

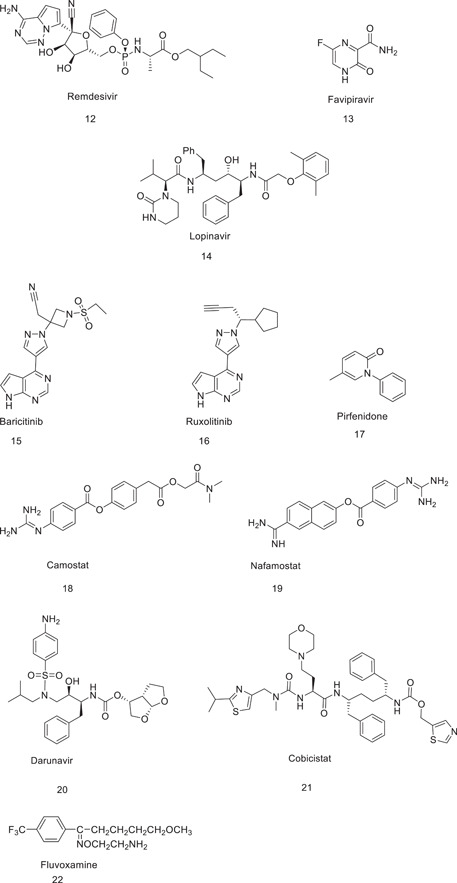
Abstract
The outbreak of the coronavirus pandemic COVID‐19 created by its severe acute respiratory syndrome corona virus‐2 (SARS‐CoV‐2) variant, known for producing a very severe acute respiratory syndrome, has created an unprecedented situation by its continual assault around the world. The crisis caused by the SARS‐CoV‐2 variant has been a global challenge, calling to mitigate this unprecedented pandemic that has engulfed the whole world. Since the outbreak and spread of COVID‐19, many researchers globally have been grappling to find new clinically trialed active drugs with anti‐COVID‐19 activity, from antimalarial drugs to JAK inhibitors, antiviral drugs, immune suppressants, and so forth. This article presents a brief discussion on the activity and synthesis of some active molecules such as favipiravir, hydroxychloroquine, pirfenidone, remdesivir, lopinavir, camostat, chloroquine, baricitinib, molnupiravir, and so forth, which are under trial.
Keywords: COVID‐19 pandemic, Delta+ variant, lopinavir, remdesivir, SARS‐CoV‐2 variant
Since the beginning of the COVID‐19 pandemic, researchers worldwide have been searching for new clinically trialed active drugs with anti‐COVID‐19 activity, from antimalarial drugs to JAK inhibitors, antiviral drugs, immune suppressants, and so forth. This article presents a brief discussion on the activities and synthesis pathways of some active molecules under trial, such as favipiravir, hydroxychloroquine, pirfenidone, remdesivir, and others.
Abbreviations
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- THF
tetrahydrofuran
1. INTRODUCTION
Human coronaviruses were first identified in the 1960s[ 1 ]; these were a broad batch of associated viruses that were linked to respiratory illnesses ranging from common colds to diseases like extreme acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS). The novel coronavirus (Figure 1) has been a previously unexplored stress caused by coronavirus, designated by the World Health Organization as severe acute respiratory syndrome corona virus‐2 (SARS‐CoV‐2),[ 2 ] and the resulting coronavirus disease‐19 (COVID‐19) has now become a major catastrophe since its discovery in Wuhan city, Hubei province of China in December 2019.[ 3 ] Before the world could prepare itself to overcome the outbreak, unexpectedly, since early 2021, a new surge of pandemic occurred in March 2021, catching the world by surprise; this lead to alarm calls raised about the onset of a second wave of the pandemic, fuelled by the spurt of another variant—the double mutants of the COVID‐19 (B 1.617) virus (the Delta+ virus). For SARS‐CoV‐2 mutation D164G enhances viral entry to the host cell as well as replication.[ 4 ] It was previously reported that the D614G mutation was present in all SARS‐CoV‐2 sequences by the summer of 2020.[ 5 ] This mutant variant has been identified to be more active, strong, and fast‐spreading, which has taken large communities and mass group in its grip within short period of time.
Figure 1.
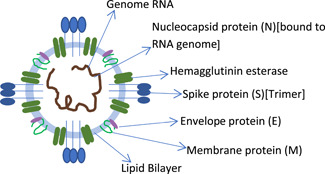
Structure of the coronavirus, showing different layers including the spike protein
There is no evidence from randomized‐controlled trials (RCTs) that any medication can enhance prognosis in people with COVID‐19, whether suspected or confirmed. There have been no clinical trial results to back up any preventive treatment. However, we have only two options that remain the best bet to fight off this crisis: (1) the vaccine option, (2) the drug option (not available so far). Many potent vaccines have been discovered so far (some of these are under trial and are in the pipelines): Covishield, Covaxin AstraZeneca, Pfizer, Johnson and Johnson's, Moderna, Novavax, Sputnik V, Zycov‐D, Gennova BiopharmNovax, Indian Immunologicals, Biological E, Baylor college of medicine and m‐RNA wax, and so forth (of these, due to severe adverse effects like blood clotting, etc., the use of Oxford Astrazeneca has been suspended in many countries). The high cost of the use of monoclonal antibodies, cell‐based therapies, interferon‐based therapies, and immunopathological treatments makes them unsuitable. Small‐molecule medications, which include pre‐existing medications to treat influenza, HBV, HCV, HIV, malaria, and filovirus medications, have piqued experts' interest due to their potential for faster development.
In this context, a recent announcement from the United States through Anthony Fauci (the Chief Medical Advisor to US President) is worth reporting. It states that the US government is planning to take another major medical step to beat the pandemic, putting in $3.2 billion to develop “antiviral pills” to treat COVID‐19 infection.
Oral therapies will be easier to produce, transport, store and administer. It could be taken at home easily during the course of the disease and therefore would be a powerful tool for battling the pandemic at the early stages and in saving lives.
The US Department of Health and Human Services revealed that 19 therapeutic agents have already been prioritized for testing in rigorous trials for outpatients and inpatients with COVID‐19 (Figure 2). Availability of Food and Drug Administration (FDA)‐authorized “antiviral pills” through these efforts would be a major breakthrough in the ongoing efforts to combat COVID‐19.
Figure 2.
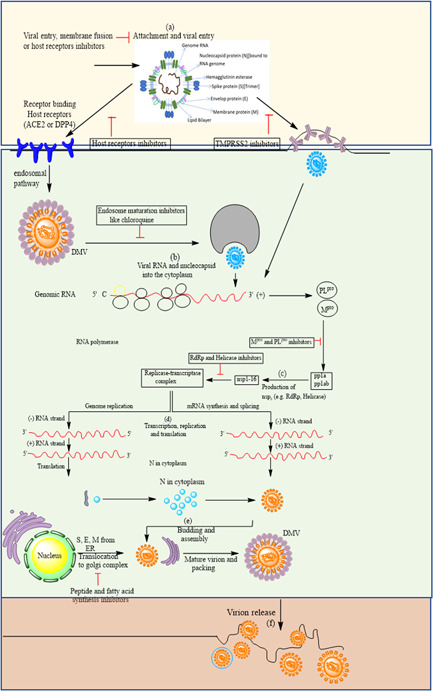
Severe acute respiratory syndrome corona virus‐2 multiplication stages inside the host cell
A review of the literature on the drugs under trial reveals that the following drugs can inhibit the key components of the coronavirus infection (Figure 3).
Figure 3.
Therapeutic molecules that are under trial for the COVID‐19 pandemic
2. INFECTION CYCLE OF THE CORONAVIRUS
The coronavirus first binds or attaches to the ACE2 (angiotensin‐converting enzyme) receptors that are present on the numosites. When it binds to the ACE2 receptors, there is a change in the S‐glycoprotein of the coronavirus; as a result, there is a cleavage or release of the fragments of S‐proteins by the transmembrane protein of the host. As a result, the virus enters the cell through the early and late endosomes. Inside the cytoplasm, there is another protease, cathepsin‐l, which further cleaves the S‐protein at low pH, and there is a fusion of the viral envelope with the phospholipid membrane of the endosomes. After this fusion, there is a release of the viral genome in the cytoplasm. The second step is the release of the viral genome. The viral genome is released into the cytoplasm, after which a positive‐strand RNA genome is converted into a negative‐strand RNA genome, which is used as signal for for messenger RNA synthesis. Ribosomes are the host and they promote the synthesis and replication of the viral genome; also, they promote the synthesis of the nonstructural proteins. Spike, membrane, and envelope proteins of the virus then enter the endoplasmic reticulum and then move to the Golgi bodies. The virus is packaged into the nucleocapsid which is transported to the Golgi bodies after replication of RNA genome to attach with other viral protein. Finally, the mature viral particles are ready to be transported out of the cells with the help of the Golgi vesicles. Once the virus is packed with the nucleocapsid, the spike proteins, the membrane proteins, and the envelope proteins, the mature viral particles move to the cells and are expelled out of the cells by the process of exocytosis (Figure 2). The drugs that are used in the various stages of COVID‐19 viral infection are listed in Table 1.
Table 1.
Involvement of drugs in various stages of COVID‐19 viral infection
| (a) | (i) Hydroxychloroquine | (a) In the viral entry inhibition of the host cell |
| (ii) Chloroquine | ||
| (iii) Mefloquine | ||
| (b) | (i) Lopinavir | (b) In the inhibition of viral replication |
| (ii) Ritonavir | ||
| (iii) Camostat | ||
| (iv) Ivermectin | ||
| (c) | (i) Tipranavir | (c) In the inhibition of 3C‐like protease |
| (d) | (i) Remdesivir | (d) In the inhibition of viral RNA synthesis by inhibiting the activity of RNA polymerase |
| (ii) Favipiravir |
There are many other established drug molecules that are used alone or in combination that may also prove to be effective for COVID‐19 therapy. This review focuses on these drugs (Figures 1 and 2 and Table 2) in terms of their activity and synthesis.
Table 2.
Drugs under trial for use against COVID‐19
| Drug | Mechanism of action | Dosage | Active against | Clinical trials | Outcomes in clinical trials |
|---|---|---|---|---|---|
| 1. Favipiravir | RNA‐dependent RNA polymerase inhibitor | 600 mg BID | Influenza | Clinical trials to severe COVID‐19 | Symptomatic improvement in mild and moderate cases |
| 2. Hydroxychloroquine | Viral entry inhibitor | Day 1 400 mg BID, followed by 200 mg BID for 5–10 days. Alternative: 200 mg TID for 10 days or 400 mg QD for 5 days | Malaria | Several clinical trials have been conducted | Less toxic option, impairs viral replication |
| 3. Pirfenidone | Inhibits apoptosis | 2403 mg orally or through a nasogastric tube as 801 mg TID, for 4 weeks | Pulmonary fibrosis | Clinical trials against severe acute respiratory syndrome corona virus‐2 (SARS‐CoV‐2) | Decreases inflammation and ameliorates oxidative stress; also protects pneumocytes |
| 4. Remdesivir | RNA‐dependent RNA polymerase inhibitor | 10‐day administration. Day 1: 200 mg QD loading dose, followed by 100 mg QD | Ebola virus, respiratory syncytial virus | Clinical case and clinical trials against SARS‐CoV‐2 | Food and Drug Administration‐approved use of remdesivir in patients aged 12 years and older. |
| 5. Lopinavir | 3C‐like (3CL) protease inhibitor | BID for up to 14 days | HIV infections | Clinical trial against SARS‐COV‐2 | Did not provide antiviral effects or improved clinical outcomes in patients with severe disease, but in early infections, clinical outcomes were improved |
| 6. Camostat | Serine protease inhibitor | 400 mg on Day 1, followed by 200 mg on Days 2–5 | Pancreatitis | Clinical trials against SARS‐CoV‐2 | Primes the spike protein of the human coronavirus and facilitates cell entry and infection |
| 7. Chloroquine | Viral entry inhibitor | 500 mg orally QD or BID for 5–10 days | Malaria | Several clinical trials have been conducted | Impairs viral replication and has anti‐inflammatory activities |
| 8. Baricitinib | Inhibits Janus kinase | 4 mg/day (check) | Rheumatoid arthritis | Clinical trials against SARS‐CoV‐2 | Improves lymphocyte count and prevents entry of the virus |
| 9. Molnupiravir | Inhibits mutation | 200–400 mg orally every 12 h for 5 days | Influenza | Clinical trials in efficiency against SARS‐CoV‐2 | Decreases damage caused to the lungs |
| 10. Ribavirin | Inhibits viral RNA synthesis | Oral, 400 mg TID (>50 ml/min), 400 mg BID (50–30 ml/min), 200 mg daily (<30 ml/min) × 10 days | Hepatitis C | Clinical trials against SARS‐CoV‐2 | Resulted in resolution of fever and lung opacities within 2 weeks |
| 11. Gemicitabine | Inhibits DNA synthesis | 1000 mg/m2, administered by a 30‐min infusion | Cancer | Clinical trials against SARS‐CoV‐2 | Result to be reported |
| 12. Fluvoxamine | Alleviates ER stress via induction of the sigma‐1 receptor | 100 mg twice daily for 10 days | Obsessive–compulsive disorders | Clinical trials against SARS‐CoV‐2 | Prevent progression of the coronavirus |
| 13. Rupintravir | Inhibits 3CL protease inhibitor | On trial | Human‐rhinovirus | Clinical trials against SARS‐CoV‐2 | Result to be reported |
3. DESCRIPTION OF DRUGS THAT ARE BEING USED DURING THE CORONA PANDEMIC
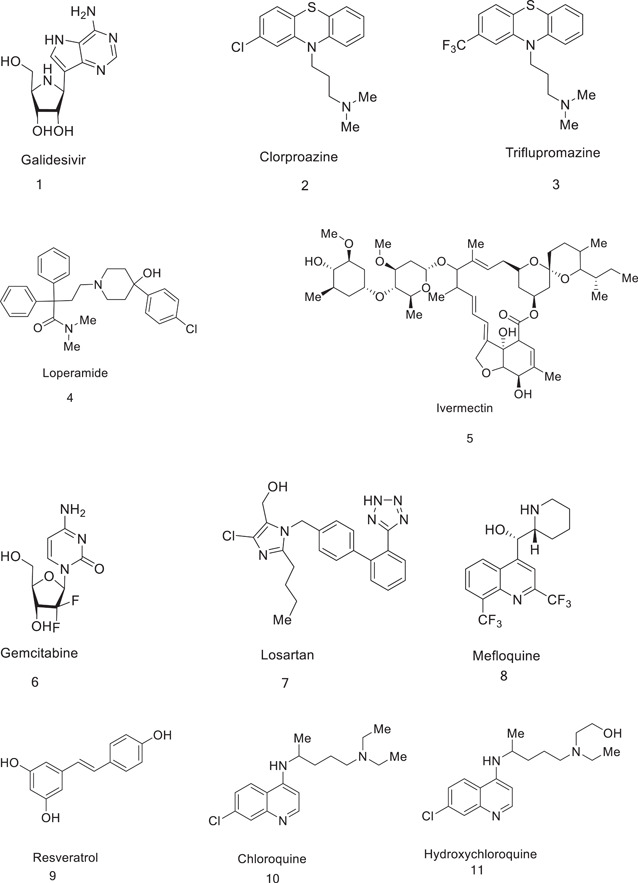
3.1. Chloroquine (CQ)
CQ, which is an antimalarial drug, was first discovered in 1934 at Bayer laboratories by Hans Andersag et al.,[ 6 ] who named it Resochin. CQ exerts an important effect that can help to deter or slow down the spread of SARS‐CoV‐2 infection. The medication could prevent the virus from infecting cells.[ 7 ] Coronaviruses use the ACE2 receptors to gain access to the cell. The compound has been shown to interact with ACE2 glycosylation.[ 8 ] This can affect the productivity of spike protein‐ACE2 binding and inhibit entry of the virus into the cells.
3.1.1. Synthesis of CQ (7)
The first description of the synthesis of CQ was provided by Andersag in 1934, and involved the condensation of 3‐chloroaniline (1) with diethyl‐2‐oxosuccinate (2), yielding diethyl (E)‐2‐[(3‐chlorophenyl)imino]succinate (3). Product 3 underwent saponification in the presence of liquid paraffin at 250°C to afford ethyl 7‐chloro‐4‐hydroxyquinoline‐2‐carboxylate (4); decarboxylation of 4 yielded 7‐chloroquinolin‐4‐ol (5). CQ (7) was obtained on chlorination of 5, followed by amination of the resulting 4,7‐dichloroquinoline (6) (Scheme 1).
Scheme 1.
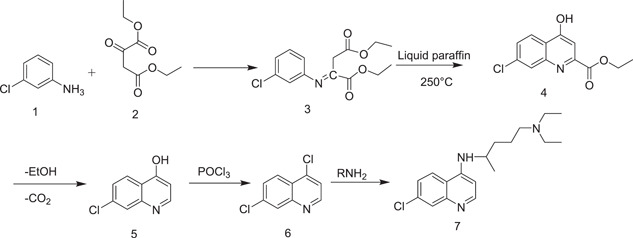
Synthesis of chloroquine by Andersag
CQ was also obtained by nucleophilic substitution of 4,7‐dichloroquinoline (6) with 2‐amino‐5‐(diethylamino)pentane (8) at 130–155°C using phenol as a solvent[ 9 ] as shown in Scheme 2.
Scheme 2.
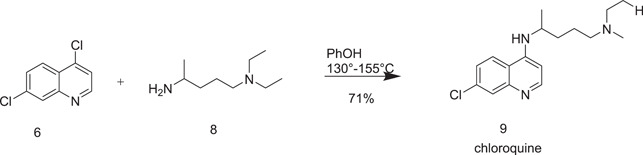
Another synthetic route to chloroquine from 6
Price and Roberts[ 10 ] described the synthesis of 4,7‐dichloroquinoline (6), which is a key ingredient for the synthesis of CQ as demonstrated in Scheme 3. Schiff base (diethyl (Z)‐2‐[(phenylimino)methyl)malonate] (11) was prepared by reaction with m‐chloroaniline (1) and ethoxy methyl enemalonic ester (10) at reflux temperature as shown in Scheme 3.
Scheme 3.
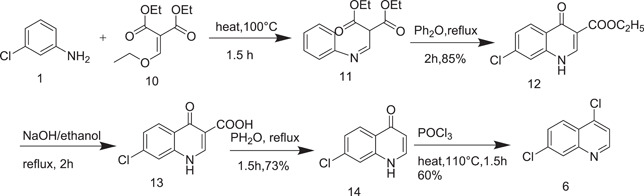
Synthetic route to 4,7‐dichloroquinoline (6)
A new route to the synthesis of 4,7‐dichloroquinoline (6) was described by Guy et al.[ 11 ] in 2004, as shown in Scheme 4. Instead of using ethoxymethylenemalonic ester, they used Meldrum's acid (15).[ 12 ] Meldrum's acid regent has the considerable advantage of producing 4,7‐dichloroquinoline in three steps instead of five.
Scheme 4.
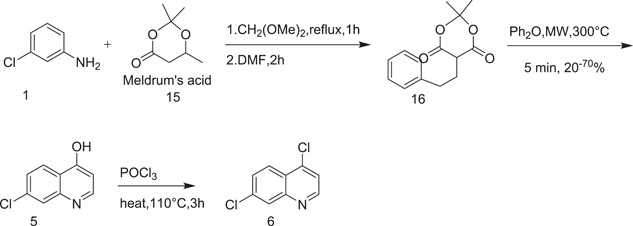
Synthesis of 4,7‐dichloroquinoline (6) using Meldrum's acid
Surrey and Hammer[ 13 ] described the synthesis of 4,7‐dichloroquinoline (6) using m‐chloroaniline (1) and diethyl ester of oxaloacetic acid (17). 4,7‐Dichloroquinoline was prepared by cyclocondensation of 18, followed by hydrolysis and decarboxylation of 19 resulted 5 which on chlorination yielded 4,7‐dichloroquinoline (Scheme 5).
Scheme 5.
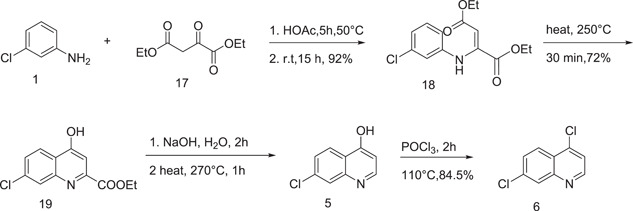
Synthesis of 6 using diethyl ester of oxaloacetic acid
3.2. Hydroxychloroquine (HCQ)
Oral medications like HCQ and CQ have long been used to treat malaria and chronic inflammatory diseases like systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA).[ 14 ] Though HCQ, a racemic combination, was found to be effective and was considered the first‐line treatment for SLE and COVID‐19 victims, HCQ and CQ are well‐tolerated medications that have been widely investigated in individuals with SLE and malaria. Both compounds, on the contrary, have severe side effects such as QTc prolongation, hypoglycaemia, retinopathy, and neuropsychiatric effects.[ 15 ]
3.2.1. Synthesis of HCQ (24)
Sterling–Winthrop discovered the first method for the synthesis of HCQ in 1950.[ 16 ] The process started with refluxing of 5‐chloropentan‐2‐one (20) and 2‐(ethylamino) ethan‐1‐ol (21) in boiling xylene for around three hours to afford 5‐(ethyl(2‐hydroxyethyl) amino)pentan‐2‐one (22), which, on reductive amination with a Raney nickel catalyst in methanolic ammonia at 1000 psi hydrogen pressure, gave 2‐[(4‐aminopentyl)(ethyl)amino]ethan‐1‐ol (23). When 23 and 6 were heated in phenol and catalytic potassium iodide for 18 h to 125–130°C and the mixture was diluted with methanol, followed by the addition of phosphoric acid, it produced diphosphate salt of HCQ (24) (Scheme 6).
Scheme 6.
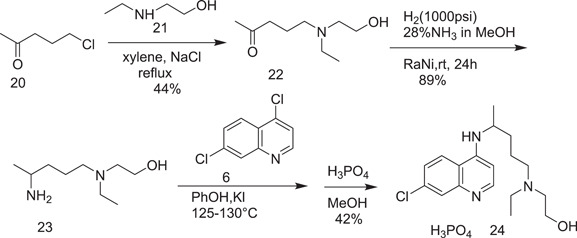
Initial route for the synthesis of hydroxychloroquine (24)
Further, an improved route was proposed for the synthesis of HCQ (24) from 5‐iodopentan‐2‐one, which was, in turn, obtained from 25 as shown in Scheme 7. When 26 was refluxed with 2‐(ethylamino)ethan‐1‐ol (21) in tetrahydrofuran (THF) at its boiling point using potassium carbonate as a base, it afforded 5‐[ethyl(2‐hydroxyethyl)amino]pentan‐2‐one (27). 2‐[(4‐Aminopentyl)(ethyl)amino]ethan‐1‐ol was obtained via the catalytic reduction of 5‐[ethyl(2‐hydroxyethyl)amino]pentan‐2‐one oxime (28). HCQ (24) was obtained on nucleophilic substitution of 6 with 29 in the presence of triethylamine.
Scheme 7.
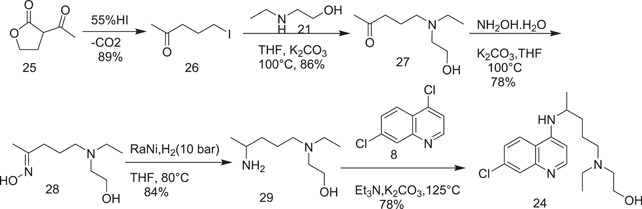
Synthesis of hydroxychloroquine (24)
Yu et al.[ 17 ] pioneered the continuous flow mechanism of synthesizing HCQ (24) by dissolving 4,7‐dichloroquinoline (6) and 5‐(N‐ethyl‐N‐2‐hydroxyethylamino)‐2‐pentylamine (30) in ethanol and then treatment with a solution of potassium carbonate and triethylamine at a high temperature. This process yielded HCQ (24) after purification with silica chromatography.
A simple method for the preparation of HCQ (24) in better yield was described by Kumar et al.[ 18 ] in 2003. They used an equimolar quantity of sodium hydroxide and a catalytic amount of sodium iodide at a higher temperature for condensation of 5‐(N‐ethyl‐N‐2‐hydroxyethylamino)‐2‐pentylamine (30) and 4,7‐dichloroquinoline (6) to afford HCQ (24) as shown in Scheme 8.
Scheme 8.
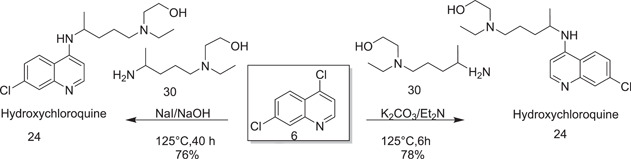
Synthesis of hydroxychloroquine (24)
3.3. Lopinavir (31)
Lopinavir (LPV) (Scheme 9) is an antiviral drug (protease inhibitor) used in HIV prevalence treatment and prevention in tandem with ritonavir (booster). In vitro testing of lopinavir's ability to inhibit the SARS‐CoV main protease (3CLpro) was performed. No studies have been done on the effectiveness of lopinavir as antiviral agents in the treatment of SARS‐CoV.[ 19 ]
Scheme 9.
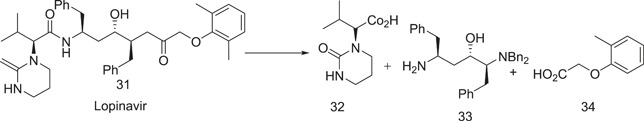
Retrosynthetic approach to lopinavir
3.3.1. Synthesis of lopinavir
In 1999 and 2000, Abbott laboratories (which are now Abbvie) described the formation of lopinavir in a patent and in two publications.[ 20 ] The first step included tribenzylation of l‐phenylalanine (35) with benzyl chloride to give benzyl dibenzyl‐l‐phenylalaninate (36). The acetonitrile anion was derived from acetonitrile treatment with a base, sodium amide was then added to the benzyl ether in the next step, and the resulting nitrile (37) was then treated with benzyl magnesium chloride in a one‐pot reaction in MTBE to give 38.
The trifluoroacetic acid (TFA) complex of enaminone (38) was reduced with NaBH4 to give amino alcohol (33) (Scheme 10).
Scheme 10.
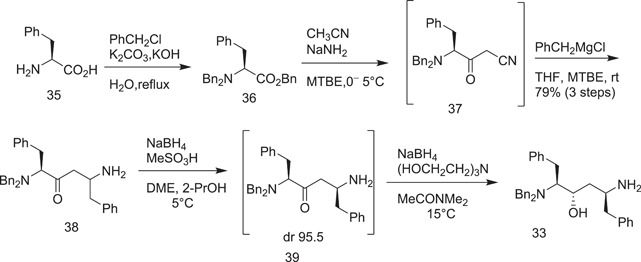
Abbott synthesis of the lopinavir chiral centre core
In the next step, l‐valine (40) was converted into (phenoxycarbonyl)‐l‐valine (41)[ 21 ] (Scheme 11) on its treatment with phenyl chloroformate in the presence of lithium chloride, which allowed the reaction to take place at a very low temperature. Neutral ammonia was also used to avoid gumming and emulsion formation. Treatment of (phenoxy‐carbonyl)‐l‐valine (41) with 3‐chloropropylamine hydrochloride and solid NaOH in THF at 10°C produced the alkylated product [(3‐chloropropyl)carbamoyl]‐l‐valine; (42) was obtained, which was cyclized by adding KO‐t‐Bu in THF at 20°C to give (S)‐3‐methyl‐2‐(2‐oxotetrahydropyrimidin‐1(2H)‐yl)butanoic acid (32).
Scheme 11.
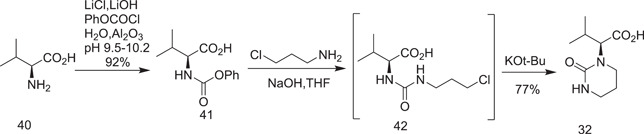
Abbott synthesis of fragment
Scheme 12 demonstrates the final step to achieve lopinavir.[ 22 ] (S)‐3‐Methyl‐2‐(2‐oxotetrahydropyrimidin‐1(2H)‐yl)butanoyl chloride (43) was coupled with an amine in EtOAc/dimethylformamide (DMF) at 30°C to give 44, which was followed by debenzylation in the presence of activated palladium and ammonium formate at 50°C to give amine (45). The diastereomeric purity was achieved by crystallizing the salt with l‐pyroglutamic acid (PGA) in 1,4‐dioxane, which yielded PGA (45) in 74% yield more than 98.5% purity. The final amide coupling of PGA 45 with acid chloride 47 was carried out at room temperature under Schotten Baumann conditions (EtOAc/aqueous NaHCO3). Lopinavir (31) was crystallized from EtOAc/heptane.
Scheme 12.
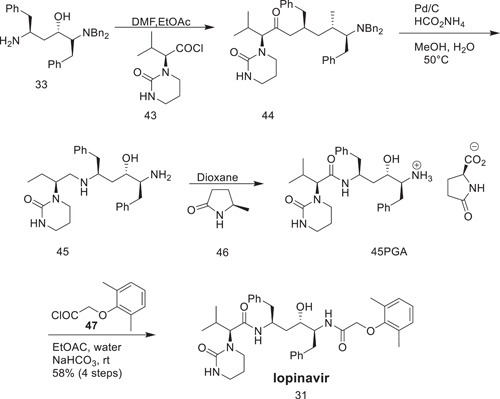
Final step to obtain lopinavir
N,N‐Carbonyldiimidazole was used to activate (2S,3S,5S)‐5‐amino‐2‐(dibenzylamino)‐1,6‐diphenylhexane‐3‐ol, which was then condensed with (2S)‐3‐methyl‐2‐(2‐oxotetrahydropyrimidine‐1‐(2H)‐yl)butanoic acid to give (2S)‐N‐[(2S,4S,5S)‐5‐(dibenzylamino)‐4‐hydroxy‐1,6‐diphenylhexan‐2‐yl]‐3‐methyl‐2‐(2‐oxotetrahydropyrimidine‐1(2H)‐yl)butanamide. This compound underwent debenzylation with ammonium formate and palladium on charcoal, giving (2S)‐N‐[(2S,4S,5S)‐5‐amino‐4‐hydroxy‐1,6‐diphenylhexan‐2‐yl]‐3‐methyl‐2‐(2‐oxotetrahydropyrimidine‐1‐(2H)‐yl)butanamide, which was then treated with PGA to give pure (2S)‐N‐[(2S,4S,5S)‐5‐amino‐4‐hydroxy‐1,6‐diphenylhexan‐2‐yl]‐3‐methyl‐2‐(2‐oxotetrahydropyrimidine‐1‐(2H)‐yl)butanamide (S)‐pyroglutamic acid salt, which, on condensation with (2,6‐dimethylphenoxy)acetic acid, gave lopinavir (Scheme 13).
Scheme 13.
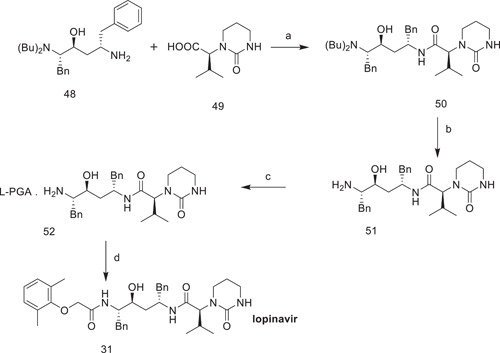
Synthesis of lopinavir (31). Reagents and conditions: (a) N,N‐carbonyldiimidazole, ethyl acetate; yield: 82.7%; (b) ammonium formate, palladium on charcoal, methanol; yield: 100%; (c) l‐pyroglutamic acid (PGA), acetone, DMF; yield: 72.4%; (d) 2,6‐dimethylphenoxyaceticacid, thionyl chloride, dichloromethane; yield: 85.7%
3.4. Camostat (57)
Ono Pharmaceutical Co., Ltd. first published the synthesis of Camostat in a patent in 1977 (US4021472A). Foipan (the mesylate salt of camostat) was approved by Japan in 1985 for the treatment of chronic pancreatitis and oesophagitis.[ 23 ] Oral squamous cell carcinoma,[ 24 ] dystrophic epidermolysis,[ 25 ] and exocrine pancreatic enzyme inhibition[ 26 , 27 ] have all been treated with camostat mesylate.
3.4.1. Synthesis of camostat (57)
4‐Hydroxyphenylacetic acid (53) was esterified with N,N‐dimethyl‐2‐bromoacetamide (54) in refluxing acetonitrile, yielding the ester intermediate (55). The ester 55 was then heated with crude acyl chloride of p‐guandinobenzoic acid. The product camostat was precipitated as camostat mesylate from a methanol/diethyl mixture (Scheme 14).
Scheme 14.
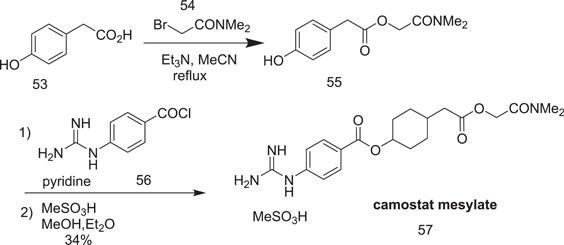
Synthesis of camostat mesylate (57)
3.5. Remdesivir (60)
Remdesivir has been considered as a potential therapeutic for COVID‐19 because of its broad‐spectrum in vitro activity against several coronavirus strains, which also includes SARS‐CoV‐2.[ 28 , 29 ] Remdesivir is administered as an intravenous injection, and its safety and pharmacokinetics have been tested in Ebola Phase I clinical trials, both as a single dose and in multiple doses. Remdesivir was found to be the most efficacious in the treatment of COVID‐19.[ 30 ] The FDA has approved remdesivir for the treatment of COVID‐19 in hospitalized adult and pediatric patients (older than 12 years of age and weighing less than 40 kg).
3.5.1. Synthesis of remdesivir (60)
In the presence of (3R,4S,5S)‐2‐(4‐aminopyrrolo[2,1‐f][1,2,4]triazin‐7‐yl)‐3,4,5‐tris(benzyloxy)tetrahydrofuran‐2‐ol (58) in dichloromethane, along with trifluoromethanesulphonic acid, trimethylsilyl trifluormethanesulphonate, and trimethylsilyl cyanide, at a temperature of −78°C, the cyanation reaction is performed (Scheme 15).
Scheme 15.
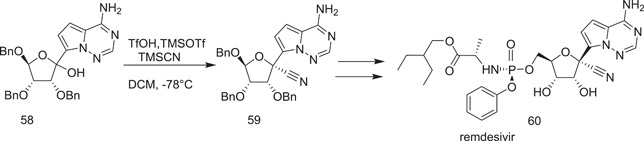
Synthetic route to remdesivir
The synthesis of remdesivir was initiated by the scientists of the Gilead Biopharmaceutical Company in 2016.[ 31 , 32 ] Synthesis was started with ribolactone (62), which was prepared from tribenzylated ribose (61) using two methods.[ 33 ] In the first method, 62 was prepared by oxidizing protected ribose (61) with dimethyl sulfoxide (DMSO) and acetic anhydride. In the second method, the preparation of 62 involved oxidation of 61 with 2,2,6,6‐tetramethylpiperidine‐1‐oxyl (TEMPO)‐catalyzed sodium hypochlorite. Condensation of 62 with 66 under the conditions given in Scheme 16 yielded 1:1 anomeric mixtures of 67 (compound 66, in turn, was prepared from 63 by the sequence of reactions shown in the scheme). 63 was converted into 64 from its reaction with hydroxylamine‐o‐sulphonic acid. 64 underwent cyclocondensation with formamide acetate to form the adenine analog 65. Selective iodination of 65 with N‐iodosuccinimide (NIS) produced its 7‐iodo derivative 66. The diastereoselective cyanation of 67 was accomplished to give the adenosine carbonitrile analog 68 on treatment of 67 with triflic acid, trimethylsilyl trifluoromethanesulphonate, and trimethylsilyl cyanide. The key intermediate, isopropylidone nucleoside 69, in the synthesis of remdesvir (60) was obtained from its debenzylation with BCl2, followed by its reaction with 2,2‐dimethoxy propane in H2SO4.
Scheme 16.
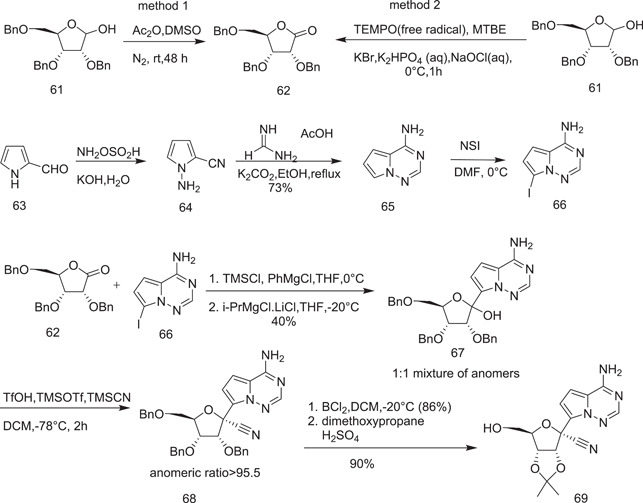
Synthesis of adenosine nucleoside
Further synthesis of remdesivir required the preparation of its precursor p‐nitrophenolate 2‐ethylbutyl‐l‐alaninate (70), which was prepared from 2‐ethylbutyl‐l‐alanine by its reaction with phosphorodichloridate (OP (OPh)Cl2) in the presence of triethylamine as a base and using dichloromethane as a solvent at –78°C, followed by reaction with 4‐nitrophenol, which afforded the diastereomeric mixture at phosphates (72a and 72b) (Schemes 17 and 18).
Scheme 17.
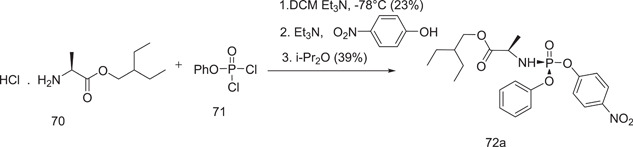
Synthesis of p‐nitrophenolate 2‐ethyl‐butyl‐l‐alaninate (72a)
Scheme 18.
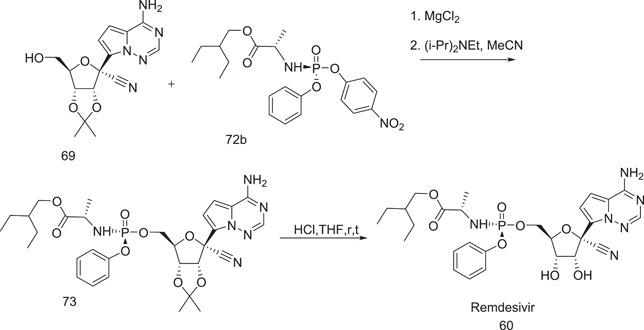
Synthesis of remdesivir (60)
Further synthesis of remdesivir (60) proceeded by coupling of nucleoside 60 with p‐nitrophenolate 2‐ethylbutyl‐l‐alaninate (72b) in the presence of MgCl2 and (i‐Pr)2–N‐Et base. Remdesivir (60) was generated by deprotecting the isopropylidene group with concentrated HCl in THF.
3.6. Favipiravir (79)
Favipiravir (79) (called T‐705, 6‐fluoro‐3‐hydroxypyrazine‐2‐carboxamide) is a medication that is used against influenza that acts by hindering the influenza virus's RNA‐dependent RNA polymerase.[ 34 ] Favipiravir also prevents a host of other pathogenic RNA viral infections, including arenavirus, bunyavirus, flavivirus, alphavirus, and norovirus.[ 35 , 36 ] Furthermore, it is considered to be a promising Ebola virus treatment candidate.[ 37 ] In the field of antiviral and antiparasitic science, favipiravir and related structures have recently received a lot of attention.[ 38 , 39 , 40 , 41 , 42 , 43 ] Favipiravir is a pyrazine derivative with three substituents (CONH2, OH, and F) at 2, 3, and 6 positions, respectively. This compound has acidic properties due to the hydroxyl group and can be tautomerized to the keto tautomers A and B[ 44 ] (Scheme 19).
Scheme 19.
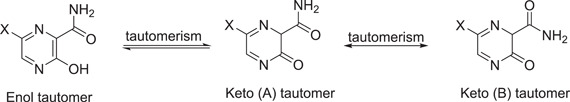
Keto–enol tautomers and keto rotational isomer for favipiravir and its analogs
3.6.1. Synthesis of favipiravir (79)
-
(A)
In one of the synthesis methods of favipiravir, methyl‐3‐amino‐6‐bromopyrazine‐2‐carboxylate (74) was subjected to diazotization, followed by alcoholysis in the presence of concentrated sulphuric acid to give methyl‐6‐bromo‐3‐methoxypyrazine‐2‐carboxylate (75) (in a yield of only 35%). The next step involved the treatment of 75 with diphenylmethanimine with a palladium catalyst, which led to the formation of methyl‐6‐amino‐3‐methoxypyrazine‐2‐carboxylate (76), whose reaction with ammonium hydroxide yielded 6‐amino‐3‐methoxypyrazine‐2‐carboxamide (77). The conversion of 77 into the corresponding fluoro derivatives (78) required treatment with the extremely corrosive Olah's reagent.[ 45 ] Given these drawbacks, this approach was not recommended for the scalable production of favipiravir (79), which finally resulted in demethylation of 78 with NaI/trimethylsilyl chloride (Scheme 20).
-
(B)
Another synthesis method of flavipiravir (79) involved the conversion of 3‐hydroxypyrazine‐2‐carboxamide (80) into 3‐hydroxy‐6‐nitropyrazine‐2‐carboxamide (81), which was obtained from its reaction with KNO3 and sulphuric acid. For the proposed strategy to move forward further (Scheme 21), it required the preparation of the intermediate 82. This resulted in one step from the treatment of 81 with phosphoryl chloride in pyridine. The conversion of the dichloro derivative 82 into the corresponding difluoro analog 83 was accomplished with the reaction of 82 with KF in the presence of Bu4NBr in DMSO.[ 46 , 47 , 48 ] The C3‐fluorine (84) is perhaps more responsive and could potentially be substituted by a hydroxyl group to produce the end product 79.[ 46 , 47 , 48 ]
Scheme 20.
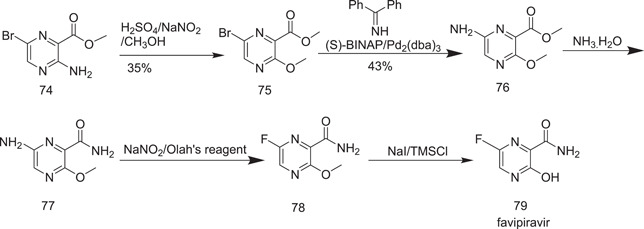
Synthesis of favipiravir
Scheme 21.
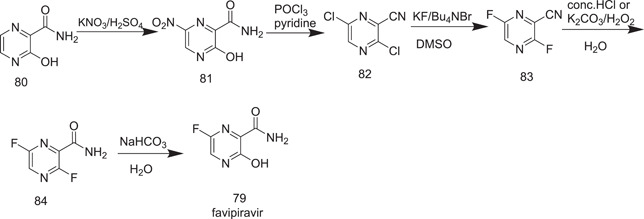
Synthesis of favipiravir
Although Scheme 21 looks inviting in terms of step count, it is not financially viable in preparing compound 3,6‐dichloropyrazine‐2‐carbonitrile (82) because of the low yield and the high cost involved in the process.
Scheme 22 was recently reported with the use of low‐cost 3‐aminopyrazine‐2‐carboxylic acid (85) as a starting material, for which 5 steps were required to obtain compound 82.
Scheme 22.
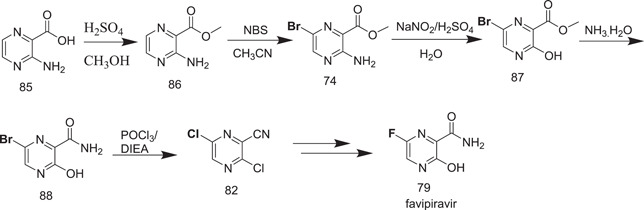
Synthesis of favipiravir
Scheme 23 shown below for the synthesis of favipiravir was described by Toyama in 2001. In this synthesis, 3‐hydroxypyrazine‐2‐carboxamide (80) was nitrated with sodium nitrate and sulphuric acid to give 3‐hydroxy‐6‐nitropyrazine‐2‐carboxamide (81). Nitropyrazine, on treatment with phosphorus oxychloride in pyridine, formed 82, whose reaction with KF and n‐Bu4NBr in DMSO at 90–100°C produced the corresponding difluoro nitrile derivative 83. Selective hydrolysis of the 3‐position fluorine atom occurred on treatment of 83 with potassium acetate in aqueous DMF to give 89; its reaction with H2O2 yielded the final product favipiravir (79).
Scheme 23.
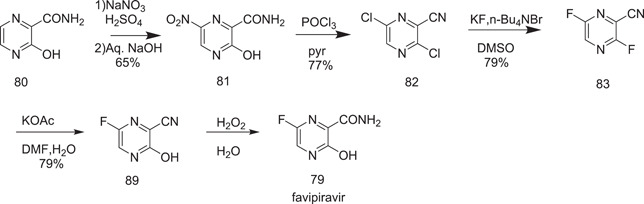
Synthesis of favipiravir
Zhang et al.[ 49‐(a) ] explored successfully the feasibility of the synthesis of favipiravir in two steps from 3‐hydroxy‐6‐nitropyrazine‐2‐carboxamide (81). The first step involved its reduction with Pd/C in acetic acid to give 6‐amino‐3‐hydroxypyrazine‐2‐carboxamide (90). Its treatment with 70% pyridinium hydrofluoride and sodium nitrite at −20°C furnished the favipiravir (79) in 70% yield (Scheme 24).
Scheme 24.
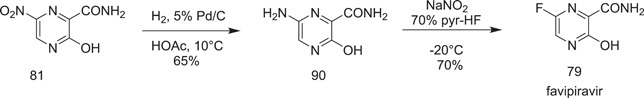
Synthesis of favipiravir
Zhang et al.[ 49‐(b) ] also reported a new method for the preparation of favipiravir (79) from 3‐hydroxypyrazine‐2‐carboxylic acid (91), which was esterified and amidated to give 3‐hydroxypyrazine‐2‐carboxamide (80). Its 6‐nitro derivative 81 was obtained on its nitration with potassium nitrate + H2SO4. A 6‐aminopyrazine 90 core was generated from 81 on its reduction in the presence of a Raney nickel catalyst. The final product favipiravir (79) was obtained on treatment of 6‐amino‐3‐hydroxypyrazine‐2‐carboxamide (90) with Olah's reagent (Scheme 25).
Scheme 25.
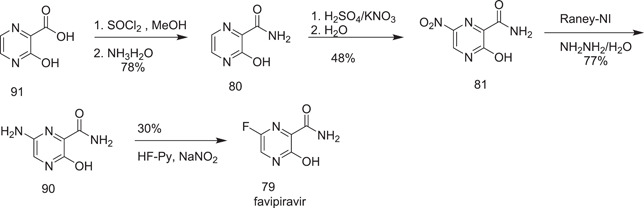
Synthesis of favipiravir
Liu et al.[ 48 ] developed a one‐pot synthesis of favipiravir (79). They avoided the isolation of volatile 3,6‐difluoro‐2‐pyrazinecarbonitrile. To obtain 83 and 84 in a one‐pot form, 3,6‐dichloropyrazine‐2‐carbonitrile (82) was treated in succession with KF and n‐Bu4NBr in DMSO at 50°C for about 3 h and with K2CO3 in DMSO, followed by aqueous hydrogen peroxide and potassium carbonate, which yielded 79 (Scheme 26).
Scheme 26.
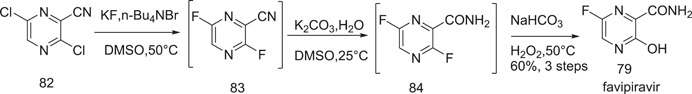
One‐pot synthesis of favipiravir
A strategy was advanced by Nippon Soda Co. Ltd.,[ 50 ] which involved the synthesis of favipiravir by treating 2‐aminopropanediamide (92) with glyoxal in alkaline medium to give sodium salt of 3‐hydroxypyrazine‐2‐carboxamide (93). This, on bromination, produced 6‐bromo‐3‐hydroxypyrazine‐2‐carboxamide (88). Favipiravir was obtained from this after chlorination, followed by fluorination and hydrolysis in the presence of hydrogen peroxide (Scheme 27).
Scheme 27.
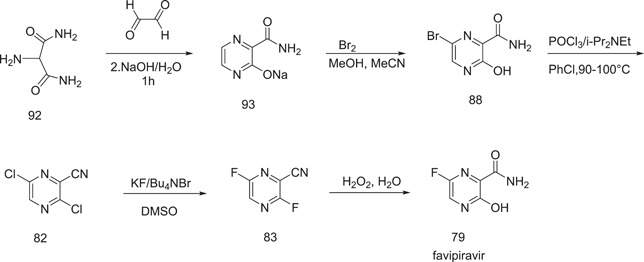
Synthesis of favipiravir according to the Nippon Soda and Toyama strategy, 2011
3.7. Pirfenidon
Pirfenidone is used to treat idiopathic pulmonary fibrosis. It works by reducing lung fibrosis by inhibiting the generation of growth factors and procollagens I and II. Following clinical trials, it was first approved in Japan for the treatment of patients with idiopathic pulmonary fibrosis. It exists in a crystalline form, designated as form A. The drug substance is filled to less than µm.[ 51 ]
3.7.1. Synthesis of pirfenidon
The synthesis begins with easily accessible 2‐amino‐5‐methylpyridine (94), which is diazotized in aqueous sulphuric acid with NaNO2 to produce 2‐hydroxy‐5‐methylpyridine (95). The scheme is based on Adams and Schrecker's initial work in 1949 on the synthesis of 2‐hydroxy‐6‐methylpyridine.[ 52 ] Conversion of 95 into pirfenidone (96) was performed by Affiliated Medical Research Inc. in 1974,[ 53 ] wherein 5‐methylpyridin‐2‐ol was reacted with potassium carbonate and zinc‐precipitated copper sulfate in refluxing iodobenzene for 18 h at 188°C (Scheme 28).
Scheme 28.
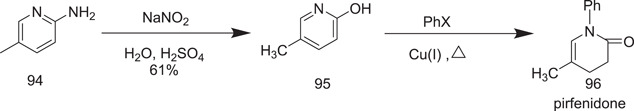
Synthesis of pirfenidon
3.8. Baricitinib
Baricitinib, with the brand name Oluminant, is used to treat RA. Lilly, in 2020, reported in their study that baricitinib can be used to treat persons with COVID‐19 due to its anti‐inflammatory property.[ 54 ] The COVID‐19 patients who were treated with placebo with remdesivir, baricitinib in combination with remdesivir minimized their recovery time of treatment.[ 55 ]
3.8.1. Synthesis of baricitinib
The first synthetic path to baricitinib was discovered by Rodgers et al.[ 56 ] They used tert‐butyl 3‐oxoazetidinel‐carboxylat as the starting material, from which neopentyl 3‐(cyanomethylene)azetidine‐1‐carboxylate (98) was obtained via the Horner–Emmons reaction, followed by deprotection under acidic conditions to give 2‐(azetidin‐3‐ylidene)acetonitrile (99). It underwent sulphonamidation with ethanesulphonyl chloride to furnish the intermediate 2‐[1‐(ethylsulphonyl)azetidin‐3‐ylidene]acetonitrile (100) (Scheme 29). The synthesis was performed further for the preparation of baricitinib from 4‐chloro‐7H‐pyrrolo[2,3‐d]pyrimidine (101), which was reacted with [2‐(chloromethoxy)ethyl]trimethylsilane (SEM‐Cl) (102) to give 4‐chloro‐7‐((2‐(trimethylsilyl)ethoxy)methyl)‐7H‐pyrrolo[2,3‐d]pyrimidine (103). It was further converted into 4‐(1H‐pyrazol‐4‐yl)‐7‐{[2‐(trimethylsilyl)ethoxy]methyl}‐7H‐pyrrolo[2,3‐d]pyrimidine (105) by reaction with 1‐(1‐ethoxyethyl)‐4‐(4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolan‐2‐yl)‐1H‐pyrazole (104). Compound 105 underwent dealkylation with aq‐HCl to give 106. Coupling of 106 with 100 (Schemes 29 and 30) in the presence of 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) produced 107. The deprotection of its SEM group under the condition furnished baricitinib.
Scheme 29.
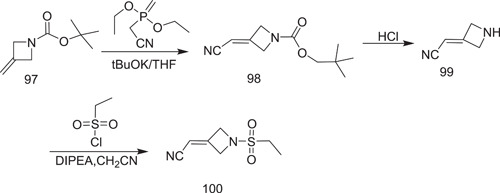
Synthesis of 2‐[1‐(ethylsulphonyl)azetidin‐3‐ylidene]acetonitrile intermediate
Scheme 30.
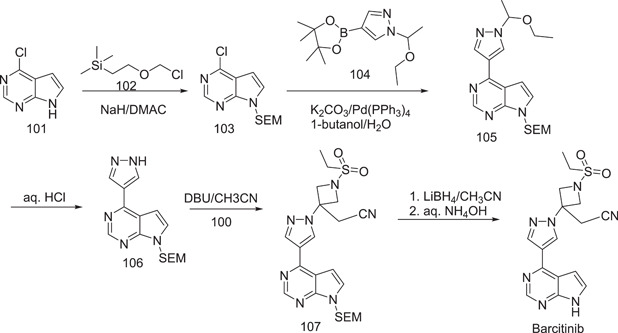
Synthesis of baricitinib
Later on, an improved procedure to synthesize baricitinib was developed by them by using tert‐butyl 3‐oxoazetidine‐1‐carboxylate (97). In this scheme (Scheme 31), 3‐ethylidine‐1‐(ethylsulphonyl)azetidine (100) was further subjected to the Horner–Emmons reaction, and instead of using t‐BuOK as a base, NaH was used.
Scheme 31.
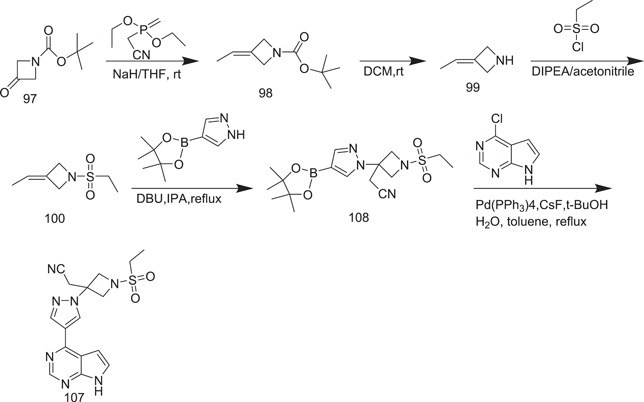
Synthesis of baricitinib
Deprotection of the N‐Boc group was carried out under TFA cleavage conditions, yielding compound 99, which was then reacted with ethanesulphonyl chloride without further purification. In the presence of DBU, the nucleophilic addition between compound 100 and 4‐(4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolan‐2‐yl)‐lH‐pyrazole was successfully performed. Finally, baricitinib was successfully obtained with a high yield of 49%.
3.9. Molnupiravir
Molnupiravir is an antiviral drug invented at Emorty University by its drug innovation company (Drug Innovation Ventures at Emory) for the treatment of influenza.[ 57 , 58 ] Molnupiravir, also referred to as EIDD‐2801, MK‐4482, is under trial for the treatment of COVID‐19.[ 59 ] It is an orally accessible, intravenous nucleotide product used against COVID‐19 that is approved by the FDA.[ 60 , 61 , 62 , 63 ] There are chances that it may amplify a synergic anti‐SARS‐CoV‐19 effect along with remdesivir.
3.9.1. Synthesis of molnupiravir
The scheme for the synthesis of molnupiravir is given below. It consists of five steps: (1) acetonide protection of 2′,3′ hydroxyl groups, (2) esterification, (3) triazole coupling, (4) hydroxyamination, and (5) deprotection.[ 64 ] Uridine (109), being the starting material, reacted with acetone to provide the protection of 2′,3′ hydroxyl groups to give 1‐((3aS,4R,6S,6aR)‐6‐hydroxy‐2,2‐dimethyltetrahydro‐4H‐cyclopenta[d][1,3]dioxol‐4‐yl)pyrimidine‐2,4(1H,3H)‐dione (110), which, on esterification with isobutyric anhydride, triethylamine and 4‐dimethylaminopyridine, produced ((3aR,4R,6R,6aS)‐6‐(2,4‐dioxo‐3,4‐dihydropyrimidin‐1(2H)‐yl)‐2,2‐dimethyltetrahydro‐4H‐ cyclopenta[d][1,3]dioxol‐4‐yl)methyl isobutyrate (111). Its coupling with triazole in the presence of phosphorus oxychloride yielded ((3aR,4R,6R,6aS)‐2,2‐dimethyl‐6‐(2‐oxo‐4‐(1H‐1,2,4‐triazol‐1‐yl)pyrimidin‐1(2H)‐yl)tetrahydro‐4H‐cyclopenta[d][1,3]dioxol‐4‐yl)methyl isobutyrate (112), which underwent hydroxyamination to give 113, whose HCOOH deprotection finally yielded molnupiravir (in low yield) (Scheme 32).
Scheme 32.
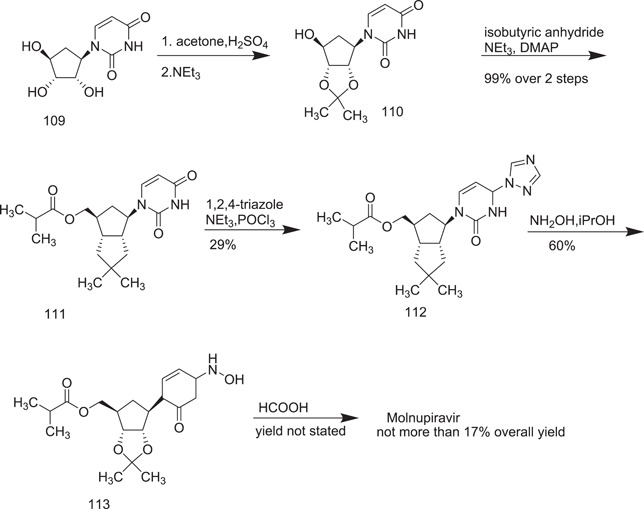
Synthesis of molnupiravir from uridine (initial route)
By using column chromatography, Vasudevan et al.[ 65 ] published a two‐step easy process of molnupiravir formation, with a total yield of 75%. Enzyme‐mediated selective esterification yielded 115, followed by direct hydroxyamination, but the high cost of the procedure restricted its industrial application. They also presented an enzyme‐free cytidine synthetic technique that was similar to the previously reported route, besides the hydroxyamination step. This synthesis yielded a higher overall yield (44%) than the previous one (Scheme 33).
Scheme 33.
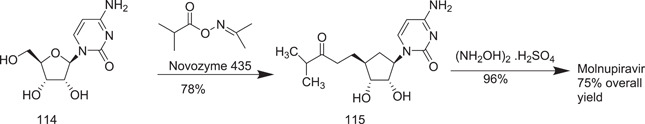
Enzyme‐mediated route to the synthesis of molnupiravir from cytidine
Vasudevan et al.[ 66 ] presented a more appropriate technique for the synthesis of molupiravir that started with either cytidine (114) or 5‐O‐butyryl derivative (118).
Steiner et al.[ 67 ] preferred uridine (109) as the starting material and changed the procedure for introducing the N‐hydroxy unit into the nucleobase by activation with triazolyl derivative (112).
Merck currently stated an eloquent multi‐enzyme method which starts with d‐ribose (118), in which after lipase‐mediated 5‐O acylation yielded ((2R,2S,4)‐3,4,5‐trihydroxytetrahydrofuran‐2‐yl)methyl isobutyrate, the uracil base was installed using a cocktail of four different enzymes to yield 1‐((2R,3R,4S,5R)‐3,4‐dihydroxy‐5‐(4‐methyl‐3‐oxopentyl)tetrahydrofuran‐2‐yl)pyrimidine‐2,4(1H,3H)‐dione. Further to this, they used a chemical process for conversion to molnupiravir, which involved in situ silylation of the product to promote product isolation and recovery.[ 68 ]
The zinc‐containing enzyme, cytidine deaminase (EC 3.5.4.5), catalyzes the conversion of cytidine into uridine.[ 69 ] As cytidine deaminase had previously been used on a large scale for synthesizing nonnatural nucleosides, it emerged as a promising candidate for industrial biocatalysts.[ 70 ] The proposed conversion of cytidine into N‐hydroxy‐cytidine catalyzed by cytidine deaminase involves the use of hydroxylamine as the nucleophile instead of water.
Wild‐type cytidine deaminase could hydrolyse N‐hydroxy‐cytidine to uridine, but only at a very low rate (about 2% activity compared to cytidine). On this basis, trials to convert cytidine into N‐hydroxy‐cytidine catalyzed by cytidine deaminase were conducted (Scheme 34).
Scheme 34.
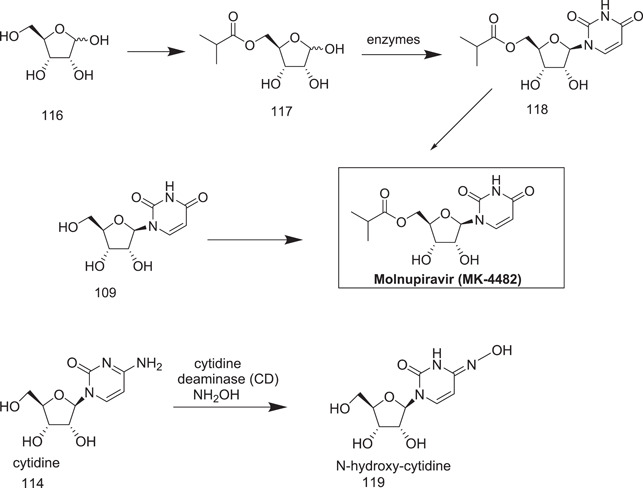
Another synthetic approach to manufacture molnupiravir
3.10. Ribavirin
Ribavirin (1‐β‐D~ribofuranosyl‐l,2,4‐triaz‐ol‐3‐carboxamide) is a highly water‐soluble nucleoside, initially synthesized in 1972.[ 71 ] The drug exerts antiviral actions after intracellular phosphorylation to mono, di, and especially, triphosphate nucleotide structures.[ 72 ] Because of its activity against both RNA and DNA viruses, it is extensively used for the treatment of influenzas, hepatitis B and C virus, SARS,[ 73 ] and syncytial virus.[ 74 ]
There are two methods for the preparation of ribavirin: a chemical method[ 75 , 76 , 77 ] and an enzymatic synthesis.[ 78 , 79 ]
3.10.1. Synthesis of ribavirin
A three‐step production procedure is standard for a chemical protocol. The condensation of 1,2,3,5‐tetra‐O‐acetyl‐β‐d‐ribofuranose (121), produced by the p‐toluenesulphonic acid catalyzed acetylation of inosine (120), with methyl 1,2,4‐triazole‐3‐carboxylate (122) yielded methyl 1‐(2,3,5‐tri‐O‐acetyl‐β‐d‐ribofuranosyl)‐1H‐1,2,4‐triazole‐3‐carboxylate, which results in ribavirin (123) via ammonolysis and deacetylation (Scheme 35).
Scheme 35.
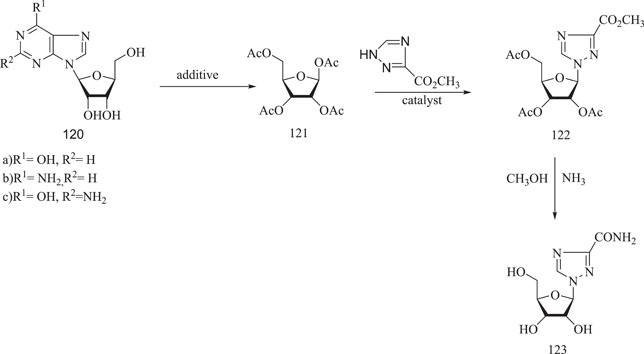
Synthesis of ribavirin
3.11. Gemcitabine
Gemcitabine is a fluorinated nucleoside analog.[ 80 ] Originally developed by Lilly, it is an anticancer drug marketed as the HCl salt under the trade name of Gemzar (Lilly). Gemcitabine is a prodrug; it undergoes intracellular phosphorylation to its active diphosphate and triphosphate forms, which inhibit DNA synthesis leading to apoptosis.[ 81 , 82 ] The drug is mainly metabolized by cytidine deaminases and almost all of it is excreted into urine as the corresponding difluorouridine species. New approaches to increase its chemotherapeutic efficiency are under investigation.[ 83 ]
3.11.1. Synthesis of gemcitabine
The first synthesis of gemcitabine was performed in the Lilly research laboratories and was published by Hertel et al.[ 84 ] in 1988 (Scheme 36).
Scheme 36.
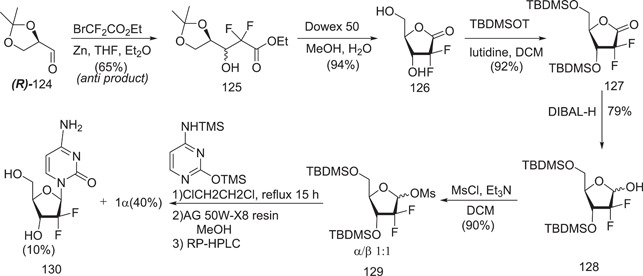
Hertel et al.[ 84 ] synthesis of gemcitabine
The synthesis begins with enantiopure d‐glyceraldehyde (R)‐124, which is easily synthesized in two stages from d‐mannitol.[ 85 ] Fluorine was introduced using a building‐block method involving ethyl bromodifluoroacetate. Under typical circumstances, the Reformatsky reaction produced a 3:1 anti/syn diastereomeric mixture, with the Felkin–Anh product as the predominant diastereomer. Following this, high‐performance liquid chromatography (SiO2) was used to separate the diastereomers syn‐ and anti‐3, yielding the required anti‐3 in a 65% yield. Following deprotection with Dowex 50, cyclization occurred simultaneously, yielding the lactone 126. The remaining free alcohol groups were subsequently protected as TBDMS ethers, and the crucial difluororibose intermediate 128 was obtained in 68% yield from anti‐3 by DIBAL‐H‐mediated reduction.
Because fluorination at the ribose‐2 position causes deactivation towards nucleobase introduction, a better anomeric leaving group was needed, such as the equivalent mesylate 129, which was produced as a 1:1 anomeric combination from lactol.
3.12. Fluvoxamine
Fluvoxamine is one of several selective serotonin reuptake inhibitors used to treat depression and anxiety disorders. The effectiveness of fluvoxamine as an antidepressant is well documented.[ 86 ] Fluvoxamine has demonstrated short‐term efficacy in the treatment of obsessive–compulsive disorders (OCD), panic disorder, social phobia, posttraumatic stress disorder, and a range of obsessive‐compulsive spectrum disorders. The drug is as effective as clomipramine in patients with OCD but appears to have a better tolerability profile.
3.12.1. Synthesis of fluvoxamine maleate
In their patent, Welle and Claassen proposed three methods for the manufacture of fluvoxamine maleate. The present production procedure is as shown in Scheme 37. Using pyridine as an acid scavenger, a combination of 5‐methoxy‐4′‐trifluoromethyl valerophenone and 2‐minooxyethylamine dihydrochloride is refluxed in 100% ethanol. The maleate salt is prepared by adding an equimolar amount of maleic acid to a fluvoxamine solution in 100% ethanol, which is then heated until it forms a clear solution. The ethanol is removed, and the acetonitrile residue is recrystallized. Scheme 38 shows the synthesis of the precursor to (O‐methyl‐~C)fluvoxamine.[ 87 , 88 ]
Scheme 37.
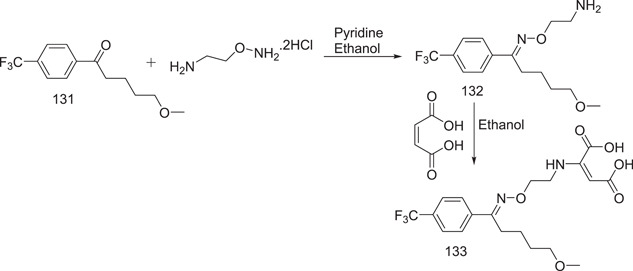
Synthesis of fluvoxamine maleate
Scheme 38.
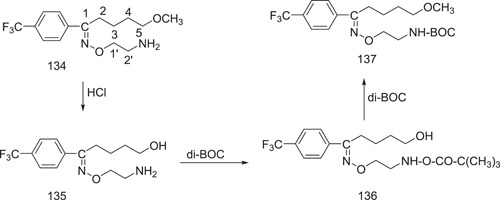
Synthesis of the precursor to (O‐methyl‐~C)fluvoxamine
3.13. Rupintrivir
Rupintrivir (AG‐7088, Rupinavir) is a peptidomimetic antiviral drug that acts as a 3C and 3CL protease inhibitor.[ 89 , 90 , 91 ] It was developed for the treatment of rhinoviruses,[ 92 , 93 ] and has subsequently been investigated for the treatment of other viral diseases including those caused by picornaviruses,[ 94 , 95 ] norovirus,[ 96 ] and coronaviruses, such as SARS and COVID‐19.[ 97 , 98 ]
3.13.1. Synthesis of rupintivir
The dianionic alkylation of N‐Boc glutamic acid dimethyl ester 137 with iodoacetonitrile was the first step in this approach. As expected, this alkylation was extremely stereoselective, yielding 138 as the end product (72%). Following this, the cyano group in 138 was hydrogenated to produce the intermediate 139. The lactam 140 was then obtained in 40% yield by in situ cyclization of intermediate 139 (from 137). The ester group in 140 was then converted into the corresponding alcohol. The conjugated ester 143 was obtained in 65% yield by oxidizing the alcohol product 141 with Dess–Martin periodinane and then performing the Wittig reaction with a suitable reagent (from 140). The protective group of 143 was removed to produce the desired lactam derivative 144 (Fragment A). Bromination and methyl esterification were used to prepare compound 146 (Part B1) from D‐4‐fluorophenylalanine. Meanwhile, Claisen condensation was used to generate the valine‐derived malonate 147. Following the SN2 reaction with bromoacetate 146 and valine‐derived malonate 147, the tert‐butoxycarbonyl (Boc) group was removed to yield the ketoester 149. Catalytic hydrogenation of 149 in the presence of di‐tert‐butyl carbonate resulted in the direct conversion of the benzyloxycarbonyl (Cbz) group into the Boc group in 150, which was followed by hydrolysis to yield the crucial ketoacid 151. (Fragment B). Rupintrivir was obtained in the final step by coupling the ketoacid 151 with the lactam derivative 144 (on the right side at first) and subsequently the isoxazole acid chloride (Fragment C) on the left side (Scheme 39).
Scheme 39.
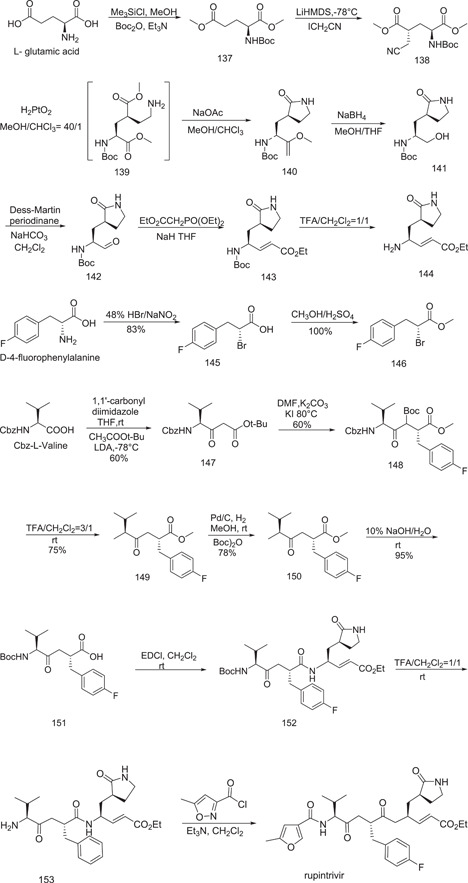
Synthesis of rupintrivir
4. CONCLUSION
The main reason for quick spreading of COVID‐19 is unavailability of proper medication. Antiviral drugs such as remdesivir or GS‐441524, as well as JAK inhibitor drugs, provide an effective treatment combination to reduce mortality before a specific medicine is available. In this review, we have updated the role of many small molecules that have been shown to be effective in inhibiting COVID‐19. This review presents a comprehensive account of CQ and HCQ favipiravir, remdesivir, and so forth, in terms of their biological activities, drug testing, and synthetic processes, for the preparation of these and many other potential drug candidates.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENT
The authors thank the Vice‐Chancellor and Department of Chemistry, Banasthali Vidyapith, Banasthali, India, for providing the necessary facilities for the compilation of this work.
Malik P., Jain S., Jain P., Kumawat J., Dwivedi J., Kishore D.. A comprehensive update on the structure and synthesis of potential drug targets for combating the coronavirus pandemic caused by SARS‐CoV‐2. Arch. Pharm. 2022;355:e2100382. 10.1002/ardp.202100382
REFERENCES
- 1. Geller C., Varbanov M., Duval R. E., Viruses 2012, 4, 3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorbalenya A. E., Baker S. C., Baric R. S., de Groot R. J., Drosten C., Gulyaeva A. A., Haagmans B. L., Lauber C., Leontovich A. M., Neuman B. W., Penzar D., Nat. Microbiol. 2020, 5, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan W., Zhao X., Ma X., Wang W., Niu P., Xu W., Gao G. F., Wu G., China CDC Wkly 2020, 2(4), 61. [PMC free article] [PubMed] [Google Scholar]
- 4. Plante J. A., Liu Y., Liu J., Xia H., Johnson B. A., Lokugamage K. G., Zhang X., Muruato A. E., Zou A. J., Fontes‐Garfias C. R., Mirchandani D., Scharton D., Bilello J. P., Ku Z., An Z., Kalveram B., Freiberg A. N., Menachery V. D., Xie X., Plante K. S., Weaver S. C., Shi P. Y., Nature 2021, 592, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spratt A. N., Kannan S. R., Woods L. T., Weisman G. A., Quinn T. P., Lorson C. L., Sönnerborg A., Byrareddy S. N., Singh K., Comput. Struct. Biotechnol. J. 2021, 19, 3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krafts K., Hempelmann E., Skórska‐Stania A., Parasitol. Res. 2012, 111, 1. [DOI] [PubMed] [Google Scholar]
- 7. Kannan S. P., Ali P. S., Sheeza A., Hemalatha K., Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2006. [DOI] [PubMed] [Google Scholar]
- 8. Singh A. K., Singh A., Shaikh A., Singh R., Misra A., Diabetes Metab. Syndr. 2020, 14, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Kenyon R. L., Wiesner J. A., Kwartler C. E., Ind. Eng. Chem. 1949, 41, 654 [Google Scholar]; (b) De D., Krogstad F. M., Byers L. D., Krogstad D. J. J., Med. Chem. 1998, 41, 4918 [DOI] [PubMed] [Google Scholar]; (c) Su T., Zhu J., Sun R., Zhang H., Huang Q., Zhang X., Du R., Qiu L., Cao R., Eur. J. Med. Chem. 2019, 178, 154 [DOI] [PubMed] [Google Scholar]; (d) Li S., Hu L., Li J., Zhu J., Zeng F., Huang Q., Qui L., Du R., Cao R., Eur. J. Med. Chem. 2019, 15(162), 666. [DOI] [PubMed] [Google Scholar]
- 10. Price C. C., Roberts R. M., J. Am. Chem. Soc. 1946, 68, 1204. [DOI] [PubMed] [Google Scholar]
- 11. Madrid P. B., Sherrill J., Liou A. P., Weisman J. L., DeRisi J. L., Guy R. K., Bioorg. Med. Chem. Lett. 2005, 15, 1015. [DOI] [PubMed] [Google Scholar]
- 12. Gaber A. E. A. M., McNab H., Synthesis 2001, 14, 2059. [Google Scholar]
- 13. Surrey A. R., Hammer H. F., J. Am. Chem. Soc. 1946, 68, 113. [DOI] [PubMed] [Google Scholar]
- 14. Savarino A., Boelaert J. R., Cassone A., Majori G., Cauda R., Lancet Infect. Dis. 2003, 3, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Kalil A. C., JAMA 2020, 323, 1897 [DOI] [PubMed] [Google Scholar]; (b) Yu E., Mangunuru H. P., Telang N. S., Kong C. J., Verghese J., Gilliland S. E. III, Ahmad S., Dominey R. N., Gupton B. F., Beilstein J. Org. Chem. 2018, 14, 583 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takla M., Jeevaratnam K., Saudi Pharm. J. 2020, 28, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Surrey A. R., United States patent US 2,546,658, 1951. ; (b) Surrey A. R., Hammer H. F., J. Am. Chem. Soc. 1950, 72, 1814. [Google Scholar]
- 17. Yu E., Mangunuru H. P., Telang N. S., Kong C. J., Verghese J., Gilliland III, S. E. , Ahmad S., Dominey R. N., Gupton B. F., Beilstein J. Org. Chem. 2018, 14, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar A., Vyas K. D., Singh D., Nanolavadekar S., Bhiae S., Jadhav A., US patent WO 2005062723, 2005.
- 19. Wu C. Y., Jan J. T., Ma S. H., Kuo C. J., Juan H. F., Cheng Y. S., Hsu H. H., Huang H. C., Wu D., Brik A., Liang F. S., Proc. Natl. Acad. Sci. 2004, 101, 10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yousefifard M., Zali A., Ali K. M., Neishaboori A. M., Zarghi A., Hosseini M., Safari S., Arch. Acad. Emerg. Med. 2020, 8, e45. [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Vardanyan R., Hruby V., Synthesis of Best‐Seller Drugs, Elsevier, New York, NY: 2016, p. 687 [Google Scholar]; (b) Ghosh A. K., Bilcer G., Schiltz G., Synthesis 2001, 2001, 2203 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Izawa K., Onishi T., Chem. Rev. 2006, 106, 2811. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., Fu S., Gao L., Cheng Z., Lu Q., Hu Y., Lancet 2020, 395, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Stoner E. J, Cooper A. J, Dickman D. A, Kolaczkowski L., Lallaman J. E., Liu J. H., Oliver‐Shaffer P. A., Patel K. M., Paterson J. B., Plata D. J., Riley D. A., Org. Process Res. Dev. 2000, 4, 264 [Google Scholar]; (b) Ramu E., Rao B. V., Tetrahedron: Asymmetry 2009, 20, 2201 [Google Scholar]; (c) Stoner E. J., Stengel P. J., Cooper A. J., Org. Process Res. Dev. 1999, 3, 145 [Google Scholar]; (d) Yao T. T., Qian J. D., Zhu WY., Wang Y., Wang G. Q., J. Med. Virol. 2020, 92, 556 [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chan K. S., Lai S. T., Chu C. M., Tsui E., Tam C. Y., Wong M. M. L., Tse M. W., Que T. L., Peiris J. S. M., Sung J., Wong V C W, Yuen K. Y., Hong Kong Med. J. 2003, 9, 399 [PubMed] [Google Scholar]; (f) Ghosh A. K., Bilcer G., Schiltz G., Synthesis 2001, 2001, 2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ohkoshi M., Fujii S., J. Natl. Cancer Inst. 1983, 71, 1053. [PubMed] [Google Scholar]
- 25. Ikeda S., Manabe M., Muramatsu T., Takamori K., Ogawa H., J. Am. Acad. Dermatol. 1988, 18, 1246. [DOI] [PubMed] [Google Scholar]
- 26.(a) Göke B., Stöckmann F., Müller R., Lankisch P. G., Creutzfeld W., Digestion 1984, 30, 171 [DOI] [PubMed] [Google Scholar]; (b) Ikeda S., Manabe M., Muramatsu T., Takarnori K., Ogawa H., J. Am. Acad. Dermatol. 1988, 18, 1246. [DOI] [PubMed] [Google Scholar]
- 27. Sai J. K., Suyama M., Kubokawa Y., Matsumura Y., Inami K., Watanabe S., J. Gastroenterol. 2010, 45, 335. [DOI] [PubMed] [Google Scholar]
- 28. Al‐Tawfiq J. A., Al‐Homoud A. H., Memish Z. A., Travel. Med. Infect. Dis. 2020, 34, 101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell Res. 2020, 30, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Grattendick K. J., Nakashima J. M., Feng L., Giri S. N., Margolin S. B., Int. Immunopharmacol. 2008, 8, 679 [DOI] [PubMed] [Google Scholar]; (b) Xue F., Zhou X., Zhou R., Zhou X., Xiao D., Gu E., Guo X., Xiang J., Wang K., Yang L., Zhong W., Org. Process Res. Dev. 2020, 24, 1772. [DOI] [PubMed] [Google Scholar]
- 31. Warren T. K., Jordan R., Lo M. K., Ray A. S., Mackman R. L., Soloveva V., Siegel D., Perron M., Bannister R., Hui H. C., Larson N., Strickley R., Wells J., Stuthman K. S., van Tongeren S. A., Garza N. L., Donnelly G., Shurtleff A. C., Retterer C. J., Gharaibeh D., Zamani R., Kenny T., Eaton B. P., Grimes E., Welch L. S., Gomba L., Wilhelmsen C. L., Nichols D. K., Nuss J. E., Nagle E. R., Kugelman J. R., Palacios G., Doerffler E., Neville S., Carra E., Clarke M. O., Zhang L., Lew W., Ross B., Wang Q., Chun K., Wolfe L., Babusis D., Park Y., Stray K. M., Trancheva I., Feng J. Y., Barauskas O., Xu Y., Wong P., Braun M. R., Flint M., McMullan L. K., Chen S. S., Fearns R., Swaminathan S., Mayers D. L., Spiropoulou C. F., Lee W. A., Nichol S. T., Cihlar T., Bavari S., Nature 2016, 531, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siegel D., Hui H. C., Doerffler E., Clarke M. O., Chun K., Zhang L., Neville S., Carra E., Lew W., Ross B., Wang Q., Wolfe L., Jordan R., Soloveva V., Knox J., Perry J., Perron M., Stray K. M., Barauskas O., Feng J. Y., Xu Y., Lee G., Rheingold A. L., Ray A. S., Bannister R., Strickley R., Swaminathan S., Lee W. A., Bavari S., Cihlar T., Lo M. K., Warren T. K., Mackman R. L., Med. Chem. 2017, 60, 1648. [DOI] [PubMed] [Google Scholar]
- 33. Barker R., Fletcher Jr., H. G. , J. Org. Chem. 1961, 26, 4605. [Google Scholar]
- 34. Furuta Y., Gowen B. B., Takahashi K., Shiraki K., Smee D. F., Barnard D. L., Antivir. Res. 2013, 100, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Furuta Y., Takahashi K., Shiraki K., Sakamoto K., Smee D. F., Barnard D. L., Gowen B. B., Julander J. G., Morrey J. D., Antivir. Res. 2009, 82, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jin Z., Tucker K., Lin X., Kao C. C., Shaw K., Tan H., Symons J., Behera I., Rajwanshi V. K., Dyatkina N., Wang G., Beigelman L., Deval J., Antimicrob. Agents Chemother. 2015, 59, 7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smither S. J., Eastaugh L. S., Steward J. A., Nelson M., Lenk R. P., Lever M. S., Antivir. Res. 2014, 104, 153. [DOI] [PubMed] [Google Scholar]
- 38. Gao J., Luo X., Li Y., Gao R., Chen H., Ji D., Chem‐Bio. Drug Des. 2018, 853, 245. [DOI] [PubMed] [Google Scholar]
- 39. Huchting J., Vanderlinden E., Winkler M., Nasser H., Naesens L., Meier C., J. Med. Chem. 2018, 61, 6193. [DOI] [PubMed] [Google Scholar]
- 40. Huchting J., Winkler M., Nasser H., Meier C., ChemMedChem 2017, 129, 652. [DOI] [PubMed] [Google Scholar]
- 41. Klejch T., Pohl R., Janeba Z., Sun M., Keough D., Guddat L., Hockova D., Tetrahedron 2018, 74, 5886. [Google Scholar]
- 42. Plebanek E., Lescrinier E., Andrei G., Snoeck R., Herdewijn P., Jonghe S. D., Eur. J. Med. Chem. 2017, 144, 93. [DOI] [PubMed] [Google Scholar]
- 43. Wang G., Wan J., Hu Y., Wu X., Prhavc M., Dyatkina N., Rajwanshi V. K., Smith D. B., Jekle A., Kinkade A., Symons J. A., J. Med. Chem. 2016, 59, 4611. [DOI] [PubMed] [Google Scholar]
- 44. El‐Nahas A., Hirao K. A., J. Mol. Struct. 1999, 4591, 229. [Google Scholar]
- 45. Furuta Y., Egawa H., European Patent Office WO 00/10569, 2000.
- 46. Beldar S. V., Jordis U., Chemistry, Vienna University of Technology, Vienna, Austria: 2009, p. 13. [Google Scholar]
- 47.(a) Hara T., Norimatsu N., Kurushima H., Kano T., United States patent US 8,835,636, 2014. ; (b) Guo Q., Xu M., Guo S., Zhu F., Xie Y., Shen J., Chem. Pap. 2019, 73, 1043 [Google Scholar]; (c) Al Bujuq N., Synthesis 2020, 52, 3735. [Google Scholar]
- 48. Liu F.‐L., Li C.‐Q., Xiang H.‐Y., Feng S., Chem. Pap. 2017, 111, 2153. [Google Scholar]
- 49.(a) Zhang R., Ding J., Yu Q., Faming Zhuanli Shenquing, CN 2012, 102675230 [Google Scholar]; (b) Shi F., Li Z., Kong L., Xie Y., Zhang T. Xu W., Drug Discov. Ther. 2014, 8, 117. [DOI] [PubMed] [Google Scholar]
- 50. Hara T., Norimatsu N., Kurushima H., Kano T., United States Patent US 8,835,636, 2014.
- 51. Pyles S. M., Cyr M., Radhakrishnan R., United States Patent US 02011003863, 2011.
- 52. Adams R., Schrecker A. W., J. Am. Chem. Soc. 1949, 71, 1186. [Google Scholar]
- 53. Gadekar S., United States Patent US 3,839,346, 1974.
- 54. Stebbing J., Nievas G. S., Falcone M., Youhanna S., Richardson P., Ottaviani S., Shen J. X., Sommerauer C., Tiseo G., Ghiadoni L., Virdis A., Sci. Adv. 2021, 7, eabe4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coronavirus (COVID‐19) Update: FDA Authorizes Drug Combination For Treatment of COVID‐19, U.S. FDA 2020.
- 56. Rodgers J. D., Shepard S., Maduskuie T. P., Wang H., Falahatpisheh N., Rafalski M., Arvanitis A. G., Sorace L., Jalluri R. K., Fridman J. S., Vaddi K., United States Patent US20070135461, 2007.
- 57. Toots M., Yoon J. J., Cox R. M., Hart M., Sticher Z. M., Makhsous N., Plesker R., Barrena A. H., Reddy P. G., Mitchell D. G., Shean R. C., Sci. Transl. Med. 2019, 11, eaax5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Toots M., Yoon J. J., Hart M., Natchus M. G., Painter G. R., Plemper R. K., Transl. Res. 2020, 218, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cox R. M., Wolf J. D., Plemper R. K., Nat. Microbiol. 2021, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Malin J. J., Suárez I., Priesner V., Fätkenheuer G., Rybniker J., Clin. Microbiol. Rev. 2020, 34, e00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pruijssers A. J., Denison M. R., Curr. Opin. Virol. 2019, 35, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jena N. R., Phys. Chem. Chem. Phys. 2020, 22, 28115. [DOI] [PubMed] [Google Scholar]
- 63. Yin W., Mao C., Luan X., Shen D. D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M., Chang S., Xie Y. C., Tian G., Jiang H. W., Tao S. C., Shen J., Jiang Y., Jiang H., Xu Y., Zhang S., Zhang Y., Xu H. E., Science 2020, 368, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Painter G. R., Perryman D., Bluemling G. R., World Patent WO1736022019, 2019.
- 65. Vasudevan N., Ahlqvist G. P., Mc Geough C. P., Paymode D. J., Cardoso F. S., Lucas T., Dietz J. P., Opatz T., Jamison T. F., Gupton F. B., Snead D. R., Chem. Commun. 2020, 56, 13363. [DOI] [PubMed] [Google Scholar]
- 66. Vasudevan N., Ahlqvist G. P., Mc Geough C. P., Paymode D. J., Cardoso F. S., Lucas T., Dietz J. P., Opatz T., Jamison T. F., Gupton F. B., Snead D. R., Chem. Commun. 2020, 56, 13363. [DOI] [PubMed] [Google Scholar]
- 67. Steiner A., Znidar D., Ötvös S. B., Snead D. R., Dallinger D., Kappe C. O., Eur. J. Org. Chem. 2020, 2020, 6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Benkovics T., McIntosh J., Silverman S., Kong J., Maligres P., Itoh T., Yang H., Huffman M., Verma D., Pan W., Ho H. I., ChemRxiv. 2020. 10.26434/chemrxiv.13472373.v1. [DOI] [Google Scholar]
- 69. Ferrero M., Gotor V., Chem. Rev. 2000, 100, 4319. [DOI] [PubMed] [Google Scholar]
- 70. Trimble R. B., Maley F., J. Bacteriol. 1971, 108, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Witkowski J. T., Robins R. K., Sidwell R. W., Simon L. N., J. Med. Chem. 1972, 15(11), 1150. [DOI] [PubMed] [Google Scholar]
- 72. Zimmerman T. P., Deeprose R. D., Biochem. Pharmacol. 1978, 27(5), 709. [DOI] [PubMed] [Google Scholar]
- 73. Birgit M., Martin M., Patrick C. B., Hans W. D., Jindrich C. J., Biochem. Biophys. Res. Commun. 2005, 326, 905.15607755 [Google Scholar]
- 74. Empey K. M., Peebles R. S., Kolls J. K., Clin. Infect. Dis. 2010, 50, 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li Y. S., Zhang J. J., Mei L. Q., Tan C. X., Org. Prep. Proced. Int. 2012, 44(4), 387. [Google Scholar]
- 76. Witkowski J. T., Robins R. K., Sidwell R. W. R. W., Simon L. N., J. Med. Chem. 1972, 15(11), 1150. [DOI] [PubMed] [Google Scholar]
- 77. Li Y. S., Zhang J. J., Mei L. Q., Tan C. X., Org. Prep. Proced. Int. 2012, 44(4), 387. [Google Scholar]
- 78. Medici R., Porro M. T., Lewkowicz E., Montserrat J., Iribarren A. M., Nucleic Acids Symp. Ser. 2009, 52(1), 541. [DOI] [PubMed] [Google Scholar]
- 79. Taverna‐Porro M., Bouvier L. A., Pereira C. A., Montserrat J. M., Iribarren A. M., Tetrahedron Lett. 2008, 49(16), 2642. [Google Scholar]
- 80.(a) Wojtowicz‐Rajchel H., J. Fluorine Chem. 2012, 143, 11 [Google Scholar]; (b) Qiu X.‐L., Xu X.‐H., Qing F.‐L., Tetrahedron 2010, 66, 789 [Google Scholar]; (c) Liu P., Sharon A., Chu C. K., J. Fluorine Chem. 2008, 129, 743 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Watts J. K., Damha M. J., Can. J. Chem. 2008, 86, 641 [Google Scholar]; (e) Meng W.‐D., Qing F.‐L., Curr. Top. Med. Chem. 2006, 6, 1499 [DOI] [PubMed] [Google Scholar]; (f) Pankiewicz K. W., Carbohydr. Res. 2000, 327, 87. [DOI] [PubMed] [Google Scholar]
- 81. Martindale. The Complete Drug Reference, 37th ed., (Ed: S. C. Sweetman), Royal Pharmaceutical Society, London, Chicago: 2011. [Google Scholar]
- 82.(a) Silvestris N., Cinieri S., La Torre I., Pezzella G., Numico G., Orlando L., Lorusso V., The Breast 2008, 17, 220 [DOI] [PubMed] [Google Scholar]; (b) Vulfovich M., Rocha‐Lima C., Expert Rev. Anticancer Ther. 2008, 8, 993 [DOI] [PubMed] [Google Scholar]; (c) Lorusso D., Di Stefano A., Fanfani F., Scambia G., Ann. Oncol. 2006, 17, V188. [DOI] [PubMed] [Google Scholar]
- 83. Maiti S., Park N., Han J. H., Jeon H. M., Lee J. H., Bhuniya S., Kang C., Kim J. S., J. Am. Chem. Soc. 2013, 135, 4567. [DOI] [PubMed] [Google Scholar]
- 84. Hertel L. W., Kroin J. S., Misner J. W., Tustin J. M., J. Org. Chem. 1988, 53(11), 2406. [Google Scholar]
- 85. Baer E., Ho F., J. Biol. Chem. 1939, 128(2), 463. [Google Scholar]
- 86. Wilde M. I., Plosker G. L., Benfield P., Drugs 1993, 46, 895. [DOI] [PubMed] [Google Scholar]
- 87. Croom K. F., Keam S. J., Drugs 2005, 65(12), 1669. [DOI] [PubMed] [Google Scholar]
- 88. Poppe S. M., Slade D. E., Chong K. T., Hinshaw R. R., Pagano P. J., Markowitz M., Ho D. D., Mo H., Gorman R. R., Dueweke T. J., Thaisrivongs S., Tarpley W. G., Antimicrob. Agents Chemother. 1997, 41(5), 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dragovich P. S., Prins T. J., Zhou R., Webber S. E., Marakovits J. T., Fuhrman S. A., Patick A. K., Matthews D. A., Lee C. A., Ford C. E., Burke B. J., Rejto P. A., Hendrickson T. F., Tuntland T., Brown E. L., Meador J. W., Ferre R. A., Harr J. E., Kosa M. B., Worland S. T., J. Med. Chem. 1999, 42(7), 1213. [DOI] [PubMed] [Google Scholar]
- 90. Santos M. M., Moreira R., Mini Rev. Med. Chem. 2007, 7(10), 1040. [DOI] [PubMed] [Google Scholar]
- 91. Yuan S., Fan K., Chen Z., Sun Y., Hou H., Zhu L., Virol. Sin. 2020, 35(4), 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Patick A. K., Binford S. L., Brothers M. A., Jackson R. L., Ford C. E., Diem M. D., Maldonado F., Dragovich P. S., Zhou R., Prins T. J., Fuhrman S. A., Antimicrob. Agents Chemother. 1999, 43(10), 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jensen L. M., Walker E. J., Jans D. A., Ghildyal R., Rhinoviruses 2015, 1221, 129. [Google Scholar]
- 94. Barnard D. L., Curr. Pharm. Des. 2006, 12(11), 1379. [DOI] [PubMed] [Google Scholar]
- 95. de Palma A. M., Vliegen I., de Clercq E., Neyts J., Med. Res. Rev. 2008, 28(6), 823. [DOI] [PubMed] [Google Scholar]
- 96. Rocha‐Pereira J., Nascimento M. S., Ma Q., Hilgenfeld R., Neyts J., Jochmans D., Antimicrob. Agents Chemother. 2014, 58(8), 4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., Hilgenfeld R., Science 2003, 300(5626), 1763. [DOI] [PubMed] [Google Scholar]
- 98. Liu C., Zhou Q., Li Y., Garner L. V., Watkins S. P., Carter L. J., Smoot J., Gregg A. C., Daniels A. D., Jervey S., Albaiu D., ACS Cent. Sci. 2020, 6(3), 315. [DOI] [PMC free article] [PubMed] [Google Scholar]