

 This work is licensed under a
This work is licensed under a Abstract
Objective
To describe clinical, laboratory, and genetic characteristics of three unrelated cases from Chile, Portugal, and Saudi Arabia with severe insulin resistance, SOFT syndrome, and biallelic pathogenic POC1A variants.
Design
Observational study.
Methods
Probands’ phenotypes, including short stature, dysmorphism, and insulin resistance, were compared with previous reports.
Results
Cases 1 (female) and 3 (male) were homozygous for known pathogenic POC1A variants: c.649C>T, p.(Arg217Trp) and c.241C>T, p.(Arg81*), respectively. Case 2 (male) was compound heterozygous for p.(Arg217Trp) variant and the rare missense variant c.370G>A, p.(Asp124Asn). All three cases exhibited severe insulin resistance, acanthosis nigricans, elevated serum triglycerides and decreased HDL, and fatty liver, resembling three previously reported cases. All three also reported severe muscle cramps. Aggregate analysis of the six known cases with biallelic POC1A variants and insulin resistance showed decreased birth weight and length mean (s.d.): −2.8 (0.9) and −3.7 (0.9) SDS, respectively), severe short stature mean (s.d.) height: −4.9 (1.7) SDS) and moderate microcephaly (mean occipitofrontal circumference −3.0 (range: −4.7 to −1.2)). These findings were similar to those reported for patients with SOFT syndrome without insulin resistance. Muscle biopsy in Case 3 showed features of muscle involvement secondary to a neuropathic process.
Conclusions
Patients with SOFT syndrome can develop severe dyslipidaemic insulin resistance, independent of the exonic position of the POC1A variant. They also can develop severe muscle cramps. After diagnosis, patients should be regularly screened for insulin resistance and muscle complaints.
Introduction
SOFT syndrome (MIM # 614813), denoting short stature, onychodysplasia, facial dysmorphism, and hypotrichosis, is the name coined for a rare primordial dwarfism syndrome encompassing severe growth failure of prenatal onset, craniofacial dysmorphism, sparse hair, and digital abnormalities (1). In 2012, two groups reported that the syndrome was caused by biallelic variants in POC1A, encoding the proteome of centrioles 1A (POC1A) protein (1, 2, 3). POC1A is an important luminal component of centrioles, playing roles in the function of centrosomes, spindle poles, and ciliary basal bodies (4, 5, 6).
Since these initial reports (1, 2, 3), 12 additional affected kindreds have been described (7, 8, 9, 10, 11, 12, 13, 14, 15, 16). In addition to the cardinal syndromic features, three of 31 patients reported to date also manifested severe dyslipidaemic insulin resistance (IR) (7, 11, 16). All 3 harboured pathogenic variants in exon 10, raising the possibility of a distinct, exon-specific ‘variant POC1A-related’ (vPOC1A) subsyndrome (11). However, an exon 9 variant in the most recently reported patient with IR (16), and variants outside exon 10 in two further individuals with early-onset type 2 diabetes (DM2) (2), suggest that IR may be part of the wider SOFT syndrome phenotype, and not uniquely associated with exon 10 variants.
We now present clinical, biochemical, and genetic characteristics of three unrelated patients carrying biallelic pathogenic POC1A variants outside exon 10 who show clinical features of SOFT syndrome plus severe dyslipidaemic IR, providing further evidence that severe IR with or without DM2 is a frequent component of SOFT syndrome. All three also suffer from severe muscle spasms and cramps, reported only in one patient to date (2).
Subjects and methods
Study approval
Patients were enrolled in genetic research projects or were referred for diagnostic genetic testing. All investigations were conducted according to the Declaration of Helsinki principles. Clinical data and images were collected with signed informed consent from participants/families. Permission was obtained to publish images in Figs 1, 2, 3, and Supplementary Fig. 1 (see section on supplementary materials given at the end of this article).
Figure 1.

Case 1. (A) Pedigree (using INVITAE Family pedigree tool). M/M indicates a biallelic POC1A variant, M/W a heterozygous carrier. (B) Growth curve (height for age) against CDC chart. (C and D) Frontal and lateral photographs aged 8.8 years. (E) Chest at 8.8 years showing the café au lait spot. (F) Hands show brachydactyly and mild fifth finger clinodactyly and broad thumbs. The nails were broad and short. (G) Feet show broad big toes. (H) Broad upper legs. (I) Muscle cramps aged 21.5 years. (J) Scalp aged 21.5 years. (K) The hand X-ray aged 8.7 years shows short phalanges, cone epiphyses of the distal phalanges, pseudo-epiphysis in the middle phalanx of the index, clinodactyly of the little finger, and a slight delay in bone maturation. (L) The pelvic X-ray aged 8.7 years shows asymmetric involvement of the femoral necks with abnormal remodelling, shortening, and deformity.
Figure 2.
Case 2. (A) Pedigree (using INVITAE Family pedigree tool). M/M indicates a biallelic POC1A variant, M/W a heterozygous carrier. (B) Height plotted against CDC charts. (C, D, E, and F) Frontal and lateral photographs aged 22.3 years.
Figure 3.

Case 3. (A) Family pedigree (using INVITAE Family pedigree tool). M/M indicates a biallelic POC1A variant, M/W a heterozygous carrier. (B) Clinical features demonstrating the abnormal findings: (i) short stature; (ii) high forehead and frontal bossing; (iii) posterior low set ear; (iv) gynaecomastia; (v) Acanthosis nigricans; (vi) hypoplastic distal phalanges and nails; (vii) wide space between big and second toes. (C) Radiological abnormalities: (i) short third metacarpal; (ii) metatarsal bone; (iii) short femoral neck; (iv) empty sella turcia.
Case reports
Detailed clinical information on the three cases is presented in the supplementary information on clinical presentations and their developmental history, clinical history, and physical examination findings are summarised in Table 1.
Table 1.
Developmental history, clinical history, and physical examination findings in the three cases.
| Features | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Development | |||
| Gender | Female | Male | Male |
| Current age | 21 years | 25 years | 32 years |
| Parents | Reportedly unrelated | Not related | First cousin consanguineous |
| Birth weight | 1520 g (−4.4 SDS) | 2450 g (−3.2 SDS) | 1800 g (−2.8 SDS) |
| Birth length | 39 cm (−5.5 SDS) | NR | 45 cm (−3.0 SDS) |
| Birth OFC | 31 cm (−2.4 SDS) | NR | 33 cm (−1.2 SDS) |
| Psychomotor development | Normal | Normal | Delayed |
| Linear growth | Severe growth failure. Adult height 120 cm (−6.6 SDS) |
Severe growth failure. Adult height 127 cm (−7.2 SDS) |
Severe growth failure. Adult height 138 cm (−5.8 SDS) |
| Clinical observations | |||
| Insulin resistance | Insulin resistance which progressed to type 2 diabetes | Insulin resistance with reactive hypoglycaemia | Insulin resistance which progressed to type 2 diabetes |
| Hypertension | Present, treated | NR | Absent |
| Hyperlipidaemia | Diagnosed at 11 years | Diagnosed at 22 years | Diagnosed at 22 years |
| Ophthalmological assessment | Astigmatism | NR | Mild non-proliferative diabetic retinopathy |
| Pubertal development | Tanner B2 at 9.8 years, menarche at 15.3 years | Tanner G2 at 11 years; G3 (testes 8 mL) at 13.5 years | Absent (G1 at 21 years), gynaecomastia |
| Muscle cramps | Onset aged 2 years | Onset aged 13 years | Onset aged 22 years |
| Alopecia | Present | Present | Present |
| Centripetal obesity | Absent (waist circumference 72 cm) | Present | Present |
| Acanthosis Nigricans | Present from 10.1 years | Present from 13.5 years | Present from 21 years |
| Hypotonia | NR | NR | Present |
| High pitched voice | Present | Present | Absent |
| Adult gonadal status | Partial ovarian failure | NR | Borderline low plasma testosterone |
| Laboratory | |||
| Insulin | Increased | Increased | Increased |
| Creatine Kinase | Increased | Increased | Increased |
| Additional findings | |||
| Empty sella turcica | NR | NR | Present |
| Diffuse fatty liver | Present | Present | Present |
| Kidney anatomy | Normal kidney ultrasonography | NR | Left ectopic kidney |
| Electromyography | Reduced recruitment of MUAPs firing at increased frequency with increased amplitude, polyphasic potentials. Spontaneous fasciculations. |
NR | Rare fibrillations and positive sharp waves. Normal MUAPs, morphology and recruitments. Muscular cramps induced by leg exercise accompanied by fasciculation |
| Colonoscopy | NR | NR | Transverse colon polyp, no dysplasia or malignancy |
MUAPs, motor unit action potentials; NR, not reported; OFC, occipitofrontal circumference; SDS, standard deviation score.
Case 1
Case 1 is a 21.5-year-old Chilean woman born to healthy parents of normal height (Fig. 1A) with an extremely low birth size and poor postnatal growth (Fig. 1B and Table 1). Further clinical features include microcephaly, bilateral hip pain, prominent forehead, deep-set eyes, hypoplastic nostrils, smooth philtrum, thin upper lip, light skin, café au lait macules, joint hyperlaxity, broad hands and feet with broad thumbs/big toes, and broad upper legs (Fig. 1C, D, E, F, G, and H). Radiographs showed short phalanges, cone epiphyses of the distal phalanges, pseudo-epiphysis in the middle phalanx of the second finger, and fifth finger clinodactyly with bone age 7.9 years (chronological age 8.7 years) (Fig. 1K). Femoral necks were asymmetrical with abnormal remodelling, shortening, and deformity (Fig. 1L). Endocrine assessment showed transient elevated serum IGF-I, increased plasma insulin concentration (Supplementary Table 1), and a normal GH response to clonidine. Breast development was relatively early but menarche was delayed and followed by oligomenorrhoea. Hair became progressively dry, sparse, and brittle (Fig. 1J), with increased scalp sensitivity.
Recombinant human growth hormone (rhGH) plus a GnRH analogue was administered from 10.1 to 11.6 years resulting in a small increase of height SDS, but was discontinued due to the poor growth response and development of acanthosis nigricans and hypertension. From 18 years onward, muscle cramps have been the major complaint, affecting limbs, abdominal muscles, tongue, and jaw. The electromyography (EMG) needle triggered painful vastus lateralis spasms, leading to prolonged continuous muscle activity (Fig. 1I). Cramps subsided with amitriptyline. Metabolic evaluation (Supplementary Table 1) showed progressive IR (treated with metformin), elevated serum triglycerides, and fatty liver.
Case 2
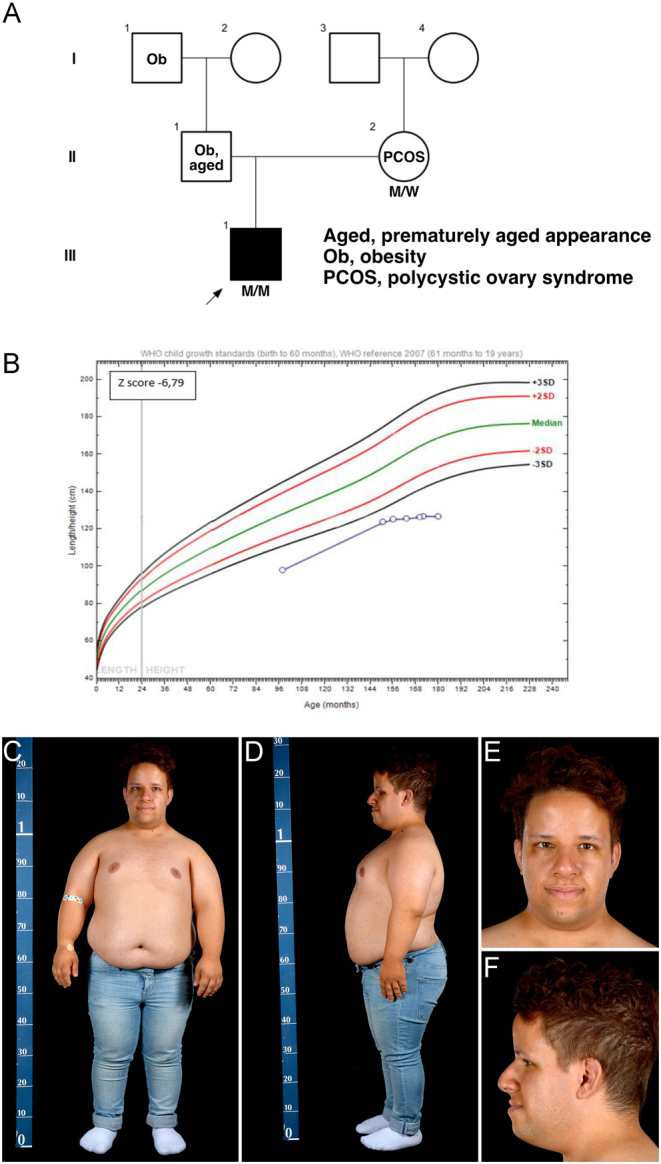
Case 2 is a 25-year-old man, the only child of unrelated Portuguese parents. His mother is healthy and normal-statured. His father is short (−2.1 SDS), with a prematurely aged appearance, hearing impairment, obesity, premature loss of dentition, but normal intellectual ability (Fig. 2A). The proband was born with a low birthweight and showed poor postnatal growth (Fig. 2B and Table 1) and centripetal adiposity (BMI 2.5 SDS) (Fig. 2C, D, E, and F). Further clinical features include brachydactyly; mild fifth finger clinodactyly with broad, short nails; scattered depigmented patches on the abdomen; irregular café au lait patches on the lower back; joint hypermobility; supernumerary teeth; and mild acanthosis nigricans. Rapid, patchy hair loss was noted at age 25 years.
rhGH therapy from 9.5 to 10.5 years yielded no benefit and was discontinued due to excessive weight gain. The metabolic assessment showed extreme fasting hyperinsulinaemia without diabetes, reactive hypoglycaemia, fatty liver, and mildly elevated serum creatine kinase (Supplementary Table 2). For 13.6 years he has intermittently complained of muscle cramps. At 25 years old, he reported severe muscular pains, significantly worse than in his teenage years. These were spasmodic, associated with paraesthesia in the fingers, and were exacerbated by cold.
Case 3
Case 3 is a 32-year-old Saudi Arab male born to parents who are first cousins and were diagnosed with DM2 at 42 years of age (Fig. 3A). The proband was born small for date (Table 1) and showed poor postnatal growth (Fig. 3B) and delayed developmental milestones (current IQ 68). Further clinical features include several facial dysmorphisms (detailed in Supplementary Information); brachydactyly; posteriorly rotated, low set ears; small, broad hands and feet with hypoplastic distal phalanges and nails; widely spaced first and second toes; single palmar creases; alopecia; and centripetal adiposity (Fig. 3B). A skeletal survey (Fig. 3C) revealed short femoral neck and phalanges, short left third metacarpal and metatarsal bone, hypoplastic distal phalanges and nails, and short, thick long bones.
GH deficiency was suspected and rhGH therapy was given from 8 years of age for 6 years, but information on serum IGF-I, GH stimulation testing, and growth response is unavailable. The metabolic assessment showed nuchal and axillary acanthosis nigricans, DM2, non-proliferative diabetic retinopathy, persisting fatty liver, hypercholesterolaemia, and hypertriglyceridaemia (Supplementary Table 3). For the borderline low plasma testosterone, no cause was found. At 26 years, muscle cramps in legs and chest on exertion and at rest were reported, with elevated serum creatinine kinase concentration. Muscle biopsy (Supplementary Fig. 1) showed nonspecific myopathic changes suggestive of a secondary neuropathic process.
Laboratory investigations
Details of genetic analysis are presented in Supplementary information on genetic analyses. Biochemical investigations were undertaken in accredited hospital laboratories. The presented reference ranges are as provided by these laboratories, except for fasting plasma insulin, triglycerides, cholesterol, HDL, and LDL. Reference ranges for fasting insulin in prepubertal children (to 11 years) were from Peplies et al. (17), for pubertal adolescents from Ballerini et al. (18), and for young adults from Tohidi et al. (19). For plasma lipids, we used the recommendations of the Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents (20).
Analysis of facial characteristics
Frontal facial photographs and clinical and genetic information of the three cases presented and eight previously reported (three from (3), single patients from (7, 9, 13, 14, 16)) were uploaded to the Face2Gene (FDNA Inc, Sunrise, FL, USA) platform. A ‘DeepGestalt‘ of the facial features of SOFT syndrome was generated, as previously reported for other syndromes (21).
Results
In Case 1 a rare homozygous POC1A missense variant (c.649C>T, p.(Arg217Trp)) was found, as previously reported in a Chilean girl with SOFT syndrome (13). In Case 2 and his mother, the same p.(Arg217Trp) variant was identified in heterozygous form. A second rare heterozygous missense variant (c.370G>A), p.(Asp124Asn)) was detected in Case 2 but not his mother. The father was unavailable for study, but based on these findings the POC1A variants in the proband were deemed highly likely to be compound heterozygous. Case 3 harboured the same homozygous truncating variant in POC1A reported by Shaheen et al. in a Saudi family (3) (c.241C>T, p.(Arg81*)). Further details on these genetic variants are shown in Supplementary Table 4.
All known cases with SOFT syndrome and IR or DM2 are summarised in Supplementary Table 5. All six fully documented cases had acanthosis nigricans, insulin resistance, elevated triglycerides, and fatty liver. Data from three members of the large Arab pedigree reported by Shalev et al. (2), two of whom were reported to have DM2, are also shown. For these cases, the evaluation of plasma insulin, acanthosis nigricans, and fatty liver was unavailable, but serum triglycerides were increased (4.6, 6.2, and 5.5 mmol/L (reference <1.3 (20)) and HDL levels were low (0.9, 1.1, and 1.1 mmol/L, reference >1.2 (20), personal communication, Dr Shalev).
Supplementary Table 6 shows the anthropometric profile of all reported cases, stratified by the presence of IR. All except two patients without IR were younger than 15 years. In contrast, all patients with IR were older than 22 years. Auxological findings were similar between groups. Birth weight and length were low except for patients with the p.(Leu171Pro) variant (2). In contrast, head occipitofrontal circumference (OFC) at birth was normal in almost all patients resulting in relative macrocephaly. The average height was −5 to −6 SDS, with a wide range (−9 to −2 SDS), while OFC was relatively spared (mean approximately −3 SDS).
Based on analysis of facial characteristics of our patients and 8 reported previously, a general facial representation of patients with SOFT syndrome (DeepGestalt) was generated (Supplementary Fig. 2), featuring a prominent nose with a broad tip and broad mouth. Subjective inspection showed a triangular face in young children, less striking in older subjects. The syndrome is not yet recognised by the algorithms, which require further images for training (21).
Discussion
This report conveys two main messages. Firstly, it solidifies dyslipidaemic IR and fatty liver as being associated with loss of POC1Afunction, showing this is not exclusive to pathogenic variants in exon 10. Second, it suggests that muscle involvement, likely secondary to neuronal dysregulation, is a novel phenotypic feature of SOFT syndrome.
Besides the dyslipidaemic IR in our three cases and three previously reported (7, 11, 16), we know of three cases with early-onset DM2 in a family reported in 2012 (2). Two of these were reported, with one further case diagnosed at 26 years old (Dr Shalev, personal communication). Nine cases of SOFT syndrome with reported dyslipidaemic IR, or 26% of all reported cases, are thus known. In most cases with IR, POC1A variants are outside exon 10, and anthropometric data do not discriminate cases with or without IR (Supplementary Table 6). We believe there is no basis to classify patients with biallelic POC1A variants and IR as having a specific subsyndrome as previously suggested (11). The prevalence of IR in SOFT syndrome would likely be higher if patients were biochemically screened from childhood onward. All but two previously reported cases without IR were younger than 15 years, while 8 of 9 cases with IR were adult at IR diagnosis (Supplementary Table 6), suggesting that IR development is age-dependent.
The mechanism linking dyslipidaemic IR to POC1A variants is unknown, but other forms of monogenic IR offer clues. Dyslipidaemia and fatty liver are common and severe in monogenic IR caused by adipose tissue defects, and the trajectory of dyslipidaemic IR in SOFT syndrome is reminiscent of lipodystrophies, where metabolic derangement commonly becomes clinically manifested peripubertally (22). In contrast, primary insulin signalling defects (in INSR or PIK3R1) do not result in dyslipidaemia or fatty liver (23, 24). Interestingly, several other genetic defects affecting the centrosome/primary cilium also feature dyslipidaemic IR, including Alström Syndrome (e.g. (25), caused by biallelic ALMS1 variants (26) and Osteodysplastic Primordial Dwarfism of Majewski Type 2 (27), caused by biallelic PCNT variants (28). This suggests a possible unifying mechanism linking certain forms of centrosome dysfunction to IR, possibly mediated by effects on adipose tissue. Addressing this experimentally will be challenging due to the numerous functions of the centrosome, but the viability of mice with Poc1a deficiency, which recapitulate skeletal manifestations of SOFT syndrome (29), will permit future studies.
Regarding the question of how the loss of POC1A causes the broad clinical phenotype, we can only speculate. POC1A protein expression is nearly ubiquitous, so the pattern of tissue involvement cannot easily be explained by expression pattern alone. Given preliminary evidence of abnormal mitotic kinetics and perhaps shorter cilia in POC1A deficiency, and given recent evidence that cilia play a key role in adipocyte development in vivo (30), inefficient adipogenesis, or deranged kinetics of a mesenchymal stem cell pool, may impair the crucial function of adipose tissue in metabolic homeostasis. A similar phenomenon could be present in other tissues such as the epiphyseal growth plate, hair follicles, muscle, and gonads.
The effect of rhGH treatment in cases 1 and 2 was minimal, and in case 3 the low adult height achieved renders a positive effect of rhGH treatment unlikely. In case 1 this treatment coincided with worsened IR and increased blood pressure and in case 2 with increasing obesity. We therefore suggest that rhGH treatment is not indicated in SOFT syndrome.
To date, muscular cramps have not been included in SOFT syndrome (MIM # 614813), although reported in one Arab case (2). After we identified them as prominent complaints in our three cases, we approached a previously described patient with IR (7). She also reported severe muscle cramps in her hands, neck, abdomen, and legs from early childhood, usually at night, and more commonly in winter. A further patient described by Giorgio et al. (11) subsequently also complained of muscle cramps (Drs E. Rubino, A. Brusco; personal communication). Sica et al. (31) (MIM %600771) reported two brothers with short stature (130–132 cm), sparse scalp and absent body hair, low set ears, large noses, high-pitched voices, enlarged cardiac ventricles, and severe ‘undulating’ painful muscle spasms from 8 to 10 years. We believe these siblings likely had SOFT syndrome on clinical grounds. We therefore speculate that muscle cramps may be a common, albeit so far unrecognised, feature of the syndrome.
Muscle cramps and pain generally increased with exercise, associated with fasciculation-like twitches in limbs and elevated blood creatine kinase concentration. Electrophysiological evaluation, and some aspects of muscle biopsy, suggested a likely neurogenic origin. Further investigation and case descriptions are needed to elucidate the pathophysiology of neuromuscular involvement. Of note, the association between IR and muscle involvement is not unique to SOFT syndrome, however. The entity ‘acanthosis nigricans with muscle cramps and acral enlargement’ (MIM 200170), was described in 1980 (32, 33) and features of severe IR with phenytoin-responsive muscle cramps have been reported (33, 34). No features clearly conforming to SOFT syndrome were described. Other conditions such as some laminopathies and congenital generalised lipodystrophy type 4, feature myopathy and lipodystrophic IR (35).
The composite image of SOFT syndrome generates a step towards automated assistance to clinicians in making diagnoses on upload of a facial image and clinical features (21). Since SOFT syndrome is rare, the database could ultimately be of value in facilitating early diagnosis and screening for complications, however further images are required to train recognition algorithms fully. Although modern diagnostic procedures in high-income countries tend to use a hypothesis-free approach (e.g. next-generation sequencing techniques like exome sequencing (ES) and whole-genome sequencing in the near future), we believe that visual recognition of a facial phenotype remains important, particularly in countries where genetic testing is not available or reimbursed.
In conclusion, patients with SOFT syndrome often manifest severe dyslipidaemic IR and muscle cramps, independent of the position of the POC1A variant. After diagnosis, patients should be regularly screened for IR and muscle complaints. Further studies are needed to clarify the pathophysiology of these clinical features of SOFT syndrome.
Supplementary Material
Declaration of interest
Prof Rob Semple is a Deputy Editor on the European Journal of Endocrinology Editorial board. Prof Rob Semple was not involved in the review or editorial process for this paper, on which he is listed as an author. The other authors declare no competing interests.
Funding
This work was supported in part by the Wellcome Trust (grant number 210752/Z/18/Z to R K S).
Author contribution statement
V M, I H-D, D A, J B, C C, K A, and R K S contributed by performing, interpreting and describing the clinical assessment of the patients. C de B advised on endocrine assessment, Y H was responsible for uploading and interpreting facial dysmorphology, and E B advised on the diagnosis of the muscle phenotype. F S A, M L, and R K S performed the genetic analyses. J M W coordinated the writing process. All authors contributed in data interpretation and various revisions of the manuscript and have approved the submitted manuscript.
Acknowledgements
Dr E Bertini is a representing HCP Member of the European Rare Disease Network for ERN-NMD and ERN-RND. The authors are grateful to Dr S Shalev for providing laboratory data encountered in the three young adults carrying a homozygous POC1A variant. The authors thank Dr S Silva Soto for informing us on laboratory results on his patient. The authors are grateful to Drs M Al-Hashem, S Silva Soto, A Jorge, V López-González and S Majore for supplying photographs of previously reported patients with SOFT syndrome which have contributed to the characteristics of the facial phenotype. The authors thank Drs E Rubino and A Brusco for providing information about muscle cramps in the patient described by Giorgio et al. (11). The authors are grateful to Dr Nicole Fleischer for advice on the Face2Gene (FDNA Inc, USA) analysis.
References
- 1.Sarig O, Nahum S, Rapaport D, Ishida-Yamamoto A, Fuchs-Telem D, Qiaoli L, Cohen-Katsenelson K, Spiegel R, Nousbeck J, Israeli Set al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. American Journal of Human Genetics 201291337–342. ( 10.1016/j.ajhg.2012.06.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shalev SA, Spiegel R, Borochowitz ZU. A distinctive autosomal recessive syndrome of severe disproportionate short stature with short long bones, brachydactyly, and hypotrichosis in two consanguineous Arab families. European Journal of Medical Genetics 201255256–264. ( 10.1016/j.ejmg.2012.02.011) [DOI] [PubMed] [Google Scholar]
- 3.Shaheen R, Faqeih E, Shamseldin HE, Noche RR, Sunker A, Alshammari MJ, Al-Sheddi T, Adly N, Al-Dosari MS, Megason SGet al. POC1A truncation mutation causes a ciliopathy in humans characterized by primordial dwarfism. American Journal of Human Genetics 201291330–336. ( 10.1016/j.ajhg.2012.05.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearson CG, Osborn DP, Giddings Jr TH, Beales PL, Winey M. Basal body stability and ciliogenesis requires the conserved component Poc1. Journal of Cell Biology 2009187905–920. ( 10.1083/jcb.200908019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venoux M, Tait X, Hames RS, Straatman KR, Woodland HR, Fry AM. Poc1A and Poc1B act together in human cells to ensure centriole integrity. Journal of Cell Science 2013126163–175. ( 10.1242/jcs.111203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li S, Fernandez JJ, Marshall WF, Agard DA. Electron cryo-tomography provides insight into procentriole architecture and assembly mechanism. eLife 20198e43434. ( 10.7554/eLife.43434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen JH, Segni M, Payne F, Huang-Doran I, Sleigh A, Adams CConsortium UK, Savage DB, O’Rahilly S, Semple RKet al. Truncation of POC1A associated with short stature and extreme insulin resistance. Journal of Molecular Endocrinology 201555147–158. ( 10.1530/JME-15-0090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koparir A, Karatas OF, Yuceturk B, Yuksel B, Bayrak AO, Gerdan OF, Sagiroglu MS, Gezdirici A, Kirimtay K, Selcuk Eet al. Novel POC1A mutation in primordial dwarfism reveals new insights for centriole biogenesis. Human Molecular Genetics 2015245378–5387. ( 10.1093/hmg/ddv261) [DOI] [PubMed] [Google Scholar]
- 9.Barraza-Garcia J, Ivan Rivera-Pedroza C, Salamanca L, Belinchon A, Lopez-Gonzalez V, Sentchordi-Montane L, del Pozo Á, Santos-Simarro F, Campos-Barros Á, Lapunzina Pet al. Two novel POC1A mutations in the primordial dwarfism, SOFT syndrome: clinical homogeneity but also unreported malformations. American Journal of Medical Genetics: Part A 2016170A210–216. ( 10.1002/ajmg.a.37393) [DOI] [PubMed] [Google Scholar]
- 10.Ko JM, Jung S, Seo J, Shin CH, Cheong HI, Choi M, Kim OH, Cho TJ. SOFT syndrome caused by compound heterozygous mutations of POC1A and its skeletal manifestation. Journal of Human Genetics 201661561–564. ( 10.1038/jhg.2015.174) [DOI] [PubMed] [Google Scholar]
- 11.Giorgio E, Rubino E, Bruselles A, Pizzi S, Rainero I, Duca S, Sirchia F, Pasini B, Tartaglia M, Brusco A. A syndromic extreme insulin resistance caused by biallelic POC1A mutations in exon 10. European Journal of Endocrinology 2017177K21–K27. ( 10.1530/EJE-17-0431) [DOI] [PubMed] [Google Scholar]
- 12.Mostofizadeh N, Gheidarloo M, Hashemipour M, Dehkordi EH. SOFT syndrome: the first case in Iran. Advanced Biomedical Research 20187 128. ( 10.4103/abr.abr_13_18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saida K, Silva S, Solar B, Fujita A, Hamanaka K, Mitsuhashi S, Koshimizu E, Mizuguchi T, Miyatake S, Takata Aet al. SOFT syndrome in a patient from Chile. American Journal of Medical Genetics: Part A 2019179338–340. ( 10.1002/ajmg.a.61015) [DOI] [PubMed] [Google Scholar]
- 14.Homma TK, Freire BL, Honjo Kawahira RS, Dauber A, Funari MFA, Lerario AM, Nishi MY, Albuquerque EV, Vasques GA, Collett-Solberg PFet al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. Journal of Pediatrics 2019215192–198. ( 10.1016/j.jpeds.2019.08.024) [DOI] [PubMed] [Google Scholar]
- 15.Al-Kindi A, Al-Shehhi M, Westenberger A, Beetz C, Scott P, Brandau O, Abbasi-Moheb L, Yuksel Z, Bauer P, Rolfs Aet al. A novel POC1A variant in an alternatively spliced exon causes classic SOFT syndrome: clinical presentation of seven patients. Journal of Human Genetics 202065193–197. ( 10.1038/s10038-019-0693-2) [DOI] [PubMed] [Google Scholar]
- 16.Majore S, Agolini E, Micale L, Pascolini G, Zuppi P, Cocciadiferro D, Morlino S, Mattiuzzo M, Valiante M, Castori Met al. Clinical presentation and molecular characterization of a novel patient with variant POC1A-related syndrome. Clinical Genetics 202199540–546. ( 10.1111/cge.13911) [DOI] [PubMed] [Google Scholar]
- 17.Peplies J, Jimenez-Pavon D, Savva SC, Buck C, Gunther K, Fraterman A, Russo P, Iacoviello L, Veidebaum T, Tornaritis Met al. Percentiles of fasting serum insulin, glucose, HbA1c and HOMA-IR in pre-pubertal normal weight European children from the IDEFICS cohort. International Journal of Obesity 201438 (Supplement 2) S39–S47. ( 10.1038/ijo.2014.134) [DOI] [PubMed] [Google Scholar]
- 18.Ballerini MG, Bergada I, Rodriguez ME, Keselman A, Bengolea VS, Pipman V, Domene HM, Jasper HG, Ropelato MG. Insulin level and insulin sensitivity indices among healthy children and adolescents. Archivos Argentinos de Pediatria 2016114329–336. ( 10.5546/aap.2016.eng.329) [DOI] [PubMed] [Google Scholar]
- 19.Tohidi M, Ghasemi A, Hadaegh F, Derakhshan A, Chary A, Azizi F. Age- and sex-specific reference values for fasting serum insulin levels and insulin resistance/sensitivity indices in healthy Iranian adults: Tehran Lipid and Glucose Study. Clinical Biochemistry 201447432–438. ( 10.1016/j.clinbiochem.2014.02.007) [DOI] [PubMed] [Google Scholar]
- 20.Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics 2011128 (Supplement 5) S213–S256. ( 10.1542/peds.2009-2107C) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, Basel-Salmon L, Krawitz PM, Kamphausen SB, Zenker Met al. Identifying facial phenotypes of genetic disorders using deep learning. Nature Medicine 20192560–64. ( 10.1038/s41591-018-0279-0) [DOI] [PubMed] [Google Scholar]
- 22.Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocrine Reviews 201132498–514. ( 10.1210/er.2010-0020) [DOI] [PubMed] [Google Scholar]
- 23.Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington Pet al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. Journal of Clinical Investigation 2009119315–322. ( 10.1172/JCI37432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang-Doran I, Tomlinson P, Payne F, Gast A, Sleigh A, Bottomley W, Harris J, Daly A, Rocha N, Rudge Set al. Insulin resistance uncoupled from dyslipidemia due to C-terminal PIK3R1 mutations. JCI Insight 20161 e88766. ( 10.1172/jci.insight.88766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geberhiwot T, Baig S, Obringer C, Girard D, Dawson C, Manolopoulos K, Messaddeq N, Bel Lassen P, Clement K, Tomlinson JWet al. Relative adipose tissue failure in Alstrom syndrome drives obesity-induced insulin resistance. Diabetes 202170364–376. ( 10.2337/db20-0647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin Met al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nature Genetics 20023174–78. ( 10.1038/ng867) [DOI] [PubMed] [Google Scholar]
- 27.Huang-Doran I, Bicknell LS, Finucane FM, Rocha N, Porter KM, Tung YC, Szekeres F, Krook A, Nolan JJ, O’Driscoll Met al. Genetic defects in human pericentrin are associated with severe insulin resistance and diabetes. Diabetes 201160925–935. ( 10.2337/db10-1334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KHet al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 2008319816–819. ( 10.1126/science.1151174) [DOI] [PubMed] [Google Scholar]
- 29.Geister KA, Brinkmeier ML, Cheung LY, Wendt J, Oatley MJ, Burgess DL, Kozloff KM, Cavalcoli JD, Oatley JM, Camper SA. LINE-1 mediated insertion into Poc1a (protein of centriole 1 A) causes growth insufficiency and male infertility in mice. PLoS Genetics 201511 e1005569. ( 10.1371/journal.pgen.1005569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hilgendorf KI, Johnson CT, Mezger A, Rice SL, Norris AM, Demeter J, Greenleaf WJ, Reiter JF, Kopinke D, Jackson PK. Omega-3 fatty acids activate ciliary FFAR4 to control adipogenesis. Cell 2019179 1289.e21–1305.e21. ( 10.1016/j.cell.2019.11.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sica RE, Espinoza R, Benavente O, Sanz OP, Molina H. Familial dwarfism and painful muscle spasms. Medicina 199555111–116. [PubMed] [Google Scholar]
- 32.Flier JS, Young JB, Landsberg L. Familial insulin resistance with acanthosis nigricans, acral hypertrophy, and muscle cramps. New England Journal of Medicine 1980303970–973. ( 10.1056/NEJM198010233031704) [DOI] [PubMed] [Google Scholar]
- 33.Minaker KL, Flier JS, Landsberg L, Young JB, Moxley RT, Kingston WJ, Meneilly GS, Rowe JW. Phenytoin-induced improvement in muscle cramping and insulin action in three patients with the syndrome of insulin resistance, acanthosis nigricans, and acral hypertrophy. Archives of Neurology 198946981–985. ( 10.1001/archneur.1989.00520450051018) [DOI] [PubMed] [Google Scholar]
- 34.Ghosh U, Thomas M, Mathai S. Syndrome of insulin resistance with acanthosis nigricans, acral hypertrophy and muscle cramps in an adolescent – a rare diagnosis revisited. Indian Journal of Pediatrics 2014811389–1391. ( 10.1007/s12098-014-1503-7) [DOI] [PubMed] [Google Scholar]
- 35.Akinci B, Sahinoz M, Oral E. Lipodystrophy syndromes: presentation and treatment. In Endotext. Eds Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, K Dungan, JM Hersh, J Hofland, S Kalra. South Dartmouth (MA), 2000. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.