

Abstract
Young age, female sex, absence of comorbidities, and prior infection or vaccination are known epidemiological barriers for contracting the new infection and/or increased disease severity. Demographic trends from the recent coronavirus disease 2019 waves, which are believed to be driven by newer severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) variants, indicate that the aforementioned epidemiological barriers are being breached and a larger number of younger and healthy individuals are developing severe disease. The new SARS‐CoV‐2 variants have key mutations that can induce significant changes in the virus‐host interactions. Recent studies report that, some of these mutations, singly or in a group, enhance key mechanisms, such as binding of the receptor‐binding domain (RBD) of the viral spike protein with the angiotensin‐converting enzyme 2 (ACE2) receptor in the host‐cells, increase the glycosylation of spike protein at the antigenic sites, and enhance the proteolytic cleavage of the spike protein, thus leading to improved host‐cell entry and the replication of the virus. The putative changes in the virus–host interactions imparted by the mutations in the RBD sequence can potentially be the reason behind the breach of the observed epidemiological barriers. Susceptibility for contracting SARS‐CoV‐2 infection and the disease outcomes are known to be influenced by host‐cell expressions of ACE2 and other proteases. The new variants can act more efficiently, and even with the lesser availability of the viral entry‐receptor and the associated proteases, can have more efficient host‐cell entry and greater replication resulting in high viral loads and prolonged viral shedding, widespread tissue‐injury, and severe inflammation leading to increased transmissibility and lethality. Furthermore, the accumulating evidence shows that multiple new variants have reduced neutralization by both, natural and vaccine‐acquired antibodies, indicating that repeated and vaccine breakthrough infections may arise as serious health concerns in the ongoing pandemic.
Keywords: COVID‐19, epidemiology, mutation, pandemic, SARS‐CoV‐2 variants, waves
Highlights
Emerging SARS‐CoV‐2 variants:
Harbor key mutations altering the virus‐host interactions.
Show more efficient host‐cell entry and greater replication resulting in higher viral loads, prolonged viral shedding, and greater tissue injury.
Show reduced neutralization by natural and vaccine acquired antibodies.
Causing symptomatic illness in increasing number of young, women, and healthy individuals.
1. INTRODUCTION
Since the onset of the coronavirus disease 2019 (COVID‐19) pandemic a large number of variants of the causative agent, Severe Acute Respiratory Syndrome Coronavirus‐2 (SARS‐CoV‐2), have arisen, of which some have raised serious epidemiological concerns. The successive COVID waves triggered by emerging variants are presenting with varying epidemiological characteristics than the first wave caused by wild‐type (WT) strain and the early mutants. 1 , 2 , 3 , 4 WT strain was known to cause greater fatality among the aged, male sex, and those with comorbidities. 5 , 6 , 7 In contrast, the data coming out from the recent preclinical/clinical and epidemiological studies are giving clear indications that multiple newer variants, more particularly the variants of concerns (VOCs), can potentially breach the set epidemiological barriers and are capable of causing significant fatality across the demographic categories. 1 , 2 , 3 , 4 Moreover, a gain of resistance against the natural and vaccine acquired, and multiple therapeutically used monoclonal antibodies have been noted in multiple variants, 8 , 9 , 10 , 11 which may make the gain of herd immunity against the SARS‐CoV‐2 infection far‐reaching goal. In this perspective, we examine the strength of the empirical evidence available for the increased transmissibility, virulence, and immune escape in emerging SARS‐CoV‐2 variants and provide assessments of their potential impacts on the epidemiological characteristics of the pandemic. Our propositions for the definitive changes in the characteristics of the COVID‐19 pandemic are primarily based on the analysis of the current epidemiological trends and findings from the preclinical and clinical studies, and evidence‐based interpretations of the lineage characterizing mutations (mainly in the spike protein regions) appearing in the emerging SARS‐CoV‐2 variants.
1.1. Wild‐type SARS‐CoV‐2
The SARS‐CoV‐2 is an enveloped positive‐sense single‐stranded RNA virus (~29.9 kB in length) belonging to the genus betacoronaviruses (BCoVs). The BcoVs also includes SARS‐CoV‐1 and the Middle East Respiratory Syndrome coronavirus (MERS‐CoV) which had caused earlier acute respiratory syndrome epidemics of SARS‐2002/2003 and MERS‐2012, respectively. 12 The SARS‐CoV‐2 has a unique spherical structure with a ribonucleic acid core and proteinaceous double‐layered envelop, the outer layer of which contains unique spike‐like features composed of glycoproteins (Figure 1). 12 , 13 To infect a host cell, SARS‐CoV‐2 requires binding to the cell‐surface receptor, angiotensin‐converting enzyme‐2 (ACE2), through the receptor‐binding domain (RBD) present on its spike (S) protein. 13 , 14 As a prerequisite to binding to ACE2, it is necessary that the viral spike protein (S) gets cleaved by a set of host proteases—an event called “priming or activation” which is considered essential for the fusion of the virus with the host cell‐membranes. 13 , 14 The known host proteases for SARS‐CoV‐2 are transmembrane serine protease 2 (TMPRSS2) and furin, are expressed in the cytoplasmic membrane, and Cathepsin B or L (CTS‐B or L) is expressed in the endosomal membranes of the host cells (Figure 1). 14 , 15 , 16 ACE2 has been a host‐cell entry receptor for a few other CoVs causing acute respiratory illness as well, such as SARS‐CoV‐1 and HCoV‐NL‐63. 13 , 14 The host‐cell entry receptor ACE2 and entry associated proteases are not limited only to the respiratory system, but are widely expressed across the human tissue types, which is a stated reason why beyond the respiratory system pathology COVID‐19 leads to multiorgan involvement and a systemic illness. 17 Notably, ACE2 is an interferon (IFN) stimulated gene 18 hinting that SARS‐CoV‐2‐receptor binding mediated dysregulation of ACE2 expression may be a likely molecular mechanism responsible for prominent IFNs‐dysregulation characteristically observed in COVID‐19 patients. 19
Figure 1.
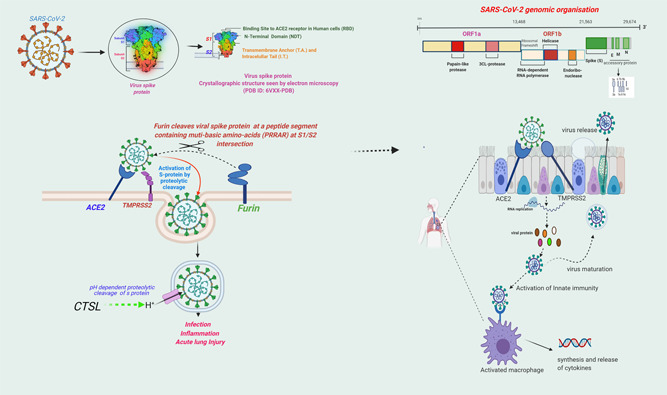
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2): host‐cell entry mechanisms. Entry of SARS‐CoV‐2 into host cell depends on binding of receptor‐binding domain (RBD) of viral spike (S) protein to the cell surface receptor angiotensin‐converting enzyme‐2 (ACE2). For a successful binding to ACE2, “S” protein is required to be cleaved by the host proteases, TMPRSS2 and Furin. Another host protease CTSL is involved in a pH‐dependent cleavage of the S protein inside the endosomes. The binding to ACE2 leads to endocytosis and allows for replication of the virus inside the host cell. The newly formed virions are released after bursting of the infected cell and spread further. Viral infection of the host‐cell subsequently leads to activation of innate host defense mechanism, recruitment of the immune cells by the infected tissue, and synthesis and release of the cytokines
SARS‐CoV‐2 has a close genomic sequence resemblance to a bat CoV RaTG3 (~96%) and to SARS‐CoV‐1 (~79%) indicating an evolutionary linkage among these viruses. 20 Of note, virus‐host interactions are very similar for the WT strains of SARS‐CoV‐1 and SARS‐CoV‐2, however, the later has gained multiple advantageous mutations within the RBD encompassing receptor binding interface. 21 , 22 The recent studies examining the strength of virus RBD:ACE2 complex have established that SARS‐CoV‐2 has a more efficient binding to ACE2 than the SARS‐CoV‐1, 21 , 23 thus imparting it higher transmissibility and virulence than the later. Interestingly, the inclusion of furin in the list of entry associated protease also seems an evolutionary gain in SARS‐CoV‐2 as it is not present in SARS‐CoV‐1 or other SARS‐related viruses. 14 The furin cleavage site (FCS) of SARS‐CoV‐2 is a small stretch of peptide (PRRAR) inserted at the intersection of spike segments S1 and S2 (681–685 aa residues), facilitating proteolytic cleavage of the viral spike protein at that point 14 (Figure 1). Noteworthy, FCS is not present in SARS‐CoV‐1 and other SARS‐related viruses (although it is present in a number of other human coronaviruses, including MERS‐CoV, HKU1‐CoV, and OC43‐CoV) and is considered as an evolutionary gain in SARS‐CoV‐2 towards imparting it higher virulence. 14 Furin cleavage also improves further proteolytic cleavage by another host‐proteases TMPRSS2 at S1‐S2 intersection—an event essential for the priming of the virus membrane for host‐cell fusion. 24 , 25 , 26
Apart from the spike protein, multiple nonstructural proteins (NSPs) have been linked to higher virulance and excessive immunological dysregulations by SARS‐CoV‐2 in comparison to SARS‐CoV‐1 and other CoVs, and influenza viruses. Of note, SARS‐CoV‐2 manipulates the host immune cells to ensue a delayed and excessive IFNs response, which is a key innate defense mechanism for the protection against new viral infections (reviewed in Kumar et al. 19 ).
2. EMERGING SARS‐COV‐2 VARIANTS
Emerging variants are classified as a variant of interest (VOI) or global VOC by WHO depending upon epidemiological characteristics of the strain. A VOI status is designated if the strain has been identified to cause community transmission/multiple COVID‐19 cases/clusters. A VOC is designated to an emergent strain when a variant is detected in multiple countries and either the strain causes an increase in transmissibility or detrimental changes in COVID‐19 epidemiology; or increase in virulence or changes in the clinical disease presentation; or decrease in the effectiveness of public health and social measures or available diagnostics, vaccines, and therapeutics. 27 The United States Centers for Disease Control and Prevention (US‐CDC) additionally, classifies Variants of High Consequence, although at present there has been no variant under this category. 28 A list of emerging variants, along with a description of key mutations, date, and country of origin, and current evidence for an increase in transmissibility, lethality, or immune escape, is presented in Table 1.
Table 1.
Emerging SARS‐CoV‐2 variants across the globe
| Name (Pango lineage external icon) | WHO label | Spike protein substitutions | Name (Next strain external icon) | First detected | Transmission* | Lethality* | Immune escape* |
|---|---|---|---|---|---|---|---|
| Global Variants of Concern (VOCs) | |||||||
| B.1.1.7 | Alpha | Spike: 69del, 70del, 144del, E484K, S494P, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H K1191N | 20I/501Y.V1 | United Kingdom | ~50% increased transmission compared to B.1 | Potential increased severity based on hospitalizations and case fatality rates | No impact on susceptibility to monoclonal antibody treatments |
| Minimal impact on neutralization by convalescent and postvaccination sera | |||||||
| B.1.351 | Beta | Spike: D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V | 20H/501.V2 | South Africa | ~50% increased transmission | More lethal | Significant decrease in susceptibility to the combination of bamlanivimab and etesevimab monoclonal antibody treatment |
| September 2020 | Reduced neutralization by convalescent and postvaccination sera | ||||||
| P.1 | Gamma | Spike: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I | 20J/501Y.V3 | Japan/Brazil | Yet not known | More lethal | Significant decrease in susceptibility to the combination of bamlanivimab and etesevimab monoclonal antibody treatment |
| December 2020 | Reduced neutralization by convalescent and postvaccination sera | ||||||
| B.1.617.2 | Delta | Spike: T19R, (G142D), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N | 20A/S:478K | India – December 2020 | ~50%–60% increased transmission compared to B.1.1.7 | Preliminary results suggest 2.61 times higher the risk of hospitalization within 14 days compared with the B.1.1.7. | Potential reduction in neutralization by some monoclonal antibody treatments |
| Sub‐lineages: B.1.617.2.1‐47 (or AY.1‐47) | Potential reduction in neutralization by postvaccination sera | ||||||
| Global Variants of Interest (VOI) | |||||||
| C.37 | Lambda | Spike: D614G, L452Q, F490S, T859N, T76I, G75V, del247/253 | 20D | Peru, Aug‐2020 | Yet not known | Yet not known | Yet not known |
| B.1.621, B.1.621.1 | Mu | Spike: T95I, Y144S, Y145N, R346K, E484K, N501Y, D614G, P681H, D950N | 21H | Colombia, Jan‐2021 | Yet not known | Yet not known | Yet not known |
| Former VOI now under monitoring # | |||||||
| B.1.427/B.1.429 | Epsilon | Spike:L452R, D614G (B.1.427) Spike:S13I, W152C, | 20C/S:452R | United States‐(California) | ~20% increased transmissibility | Yet not known | Modest decrease in susceptibility to the combination of bamlanivimab and etesevimab; however. |
| L452R, D614G (B.1.429) | September 2020 | Reduced neutralization by convalescent and postvaccination sera | |||||
| B.1.525 | Eta | Spike: A67V, 69del, 70del, 144del, E484K, D614G, Q677H, F888L | 20 A/S:484 K | United Kingdom/Nigeria – December 2020 | Yet not known | Yet not known | Potential reduction in neutralization by some monoclonal antibody treatments |
| Potential reduction in neutralization by convalescent and postvaccination sera | |||||||
| B.1.526 | Iota | Spike: L5F, T95I, D253G, S477N, E484K, D614G, A701V | 20C/S:484K | United States (New York) – November 2020 | Reduced susceptibility to the combination of bamlanivimab and etesevimab monoclonal antibody treatment. | ||
| Reduced neutralization by convalescent and postvaccination sera | |||||||
| Yet not known | Yet not known | ||||||
| B.1.617.1 | Kappa | Spike: T95I, G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H | 20 A/S:154 K | India – December 2020 | More transmissible | Increased lethality in animal model. In humans yet not known | Potential reduction in neutralization by some monoclonal antibody treatments |
| Potential reduction in neutralization by postvaccination sera |
The SARS‐CoV‐2 strain with D614G (B.1) was the first noticeable variant having a significant edge over the WT strain. 29 The variant was found to be more transmissible 29 and by the end of 2020 it almost replaced the WT strain globally. However, there had been no substantial evidence suggesting that it had increased virulence. Following B.1 multiple variants have emerged, most are its descendants bearing D614G: 23330/A→G) as the key mutation. Post dominance of B.1 over the WT strain, successive variants are showing faster global spread and gain of dominance against the existing strains (Figure 2).
Figure 2.
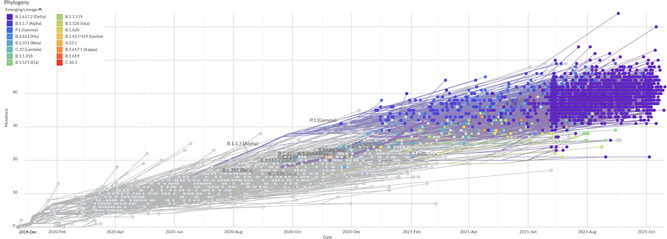
Phylodynamics of emerging SARS‐CoV‐2 lineages across the globe. (Data source: GISAID Initiative (www.gisaid.org, accessed on November 1, 2021. The image is created using EpiCoV™ application using 3572 SARS‐CoV‐2 genomes sampled between December 2019 and October 2021.). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
B.1.1.7, a VOC, in a short time period has become a globally dominant strain replacing the B.1. Some of the variants have been found to have significantly higher transmissibility and virulence than the wild and B.1 strains and have triggered subsequent waves of COVID‐19 in multiple countries, primarily B.1.1.7, B.1.351, P.1, and B.1.617.2, which have been currently designated as the VOCs by World Health Organization (WHO). 27 The most recent of the VOCs have been B.1.617.2, which is thought to be responsible for triggering the recent devastating second COVID‐19 wave in India. 30 , 31 B.1.617 lineage is of special interest being the latest variant in the list. 27 Preliminary evidence suggests higher transmissibility and perhaps more lethality from this lineage. First detected in India in October 2020, this lineage soon developed into three sub‐lineages: B.1.617.1‐3. Key mutations are common among these three sublineages, however, surprisingly, B.1.617.2, which was first detected in December 2020, is spreading much faster than the others and has become the dominant strain in India and in parts of UK, and also has been reported from at least 161 countries across the globe (as of November 1, 2021). 27 , 30 More recently, another variant with closest phylogenetic linkage with B.1.617.2 variant is reported from Vietnam. 32 The new variant has a characteristic 144Y: 21991‐21993 deletion (also present in B.1.1.7) in spike protein sequence, however, other key mutations are shared with B.1.617.2. It has shown higher transmissibility in situ and possibly fueled a recent wave of infections in the country. However, there is no evidence whether it had increased lethality. 32 Furthermore, a newer mutation (K417N: 22813, G→T), which is known to be present in B.1.351 and P.1, has appeared in B.1.617.2. The new variant has been named B.1.617.2.1 or ‘Delta plus K417N' (a.k.a. AY.1) and it has been currently reported from Nepal, India, UK, USA, and a few other countries. 33 , 34 A further sub‐lineage of Delta plus K417N with new spike mutations as V70F (69), A222V, T299I, and A958S, and exclusion of G504Q has been reported from these countries and has been named as B.1.617.2.2 or AY.2. 33 , 35 Furthermore, multiple other variants have been adding up to the list of Delta plus lineages—AY.3‐47. 33 Interestingly, the K417N spike mutation has been reverted back in AY.3‐47. 33 Of note, WHO still considers AY strains as the sub‐lineages of VOC Delta, 27 and the studies are still awaited which would establish whether they have an advantage over Delta variant in reference of the key epidemiological characteristics. A study comparing genomic sequences of the Delta and Delta plus (AY.1) variants showed that the high prevalence mutations (more than 20%) were greater in Delta plus than in Delta (40 vs. 29). 36
Theoretically, based on the genomic location of the characterizing mutations, it can be assumed that this variant, i.e. Delta plus (AY.1), may have an advantage against the neutralization by natural or acquired antibodies 37 ; however, currently, there is not much knowledge about it. As per the latest epidemiological surveillance, from Public Health England (PHE), UK, in the preliminary analysis of the limited data, AY.1 was found capable of doing breakthrough infections (27/36) however only very few cases (2/36) were infected after 14 days of the second dose of vaccine. Notably, the majority of the infected were from the younger age group (<60 year) (34/36). 35
3. KEY MUTATIONS IN EMERGING SARS‐COV‐2 VARIANTS
Each of the emerging variants harbors the key lineage identifying mutations within the spike as well as non‐spike protein regions (Figures 3, 4). Many of the variants show common mutations, primarily in the spike region. The shared mutations can have a founder effect, however, some of these are shared among the strains which are phylogenetically not very close indicating their selection against the evolutionary adaptive pressure.
Figure 3.
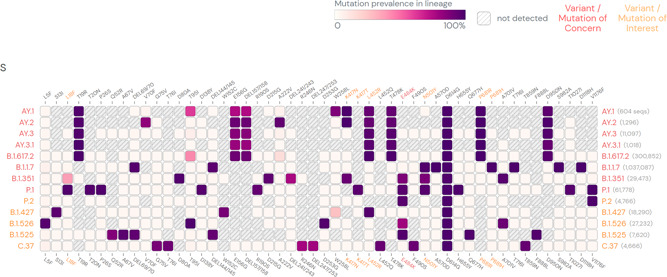
SARS‐CoV‐2 lineage specific mutations in spike protein regions. (Mutations with > 75% prevalence in at least one lineage are shown. Data source: www.outbreak.info, accessed on August 18, 2021). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Figure 4.
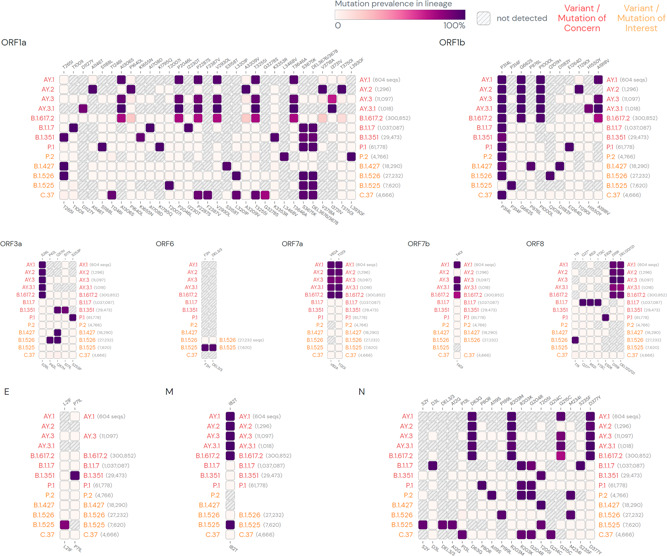
SARS‐CoV‐2 lineage‐specific mutations in non‐spike protein regions. (Mutations with > 75% prevalence in at least one lineage are shown. Data source: www.outbreak.info, accessed on August 18, 2021).SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
3.1. Spike mutations
The spike mutations are enriched at the regions which bear epitopes for the natural, vaccine acquired, and monoclonal antibodies, primarily at the receptor‐binding domain (RBD) and N‐ terminal of spike (S) protein (Figure 3). It appears that these mutations are imparting the emerging variants a selection advantage over the WT strain (and early variants), making them more suitable for the spread in a population that has developed only partial herd immunity. 37 The selective mutations which are either present at (N501Y: 22991/A→T) or in the vicinity of the receptor‐binding motif (RBM) (L452R:22917:T→G, E484K: 22940/G→A, and T478K:22995/C→A) are believed to stabilize binding with ACE2 by reducing binding energy and creating stronger electrostatic interactions or new hydrogen bonds. 10 , 37 , 38 , 39 Interestingly, E484K and N501Y, the lineage characterizing mutations for B.1.351 and P.1, can potentially compensate for the RBD:ACE2 complex stability reduction, putatively caused by K417N/T. 37 , 40 It is currently not known whether a similar advantage for the emerging variants in B.1.617.2 lineage—AY.1‐2, which contains L452R and T478K, and K417N as the key spike mutations at RBD:ACE2 interface. A comparative analysis of the prevalence of key spike mutations (T95I, G142D, R158G, L452R, T478K, and K417N) at different time points show that these mutations have increased over time in Delta, and all mutations had a significantly higher prevalence in the Delta plus variant (AY.1). A progressive increase in the prevalence of key spike mutations from Delta to Delta plus indicates that not only K417N but other spike mutations too may have significant role in determining epidemiological properties of this emerging variant. 36
The epistasis interactions between the spike mutations in the emerging variants are indicating that continued mutagenesis may have a mechanistic role in the maintenance of the evolutionarily achieved stability for RBD:ACE2 complex. 41
Furthermore, there is concrete proof that a key mutation occurring at amino acid position 681 in the spike protein of B.1.1.7 and B.1.617 lineage variants (L—H/R: 23604/C→G) improves the infectivity of these variants, 42 , 43 , 44 which we discuss later in this article.
3.2. Non‐spike mutations
Each of the emerging variant has multiple mutations in the non‐spike protein regions (Figure 4). Notably, many of the non‐spike mutations are also shared across the variants. Most common of the shared mutations is P314L in the NSP12b region encoding for RNA‐dependent RNA polymerase (RdRp). 31 , 45 The mutation, first appeared in B.1 strain and thereafter seems to be persisting in the subsequently emerging variants. 46 A mutation that introduces a premature stop codon at position 27 of ORF8 protein is characteristically noted in the sequence of Alpha variant (B.1.1.7). 47 An inactivating mutation in ORF8 protein sequence is important as this is an immune‐evasive protein involved in downregulation of major histocompatibility complex class I (MHC‐I) in host cells. 48 However, currently, there is little evidence available that whether this mutation in the viral sequence influences host interactions with the Alpha variant. Differential distribution of the multiple non‐spike mutations has been noted between Delta and Delta plus variants. For example, nsp3:P822L (ORF1a:P1604L), nsp4:A446V (ORF1a:A3209V), nsp6:V149S (ORF1a: V3718S), and nsp6:T181I (ORF1a:T3750I) are present at 16% in Delta, but at 58% in Delta plus (AY.1). 36
Non‐spike mutations in the emerging variants have been yet less studied for their biological significance, albeit, their possible contributions in altered interactions with the host and the further impact on epidemiological characteristics of the emerging variant cannot be denied.
4. IMPLICATIONS OF EMERGING SARS‐COV‐2 VARIANTS
4.1. Increased transmissibility and virulence
Increased transmissibility and virulence have been speculated for nearly all of the VOCs based on the analysis of the structural and functional changes imparted by the mutations. 44 The predictions have been further validated in recent animal model studies demonstrating increased lethality for some of these variants, primarily B.1.1.7 and B.1.617.1. 49 , 50 Multiple epidemiological studies have indicated increased lethality of emerging variants, however, most of the available data is for B1.1.7. Emerging evidence is substantiating that increased transmissibility is a characteristic feature for other multiple new variants including B.1.351, and P1 and B.1.617.2. 31 , 51 , 52
The emerging variants are accumulating mutations in the spike protein— which encompasses the binding site for the host‐cell entry‐receptor and is also the most antigenic region of the virus, towards which natural and acquired antibodies are targeted. 44 Key mutations in the spike protein (Table 1), primarily in the RBD are believed to have introduced conformational changes in it leading to stronger binding to the key host‐cell entry‐receptor ACE2. 37 , 44 Few of the newer mutations have particularly occurred at the receptor‐binding motif (RBM) of the RBD that creates newer contact sites with ACE2. 37 , 44 Certain other mutations have also resulted in strong electrostatic interactions or newer hydrogen bonds. 37 This was earlier observed for the D614G variant (B.1). 37 Multiple recent in situ and animal model studies have confirmed that emerging variants indeed cause higher viral load and prolong viral shedding, however, whether these properties are attributed to the mutations linked to RBD:ACE2 binding is still not clear. 37 , 44 , 49 , 50 , 53 , 54 Of interest, multiple studies have demonstrated that the WT strain of SARS‐CoV‐2 had gained instability for RBD:ACE2 binding when it added new mutations at the binding motif present in SARS‐CoV‐1, which uses the same entry‐receptor ACE2 and had caused the first epidemic of SARS in 2002. SARS‐CoV‐2 RBD:ACE2 binding stability was further improved with the D614G mutation in WT SARS‐CoV‐2, which stabilized the trimeric structure and created a more open conformation of the RBD allowing a stronger binding with ACE2, and also enhanced furin mediated proteolytic cleavage of the spike protein at the S1/S2 junction. It is noteworthy that the emerging variants are mostly descendants of the B.1 lineage and carry forward the D614G mutation. Emerging evidence indicate that the newer mutations are further improving the RBD:ACE2 binding in the sub‐lineages of B.1. 38 , 41 , 43 , 55 , 56 , 57 However, whether that makes any clinical difference is still not known.
Interestingly, increasing evidence from in situ studies substantiates the claim that the mutations in the spike protein of B.1.1.7 and B.1.617 lineage variants occurring at amino acid position 681 (P—H/R), which falls in the furin cleavage site (FCS), improve proteolytic cleavage of the spike protein and strengthening the fusion of the viral membrane with the host cell. 24 , 42 , 50 , 58
Recent evidence from in situ studies has demonstrated enhanced virus‐host membrane fusion for the B.1.617 lineage variants which contain P681R. 58 , 59 An improved viral–host cell fusion results in larger syncytia formation. 60 Syncytia formation—fusion of the infected host‐cell with others facilitating viral spread— has been a distinctive characteristic of SARS‐CoV‐2 when compared to SARS‐CoV‐1—believed to impart higher transmissibility. 25 , 60 , 61 Recent reports have shown that B.1.617 lineage variants form larger syncytia, however, whether that contributes to the increased transmissibility and virulence remains to be established. 58 , 59 A more efficient virus‐host membrane fusion may not only impart increased transmissibility but also may be providing the variants increased virulence by facilitating higher tissue tropism, which, in turn, induces a stronger immunogenic response by the host and consequently widespread tissue injury leading to more severe disease.
A recent study traced the index cases and analyzed viral loads in the patients within a community outbreak of B.1.617.2 in China. The authors noted that the incubation period for developing COVID‐19 is reduced to 4 days with B.1.617.2 variant, in comparison to average of 6 days for the WT strains. Moreover, the viral loads were observed, on average, approximately 1000 times more for this variant. 53 Another recent study conducted in the UK population further noted that risk of hospitalization and emergency visits doubled with B.1.617.2 in comparison to B.1.1.7 infection (Hazard ratio [HR], 95% confidence interval [CI]: 2.26 [1.32–3.89]). 62
Currently, the studies examining FCS mutation gain for the other emerging variants, such as B.1.617.2, are lacking. If novel mutations in the SARS‐CoV‐2 FCS contribute to increasing virulence, this is an indication that further mutations may appear within/nearby this site.
Notably, B.1.617. 2 reflects to have the highest transmissibility among the sub‐lineages of B.1.617, and it is also 50%–60% more transmissible than B.1.1.7, for which reasons are currently very little explained. 31 , 52 , 63 The higher transmissibility of B.1.617. 2 may be attributed to the unique set of spike mutations it bears. 64 Compared with B.1.617 (S: L452R, E484Q, D614G, del681, and del1072), B.1.617.1 (S: T95I, G142D, E154K, L452R, E484Q, D614G, P681R, and Q1071H), and B.1.617.3 (S: T19R, L452R, E484Q, D614G, and P681R), B.1.617.2 has more number of spike mutations (S: T19R, G142D, del156‐157, R158G, L452R, T478K, D614G, P681R, and D950N) (Table 1). It has also a greater number of spike mutations at or in vicinity of the RBM (L452R, E484K, and T478K) of spike protein, which suggests it may have a greater advantage in binding to host ACE2. 64
Of note, the emerging variants have multiple mutations in the non‐spike region (Figure 4). The non‐spike proteins have proven role in determining the virulence of the SARS‐CoV‐2 wild strain, primarily NSP1, 3, 10, 16, and ORF8 14 ; however, at present, there is very little evidence available in the support of any of the non‐spike protein region mutations having a distinctive influence in increasing the virulence of the variants. Moreover, how the new mutations in the emerging variants are affecting the host‐cell metabolism, will greatly affect their success in gaining the entry and multiplication inside the cells. 65 This also requires to be explored in‐depth to understand the evolving host‐virus interactions in the emerging variants.
4.2. Increasing immune escape
Recent studies have shown that most of the emerging variants, primarily VOCs, have gained a certain level of resistance against the natural and vaccine‐acquired antibodies (Table 1). 9 , 58 , 66 Resistance has been also reported against the multiple monoclonal antibodies currently being used in the treatment of COVID‐19. 8 , 67 , 68 A significant fall in antibody‐mediated neutralization has been observed in most of the variants (Table 1). The most likely mechanisms for the gain of immune escape by variants are (i) inclusion or deletion of amino‐acid residues at immunogenic epitopes, thus bringing conformational changes at the binding interface, (ii) remodeling of the electrostatic surface potential, and (iii) gain of additional glycosylation sites thus shielding the binding site for the neutralizing antibodies. 10 , 37 , 39 , 44 , 56 , 69 Gain of an extra glycosylation site at spike region has been reported for P1 variant in a preprint study. 70 In another recent study, Liu et al analyzed the crystal structures of viral RBD‐Fab (antigen‐binding fragment) complexes in an attempt to elucidate the mechanism of reduced antibody potency to B.1.617 lineage variants. The authors noted that a mutation at the residue position 452 (leucine to arginine) in the variants has led to an increase in the RBD side‐chain interacting with the 16‐residue‐long heavy chain (HC) complementarity determining region (CDR) 3 of the Fab, thus interrupting antibody binding (mAb 278). 71
Clinical evidence of the gain of immune escape against natural and acquired infections has been continuously reflected in increasing trends of re‐infections and vaccine breakthrough infections, which supposedly are being caused by the emerging variants. 3 , 31 , 59 , 72 Most recent variants, as the emerging evidence suggests, particularly B.1.617.2, seem to be more capable of establishing a re‐infection and the vaccine breakthrough infections. 31 , 73 The mutations which are present in NTD (T19R, R158G, del156‐157) of the spike protein of B.1.617.2 occur on the prominent monoclonal antibody recognition site, which indicates a likely reason why this strain is more efficient in evading antibody recognition. 8 The latest variants in B.1.617.2 lineage—or Delta plus K417N or AY.1 and AY.2 (Figure 5)—are also found to be capable of doing breakthrough infections, however, the data is currently limited. 35 , 73 Notably, K417 locus on SARS‐CoV‐2 RBM is a known binding site for the monoclonal antibodies in therapeutic use, such as CB6 37 and its inclusion may have increased the immune escape capability of the AY lineage variants. 74 A reversal of characteristic Delta plus lineage mutation K417N in later AY sub‐lineages, AY.3‐47, is astonishing (Figure 5), although the evolutionary reasons for regression of this mutation are not well understood yet, a destabilizing effect of this mutation on RBD:ACE2 binding may be a plausible explanation. 74 Currently, the biological evidence is limited which can explain the clinical significance of this reversal. 33
Figure 5.
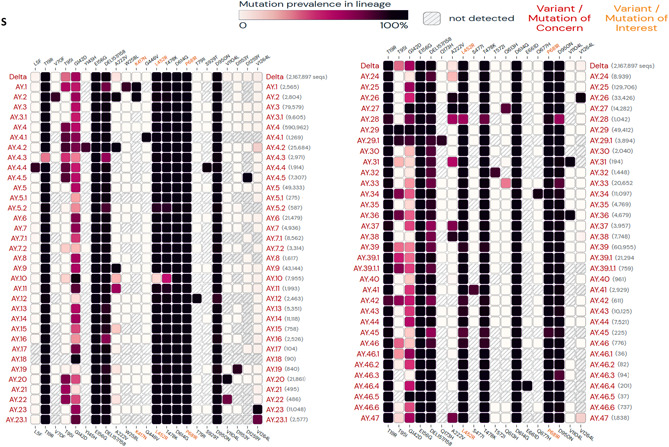
Lineage specific mutations in spike protein regions of SARS‐CoV‐2 Delta plus variants. (Mutations with > 75% prevalence in at least one lineage are shown. Data source: www.outbreak.info, accessed on November 1, 2021). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
4.3. Clinical and epidemiological significance
Increased transmissibility of the emerging variants, primarily B.1.351, P1, and B.1.617.2 have predicted their prevalence in the population in comparison to the WT strain and early variants 31 , 51 , 52 , 75 (Figure 6). Accumulating spike mutations imparting greater transmissibility has been reflected first in a gain of global dominance for B.1 strain with D614G mutation, followed by its recent replacement by B.1.1.7 which shows greater transmissibility, 63 perhaps arising because of multiple new mutations within this strain. 38 B.1.1.7 is uniquely missed for detection by the PCR kits targeting S gene due to the characteristic deletion of amino acids 69–70 in this strain. 76 More recent variants have now either taken over or outpacing the B.1.1.7 strain in large parts of the world. The global spread of B.1.617.2 has been surprisingly fast (Figure 6), 77 thus indicating a very high level of adaptive selection for this strain.
Figure 6.
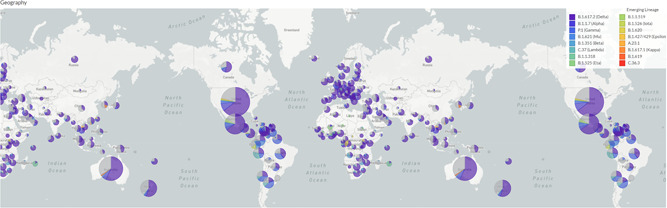
The global spread of emerging SARS‐CoV‐2 variants. (Data source: GISAID Initiative (www.gisaid.org, accessed on Nov 1, 2021. The image is created using EpiCoV™ application using 3572 SARS‐CoV‐2 genomes sampled between December 2019 and October 2021.). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Rapidly spreading newer variants can potentially create new waves of the pandemic which has been recently observed in many countries across the globe. The most recent has been a devastating second wave in India, which is believed to be triggered by B.1.617 lineage variants, primarily B.1.617.2. 30 , 31 , 78 Increased transmissibility and virulence of the emerging variants may cause a breach of the known epidemiological barriers, like age and sex, for contracting the infection and disease severity and outcomes known for the WT strain. 1 , 2 , 35 , 51 , 52 , 79 A large body of the studies since the first COVID‐19 wave has established that old aged (>60 year) 7 , 80 and male sex 81 , 82 are risk factors for developing severe COVID‐19 and higher mortality following infection with WT strain. In contrast, recent epidemiological data from the countries reporting new waves have shown a deviation from the established pattern. An incremental increase in transmissibility of the emerging variants from B.1 to B.1.617.2 suggests that more numbers of younger age individuals, including pediatric age group, may get involved in the pandemic as the strains further evolve. 1 , 2 , 4 , 35 , 79 Also, early indications for narrowing the sex‐based differences in patient outcomes is an important epidemiological concern. 83 , 84
In a recent study, among the Indian population, using data from the national clinical registry for COVID‐19 authors have compared clinical profiles of the hospitalized patients during two successive COVID‐19 waves (N = 12 059 and 6903). The authors noted that the mean age of the patients was significantly lower in the second wave (48.7 [18.1] year vs. 50.7 [18.0] year, p < 0.001) with more numbers of patients in the younger age groups intervals (<20, and 20–39 year). The proportion of men were lower in second wave (4400 [63.7%] vs. 7886 [65.4%], p = 0.02). A significantly higher proportion (2625 [48.6%] vs. 4420 [42.8%], p < 0.003) complained of severe symptoms, such as shortness of breath, developed acute respiratory distress syndrome (ARDS) (422 [13%] vs. 880 [7.9%], p < 0.001), and required supplemental oxygen (1637 [50.3%] vs. 4771 [42.7%], p < 0.001) and mechanical ventilation (260 [15.9%] vs. 530 [11.1%], p < 0.001). Mortality also is obseved to be significantly higher (odds ratio [OR]: 1.35 [95% CI: 1.19, 1.52]) in all age groups except in < 20 year. The observations of this study validate that the newer variants driven by successive COVID‐19 waves have significantly altered epidemiological characteristics. 78 Notably, the first and second COVID‐19 waves in India were most likely driven by WT strains and B.1.617.2 (Delta variant), respectively. 30 , 31 More recently, Kumar et al., 84 conducted a cross‐sectional study of COVID‐19 cases in the Indian population caused by D614G variant (B.1) (which shares close epidemiological similarity to WT strain) and Delta variant of SARS‐CoV‐2 (N = 9500, N Delta = 6238, N WT = 3262]. 84 The authors noted that in comparison to B.1 higher proportion of young individuals (<20 year) were infected (0–9 year: 4.47% vs. 2.3%, 10–19 year: 9% vs. 7%). Further, a higher proportion of total young population (10% vs. 4%) had developed symptomatic illness and was hospitalized. The proportions of women contracting an infection (41% vs. 36%) and developing symptomatic illness and hospitalized (<20 year, 14% vs. 3%, 20–59 year, 75% vs. 55%) were also increased. Notably, the mean age was significantly lower for contracting infection (men = 37.9 [±17.2] vs. 39.6 [±16.9] year, women = 36.6 [±17.6] vs. 40.1 [±17.4] year (p < 0.001]) as well as developing symptoms/hospitalization (men = 39.6 [± 17.4] vs. men = 47 [±18] year, women = 35.6 [±16.9] vs. 49.5 [±20.9] year, [p < 0.001]). The total mortality was about 1.8 times higher and the risk of death is increased irrespective of the sex (OR: 3.034, 95% CI: 1.7–5.2, p < 0.001). Interestingly, although, the proportion of mortality was still higher in men than women, an increased number of women (32% with Delta vs. 25% with B.1) died. 84
Moreover, increasing incidences of severe disease and poor outcomes in individuals with no significant co‐morbidity are concerning and should be considered alarming. 2 , 35 , 51 Also, repeated and vaccine breakthrough infections have been reported frequently with the new variants. 3 , 31 , 35
The changing epidemiological characteristics of the COVID‐19 pandemic with the emergence of more transmissible and virulent variants give clear indications that an increasing number of younger and healthy individuals, irrespective of sex, may develop severe COVID‐19 as these variants dominate over the global population.
Increasing immune escape against natural, vaccine acquired and therapeutically administered monoclonal antibodies in the emerging variants is a grave public health concern threatening of possible future COVID‐19 waves (Table 1). 85 High transmissibility of the newer variants, including within the earlier infected and vaccinated, can not only have a serious impact on the treatment and vaccination strategies but this can potentially fail the ongoing pandemic containment measures and may make reaching the post‐immunization herd immunity improbable. Notably, most of the world's population is still unvaccinated. Hence, there is strong plausibility that key mutations in the newer variants may get further selected or give way to the more adaptive mutations, as they encounter partially immunized hosts globally. This may not only prolong the duration of the pandemic but also can tremendously increase the burden of globally active cases.
5. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Increasing numbers of the studies have now indicated higher transmissibility and virulence, and gain of immune escape by the emerging SARS‐CoV‐2 variants. There is compelling evidence to reason that these variants can potentially breach key epidemiological barriers set by the WT strain and the variants that emerged in the earlier days of the pandemic. These newer developments at the population level viral dynamics caused by the selection of adaptive mutations may render the control measures ineffective. Immediate strong measures need to be taken to put a brake on the spread of the more threatening variants. A population‐matched local and global surveillance of the further changes in the SARS‐CoV‐2 genome is the need of the hour to ensure that newly emerging variants are timely identified and contained. Further, intensive clinical and epidemiological studies will be necessary to accurately assess the health impacts of the newer variants, which we still know very little about. Using variant‐specific PCR kits will help in the rapid detection of these variants, and hence it presents an immediate necessity. Moreover, the development of the monoclonal antibodies and vaccines targeting more conserved regions of the spike protein, immunizing the population against currently dominating variants, giving booster doses to those who are vaccinated, and vaccinating the unexposed and vulnerable groups on priority, are other key necessary measures, which needs to be taken on, in priority.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
The concept was developed by Ashutosh Kumar who also wrote the first draft, and Rakesh Parashar, Muneeb A. Faiq, Sujeet Kumar, Chiman Kumari, Maheswari Kulandhasamy, Ravi K. Narayan, Rakesh K. Jha, Himanshu N. Singh, Pranav Prasoon, Sada N. Pandey, and Kamla Kant revised it. All authors consented for the final draft.
ACKNOWLEDGMENTS
Authors express their sincere gratitude to GISAID (https://www.gisaid.org/) and Outbreak.info (https://outbreak.info/) databases, which were utilized for generating data graphs and/or mutation landscapes for SARS‐CoV‐2 variants.
Kumar A, Parashar R, Kumar S, et al. Emerging SARS‐CoV‐2 variants can potentially break set epidemiological barriers in COVID‐19. J Med Virol. 2022;94:1300‐1314. 10.1002/jmv.27467
DATA AVAILABILITY STATEMENT
The study has used publically available databases GISAID (https://www.gisaid.org/) and Outbreak. info (https://outbreak.info/) for generating data graphs and/or mutation landscapes for SARS‐CoV‐2 variants (accessed on date 18/08/2021). The relevant data can be downloaded from these publically accessible sites.
REFERENCES
- 1. Brookman S, Cook J, Zucherman M, Broughton S, Harman K, Gupta A. Effect of the new SARS‐CoV‐2 variant B.1.1.7 on children and young people. Lancet Child Adolesc Heal. 2021;5(4):e9‐e10. 10.1016/S2352-4642(21)00030-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taylor L. COVID‐19: Brazil's spiralling crisis is increasingly affecting young people. BMJ. 2021;373:n879. 10.1136/bmj.n879 [DOI] [PubMed] [Google Scholar]
- 3. Kustin T, Harel N, Finkel U, et al. Evidence for increased breakthrough rates of SARS‐CoV‐2 variants of concern in BNT162b2‐mRNA‐vaccinated individuals. Nat Med. 2021;27:1‐6. 10.1038/s41591-021-01413-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Made CI, Simons A, Schuurs‐Hoeijmakers J, et al. Presence of genetic variants among young men with severe COVID‐19. JAMA. 2020;324:663‐673. 10.1001/jama.2020.13719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao Q, Hu Y, Dai Z, Xiao F, Wang J, Wu J. The epidemiological characteristics of 2019 novel coronavirus diseases (COVID‐19) in Jingmen, Hubei, China. Med (United States). 2020;99(23):20605. 10.1097/MD.0000000000020605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang X, Tan Y, Ling Y, et al. Viral and host factors related to the clinical outcome of COVID‐19. Nature. 2020;583:1‐7. 10.1038/s41586-020-2355-0 [DOI] [PubMed] [Google Scholar]
- 7. Kumar A, Sesham K, Narayan RK, et al. Host vulnerability factors affecting patient outcomes in COVID‐19: an update. SSRN Electron J. Published online January 26, 2021. 10.2139/ssrn.3769784 [DOI]
- 8. Hoffmann M, Hofmann‐Winkler H, Krüger N, et al. SARS‐CoV‐2 variant B.1.617 is resistant to bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021;36(3):109415. 10.1016/J.CELREP.2021.109415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yadav PD, Sapkal GN, Abraham P, et al. Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccinees. Clin Infect Dis. 2021. 10.1093/cid/ciab411 [DOI] [PubMed] [Google Scholar]
- 10. Gobeil SM, Janowska K, McDowell S, et al. Effect of natural mutations of SARS‐CoV‐2 on spike structure, conformation, and antigenicity. Science. 2021;373(6555). 10.1126/SCIENCE.ABI6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann M, Arora P, Groß R, et al. SARS‐CoV‐2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell. 2021;184(9):2384‐2393. 10.1016/j.cell.2021.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mittal A, Manjunath K, Ranjan RK, Kaushik S, Kumar S, Verma V. COVID‐19 pandemic: insights into structure, function, and hACE2 receptor recognition by SARS‐CoV‐2. PLoS Pathog. 2020;16(8):e1008762. 10.1371/JOURNAL.PPAT.1008762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181:281‐292. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kumar A, Prasoon P, Kumari C, et al. SARS‐CoV‐2‐specific virulence factors in COVID‐19. J Med Virol. 2021;93:1343‐1350. 10.1002/jmv.26615 [DOI] [PubMed] [Google Scholar]
- 15. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao M‐M, Yang W‐L, Yang F‐Y, et al. Cathepsin L plays a key role in SARS‐CoV‐2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther. 2021;6(1):1‐12. 10.1038/s41392-021-00558-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar A, Narayan RK, Prasoon P, et al. COVID‐19 mechanisms in the human body—what we know so far. Front Immunol. 2021;1AD 0:4500. 10.3389/FIMMU.2021.693938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ziegler C, Allon S, Nyquist S, et al. SARS‐CoV‐2 receptor ACE2 is an interferon‐stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues. Cell. 2020;181(5):1016‐1035. 10.1016/j.cell.2020.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumar A, Prasoon P, Sekhawat PS, et al. Pathogenesis guided therapeutic management of COVID‐19: an immunological perspective. Int Rev Immunol. 2021;40:54‐71. 10.1080/08830185.2020.1840566 [DOI] [PubMed] [Google Scholar]
- 20. Jaimes JA, André NM, Chappie JS, Millet JK, Whittaker GR. Phylogenetic analysis and structural modeling of SARS‐CoV‐2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J Mol Biol. 2020;432(10):3309‐3325. 10.1016/J.JMB.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nat 2020 5817807. 2020;581(7807):221‐224. 10.1038/s41586-020-2179-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jawad B, Adhikari P, Podgornik R, Ching W‐Y. Key Interacting residues between RBD of SARS‐CoV‐2 and ACE2 receptor: combination of molecular dynamics simulation and density functional calculation. J Chem Inf Model. 2021;61(9):4425‐4441. 10.1021/ACS.JCIM.1C00560 [DOI] [PubMed] [Google Scholar]
- 23. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science (80‐). 2020;367(6485):1444‐1448. 10.1126/SCIENCE.ABB2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pohl MO, Busnadiego I, Kufner V, et al. SARS‐CoV‐2 variants reveal features critical for replication in primary human cells. PLoS Biol. 2021;19(3):e3001006. 10.1371/journal.pbio.3001006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoffmann M, Kleine‐Weber H, Pöhlmann S. A Multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell. 2020;78(4):779‐784. 10.1016/j.molcel.2020.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaimes JA, Millet JK, Whittaker GR. Proteolytic cleavage of the SARS‐CoV‐2 spike protein and the role of the novel S1/S2 Site. iScience. 2020;23(6):101212. 10.1016/j.isci.2020.101212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tracking SARS‐CoV‐2 variants . Accessed November 1, 2021. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
- 28. SARS‐CoV‐2 Variant Classifications and Definitions . Accessed November 1, 2021. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html
- 29. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS‐CoV‐2 spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 2020;182(4):812‐827. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar A, Dwivedi P, Kumar G, et al. Second wave of COVID‐19 in India could be predicted with genomic surveillance of SARS‐CoV‐2 variants coupled with epidemiological data: a tool for future. medRxiv. Published online June 13, 2021. 10.1101/2021.06.09.21258612 [DOI]
- 31. Dhar MS, Marwal R, Vs R, et al. Genomic characterization and epidemiology of an emerging SARS‐CoV‐2 variant in Delhi, India. Science. 2021;374:995‐999. 10.1126/SCIENCE.ABJ9932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Is a hybrid COVID strain behind Vietnam's latest wave? Not exactly . Accessed June 28, 2021. https://theconversation.com/is-a-hybrid-covid-strain-behind-vietnams-latest-wave-not-exactly-161879
- 33. Alaa AL, Julia LM, Manar A, et al. Accessed November 1, 2021. https://outbreak.info/situation-reports
- 34. Health England P . SARS‐CoV‐2 variants of concern and variants under investigation, Technical Briefing 17, 25 June 2021. Accessed August 23, 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1001354/Variants_of_Concern_VOC_Technical_Briefing_17
- 35. Health England P . SARS‐CoV‐2 variants of concern and variants under investigation in England, Technical Briefing 15, 11 June, 2021. Accessed August 23, 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/993879/Variants_of_Concern_VOC_Technical_Briefing_15
- 36. Kannan SR, Spratt AN, Cohen AR, et al. Evolutionary analysis of the delta and Delta Plus variants of the SARS‐CoV‐2 viruses. J Autoimmun. 2021;124:102715. 10.1016/J.JAUT.2021.102715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luan B, Huynh T. Insights into SARS‐CoV‐2's mutations for evading human antibodies: sacrifice and survival. J Med Chem. Published online April 9, 2021. 10.1021/acs.jmedchem.1c00311 [DOI] [PubMed]
- 38. Sk R, Islam D, Prusty S, Kanti M. Structural basis of fitness of emerging SARS‐COV‐2 variants and considerations for screening, testing and surveillance strategy to contain their threat. medRxiv. Published online January 31, 2021. 10.1101/2021.01.28.21250666 [DOI]
- 39. Cai Y, Zhang J, Xiao T, et al. Structural basis for enhanced infectivity and immune evasion of SARS‐CoV‐2 variants. Science. 2021;373(6555):642‐648. 10.1126/SCIENCE.ABI9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khan A, Zia T, Suleman M, et al. Higher infectivity of the SARS‐CoV‐2 new variants is associated with K417N/T, E484K, and N501Y mutants: an insight from structural data. J Cell Physiol. 2021;236:7045‐7057. 10.1002/jcp.30367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thomson EC, Rosen LE, Shepherd JG, et al. Circulating SARS‐CoV‐2 spike N439K variants maintain fitness while evading antibody‐mediated immunity. Cell. 2021;184(5):1171‐1187. 10.1016/j.cell.2021.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lubinski B, Tang T, Daniel S, Jaimes JA, Whittaker GR. Functional evaluation of proteolytic activation for the SARS‐CoV‐2 variant B.1.1.7: role of the P681H mutation. bioRxiv Prepr Serv Biol. Published online April 8, 2021. 10.1101/2021.04.06.438731 [DOI]
- 43. Cherian S, Potdar V, Jadhav S, et al. Convergent evolution of SARS‐CoV‐2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID‐19 in Maharashtra, India. bioRxiv. Published online May 3, 2021. 10.1101/2021.04.22.440932 [DOI] [PMC free article] [PubMed]
- 44. Harvey WT, Carabelli AM, Jackson B, et al. SARS‐CoV‐2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409‐424. 10.1038/s41579-021-00573-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar S, Tao Q, Weaver S, et al. Mutational analysis unveils the temporal and spatial distribution of G614 genotype of SARS‐CoV‐2 in different Indian states and its association with case fatality rate of COVID‐19. bioRxiv. Published online July 31, 2021. 10.1101/2020.07.27.222562 [DOI]
- 46. Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS‐CoV‐2 mutations. Front Microbiol. 2020;0:1800. 10.3389/FMICB.2020.01800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Preliminary genomic characterisation of an emergent SARS‐CoV‐2 lineage in the UK defined by a novel set of spike mutations ‐ SARS‐CoV‐2 coronavirus/nCoV‐2019 Genomic Epidemiology ‐ Virological. Accessed August 23, 2021. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- 48. Khateeb J, Li Y, Zhang H. Emerging SARS‐CoV‐2 variants of concern and potential intervention approaches. Crit Care 2021 251. 2021;25(1):1‐8. 10.1186/S13054-021-03662-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horspool AM, Ye C, Wong TY, et al. SARS‐CoV‐2 B.1.1.7 and B.1.351 variants of concern induce lethal disease in K18‐hACE2 1 transgenic mice despite convalescent plasma therapy 2. bioRxiv. Published online May 5, 2021. 10.1101/2021.05.05.442784 [DOI]
- 50. Yadav PD, Mohandas S, Shete AM, et al. SARS CoV‐2 variant B.1.617.1 is highly pathogenic in hamsters than B.1 variant. bioRxiv. Published online May 5, 2021. 10.1101/2021.05.05.442760 [DOI]
- 51. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of the P.1 SARS‐CoV‐2 lineage in Manaus, Brazil. Science (80‐). 2021;372(6544):815‐821. 10.1126/science.abh2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Campbell F, Archer B, Laurenson‐Schafer H, et al. Increased transmissibility and global spread of SARS‐CoV‐2 variants of concern as at June 2021. Euro Surveill. 2021;26(24). 10.2807/1560-7917.ES.2021.26.24.2100509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li B, Deng A, Li K, et al. Viral infection and transmission in a large, well‐traced outbreak caused by the SARS‐CoV‐2 Delta variant. medRxiv. Published online July 23, 2021. 10.1101/2021.07.07.21260122 [DOI] [PMC free article] [PubMed]
- 54. Montagutelli X, Prot M, Levillayer L, et al. The B1.351 and P.1 variants extend SARS‐CoV‐2 host range to mice. bioRxiv. Published online March 18, 2021. 10.1101/2021.03.18.436013 [DOI]
- 55. Deng X, Garcia‐Knight MA, Khalid MM, et al. Transmission, infectivity, and antibody neutralization of an emerging SARS‐CoV‐2 variant in California carrying a L452R spike protein mutation. medRxiv Prepr Serv Heal Sci. Published online March 9, 2021. 10.1101/2021.03.07.21252647 [DOI]
- 56. Li Q, Wu J, Nie J, et al. The Impact of Mutations in SARS‐CoV‐2 Spike on Viral Infectivity and Antigenicity. Cell. 2020;182(5):1284‐1294. 10.1016/j.cell.2020.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tchesnokova V, Kulakesara H, Larson L, et al. Acquisition of the L452R mutation in the ACE2‐binding interface of Spike protein triggers recent massive expansion of SARS‐Cov‐2 variants. bioRxiv Prepr Serv Biol. Published online March 11, 2021. 10.1101/2021.02.22.432189 [DOI] [PMC free article] [PubMed]
- 58. Planas D, Veyer D, Baidaliuk A, et al. Reduced sensitivity of SARS‐CoV‐2 variant Delta to antibody neutralization. Nat 2021. 2021;596:1‐7. 10.1038/s41586-021-03777-9 [DOI] [PubMed] [Google Scholar]
- 59. Mlcochova P, Kemp S, Dhar MS, et al. SARS‐CoV‐2 B.1.617 emergence and sensitivity to vaccine‐elicited antibodies. bioRxiv. Published online May 18, 2021. 10.1101/2021.05.08.443253 [DOI]
- 60. Tang T, Bidon M, Jaimes JA, Whittaker GR, Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020;178:104792. 10.1016/j.antiviral.2020.104792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xia S, Lan Q, Su S, et al. The role of furin cleavage site in SARS‐CoV‐2 spike protein‐mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. 2020;5(1):92. 10.1038/s41392-020-0184-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Twohig KA, Nyberg T, Zaidi A, et al. Hospital admission and emergency care attendance risk for SARS‐CoV‐2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study. Lancet Infect Dis. 2021;0(0). 10.1016/S1473-3099(21)00475-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.SARS‐CoV‐2 variants of concern as of 3 June 2021. Accessed June 16, 2021. https://www.ecdc.europa.eu/en/covid-19/variants-concern
- 64. Sanches PRS, Charlie‐Silva I, Braz HLB, et al. Recent advances in SARS‐CoV‐2 Spike protein and RBD mutations comparison between new variants Alpha (B.1.1.7, United Kingdom), Beta (B.1.351, South Africa), Gamma (P.1, Brazil) and Delta (B.1.617.2, India). J Virus Erad. 2021;7(3):100054. 10.1016/J.JVE.2021.100054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cluzel N, Lambert A, Maday Y, Turinici G, Danchin A. Biochemical and statistical lessons from the evolution of the SARS‐CoV‐2 virus: paths for novel antiviral warfare. C R Biol. 2020;343(2):177‐209. 10.5802/CRBIOL.16 [DOI] [PubMed] [Google Scholar]
- 66. Wang R, Chen J, Gao K, Wei G‐W. Vaccine‐escape and fast‐growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID‐19‐devastated countries. Genomics. 2021;113(4):2158‐2170. 10.1016/j.ygeno.2021.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deng X, Garcia‐Knight MA, Khalid MM, et al. Transmission, infectivity, and neutralization of a spike L452R SARS‐CoV‐2 variant. Cell. 2021;184:3426‐3437. 10.1016/j.cell.2021.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang P, Nair MS, Liu L, et al. Antibody resistance of SARS‐CoV‐2 variants B.1.351 and B.1.1.7. Nature. 2021;593(7857):130‐135. 10.1038/s41586-021-03398-2 [DOI] [PubMed] [Google Scholar]
- 69. McCallum M, Bassi J, De Marco A, et al. SARS‐CoV‐2 immune evasion by the B.1.427/B.1.429 variant of concern. Science. 2021;373(6555):648‐654. 10.1126/SCIENCE.ABI7994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ge A, Rioux M, Kelvin AA, Ca AK, Kelvin A. Computational assessment of the spike protein antigenicity reveals diversity in B cell epitopes but stability in T cell epitopes across SARS‐CoV‐2 variants. bioRxiv. Published online March 25, 2021. 10.1101/2021.03.25.437035 [DOI]
- 71. Liu C, Ginn HM, Dejnirattisai W, et al. Reduced neutralization of SARS‐CoV‐2 B.1.617 by vaccine and convalescent serum. Cell. 2021;184(16):4220‐4236. 10.1016/J.CELL.2021.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hacisuleyman E, Hale C, Saito Y, et al. Vaccine breakthrough infections with SARS‐CoV‐2 variants. N Engl J Med. 2021;384(23):2212‐2218. 10.1056/nejmoa2105000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gupta S, Nakabo S, Chu J, Hasni S, Kaplan MJ. Clinical characterization and Genomic analysis of COVID‐19 breakthrough infections during second wave in different states of India. medRxiv. Published online July 15, 2020. 10.1101/2021.07.13.21260273 [DOI]
- 74. Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA. Effects of common mutations in the SARS‐CoV‐2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife. 2021;10:10. 10.7554/ELIFE.70658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Volz E, Mishra S, Chand M, et al. Assessing transmissibility of SARS‐CoV‐2 lineage B.1.1.7 in England. Nature. 2021;593(7858):266‐269. 10.1038/s41586-021-03470-x [DOI] [PubMed] [Google Scholar]
- 76. Galloway SE, Paul P, MacCannell DR, et al. Emergence of SARS‐CoV‐2 B.1.1.7 lineage — United States, December 29, 2020–January 12, 2021. MMWR Morb Mortal Wkly Rep. 2021;70(3):95‐99. 10.15585/mmwr.mm7003e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Alaa AL, Julia LM, Manar A, et al. B.1.617.2 Lineage report. outbreak.info. Accessed November 1, 2021. https://outbreak.info/situation-reports
- 78. Kumar G, Mukherjee A, Sharma RK, et al. Clinical profile of hospitalized COVID‐19 patients in first & second wave of the pandemic: Insights from an Indian registry based observational study. Indian J Med Res. 2021;153:619‐628. 10.4103/IJMR.IJMR_1628_21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nyberg T, Twohig KA, Harris RJ, et al. Risk of hospital admission for patients with SARS‐CoV‐2 variant B.1.1.7: cohort analysis. BMJ. 2021;373:n1412. 10.1136/bmj.n1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pijls BG, Jolani S, Atherley A, et al. Demographic risk factors for COVID‐19 infection, severity, ICU admission and death: A meta‐analysis of 59 studies. BMJ Open. 2021;11(1):44640. 10.1136/bmjopen-2020-044640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gadi N, Wu SC, Spihlman AP, Moulton VR. What's sex got to do with COVID‐19? Gender‐based differences in the host immune response to coronaviruses. Front Immunol. 2020;11:2147. 10.3389/fimmu.2020.02147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kumar A, Narayan RK, Kulandhasamy M, et al. COVID‐19 pandemic: insights into molecular mechanisms leading to sex‐based differences in patient outcomes. Expert Rev Mol Med. 2021;23:e7. 10.1017/ERM.2021.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Health England P . SARS‐CoV‐2 variants of concern and variants under investigation in England, Technical Briefing 14, 3 June, 2021. Accessed on August 28, 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/991343/Variants_of_Concern_VOC_Technical_Briefing_14
- 84. Kumar A, Asghar A, Raza K, et al. Demographic characteristics of SARS‐CoV‐2 B.1.617.2 (Delta) variant infections in Indian population. medRxiv. Published online September 26, 2021. 10.1101/2021.09.23.21263948 [DOI]
- 85. Dyson L, Hill EM, Moore S, et al. Possible future waves of SARS‐CoV‐2 infection generated by variants of concern with a range of characteristics. medRxiv. Published online June 10, 2021. 10.1101/2021.06.07.21258476 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The study has used publically available databases GISAID (https://www.gisaid.org/) and Outbreak. info (https://outbreak.info/) for generating data graphs and/or mutation landscapes for SARS‐CoV‐2 variants (accessed on date 18/08/2021). The relevant data can be downloaded from these publically accessible sites.