TCA cycle enzyme, succinate dehydrogenase, in the mitochondrion [42,43], influence HIF-1α, NRF2, and the inflammasome in the cytosol/nucleus [7,27,28,36,37], and exert a bactericidal function in the extracellular space [19]. Therefore, processes which control metabolite subcellular distribution also have significant impact on immune functions. An important example is the distribution of acetyl-CoA. Acetyl-CoA is largely produced in mitochondria but acts as an important substrate for histone acetylation in the nucleus and for fatty acid and cholesterol synthesis in the cytosol. Export of acetyl-CoA from the mitochondrion requires its conversion to citrate, export of citrate to the cytosol, and subsequent conversion back to acetyl-CoA by ATP citrate lysate (ACLY). During the early response to LPS stimulation, increased mitochondrial production of acetyl-CoA [9], citrate export [44] and ACLY activity [9, 45, 46] promote the production of NO, ROS and prostaglandins [44, 46] and specific inflammatory cytokines [9, 45]. The availability of cytosolic acetyl-CoA can support inflammatory functions via multiple mechanisms [47], including supporting histone acetylation at the promoters of inflammatory genes [9, 45].
Macrophage metabolism is dynamically rewired in coordination with functional transitions
Dynamic functional changes during macrophages’ immune response
When exposed to PAMPs, macrophages rapidly develop a pro-inflammatory phenotype, turning on functions such as phagocytosis and inflammatory cytokine production. Over time, however, they transition to a less inflammatory and more pro-resolution state. Following the resolution of the initial responses, macrophages can additionally develop innate immune memory, characterized by the response to subsequent stimulation being either enhanced (“training”) or dampened (“tolerance”) compared to the initial response [10,48,49] (Box 2).
Box 2: Metabolic regulation of epigenetics in macrophage innate immune memory.
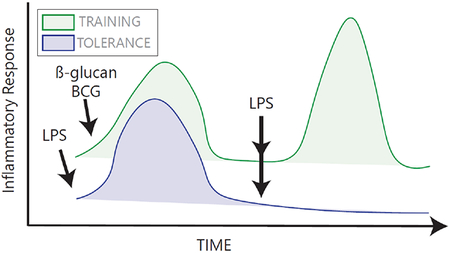
It was long known that the adaptive immune system has the ability to develop memory. It is becoming increasingly clear that innate immune cells can also develop memory in response to certain stimuli [10,48,49]. These memory responses are typically classified as “tolerance”, which is characterized by a general dampening of the response upon re-stimulation, or “training”, which typically refers to an enhanced inflammatory response upon re-stimulation (See Figure). Training does not have antigen specificity: priming with one training stimulus (for example, β-glucan, the tuberculosis vaccine Bacille Calmette-Guérin (BCG), or endogenously derived uric acid or oxidized low density lipoprotein [86]) can lead to enhanced reactivity to a different pathogen. Tolerance, on the other hand, is usually observed in the context of repeated stimulation with LPS. Interestingly, tolerance does not lead to a complete shutdown of all inflammatory mediators, as some “non-tolerizable” genes can be expressed upon LPS restimulation, resulting in gene-specific tolerance [49].
Epigenetics are implicated in both forms of memory. Training leads to a persistent enrichment of promoters with H3K4me3 and enhancers with H3K4me1, allowing for quicker recruitment of transcription factors upon second stimulation [86]. Tolerance is associated with a loss of transcription permissive histone acetylation and methylation marks at specific genes and/or limited ability to regain these features upon re-stimulation [49].
Histone acetylation and methylation can be influenced by changes in cellular metabolism, as metabolites act as substrates, inhibitors, or activators of enzymes responsible for “writing” or “erasing” these epigenetic modifications [87,88]. As such, changes in metabolism upon initial stimulation can regulate the adoption of these memory epigenetic states [86,89]. Training of monocytes with β-glucan leads to high rates of glycolysis and buildup of the TCA cycle metabolite fumarate. Fumarate can inhibit histone demethylases and thus contributes to the methylation state required for training [90]. Tolerance has been linked to α-KG production during LPS stimulation, which can promote α-KG dependent histone demethylase activity [41]; to high itaconate production, which has numerous anti-inflammatory effects [41,70]; and to the impairment of the TCA cycle, which in part controls the availability of acetyl-CoA for histone acetylation upon restimulation [9]. Although we are learning more about innate immune memory, there is still much to discover about how metabolism may influence the duration and phenotypes of memory states, and how to target metabolism to influence these processes in the context of disease. For a broader discussion on this topic, see excellent recent reviews [8,48,49,86,89].
These dynamic functional transitions are coordinated by a sequence of signaling and transcriptional events [10,50]. The timing of these events is partially determined by the rates of regulatory mechanisms. For example, post-translational activation of proteins and transcription of genes with a primed chromatin structure can happen more quickly than processes which require de novo protein synthesis or remodeling of chromatin [50]. Another key determinant of the temporal order is the dependence of one mechanism on the activation or suppression of a preceding event. An example is the production of the anti-inflammatory cytokine IL10, which occurs during the later response to LPS stimulation. IL10 production is dependent on type-1 interferon autocrine signaling. Due to the time required to synthesize and release interferons, IL10 expression peaks several hours after inflammatory cytokine expression. This ensures delayed, but eventual, IL10 production, which acts to suppress inflammatory functions and promote resolution [51].
Dynamic metabolic changes during macrophages’ immune response
While many investigations have illustrated a temporal structure to macrophage signaling and functional transitions, most research on macrophage metabolism thus far has focused on single timepoints post stimulation. Given that many molecular mechanisms, as reviewed above, tightly link the metabolic state of macrophages to their functions, it is logical that metabolic rewiring is also dynamic. Indeed, studies using high-throughput techniques to characterize transcriptomic or proteomic programming across time indicate that metabolic enzymes are among the many dynamically altered during the immune response [52-57]. Recent investigations, more specifically focused on metabolism, have confirmed that the metabolome and lipidome also change in a time-dependent manner during the immune response [7,11,16,58] with substantial and functionally relevant metabolic changes occurring as soon as minutes, to as late as days, after stimulation.
Earlier lipidomic work showed that macrophages stimulated with Kdo2-Lipid A (KLA, an active component of LPS) display dynamic changes in both lipid species and expression of lipid synthesis enzymes across a 24h time-course. Very early timepoints were characterized by increases in pro-inflammatory eicosanoid synthesis, which was followed by later changes in sphingolipid, glycerophospholipid, and sterol synthesis [58]. More recent analysis of this data as well as independent lipidomic experiments reveal an accumulation of polyunsaturated fatty acids and specific pro-resolving eicosanoid species during the later response to LPS and KLA stimulation [11,59-61]. In this way, the dynamic changes in lipid profile act, in part, to coordinate a switch in production of pro-inflammatory to pro-resolving lipid species over time.
The metabolism of polar metabolites also changes dynamically in response to PAMPs. Particularly, several recent studies have revealed substantial remodeling of the TCA cycle at multiple steps, which is a key feature associated with macrophage activation [5]. Together, an interesting timeline has emerged. Very early in response to LPS stimulation, oxidative metabolism via the TCA cycle is enhanced. This is supported by an early increase in glycolysis which supplies carbon to the TCA cycle via pyruvate dehydrogenase (PDH) [9,45,62,63]. However, after several hours, TCA cycle flux starts to shut down at multiple points. One of the earliest blockages occurs at isocitrate dehydrogenase (IDH), which converts isocitrate to α-KG [64]. IDH inhibition contributes to a buildup of the upstream metabolite citrate, and together with the transcriptional upregulation of IRG1 (which converts cis-aconitate to itaconate) leads to accumulation of itaconate [64,65]. As revealed by recent studies, itaconate can competitively inhibit the TCA cycle enzyme succinate dehydrogenase (SDH), leading to concurrent succinate accumulation as well [42,43]. The large reduction in flux through these parts of the TCA cycle contribute to an intermediate state, characterized by high levels of the immunomodulatory metabolites, itaconate, succinate and citrate [7]. As time progresses (over a couple of days), however, the levels of these metabolites subside. This is largely due to simultaneous blockages in fluxes through two enzymes in the α-ketoacid dehydrogenase family: PDH and oxoglutarate dehydrogenase (OGDH) [7,66]. Their inhibition causes their products acetyl-CoA and succinyl-CoA to deplete, and greatly restricts carbon flux to citrate, itaconate and succinate. An impairment in the flux through aconitase, which likely occurs at a similar time, also can contribute to the reduction in itaconate levels at this time [66]. In conjunction with these changes, activity of the electron transport chain (ETC) proteins is impaired [9,66-69]. This later metabolic state is therefore characterized by lower levels of immunomodulatory metabolites and impaired oxidative metabolism. Some of these metabolic alterations can last for an extended period but are eventually reversible. For example, upon acute stimulation (2h) with LPS+IFN-γ, impaired PDH and OGDH activity is observed as late as 72h post stimulation but starts recover after 96h [7]. This sequence of changes in TCA cycle enzymes’ activities leads to waves of dynamic alterations in the abundance of TCA cycle metabolites and their derivatives (Fig 3).
Figure 3. LPS induced rewiring in central carbon metabolism leads to a dynamic alteration in immunoregulatory metabolites.

During the early response (“Early”) to LPS (± IFNγ) stimulation, increased glycolysis and oxidative respiration, sustained flux through pyruvate dehydrogenase (PDH), and activation of ATP citrate lyase (ACLY) supports production of cytosolic acetyl-CoA. After some time (“Mid” stage), when reactive nitrogen species (RNS) start to accumulate, isocitrate dehydrogenase (IDH) is inhibited by nitrosation and RNS-independent transcriptional suppression. This promotes accumulation of the upstream metabolite citrate. At a similar time, increased immune responsive gene 1 (IRG1) expression causes an accumulation of itaconate, which inhibits succinate dehydrogenase (SDH), leading to a concurrent increase in succinate. This “Mid” stage is therefore characterized by accumulation of the immunomodulatory metabolites, itaconate, succinate, and citrate and corresponds with high levels of HIF-1α, TNF-α and IL6. As time continues (“Late” stage), RNS and ROS accumulate, and oxidative metabolism continues to shut down. This is driven by blockages in flux through PDH, oxoglutarate dehydrogenase (OGDH), and aconitase. Aconitase is inhibited by RNS disruption of its Fe-S cluster. PDH inhibition is mediated by multiple mechanisms targeting each of its subunits. OGDH inhibition is due to loss of catalytic lipoylation of its E2 subunit. All this leads to a decrease in itaconate, succinate, citrate, acetyl-CoA, and succinyl-CoA, which correlates with a decrease in HIF-1α, TNF-α and IL6. Inhibition of the electron transport chain also contributes to the sustained low respiration rate during the late response. The figure shows a timeline synthesized from multiple studies in recent literature, to the best of our knowledge. As there are variations among these studies, comparing exact timing across different studies can be challenging.
Functional relevance of the dynamics in macrophage metabolism
The field is beginning to uncover some of the functional implications of the dynamic changes in macrophage cellular metabolism. Although the functional relevance of the expansive LPS/KLA-induced lipidome remodeling requires further investigation, the observed switch in the production of bioactive lipid species over time likely plays an important role in the functional transition towards a pro-resolution phenotype. Recent work reported that knocking out the transcriptional regulator, SREBP1 (sterol regulatory element-binding protein 1), which is responsible for the accumulation of anti-inflammatory unsaturated fatty acids during the late response, impaired inflammation resolution. This impaired ability to fully suppress inflammatory mediators, such as IL1α and IL6, during later time-points could be rescued in vitro and in vivo by supplementing with the polyunsaturated fatty acids DHA or EPA (docosahexaenoic or eicosapentaenoic acid) [11].
The dynamics observed in the TCA cycle also have important functional implications, particularly because, as reviewed above, many TCA cycle intermediates or compounds derived from TCA cycle, including succinate, itaconate, α-KG, acetyl-CoA, and mitochondrial ROS, have been shown to have major immunoregulatory functions [5]. One metabolite in the spotlight over the last five years is itaconate [41]. Over the course of response to LPS and the cytokine interferon-γ, the rise-and-fall in itaconate level correlates with the changing production profile of various cytokines [7]. The accumulation of itaconate in the middle of response time course is likely important for the transition to a more immunosuppressive phenotype, as multiple works, as discussed above, have identified itaconate’s role in promoting anti-inflammatory functions and in the establishment of tolerance [28,36,70]. Another example is the impact of dynamic changes in acetyl-CoA availability on histone acetylation. Early increases in glycolysis and intact oxidative metabolism support production of acetyl-CoA, which promotes de novo histone acetylation and transcription of inflammatory cytokines [9]. However, as time progresses the TCA cycle begins to shut down, restricting acetyl-CoA production [7,9]. This limits the ability to induce expression of IL6 and IL1β upon restimulation with LPS by limiting histone acetylation at their promoters [9]. In this way an impaired TCA cycle can contribute to immune tolerance. Multiple other studies have also indicated that the suppression of oxidative metabolism can be persistent and contribute to tolerance or an impaired ability to repolarize to an anti-inflammatory state [67,71] (Box 2).
Dynamics in other metabolic pathways are also likely to contribute to the macrophage functional transitions. For example, glycolytically derived lactate accumulation, which is high during the later response to PAMP exposure, has been proposed to contribute to the transition to an anti-inflammatory phenotype, by increasing a newly identified histone modification, lactylation, at anti-inflammatory genes such as Arg-1 and contributing to their increased expression [72].
Mechanisms controlling the temporal structure of macrophage metabolic rewiring
Like the functional transitions, the sequence of metabolic alterations in macrophages throughout an immune response is determined by the sequential hierarchy among the regulatory events and the different rates of action of specific regulatory mechanisms. Small molecule driven regulation such as competitive inhibition and allosteric regulation of enzymes can take effect immediately, transcriptional regulation can take hours and have effects which persist for even longer, and the rate and persistence of post-translational modification-based regulation can vary greatly depending on the specific type of modification. Therefore, the same signal or perturbation can influence different metabolic activities on different timescales.
This is exemplified by the orderly remodeling of the TCA cycle. RNS, which macrophages rapidly produce in large amounts upon PAMP stimulation, contribute to the inhibition of multiple TCA cycle enzymes at different times [66,68,73] (Figure 3). The inhibition of IDH, which occurs relatively early and persists over time, is mediated by rapid RNS-dependent nitrosylation, as well as RNS-independent transcriptional down regulation, which likely contributes to its inhibition at later time points [64,68]. Loss of flux through aconitase is caused by RNS disrupting its [Fe-S] cluster [66]. The inhibition of PDH, which occurs gradually over time, is mediated by the combination of multiple mechanisms including; inhibitory phosphorylation of its E1 subunit, loss of the active lipoic group on its E2 subunit, and the nitrosylation of its E3 subunit [7,66]. RNS contribute to multiple of these mechanisms. Loss of the catalytically active lipoic group on the E2 subunit is reported to be caused by RNS-mediated modifications of the thiols on the lipoic arm [74], as well as RNS-mediated inhibition of the [Fe-S] cluster enzymes required for the synthesis of lipoic acid [75]. RNS also cause the inhibitory nitrosylation of the E3 subunit [66]. The RNS mediated thiol modifications of E2 subunit’s lipoic arm and E3 subunit’s cysteine residue can occur more rapidly and act as the major mechanism driving the initial inhibition of PDH and OGDH. The inhibition of lipoic acid synthesis requires the much slower process of PDH and OGDH protein turnover, and thus likely contributes more to inhibition of these enzymes at later timepoints. In addition to inhibition of these TCA cycle enzymes, RNS also contributes to loss of ETC activity and protein levels [66,68,73].
In addition to the inherent rate of different regulatory mechanisms, the timing of these metabolic alterations would be further impacted by the localization of RNS to the specific target enzyme and the concentration of RNS required for the specific mechanism to take effect. iNOS expression and RNS production starts relatively quickly after LPS and interferon-γ stimulation and continues to accumulate over time [7]. This gradual accumulation contributes to the sequential shut down of oxidative metabolism starting with IDH, followed by PDH and OGDH. The specific mechanism by which RNS influence these enzymes can also determine the persistence and reversibility of their inhibition. For example, cysteine nitrosylation is more reversible, while a decrease in protein abundance or loss of PDH lipoylation can be more stable [7,67]. These tight connections between NO production and TCA cycle remodeling demonstrate an example of how early metabolic alterations (shifted arginine metabolism) can lead to important subsequent changes in other metabolic pathways (TCA cycle). The metabolic network is highly interconnected by numerous other regulatory mechanisms, which also likely influence the temporal structure of the metabolic response to stimulation, as discussed in Box 3.
Box 3. Connections between glycolysis and oxidative metabolism in macrophages.
An increasing number of studies have identified a network of regulatory interactions among metabolic pathways known to be important for macrophage functions. For instance, many mechanisms have been identified which control communication between the mitochondrial metabolism and glycolysis in macrophages (see Figure). Glycolysis provides substrate for mitochondrial metabolism. HIF-1α, whose targets include numerous glycolytic enzymes, is stabilized by the TCA cycle derived metabolites succinate, itaconate and mtROS [7,32,33]. Itaconate has also been shown able to impair glycolytic flux directly through post-translational modification of multiple glycolytic enzymes [91]. Recent work showed that malonyl-CoA, a metabolite produced from acetyl-CoA, accumulates in response to LPS stimulation and is capable of post-translationally modifying the glycolytic enzyme, GAPDH. Malonylation not only increases the glycolytic activity of GAPDH but also inhibits GAPDH’s role as an inhibitor of TNF-α mRNA translation [92]. These few examples illustrate the interconnected nature of metabolic pathways. Such regulatory relationships likely contribute to the orderly time-dependent changes in different parts of the metabolic network in macrophages during the immune response.
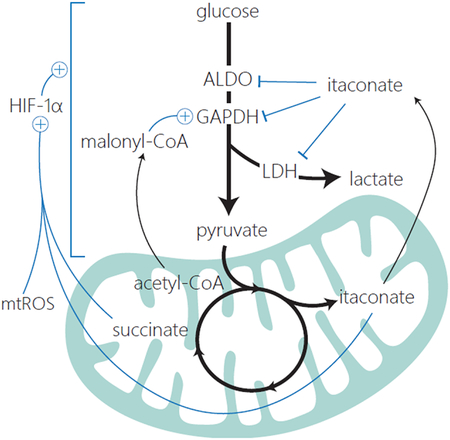
In addition to the regulatory interactions connecting different metabolic pathways, the interplay between signaling and metabolism can also control the temporal sequence of metabolic events. An example is the recent findings that autocrine signaling by IL10, which is released during the later response to PAMP stimulation, suppresses glycolysis and RNS production, and promotes oxidative metabolism [76,77].
The regulatory connections between metabolism and signaling are bidirectional, and thus can form complex regulatory loops. An example is the dynamic metabolic regulation of the transcription factor HIF-1α. HIF-1α is stabilized by succinate and itaconate and the transient accumulation of these metabolites at the intermediate stage of the inflammatory response causes HIF-1α levels to increase [7,33]. Interestingly, this HIF-1α stabilization promotes transcription of pyruvate dehydrogenase kinase (PDK), which phosphorylates PDH’s E1 subunit and contributes to its inhibition. PDH inhibition then limits flux into succinate and itaconate, causing their eventual subsidence during the later stage of response. The feedback loop continues as the decrease in itaconate and succinate de-inhibit HIF-1α degradation, leading to HIF-1α’s eventual reduction [7]. As discussed above, a variety of metabolites can influence numerous other transcription factors, signaling pathways, and epigenetic mechanisms, which likely lead to similar feedforward and feedback loops that shape the temporal structure of metabolic and functional transitions.
Concluding Remarks and Future Perspectives
A body of work, developed over the past decade, has clearly established metabolism as an important regulator of macrophage function. With the help of various cutting-edge technologies, the field has revealed detailed mechanisms underlying this metabolism-function connection. While macrophage metabolism is often studied at a single snapshot post-stimulation, recent work has highlighted that metabolic rewiring in macrophages is quite dynamic and has underscored the need to consider time as a key parameter when studying macrophage metabolism. The consideration of time will bring important new insights in a few ways. First, systematic characterization of the metabolic and functional changes across time provides rich information on their temporal coordination, revealing potential causal relationships between metabolism and function. Second, investigation of time dependent metabolic changes and integration with parallel transcriptomic and proteomic studies can uncover novel mechanisms regulating macrophage metabolism, particularly within a specific temporal context. For example, recent work suggested that the persistence of a single metabolic feature, such as enhanced glycolysis, can be supported by different regulatory mechanisms during the very early (Myc) and later (HIF-1α) response to stimulation [78]. Third, considering time can potentially lead to reconciliation of seemingly conflicting observations, and ultimately lead to a more holistic understanding of the involvement of a metabolic process over the course of the immune response. For example, NAPDH production can play dual time-dependent roles in RNS/ROS metabolism: Early after stimulation, NADPH is required by NOX and iNOS to drive the production of ROS and RNS for pathogen killing; however, later in the response, NADPH may play a more important role in controlling ROS/RNS and repairing oxidative damage via antioxidant systems [79]. Finally, the dynamics of macrophage metabolism has great relevance in health and disease. The timing of metabolic rewiring plays a part in the onset and resolution of inflammation and can impact the development of innate immune memory. Modulating these critical functional transitions with metabolic interventions has potential applications in a broad range of pathologies.
The appreciation for the dynamic nature of macrophage metabolism is relatively new, and there is still a lot to learn about the macrophage immune response, as outlined in “Outstanding Questions”.
Outstanding questions:
Only a few studies have investigated changes in cellular metabolism across the full time-course following stimulation, and those that have, have mainly focused on specific pathways and the response to LPS. What other pathways are dynamically rewired? How does macrophage metabolism change over time in response to diverse stimuli?
What metabolic changes are reversible, and which are persistent? The development of innate immune memory can be mediated by epigenetic or metabolic features. Does the persistence/reversibility of the metabolic alterations influence the duration and phenotype of the memory state?
Macrophages play an important role in modifying the local milieu. How does the macrophage response influence the dynamics of systemic metabolism and inflammation? Vice versa, macrophage function and metabolism can be substantially influenced by environmental factors. How does the microenvironment during the immune response influence the temporal structure of metabolic changes?
In a population of macrophages, particularly under complex physiological conditions, to what extent is the dynamic metabolic response synchronized and what may control any temporal heterogeneity?
As investigation into the temporal structure of metabolic rewiring in macrophages is relatively young, the therapeutic potential of targeting the metabolic regulators of functional transitions requires further study. Can metabolism or metabolic regulators be targeted to modulate the dynamics of the immune response? Does timing of these interventions relative to the metabolic or functional state of the macrophages influence their efficacy?
Another important factor closely connected with time is location. Here we discussed how subcellular localization of key metabolites influences the dynamics and functional roles of metabolic remodeling. While this review focuses on cellular metabolism of macrophages, the concept of time-dependent metabolic rewiring can extend to systemic metabolism. Emerging work has revealed that the immune response is associated with re-allocation of nutrients and rewiring of metabolism across tissues. Signals (such as cytokines, hormones, signaling metabolites) released by macrophages and other immune “sensor” cells, can regulate these systemic metabolic changes [80-83]. The dynamics of systemic metabolic changes during infection or inflammation, similar to cellular metabolic rewiring, are likely influenced by the timing of the release, the spatial distribution, and the mechanism of action of these signals. An important future direction is to gain a holistic temporal-spatial-specific understanding of the metabolic rewiring during immune response at both the cellular and organismal level.
Highlights:
Growing research shows that metabolic rewiring plays a critical role in the immune response. Metabolism supports and regulates macrophage functions by producing bioactive and signaling molecules, communicating to the inflammasome, and regulating transcriptional and epigenetic remodeling.
Over the course of response to stimulation, macrophages transition through a sequence of functional states, which coordinate the timely adoption and resolution of the inflammatory response. It is becoming clear that during this process, cellular metabolism also undergoes coordinated waves of time-dependent changes.
Multiple mechanisms control the temporal structure of metabolic reprogramming during the immune response. Regulatory interactions among metabolic and signaling pathways connect earlier events to subsequent changes, driving a structured sequence of metabolic and functional alterations.
Acknowledgement
We acknowledge the support from the following funding sources: R56AI158958 (J.F.), NRSA Individual Predoctoral Fellowship F31AI152280 (G.L.S.).
Glossary
- Autocrine signaling:
A type of cell signaling in which a signal released from a cell, binds to a receptor on that same cell, in a form of self-regulation.
- Eicosanoids
A large family of signaling lipids formed by oxidation of arachidonic acid or other polyunsaturated fatty acids by cyclooxygenase, lipoxygenase and cytochrome P450 enzymes, or via non-enzymatic mechanisms. Eicosanoids, depending on the species and cell type of action, can have a wide range of physiological effects, acting to promote both pro-inflammatory and pro-resolution functions.
- Epigenetic modifications
Typically refers to covalent modifications of DNA and histones, which can regulate gene expression by influencing gene accessibility and the specific interactions with various chromatin readers. DNA methylation generally acts to suppress expression, histone acetylation generally leads to a more open and active chromatin, and histone methylation can both promote and repress transcription depending on the specific site and methylation state. The addition or removal of epigenetic modifications is influenced by the level of many key metabolites closely involved in these reactions, including acetyl-CoA (acetylation substrate), NAD+ (deacetylation substrate), SAM (methylation substrate), αKG (demethylation substrate), succinate (demethylation inhibitor), and many others.
- Gasdermin D
A protein, which upon N-terminal processing by the inflammasome forms pores on the plasma membrane. Pores formed by gasdermin are important for the excretion of IL-1 cytokines and also promote a lytic form of cell death called pyroptosis.
- Inflammasomes
Protein complexes that act as innate immune sensors. Once assembled and activated, inflammasomes act to process pro-IL1β and pro-IL18 as well as gasdermin D to their mature active forms. There are multiple types of inflammasome, such as the NLR family and the AIM2 inflammasome, which are activated by a variety of host and pathogen derived signals.
- LPS (Lipopolysaccharide)
a component of the membrane of Gram-negative bacteria which acts as a pro-inflammatory stimulus by interacting with the toll-like receptor 4 (TLR4) on macrophages.
- Mitochondrial α-ketoacid dehydrogenase complex family
A family of structurally related multi-subunit mitochondrial enzyme complexes, including pyruvate dehydrogenase (PDH), oxoglutarate dehydrogenase (OGDH), and branched chain keto-acid dehydrogenase (BCKDH). They use similar catalytic mechanisms involving coupled reactions with 3 subunits. The E1 subunit decarboxylates the substrate and transfers the acyl groups to thiamin pyrophosphate (TPP). Using a prosthetic lipoic arm covalently attached to a lysine residue, the E2 subunit transfers the acyl groups to coenzyme A (CoA) to produce corresponding CoA-compounds (e.g. acetyl-CoA for PDH, succinyl-CoA for OGDH). The E3 units re-oxidize the reduced lipoic arm of E2 subunit, coupled to the production of NADH.
- Nuclear factor E2-related factor 2 (NRF2)
A transcription factor which plays a central role in suppressing pro-inflammatory cytokine production in macrophages and in the response to cellular oxidative stress by promoting the transcription of antioxidant genes.
- PAMP (Pathogen Associated Molecular Pattern)
Conserved motifs on microbes which are recognized by various receptors on immune cells leading to their activation.
- Purinergic receptors
A family of membrane receptors that sense signaling by nucleotides and nucleosides. They include P1 receptors, which mainly sense adenosine and promote anti-inflammatory phenotypes, and P2 receptors, which can sense ATP and other nucleotides, and mainly promote pro-inflammatory phenotypes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murray PJ (2016) Macrophage Polarization. Annu. Rev. Physiol 79, 541–566 [DOI] [PubMed] [Google Scholar]
- 2.He W et al. (2021) Complexity of macrophage metabolism in infection. Curr. Opin. Biotechnol 68, 231–239 [DOI] [PubMed] [Google Scholar]
- 3.Langston PK et al. (2017) Metabolism Supports Macrophage Activation. Front. Immunol 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakker NT and Pearce EJ (2020) Cell-intrinsic metabolic regulation of mononuclear phagocyte activation: Findings from the tip of the iceberg. Immunol. Rev 295, 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan DG and O’Neill LAJ (2020) Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol 38, 1–25 [DOI] [PubMed] [Google Scholar]
- 6.Wang Y et al. (2021) Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem 297, 100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seim GL et al. (2019) Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab 1, 731–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luan H and Horng T (2021) Dynamic changes in macrophage metabolism modulate induction and suppression of Type I inflammatory responses. Curr. Opin. Immunol 73, 9–15 [DOI] [PubMed] [Google Scholar]
- 9.Langston PK et al. (2019) Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol 20, 1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivashkiv LB (2011) Inflammatory signaling in macrophages: Transitions from acute to tolerant and alternative activation states. Eur. J. Immunol 41, 2477–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oishi Y et al. (2017) SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab. 25, 412–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herb M and Schramm M (2021) Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 10, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rath M et al. (2014) Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol 5, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munder M et al. (1998) Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J. Immunol 160, 5347–5354 [PubMed] [Google Scholar]
- 15.Dennis EA and Norris PC (2015) Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol 15, 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castoldi A et al. (2020) Triacylglycerol synthesis enhances macrophage inflammatory function. Nat. Commun 11, 4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cekic C and Linden J (2016) Purinergic regulation of the immune system. Nat. Rev. Immunol 16, 177–192 [DOI] [PubMed] [Google Scholar]
- 18.Zasłona Z and O’Neill LAJ (2020) Cytokine-like Roles for Metabolites in Immunity. Mol. Cell 78, 814–823 [DOI] [PubMed] [Google Scholar]
- 19.McFadden BA et al. (1971) Mechanism of action of isocitrate lyase from Pseudomonas indigofera. Biochemistry 10, 1384–1390 [DOI] [PubMed] [Google Scholar]
- 20.Krzak G et al. (2020) Succinate Receptor 1: An Emerging Regulator of Myeloid Cell Function in Inflammation. Trends Immunol. 42, 45–58 [DOI] [PubMed] [Google Scholar]
- 21.Munn DH and Mellor AL (2013) Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 34, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swanson KV et al. (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Próchnicki T and Latz E (2017) Inflammasomes on the crossroads of innate immune recognition and metabolic control. 26, 71–93 [DOI] [PubMed] [Google Scholar]
- 24.Zhong Z et al. (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez-Lopez E et al. (2019) Choline Uptake and Metabolism Modulate Macrophage IL-1β and IL-18 Production. Cell Metab. 29, 1350–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang EV et al. (2017) Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 171, 1057–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hooftman A et al. (2020) The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab. 32, 468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bambouskova M et al. (2021) Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep. 34, 108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyers AK and Zhu X (2020) The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 9, 1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes MM and O’Neill LAJ (2018) Metabolic regulation of NLRP3. Immunol. Rev 281, 88–98 [DOI] [PubMed] [Google Scholar]
- 31.Knight M and Stanley S (2019) HIF-1α as a central mediator of cellular resistance to intracellular pathogens. Curr. Opin. Immunol 60, 111–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills EL et al. (2016) Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frezza C et al. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palsson-McDermott EM et al. (2015) Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 21, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed SMU et al. (2017) Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta - Mol. Basis Dis 1863, 585–597 [DOI] [PubMed] [Google Scholar]
- 36.Mills E et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nat. Publ. Gr 556, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akira S et al. (2018) Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 556, 501–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivashkiv LB (2013) Epigenetic regulation of macrophage polarization and function. Trends Immunol. 34, 216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu W et al. (2019) One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 75, 1147–1160 [DOI] [PubMed] [Google Scholar]
- 40.Liu PS et al. (2017) α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 18, 985–994 [DOI] [PubMed] [Google Scholar]
- 41.O’Neill LAJ and Artyomov MN (2019) Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol 19, 273–281 [DOI] [PubMed] [Google Scholar]
- 42.Cordes T et al. (2016) Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem 291, 14274–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lampropoulou V et al. (2016) Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 24, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Infantino V et al. (2011) The mitochondrial citrate carrier: a new player in inflammation. Biochem. J 438, 433–436 [DOI] [PubMed] [Google Scholar]
- 45.Lauterbach MA et al. (2019) Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 51, 997–1011 [DOI] [PubMed] [Google Scholar]
- 46.Infantino V et al. (2013) ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Bioph. Res. Co 440, 105–111 [DOI] [PubMed] [Google Scholar]
- 47.Williams NC and O’Neill LAJ (2018) A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol 9, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riksen NP and Netea MG (2020) Immunometabolic control of trained immunity. Mol. Aspects Med 77, 100897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seeley JJ and Ghosh S (2017) Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol 101, 107–119 [DOI] [PubMed] [Google Scholar]
- 50.Medzhitov R and Horng T (2009) Transcriptional control of the inflammatory response. Nat. Rev. Immunol 9, 692–703 [DOI] [PubMed] [Google Scholar]
- 51.Chang EY et al. (2007) Cutting Edge: Involvement of the Type I IFN Production and Signaling Pathway in Lipopolysaccharide-Induced IL-10 Production. J. Immunol 178, 6705–6709 [DOI] [PubMed] [Google Scholar]
- 52.Li L et al. (2020) Data-Independent Acquisition-Based Quantitative Proteomics Analysis Reveals Dynamic Network Profiles during the Macrophage Inflammatory Response. Proteomics 20, 1900203. [DOI] [PubMed] [Google Scholar]
- 53.Butcher SK et al. (2018) Toll-Like Receptors Drive Specific Patterns of Tolerance and Training on Restimulation of Macrophages. Front. Immunol 9, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mulvey CM et al. (2021) Spatiotemporal proteomic profiling of the pro-inflammatory response to lipopolysaccharide in the THP-1 human leukaemia cell line. Nat. Commun 12, 5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levenson EA et al. (2018) Comparative Transcriptomic Response of Primary and Immortalized Macrophage to Murine Norovirus Infection. J. Immunol 200, 4157–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varga T et al. (2016) Highly Dynamic Transcriptional Signature of Distinct Macrophage Subsets during Sterile Inflammation, Resolution, and Tissue Repair. J. Immunol 196, 4771–4782 [DOI] [PubMed] [Google Scholar]
- 57.Das A et al. (2018) High-resolution mapping and dynamics of the transcriptome, transcription factors, and transcription co-factor networks in classically and alternatively activated macrophages. Front. Immunol 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dennis EA et al. (2010) A Mouse Macrophage Lipidome. J. Biol. Chem 285, 39976–39985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.von Hegedus JH et al. (2020) Toll-like receptor signaling induces a temporal switch towards a resolving lipid profile in monocyte-derived macrophages. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1865, 158740. [DOI] [PubMed] [Google Scholar]
- 60.Norris PC et al. (2014) Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Nat. Immunol 111, 12746–12751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levy BD et al. (2001) Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol 2, 612–619 [DOI] [PubMed] [Google Scholar]
- 62.Meiser J et al. (2016) Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J. Biol 291, 3932–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cameron AM et al. (2019) Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat. Immunol 20, 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jha AK et al. (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 [DOI] [PubMed] [Google Scholar]
- 65.Michelucci A et al. (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci 110, 7820–7825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Palmieri EM et al. (2020) Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun 11, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.den Bossche VJ et al. (2016) Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 17, 684–696 [DOI] [PubMed] [Google Scholar]
- 68.Bailey JD et al. (2019) Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 28, 218–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hard GC (1970) Some biochemical aspects of the immune macrophage. Br. J. Exp. Pathol 51, 97–105 [PMC free article] [PubMed] [Google Scholar]
- 70.Domínguez-Andrés J et al. (2019) The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab. 29, 211–220.e5 [DOI] [PubMed] [Google Scholar]
- 71.Timblin GA et al. (2021) Mitohormesis reprogrammes macrophage metabolism to enforce tolerance. Nat. Metab 3, 618–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang D et al. (2019) Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palmieri EM et al. (2020) Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 10, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seim GL et al. (2022). Modifications of lipoic arm by reactive nitrogen species regulate α-ketoacid dehydrogenases. Biorxiv 2022.January.31. doi: 10.1101/2022.01.31.478543. [DOI] [Google Scholar]
- 75.Tong WH et al. (2018) TLR-activated repression of Fe-S cluster biogenesis drives a metabolic shift and alters histone and tubulin acetylation. Blood Adv. 2, 1146–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baseler WA et al. (2016) Autocrine IL-10 functions as a rheostat for M1 macrophage glycolytic commitment by tuning nitric oxide production. Redox Biol. 10, 12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ip EWK et al. (2017) Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bae S et al. (2021) MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. 35, 109264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muri J and Kopf M (2021) Redox regulation of immunometabolism. Nat. Rev. Immunol 21, 363–381 [DOI] [PubMed] [Google Scholar]
- 80.Luan HH et al. (2019) GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 178, 1231–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ganeshan K et al. (2019) Energetic Trade-Offs and Hypometabolic States Promote Disease Tolerance. Cell 177, 399–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Troha K & Ayres JS (2020) Metabolic Adaptations to Infections at the Organismal Level. Trends Immunol. 41, 113–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ye J & Medzhitov R (2019) Control strategies in systemic metabolism. Nat. Metab 1, 947–957 [DOI] [PubMed] [Google Scholar]
- 84.Garaude J et al. (2016) Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol 17, 1037–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Canton M et al. (2021) Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol 12, 734229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fanucchi S et al. (2020) The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 54, 32–43 [DOI] [PubMed] [Google Scholar]
- 87.Reid MA et al. (2017) The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol 19, 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Su X et al. (2016) Metabolic control of methylation and acetylation. Curr. Opin. Chem. Biol 30, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Britt EC et al. (2020) Metabolic regulation of epigenetic remodeling in immune cells. Curr. Opin. Biotechnol 63, 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arts RJW et al. (2016) Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 24, 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qin W et al. (2020) Chemoproteomic Profiling of Itaconation by Bioorthogonal Probes in Inflammatory Macrophages. J. Am. Chem. Soc 142, 10894–10898 [DOI] [PubMed] [Google Scholar]
- 92.Galván-Peña S et al. (2019) Malonylation of GAPDH is an inflammatory signal in macrophages. Nat. Commun 10, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]