

Abstract
Osteoarthritis (OA) is the most common joint disease; thus, understanding the pathological mechanisms of OA initiation and progression is critical for OA treatment. MicroRNAs (miRNAs) have been shown to be involved in the progression of osteoarthritis, one candidate is microRNA-378 (miR-378), which is highly expressed in the synovium of OA patients during late-stage disease, but its function and the underlying mechanisms of how it contributes to disease progression remain poorly understood. In this study, miR-378 transgenic (TG) mice were used to study the role of miR-378 in OA development. miR-378 TG mice developed spontaneous OA and also exaggerated surgery-induced disease progression. Upon in vitro OA induction, miR-378 expression was upregulated and correlated with elevated inflammation and chondrocyte hypertrophy. Chondrocytes isolated from articular cartilage from miR-378 TG mice showed impaired chondrogenic differentiation. The bone marrow mesenchymal stem cells (BMSCs) collected from miR-378 TG mice also showed repressed chondrogenesis compared with the control group. The autophagy-related protein Atg2a, as well as chondrogenesis regulator Sox6, were identified as downstream targets of miR-378. Ectopic expression of Atg2a and Sox6 rescued miR-378-repressed chondrocyte autophagy and BMSC chondrogenesis, respectively. Anti-miR-378 lentivirus intra-articular injection in an established OA mouse model was shown to ameliorate OA progression, promote articular regeneration, and repress hypertrophy. Atg2a and Sox6 were again confirmed to be the target of miR-378 in vivo. In conclusion, miR-378 amplified OA development via repressing chondrocyte autophagy and by inhibiting BMSCs chondrogenesis, thus indicating miR-378 may be a potential therapeutic target for OA treatments.
Keywords: miR-378, osteoarthritis, chondrocytes, autophagy, mesenchymal stem cells, chondrogenesis
Graphical abstract
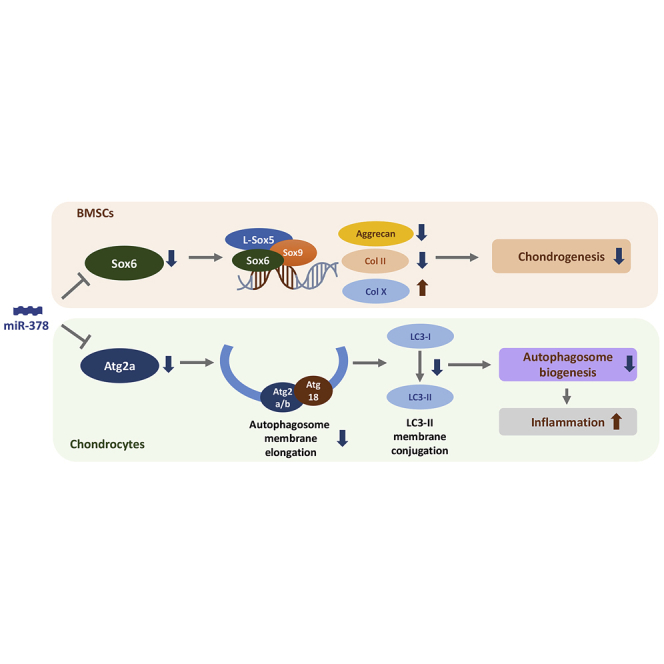
miR-378 overexpression in mice induced spontaneous osteoarthritis (OA) and exaggerated surgery-induced OA. miR-378 repressed OA chondrocyte autophagy and BMSC chondrogenesis via directly downregulating Atg2a and Sox6, respectively. Anti-miR-378 lentivirus treatment ameliorated OA progression, which indicated miR-378 as a potential therapeutic target for OA treatment.
Introduction
Osteoarthritis (OA) is a common chronic joint disease with an incidence rate around 10%–15% of adults worldwide over the age of 60 years.1 OA progression causes joint pain, deformity, and dysfunction that significantly reduces the patient’s quality of life. Its high incidence also places significant economic burden on all societies.2 The pathophysiology of OA mainly includes matrix loss, chondrocyte apoptosis, and inflammation in the cartilage and synovium.3 Unfortunately there are no effective therapies for OA other than pain and inflammation management. Non-steroidal anti-inflammatory drugs (NSAIDs) are the most common choice for OA management owing to their pain relief and anti-inflammation effects, but there is no disease-modifying benefit of these drugs and they have side effects such as gastro-enteropathy.4 Thus, new therapeutic approaches are needed that target the dysfunctional chondrocytes in OA.
MicroRNAs (miRNAs) are a class of small non-coding RNA molecules with the length of between 22 and 25 nucleotides. They have been shown to be gene silencers that bind to the coding or non-coding regions of specific mRNAs and suppress their expression at the post-transcriptional level.5 Several miRNAs were found to be involved in chondrocyte inflammation and homeostasis, including miR-146,6 miR-193,7 miR-335,8 miR-375,9 and miR-411,10 as well as bone marrow mesenchymal stem cell (BMSC) chondrogenesis such as miR-29,11 miR-140,12 and miR-1937 during OA progression. However, the full spectrum of miRNAs and their role in OA development is still emerging.
MicroRNA-378 (miR-378) is a conserved miRNA that was shown to be involved in the inflammation process. miR-378 was found to regulate the nuclear factor (NF)-κB/tumor necrosis factor alpha (TNFα) signaling pathway and was shown to play a significant role in the development of hepatic inflammation and fibrosis.13 Brettfeld et al. found that miR-378 was directly involved in adipose tissue inflammation as well as obesity-related disease.14 Finally, it was established that miR-378 is abundantly expressed in synovial fluid in late-stage OA.15 However, the exact function and mechanism of miR-378 during OA development remains speculative. In our previous studies, we found that miR-378 repressed BMSCs osteogenesis and bone regeneration.16 In this study, we aimed to investigate the role of miR-378 in OA progression and its potential as a target of OA treatment.
Result
miR-378 overexpression induced spontaneous OA development and enhanced surgery-induced OA
To study whether miR-378 overexpression could induce OA spontaneously, both wild-type (WT) and miR-378 transgenic (TG) mice at the ages of 3 and 12 months were terminated and the articular cartilage collected for histological and immunohistochemical analysis. At 3 months, the cartilage structure of miR-378 TG mice showed no big differences compared with WT mice, revealed by safranin O staining of the knee joints, which showed integrated articular cartilage and high glycosaminoglycan (GAG) content. At the age of 12 months, miR-378 TG mice showed cartilage thinning and degradation as well as significant GAG loss compared with WT mice (Figure 1A and S1). miR-378 TG mice also displayed an increased Osteoarthritis Research Society International (OARSI) score compared with that of WT mice at 12 months old (Figure 1B). The percentages of Col X- and Mmp13-positive chondrocytes were significantly higher in the 12-month-old 378 TG mice than the WT mice. Furthermore, at 3 months, the Col X-positive-stained miR-378 TG chondrocytes were highly hypertrophied. At 12 months, both Col X and Mmp13-positive-stained miR-378 TG chondrocytes showed hypertrophy morphology. At 12 months, Col X and Mmp13 were highly expressed in the chondrocytes of miR-378 TG mice and were also found secreted into the extracellular matrices of the cartilage, leading to hypertrophy (Figures 1C–1E and S2). The 12-month-old miR-378 TG mice also exhibited subchondral bone abnormalities compared with the WT mice (Figure S3), further aggravating OA progression. These finding are in agreement with our previous reports showing that miR-378 overexpression impaired bone formation.16 Next, we looked at the effect of anterior cruciate ligament transection (ACLT) combined with destabilization of the medial meniscus (DMM) in the miR-378 TG mice (Figure S4). The safranin O and fast green staining of the joints showed more serious cartilage degeneration and higher OARSI scores in the miR-378 TG mice compared with that of WT mice both at week 3 and week 6 post surgery (Figure 2A and 2B). Immunohistochemistry (IHC) staining showed higher expression level of Mmp13 and lower level of Col II in the miR-378 TG group compared with the WT group (Figures 2C–2E) indicating that miR-378 overexpression exacerbates surgery-induced disease development.
Figure 1.

miR-378 overexpression induced spontaneous OA
(A and B) Safranin O and fast green staining (A) and OARSI scores (B) of knee joint cartilage collected from both WT and miR-378 TG mice at the ages of 3 and 12 months. Scale bars: A1 and A2, A5 and A6, 200 μm; A3 and A4, A7 and A8, 100 μm (n = 8; ∗∗∗p < 0.001). (C–E) IHC staining and semi-quantitative analysis of the percentage of Col X- (C and D) and Mmp13 (C and E)-positive chondrocytes in articular cartilage from two group of mice at the ages of 3 and 12 months. Sections were counterstained with hematoxylin. Scale bar, 200 μm (n = 8; ∗∗p < 0.01).
Figure 2.

miR-378 overexpression exaggerated ACLT-induced OA
(A and B) Safranin O and fast green staining (A) and OARSI scores (B) of knee joint cartilage collected from both WT and miR-378 TG mice at weeks 3 and 6 post ACLT + DMM surgery (n = 8, ∗∗∗p < 0.001). Scale bars: A1 and A2, A5 and A6, 200 μm; A3 and A4, A7 and A8, 100 μm. (C–E) IHC staining and semi-quantitative analysis (integrated optical density[IOD])/μm2)] of the percentage of Mmp13- (C and D) and Col II (C and E)-positive chondrocytes in articular cartilage from two group of mice at weeks 3 and 6 post ACLT + DMM surgery. Sections were counterstained with hematoxylin. Scale bar, 100 μm (n = 8; ∗∗∗p < 0.001).
miR-378 enhances the inflammatory response in IL-1β-induced chondrocytes by repressing autophagy
We next studied whether miRNA-378 overexpression could amplify chondrocyte inflammation. Following interleukin (IL)-1β stimulation, both WT and miR-378 TG isolated chondrocytes showed an increase in miR-378 expression levels (Figure S5). The chondrocytes from miR-378 TG mice showed a significant increase in the expression of inflammatory markers such as iNos and Cox2 compared with WT chondrocytes (Figures 3A and 3B). The mRNA levels of the chondrogenic marker Col II were downregulated, while a marker of hypertrophy, mmp13, was significantly upregulated in the miR-378 TG chondrocytes versus the WT chondrocytes either with or without IL-1β stimulation (Figures 3C and 3D). Western blot results also indicated that miR-378 overexpression downregulated the protein expression of Col II and upregulated iNos, Cox2, and Mmp13 expression (Figures 3E and S6). These results indicated that miRNA-378 overexpression exaggerates inflammation, inhibits chondrogenesis, and promotes the hypertrophy of chondrocytes. Notably, the mRNA and protein expression levels of the autophagy marker Beclin-1 was downregulated in miR-378 TG chondrocytes with or without IL-1β stimulation (Figures 3F, 3G, and S6). The LC3 conversion from LC3-I to LC3-II was also reduced as revealed by western blot, indicating repressed autophagic signaling in the miR-378 chondrocytes under inflammatory conditions (Figures 3G and S6). In addition, the Cyto-ID green dye analysis showed a significant decrease of autophagic flow (decrease of green fluorescence intensity) in miR-378 chondrocytes under IL-1β stimulation (Figures 3H and S7). Collectively, these results demonstrate that miRNA-378 exacerbates chondrocyte inflammation and cartilage degeneration, likely by repressing chondrocyte autophagy during OA development.
Figure 3.

miR-378 exaggerates inflammatory response by repressing autophagy in OA chondrocytes
(A–E) The mRNA expression levels of inflammatory makers iNos (A) and Cox2 (B); chondrogenesis marker Col2a1 (C), chondrocyte hypertrophy marker Mmp13 (D), and autophagy marker Beclin-1 (E) of WT and miR-378 chondrocytes under normal or inflammatory condition were assessed by real-time PCR (n = 6; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (F and G) The protein expression levels of iNos, Cox2, Col II, and Mmp13 (F) as well as Beclin-1, LC3-I, and LC3-II (G) of WT and miR-378 chondrocytes under normal or inflammatory conditions were determined by western blot analysis. (H) Fluorescence images of Cyto-ID dye-stained WT and miR-378 chondrocytes under normal and inflammation conditions. Scale bar, 100 μm (n = 6).
miR-378 represses BMSC chondrogenesis
We also investigated whether miR-378 could regulate the chondrogenesis of BMSCs. BMSCs isolated from the femurs of both WT and miR-378 TG mice were micromass cultured, and chondrogenesis was monitored under defined conditions for 28 days. Safranin O and Alcian blue staining revealed higher contents of GAGs and collagens in the cell micromass of WT BMSCs compared with miR-378 TG. Col II staining also indicated a higher Col II protein expression in WT BMSCs versus miR-378 (Figure 4A and S8). Real-time PCR analysis showed lower mRNA expression levels of chondrogenesis markers, including Sox9 (Figure 4B), Col2a1 (Figure 4C), and Acan (Figure 4D), and higher levels of chondrocyte hypertrophy markers, including Col X (Figure 4E) and Mmp13 (Figure 4F), in the miR-378 TG group versus WT during chondrogenesis. Furthermore, overexpression of miR-378 in BMSCs decreased protein expression of Col II and Acan, while an increase of Mmp13 at day 21 post chondro-induction was observed (Figures 4G and S6).
Figure 4.

miR-378 repressed BMSC chondrogenesis activity
(A) Safranin O, Alcian blue, and IHC staining using COL II antibody of WT and miR-378 BMSC micromass after chondrogenesis induction. Scale bar, 400 μm. (B–F) The mRNA expression levels of chondrogenesis marker Sox9 (B), Col2a1 II (C), Acan (D), chondrocyte hypertrophy marker Col X (E), and Mmp13 (F) in WT and miR-378 BMSCs during chondrogenesis were evaluated by real-time PCR (n = 6; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (G) Protein expression levels of Col II, Acan, and Mmp13 in BMSCs after 21-day chondro-induction were revealed by western blot analysis.
Atg2a is involved in miR-378-regulated chondrocyte autophagy
To understand what downstream genes are mediated/affected by miR-378 and involved in chondrocyte autophagy, two on-line miRNA analysis tools, TargetScan (http://www.targetscan.org/) and ENCORI (http://starbase.sysu.edu.cn/), were employed to predict the target genes that are associated with miR-378 on their 3′ UTR. Candidate genes containing conserved miR-378-binding sites in both human and mouse were analyzed to increase the prediction accuracy. Overlapping candidate genes are listed in Figure S9 and were further evaluated based on their known biological functions as well as the degree of 3′ UTR conservation. Based on full analysis of the data, Atg2a, a gene highly involved in autophagy, is predicted to bind with miR-378-3p at the mRNA level. The conservation of the binding motif of Atg2a with miR-378 among human, mouse, rat, and pig is 100%. Based on these findings, a WT and a mutant (mut) form of mouse Atg2a 3′ UTR luciferase reporter vectors were constructed (Figure 5A) and studied in vitro. miR-378-3p transfection significantly repressed the luciferase activity of Atg2a 3′ UTR, while the luciferase activity of mutated Atg2a 3′ UTR was not affected by transfection with miR-378-3p, suggesting that miR-378-3p binds with Atg2a 3′ UTR at the predicted site (Figure 5B). As expected, Atg2a mRNA and protein expression levels were significantly reduced in miR-378 TG chondrocytes (Figures 5C and S6). The Atg2a overexpression vector was then transfected into chondrocytes isolated from WT and miR-378 TG mice, respectively. The mRNA expression levels of inflammatory markers including iNos and Cox2 were both reduced by the presence of Atg2a in both WT and miR-378 chondrocytes under IL-1β stimulation (Figures 5D and 5E). The chondrogenesis marker Col2a1 was increased, while a marker of hypertrophy, Mmp13, was decreased following Atg2a overexpression in both WT and miR-378 chondrocytes (Figures 5F and 5G). The protein expression levels among the different treatment groups correlated with mRNA expression patterns (Figures 5I and S6). Beclin-1 mRNA/protein levels as well as LC3-I/II shift all increased after Atg2a overexpression (Figures 5H, 5J, and S6). Finally, it was shown that Atg2a overexpression significantly enhanced autophagic puncta in chondrocytes detected by fluorescent probing both in chondrocytes from WT and miR-378 TG mice (Figures 5K and S6).
Figure 5.

Atg2a was involved in miR-378-regulated inflammatory response and autophagy of chondrocytes
(A) Conservation of the miR-378-3p-binding site on Atg2a 3′ UTR (shaded region) among different species. The wild-type (WT) and mutation (mut) forms of Atg2a 3′ UTR luciferase reporter vector are shown. (B) Effects of miR-378-3p on the luciferase activity of pmiRGLO vectors incorporated with Atg2a-wt or Atg2a-mut sequence were measured (n = 6; ∗∗∗p < 0.001). (C) The mRNA and protein expression level of Atg2a in WT and miR-378 chondrocytes were measured respectively (n = 3; ∗p < 0.05). (D–H) The mRNA expression levels of iNos (D), Cox2 (E), Col2a1 (F), Mmp13 (G), and Beclin-1 (H) of WT and miR-378 chondrocytes upon Atg2a overexpression under inflammatory conditions were assessed by real-time PCR (n = 6; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (I and J) The protein expression level of iNos, Cox2, Col II and Mmp13 (I) as well as Beclin-1 and LC3 (J) of WT and miR-378 chondrocytes upon Atg2a overexpression under inflammatory conditions were determined by western blot analysis. (K) Fluorescence images of Cyto-ID dye-stained WT and miR-378 chondrocytes upon Atg2a overexpression under inflammation conditions. Scale bar, 100 μm.
Sox6-mediated miR-378 repressed BMSC chondrogenesis
In silico analysis was performed as described previously, and Sox6, a gene shown to be important in chondrogenesis, was also identified as another target for miR-378-3p. The miR-378 Sox6-binding motif was shown to be 100% conserved in mouse, human, rat, and pig. We then constructed both WT and a mut form of the Sox6 3′ UTR luciferase reporter vector (Figure 6A). miR-378-3p mimics effectively reduced the luciferase activity of Sox6 3′ UTR, while the corresponding mut form showed no suppressive effects by miR-378-3p (Figure 6B). The mRNA and protein expression levels of Sox6 were both downregulated in BMSCs from miR-378 TG versus WT mice (Figures 6C and S6). A Sox6 overexpression vector and the corresponding control vector were then transfected into BMSCs from both WT and miR-378 TG mice, respectively. Real-time PCR analysis demonstrated that the mRNA levels of the chondrogenesis markers Col II and Acan were upregulated (Figures 6D and 6E), while the hypertrophic markers Col X and Mmp13 (Figures 6F and 6G) were decreased in WT and miR-378 BMSCs following chondro-induction. Furthermore, safranin O and Alcian blue staining indicated that Sox6 overexpression could elevate GAGs and collagen formation of BMSCs from WT and miR-378 TG mice. IHC analysis demonstrated that Col II protein expression levels were also activated both in WT and miR-378 BMSCs after Sox6 overexpression (Figures 6H–6J). Overall, Sox6 rescued miR-378-repressed BMSC chondrogenesis.
Figure 6.

Sox6-mediated miR-378 repressed BMSC chondrogenesis
(A) Conservation of the miR-378-3p-binding site on Sox6 3′ UTR (shaded region) among different species. The WT and mut forms of Sox6 3′ UTR luciferase reporter vector are shown. (B) Effects of miR-378-3p on the luciferase activity of pmiRGLO vectors incorporated with Sox6-wt or Sox6-mut sequence were measured (n = 6; ∗∗∗p < 0.001). (C) The mRNA and protein expression level of Sox6 in WT and miR-378 BMSCs were measured (n = 3; ∗p < 0.05). (D–G) The mRNA expression level of Col2a1 (D), Acan (E), Col X (F), and Mmp13 (G) in WT and miR-378 BMSCs upon Sox6 overexpression during chondrogenesis were evaluated by real-time PCR (n = 6; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (H–J) Safranin O (H), Alcian blue(I), and IHC staining using COL II antibody (J) of WT and miR-378 BMSCs micromass upon Sox6 overexpression after chondrogenesis induction. Scale bar, 400 μm.
Lentivirus injection of anti-miR-378 ameliorates OA progression
To investigate whether suppression of miR-378 could ameliorate OA progression, an anti-miR-378-3p-mediated lentivirus was intra-articularly (IA) injected in ACLT + DMM induced chondrocytes. Anti-miR-378-3p or the corresponding control lentivirus was injected weekly for 3 weeks beginning from 7 days after surgery. The successful delivery of lentivirus into chondrocytes after IA injection was confirmed by immunofluorescence of the lentivirus-integrated GFP as well as real-time PCR analysis of miR-378 expression (Figure S10). It was shown that cartilage degeneration in miR-378 TG mice was significantly alleviated after treatment with anti-miR-378-3p lentivirus compared with control virus, as determined by safranin O and fast green staining and OARSI scores (Figure 7A and 7B). Moreover, the protein expression of Col II was increased, while Mmp13 was decreased by miR-378 inhibition (Figures 7C–7E). Finally, upon anti-miR-378 lentivirus application, the protein expression levels of Sox6 and Atg2a were markedly increased, while inflammation markers iNos and Cox2 were decreased in the articular cartilage of miR-378 TG mice that underwent ACLT + DMM surgery (Figures 7C, 7F, 7G, and S11). These results indicate that miR-378 antagonists can ameliorate OA progression in an animal model.
Figure 7.

Anti-miR-378-mediated BMSCs ameliorated ACLT-induced OA
(A and B) Safranin O and fast green staining (A) and OARSI scores (B) of knee joint cartilage collected from miR-378 TG mice received anti-miR-378-3p incorporated or negative control lentivirus injection at week 4 post ACLT + DMM surgery (n = 8; ∗∗p < 0.01). Scale bars, A1 and A3, 200 μm; A3 and A4, 100 μm (A). (C–G) IHC staining (C) and quantitative analysis of the percentage of Col II- (D), Mmp13- (E), Atg2a- (F), and Sox6 (G)-positive chondrocytes in articular cartilage from two groups of mice at week 4 post ACLT + DMM surgery (n = 8; ∗p < 0.05, ∗∗p < 0.01). Sections were counterstained with hematoxylin. Scale bars, 50 μm.
miR-378 expression is elevated in human cartilage from OA patients and negatively correlates with Atg2a and Sox6 expression
To study the expression pattern of miR-378 and its target genes in clinical samples, cartilage from OA patients was collected following total knee replacement surgery. Relatively normal (RN) areas versus abraded (OA) cartilage were carefully dissected, separated, and weighed. Next, total RNA analysis was performed on the tissue comparing normal versus diseased cartilage. RNA levels were normalized using the housekeeping genes miR-103a and YWHAZ. It was found that miR-378 was significantly upregulated in the cartilage designated abraded versus cartilage designated RN (Figure 8A) and correlated with the downregulation of ATG2A and SOX6 in the abraded cartilage compared with the RN cartilage (Figures 8B, 8C, 8D, and 8E). These results may suggest the clinical significance of miR-378 in OA progression, as well as its in vivo suppression of ATG2A and SOX6.
Figure 8.

miR-378 expression was increased in OA cartilage and negatively correlated with Atg2a and Sox6
(A) miR-378 expression in RN and OA cartilages was detected by real-time PCR (n = 18; ∗p < 0.05, ∗∗p < 0.01). (B and C) The mRNA expression levels of ATG2A (D) and SOX6 (E) are negatively corelated with miR-378 expression level in an OA cartilage fragment.
Discussion
In this study, we demonstrated that older miRNA-378 TG mice develop OA spontaneously. miR-378 also exaggerated the OA progression in a surgically induced mouse model of OA. Overexpression of miR-378 inhibited autophagy in OA chondrocytes and reduced BMSC chondrogenesis. Moreover, Atg2a and Sox6 were identified as targets of miR-378, which are important in chondrocyte autophagy and BMSC chondrogenesis. The suppression of these two genes by miR-378 could be rescued by using anti-miR-378 treatment in a mouse model of OA. Furthermore, miR-378 was discovered to be upregulated in clinical OA cartilage samples and its expression level negatively correlated with Atg2a and Sox6. These studies suggest that miR-378 contributes to chondrocyte autophagy and the impairment of BMSC chondrogenesis during OA, bringing up the possibility that miR-378 is a potential therapeutic target for OA treatment.
Several miRNAs have been shown to be involved in OA pathology. miRNAs have been reported to participate in OA-related biological processes, including chondrocyte proliferation, differentiation, inflammation, cartilage degeneration, and osteophyte degradation.17 The present study revealed that miR-378 TG mice spontaneously developed age-related OA, and this was even more severe (amplified) following surgery-induced OA compared with the WT control group. This suggests a significant role for miR-378 in OA development. After IL-1β stimulation, chondrocytes from both WT and miR-378 TG mice displayed elevated miR-378 expression, suggesting that miR-378 is an inflammation-induced miRNA. Other studies have also indicated that miR-378 is elevated upon treatment of inflammatory mediators such as IL-6 and TNFα.18,19 Following miRNA-378 overexpression, the inflammatory factors iNos and Cox2 as well as the hypertrophic marker Mmp13 were all upregulated, while the chondrogenesis marker Col II was downregulated in OA chondrocytes, indicating that miR-378 impairs chondrogenic homeostasis and activates chondrocyte inflammation and hypertrophy. Interestingly, chondrocyte autophagy also decreased with miR-378 overexpression. Autophagy is a highly conserved system of degradation of proteins. When cells are under stress, including external pressure, hypoalimentation, hypoxia, or endoplasmic reticulum stress (ERS), autophagy is activated to transport malfunctioning cytoplasmic macromolecules, membranes, and organelles to lysosomes for degradation and reusing. Autophagy has been shown to have beneficial effects on preventing OA-like changes in cartilage, such as apoptosis and degeneration. Dysfunction of autophagy has been reported to occur often in OA cartilage.20 Inflammatory-mediated induction of human chondrocytes has been shown to significantly increase the expression of mammalian target of rapamycin (mTOR), which is a key negative regulator of autophagy, and mediates the inhibition of autophagy signal transduction.21 In our study, we revealed that the expression of the repressed autophagy marker Beclin-1 reduced LC3 conversion and autophagy flux in miR-378 chondrocytes following inflammatory stimulation. Beclin-1 is a key player in autophagy and is involved in autophagosome synthesis and maturation. LC3 is a stable marker of autophagosomes and it exists in two forms: LC3-I, located in the cytoplasm, and LC3-II, which is membrane bound, converted from LC3-I to initiate autophagosome formation.22 Deregulation of chondrocyte autophagy caused by miRNA signaling was reported to occur in injured articular cartilage during OA. Yang et al. found that miR-411 is able to promote chondrocyte autophagy through repression of HIF-1α and proposed HIF-1α as a therapeutic target for OA treatment.10 Mir-355 was reported to ameliorate chondrocyte inflammation via the induction of autophagy, although the precise mechanism at this time is unknown.8 In agreement with the previous studies, our results reveal that autophagy impaired by miR-378 overexpression in chondrocytes accelerates cartilage destruction in mice under OA-like conditions.
Furthermore, we demonstrated that miR-378 could directly bind to the 3′ UTR of Atg2a, which transcribes a peripheral membrane protein that is required for autophagic vesicle formation. Atg2 can form a heterotrimer with Atg9 and Atg18, which is essential for autophagosome synthesis.23 ATG2 was also reported to associate with WIPI4, an Atg18 homolog that binds and bridges the membrane acting as a tether for autophagy.24 The deficiency of Atg2 homologs Atg2a and Atg2b blocks autophagic flux and causes incomplete autophagosome accumulation. Previous reports indicated that another anabolic miRNA, miR-375, directly associates with Atg2b, effectively repressing its expression, further inhibiting autophagy, and hindering chondrocyte homeostasis.9 Similarly, miR-128a was shown to directly repress Atg12 and block chondrocyte autophagy, which accelerated OA in vivo.25 In these studies, miR-378 was confirmed to be the direct target of Atg2a in OA chondrocytes. Overexpression of Atg2a in miR-378 chondrocytes could rescue the deregulation of autophagy as well as ameliorate chondrocyte inflammation. Our study provides evidence that miRNA-378 alleviates chondrocyte-mediated inflammation at least partially through the de-activation of autophagy.
OA is mainly characterized by cartilage matrix degradation and chondrocyte death. BMSCs possess pluripotency and are capable of differentiating into multiple cell types, including chondrocytes and osteoblasts. OA treatment with BMSCs via IA transplantation has been shown to facilitate cartilage regeneration, likely due to their chondrogenic differentiation capabilities and immune-modulatory properties. Sox genes play crucial roles in the chondrogenic differentiation of mesenchymal stem cells (MSCs). Two Sox family members in particular, Sox5 and Sox6, were found to form a heterotrimer with Sox9 upregulating the expression of chondrogenic genes. Sox5 and Sox6 are required for Sox9 binding to the enhancer region of Acan, Col2a1, and Col11a1 and elevates their expression during chondrogenesis.26 The Sox trimer (Sox trio) helps maintain cartilage permanence through suppressing hypertrophic and osteogenic genes, including Col X and Runx2.27 The total abolishment of Sox9 in mouse embryonic stem cells results in the inhibition of cartilage development,28 while Sox5 and Sox6 double-knockout mice display failure of chondrogenic differentiation and cartilage maintenance.29 Given the important role that Sox genes play during chondrogenesis, their expression levels are tightly regulated for maintaining cartilage hemostasis. OA chondrocytes were found to have reduced levels of Sox6 and Sox9.30
miRNAs were reported to regulate Sox members. Budd et al. found that miR-146 acts as a negative regulator of SOX5 during chondrogenic differentiation, and miR-146 expression was increased in OA chondrocytes.31 Similarly, Sox5 has been verified to be a downstream target of miR-194 in chondrogenesis of human adipose-derived stem cells.32 We also reported that miR-145 could directly target SOX9 and repress chondrogenic differentiation.33 Our current data offer novel molecular insights into how miR-378 regulates Sox6 during chondrogenesis. Duan’s group reported that miR-103 directly regulates Sox6 expression and reduces chondrocyte proliferation and maturation, suggesting an alternative mechanism of how miRNA contributes to OA development.34 Taken together, because miR-378 regulates Sox6, it is a potential target for OA treatment as inhibition of miR-378 would induce chondrogenesis and cartilage regeneration.
In our study, we showed that miR-378 overexpression exaggerated disease progression in a surgically induced mouse model of OA. Previous studies have reported an abundance of miR-378 in the synovial fluid of late-stage OA patients. As discussed before, OA progression may lead to chondrocytes releasing extracellular vehicles (EVs) containing various miRNAs that will be absorbed by surrounding cells. Several miRNAs are differentially expressed in EVs in the synovial fluid and may serve as OA biomarkers. However, the limitation of our current study is that we did not quantify the level of miR-378 in synovial fluid EVs both in WT and miR-378 TG OA mice. Also, we do not know which cell type, chondrocytes, synoviocytes, or other cells, is responsible for miR-378 production. Future studies are needed to identify which cell type is the main producer and receiver for miR-378 during OA.
Finally, these studies suggests that miR-378 may be a potential biomarker and target for OA diagnosis and treatment, respectively. Upregulation of miR-378 in OA cartilage, and the expression of Atg2a and Sox6, negatively correlated with miR-378, indicating the repressive effects of miR-378 in cartilage health. miR-378 lentiviral inhibition ameliorated OA development and progression in vivo, increased anabolic activities, and repressed catabolic activities in cartilage in vitro.
In conclusion, the current study demonstrated that aging miRNA-378 TG mice develop spontaneous OA and disease progression in a surgically induced OA model. Overexpression of miR-378 was found to inhibit autophagy in OA chondrocytes and reduce BMSC chondrogenic differentiation (Figure S13). Interestingly, autophagy was recently discovered to be involved in the regulation of chondrogenesis. Autophagy deficiency may trigger ERS in growth plate chondrocytes and impair chondrogenesis.35 Carbonare et al. also discovered that physical exercise increased the expression of chondrogenic transcription factors SOX9 in circulating MSCs via promoting autophagy in response to oxidative stress.36 Thus, the precise mechanism by which miRNA-378 orchestrates autophagy and chondrogenesis during OA development needs to be further investigated. The potential of developing miRNA-378 inhibitors as novel diagnostics and blockers as therapeutics is also worth exploring.
Materials and methods
miR-378 TG mouse and mouse OA model
All animal experiments were approved by the Ethics Committee of Chinese University of Hong Kong and performed in accordance with the Code of Ethics of the World Medical Association. The miR-378 TG mice were obtained from Prof. Dahai Zhu’s laboratory (Chinese Academy of Medical Sciences).37 The genotyping characterization of miR-378 TG mice was performed as described in our previous study.16 C57BL/6J male mice were used as WT control. The joint articular cartilage was collected and the TG efficiency was confirmed by real-time PCR (Figure S5). The right knee joints of WT and miR-378 TG male mice were harvested at 3 or 12 months old for further analysis. For the experimental OA model, the right knee joints of 3-month-old male WT and miR-378 TG mice were treated with ACLT + DMM surgery following the previously reported procedures.38 Briefly, the mice were anesthetized under standard conditions (with intraperitoneal injection of 0.2% xylazine and 1% ketamine in PBS), and the anterior cruciate plus medial meniscotibial ligaments were transected in the right knee joint. Sham operations were also performed as negative control. Mice were sacrificed 3 or 6 weeks after surgery. The right knee joints were collected and processed for histological and histochemical analyses. For the therapeutic experiment, the anti-miR-378 lentivirus was designed and constructed by GenePharma Company (GenePharma, China). Two groups of 3-month-old miR-378 TG mice were treated with ACLT + DMM surgery. One week after surgery, one group of mice were treated with IA injection of lentivirus-incorporated miR-378-3p inhibitor with the dose of 1 × 108 particles resuspended in 5 μL of PBS, and the other group was treated with corresponding control lentivirus once every week for 3 weeks. Mice were sacrificed 3 weeks after the first lentivirus injection.
Histological analysis
Collected cartilages were fixed with 4% formaldehyde for 24 h and decalcified with 10% EDTA, pH 7.4 at 37°C for 2 weeks. The cartilages were then dehydrated, embedded with paraffin, and sectioned at 5-μm thickness by a rotary microtome (HM 355S; Thermo Fisher Scientific, USA). After deparaffinization and rehydration, safranin O and fast green staining was performed to evaluate the OA status of the cartilage. The OARSI scoring was performed following the OA cartilage pathology assessment system presented by OARSI 2000 and described in Figure S1.39 The IHC staining was performed as described in the previous studies.40 The primary antibodies, including rabbit anti-Col II (1:100, Ab34712, Abcam, USA), rabbit anti-Mmp13 (1:100, Ab39012, Abcam, USA), mouse anti-Sox6 (1:50, 393,314, Santa Cruz, USA), and mouse anti-Atg2a (1:50, 514,207, Santa Cruz, USA) were applied to the sections at 4°C overnight. The sections were then applied with horseradish peroxidase-streptavidin system (K500711, Dako, USA) IHC signal detection. The IHC images were captured by using a light microscope (Leica DM5500, Wetzlar, Germany). The semi-quantitative analysis of positive-stained cell intensity was performed using ImageJ software following the previous protocol.41 Briefly, the region of interest (ROI) was chosen at cartilage sites. The threshold for signal detection was set the same for all images. One unbiased representative image each was selected from five samples in one group. A 600 μm × 400 μm rectangle tool was applied in 100× magnification photos to select five areas randomly. The value of positive cell percentage or the average value of integrated optical density (IOD) divided by a specific area (IOD/μm2) were calculated accordingly.
Cell isolation and culture
Human embryonic kidney 293 (HEK293) cells as well as BMSCs from both WT and miR-378 mice were kept or generated in our laboratory as previously described.16 Primary chondrocytes were isolated from WT and miR-378 TG mice following the previous protocol.38 Briefly, the mice were sacrificed under anesthesia. The mice articular cartilages were collected by surgery and the attached muscle and connective tissue were removed. The cartilages were minced by scissor and washed three times with PBS. Tissues were digested by 0.25% trypsin (Thermo Fisher, USA) for 30 min, PBS washed three times, and further digested with collagenase type II (200 unit/mL) for 10 min. The chondrocytes were collected and cultured in DMEM containing 10% fetal bovine serum (FBS, Gibco, USA), 100 units/mL of penicillin, and 100 mg/mL of streptomycin (Life Technologies, USA) and cultured at 37°C in 5% CO2 atmosphere. The primary chondrocytes were identified by toluidine blue O staining and IHC staining of collagen type II (Figure S12). Chondrocytes at passage 1 to 3 were used in the experiment.
Cloning construction and transfection
Mouse Atg2a and Sox6 3′ UTR sequences were subcloned into pmiRGLO vector. The cDNA sequence with full open reading frame (ORF) of mouse Atg2a and Sox6 were subcloned into pcDNA3 vector. The construction of pcDNA3-Atg2a, and pcDNA3-Sox6 cloning vectors as well as pmiRGLO-Atg2a and pmiRGLO-Sox6 WT and mutated luciferase reporter vectors were performed by Genscript Biotech Company (Nanjing, China). All the cDNA sequences were obtained from the NCBI database (http://www.ncbi.nlm.nih.gov/). Mouse miR-378-3p mimics were purchased from GenePharma Company (Shanghai, China). miRNA mimics and/or recombinant vectors were transfected using transfection reagent Lipofectamine 3000 (Invitrogen, USA) following the manufacturer’s instruction.
BMSC in vitro chondrogenesis induction
To investigate the influence of miR-378 overexpression on BMSC chondrogenesis activity, the BMSC micromass culture method was applied. Briefly, the BMSCs were trypsinized and resuspended in PBS at a concentration of 2 × 105 cells/10 μL. After that, 10 μL of concentrated BMSCs were pipetted down into the center of the 24-well plates to form a hemispherical droplet. After 1 h of condensation, 500 μL of chondrogenic induction medium (Life Technologies, USA) was added into the well to let the medium merge the cell droplets. The cell droplet formed a spherical aggregate at the bottom of the well after 1 day. The chondrogenesis induction medium was replaced every 3 days. The cell micromasses were harvested at days 7, 14, 21, and 28 upon chondro-induction.
Histological and immunostaining
For the chondro-induced micromass-cultured BMSCs, the cell micromass was embedded with Tissue-Tek OCT compound (OCT, Sakura Finetech, USA) and stored at −80°C. OCT blocks were cut into 5-μm sections in the cryostat (Life Technologies, USA) at −20°C. The cryo-sections were stained with safranin O and Alcian blue to visualize the distribution of major constituents of extracellular matrix (ECM), including glycosaminoglycan (GAG) and collagens. IHC staining using rabbit anti-Col II (1:100, Ab34712, Abcam, USA) was performed as described previously.
Autophagy flux detection
Autophagy flux in chondrocytes was assessed using the Cyto-ID Autophagy Detection Kit (Enzo Life Sciences, USA) following the manufacturer’s protocol. The chondrocytes were seeded on the coverslips and grew up to 70% confluency. The slips with attached chondrocytes were washed with PBS and then incubated with Cyto-ID and Hoechst 33258 in the dark for 15 min. The excess dye was removed by PBS washing. The images were taken using a Leica DM5500B fluorescence microscope (Leica, USA).
Luciferase assay
Dual-luciferase assay was performed following the previous protocol.16 HEK293 cells were transfected with pmiRGLO-Atg2a or pmiRGLO-Sox6 WT and site-mutation (mut) recombinant vectors together with miR-378-3p mimics. The renilla luciferase vector was co-transfected as normalization control. The luciferase activities were measured using PerkinElmer Victor X2 2030 multilabel reader (Waltham, USA). The renilla luciferase activity was also measured for normalization.
Human cartilage sample collection
The study was performed under the approval of the Ethics Committee with the patients’ full consent. A total of 18 knee OA patients (n = 18, 11 men and seven women) who underwent total knee replacement surgery were recruited as study subjects. The inner knee cartilage was usually corroded more severely compared with outer knee cartilage and was defined as the osteoarthritic (OA) side. The less abraded fragments from the other side were grouped as the RN side. All cartilage samples were harvested from tibial plateau and preserved in liquid nitrogen until use. To collect tissue RNA, the cartilage samples were put into a mortar, 1 mL of RNAiso (Takara, Japan) was added, and they were ground with a pestle. All the manipulations were performed with the continuous supplement of liquid nitrogen. After homogenization, the cartilage fragment lysates in RNAiso were collected for further analysis.
Real-time PCR
For RNA extraction of chondrocytes or micromass-cultured BMSCs, the cultured cells or cell micromass were homogenized with RNAiso reagent (Takara, Japan). After homogenization, RNA was extracted following the manufacturer’s guidance. The extracted RNA was then quantified and reverse transcribed into cDNA using PrimeScript RT Master Mix (TaKaRa, Japan). Quantitative real-time PCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, USA). The relative gene expression was calculated, normalized, and compared using the 2−ΔΔCT method following the previous protocol.16 The miRNA reverse transcription and real-time PCR was performed with miRCURY LNA miRNA PCR System (Qiagen, German). The relative miRNA expression was normalized to hsa-miR-103a-3p or mmu-miR-103a-3p. The primers used for cloning construction are listed in Table S1.
Western blot
The BMSCs or chondrocytes in tissue culture dish were in situ lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (Sigma-Aldrich, USA) with 1 mM phenylmethylsulfonyl fluoride (PMSF, Roche, USA). The protein samples were prepared, electrophoresed, and electroblotted following the previous studies.42 The primary antibodies applied for signal detection included rabbit anti-iNos (1:3,000, Ab15323, Abcam, Cambridge, UK), goat anti-Cox2 (1:1,000, Sc-1745, Santa Cruz, USA), rabbit anti-Col II (1:3,000, Ab34712, Abcam, USA), rabbit anti-Aggrecan (1:3,000, Ab36861, Abcam, USA), rabbit anti-Mmp13 (1:3,000, Ab39012, Abcam, USA), rabbit anti-Beclin 1 (1:1,000, 3495P, Cell Signaling Technology, USA), rabbit anti-LC3B (1:1,000, 2775, Cell Signaling Technology, USA) and mouse anti-β-actin (1:3,000, Sc-8432, Santa Cruz, USA).
Statistical analysis
All data are presented as mean ± standard deviation or mean ± medians (minimum-maximum) according to graph types applied. The data from clinical studies are presented as before-after plot. The OARSI scoring was presented as scattered dot plot. Experiments were repeated independently at least three times, and representative data are shown. The statistical significance between two groups was calculated by unpaired, two-tailed Student’s t test for parametric data and by Mann-Whitney U test for non-parametric data analysis. The two-paired sample t test was applied for clinical sample analysis. The analysis was performed with GraphPad Prism 8 (GraphPad Software, USA), and p < 0.05 was considered a significant difference.
Acknowledgments
This work was partially supported by grants from the National Natural Science Foundation of China (81772322); University Grants Committee, Research Grants Council of the Hong Kong Special Administrative Region, China (14120118, 14108720, C7030-18G, T13-402/17-N, and AoE/M-402/20); Hong Kong Innovation Technology Commission Funds (PRP/050/19FX); and Hong Kong Medical Research Funds (16170951 and 17180831). This study also received support from the research funds from the Health@InnoHK program launched by Innovation Technology Commission of the Hong Kong SAR, PR China.
Author contributions
G.L., X.H.P., and L.F. designed the study. L.F., Z.Y.M., Y.L., Q.P., X.T.Z., X.M.W., H.T.L., H.X.W., X.L., M.W., and X.L. performed the experiments and acquired the data. L.F., and Z.Y. analyzed and interpreted the data and drafted the manuscript. G.L. and X.H.P. finalized and approved the manuscript for submission.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.03.016.
Contributor Information
Xiaohua Pan, Email: szpxh4141@foxmail.com.
Gang Li, Email: gangli@cuhk.edu.hk.
Supplemental information
References
- 1.Wittenauer R., Smith L., Aden K. Priority Medicines for Europe and the World “A Public Health Approach to Innovation” Update on 2004 background paper, January 28th 2013. World Health Organization; 2013. Background Paper 6.12 Osteoarthritis.https://www.who.int/medicines/areas/priority_medicines/BP6_12Osteo.pdf [Google Scholar]
- 2.Song Y., Zhang J., Xu H., Lin Z., Chang H., Liu W., Kong L. Mesenchymal stem cells in knee osteoarthritis treatment: a systematic review and meta-analysis. J. Orthop. Translat. 2020;24:121–130. doi: 10.1016/j.jot.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen D., Kim D.J., Shen J., Zou Z., O'Keefe R.J. Runx2 plays a central role in Osteoarthritis development. J. Orthop. Translat. 2020;23:132–139. doi: 10.1016/j.jot.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bindu S., Mazumder S., Bandyopadhyay U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem. Pharmacol. 2020;180:114147. doi: 10.1016/j.bcp.2020.114147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shukla G.C., Singh J., Barik S. MicroRNAs: processing, maturation, target recognition and regulatory functions. Mol. Cell Pharmacol. 2011;3:83–92. [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X., Wang C., Zhao J., Xu J., Geng Y., Dai L., Huang Y., Fu S.C., Dai K., Zhang X. miR-146a facilitates osteoarthritis by regulating cartilage homeostasis via targeting Camk2d and Ppp3r2. Cell Death Dis. 2017;8:e2734. doi: 10.1038/cddis.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng F., Li Z., Zhang Z., Yang Z., Kang Y., Zhao X., Long D., Hu S., Gu M., He S., et al. MicroRNA-193b-3p regulates chondrogenesis and chondrocyte metabolism by targeting HDAC3. Theranostics. 2018;8:2862–2883. doi: 10.7150/thno.23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong G., Long H., Ma S., Shunhan Y., Li J., Yao J. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. 2019;226:164–172. doi: 10.1016/j.lfs.2019.03.071. [DOI] [PubMed] [Google Scholar]
- 9.Li H., Li Z., Pi Y., Chen Y., Mei L., Luo Y., Xie J., Mao X. MicroRNA-375 exacerbates knee osteoarthritis through repressing chondrocyte autophagy by targeting ATG2B. Aging (Albany NY) 2020;12:7248–7261. doi: 10.18632/aging.103073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang F., Huang R., Ma H., Zhao X., Wang G. miRNA-411 regulates chondrocyte autophagy in osteoarthritis by targeting hypoxia-inducible factor 1 alpha (HIF-1alpha) Med. Sci. Monit. 2020;26:e921155. doi: 10.12659/MSM.921155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le L.T., Swingler T.E., Crowe N., Vincent T.L., Barter M.J., Donell S.T., Delany A.M., Dalmay T., Young D.A., Clark I.M. The microRNA-29 family in cartilage homeostasis and osteoarthritis. J. Mol. Med. (Berl) 2016;94:583–596. doi: 10.1007/s00109-015-1374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan L., Liang Y., Xu X., Xiao Y., Wang D. Recent progress on the role of miR-140 in cartilage matrix remodelling and its implications for osteoarthritis treatment. Arthritis Res. Ther. 2020;22:194. doi: 10.1186/s13075-020-02290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang T., Hu J., Wang X., Zhao X., Li Z., Niu J., Steer C.J., Zheng G., Song G. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-kappaB-TNFalpha pathway. J. Hepatol. 2019;70:87–96. doi: 10.1016/j.jhep.2018.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brettfeld C., Maver A., Aumuller E., Peterlin B., Haslberger A.G. MicroRNAs responsible for inflammation in obesity. J. Endocrinol. Metab. 2017;7:77–85. [Google Scholar]
- 15.Li Y.H., Tavallaee G., Tokar T., Nakamura A., Sundararajan K., Weston A., Sharma A., Mahomed N.N., Gandhi R., Jurisica I., Kapoor M. Identification of synovial fluid microRNA signature in knee osteoarthritis: differentiating early- and late-stage knee osteoarthritis. Osteoarthritis Cartilage. 2016;24:1577–1586. doi: 10.1016/j.joca.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 16.Feng L., Zhang J.F., Shi L., Yang Z.M., Wu T.Y., Wang H.X., Lin W.P., Lu Y.F., Lo J.H.T., Zhu D.H., Li G. MicroRNA-378 suppressed osteogenesis of MSCs and impaired bone formation via inactivating Wnt/beta-catenin signaling. Mol. Ther. Nucleic Acids. 2020;21:1017–1028. doi: 10.1016/j.omtn.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y.P., Wei X.C., Li P.C., Chen C.W., Wang X.H., Jiao Q., Wang D.M., Wei F.Y., Zhang J.Z., Wei L. The role of miRNAs in cartilage homeostasis. Curr. Genomics. 2015;16:393–404. doi: 10.2174/1389202916666150817203144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H., Song Y., Wu Y., Kumar V., Mahato R.I., Su Q. Activation of dsRNA-dependent protein kinase R by miR-378 sustains metabolic inflammation in hepatic insulin resistance. Diabetes. 2021;70:710–719. doi: 10.2337/db20-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L.L., Shi C.M., Xu G.F., Chen L., Zhu L.L., Zhu L., Guo X.R., Xu M.Y., Ji C.B. TNF-alpha, IL-6, and leptin increase the expression of miR-378, an adipogenesis-related microRNA in human adipocytes. Cell Biochem. Biophys. 2014;70:771–776. doi: 10.1007/s12013-014-9980-x. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki H., Takayama K., Matsushita T., Ishida K., Kubo S., Matsumoto T., Fujita N., Oka S., Kurosaka M., Kuroda R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64:1920–1928. doi: 10.1002/art.34323. [DOI] [PubMed] [Google Scholar]
- 21.Vinatier C., Dominguez E., Guicheux J., Carames B. Role of the inflammation-autophagy-senescence integrative network in osteoarthritis. Front. Physiol. 2018;9:706. doi: 10.3389/fphys.2018.00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Malta C., Cinque L., Settembre C. Transcriptional regulation of autophagy: mechanisms and diseases. Front. Cell Dev. Biol. 2019;7:114. doi: 10.3389/fcell.2019.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotani T., Kirisako H., Koizumi M., Ohsumi Y., Nakatogawa H. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc. Natl. Acad. Sci. U S A. 2018;115:10363–10368. doi: 10.1073/pnas.1806727115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graef M. Membrane tethering by the autophagy ATG2A-WIPI4 complex. Proc. Natl. Acad. Sci. U S A. 2018;115:10540–10541. doi: 10.1073/pnas.1814759115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lian W.S., Ko J.Y., Wu R.W., Sun Y.C., Chen Y.S., Wu S.L., Weng L.H., Jahr H., Wang F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018;9:919. doi: 10.1038/s41419-018-0994-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han Y., Lefebvre V. L-Sox5 and Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol. Cell Biol. 2008;28:4999–5013. doi: 10.1128/MCB.00695-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda T., Kamekura S., Mabuchi A., Kou I., Seki S., Takato T., Nakamura K., Kawaguchi H., Ikegawa S., Chung U.I. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004;50:3561–3573. doi: 10.1002/art.20611. [DOI] [PubMed] [Google Scholar]
- 28.Hargus G., Kist R., Kramer J., Gerstel D., Neitz A., Scherer G., Rohwedel J. Loss of Sox9 function results in defective chondrocyte differentiation of mouse embryonic stem cells in vitro. Int. J. Dev. Biol. 2008;52:323–332. doi: 10.1387/ijdb.072490gh. [DOI] [PubMed] [Google Scholar]
- 29.Smits P., Li P., Mandel J., Zhang Z., Deng J.M., Behringer R.R., de Crombrugghe B., Lefebvre V. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev. Cell. 2001;1:277–290. doi: 10.1016/s1534-5807(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 30.Haag J., Gebhard P.M., Aigner T. SOX gene expression in human osteoarthritic cartilage. Pathobiology. 2008;75:195–199. doi: 10.1159/000124980. [DOI] [PubMed] [Google Scholar]
- 31.Budd E., de Andres M.C., Sanchez-Elsner T., Oreffo R.O.C. MiR-146b is down-regulated during the chondrogenic differentiation of human bone marrow derived skeletal stem cells and up-regulated in osteoarthritis. Sci. Rep. 2017;7:46704. doi: 10.1038/srep46704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J., Kang Y., Liao W.M., Yu L. MiR-194 regulates chondrogenic differentiation of human adipose-derived stem cells by targeting Sox5. PLoS One. 2012;7:e31861. doi: 10.1371/journal.pone.0031861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng L., Yang Z.M., Li Y.C., Wang H.X., Lo J.H.T., Zhang X.T., Li G. Linc-ROR promotes mesenchymal stem cells chondrogenesis and cartilage formation via regulating SOX9 expression. Osteoarthritis Cartilage. 2021;29:568–578. doi: 10.1016/j.joca.2020.12.020. [DOI] [PubMed] [Google Scholar]
- 34.Chen J., Wu X. MicroRNA-103 contributes to osteoarthritis development by targeting Sox6. Biomed. Pharmacother. 2019;118:109186. doi: 10.1016/j.biopha.2019.109186. [DOI] [PubMed] [Google Scholar]
- 35.Kang X., Yang W., Feng D., Jin X., Ma Z., Qian Z., Xie T., Li H., Liu J., Wang R., et al. Cartilage-specific autophagy deficiency promotes ER stress and impairs chondrogenesis in PERK-ATF4-CHOP-dependent manner. J. Bone Miner Res. 2017;32:2128–2141. doi: 10.1002/jbmr.3134. [DOI] [PubMed] [Google Scholar]
- 36.Dalle Carbonare L., Mottes M., Cheri S., Deiana M., Zamboni F., Gabbiani D., Schena F., Salvagno G.L., Lippi G., Valenti M.T. Increased gene expression of RUNX2 and SOX9 in mesenchymal circulating progenitors is associated with autophagy during physical activity. Oxid. Med. Cell Longev. 2019;2019:8426259. doi: 10.1155/2019/8426259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Li C., Li H., Song Y., Zhao Y., Zhai L., Wang H., Zhong R., Tang H., Zhu D. miR-378 activates the pyruvate-PEP futile cycle and enhances lipolysis to ameliorate obesity in mice. EBioMedicine. 2016;5:93–104. doi: 10.1016/j.ebiom.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z., Feng L., Huang J., Zhang X., Lin W., Wang B., Cui L., Lin S., Li G. Asiatic acid protects articular cartilage through promoting chondrogenesis and inhibiting inflammation and hypertrophy in osteoarthritis. Eur. J. Pharmacol. 2021;907:174265. doi: 10.1016/j.ejphar.2021.174265. [DOI] [PubMed] [Google Scholar]
- 39.Pritzker K.P., Gay S., Jimenez S.A., Ostergaard K., Pelletier J.P., Revell P.A., Salter D., van den Berg W.B. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14:13–29. doi: 10.1016/j.joca.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Chen Y., Lin S., Sun Y., Pan X., Xiao L., Zou L., Ho K.W., Li G. Translational potential of ginsenoside Rb1 in managing progression of osteoarthritis. J. Orthop. Translat. 2016;6:27–33. doi: 10.1016/j.jot.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi L., Liu Y., Yang Z., Wu T., Lo H.T., Xu J., Zhang J., Lin W., Zhang J., Feng L., Li G. Vasoactive intestinal peptide promotes fracture healing in sympathectomized mice. Calcif. Tissue Int. 2021;109:55–65. doi: 10.1007/s00223-021-00820-9. [DOI] [PubMed] [Google Scholar]
- 42.Shi L., Feng L., Zhu M.L., Yang Z.M., Wu T.Y., Xu J., Liu Y., Lin W.P., Lo J.H.T., Zhang J.F., Li G. Vasoactive intestinal peptide stimulates bone marrow-mesenchymal stem cells osteogenesis differentiation by activating Wnt/beta-catenin signaling pathway and promotes rat skull defect repair. Stem Cells Dev. 2020;29:655–666. doi: 10.1089/scd.2019.0148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.