

Abstract
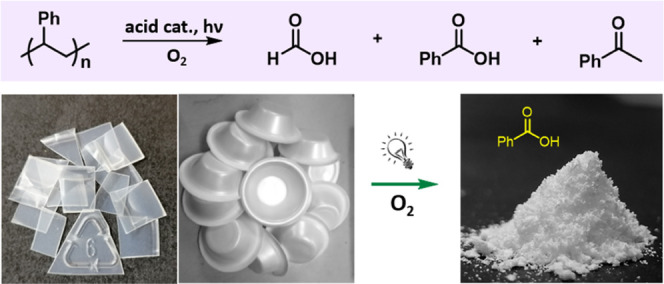
Chemical recycling is one of the most promising technologies that could contribute to circular economy targets by providing solutions to plastic waste; however, it is still at an early stage of development. In this work, we describe the first light-driven, acid-catalyzed protocol for chemical recycling of polystyrene waste to valuable chemicals under 1 bar of O2. Requiring no photosensitizers and only mild reaction conditions, the protocol is operationally simple and has also been demonstrated in a flow system. Electron paramagnetic resonance (EPR) investigations and density functional theory (DFT) calculations indicate that singlet oxygen is involved as the reactive oxygen species in this degradation process, which abstracts a hydrogen atom from a tertiary C–H bond, leading to hydroperoxidation and subsequent C–C bond cracking events via a radical process. Notably, our study indicates that an adduct of polystyrene and an acid catalyst might be formed in situ, which could act as a photosensitizer to initiate the formation of singlet oxygen. In addition, the oxidized polystyrene polymer may play a role in the production of singlet oxygen under light.
Introduction
Since the 1950s, synthetic plastics derived from petroleum have been widely used to improve the quality of people’s lives through clothing, food preservation, and medical applications, among many other domestic and industrial applications. Over the past 70 years, their production has risen sharply, from 1.5 million tonnes in 1950 to 368 million tonnes in 2019,1,2 and the production is projected to double again within the next 20 years.3 However, once these plastics serve their designated purpose, they pose a serious problem, as most of them are not recycled and do not degrade.4−6 Globally, 58% of discarded plastics end up in landfills or are incinerated,2,4,7,8 causing severe environmental pollution, including soil contamination, water contamination, air pollution, microplastic pollution, etc.9−16 Over the past several decades, some successes have been made to develop closed-loop life cycles for synthetic plastics via collection, separation, and mechanical recycling.17,18 Nevertheless, those successes are limited, as the recycled plastics can only be used for downgraded applications19−21 and eventually end up in landfills or used for energy recovery after a single recycle. In this regard, chemical recycling is considered one of the most promising solutions to the challenge posed by plastic waste,22,23 since this recycling method is able to retain the value of postconsumer polymers by converting them into their original monomers, fuels, or valuable chemicals with potential for upcycled applications.24−27 However, to date, chemical recycling is often more energy-intensive and expensive to implement, in comparison to mechanical recycling and incineration.8,19,28−30 Therefore, developing more efficient, low-cost methods for the chemical recycling of plastics has become a critical area of research in both chemistry and chemical engineering.31 In particular, the search for industrially applicable methods capable of selectively converting plastic wastes to valuable and isolable chemicals with narrow distribution is of utmost interest.
Polystyrene (PS), one of the most important materials in the modern plastic industry, has been widely used in our daily life from building materials, electronics, protective packaging, to food containers. Tens of millions of tonnes are produced annually, accounting for about 6% of the current global plastic market share.32 Since all atoms of PS are connected by strong C–C and C–H bonds, PS is remarkably inert and difficult to degrade without special treatment. Thermal and catalytic pyrolysis has been developed for chemical recycling of PS under an inert or hydrogen atmosphere (Scheme 1, method A).33−39 Generally, this technique requires high temperature (typically >300 °C), appropriate reactors, and catalysts to produce hydrocarbons with a narrow distribution, which lead to high costs.40−42 In the last two decades or so, there have been only three examples of catalytic oxidative degradation of PS, reporting aerobic oxidation of PS to form benzoic acid, but the reaction conditions tend to be harsh or complex with a long degradation time,43−45 which limits its practical application (Scheme 1, method B). Recently, a new method for postpolymerization modification of PS to the corresponding fluoroalkyl polymers has been developed by Leibfarth and co-workers (Scheme 1, method C).46 These modifications could lead to upcycled applications of PS waste, even though the upgraded polymer products are not likely to be further recyclable after use. It is clear that, until now, there appear to be no known efficient methods capable of chemically recycling PS under mild conditions. Herein, following on from our recent studies of dioxygen (O2) activation and aerobic oxidation reactions,47−52 we report a novel, simple selective degradation method that enables the oxidative cleavage of PS to benzoic acid, formic acid, and acetophenone by singlet oxygen (1O2) under ambient temperature and pressure with cheap, readily available inorganic or organic acids as a simple catalyst (Scheme 1).
Scheme 1. Methods for the Chemical Recycling of PS.
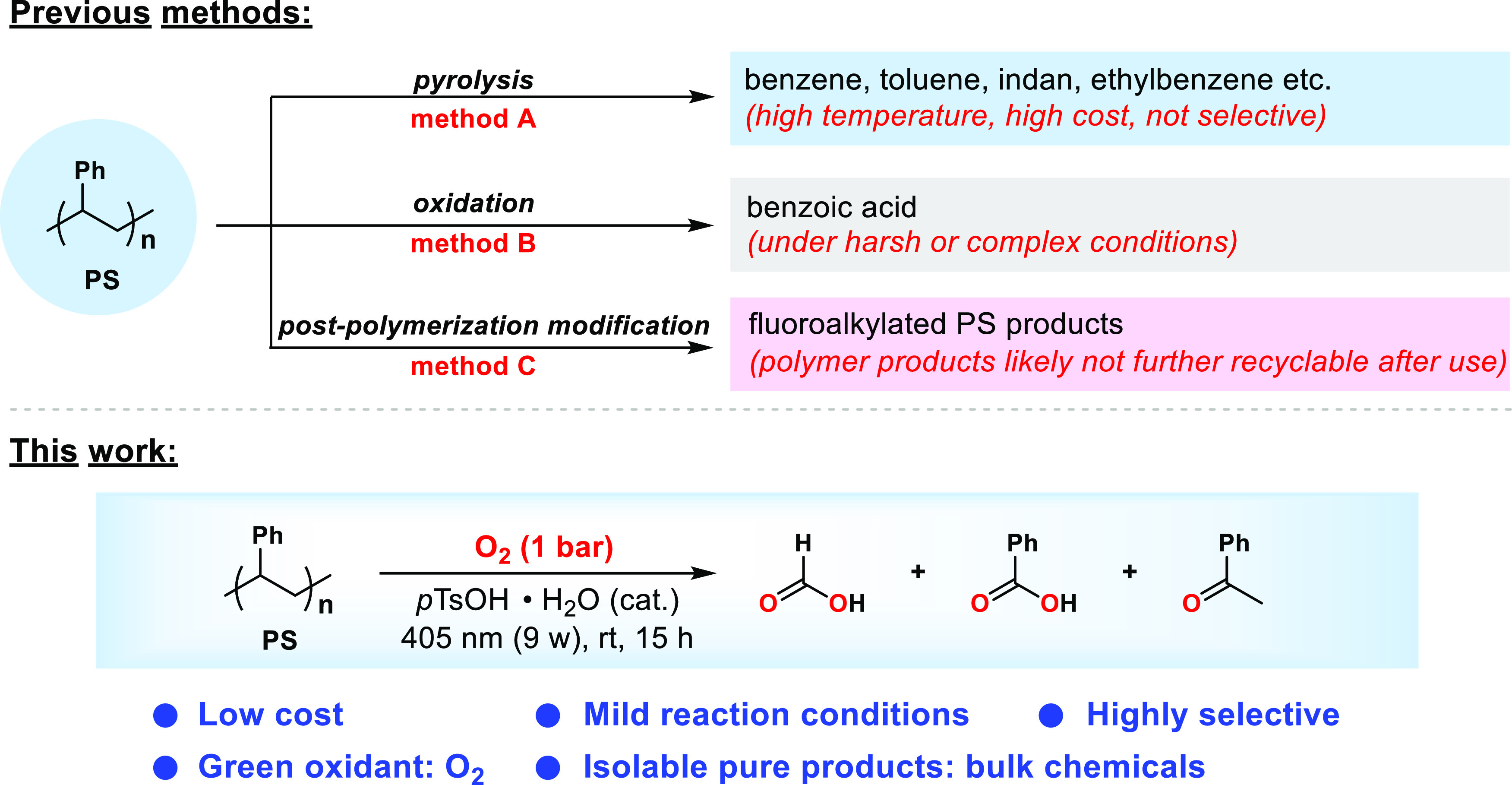
Singlet oxygen is a well-known reactive oxygen species (ROS), which has a relatively high energy of about 94 kJ/mol compared to the ground-state molecular O2.53 Therefore, 1O2 is able to initiate low-temperature oxidation of various organic molecules, inspiring a wide array of applications in chemical and biochemical reactions as well as treatment of organic wastes and contaminants.53−58 Based on this knowledge, we envisaged that 1O2 may be able to abstract hydrogen at the weak tertiary benzylic C–H bond in PS and thereby induce the chemical degradation of PS. Indeed, there are some methods in the literature regarding the 1O2-mediated degradation of PS under light irradiation.59−62 However, there are two main disadvantages present in these processes, limiting their practical application. One is that none of these 1O2-mediated degradation processes could selectively produce pure, valuable chemicals, as they often occur through an uncontrollable radical pathway,63,64 while the other is that expensive and/or toxic photosensitizers or initiators are required to produce 1O2 for the degradation.65,66 Hence, developing more practically applicable methods capable of producing 1O2 and subsequently achieving the degradation of PS in a highly selective manner is urgent. Our findings are presented below.
Results and Discussion
We commenced our exploration of selective aerobic degradation by employing commercial PS (F.W.: 192,000) as a model substrate. During the search for an able catalyst, we surprisingly found that triflic acid (5 mol %, HOTf) can catalyze the selective degradation of PS using O2 (1 bar) as an oxidant under the irradiation of violet-blue light (405 nm), affording isolable formic acid (1, 72%), benzoic acid (2, 40%), and benzophenone (3, 2%) products (Table 1, entry 1). Acid is an essential catalyst for this degradation reaction, without which the chemistry could not proceed (Table 1, entry 2). Further examinations also indicate that light irradiation plays an important role during this aerobic degradation, as no desired products were obtained without irradiation (Table 1, entry 3), and only a small amount of the corresponding products were detected under the irradiation at 475 nm (Table 1, entry 5). The yield achieved under 365 nm irradiation is slightly lower than that of 405 nm (entry 4). We then tested several acid catalysts under irradiation at 405 nm (Table 1, entries 6–9 and 17). The results show that trifluoroacetic acid (CF3COOH) and nitric acid (HNO3) are inactive, while, in addition to triflic acid, methanesulfonic acid (CH3SO3H), sulfuric acid (H2SO4), and p-toluenesulfonic acid monohydrate (pTsOH·H2O) are all good to afford the corresponding products. Several Lewis acids were also examined. Among them, only Sc(OTf)3 and La(OTf)3 afforded compounds 1–3 but in significantly lower yields (entries 27–30). Thereafter, the influence of the quantities of acid catalysts on the yield was investigated. The results showed that 5–10 mol % of an acid catalyst gave better yields (entries 10, 18, and 19), and a larger amount led to a yield drop for 1 (entries 11 and 20). This is likely a result of formic acid decomposition rather than suppression of the reaction by the acid formed (see Section 3.1 in SI). The choice of the reaction solvent revealed a great influence, as no or only trace amounts of target products were obtained when benzene, CH3CN, acetone, or EtOAc were employed (entries 12–15), and lower yields were obtained when DCE was used (entry 16). The effect of reaction time, concentration, as well as a solvent on product yields were further examined using pTsOH·H2O as a catalyst (entries 21–25). The combination of pTsOH·H2O (5 mol %) and benzene/CH3CN (1/1, 1 mL) for 15 h was able to afford optimum product yields, while benzene could be replaced by DCE or EtOAc to afford the desired products in slightly lower yields (see SI for more details).67 Note that the reaction was much less efficient when carried out in air (entry 26). Compared to the cheap inorganic acid H2SO4, the industrial-scale and milder pTsOH·H2O is much easier to handle. Therefore, our subsequent investigation was centered around using pTsOH·H2O (5 mol %) as a catalyst and O2 (1 bar) as an oxidant in benzene/CH3CN (1/1, 1 mL) with continuous violet-blue light irradiation at room temperature for 15 h. Note that under such conditions, the main byproduct resulting from the aerobic degradation is oxidized PS of a smaller average molecular weight. An example is seen in the reaction of PS (Mw = 172,389; Mz = 329,367; Mn = 26,934; Mp = 144,697; Mw/Mn = 6.400), in which oxidized PS of a considerably reduced molecular weight was observed (Mw = 41,014; Mz = 92,054; Mn = 15,378; Mp = 16,451; Mw/Mn = 2.667) (see Figures S2 and S3 for the detailed GPC results).
Table 1. Optimization of the Reaction Conditionsabcdefghij.

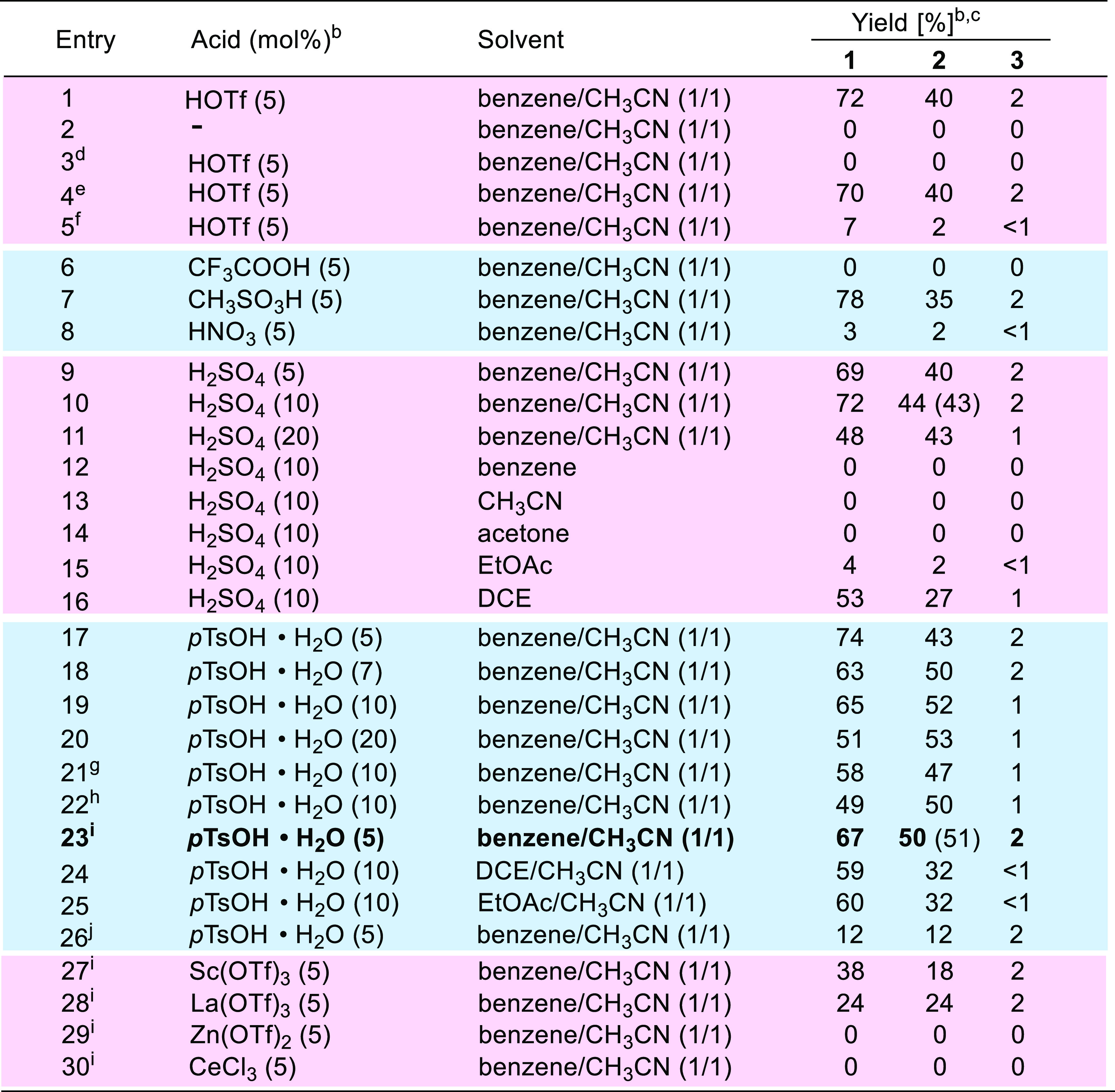
Reaction was carried out with 104 mg of PS in the presence of an acid catalyst in 2 mL of a solvent under O2 (1 bar) and violet-blue light (405 nm, 9 W) for 15 h.
Catalytic amount of acid and the yield of products are based on the single repeat unit (1 mmol) of polystyrene.
Yield determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard; isolated yield in parentheses.
Without light.
365 nm.
475 nm.
10 h.
24 h.
Benzene/CH3CN (1/1, 1 mL).
Under air.
With our optimized reaction conditions established, the aerobic degradation of commercial, pure PS, and PS waste from our daily life was investigated. As shown in Table 2, all PS materials were readily cleaved by O2 to the desired acid products in a highly selective manner. Thus, commercial pure PS of different average molecular weights could be oxidatively cleaved to afford 1 in a 57–67% NMR yield, 2 in a 36–51% isolated yield, and 3 in a 2–5% NMR yield (entries 1–3). Moreover, it seems that PS with a higher molecular weight could give better product yields. Remarkably, PS waste from cup lids, yogurt containers, loose-fill chips, EPS foam, food boxes, as well as laboratory weighing boats are all suitable, producing 1 in a 58–64% NMR yield, 2 in a 38–48% isolated yield, and 3 in a 2–3% NMR yield (entries 4-9). Poly(4-tert-butylstyrene) (F.W.: 50,000–100,000) was also tested, which could be selectively degraded to 1 in a 49% NMR yield, 4-tert-butylbenzoic acid 4 in a 50% isolated yield, and 4′-tert-butylacetophenone 5 in a 3% NMR yield (entry 10).
Table 2. Aerobic Degradation of Commercial Pure PS or PS Waste from Our Daily Lifeabcd.


Reaction was carried out with PS (104 mg) in the presence of pTsOH·H2O (9.5 mg) as the catalyst in 1 mL of benzene/CH3CN (1/1) under O2 (1 bar) and violet-blue light (405 nm, 9 W) for 15 h.
Catalytic amount of acid and the yield of products are based on the single repeat unit (1 mmol) of PS.
Yield determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard.
Isolated yield in parentheses.
It is worth noting that benzoic acid 2 could be isolated as a pure white crystalline powder from the above-mentioned degradation reactions, as shown in the images in Table 2. Meanwhile, the resulting formic acid could be converted to the isolable pure formanilide by the addition of 1.5 equiv of aniline or p-toluidine to the reaction mixture after oxidation. Examples are shown in Scheme 2, where a 55% yield of formanilide 6a and a 44% yield of 2 were isolated after the addition of aniline, while a 56% yield of 4′-methylformanilide 6b and a 48% yield of 2 were isolated after the addition of p-toluidine.
Scheme 2. Conversion of the Resulting Formic Acid to Isolable Pure Formanilide.

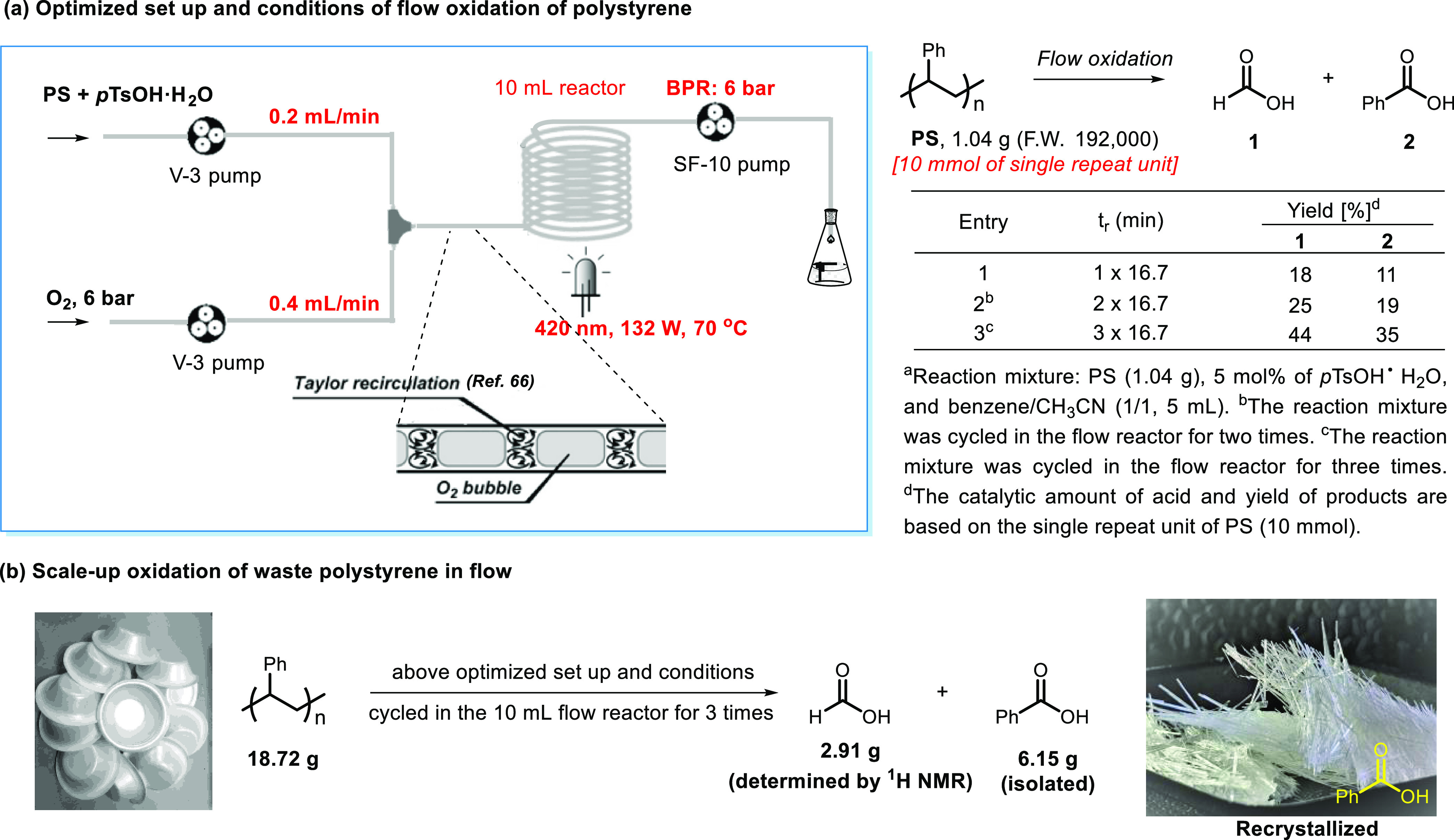
The practical applicability of this photo-acid-enabled protocol was further enhanced using the continuous-flow microreactor technology, which has been hailed as an enabling technology to scale-up operationally photochemical transformations.68,69 Pleasingly, after a systematic optimization (see the SI for more details), the flow degradation of PS was able to afford the desired products smoothly when a solution of PS and pTsOH·H2O was mixed with oxygen gas under 6 bar of pressure (BPR) at 70 °C in the presence of violet-blue light (Scheme 3a, entry 1). Note that the 405 nm light is normally more efficient than 420 nm for the aerobic degradation of PS; however, we chose a 420 nm, 132 W high power lamp because it is more easily available and offers acceptable yields (see Table S1 in the SI). Recycling the resulting reaction solution in the flow reactor one or two more times further increased the product yields (Scheme 3a, entries 2 and 3). With the established setup and flow conditions, a scale-up degradation of the waste PS food box was carried out, which produced pure crystalline benzoic acid and formic acid at a gram scale (Scheme 3b).
Scheme 3. Degradation of PS Enabled by Photocatalysis in Flow: (a) Optimized Setup and Conditions and (b) Gram-Scale Reaction (Note: the E-Series System from Vapourtec Was Used for This Transformation).
To shed light on possible reaction pathways, a range of control experiments were carried out. As singlet oxygen (1O2) was proposed as the ROS for the photodegradation of PS,63,64 we thought that it is important to first determine whether 1O2 indeed plays a role in our degradation system. When PS was subjected to the standard oxidation conditions but in the presence of DPA or NaN3 as a 1O2 trap or scavenger,70,71 no conversion or expected degradation products were observed (Scheme 4, eq 1). These observations indicate that 1O2 is likely the ROS involved in our photo-acid-enabled degradation reaction. Meanwhile, a radical trapping experiment was also conducted. As shown in Scheme 4, eq 2, no PS was converted to the target products under the standard oxidation conditions in the presence of TEMPO, suggesting that the degradation might occur via radical pathways, which is consistent with the reaction mechanism of 1O2.63,64,72
Scheme 4. Controlled Experiments in the Presence of an 1O2 Trap, a Scavenger (1), or a Radical Trap (2).

Further evidence on the generation of 1O2 was obtained by in situ electron paramagnetic resonance (EPR) spectroscopy measurements with 4-oxo-TMP (2,2,6,6-tetramethyl-4-piperidone), a well-known 1O2 trap that generates the nitroxyl radical 4-oxo-TEMPO (2,2,6,6-tetramethyl-4-piperidone-N-oxyl). Upon 405 nm irradiation of the reaction solution containing 4-oxo-TMP, the characteristic three-line spectrum is observed immediately but is not observed in an identical experiment under a N2 atmosphere (Figure S6A). Furthermore, control experiments show that this signal is enhanced by a known 1O2 photosensitizer (H2TPP; tetraphenyl porphyrin; Figure S6B) and is diminished by a 1O2 quencher (β-carotene; Figure S6B).
The subsequent formation of radicals in the depolymerization reaction was probed by in situ EPR reactions also in the presence of the radical trap DMPO (5,5-dimethyl-1-pyrroline N-oxide), although this is complicated because DMPO and TEMPO can inhibit reactivity and possibly undergo other reactions under the experimental conditions (see the SI). In experiments with 4-oxo-TMP and DMPO spin traps (1:4, excess DMPO is necessary), we observe complex spectra that can be deconvoluted into four components (identified by the observed hyperfine coupling patterns): a three-line nitroxyl spectrum (presumably 4-oxo-TEMPO and an unidentified DMPO-nitroxide, but not DMPO-X); an oxygen-centered DMPO adduct, most likely DMPO-OR (although DMPO-O2H and DMPO-OR have similar spectra, the former is not very persistent, and DMPO-OR forms more readily than DMPO-O2R);73 and a carbon-centered DMPO-R adduct (Figure 1). The nitroxyl and DMPO-OR adducts are detected initially; then, as they peak, the DMPO-R signal develops (Figure S10). Although we cannot unambiguously identify the radicals, O- and C-centered DMPO-radical adducts are quite distinct,74 and control experiments show that the formation of all three types of radical is much quicker in the presence of a PS substrate and an acid catalyst cf. one or neither (Figure S10).
Figure 1.

cw EPR spectra from in situ irradiation at 405 nm (1 mW LED) of PS and a pTsOH·H2O solution with 4-oxo-TMP and DMPO spin traps (1:4). Center: experimental spectra measured after 10 min (red) and 50 min (blue) irradiation. Left and right: simulated spectra of three separate components of a nitroxyl [g = 2.0056, aiso(14N) = 1. 5 mT], a carbon-centered DMPO adduct [g = 2.0055, aiso(14N) = 1.45 mT, aiso(β-1H) = 2.11 mT], and an oxygen-centered DMPO adduct [g = 2.0057, aiso(14N) = 1.29 mT, aiso(β-1H) = 1.03 mT, aiso(γ-1H) = 0.13 mT]. Simulations (red) of the experimental spectra (black) after 10 and 50 min (left and right, respectively) are weighted sums of these three components. The magnetic field positions marked in the central panel were used to monitor the separate components as function of time (Figure S10).
The formation of 1O2 under the current conditions raised another question, i.e., how is it formed in the absence of a photosensitizer? To answer the question, UV–vis experiments were performed. As shown in Figure 2A, no absorption band was observed at about 405 nm for both PS and pTsOH·H2O; however, when they were mixed together, an obvious absorption was detected at around 408 nm. A similar absorption was found in the mixture of PS and H2SO4 (or HOTf). All of these observations suggested that a [PS---H+] adduct resulting from the interaction of PS with the acid might be the photosensitizer that initiates the formation of 1O2 under the irradiation of violet-blue light.37 Meanwhile, the UV–vis spectrum of the isolated byproduct (oxidized PS, Mw: 41,014) was measured, which shows a strong absorption at 405 nm (Figure 2B). This result indicates that during the photochemical degradation, the in situ-formed oxidized PS byproduct could further boost the formation of 1O2.
Figure 2.

UV–vis spectra of PS, pTsOH·H2O, the mixture of PS and acid (pTsOH·H2O, H2SO4, or HOTf), and the byproduct ([PS]: 10 mM (based on single repeat unit); [acid]: 10 mM; [byproduct]: 1 mg/mL, in DCE).
To simplify the mechanistic investigation and gain further insight into the mechanistic possibilities, a styrene dimer, 1,3-diphenylbutane 7, was employed to replace PS as a model substrate. As shown in Scheme 5, 0.5 mmol of 7 could be photochemically oxidized to 0.55 mmol of 1, 0.39 mmol of 2, and 0.33 mmol of 3 under the standard oxidation conditions. According to this quantitative data, we suspected that there may be at least two pathways for this photochemical oxidation of 7, i.e., the main reaction pathway that affords 1 equiv of 1, 2, and 3, respectively, and some side reactions, e.g., one that leads to the formation of 2 equiv of 1 and 2, respectively.
Scheme 5. Oxidative Cleavage of 1,3-Diphenylbutane.

Following on from these experimental studies, density functional theory (DFT) calculations were carried out to investigate the oxidative cleavage of 7 with various ROS, including singlet oxygen (1O2), hydroxyl radical (•OH), superoxide ion (O2•–), and triplet oxygen (3O2). These ROS could initiate the oxidation of 7 by abstracting the hydrogen atom from the tertiary C–H bond and forming a stable benzyl radical intermediate 8. As shown in Scheme 6, the 1O2 involved hydrogen atom transfer (HAT) through the transition state 1TS-1 has the lowest energy barrier (ΔG‡ = 4.8 kcal/mol), which indicates that 1O2 is the most likely ROS to initial PS degradation. This computational result is consistent with the 1O2 quenching experiments as well as EPR investigations that support 1O2 to be the real ROS for the degradation of PS.
Scheme 6. Computational Study of the Hydrogen Atom Transfer (HAT) Reaction of 1,3-Diphenylbutane 7 with Various Oxygen Species.

All energies were calculated at the B3LYP-D3/6-311+G(d,p)/SMD(acetonitrile75)//B3LYP -D3/6-31G(d) level of theory.
Subsequently, the rebound of a hydroperoxyl radical (•OOH) generates the peroxide compound 9 irreversibly (Scheme 6). The formation of 9 is exergonic by 44.9 kcal/mol with respect to 7, which indicates that this stable intermediate is most likely an active intermediate for the following C–C bond cleavage.76 Indeed, this speculation was then verified by experimental studies. As shown in Scheme 7, when 9 was subjected to the standard conditions but in the absence of O2, ethylbenzene (resulting from 2-phenylethyl radical 10) and acetophenone were obtained as the main products. Meanwhile, formic acid, benzoic acid, and acetophenone were also observed when the decomposition of 9 was performed under O2. In a recent study,1O2 has been shown to easily insert into α-ethereal C–H bonds, forming hydroperoxides.56
Scheme 7. Decomposition of Peroxide 9 under N2 or O2.

Computational studies of the decomposition of 9 suggest that the homolytic cleavage of the O–O bond through an open-shell singlet transition state TS-4 appears less likely, requiring an activation free energy of 30.0 kcal/mol (Scheme 8). Surprisingly somehow, the radical substitution pathway with •OH through a doublet transition state TS-5 entails a significantly lower energy barrier (ΔG‡ = 20.6 kcal/mol). This step irreversibly generates a key O-centered radical 11, which can undergo the β-scission facilely (via TS-6, ΔG‡ = 4.0 kcal/mol), leading to the C–C bond cleavage. The formation of acetophenone and alkyl radical 10 is also highly exergonic. The following radical substitution between alkyl radical 10 and hydrogen peroxide gives rise to phenethyl alcohol 12 and regenerates the hydroxyl radical (•OH), thereby completing the catalytic cycle. Because phenethyl alcohol 12 also contains the benzyl C–H bond, analogous pathways that consist of HAT, radical rebound, radical substitution, and β-scission of an O-centered radical will result in the decomposition of 12 as well. The generated benzaldehyde and alkyl radical can be finally oxidized to benzoic acid 2 and formic acid 1 under the standard oxidation conditions.
Scheme 8. Computational Study of the Decomposition of Peroxide 9.
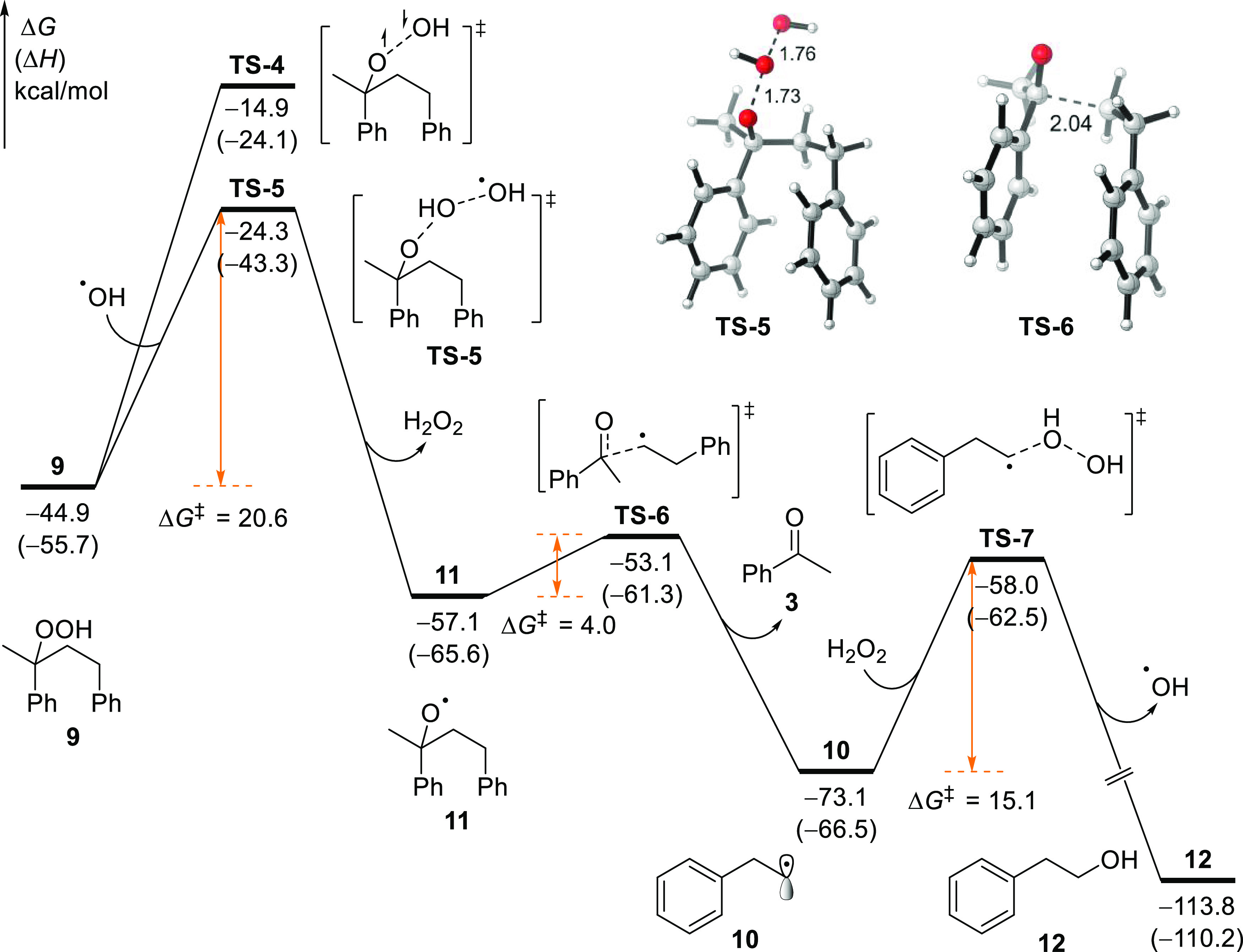
All energies were calculated at the B3LYP-D3/6-311+G(d,p)/SMD(acetonitrile)//B3LYP-D3/6-31G(d) level of theory.
Conclusions
In summary, a novel photo-acid-enabled protocol has been established for the selective degradation of polystyrene wastes by molecular oxygen for the first time. Featuring photosensitizer-free and mild reaction conditions, the protocol is operationally simple for the chemical recycling of polystyrene waste to valuable chemicals, such as formic acid, benzoic acid, and acetophenone. Flow degradation of polystyrene has also been demonstrated, providing support toward its potential application in industry. Mechanistic investigations indicate that singlet oxygen is formed as the key ROS in the degradation process. Notably, a possible [polystyrene---acid] adduct plays a vital role in the formation of 1O2 under violet-blue light, the concentration of which is likely to be boosted by the in situ-formed oxidized polystyrene polymer acting as a photosensitizer. These findings may open new photosensitizer-free pathways that were previously considered impossible for the aerobic degradation of polystyrene or other polymers featuring weak C–H bonds. DFT calculations suggest that the 1O2-mediated selective C–H bond hydroperoxidation is the key process for the subsequent C–C bond cracking of polystyrene, although many radical pathways may well follow. Spin-trapping EPR experiments support the involvement of O- and C-centered radicals in this degradation process. Last but not least, this type of chemical recycling can result in the displacement of fossil carbon-based feedstocks while incentivizing better management of polystyrene waste by recovering a considerable material value that can be recirculated into the global economy.
Acknowledgments
The authors are grateful to the EPSRC (EP/R009694/1 and EP/R511729/1) for funding, including the EPR National Facility at Manchester (EP/V035231/1 and EP/S033181/1), the China Scholarship Council and the University of Liverpool for a PhD studentship (RPG), the Royal Society for a University Research Fellowship (AGS), the Analytical Services of the Department of Chemistry of the University of Liverpool for product analysis, and Vapourtec Ltd. for the free loan of E-series flow chemistry equipment. They also thank Dr. Steven Robinson (Technical Support Officer in the Materials Innovation Factory, University of Liverpool) and Mr. Henry Morris (Department of Chemistry, University of Liverpool) for technical assistance and Mark Norman and his team in the EEE Department Electronics Workshop and Mr. Gordon Bostock and Chemistry Electronics Workshop for help with the design and fabrication of our photoreactors. This work made use of equipment from the Analytical Services/Department of Chemistry at the University of Liverpool as well as the shared equipment at the Materials Innovation Factory (MIF) created as part of the U.K. Research Partnership Innovation Fund (Research England) and cofounded by the Sir Henry Royce Institute. The theoretical calculations were performed on the supercomputing system in the Supercomputing Center of Wuhan University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c01410.
Experimental details and procedures, optimization studies, mechanistic experiments, and spectral data for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Simon N.; Raubenheimer K.; Urho N.; Unger S.; Azoulay D.; Farrelly T.; Sousa J.; van Asselt H.; Carlini G.; Sekomo C.; Schulte M. L.; Busch P.-O.; Wienrich N.; Weiand L. A binding global agreement to address the life cycle of plastics. Science 2021, 373, 43–47. 10.1126/science.abi9010. [DOI] [PubMed] [Google Scholar]
- Geyer R.; Jambeck J. R.; Law K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782 10.1126/sciadv.1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton L.; Andrady A. Future scenarios of global plastic waste generation and disposal. Palgrave Commun. 2019, 5, 6 10.1057/s41599-018-0212-7. [DOI] [Google Scholar]
- Chamas A.; Moon H.; Zheng J.; Qiu Y.; Tabassum T.; Jang J. H.; Abu-Omar M.; Scott S. L.; Suh S. Degradation rates of plastics in the environment. ACS Sustainable Chem. Eng. 2020, 8, 3494–3511. 10.1021/acssuschemeng.9b06635. [DOI] [Google Scholar]
- Andrady A. L. Assessment of environmental biodegradation of synthetic polymers. J. Macromol. Sci. Polymer Rev. 1994, 34, 25–76. 10.1080/15321799408009632. [DOI] [Google Scholar]
- United Nations Environment Programme, Single-Use Plastics: A Roadmap for Sustainability, 2018.
- Barnes D. K. A.; Galgani F.; Thompson R. C.; Barlaz M. Accumulation and fragmentation of plastic debris in global environments. Phil. Trans. R. Soc. B 2009, 364, 1985–1998. 10.1098/rstb.2008.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; von Jouanne A.; Yokochi A. Current technologies in depolymerization process and the road ahead. Polymers 2021, 13, 449. 10.3390/polym13030449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambeck J. R.; Geyer R.; Wilcox C.; Siegler T. R.; Perryman M.; Andrady A.; Narayan R.; Law K. L. Plastic waste inputs from land into the ocean. Science 2015, 347, 768–771. 10.1126/science.1260352. [DOI] [PubMed] [Google Scholar]
- Thompson R. C.; Swan S. H.; Moore C. J.; vom Saal F. S. Our plastic age. Phil. Trans. R. Soc. B 2009, 364, 1973–1976. 10.1098/rstb.2009.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod M.; Arp H. P. H.; Tekman M. B.; Jahnke A. The global threat from plastic pollution. Science 2021, 373, 61–65. 10.1126/science.abg5433. [DOI] [PubMed] [Google Scholar]
- Santos R. G.; Machovsky-Capuska G. E.; Andrades R. Plastic ingestion as an evolutionary trap: Toward a holistic understanding. Science 2021, 373, 56–60. 10.1126/science.abh0945. [DOI] [PubMed] [Google Scholar]
- Stubbins A.; Law K. L.; Muñoz S. E.; Bianchi T. S.; Zhu L. Plastics in the Earth system. Science 2021, 373, 51–55. 10.1126/science.abb0354. [DOI] [PubMed] [Google Scholar]
- Weiss L.; Ludwig W.; Heussner S.; Canals M.; Ghiglione J.-F.; Estournel C.; Constant M.; Kerhervé P. The missing ocean plastic sink: Gone with the rivers. Science 2021, 373, 107–111. 10.1126/science.abe0290. [DOI] [PubMed] [Google Scholar]
- Eriksson O.; Finnveden G. Plastic waste as a fuel - CO2-neutral or not?. Energy Environ. Sci. 2009, 2, 907–914. 10.1039/b908135f. [DOI] [Google Scholar]
- Bucci K.; Tulio M.; Rochman C. M. What is known and unknown about the effects of plastic pollution: A meta-analysis and systematic review. Ecol. Appl. 2020, 30, e02044 10.1002/eap.2044. [DOI] [PubMed] [Google Scholar]
- Awoyera P. O.; Adesina A. Plastic wastes to construction products: Status, limitations and future perspective. Case Stud. Constr. Mater. 2020, 12, e00330 10.1016/j.cscm.2020.e00330. [DOI] [Google Scholar]
- Hopewell J.; Dvorak R.; Kosior E. Plastics recycling: Challenges and opportunities. Phil. Trans. R. Soc. B 2009, 364, 2115–2126. 10.1098/rstb.2008.0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi A.; García J. M. Chemical recycling of waste plastics for new materials production. Nat. Rev. Chem. 2017, 1, 0046 10.1038/s41570-017-0046. [DOI] [Google Scholar]
- Kartalis C. N.; Papaspyrides C. D.; Pfaendner R.; Hoffmann K.; Herbst H. Mechanical recycling of post-used HDPE crates using the restabilization technique. II: Influence of artificial weathering. J. Appl. Polym. Sci. 2000, 77, 1118–1127. . [DOI] [Google Scholar]
- Torres N.; Robin J. J.; Boutevin B. Study of thermal and mechanical properties of virgin and recycled poly(ethylene terephthalate) before and after injection molding. Eur. Polym. J. 2000, 36, 2075–2080. 10.1016/S0014-3057(99)00301-8. [DOI] [Google Scholar]
- Kosloski-Oh S. C.; Wood Z. A.; Manjarrez Y.; de los Rios J. P.; Fieser M. E. Catalytic methods for chemical recycling or upcycling of commercial polymers. Mater. Horiz. 2021, 8, 1084–1129. 10.1039/D0MH01286F. [DOI] [PubMed] [Google Scholar]
- Garcia J. M.; Robertson M. L. The future of plastics recycling. Science 2017, 358, 870–872. 10.1126/science.aaq0324. [DOI] [PubMed] [Google Scholar]
- Plastic upcycling. Nat. Catal. 2019, 2, 945–946. 10.1038/s41929-019-0391-7. [DOI] [Google Scholar]
- Korley L. T. J.; Epps T. H.; Helms B. A.; Ryan A. J. Toward polymer upcycling-adding value and tackling circularity. Science 2021, 373, 66–69. 10.1126/science.abg4503. [DOI] [PubMed] [Google Scholar]
- Britt P.; Coates G.; Winey K.; Nasskar A.; Saito T.; Wu Z.. Roundtable on Chemical Upcycling of Polymers; US Department of Energy Office of Science: Maryland, 2019. [Google Scholar]
- Thiounn T.; Smith R. C. Advances and approaches for chemical recycling of plastic waste. J. Polym. Sci. 2020, 58, 1347–1364. 10.1002/pol.20190261. [DOI] [Google Scholar]
- Dimitris S.; Achilias L.. Recent advances in the chemical recycling of polymers (PP, PS, LDPE, HDPE, PVC, PC, Nylon, PMMA). Material Recycling - Trends and Perspectives; IntechOpen, 2012; Vol. 3, p 64. [Google Scholar]
- Patel M.; von Thienen N.; Jochem E.; Worrell E. Recycling of plastics in Germany. Resour., Conserv. Recycl. 2000, 29, 65–90. 10.1016/S0921-3449(99)00058-0. [DOI] [Google Scholar]
- Ignatyev I. A.; Thielemans W.; Vander Beke B. Recycling of polymers: A review. ChemSusChem 2014, 7, 1579–1593. 10.1002/cssc.201300898. [DOI] [PubMed] [Google Scholar]
- Vollmer I.; Jenks M. J. F.; Roelands M. C. P.; White R. J.; van Harmelen T.; de Wild P.; van der Laan G. P.; Meirer F.; Keurentjes J. T. F.; Weckhuysen B. M. Beyond mechanical recycling: Giving new life to plastic waste. Angew. Chem., Int. Ed. 2020, 59, 15402–15423. 10.1002/anie.201915651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw P.; Turra A.; Galgani F., Guidelines for the Monitoring and Assessment of Plastic Litter in the Ocean; GESAMP, 2019. [Google Scholar]
- Maharana T.; Negi Y. S.; Mohanty B. Review Article: Recycling of Polystyrene. Polym.-Plast. Technol. Eng. 2007, 46, 729–736. 10.1080/03602550701273963. [DOI] [Google Scholar]
- Zhang Z.; Hirose T.; Nishio S.; Morioka Y.; Azuma N.; Ueno A.; Ohkita H.; Okada M. Chemical recycling of waste polystyrene into styrene over solid acids and bases. Ind. Eng. Chem. Res. 1995, 34, 4514–4519. 10.1021/ie00039a044. [DOI] [Google Scholar]
- Ukei H.; Hirose T.; Horikawa S.; Takai Y.; Taka M.; Azuma N.; Ueno A. Catalytic degradation of polystyrene into styrene and a design of recyclable polystyrene with dispersed catalysts. Catal. Today 2000, 62, 67–75. 10.1016/S0920-5861(00)00409-0. [DOI] [Google Scholar]
- Kim J.-S.; Lee W.-Y.; Lee S.-B.; Kim S.-B.; Choi M.-J. Degradation of polystyrene waste over base promoted Fe catalysts. Catal. Today 2003, 87, 59–68. 10.1016/j.cattod.2003.10.004. [DOI] [Google Scholar]
- Marczewski M.; Kamińska E.; Marczewska H.; Godek M.; Rokicki G.; Sokołowski J. Catalytic decomposition of polystyrene. The role of acid and basic active centers. Appl. Catal., B 2013, 129, 236–246. 10.1016/j.apcatb.2012.09.027. [DOI] [Google Scholar]
- Achilias D. S.; Kanellopoulou I.; Megalokonomos P.; Antonakou E.; Lappas A. A. Chemical recycling of polystyrene by pyrolysis: Potential use of the liquid product for the reproduction of polymer. Macromol. Mater. Eng. 2007, 292, 923–934. 10.1002/mame.200700058. [DOI] [Google Scholar]
- Tiwary P.; Guria C. Effect of metal oxide catalysts on degradation of waste polystyrene in hydrogen at elevated temperature and pressure in benzene solution. J. Polym. Environ. 2010, 18, 298–307. 10.1007/s10924-010-0235-7. [DOI] [Google Scholar]
- Dong D.; Tasaka S.; Inagaki N. Thermal degradation of monodisperse polystyrene in bean oil. Polym. Degrad. Stab. 2001, 72, 345–351. 10.1016/S0141-3910(01)00031-3. [DOI] [Google Scholar]
- Nisar J.; Ali G.; Shah A.; Shah M. R.; Iqbal M.; Ashiq M. N.; Bhatti H. N. Pyrolysis of expanded waste polystyrene: Influence of nickel-doped copper oxide on kinetics, thermodynamics, and product distribution. Energy Fuels 2019, 33, 12666–12678. 10.1021/acs.energyfuels.9b03004. [DOI] [Google Scholar]
- Woo O. S.; Ayala N.; Broadbelt L. J. Mechanistic interpretation of base-catalyzed depolymerization of polystyrene. Catal. Today 2000, 55, 161–171. 10.1016/S0920-5861(99)00235-7. [DOI] [Google Scholar]
- Pifer A.; Sen A. Chemical recycling of plastics to useful organic compounds by oxidative degradation. Angew. Chem., Int. Ed. 1998, 37, 3306–3308. . [DOI] [PubMed] [Google Scholar]
- Sikkenga D. L.Conversion of polystyrene to benzoic acid. U.S. Patent US20200140367A12020.
- Zhang G.; Zhang Z.; Zeng R. Photoinduced FeCl3-catalyzed alkyl aromatics oxidation toward degradation of polystyrene at room temperature. Chin. J. Chem. 2021, 39, 3225–3230. 10.1002/cjoc.202100420. [DOI] [Google Scholar]
- Lewis S. E.; Wilhelmy B. E.; Leibfarth F. A. Upcycling aromatic polymers through C–H fluoroalkylation. Chem. Sci. 2019, 10, 6270–6277. 10.1039/C9SC01425J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Guan R.; Shanmugam M.; Bennett E. L.; Robertson C. M.; Brookfield A.; McInnes E. J. L.; Xiao J. Oxidative cleavage of alkenes by O2 with a non-heme manganese catalyst. J. Am. Chem. Soc. 2021, 143, 10005–10013. 10.1021/jacs.1c05757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-de-Castro A.; Xiao J. Green and efficient: Iron-catalyzed selective oxidation of olefins to carbonyls with O2. J. Am. Chem. Soc. 2015, 137, 8206–8218. 10.1021/jacs.5b03956. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang C.; Xue D.; Xiao M.; Liu J.; Li C.; Xiao J. Reactions catalysed by a binuclear copper complex: Relay aerobic oxidation of N-aryl tetrahydroisoquinolines to dihydroisoquinolones with a vitamin B1 analogue. Chem. Eur. J. 2017, 23, 3062–3066. 10.1002/chem.201604750. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang C.; Liu Y.; Xiao J. Acceptorless dehydrogenation and aerobic oxidation of alcohols with a reusable binuclear rhodium(ii) catalyst in water. Green Chem. 2016, 18, 4605–4610. 10.1039/C6GC01272H. [DOI] [Google Scholar]
- Gonzalez-de-Castro A.; Robertson C. M.; Xiao J. Dehydrogenative α-oxygenation of ethers with an iron catalyst. J. Am. Chem. Soc. 2014, 136, 8350–8360. 10.1021/ja502167h. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yan Y.; Xue D.; Wang Z.; Xiao J.; Wang C. Highly efficient binuclear copper-catalyzed oxidation of N,N-dimethylanilines with O2. ChemCatChem 2020, 12, 2221–2225. 10.1002/cctc.201901962. [DOI] [Google Scholar]
- Al-Nu’airat J.; Oluwoye I.; Zeinali N.; Altarawneh M.; Dlugogorski B. Z. Review of chemical reactivity of singlet oxygen with organic fuels and contaminants. Chem. Rec. 2021, 21, 315–342. 10.1002/tcr.202000143. [DOI] [PubMed] [Google Scholar]
- Ghogare A. A.; Greer A. Using singlet oxygen to synthesize natural products and drugs. Chem. Rev. 2016, 116, 9994–10034. 10.1021/acs.chemrev.5b00726. [DOI] [PubMed] [Google Scholar]
- Ossola R.; Jönsson O. M.; Moor K.; McNeill K. Singlet oxygen quantum yields in environmental waters. Chem. Rev. 2021, 121, 4100–4146. 10.1021/acs.chemrev.0c00781. [DOI] [PubMed] [Google Scholar]
- Sagadevan A.; Hwang K. C.; Su M.-D. Singlet oxygen-mediated selective C–H bond hydroperoxidation of ethereal hydrocarbons. Nat. Commun. 2017, 8, 1812 10.1038/s41467-017-01906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q.; Zhang J.; Zhao G.; Qi Y.; Wang H.; Lei A. Dioxygen-triggered oxidative radical reaction: Direct aerobic difunctionalization of terminal alkynes toward β-keto sulfones. J. Am. Chem. Soc. 2013, 135, 11481–11484. 10.1021/ja4052685. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Wu P.; Yang Y.; Zeng Z.; Liu J.; Yi H.; Lei A. Room-temperature copper-catalyzed oxidation of electron-deficient arenes and heteroarenes using air. Angew. Chem., Int. Ed. 2012, 51, 4666–4670. 10.1002/anie.201200750. [DOI] [PubMed] [Google Scholar]
- Yousif E.; Haddad R. Photodegradation and photostabilization of polymers, especially polystyrene: Review. SpringerPlus 2013, 2, 398 10.1186/2193-1801-2-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C. P.; Armstrong C. J.; Walsh A. N.; Jackson J. H.; Reddy C. M. Sunlight converts polystyrene to carbon dioxide and dissolved organic carbon. Environ. Sci. Technol. Lett. 2019, 6, 669–674. 10.1021/acs.estlett.9b00532. [DOI] [Google Scholar]
- Shang J.; Chai M.; Zhu Y. Photocatalytic degradation of polystyrene plastic under fluorescent light. Environ. Sci. Technol. 2003, 37, 4494–4499. 10.1021/es0209464. [DOI] [PubMed] [Google Scholar]
- Khaled A.; Rivaton A.; Richard C.; Jaber F.; Sleiman M. Phototransformation of plastic containing brominated flame retardants: Enhanced fragmentation and release of photoproducts to water and air. Environ. Sci. Technol. 2018, 52, 11123–11131. 10.1021/acs.est.8b03172. [DOI] [PubMed] [Google Scholar]
- Ranby B.; Lucki J. New aspects of photodegradation and photooxidation of polystyrene. Pure Appl. Chem. 1980, 52, 295–303. 10.1351/pac198052020295. [DOI] [Google Scholar]
- Rabek J. F.; Rånby B. Studies on the photooxidation mechanism of polymers. I. Photolysis and photooxidation of polystyrene. J. Polym. Sci., Part A: Polym. Chem. 1974, 12, 273–294. 10.1002/pol.1974.170120203. [DOI] [Google Scholar]
- Ogilby P. R. Singlet oxygen: There is indeed something new under the sun. Chem. Soc. Rev. 2010, 39, 3181–3209. 10.1039/b926014p. [DOI] [PubMed] [Google Scholar]
- DeRosa M. C.; Crutchley R. J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233–234, 351–371. 10.1016/S0010-8545(02)00034-6. [DOI] [Google Scholar]
- The mixed benzene/CH3CN solvent offered the best yield, which might be due to the good solubility of polystyrene and oxygen and the stability of relatively long-lived singlet oxygen in this solvent. See references:; a García M. T.; Gracia I.; Duque G.; de Lucas A.; Rodríguez J. F. Study of the solubility and stability of polystyrene wastes in a dissolution recycling process. Waste Manage. 2009, 29, 1814–1818. 10.1016/j.wasman.2009.01.001. [DOI] [PubMed] [Google Scholar]; b Franco C.; Olmsted J. Photochemical determination of the solubility of oxygen in various media. Talanta 1990, 37, 905–909. 10.1016/0039-9140(90)80251-A. [DOI] [PubMed] [Google Scholar]; c Sato T.; Hamada Y.; Sumikawa M.; Araki S.; Yamamoto H. Solubility of oxygen in organic solvents and calculation of the Hansen solubility parameters of oxygen. Ind. Eng. Chem. Res. 2014, 53, 19331–19337. 10.1021/ie502386t. [DOI] [Google Scholar]; d Ogilby P. R.; Foote C. S. Chemistry of singlet oxygen. 42. Effect of solvent, solvent isotopic substitution, and temperature on the lifetime of singlet molecular oxygen (1Δg). J. Am. Chem. Soc. 1983, 105, 3423–3430. 10.1021/ja00349a007. [DOI] [Google Scholar]
- Noël T., Photochemical Processes in Continuous-Flow Reactors. World Scientific: Singapore, 2017. [Google Scholar]
- Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noël T. Applications of continuous-flow photochemistry in organic synthesis, material science, and water treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- You Y. Chemical tools for the generation and detection of singlet oxygen. Org. Biomol. Chem. 2018, 16, 4044–4060. 10.1039/C8OB00504D. [DOI] [PubMed] [Google Scholar]
- Bancirova M. Sodium azide as a specific quencher of singlet oxygen during chemiluminescent detection by luminol and Cypridina luciferin analogues. Luminescence 2011, 26, 685–688. 10.1002/bio.1296. [DOI] [PubMed] [Google Scholar]
- Heitner C.; Dimmel D.; Schmidt J., Lignin and Lignans: Advances in Chemistry; CRC press, 2019. [Google Scholar]
- Dikalov S. I.; Mason R. P. Reassignment of organic peroxyl radical adducts. Free Rad. Biol. Med. 1999, 27, 864–872. 10.1016/S0891-5849(99)00134-3. [DOI] [PubMed] [Google Scholar]
- Buettner G. R. Spin trapping: ESR parameters of spin adducts 1474 1528V. Free Rad. Biol. Med. 1987, 3, 259–303. 10.1016/S0891-5849(87)80033-3. [DOI] [PubMed] [Google Scholar]
- The model compound reacts in acetonitrile. Thus, when the photochemical oxidation of 7 was performed under the standard conditions but in CH3CN, 0.36 mmol of 1, 0.23 mmol of 2, and 0.21 mmol of 3 were obtained. Therefore, the DFT calculations were performed in CH3CN. Additional DFT calculations were performed in the 1,2-dichloroethane solvent, revealing energy barriers close to those calculated in acetonitrile (See SI for details).
- Rahimi A.; Azarpira A.; Kim H.; Ralph J.; Stahl S. S. Chemoselective metal-free aerobic alcohol oxidation in lignin. J. Am. Chem. Soc. 2013, 135, 6415. 10.1021/ja401793n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.