

Abstract
Investigating basic biological mechanisms underlying human diseases relies on the availability of sufficient quantities of patient cells. As most primary somatic cells have a limited lifespan, obtaining sufficient material for biological studies has been a challenge. The development of induced pluripotent stem cell (iPSC) technology has been a game changer, especially in the field of rare genetic disorders. iPSC are essentially immortal, can be stored indefinitely, and can thus be used to generate defined somatic cells in unlimited quantities. Further, the availability of genome editing technologies, such as CRISPR/CAS, has provided us with the opportunity to create “designer” iPSC lines with defined genetic characteristics. A major advancement in biological research stems from the development of methods to direct iPSC differentiation into defined cell types. In this article, we provide the basic protocol for the generation of human iPSC‐derived keratinocytes (iPSC‐K). These cells have the characteristics of basal epidermal keratinocytes and represent a tool for the investigation of normal epidermal biology, as well as genetic and acquired skin disorders. © 2022 The Authors. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol: Directed differentiation of human iPSC into keratinocytes
Support Protocol 1: Coating cell culture dishes or plates with Vitronectin XF™
Support Protocol 2: Freezing iPSC
Support Protocol 3: Preparing AggreWell™400 6‐well plates for EB formation
Support Protocol 4: Coating cell culture dishes or plates with Collagen IV
Support Protocol 5: Immunofluorescence staining of cells
Keywords: epidermal biology, induced pluripotent stem cell, iPSC, iPSC‐derived keratinocytes, keratinocytes, skin diseases
INTRODUCTION
Investigating the mechanisms underlying human genetic or acquired skin disorders relies on the use of keratinocytes derived from affected individuals. Although primary keratinocytes are relatively easy to isolate from skin biopsies, these cells have a limited lifespan (Chapman, Liu, Meyers, Schlegel, & McBride, 2010; Green, Rheinwald, & Sun, 1977). Thus, these cells are not of practical use for experiments that require large numbers of cells or experiments that take place over a long period of time. To overcome this issue, investigators can generate keratinocytes from induced pluripotent stem cells (iPSC). iPSC have an unlimited lifespan and can be differentiated into many somatic cell types. As an additional advantage, iPSC are amenable to gene editing, allowing investigators to generate conisogenic iPSC lines. Further, as iPSC can be stored indefinitely in liquid nitrogen storage, keratinocytes can be generated at any time from the same source material. In this protocol, we provide detailed instructions on generating keratinocytes from human iPSC. These so‐called iPSC‐K have similar characteristics to normal human keratinocytes as determined by RNAseq analyses, protein expression, and their potential to differentiate into keratinocytes representing the different layers of the epidermis. Thus, they can be used for downstream applications, such as the analysis of cell characteristics and behavior, various ‐omics applications, and 3D cultures (Dinella et al., 2018; Itoh et al., 2013; Jackow et al., 2019; Koch, Dinella, Fete, Siegfried, & Koster, 2014; Sebastiano et al., 2014).
Here, we describe the overall method for generating keratinocytes from human iPSC in one Basic Protocol and five Support Protocols. The Basic Protocol provides experimental details on the generation of keratinocytes from human iPSC. Support Protocol 1 describes the procedure for freezing iPSC for future use. Support Protocols 2‐4 describe the preparation of materials necessary for the successful completion of the Basic Protocol. Finally, Support Protocol 5 describes the use of immunofluorescence staining to determine the proper characteristics of the resulting keratinocyte cultures. We also describe and show morphological benchmarks at different times during the differentiation program.
STRATEGIC PLANNING
This protocol requires reagents that are not always reliably available from suppliers. We strongly recommend that investigators secure all reagents in sufficient quantities before starting the differentiation program. In addition, investigators should carefully consider the iPSC line selected for their studies. Relevant iPSC characteristics to be evaluated include a normal karyotype and expression of pluripotency markers such as OCT4 and NANOG (Takahashi et al., 2007; Yu et al., 2009). Please refer to the “Critical Parameters” section for additional details.
DIFFERENTIATION OF HUMAN iPSC INTO KERATINOCYTES
In this protocol, we describe in detail the steps for the guided differentiation of human iPSC into keratinocytes. As outlined in Figure 1, key steps include the generation of embryoid bodies (EB), the initiation of differentiation into the keratinocyte lineage through exposure of the cells to retinoic acid (RA) and Bone Morphogenetic Protein 4 (BMP4), and the selection and expansion of keratinocytes. Please note that Rock inhibitor is used at different stages during this protocol. Rock inhibitor facilitates EB formation and increases the plating efficiency of iPSC and iPSC‐K. Optimal concentrations of Rock inhibitor for each of these steps were determined empirically. Prior to starting this protocol, users are encouraged to familiarize themselves with the processes of culturing and passaging human iPSC (Rivera, Zhao, Ni, & Wang, 2020). It is important to note that, even when conducted properly, a subset of cultures will fail to differentiate into keratinocytes (see “Critical Parameters”). Although the underlying reason for this is not always clear, it is important that investigators observe cultures regularly (at least every other day) to ensure they are progressing as expected. Morphological criteria are described in the protocol and shown in later figures. Investigators should also analyze expression of keratinocyte markers to ensure iPSC differentiation into keratinocytes.
Figure 1.
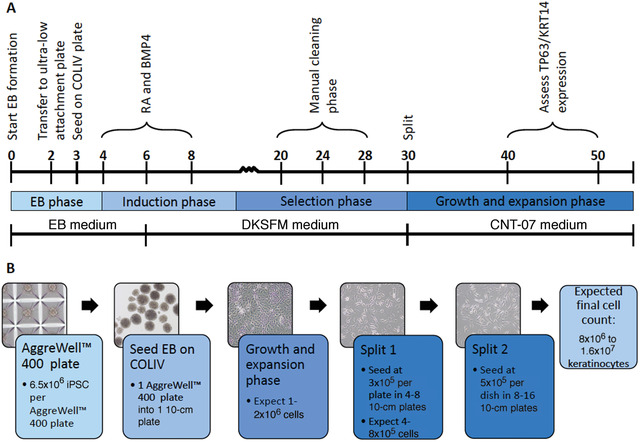
Timeline for iPSC‐K generation. (A) Annotated timeline for the generation of iPSC‐K. Numbers on the timeline reflect days. (B) Morphology as well as numbers of cells and plates generated using this protocol.
IMPORTANT NOTES
-
1.
Prewarm all media and solutions before use:
-
a.
Unless otherwise indicated, prewarm all media and solutions in a 37°C water bath before use.
-
b.
Spray all bottles with 70% EtOH before placing in biosafety cabinet.
-
a.
-
2.
Only use sterilized materials for use in cell culture.
-
3.
Cell culture:
-
a.
Culture cells in a humidified cell culture incubator at 37°C with 5% C02.
-
b.
Change media every other day unless otherwise indicated.
-
c.
To wash cells, aspirate media, add Mg2+/Ca2+‐free PBS to the well/dish (∼2 ml for a well of a 6‐well plate), gently swirl plate/dish, and aspirate PBS.
-
d.
Use only Mg2+/Ca2+‐free PBS for washing cells.
-
a.
-
4.
Record the date and passage number on all dishes and tubes.
-
5.
Use of Rock inhibitor (Y‐27632):
-
a.
Use Rock inhibitor at a concentration of 100 µM for culture of embryoid bodies (EBs).
-
b.
Use Rock inhibitor at a concentration of 10 µM for culture of iPSC and iPSC‐derived keratinocytes.
-
c.
Media are supplemented with Rock inhibitor to facilitate attachment of the cells to the tissue culture dish. Rock inhibitor is not added for subsequent media changes.
-
a.
Materials
Vitronectin XF™ (Stemcell Technologies, cat. no. 100‐0763)
mTESR™ Plus medium kit (Stemcell Technologies, cat. no. 100‐0276; see recipe)
Human iPSC (ATCC, cat. no. ATCC‐DYS0100, or equivalent, or custom‐designed patient‐specific iPSC)
Rock inhibitor (BD Biosciences, cat no. 562822)
Phosphate‐buffered saline (PBS) Ca2+/Mg2+‐free (ThermoFisher Scientific, cat. no.10010023)
AccutaseTM (Stemcell Technologies, cat. no. 07922)
hEB medium (see recipe)
Trypan blue (Invitrogen, cat. no. T10282)
Collagen from human placenta (Sigma, cat. no. C5533)
Embryonic stem cell‐qualified FBS (R&D Systems, cat. no. S10250)
BMP4 (R&D Systems, cat. no. 314BP010)
Retinoic acid (Sigma, cat. no. R2625)
DKSFM medium (see recipe)
CNT‐07 medium (see recipe)
p63‐α (D2K8X) XP Rabbit mAb (Cell Signaling, cat. no. 13109)
Anti‐Keratin K14 guinea pig polyclonal (Progen, cat. no. GP‐CK14)
Biosafety cabinet (Thermo Scientific Hera‐Safe 2030i, or equivalent)
Fisher 6‐well plates (Fisher Scientific, cat. no. 09‐761‐146)
15‐ml polypropylene tubes (Fisher Scientific, cat. no. 22‐010‐072)
Water bath
10‐ml serological pipettes (Fisher Scientific, cat. no. 02‐923‐204)
Centrifuge with rotor appropriate for 15‐ml tubes (Eppendorf 5804 rotor A‐4‐4, or equivalent)
Glass aspirators (Fisher Scientific, cat. no. 13‐678‐20A)
Cell culture incubator Thermo Scientific Forma series II water‐jacketed (Fisher Scientific, cat. no. 13‐998074)
CELLTREAT Scientific Products 10‐cm Tissue Culture Treated Dish (Fisher Scientific, cat. no. 50202029)
Inverted light microscope (Nikon TS100, or equivalent)
5‐ml serological pipettes (Fisher Scientific, cat. no. 02‐923‐203)
200‐µl filter tips (ThermoFisher Scientific, cat. no. AM12655)
Laminar flow hood (Nuaire model no. NU‐201‐430, or equivalent)
Cell counter (Bio‐Rad, TC Automated Cell Counter, or equivalent)
AggreWell™400 (Stemcell Technologies, cat. no. 34421)
Centrifuge with plate spinner (Eppendorf 5804 rotor A2‐DW, or equivalent)
6‐well Clear Flat Bottom Ultra‐Low Attachment Plates (Corning, cat. no. 3471)
Thermo Scientific NuncLab‐TekII Chamber Slide System (Fisher Scientific, cat. no. 12‐565‐8)
Upright fluorescent microscope (Nikon, or equivalent)
Thaw and plate iPSC
-
1
Prior to starting this step, coat one well of a 6‐well plate with Vitronectin XF™ (see Support Protocol 1).
-
2
Prepare a 15‐ml tube with 5 ml prewarmed mTESR™ Plus medium.
-
3
Thaw one vial of frozen iPSC in a 37°C water bath until there is a small cube of ice left (∼2 min).
-
4
Transfer the contents of the vial into the 15‐ml tube prefilled with 5 ml prewarmed mTESR™ Plus medium.
-
5
Gently invert the 15‐ml tube 2‐3 times to mix.
A 10‐ml serological pipette can be used for mixing, but do not use anything with a smaller diameter as this will break up the colonies.
-
6
Centrifuge for 5 min at 150 × g, room temperature.
-
7
Aspirate media, being careful to leave the cell pellet intact.
-
8
Add 2 ml mTESR™ Plus medium supplemented with 10 µM Rock inhibitor.
-
9
Gently resuspend the cell pellet using a 10‐ml serological pipette.
-
10
Transfer the cell suspension into one well of a 6‐well plate precoated with Vitronectin XF™.
-
11
Culture overnight in a cell culture incubator at 37°C.
-
12
Change media to mTESR™ Plus without Rock inhibitor.
-
13
Culture for 4‐5 days in mTESR™ Plus medium. Change the medium every other day until the culture is ∼60% confluent (Fig. 2A).
Figure 2.
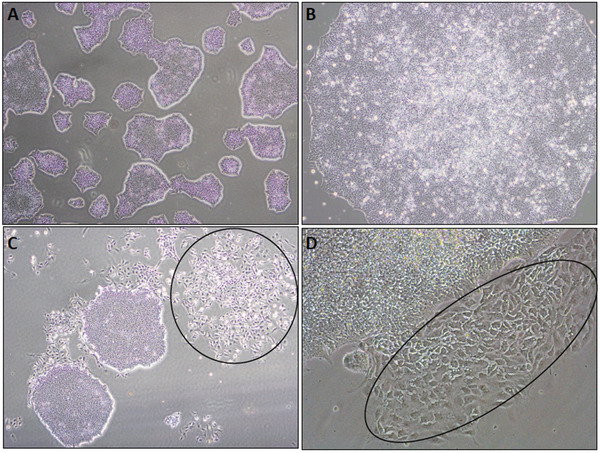
Phase‐contrast images showing morphology of human iPSC colonies. (A) Density at which iPSC colonies are passaged (approximately 60% colony density). (B) Higher magnification of a normal iPSC colony. Note the smooth edge of the colony and the high nuclear‐to‐cytoplasmic ratio. Both characteristics are signs of an undifferentiated healthy iPSC colony. (C,D) Normal iPSC colonies surrounded by scattered spontaneously differentiating cells (circles).
Expand hiPSC
-
14
Prior to starting this step, coat two 10‐cm cell culture dishes with Vitronectin XF™ and add 9 ml mTESR™ Plus medium supplemented with 10 µM Rock inhibitor per dish (see Support Protocol 1).
-
15
Aspirate the medium from the wells containing ∼60% confluent iPSC (step 13).
-
16
Wash the wells with 3 ml Mg2+/Ca2+‐free PBS.
-
17
Add 1 ml prewarmed Accutase™.
Accutase™ is the preferred reagent to release cells from the culture dish as this will maintain cells in small clumps.
-
18
Incubate the plates in the cell culture incubator for 3‐5 min until all colonies have lifted off the plate.
Check under inverted light microscope to ensure all colonies have lifted off the plate.
Ensure that cells are maintained in small clumps of ∼50‐200 µm in diameter. Avoid generating a single‐cell suspension as this will reduce cell viability. Loss of cell viability is judged by a strongly diminished ability to attach to the culture plate and proliferate.
-
19
Collect cell clusters with a 5‐ml serological pipette and transfer into a 15‐ml tube prefilled with 5 ml prewarmed mTESR™ Plus medium.
-
20
Invert the tubes 2‐3 times to mix.
-
21
Centrifuge at 150 × g for 5 min, room temperature.
-
22
Aspirate media, leaving the cell pellet intact.
-
23
Resuspend the pellet in 2 ml mTESR™ Plus medium supplemented with 10 µM Rock inhibitor.
-
24
Add 1 ml cell suspension to each 10‐cm dish that was precoated with Vitronectin XF™ and prefilled with mTESR™ Plus medium in step 14.
-
25
Culture overnight in a cell culture incubator.
-
26
Change the medium to mTESR™ Plus without Rock inhibitor.
-
27
Culture to ∼60% confluency changing mTESR™ Plus medium every other day (4‐5 days total).
During this time, make sure to observe cultures regularly and remove cells that do not form tight clusters. Remove cells by scraping with a 200‐µl filtered pipette tip. This step should be performed using an inverted light microscope placed in a laminar flow hood to maintain sterility of the cells. Wash with Mg2+/Ca2+‐free PBS and add fresh medium after cleaning.
Examples of iPSC colonies with typical morphology are shown in Figures 2A‐B
Freeze excess iPSC using Support Protocol 2
Generate single‐cell iPSC suspension in preparation for embryoid body (EB) formation
In our experience, forming embryoid bodies as the starting point for the generation of iPSC‐K is the most efficient and reproducible method.
Note that this is the only step in the protocol where single iPSC are desired.
-
28
Feed the cells with fresh mTESR™ Plus medium 1‐2 hr prior to starting EB formation.
-
29
Aspirate the medium and wash iPSC with 3‐5 ml Mg2+/Ca2+‐free PBS.
-
30
Add 3 ml prewarmed Accutase™.
-
31
Incubate for 5 min at 37°C.
-
32
Add 3 ml Mg2+/Ca2+‐free PBS and gently mix the cells by pipetting using a 10‐ml serological pipette. It is essential to perform this step gently but thoroughly to generate a single‐cell suspension for EB formation.
-
33
Collect the cell suspension in a 15‐ml tube.
-
34
Centrifuge for 5 min at 150 × g, room temperature.
-
35
Aspirate the medium and resuspend the cell pellet in 3‐5 ml hEB medium.
-
36Count live cells as follows:,
- Mix 10 µl cell suspension with 10 µl trypan blue.
- Count unstained (not blue) cells using a manual or automatic cell counter.
Generate EBs (day 0)
-
37
Prepare an AggreWell™400 plate prior to starting this step (see Support Protocol 3).
-
38
Prepare cell suspension of 6.5 × 106 cells in 3 ml hEB medium.
-
39
Add cell suspension to 1 well of a prepared 6‐well AggreWell™400 plate.
As 2 ml hEB medium are added to each well during plate preparation, the total volume per well will be 5 ml.
A 6‐well AggreWell™400 plate contains 5900 microwells per well. Therefore, each microwell will contain ∼900 cells.
The recommended cell number per microwell is 50‐3,000. In our experience, ∼900 cells per microwell leads to the formation of a homogeneous population of EBs with an optimal potential to differentiate into iPSC‐K.
-
40
Centrifuge the AggreWell™400 plate for 5 min at 130 × g, room temperature using a plate spinner in the centrifuge.
-
41
Incubate the plates for 2 days in a cell culture incubator to generate EBs. Do not disturb the plates during this time.
Note that the EBs will not attach to the AggreWell™400 plate.
Transfer EBs to ultra‐low attachment plate (day 2)
The purpose of this step is to ensure expansion of the EBs without them attaching to the plate, as this could cause differentiation into unintended cell types.
-
42
Create a homogeneous EB suspension by gently pipetting the medium containing the EBs using a 5‐ml serological pipette.
-
43
Transfer the suspension into a 15‐ml tube.
-
44
Gently wash each well with 5 ml Mg2+/Ca2+‐free PBS and add this to the 15‐ml tube.
This step ensures that any remaining EBs from the AggreWell™400 plate are collected.
-
45
Gently mix the EB suspension in the 15‐ml tube by pipetting up and down with a 5‐ml serological pipette.
Ensure that EBs remain intact during this time.
-
46
Allow EBs to settle down to the bottom of the 15‐ml tube for 5‐10 min (do not centrifuge the tubes at this step). You should see a large soft pellet at the bottom of the tube.
-
47
Carefully aspirate media leaving about 0.5 ml of media containing the EBs.
-
48
Add 5 ml of prewarmed hEB media plus 100 µM Rock inhibitor and transfer the suspension to 1 well of a 6‐well ultra‐low attachment microplate.
Note that the concentration of Rock inhibitor is 100 µM for EB formation and 10 µM for iPSC culture and differentiation.
-
49
Culture overnight in a cell culture incubator.
Note that the EBs will not attach to the ultra‐low attachment plates.
Seed EBs on Collagen IV‐coated plates (day 3)
Collagen IV will facilitate the differentiation of iPSC into keratinocytes.
-
50
Prepare Collagen IV‐coated 10‐cm plates prior to starting this step (see Support Protocol 4).
-
51Wash EBs three times, with Mg2+/Ca2+‐free PBS as follows:
- Collect EBs into a 15‐ml tube and add 5 ml Mg/Ca‐free PBS. Allow EBs to settle at the bottom of the tube (∼5‐10 min). Do not centrifuge the tubes.
- Carefully aspirate the PBS, leaving about 1 ml of PBS with EBs at the bottom of the tube.
- Repeat step 51a and 51b two more times for a total of three washes.
-
52
Plate EBs in one Collagen IV‐coated 10‐cm plate in 10 ml hEB medium supplemented with 10 µM Rock inhibitor and 2% ES‐qualified FBS.
Adding FBS facilitates adhesion of the EBs to the cell culture dish.
-
53
Culture EBs overnight in a cell culture incubator.
Do not disturb the plates during this time to allow the EBs to attach to the Collagen IV‐coated plate.
Start of keratinocyte differentiation (day 4‐10)
Day 4
-
54
Aspirate media to remove unattached EBs
-
55
Add 10 ml hEB medium supplemented with 25 ng/ml bone morphogenetic protein 4 (BMP4), 1 µM Retinoic Acid (RA), and 10 µM Rock inhibitor
Figures 3A‐B show examples of EBs at day 5 of differentiation. EBs with a high potential to differentiate into iPSC‐K are characterized by a smooth edge and are composed of large cells with a clearly visible nucleus (arrowheads in Fig. 3A‐B). Dashed arrows in Figure 3B point to EBs that did not attach as a monolayer and will likely die. Solid arrows point to EBs that may survive and differentiate correctly.
Figure 3.
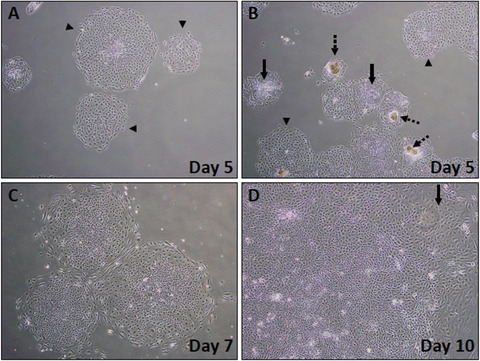
Phase‐contrast images showing early stages of iPSC‐K differentiation (days 5‐10). (A) Healthy colonies at day 5 after start of iPSC‐K differentiation (arrowheads). (B) Mix of healthy and unhealthy colonies at day 5 of differentiation. Dashed arrows point to colonies that did not properly attach to the plate. Note the darker (brown) cell clumps. These colonies will die. Solid arrows point to colonies that do not look ideal, but that may differentiate into keratinocytes. Arrowheads point to colonies with the expected epithelial morphology. (C) Expanding early‐stage differentiating colony at day 7. (D) Higher magnification of a day 10 colony. Note the emerging cobblestone appearance, a typical feature of epithelial cells. Arrow points to cells that have acquired the expected cobblestone morphology.
Day 6 and day 8
-
56
Replace the medium with 10 ml fresh DKSFM plus 25 ng/ml BMP4, 1 µM RA, and 10 µM Rock inhibitor.
Figure 3C shows examples of EBs with high potential to differentiate into iPSC‐K at day 7. Note the increase in cell size and pronounced cell nuclei in the cells at the edges of the EBs.
Day 10
-
57
Change the medium to DKSFM without BMP4, RA, or Rock inhibitor.
Figure 3D shows an example of an EB with high potential to differentiate into iPSC‐K at Day 10. Note the keratinocyte‐like morphology of cells at the edges of the EB. The arrow in Figure 3D points to the keratinocyte‐like cells. Note that these cells are larger and have acquired a cobblestone morphology.
Selection phase for keratinocytes (day 10‐30)
The purpose of this phase is to enrich for keratinocytes. This is accomplished by removing cells that fail to display the expected cobblestone morphology. Frequently encountered cell types that need to be removed are cells with a spindle‐like shape.
-
58
Feed cells with fresh DKSFM (without BMP4, RA, or Rock inhibitor) every other day between day 10 and day 30.
Do not split the cells during this phase.
Monitor cultures regularly during this period. Some cell death will occur, and cultures need to be washed with Mg2+/Ca2+‐free PBS periodically to remove dead cells.
Observe colony growth and expansion daily to determine whether cleaning (removal of cells with non‐epithelial morphology) is required.
-
59
Cultures may need cleaning to remove non‐keratinocytes. Cleaning should be performed by scratching non‐epithelial cells off the plate using a 200‐µl filtered pipette tip.
Figures 4A‐D show cultures that have acquired the cobblestone morphology as expected of keratinocytes, as well as colonies that contain cells with different morphologies and that require cleaning (see figure legend for details).
Figure 4.
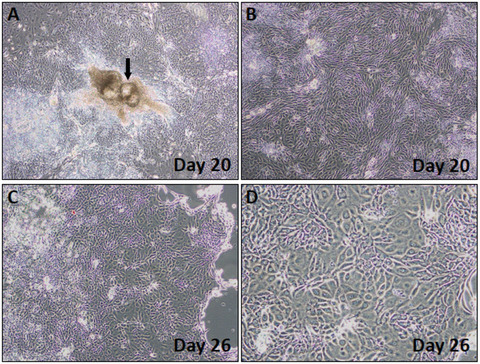
Phase‐contrast images showing colony morphologies observed during the selection phase. (A,B) Day 20 colonies. Arrow in (A) points to a colony with abnormal morphology. (B) Day 20 colony with the expected cobblestone cell morphology (C,D) Day 26 cells with emerging keratinocyte morphology. Note the shiny edge in the colony shown in (C), a sign that the cells are ready to be passaged.
Growth and expansion phase (day 30‐50)
-
60Split the cells around day 30 when the colonies appear bright at the edges (Fig. 4C; higher magnification shown in Fig. 4D) as follows:
- Aspirate the medium and wash with 3 ml Mg2+/Ca2+‐free PBS.
- Add 3 ml prewarmed Accutase™ and incubate for 5 min in a 37°C incubator.
- Use an inverted microscope to check if the cells are detached. If not, incubate for another 3 min at 37°C.
- Collect all detached keratinocytes into a 15‐ml tube.
- Bring volume up to 15 ml with CNT‐07 medium.
- Centrifuge cells for 5 min at 150 × g, room temperature.
- Carefully aspirate the medium leaving the cell pellet intact.
- Bring volume up to 5 ml with CNT‐07 and pipet up and down to produce a single‐cell suspension.
- Count live cells as in step 36.
- Plate keratinocytes on Collagen IV‐coated plates at a density of 300,000 cells per 10‐cm dish (∼5000 cells per cm2) in CNT‐07 supplemented with 10 µM Rock inhibitor.
-
Replace the medium every other day with CNT‐07 (without Rock inhibitor).Figure 5A shows cells that have the expected keratinocyte morphology.
Figure 5.
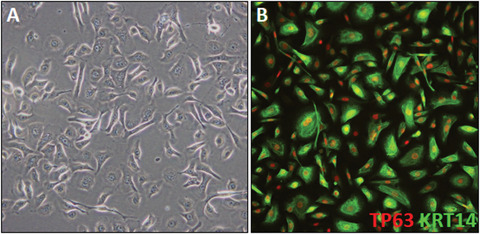
Fully differentiated iPSC‐K. (A) Phase‐contrast image of day 45 iPSC‐K. The cells show typical cobblestone keratinocyte morphology. A few cells appear to differ in morphology. These cells are mostly likely migratory leading to the elongated appearance. (B) Example of a day 43 culture labelled with TP63 and KRT14 antibodies.
-
61Split cells as in step 60 when keratinocytes are ∼60% confluent (5‐7 days). Repeat when cells reach 60% confluency. Expect to split cells a total of 2‐3 times.
- During these splits, plate keratinocytes on Collagen IV‐coated plates at a density of 500,000 cells per 10‐cm dish.
- At each passage, plate 20,000‐30,000 keratinocytes in one well of a Collagen IV‐ coated 8‐well chamber slide.
-
Perform immunofluorescence staining for TP63/KRT14 to assess the extent to which cells have acquired a keratinocyte fate (Fig. 5B and Support Protocol 5)Note that keratinocyte proliferation will start to slow around day 45‐50. Cells should be used immediately if a significant reduction in proliferation is observed.In our experience freezing iPSC‐K leads to a drastic loss of viability. We recommend using cells when they meet the pre‐determined quality control criteria (i.e., >95% TP63/KRT14‐ positive).
Support Protocol 1. COATING CELL CULTURE DISHES OR PLATES WITH VITRONECTIN XF™
This protocol describes the coating of dishes or plates with Vitronectin XF™ for the plating of human iPSC.
Materials
Vitronectin XF™ (Stemcell Technologies, cat. no. 100‐0763)
CellAdhere™ dilution buffer (Stemcell Technologies, cat. no. 07183)
PBS Ca2+/Mg2+‐free (ThermoFisher Scientific, cat. no. 10010023)
Biosafety cabinet (Thermo Scientific Hera‐Safe 2030i, or equivalent)
15‐ml polypropylene tubes (Fisher Scientific, cat. no. 22‐010‐072)
Fisher 6‐well plates (Fisher Scientific, cat. no. 09‐761‐146)
CELLTREAT Scientific Products 10cm Tissue Culture Treated Dish (Fisher Scientific, cat. no. 50202029)
-
1
Dilute Vitronectin XF™ in CellAdhere™ dilution buffer to a concentration of 10 µg/ml by adding 40 µl Vitronectin XF™ to 1 ml CellAdhere™ dilution buffer in a 15‐ml tube. Gently mix by pipetting up and down a few times.
Diluted Vitronectin XF™ should be prepared fresh prior each use.
-
2
Add 1 ml diluted Vitronectin XF™ per well of a 6‐well plate or 6 ml per 10 cm plate. Gently rock plates for even coating.
-
3
Incubate for 1 hr at room temperature.
-
4
Aspirate Vitronectin XF™ and wash with 2 ml Mg2+/Ca2+‐free PBS.
-
5
Add iPSC to freshly coated plates.
Do not allow Vitronectin XF™ to dry out before adding cells.
Unused Vitronectin XF™ stock can be stored at 4°C for up to 2 weeks or frozen in aliquots and stored at −20°C. Avoid repeated freeze and thaw cycles.
Support Protocol 2. FREEZING iPSC
This protocol describes the freezing of human iPSC for future use.
Materials
2× freezing medium (see recipe)
mTESR™ Plus medium kit (Stemcell Technologies, cat. no. 100‐0276)
Phosphate‐buffered saline (PBS) Ca2+/Mg2+‐free (ThermoFisher Scientific, cat. no. 10010023)
Accutase™ (Stemcell Technologies, cat. no. 07922)
Isopropanol (Fisher Scientific, cat. no. HC‐500‐1Gal)
Liquid nitrogen
Biosafety cabinet (Thermo Scientific Hera‐Safe 2030i, or equivalent)
Water bath
0.2‐µm syringe filter (Fisher Scientific, cat no. 09‐754‐29)
Cell culture incubator
Glass aspirators (Fisher Scientific, cat. no. 13‐678‐20A)
15‐ml polypropylene tubes (Fisher Scientific, cat. no. 22‐010‐072)
Centrifuge with rotor appropriate for 15‐ml tubes (Eppendorf 5804 rotor A‐4‐4, or equivalent)
Externally and Internally Threaded Cryogenic Storage Vials (Fisher Scientific, cat. no. 12‐567‐500)
Thermo Scientific™ Mr. Frosty™ Freezing Container (Fisher Scientific, cat. no. 5100‐0050)
−80°C freezer
NOTE: iPSC should be ∼60% confluent prior to freezing (Fig. 2A).
-
1
Prepare 2× freezing medium, sterile filter using a 0.2‐µm syringe filter, and place on ice for at least 20 min.
The freezing medium should be ice‐cold throughout to prevent cell death.
-
2
Replace medium with fresh mTESR™ Plus medium 2 hr prior to freezing.
-
3
Aspirate medium from iPSC.
-
4
Wash the cells with ∼2 ml Mg2+/Ca2+‐free PBS per 10‐cm dish.
-
5
Add 3 ml Accutase™ per 10‐cm dish.
-
6
Incubate in cell culture incubator for 5 min.
-
7
Add 5 ml mTESR™ Plus medium and gently transfer the cell suspension into a 15‐ml conical tube.
Keep the cells in small clusters of about 50‐200 µm in diameter during this step.
Avoid generating a single‐cell suspension as this will reduce cell viability upon thawing.
-
8
Centrifuge for 5 min at 150 × g, room temperature. While centrifuging, label six cryovials per 10‐cm plate.
-
9
Aspirate the medium and resuspend the cell pellet in 3 ml mTESR™ Plus medium.
-
10
Add 3 ml of 2× freezing medium in a careful dropwise manner.
-
11
Gently mix the cell suspension by inverting the tube ∼5 times.
-
12
Transfer 1 ml of cell suspension into each cryovial.
-
13
Transfer the vials to Thermo Scientific™ Mr. Frosty™ Freezing Container filled with isopropanol (at room temperature).
-
14
Transfer the Thermo Scientific™ Mr. Frosty™ Freezing Container to −80°C freezer.
-
15
Transfer the vials to liquid nitrogen storage after 24 hr.
Support Protocol 3. Preparing AggreWell™400 6‐WELL PLATES FOR EB FORMATION
This protocol describes the preparation of AggreWell™400 6‐well plates for the formation of embryoid bodies (EB)
Materials
Anti‐adherence rinsing solution (Stemcell Technologies, cat. no. 07010)
Phosphate‐buffered saline (PBS) Ca2+/Mg2+‐free (ThermoFisher Scientific, cat. no. 10010023)
KnockOut DMEM (ThermoFisher Scientific, cat. no. 10829018)
hEB medium (see recipe)
Biosafety cabinet (Thermo Scientific Hera‐Safe 2030i, or equivalent)
AggreWell™400 (Stemcell Technologies, cat. no. 34421)
Centrifuge with plate spinner (Eppendorf 5804 rotor A2‐DW, or equivalent)
Inverted light microscope (Nikon TS100, or equivalent)
Glass aspirators (Fisher Scientific, cat. no. 13‐678‐20A)
5‐ml serological pipettes (Fisher Scientific, cat. no. 02‐923‐203)
-
1
Add 1.5 ml anti‐adherence rinsing solution to each well of an AggreWell™400 6‐well plate using a 5‐ml serological pipette.
-
2
Rock the plate to fully coat the well.
-
3
Centrifuge the plate for 5 min at 130 × g, room temperature, in a swinging‐bucket rotor fitted with plate holders.
Plates must be well‐balanced. This step will help remove air bubbles within the microwells.
-
4
Observe the plate under an inverted microscope to ensure that bubbles have been removed from microwells.
If bubbles remain trapped in any microwells, centrifuge at 130 × g, room temperature, for an additional 5 min.
-
5
Aspirate anti‐adherence rinsing solution.
-
6
Rinse each well twice, each time with 3 ml Mg2+/Ca2+‐free PBS.
-
7
Aspirate PBS and perform final wash with 2 ml Knockout‐DMEM medium. Leave some medium in the well after aspirating.
-
8
Add 2 ml prewarmed hEB medium to each well.
-
9
Prepare the cells according to the Basic Protocol and add 3 ml of the cell suspension prepared in step 38 of the Basic Protocol. The total volume per well will be 5 ml.
Support Protocol 4. COATING CELL CULTURE DISHES OR PLATES WITH COLLAGEN IV
This protocol describes the coating of dishes or plates with Collagen IV for the plating and culture of EBs and iPSC‐K.
Materials
Type IV collagen from human placenta (Sigma, cat. no. C5533)
Acetic acid (Fisher Scientific, cat. no. A38‐500)
Phosphate‐buffered saline (PBS) Ca2+/Mg2+‐free (ThermoFisher Scientific, cat. no. 10010023)
Biosafety cabinet (Thermo Scientific Hera‐Safe 2030i, or equivalent)
50‐ml polypropylene tubes (Fisher Scientific, cat. no. 05‐539‐9)
10‐ml serological pipettes (Fisher Scientific, cat. no. 02‐923‐204)
Fisher 6‐well plates (Fisher Scientific, cat. no. 09‐761‐146)
CELLTREAT Scientific Products 10‐cm Tissue Culture Treated Dish (Fisher Scientific, cat. no. 50202029
Thermo Scientific NuncLab‐TekII Chamber Slide System (Fisher Scientific, cat. no. 12‐565‐8)
Glass aspirators (Fisher Scientific, cat. no. 13‐678‐20A)
-
1
Dissolve collagen IV in cold 0.25% acetic acid at a concentration of 1 mg/ml overnight in a 4°C refrigerator.
Overnight incubation is essential to ensure all Collagen IV is in solution.
-
2
Make 400‐µl aliquots and store up to 1 year at −20°C.
-
3
To use, thaw one aliquot of Collagen IV and add to 39.6 ml sterile cold Mg2+/Ca2+‐free PBS in a 50‐ml conical tube.
-
4
Coat wells/dishes by adding diluted Collagen IV.
Use 3 ml for a 10‐cm plate, 1 ml per well of a 6‐well plate, or 200 µl per well of an 8‐well chamber slide.
-
5
Incubate for 1 hr at room temperature.
-
6
Aspirate Collagen IV solution before adding cells.
Coated plates can be stored up to 2 weeks at 4°C.
Support Protocol 5. IMMUNOFLUORESCENCE STAINING OF CELLS
This protocol describes the immunofluorescence staining of iPSC‐K with antibodies against TP63 and KRT14. This is necessary to determine the percentage of keratinocytes (i.e., cells that express both TP63 and KRT14) in iPSC‐K cultures.
Materials
Keratinocytes (generated in this protocol; also see step 61 of the Basic Protocol)
CNT‐07 media 500 ml kit (ZenBio, cat. no. Cnt‐07)
Rock inhibitor (BD Biosciences, cat. no. 562822)
Embryonic stem cell‐qualified FBS (R&D Systems, cat. no. S10250)
Phosphate‐buffered saline (PBS) Ca2+/Mg2+‐free (ThermoFisher Scientific, cat. no. 10010023)
Methanol (Sigma, cat. no. 179337)
Protease‐free bovine serum albumin (BSA; ThermoFisher Scientific, cat. no. BP9706100)
p63‐α (D2K8X) XP Rabbit mAb (Cell Signaling, cat. no. 13109)
Anti‐Keratin K14 guinea pig polyclonal (Progen, cat. no. GP‐CK14)
Goat anti‐Rabbit IgG (H+L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 594 (ThermoFisher Scientific, cat. no. A11037)
Goat anti‐Guinea Pig IgG (H+L) Highly Cross‐Adsorbed Secondary Antibody, Alexa Fluor 488 (ThermoFisher Scientific, cat. no. A11073)
DAPI Fluoromount‐G (Southern Biotech, cat. no. 010020)
Biosafety cabinet (Thermo Scientific Hera‐Safe 2030i, or equivalent)
Water bath
Thermo Scientific NuncLab‐TekII Chamber Slide System (Fisher Scientific, cat. no. 12‐565‐8)
Cell culture incubator Thermo Scientific Forma series II water‐jacketed (Fisher Scientific, cat. no. 13‐998074)
Humidifier chamber (Tedpella cat. no. 21053)
Upright fluorescence microscope with 10×, 20×, and 40× objectives (Nikon, or equivalent)
Glass aspirators (Fisher Scientific, cat. no. 13‐678‐20A)
Fisherbrand Microcentrifuge tubes (Fisher Scientific, cat. no. 05‐408‐129)
Glass coverslips (Fisher Scientific, cat. no. 12‐544‐18)
-
1
Plate 20,000‐30,000 keratinocytes in a well of a Collagen IV‐coated 8‐well chamber slide (step 61 of Basic Protocol). Plate cells in 200 μl CNT‐07 supplemented with 10 μM Rock inhibitor and 2% ES cell‐qualified FBS.
ES cell‐qualified FBS improves the plating efficiency of keratinocytes in chamber slides.
-
2
Culture chamber slide overnight in a cell culture incubator.
-
3
Two hours before starting the immunofluorescence staining, change the media to fresh CNT‐07 medium (without Rock inhibitor or FBS).
The following steps are performed at room temperature unless otherwise indicated.
-
4
Gently wash the wells 3 times, each time with ∼500 µl Mg2+/Ca2+‐free PBS.
-
5
Fix cells in 500 µl pre‐cooled (stored in −20°C freezer) methanol for 4 min.
-
6
Aspirate methanol using a glass aspirator and wash the wells 3 times, each time with ∼500 µl PBS.
-
7
Block with 100 µl of 10% protease‐free BSA (in PBS) for 30 min at room temperature.
-
8
While blocking, prepare 100 µl primary antibody dilution in 0.1% protease‐free BSA per well in a 1.5‐ml microcentrifuge tube.
Primary antibodies are rabbit‐anti‐TP63 antibody (1:100) and guinea pig‐anti‐KRT14 (1:100).
-
9
Aspirate excess blocking buffer.
-
10
Add 100 µl primary antibody mix per well.
-
11
Incubate overnight at 4°C in a humidified chamber.
A humidified chamber can be produced by placing wet paper towels on the bottom of the chamber. Ensure that the wet paper towels do not touch the slides.
-
12
Wash the cells 3 times, each time with 200 µl PBS for 5 min each.
-
13
While washing, prepare 100 µl secondary antibody dilution in 0.1% protease‐free BSA per well: in a 1‐5 ml microcentrifuge tube
Secondary antibodies are Alexa fluor 594 goat‐anti‐rabbit (1:200) and Alexa fluor 488‐ goat‐anti‐guinea pig (1:200).
Note that these are examples of appropriate secondary antibodies. Other species‐ appropriate secondary antibodies may be substituted.
-
14
Add 100 µl secondary antibody per well.
-
15
Incubate 1‐2 hr in a dark humidified chamber.
-
16
Wash the cells three times, each time with 200 µl PBS for 5 min each.
-
17
Remove the chambers using the tool provided with the chamber slides and scrape off any remaining glue.
-
18
Air dry the slides.
-
19
Mount the slides in Fluoromount‐G mounting medium with DAPI using glass coverslips.
-
20
Store the slides in the dark at room temperature.
-
21
Using a fluorescence microscope, count the number of TP63/KRT14 expressing cells.
Cells can be used if cultures are >95% TP63/KRT14‐positive (Fig. 5B).
REAGENTS AND SOLUTIONS
CNT‐07
CNT‐BM.1 (ZenBio, cat. no. Cnt‐07)
1 vial Supplement A
1 vial Supplement B
1 vial Supplement C
1% penicillin‐Streptomycin (5,000 U/ml; ThermoFisher Scientific, cat. no. 15070063)
Store up to 1 month at 4°C
Defined K SFM (DKSFM)
1 vial Defined Keratinocyte‐SFM Growth Supplement (ThermoFisher Scientific, cat. no. 10744019
1% Penicillin‐Streptomycin (5,000 U/ml; ThermoFisher Scientific, cat. no. 15070063)
Store up to 1 month at 4°C
Freezing medium, 2×
80% Embryonic stem cell‐qualified FBS (R&D Systems, cat. no. S10250)
20% dimethyl sulfoxide (DMSO; Sigma, cat. no. D2650)
Make fresh each time and cool on ice for at least 20 min before use.
hEB (human embryoid body) medium
75.9 ml KnockOut‐DMEM (ThermoFisher Scientific, cat. no. 10829018)
20 ml KnockOut‐Serum Replacement (ThermoFisher Scientific, cat. no. 10828028)
1 ml l‐glutamine (200 mM) (ThermoFisher Scientific, cat. no. 25030081) or glutaMAX
1 ml Penicillin/Streptomycin (5,000 U/ml) (ThermoFisher Scientific, cat. no. 15070063)
1 ml Non‐essential amino acids (NEAA; 100 ×; ThermoFisher Scientific, cat. no. 11140050)
1 ml 10 mM Rock Inhibitor (100 µM final)
0.1 ml 2‐mercaptoethanol (Gibco, cat. no. 21985023)
Store up to 1 week at 4°C
mTeSR™ Plus
mTeSR™ Plus Basal Medium
1 bottle mTeSR™ Plus 5× Supplement
Store up to 2 weeks at 4°C
COMMENTARY
Background Information
One of the obstacles encountered by investigators working on rare genetic diseases is the limited availability of patient material (Koch & Koster, 2021). This limitation has hampered research into human genetic skin disorders, many of which are extremely rare but dramatically impact the quality of life of affected individuals. Because of the paucity of patient material, it is often not possible to conduct the extensive experiments necessary to identify disease mechanisms and therapeutic targets. iPSC technology has opened the door to generating an unlimited source of genetically defined stem cells that can be differentiated into somatic cells, such as epidermal keratinocytes. Further, genome editing techniques can be applied to correct disease‐causing mutations, thereby creating pairs of genetically identical cell lines that differ only with respect to the presence of a mutation. These conisogenic pairs of cells are ideal tools to analyze disease mechanisms (Dinella et al., 2018).
This protocol will yield a potentially unlimited amount of starting material for cell biological experiments. Further, this approach allows for transcriptome and proteome studies using genetically defined patient‐derived cells. These studies would be essentially impossible to conduct if dependent on primary patient material.
The present protocol utilizes exposure to retinoic acid (RA) and bone morphogenetic protein 4 (BMP4) to drive keratinocyte differentiation of fibroblast‐derived iPSC (Dinella et al., 2018; Itoh, Kiuru, Cairo, & Christiano, 2011; Koch et al., 2014; Petrova et al., 2014). The protocol will yield approximately 8 × 106‐ 1.6 × 107 keratinocytes after 45‐55 days. As outlined below, the yield of appropriate cells and duration of the differentiation protocol depend on various factors, including cell culture media and extracellular matrices used to generate iPSC‐K.
Critical Parameters
A prerequisite for the successful generation of iPSC‐derived somatic cells is the availability of high‐quality iPSC cell lines. iPSC lines are routinely produced by individual laboratories, academic core facilities, and commercial entities. Each of these might use a different approach to generate iPSC and researching the protocols used to generate iPSC can be challenging. Initially, many iPSC lines were generated using viral vectors that integrate into the DNA (Park, Lerou, Zhao, Huo, & Daley, 2008; Takahashi et al., 2007). Although effective, these techniques generally leave a genetic footprint in the genome. This genetic footprint can affect the ability of iPSC to properly differentiate into somatic cells as well as the cellular and molecular characteristics of the resulting somatic cells. Newer approaches utilize non‐integrating vector systems or RNA reprogramming technologies that do not result in permanent modifications of the genomic DNA (Brouwer, Zhou, & Nadif Kasri, 2016; Scesa, Adami, & Bottai, 2021; Schambach, Cantz, Baum, & Cathomen, 2010). Another factor to consider is the original cell type used to generate the iPSC. As a rule of thumb, it is advantageous to generate iPSC from cells in the same lineage as the cells that will be generated through directed iPSC differentiation (e.g., use of hematopoietic stem cells for iPSC generation if these cells are later used to produce lymphocytes; (Scesa et al., 2021)). Finally, it is of importance to note that even iPSC clones derived from the same cell source might differ in their ability to differentiate into the desired somatic lineage. Underlying reasons for this include genetic heterogeneity, for example if the source of cells used for reprogramming was isolated from sun‐exposed skin, and the accumulation of mutations during extended iPSC culture (Kuijk et al., 2020). It is therefore useful to conduct preliminary tests to determine if the iPSC lines used can generate cells representing mesodermal, endodermal, and ectodermal cell lineages (for example by using the StemCell Technologies STEMdiffTM trilineage differentiation kit, cat. no. 05230). This is of particular importance if different somatic lineages will be generated from the same iPSC line (e.g., keratinocytes and fibroblasts).
One of the persistent challenges in the field has been the development of protocols that reliably generate a homogeneous population of the desired somatic cell type. As outlined below, identifying, and removing undesired iPSC‐derived cell populations during differentiation is essential for the successful generation of keratinocytes. Learning to identify undesired cells based on morphological criteria requires experience and a good eye. Therefore, it is essential that quality control criteria are established to define the cell population used for experimentation (e.g., percentage of TP63 and KRT14 double‐positive cells in this protocol). Additional markers, such as ITGA6, ITGB4, DSC3, and DSG3, can be used to further define the desired cell population. Further, FACS is a useful tool to obtain near homogeneous populations of the desired cell types if dealing with heterogenous cell cultures.
The recent supply chain disruptions have posed a significant obstacle in maintaining and differentiating iPSC. For example, media and extracellular matrices have not been consistently available from suppliers. As the use of different reagents will lead to the generation of iPSC‐K with different morphology and gene expression patterns, they cannot be used interchangeably. It is therefore essential to plan these experiments well and obtain all reagents in sufficient amounts prior to starting the differentiation program.
iPSC‐K differentiation will yield keratinocytes with a limited lifespan. Prolonged culture of these cells will lead to a reduction in the proliferation rate and eventually a loss of the culture. In our experience, iPSC‐ K should be used at around 45‐55 days, when >95% of the cells are TP63/KRT14‐positive. Cultures maintained longer will show reduced cell proliferation, reduced plating efficiency, and increased cell death.
Lastly, iPSC cell culture and differentiation, especially in terms of “cleaning” cultures by removing undesired cells and colonies during directed differentiation, require considerable experience. Expect a steep learning curve to achieve optimal results.
Troubleshooting
Please see Table 1 for a list of common problems, their possible causes, and recommended solutions.
Table 1.
Troubleshooting Guide for iPSC Differentiation into Keratinocytes
| Problem | Possible Cause | Solution |
|---|---|---|
| iPSC do not attach to plate | Insufficient coating of plate | Ensure plates are coated for at least 1 hr with Vitronectin XF™ |
| Inappropriate media | Ensure Rock inhibitor has been added to media | |
| Inappropriate Accutase™ treatment | Ensure iPSC were passaged as small clumps, not as single cells | |
| Equipment failure | Check appropriate temperature and CO2 concentration of cell culture incubator | |
| Poor detachment of iPSC during dissociation | Incomplete Accutase™ treatment |
Wash cells with PBS (Ca2+ and Mg2+‐free) Extend Accutase™ treatment up to 10 min. Ensure Accutase™ is prewarmed. |
| EBs do not attach to Collagen IV‐coated plate | Inappropriate media | Ensure 2% FBS and Rock inhibitor were added to the media |
| Inappropriate plate coating | Ensure plates were coated for at least 1 hr | |
| Large number of non‐ keratinocyte colonies in day 20‐26 cultures | Media issues | Ensure BMP4 and RA are not expired and at the correct concentration |
| Insufficient cleaning of cultures |
Manually remove non‐keratinocyte colonies and cells. Remove non‐keratinocytes by incubating with Accutase™ for 2 min and discarding cells that detach. After that, a normal split can be performed. |
|
| Poor attachment of keratinocytes to Collagen IV‐coated plates | Media issue | Add 10 μM Rock inhibitor to media |
| Plate coating issue | Coat plates at least 1 hr with Collagen IV | |
| Heterogeneous keratinocyte cultures | Ineffective differentiation | Enrich keratinocytes by FACS using cell surface antigens such as α6 integrin and β4 integrin |
Understanding Results
The procedure detailed in this protocol is expected to result in the generation of ∼8 × 106‐1.6 × 107 keratinocytes in 45‐55 days. To confirm that the cells generated through this protocol represent normal keratinocytes, several experiments can be performed. In our laboratory, we routinely determine the expression and localization of well‐established keratinocyte markers in iPSC‐K. For example, exposure of normal human keratinocytes to Ca2+ (1.3 mM) leads to well‐documented changes in cell morphology, gene and protein expression, and protein localization (Bikle, Xie, & Tu, 2012; Kowalczyk & Green, 2013). In Figure 6, we show examples of immunofluorescence staining and Western blot analysis of normal human keratinocytes (NHEK) and iPSC‐K. Note the robust expression and localization of the cell junction proteins DSC3, DSG3, and β‐catenin to the cell‐cell borders as expected (Fig. 6A‐G). Other approaches to determine that keratinocytes were derived could include a comparative transcriptome analysis with primary human keratinocytes.
Figure 6.
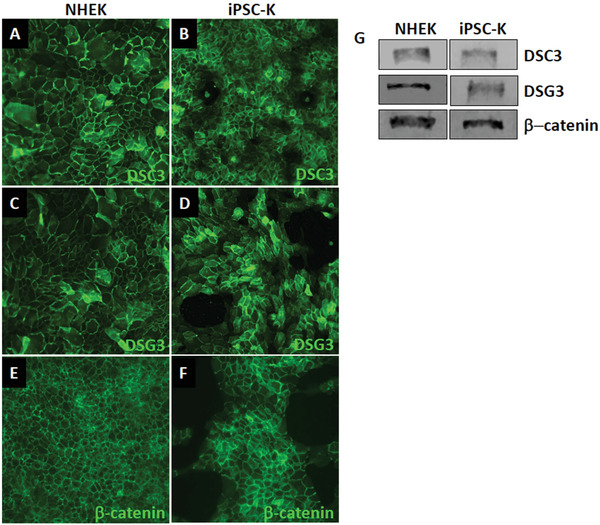
Expression of keratinocyte markers in iPSC‐K. iPSC‐K and primary human epidermal keratinocytes were exposed to 1.3 mM Ca2+ for 48 hr. (A‐F) Immunofluorescence staining and (G) Western blot analysis demonstrates normal expression and localization of desmosomal proteins (DSC3, DSG3) and an adherens junction protein (β‐catenin). Antibodies used for immunofluorescence staining are: DSG3 (clone 5H10; courtesy of Dr. James K Wahl III, PhD, University of Nebraska Medical Center, Lincoln, NE), DSC3 (Progen cat. no. 61093), and β‐catenin (Santa Cruz cat. no. sc‐7963). Antibodies used forlotting are: DSG3 (Invitrogen cat. no. MA5‐16025), DSC3 (Progen cat. no. 61093), and β‐ catenin (Santa Cruz cat. no. sc‐7963).
Time Considerations
Starting from EB formation, the protocol will take approximately 45‐55 days to complete. Preparing the iPSC prior to the start of EB formation will take approximately 5 days. Figure 1A shows a detailed timeline of the critical steps of the differentiation program.
Author Contributions
Peter Koch: Conceptualization, Formal analysis, Funding acquisition, Methodology, Supervision, Writing — original draft, Writing — review & editing; Saiphone Webb: Investigation, Methodology, Writing — original draft, Writing — review & editing; Jessica Gugger: Investigation, Methodology, Writing — review & editing; Maddison Salois: Investigation, Methodology, Writing — original draft, Writing — review & editing; Maranke Irene Koster: Conceptualization, Formal analysis, Funding acquisition, Methodology, Supervision, Writing — original draft, Writing — review & editing.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
This work has been supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and the National Eye Institute (NEI) of the National Institutes of Health (NIH) under award numbers R01AR072621 and R21EY029081 (awarded to PJK and MIK). This work has also been supported by the National Foundation for Ectodermal Dysplasias (NFED; awarded to PJK and MIK).
Koch, P. J. , Webb, S. , Gugger, J. A. , Salois, M. N. , & Koster, M. I. (2022). Differentiation of human induced pluripotent stem cells into keratinocytes. Current Protocols, 2, e408. doi: 10.1002/cpz1.408
Data Availability Statement
The data that support the protocol are available from the corresponding author upon reasonable request.
Literature Cited
- Bikle, D. D. , Xie, Z. , & Tu, C. L. (2012). Calcium regulation of keratinocyte differentiation. Expert Review of Endocrinology & Metabolism, 7(4), 461–472. doi: 10.1586/eem.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer, M. , Zhou, H. , & Nadif Kasri, N. (2016). Choices for induction of pluripotency: Recent developments in human induced pluripotent stem cell reprogramming strategies. Stem Cell Reviews and Reports, 12(1), 54–72. doi: 10.1007/s12015-015-9622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, S. , Liu, X. , Meyers, C. , Schlegel, R. , & McBride, A. A. (2010). Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. The Journal of Clinical Investigation, 120(7), 2619–2626. doi: 10.1172/jci42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinella, J. D. , Chen, J. , Webb, S. , Siegfried, E. , Bree, A. F. , Lakshmanachetty, S. , … Koch, P. J. (2018). A human stem cell‐based system to study the role of TP63 mutations in ectodermal dysplasias. Journal of Investigative Dermatology, 138(7), 1662–1665. doi: 10.1016/j.jid.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, H. , Rheinwald, J. G. , & Sun, T. T. (1977). Properties of an epithelial cell type in culture: The epidermal keratinocyte and its dependence on products of the fibroblast. Progress in Clinical and Biological Research, 17, 493–500. [PubMed] [Google Scholar]
- Itoh, M. , Kiuru, M. , Cairo, M. S. , & Christiano, A. M. (2011). Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa‐induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America, 108(21), 8797–8802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, M. , Umegaki‐Arao, N. , Guo, Z. , Liu, L. , Higgins, C. A. , & Christiano, A. M. (2013). Generation of 3D skin equivalents fully reconstituted from human induced pluripotent stem cells (iPSCs). PLoS One, 8(10), e77673. doi: 10.1371/journal.pone.0077673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackow, J. , Guo, Z. , Hansen, C. , Abaci, H. E. , Doucet, Y. S. , Shin, J. U. , … Christiano, A. M. (2019). CRISPR/Cas9‐based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proceedings of the National Academy of Sciences of the United States of America, 116(52), 201907081. doi: 10.1073/pnas.1907081116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, P. J. , Dinella, J. , Fete, M. , Siegfried, E. C. , & Koster, M. I. (2014). Modeling AEC‐New approaches to study rare genetic disorders. American Journal of Medical Genetics Part A, 164A(10), 2443–2454. doi: 10.1002/ajmg.a.36455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, P. J. , & Koster, M. I. (2021). Rare genetic disorders: Novel treatment strategies and insights into human biology. Frontiers in Genetics, 12, 714764. doi: 10.3389/fgene.2021.714764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk, A. P. , & Green, K. J. (2013). Structure, function, and regulation of desmosomes. Progress in Molecular Biology and Translational Science, 116, 95–118. doi: 10.1016/B978-0-12-394311-8.00005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijk, E. , Jager, M. , van der Roest, B. , Locati, M. D. , Van Hoeck, A. , Korzelius, J. , … Cuppen, E. (2020). The mutational impact of culturing human pluripotent and adult stem cells. Nature Communication, 11(1), 2493. doi: 10.1038/s41467-020-16323-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, I. H. , Lerou, P. H. , Zhao, R. , Huo, H. , & Daley, G. Q. (2008). Generation of human‐induced pluripotent stem cells. Nature Protocol, 3(7), 1180–1186. doi: 10.1038/nprot.2008.92. [DOI] [PubMed] [Google Scholar]
- Petrova, A. , Celli, A. , Jacquet, L. , Dafou, D. , Crumrine, D. , Hupe, M. , … Ilic, D. (2014). 3D In vitro model of a functional epidermal permeability barrier from human embryonic stem cells and induced pluripotent stem cells. Stem Cell Reports, 2(5), 675–689. doi: 10.1016/j.stemcr.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scesa, G. , Adami, R. , & Bottai, D. (2021). iPSC preparation and epigenetic memory: Does the tissue origin matter? Cells, 10(6), 1470. doi: 10.3390/cells10061470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambach, A. , Cantz, T. , Baum, C. , & Cathomen, T. (2010). Generation and genetic modification of induced pluripotent stem cells. Expert Opinion on Biological Therapy, 10(7), 1089–1103. doi: 10.1517/14712598.2010.496775. [DOI] [PubMed] [Google Scholar]
- Sebastiano, V. , Zhen, H. H. , Haddad, B. , Bashkirova, E. , Melo, S. P. , Wang, P. , … Oro, A. E. (2014). Human COL7A1‐corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Science Translational Medicine, 6(264), 264ra163. doi: 10.1126/scitranslmed.3009540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K. , Tanabe, K. , Ohnuki, M. , Narita, M. , Ichisaka, T. , Tomoda, K. , & Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131(5), 861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Rivera, T. , Zhao, Y. , Ni, Y. , & Wang, J. (2020). Human‐induced pluripotent stem cell culture methods under cGMP conditions. Current Protocols in Stem Cell Biology, 54(1), e117. doi: 10.1002/cpsc.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Hu, K. , Smuga‐Otto, K. , Tian, S. , Stewart, R. , Slukvin, I. I. , & Thomson, J. A. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science, 324(5928), 797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the protocol are available from the corresponding author upon reasonable request.