

Abstract
Introduction
The mechanisms that lead to cognitive impairment associated with COVID‐19 are not well understood.
Methods
Brain lysates from control and COVID‐19 patients were analyzed for oxidative stress and inflammatory signaling pathway markers, and measurements of Alzheimer’s disease (AD)‐linked signaling biochemistry. Post‐translational modifications of the ryanodine receptor/calcium (Ca2+) release channels (RyR) on the endoplasmic reticuli (ER), known to be linked to AD, were also measured by co‐immunoprecipitation/immunoblotting of the brain lysates.
Results
We provide evidence linking SARS‐CoV‐2 infection to activation of TGF‐β signaling and oxidative overload. The neuropathological pathways causing tau hyperphosphorylation typically associated with AD were also shown to be activated in COVID‐19 patients. RyR2 in COVID‐19 brains demonstrated a “leaky” phenotype, which can promote cognitive and behavioral defects.
Discussion
COVID‐19 neuropathology includes AD‐like features and leaky RyR2 channels could be a therapeutic target for amelioration of some cognitive defects associated with SARS‐CoV‐2 infection and long COVID.
Keywords: Alzheimer's disease, calcium, COVID‐19, ryanodine receptor, tau pathology
1. NARRATIVE
1.1. Contextual background
Patients suffering from COVID‐19 exhibit multi‐system organ failure involving not only pulmonary 1 but also cardiovascular, 2 neural, 3 and other systems. The pleiotropy and complexity of the organ system failures both complicate the care of COVID‐19 patients and contribute, to a great extent, to the morbidity and mortality of the pandemic. 4 Severe COVID‐19 most commonly manifests as viral pneumonia‐induced acute respiratory distress syndrome (ARDS). 5 Respiratory failure results from severe inflammation in the lungs, which arises when SARS‐CoV‐2 infects lung cells. Cardiac manifestations are multifactorial and include hypoxia, hypotension, enhanced inflammatory status, angiotensin‐converting enzyme 2 (ACE2) receptor downregulation, endogenous catecholamine adrenergic activation, and direct viral‐induced myocardial damage. 6 , 7 Moreover, patients with underlying cardiovascular disease or comorbidities, including congestive heart failure, hypertension, diabetes, and pulmonary diseases, are more susceptible to infection by SARS‐CoV‐2, with higher mortality. 6 , 7
In addition to respiratory and cardiac manifestations, it has been reported that approximately one‐third of patients with COVID‐19 develop neurological symptoms, including headache, disturbed consciousness, and paresthesias. 8 Brain tissue edema, stroke, neuronal degeneration, and neuronal encephalitis have also been reported. 2 , 8 , 9 , 10 In a recent study, diffuse neural inflammatory markers were found in >80% of COVID‐19 patient brains, processes which could contribute to the observed neurological symptoms. 11 Furthermore, another pair of frequent symptoms of infection by SARS‐CoV‐2 are hyposmia and hypogeusia, the loss of the ability to smell and taste, respectively. 3 Interestingly, hyposmia has been reported in early‐stage Alzheimer's disease (AD), 3 and AD type II astrocytosis has been observed in neuropathology studies of COVID‐19 patients. 10
Systemic failure in COVID‐19 patients is likely due to SARS‐CoV‐2 invasion via the ACE2 receptor, 9 which is highly expressed in pericytes of human heart 8 and epithelial cells of the respiratory tract, 12 kidney, intestine, and blood vessels. ACE2 is also expressed in the brain, especially in the respiratory center and hypothalamus in the brain stem, the thermal center, and cortex, 13 which renders these tissues more vulnerable to viral invasion, although it remains uncertain whether SARS‐CoV‐2 virus directly infects neurons in the brain. 14 The primary consequences of SARS‐CoV‐2 infection are inflammatory responses and oxidative stress in multiple organs and tissues. 15 , 16 , 17 Recently it has been shown that the high neutrophil‐to‐lymphocyte ratio observed in critically ill patients with COVID‐19 is associated with excessive levels of reactive oxygen species (ROS) and ROS‐induced tissue damage, contributing to COVID‐19 disease severity. 15
Recent studies have reported an inverse relationship between ACE2 and transforming growth factor‐β (TGF‐β). In cancer models, decreased levels of ACE2 correlated with increased levels of TGF‐β. 18 In the context of SARS‐CoV‐2 infection, downregulation of ACE2 has been observed, leading to increased fibrosis formation, as well as upregulation of TGF‐β and other inflammatory pathways. 19 Moreover, patients with severe COVID‐19 symptoms had higher blood serum TGF‐β concentrations than those with mild symptoms, 20 thus further implicating the role of TGF‐β and warranting further investigation.
Interestingly, reduced angiotensin/ACE2 activity has been associated with tau hyperphosphorylation and increased amyloid beta (Aβ) pathology in animal models of AD. 21 , 22 The link between reduced ACE2 activity and increased TGF‐β and tau signaling in the context of SARS‐CoV‐2 infection needs further exploration.
Our laboratory has shown that stress‐induced ryanodine receptor (RyR)/intracellular calcium release channel post‐translational modifications, including oxidation and protein kinase A (PKA) hyperphosphorylation related to activation of the sympathetic nervous system and the resulting hyper‐adrenergic state, deplete the channel stabilizing protein (calstabin) from the channel complex, destabilizing the closed state of the channel and causing RyR channels to leak Ca2+ out of the endoplasmic/sarcoplasmic reticulum (ER/SR) in multiple diseases. 23 , 24 , 25 , 26 , 27 , 28 , 29 Increased TGF‐β activity can lead to RyR modification and leaky channels, 30 and SR Ca2+ leak can cause mitochondrial Ca2+ overload and dysfunction. 29 Increased TGF‐β activity 31 and mitochondrial dysfunction 32 are also associated with SARS‐CoV‐2 infection.
Here we show that SARS‐CoV‐2 infection is associated with adrenergic and oxidative stress and activation of the TGF‐β signaling pathway in the brains of patients who have succumbed to COVID‐19. One consequence of this hyper‐adrenergic and oxidative state is the development of tau pathology normally associated with AD. In this article, we investigate potential biochemical pathways linked to tau hyperphosphorylation. Based on recent evidence that has linked tau pathology to Ca2+ dysregulation associated with leaky RyR channels in the brain, 3 , 33 we investigated RyR2 biochemistry and function in COVID‐19 patient brains.
RESEARCH‐IN‐CONTEXT
Systematic review: The authors reviewed the literature using PubMed. While the mechanisms that lead to cognitive impairment associated with COVID‐19 are not well understood, there have been recent reports studying SARS‐CoV‐2 infection and brain biochemistry and neuropathology. These relevant citations are appropriately cited.
Interpretation: Our findings link the inflammatory response to SARS‐CoV‐2 infection with the neuropathological pathways causing tau hyperphosphorylation typically associated with Alzheimer's disease (AD). Furthermore, our data indicate a role for leaky ryanodine receptor 2 (RyR2) in the pathophysiology of SARS‐CoV‐2 infection.
Future directions: The article proposes that the alteration of cellular calcium dynamics due to leaky RyR2 in COVID‐19 brains is associated with the activation of neuropathological pathways that are also found in the brains of AD patients. Both the cortex and cerebellum of SARS‐CoV‐2–infected patients exhibited a reduced expression of the Ca2+ buffering protein calbindin. Decreased calbindin could render these tissues more vulnerable to cytosolic Ca2+ overload. Ex vivo treatment of the COVID‐19 brain using a Rycal drug (ARM210) that targets RyR2 channels prevented intracellular Ca2+ leak in patient samples. Future experiments will explore calcium channels as a potential therapeutic target for the neurological complications associated with COVID‐19.
1.2. Study conclusions and disease implications
Our results indicate that SARS‐CoV‐2 infection activates inflammatory signaling and oxidative stress pathways resulting in hyperphosphorylation of tau, but normal amyloid precursor protein (APP) processing in COVID‐19 patient cortex and cerebellum. There was reduced calbindin expression in both cortex and cerebellum rendering both tissues vulnerable to Ca2+‐mediated pathology. Moreover, COVID‐19 cortex and cerebellum exhibited RyR Ca2+ release channels with the biochemical signature of ‘‘leaky’’ channels and increased activity consistent with pathological intracellular Ca2+ leak. RyR2 were oxidized, associated with increased NADPH oxidase 2 (NOX2), and were PKA hyperphosphorylated on serine 2808, both of which cause loss of the stabilizing subunit calstabin2 from the channel complex promoting leaky RyR2 channels in COVID‐19 patient brains. Furthermore, ex vivo treatment of COVID‐19 patient brain samples with the Rycal drug ARM210, which is currently undergoing clinical testing at the National Institutes of Health for RyR1‐myopathy (ClinicalTrials.gov Identifier: NCT04141670), fixed the channel leak. Thus, our experiments demonstrate that SARS‐CoV‐2 infection activates biochemical pathways linked to the tau pathology associated with AD and that leaky RyR Ca2+ channels may be a potential therapeutic target for the neurological complications associated with COVID‐19.
The molecular basis of how SARS‐CoV‐2 infection results in ‘‘long COVID’’ is not well understood, and questions regarding the role of defective Ca2+ signaling in the brain in COVID‐19 remain unanswered. A recent comprehensive molecular investigation revealed extensive inflammation and degeneration in the brains of patients that died from COVID‐19, 34 including in patients with no reported neurological symptoms. These authors also reported overlap between marker genes of AD and genes that are upregulated in COVID‐19 infection, consistent with the findings of increased tau pathophysiology reported in the present study. We propose a potential mechanism that may contribute to the neurological complications caused by SARS‐CoV‐2: defective intracellular Ca2+ regulation and activation of AD‐like neuropathology.
TGF‐β belongs to a family of cytokines involved in the formation of cellular fibrosis by promoting epithelial‐to‐mesenchymal transition, fibroblast proliferation, and differentiation. 35 TGF‐β activation has been shown to induce fibrosis in the lungs and other organs by activation of the SMAD‐dependent pathway. We have previously reported that TGF‐β/SMAD3 activation leads to NOX2/4 translocation to the cytosol and its association with RyR channels, promoting oxidization of the channels and depletion of the stabilizing subunit calstabin in skeletal muscle and in heart. 28 , 30 Alteration of Ca2+ signaling may be particularly crucial in COVID‐19‐infected patients with cardiovascular/neurological diseases due, in part, to the multifactorial RyR2 remodeling after the cytokine storm, increased TGF‐β activation, and increased oxidative stress. Moreover, SARS‐CoV‐2–infected patients exhibited a hyperadrenergic state. The elevated expression of glutamate carboxypeptidase 2 (GCPII) in COVID‐19 brains reported in the present study could also contribute directly to increased PKA signaling of RyR2 by reducing PKA inhibition via metabotropic glutamate receptor 3 (mGluR3). 36 Hyperphosphorylation of RyR2 channels can promote pathological remodeling of the channel and exacerbate defective Ca2+ regulation in these tissues. The increased Ca2+/cAMP/PKA signaling could also open nearby K+ channels which could potentially weaken synaptic connectivity, reduce neuronal firing, 36 and could activate Ca2+ dependent enzymes.
Interestingly, both the cortex and cerebellum of SARS‐CoV–2‐infected patients exhibited a reduced expression of the Ca2+ buffering protein calbindin. Decreased calbindin could render these tissues more vulnerable to the cytosolic Ca2+ overload. This finding is in accordance with previous studies showing reduced calbindin expression levels in Purkinje cells and the CA2 hippocampal region of AD patients 37 , 38 , 39 and in cortical pyramidal cells of aged individuals with tau pathology. 33 , 40 In contrast to the findings in the brains of COVID‐19 patients in the present study, calbindin was not reduced in the cerebellum of AD patients, possibly protecting these cells from AD pathology. 39 , 41
Leaky RyR channels, leading to increased mitochondrial Ca2+ overload and ROS production and oxidative stress, have been shown to contribute to the development of tau pathology associated with AD. 3 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 33 Recent studies of the effects of COVID‐19 on the central nervous system have found memory deficits and biological markers similar to those seen in AD patients. 42 , 43 Our data demonstrate increased activity of enzymes responsible for phosphorylating tau (pAMPK, pGSK3β), as well as increased phosphorylation at multiple sites on tau in COVID‐19 patient brains. The tau phosphorylation observed in these samples exhibited some differences from what is typically observed in AD, occurring in younger patients and in areas of the brain, specifically the cerebellum, that usually do not demonstrate tau pathology in AD patients. Taken together, these data suggest a potential contributing mechanism to the development of tau pathology in COVID‐19 patients involving oxidative overload‐driven RyR2 channel dysfunction. Furthermore, we propose that these pathological changes could be a significant contributing factor to the neurological manifestations of COVID‐19 and in particular the “brain fog” associated with long COVID, and represent a potential therapeutic target for ameliorating these symptoms. For example, tau pathology in the cerebellum could explain the recent finding that 74% of hospitalized COVID‐19 patients experienced coordination deficits. 44 The data presented also raise the possibility that prior COVID‐19 infection could be a potential risk factor for developing AD in the future.
The present study was limited to the use of existing autopsy brain tissues at the Columbia University Biobank from SARS‐CoV‐2–infected patients. The number of subjects is small and information on their cognitive function as well as their brain histopathology and levels of Aβ in cerebrospinal fluid and plasma are lacking. Furthermore, we did not have access to a suitable animal model of SARS‐CoV‐2 infection in which to test whether the observed biochemical changes in COVID‐19 brains and potential cognitive and behavioral deficits associated with the brain fog of long COVID could be reversed or attenuated by therapeutic interventions. The design of future studies should include larger numbers of subjects that are age‐ and sex‐matched. The cognitive function of SARS‐CoV‐2–infected patients who presented cognitive symptoms should be assessed and regularly monitored. Moreover, it is important to know whether the observed neuropathological signaling is unique to SARS‐CoV‐2 infection or are common to all other viral infections. Previous studies have reported cognitive impairment in Middle East respiratory syndrome 45 as well as Ebola 46 , 47 patients. Retrospective studies comparing the incidence and the magnitude of cognitive impairments caused by these different viral infections would improve our understanding of these neurological complications of viral infections.
2. CONSOLIDATED RESULTS AND STUDY DESIGN
There were increased markers of oxidative stress (glutathione disulfide [GSSG]/ glutathione [GSH]) in the cortex (mesial temporal lobe) and cerebellum (cerebellar cortex, lateral hemisphere) of COVID‐19 tissue. Kynurenic acid, a marker of inflammation, was increased in COVID‐19 cortex and cerebellum brain lysates compared to controls, is in accordance with recent studies showing a positive correlation between kynurenic acid and cytokines and chemokine levels in COVID‐19 patients. 48 , 49 , 50
To determine whether SARS‐CoV‐2 infection also increases tissue TGF‐β activity, we measured SMAD3 phosphorylation, a downstream signal of TGF‐β, in control and COVID‐19 tissue lysates. Phosphorylated SMAD3 (pSMAD3) levels were increased in COVID‐19 cortex and cerebellum brain lysates compared to controls, indicating that SARS‐CoV‐2 infection increased TGF‐β signaling in these tissues. Interestingly, brain tissues from COVID‐19 patients exhibited activation of the TGF‐β pathway, despite the absence of the detectable (by immunohistochemistry and polymerase chain reaction, data not shown) virus in these tissues. These results suggest that the TGF‐β pathway is activated systemically by SARS‐CoV‐2, resulting in its upregulation in the brain, as well as other organs. In addition to oxidative stress, COVID‐19 brain tissues also demonstrated increased PKA and calmodulin‐dependent protein kinase II association domain (CaMKII) activity, most likely associated with increased adrenergic stimulation. Both PKA and CaMKII phosphorylation of tau have been reported in tauopathies. 51 , 52
The hallmarks of AD brain neuropathology are the formation of Aβ plaques from abnormal APP processing by BACE1, as well as tau ‘‘tangles’’ caused by tau hyperphosphorylation. 53 Brain lysates from COVID‐19 patients’ autopsies demonstrated normal BACE1 and APP levels compared to controls. The patients analyzed in the present study were grouped by age (young ≤ 58 years old, old ≥ 66 years old) to account for normal, age‐dependent changes in APP and tau pathology. Abnormal APP processing was only observed in brain lysates from patients diagnosed with AD. However, AMPK and GSK3β phosphorylation were increased in both the cortex and cerebellum in COVID‐19 brains. Activation of these kinases in SARS‐CoV‐2–infected brains leads to a hyperphosphorylation of tau consistent with AD tau pathology in the cortex. COVID‐19 brain lysates from older patients showed increased tau phosphorylation at S199, S202, S214, S262, and S356. Lysates from younger COVID‐19 patients showed increased tau phosphorylation at S214, S262, and S356, but not at S199 and S202, demonstrating increased tau phosphorylation in both young and old individuals and suggesting a tau pathology similar to AD in COVID‐19–affected patients. Interestingly, both young and old patient brains demonstrated increased tau phosphorylation in the cerebellum, which is not typical of AD.
RyR channels may be oxidized due to the activation of the TGF‐β signaling pathway. 30 NOX2 binding to RyR2 causes oxidation of the channel, which activates the channel, manifested as an increased open probability that can be assayed using 3[H]ryanodine binding. 54 When the oxidization of the channel is at pathological levels, there is destabilization of the closed state of the channel, resulting in spontaneous Ca2+ release or leak. 27 , 30 To determine the effect of the increased TGF‐β signaling associated with SARS‐CoV‐2 infection on NOX2/RyR2 interaction, RyR2 and NOX2 were co‐immunoprecipitated from brain lysates of COVID‐19 patients and controls. NOX2 associated with RyR2 in brain tissues from SARS‐CoV‐2–infected individuals were increased compared to controls.
Given the increased oxidative stress and increased NOX2 binding to RyR2 seen in COVID‐19 brains, RyR2 post‐translational modifications were investigated. Immunoprecipitated RyR2 from brain lysates demonstrated increased oxidation, PKA phosphorylation on serine 2808, and depletion of the stabilizing protein subunit calstabin2 in SARS‐CoV‐2–infected tissues compared to controls. This biochemical remodeling of the channel is known as the ‘‘biochemical signature’’ of leaky RyR2 23 , 55 , 56 that is associated with destabilization of the closed state of the channel. This leads to SR/ER Ca2+ leak, which contributes to the pathophysiology of a number of diseases including AD. 23 , 24 , 26 , 30 , 55 , 56 , 57 RyR channel activity was determined by binding of 3[H]ryanodine, which binds only to the open state of the channel. RyR2 was immunoprecipitated from tissue lysates and ryanodine binding was measured at both 150 nM and 20 μM free Ca2+. RyR2 channels from SARS‐CoV‐2–infected brain tissue demonstrated abnormally high activity (increased ryanodine binding) compared to channels from control tissues at physiologically resting conditions (150 nM free Ca2+), when channels should be closed. Interestingly, cortex and cerebellum of SARS‐CoV‐2–infected patients also exhibited a reduced expression of the Ca2+ binding protein calbindin. Calbindin is typically not reduced in the cerebellum of AD patients, possibly providing some protection against AD pathology. The low calbindin levels in the cerebellum of COVID‐19 brains could contribute to the observed tau pathology in this brain region. An additional atypical finding in the COVID‐19 brains studied in this investigation is an increased level of GCPII. This could contribute to the observed RyR PKA phosphorylation by increasing cAMP and inhibiting the metabotropic glutamate receptor type 3. 36
3. DETAILED METHODS AND RESULTS
3.1. Methods
3.1.1. Human samples
De‐identified human heart, lung, and brain tissue were obtained from the COVID BioBank at Columbia University. The cortex samples were from the mesial temporal lobe and the cerebellum samples were from the cerebellar cortex, lateral hemisphere. The Columbia University BioBank functions under standard operating procedures, quality assurance, and quality control for sample collection and maintenance. Age‐ and sex‐matched controls exhibited absence of neurological disorders and cardiovascular or pulmonary diseases. Sex, age, and pathology of patients are listed in Table 1.
TABLE 1.
Sex, age, and pathology of COVID‐19 patients
| Patient Number | Sex | Age | Pathology |
|---|---|---|---|
| 1 | Male | 57 | Acute hypoxic‐ischemic injury in the hippocampus, pons, and cerebellum. |
| 2 | Female | 38 | Hypoxic ischemic encephalopathy, severe, global. |
| 3 | Male | 58 | Hypoxic/ischemic injury, global, widespread astrogliosis/microgliosis. |
| 4 | Male | 84 | Dementia. Beta‐amyloid plaques are noted in cortex and cerebellum. |
| 5 | Female | 80 | Severe hypoxic ischemic encephalopathy, severe. Global astrogliosis and microgliosis. Mild Alzheimer‐type pathology. |
| 6 | Female | 74 | Acute hypoxic‐ischemic encephalopathy, global, moderate to severe. Arteriolosclerosis, mild. Metabolic gliosis, moderate |
| 7 | Male | 66 | Left frontal subacute hemorrhagic infarct. Multifocal subacute infarcts in pons and left cerebral peduncle. Global astrogliosis and microgliosis (see microscopic description). Alzheimer's pathology. |
| 8 | Female | 76 | Hypoxic ischemic encephalopathy, moderate. Alzheimer's pathology. Atherosclerosis, moderate. Arteriolosclerosis, moderate |
| 9 | Male | 72 | Hypoxic/ischemic injury, acute to subacute, involving hippocampus, medulla and cerebellum. Mild atherosclerosis. Mild arteriolosclerosis |
| 10 | Male | 71 | Hypoxic‐ischemic encephalopathy, acute, global, mild to moderate. Diffuse Lewy body disease, neocortical type, consistent with Parkinson disease dementia. Atherosclerosis, severe. Arteriolosclerosis, mild. |
Lysate preparation and Western blots
Tissues (50 mg) were isotonically lysed using a Dounce homogenizer in 0.25 ml of 10 mM Tris maleate (pH 7.0) buffer with protease inhibitors (Complete inhibitors from Roche). Samples were centrifuged at 8000 × g for 20 minutes and the protein concentrations of the supernatants were determined by Bradford assay. To determine protein levels in tissue lysates, tissue proteins (20 μg) were separated by 4% to 20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and immunoblots were developed using the following antibodies: pSMAD3 (Abcam, 1:1000), SMAD3 (Abcam, 1:1000), AMPK (Abcam, 1:1000), tau (Thermo Fisher, 1:1000), pTauS199 (Thermo Fisher, 1:1000), pTauS202/T205 (Abcam, 1:1000), pTauS262 (Abcam, 1:1000), GSK3β (Abcam, 1:1000), pGSK3βS9 (Abcam, 1:1000), pGSK3βT216 (Abcam, 1:1000), APP (Abcam, 1:1000), BACE1 (Abcam, 1:1000), GAPDH (Santa Cruz Biotech, 1:1000), CTF‐β (Santa Cruz Biotechnology, Inc., 1:1000), Calbindin (Abcam, 1:1000), and GCPII (Thermo Fisher, 1:4000).
Analyses of ryanodine receptor complex
Tissue lysates (0.1 mg) were treated with buffer or 10 μM Rycal (ARM210) at 4°C. RyR2 was immunoprecipitated from 0.1 mg lung, heart, and brain using an anti‐RyR2 specific antibody (2 μg) in 0.5 ml of a modified radioimmune precipitation assay buffer (50 mm Tris‐HCl, pH 7.2, 0.9% NaCl, 5.0 mm NaF, 1.0 mm Na3VO4, 1% Triton X‐100, and protease inhibitors; RIPA) overnight at 4°C. RyR2‐specific antibody was an affinity‐purified polyclonal rabbit antibody using the peptide CKPEFNNHKDYAQEK corresponding to amino acids 1367–1380 of mouse RyR2 with a cysteine residue added to the amino terminus. The immune complexes were incubated with protein A‐Sepharose beads (Sigma) at 4°C for 1 hour, and the beads were washed three times with RIPA. The immunoprecipitates were size‐fractionated on SDS‐PAGE gels (4%–20% for RyR2, calstabin2, and NOX2) and transferred onto nitrocellulose membranes for 1 hour at 200 mA. Immunoblots were developed using the following primary antibodies: anti‐RyR2 (Affinity BioReagents, 1:2500), anti‐phospho‐RyR‐Ser(pS)‐2808 (Affinity BioReagents 1:1000), anti‐ calstabin2 (FKBP12 C‐19, Santa Cruz Biotechnology, Inc., 1:2500), and anti‐NOX2 (Abcam, 1:1000). To determine channel oxidation, the carbonyl groups in the protein side chains were derivatized to DNP by reaction with 2,4‐dinitrophenylhydrazine. The DNP signal associated with RyR2 was determined using a specific anti‐DNP antibody according to the manufacturer using an Odyssey system (LI‐COR Biosciences) with infrared‐labeled anti‐mouse and anti‐rabbit immunoglobulin G (IgG; 1:5000) secondary antibodies.
Ryanodine binding
RyR2 was immunoprecipitated from 1.5 mg of tissue lysate using an anti‐RyR2 specific antibody (25 μg) in 1.0 ml of a modified RIPA buffer overnight at 4°C. The immune complexes were incubated with protein A‐Sepharose beads (Sigma) at 4°C for 1 hour, and the beads were washed three times with RIPA buffer, followed by two washes with ryanodine binding buffer (10 mM Tris‐HCl, pH 6.8, 1 M NaCl, 1% CHAPS, 5 mg/ml phosphatidylcholine, and protease inhibitors). Immunoprecipitates were incubated in 0.2 ml of binding buffer containing 20 nM [3H] ryanodine and either of 150 nM and 20 μm free Ca2+ for 1 hour at 37°C. Samples were diluted with 1 ml of ice‐cold washing buffer (25 mm Hepes, pH 7.1, 0.25 m KCl) and filtered through Whatman GF/B membrane filters pre‐soaked with 1% polyethyleneimine in washing buffer. Filters were washed three times with 5 ml of washing buffer. The radioactivity remaining on the filters is determined by liquid scintillation counting to obtain bound [3H] ryanodine. Nonspecific binding was determined in the presence of 1000‐fold excess of non‐labeled ryanodine.
GSSG/GSH ratio measurement and SMAD3 phosphorylation
Approximately 20 mg of tissue suspended in 200 μL of ice‐cold phosphate‐buffered saline/0.5% NP‐40, pH6.0 was used for lysis. Tissue was homogenized with a Dounce homogenizer with 10 to 15 passes. Samples were centrifuged at 8000 × g for 15 minutes at 4°C to remove any insoluble material. Supernatant was transferred to a clean tube. Deproteinizing of the samples was accomplished by adding 1 volume ice‐cold 100% (w/v) trichloroacetic acid (TCA) into five volumes of sample and vortexing briefly to mix well. After incubating for 5 minutes on ice, samples were centrifuged at 12,000 × g for 5 minutes at 4°C and the supernatant was transferred to a fresh tube. The samples were neutralized by adding NaHCO3 to the supernatant and vortexing briefly. Samples were centrifuged at 13,000 × g for 15 minutes at 4°C and supernatant was collected. Samples were then deproteinized, neutralized, TCA was removed, and they were ready to use in the assay. The GSSG/GSH was determined using a ratio detection assay kit (Abcam, ab138881). Briefly, in two separate assay reactions, GSH (reduced) was measured directly with a GSH standard and Total GSH (GSH + GSSG) was measured by using a GSSG standard. A 96‐well plate was set up with 50 μL duplicate samples and standards with known concentrations of GSH and GSSG. A Thiol green indicator was added, and the plate was incubated for 60 minutes at room temperature (RT). Fluorescence at Ex/Em = 490/520 nm was measured with a fluorescence microplate reader and the GSSG/GSH for samples were determined comparing fluorescence signal of samples with known standards.
FIGURE 1.
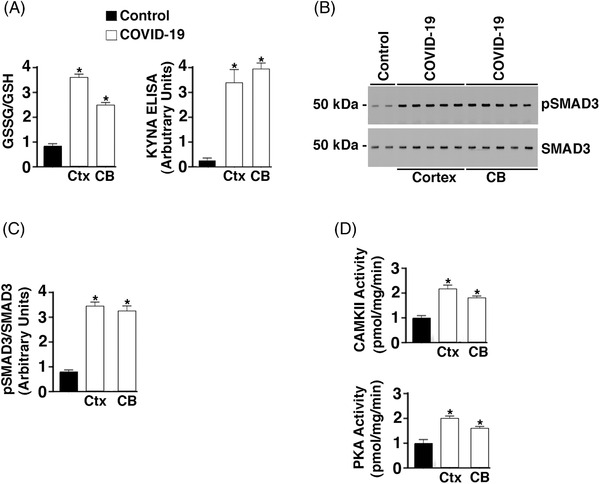
Increased oxidative stress, inflammatory and adrenergic signaling in brains of COVID‐19 patients. A, Bar graph depicting the glutathione disulfide (GSSG)/ glutathione (GSH) ratio and kynurenic acid (KYNA) enzyme‐linked immunsorbent assay signal from control (n = 6) and COVID‐19 (n = 6) tissue lysates. CB, cerebellum; Ctx, cortex. Data are mean ± standard deviation (SD). *P < .05 control versus COVID‐19. B, Western blots showing phospho‐SMAD3 and total SMAD3 from control (n = 4) and COVID‐19 (n = 7) brain lysates. C, Bar graphs depicting quantification of pSMAD3/SMAD3 from Western blot signals in B. D, Calmodulin‐dependent protein kinase II association domain (CaMKII) and protein kinase A (PKA) activity of brain tissue lysates. Data are mean ± SD. *P < .05 control versus COVID‐19
Kynurenic acid assay
Kynurenic acid (KYNA) concentration in brain lysates was determined using an enzyme‐linked immunosorbent assay (ELISA) kit for KYNA (ImmuSmol). Briefly, samples (50 μl) were added to a microtiter plate designed to extract the KCNA from the samples. An acylation reagent was added for 90 minutes at 37°C to derivatize the samples. After derivatization, 50 μl of the prepared standards and 100 μl samples were pipetted into the appropriate wells of the KYNA microtiter plate. KYNA Antiserum was added to all wells and the plate was incubated overnight at 4°C. After washing the plate four times, the enzyme conjugate was added to each well. The plate was incubated for 30 minutes at RT on a shaker at 500 rpm. The enzyme substrate was added to all wells and the plate was incubated for 20 minutes at RT. Stop solution was added to each well. A plate reader was used to determine the absorbance at 450 nm. The sample signals were compared to a standard curve.
PKA activity assay
PKA activity in brain lysates was determined using , a PKA activity kit (Thermo Fisher, EIAPKA). Briefly, samples were added to a microtiter plate containing an immobilized PKA substrate that is phosphorylated by PKA in the presence of ATP. After incubating the samples with ATP at RT for 2 hours, the plate was incubated with the phospho‐PKA substrate antibody for 60 minutes. After washing the plate with wash buffer, goat anti‐rabbit IgG horseradish peroxidase (HRP) conjugate was added to each well. The plate was aspirated, washed, and TMB substrate was added to each well, which was then incubated for 30 minutes at RT. A plate reader was used to determine the absorbance at 450 nm. The sample signals were compared to a standard curve.
FIGURE 2.
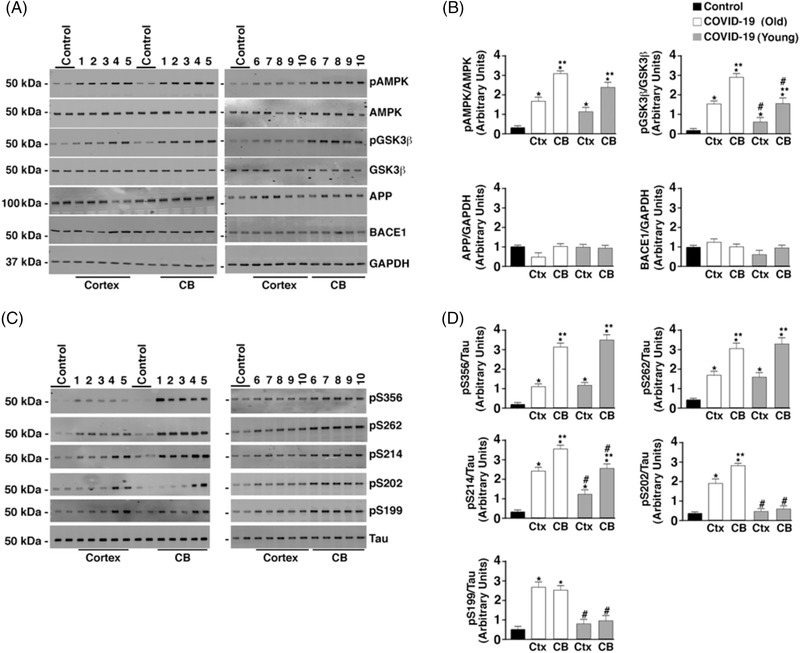
Hyperphosphorylation of tau but normal amyloid precursor protein (APP) processing in COVID‐19 brains. A, Brain (CB, cerebellum; Ctx, cortex) lysates were separated by 4% to 20% polyacrylamide gel electrophoresis. Immunoblots were developed for pAMPK, AMPK, GSK3β, pGSK3β (T216), APP, BACE1, and GAPDH loading control. The numbers (1–10) above immunoblots refer to patient numbers listed in Table 1. B, Bar graphs showing quantification of pAMPK, pGSK3β, APP/GAPDH, and BACE1/GAPDH from Western blots in (A). Data are mean ± standard deviation (SD). *P < .05 control versus COVID‐19; **P < .05 CB versus Ctx; #P < .05 COVID (Young) versus COVID (Old). C, Immunoblots of brain lysates showing total tau and tau phosphorylation on residues S199, S202/T205, S214, S262, and S356. D, Bar graphs showing quantification phosphorylated tau at the residues shown on Western blots in (C). Data are mean ± SD. *P < .05 control versus COVID‐19; **P < .05 CB versus Ctx; #P < .05 COVID (Young) versus COVID (Old)
FIGURE 3.
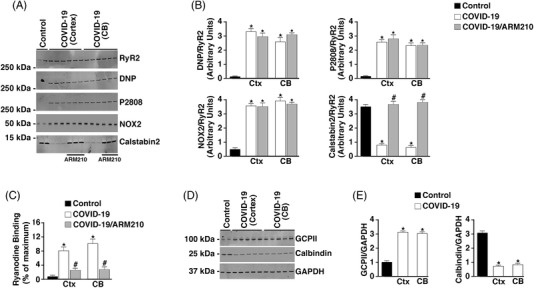
Dysregulation of calcium‐handling proteins in COVID‐19 brains. A, Western blots depicting ryanodine receptor 2 (RyR2) oxidation, protein kinase A (PKA) phosphorylation, and calstabin2 or NADPH oxidase 2 (NOX2) bound to the channel from brain (CB, cerebellum; Ctx, cortex) lysates. B, Bar graphs quantifying DNP/RyR2, pS2808/RyR2, and calstabin2 and NOX2 bound to the channel from the Western blots. Data are mean ± standard deviation (SD). *P < .05 control versus COVID‐19; # P < .05 COVID‐19 versus COVID‐19+ARM210. C, 3[H]ryanodine binding from immunoprecipitated RyR2. Bar graphs show ryanodine binding at 150 nM Ca2+ as a percent of maximum binding (Ca2+ = 20 μM). Data are mean ± SD. *P < .05 control versus COVID‐19; #P < .05 COVID‐19 versus COVID‐19+ARM210. D, Western blots showing the levels of glutamate carboxypeptidase 2 (GCPII), calbindin, and GAPDH loading control in brain (Ctx, CB). E, Bar graphs quantifying GCPII/GAPDH and calbindin/GAPDH from the western blots. Data are mean ± SD. *P < .05 control versus COVID‐19
CaMKII activity assay
CaMKII activity in brain lysates was determined using the CycLex CaM kinase II Assay Kit (MBL International). Briefly, samples were added to a microtiter plate containing an immobilized CaMKII substrate that is phosphorylated by CaMKII in the presence of Mg2+ and ATP. After incubating the samples in kinase buffer containing Mg2+ and ATP at RT for 1 hour, the plate was washed and incubated with the HRP conjugated anti‐phospho‐CaMKII substrate antibody for 60 minutes. The plate was aspirated, washed, and TMB substrate was added to each well, which was then incubated for 30 minutes at RT. A plate reader was used to determine the absorbance at 450 nm. The sample signals were compared to a standard curve.
Statistics
Group data are presented as mean ± standard deviation. Statistical comparisons between the two groups were determined using an unpaired t‐test. Values of P < .05 were considered statistically significant. All statistical analyses were performed with GraphPad Prism 8.0.
3.2. Results
3.2.1. Oxidative stress and TGF‐β, PKA, and CaMKII activation
Oxidative stress levels were determined in brain tissues (cortex, cerebellum) from COVID‐19 patient autopsy tissues and controls by measuring the ratio of GSSG to GSH by an ELISA kit. COVID‐19 patients exhibited significant oxidative stress with a 3.8‐ and 3.2‐fold increase in GSSG/GSH ratios in cortex (Ctx) and cerebellum (CB) compared to controls, respectively (Figure 1A). High circulating levels of kynurenine have been reported in COVID‐19. 48 , 49 , 50 However, the expression of KYNA in COVID‐19 brain tissue has not been examined. Levels in the Ctx and CB were measured using an ELISA kit. COVID‐19 brains had a significant increase in the Ctx and CB compared to controls (Figure 1A). An additional marker of tissue inflammation is increased cytokine expression. SMAD3 phosphorylation, a downstream signal of TGF‐β, was increased in COVID‐19 Ctx and CB tissue lysates compared to controls (Figure 1B and 1C). Increased adrenergic activation in the brain of patients infected with SARS‐CoV‐2 was also demonstrated by measuring PKA activity in the Ctx and CB and CaMKII activity was increased as well (Figure 1D).
Activation of AD‐linked signaling
Both PKA and CaMKII have been directly implicated in the increased phosphorylation of tau associated with AD. 51 , 52 Because COVID‐19 brain lysates had increased PKA and CaMKII activity, AD‐linked biochemistry was evaluated in the COVID‐19 brain lysates. Normal APP processing was observed in COVID‐19 brain lysates as demonstrated by normal BACE1 and APP levels compared to controls (Figure 2A and B). Abnormal APP processing was only observed in brain lysates from patients diagnosed with AD (see Table 1 for patient details). However, phosphorylation/activation of AMPK and GSK3β was observed in SARS‐CoV‐2–infected patient brain lysates. Activation of these kinases along with the activation of PKA and CaMKII (Figure 1) leads to a hyperphosphorylation of tau at multiple residues (Figure 2C and D). Tau hyperphosphorylation in the cerebellum is not typical of AD pathology. The CB tau pathology demonstrated in COVID‐19 warrants further investigation.
RyR2 channel oxidation and leak
RyR2 biochemistry was investigated to determine whether RyR2 in COVID‐19 brain tissues demonstrated a “leaky” phenotype. Increased NOX2/RyR2 binding was shown in Ctx and CB lysates from SARS‐CoV‐2–infected individuals compared to controls using co‐immunoprecipitation (Figure 3A and B). In addition, RyR2 from SARS‐CoV‐2–infected brains had increased oxidation, increased serine 2808 PKA phosphorylation, and depletion of the stabilizing protein subunit calstabin2 compared to controls (Figure 3A and B). RyR channels exhibiting these characteristics can be inappropriately activated at low cytosolic Ca2+ concentrations resulting in a pathological ER/SR Ca2+ leak. 3[H]Ryanodine binding to immunoprecipitated RyR2 was measured at both 150 nM and 20 μM free Ca2+. Because ryanodine binds only to the open state of the channel under these conditions, 3[H]Ryanodine binding may be used as a surrogate measure of channel open probability. The total amount of RyR immunoprecipitated was the same for control and COVID‐19 samples (data not shown). Increased RyR2 channel activity at resting conditions (150 nM free Ca2+) was observed in COVID‐19 channels compared to controls (Figure 3C). Under these conditions, RyR channels should be closed. Rebinding of calstabin2 to RyR2, using a Rycal, has been shown to reduce SR/ER Ca2+ leak, despite the persistence of the channel remodeling. Indeed, calstabin2 binding to RyR2 was increased when COVID‐19 patient brain tissue lysates were treated ex vivo with the Rycal drug ARM210 (Figure 3A and B). Abnormal RyR2 activity observed at resting Ca2+ concentration was also decreased by Rycal treatment (Figure 3C).
FIGURE 4.
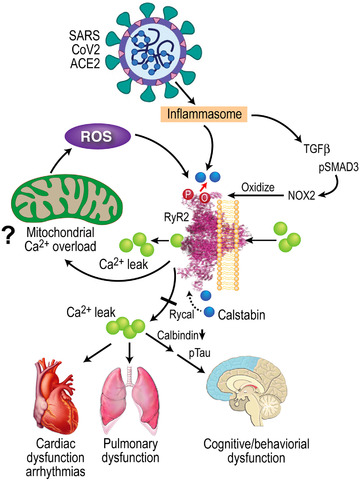
SARS‐CoV‐2 infection results in leaky ryanodine receptor 2 (RyR2) that may contribute to cardiac, pulmonary, and cognitive dysfunction. SARS‐CoV‐2 infection targets cells via the angiotensin‐converting enzyme 2 (ACE2) receptor, inducing inflammasome stress response/activation of stress signaling pathways. This results in increased transforming growth factor‐β (TGF‐β) signaling, which activates SMAD3 (pSMAD) and increases NADPH oxidase 2 (NOX2) expression and the amount of NOX2 associated with RyR2. Increased NOX2 activity at RyR2 oxidizes the channel, causing calstabin2 depletion from the channel macromolecular complex, destabilization of the closed state, and ER/SR calcium leak that is known to contribute to cardiac dysfunction, 55 arrhythmias, 61 pulmonary insufficiency, 23 , 25 and cognitive and behavioral abnormalities associated with neurodegenreation. 24 , 26 Decreased calbindin in COVID‐19 may render brain more susceptible to tau pathology. Rycal drugs fix the RyR2 channel leak by restoring calstabin2 binding and stabilizing the channel closed state. Fixing leaky RyR2 may improve cardiac, pulmonary, and cognitive function in COVID‐19.
An interesting finding concerning the tau phosphorylation in brain lysates from SARS‐CoV‐2 patients was the increase of phosphorylation at multiple sites in the cerebellum. This is atypical of AD. One potential mechanism to explain this finding is the significantly decreased levels of calbindin expressed in COVID‐19 cerebellum (Figure 3D, 3E). The decreased cerebellar calbindin levels could make this area of the brain more susceptible to Ca2+‐induced activation of enzymes upstream of tau phosphorylation. Moreover, increased GCPII expression was observed in COVID‐19 cortex and cerebellar lysates (Figure 3D, 3E), which would reduce mGluR3 inhibition of PKA signaling and could contribute to the PKA hyperphosphorylation of RyR2.
Model for the role for leaky RyR2 in the pathophysiology of SARS‐CoV‐2 infection
Our data indicate a role for leaky RyR2 in the pathophysiology of SARS‐CoV‐2 infection (Figure 4). In addition to the brain of COVID‐19 patients, we observed increased systemic oxidative stress and activation of the TGF‐β signaling pathway in lung, and heart, which correlates with oxidation‐driven biochemical remodeling of RyR2 (Figure 3 and S1 in supporting inormation). This RyR2 remodeling results in intracellular Ca2+ leak, which can play a role in heart failure progression, pulmonary insufficiency, as well as cognitive dysfunction. 23 , 24 , 25 , 26 , 28 The alteration of cellular Ca2+ dynamics has also been implicated in COVID‐19 pathology. 58 , 59 Taken together, the present data suggest that leaky RyR2 may play a role in the long‐term sequelae of COVID‐19, including the “brain fog” associated with SARS‐CoV‐2 infection which could be a forme fruste of AD, 60 and could predispose long COVID patients to developing AD later in life. Leaky RyR2 channels may be a therapeutic target for amelioration of some of the persistent cognitive deficits associated with long COVID.
AUTHOR CONTRIBUTIONS
Andrew R. Marks conceived the study; Steve Reiken, Leah Sittenfeld, Haikel Dridi, Yang Liu, Xiaoping Liu, and Andrew R. Marks designed experiments, analyzed data, and edited/wrote the paper.
CONFLICTS OF INTERESTS
Columbia University and Andrew Marks own stock in ARMGO Pharma, Inc., a company developing compounds targeting RyR and have patents on Rycals. Steven Reiken has consulted for ARMGO Pharma, Inc. in the last 36 months. All other authors declare no competing interests or conflicts.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The Authors would like to acknowledge the COVID BioBank at Columbia University for providing human heart, lung, and brain tissue for this study.
Reiken S, Sittenfeld L, Dridi H, Liu Y, Liu X, Marks AR. Alzheimer's‐like signaling in brains of COVID‐19 patients. Alzheimer's Dement. 2022;18:955–965. 10.1002/alz.12558
REFERENCES
- 1. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shi S, Qin M, Shen B, et al. Association of cardiac injury with mortality in hospitalized patients with COVID‐19 in Wuhan, China. JAMA Cardiol. 2020;5:802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meinhardt J, Radke J, Dittmayer C, et al. Olfactory transmucosal SARS‐CoV‐2 invasion as a port of central nervous system entry in individuals with COVID‐19. Nat Neurosci. 2021;24:168‐175. [DOI] [PubMed] [Google Scholar]
- 4. Guan W‐J, Liang W‐H, Zhao Y, et al. Comorbidity and its impact on 1590 patients with COVID‐19 in China: a nationwide analysis. Eur Respir J. 2020;55:2000547. 10.1183/13993003.00547-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Torres Acosta MA, Singer BD. Pathogenesis of COVID‐19‐induced ARDS: implications for an ageing population. Eur Respir J. 2020;56:2002049. 10.1183/13993003.02049-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arentz M, Yim E, Klaff L, et al. Characteristics and outcomes of 21 critically ill patients with COVID‐19 in Washington State. JAMA. 2020;323:1612–1614. 10.1001/jama.2020.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng Y‐Y, Ma Y‐T, Zhang J‐Y, Xie X. COVID‐19 and the cardiovascular system. Nat Rev Cardiol. 2020;17:259‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu Y, Xu X, Chen Z, et al. Nervous system involvement after infection with COVID‐19 and other coronaviruses. Brain Behav Immun. 2020;87:18‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581:215‐220. [DOI] [PubMed] [Google Scholar]
- 10. Lou JJ, Movassaghi, M & Gordy, D , et al. Neuropathology of COVID‐19 (neuro‐COVID): clinicopathological update. Free Neuropathol. 2021;Jan 18;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thakur KT, Miller EH, Glendinning MD, et al. COVID‐19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain. 2021;144:2696‐2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jia HP, Look DC, Shi L, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79:14614‐14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abiodun OA, Ola MS. Role of brain renin angiotensin system in neurodegeneration: an update. Saudi J Biol Sci. 2020;27:905‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Farhadian SF, Seilhean D, Spudich S. Neuropathogenesis of acute coronavirus disease 2019. Curr Opin Neurol. 2021;34:417‐422. [DOI] [PubMed] [Google Scholar]
- 15. Laforge M, Elbim C, Frère C, et al. Tissue damage from neutrophil‐induced oxidative stress in COVID‐19. Nat Rev Immunol. 2020;20:515‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merad M, Martin JC. Pathological inflammation in patients with COVID‐19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Y, Shen C, Li J, et al. Plasma IP‐10 and MCP‐3 levels are highly associated with disease severity and predict the progression of COVID‐19. J Allergy Clin Immunol. 2020;146:119‐127.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bao R, Hernandez K, Huang L, Luke JJ. ACE2 and TMPRSS2 expression by clinical, HLA, immune, and microbial correlates across 34 human cancers and matched normal tissues: implications for SARS‐CoV‐2 COVID‐19. J Immunother Cancer. 2020;8:e001020 10.1136/jitc-2020-001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delpino MV, Quarleri J. SARS‐CoV‐2 Pathogenesis: imbalance in the renin‐angiotensin system favors lung fibrosis. Front Cell Infect Microbiol. 2020;10:340 10.3389/fcimb.2020.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Agrati C, Sacchi A, Bordoni V, et al. Expansion of myeloid‐derived suppressor cells in patients with severe coronavirus disease (COVID‐19). Cell Death Differ. 2020;27:3196‐3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang T, Zhang Y‐D, Zhou J‐S, et al. Angiotensin‐(1‐7) is reduced and inversely correlates with tau hyperphosphorylation in animal models of Alzheimer's disease. Mol Neurobiol. 2016;53:2489‐2497. [DOI] [PubMed] [Google Scholar]
- 22. Kehoe PG, Wong S, Al Mulhim N, Palmer LE, Miners JS. Angiotensin‐converting enzyme 2 is reduced in Alzheimer's disease in association with increasing amyloid‐beta and tau pathology. Alzheimers Res Ther. 2016;8:50. 10.1186/s13195-016-0217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matecki S, Dridi H, Jung B, et al. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc Natl Acad Sci USA. 2016;113:9069‐9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lacampagne A, Liu X, Reiken S, et al. Post‐translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease‐like pathologies and cognitive deficits. Acta Neuropathol. 2017;134:749‐767. [DOI] [PubMed] [Google Scholar]
- 25. Dridi H, Liu X, Yuan Q, et al. Role of defective calcium regulation in cardiorespiratory dysfunction in Huntington's disease. JCI Insight. 2020;5:140614. 10.1172/jci.insight.140614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X, Betzenhauser MJ, Reiken S, et al. Role of leaky neuronal ryanodine receptors in stress‐induced cognitive dysfunction. Cell. 2012;150:1055‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dridi H, Kushnir A, Zalk R, Yuan Q, Melville Z, Marks AR. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat Rev Cardiol. 2020;17:732‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dridi H, Wu W, Reiken SR, et al. Ryanodine receptor remodeling in cardiomyopathy and muscular dystrophy caused by lamin A/C gene mutation. Hum Mol Genet. 2021;29:3919‐3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie W, Santulli G, Reiken SR, et al. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Waning DL, Mohammad KS, Reiken S, et al. Excess TGF‐beta mediates muscle weakness associated with bone metastases in mice. Nat Med. 2015;21:1262‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao X, Nicholls JM, Chen Y‐G. Severe acute respiratory syndrome‐associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor‐beta signaling. J Biol Chem. 2008;283:3272‐3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saleh J, Peyssonnaux C, Singh KK, Edeas M. Mitochondria and microbiota dysfunction in COVID‐19 pathogenesis. Mitochondrion. 2020;54:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Datta D, Leslie SN, Wang M, et al. Age‐related calcium dysregulation linked with tau pathology and impaired cognition in non‐human primates. Alzheimers Dement. 2021;17:920‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang AC, Kern F, Losada PM, et al. Dysregulation of brain and choroid plexus cell types in severe COVID‐19. Nature. 2021;595:565‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khalil N. Regulation of the effects of TGF‐beta 1 by activation of latent TGF‐beta 1 and differential expression of TGF‐beta receptors (T beta R‐I and T beta R‐II) in idiopathic pulmonary fibrosis. Thorax. 2001;56:907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arnsten AFT, Wang M. The evolutionary expansion of mGluR3‐NAAG‐GCPII signaling: relevance to human intelligence and cognitive disorders. Am J Psychiatry. 2020;177:1103‐1106. [DOI] [PubMed] [Google Scholar]
- 37. Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol. 1991;111:293‐301. [DOI] [PubMed] [Google Scholar]
- 38. Bastianelli E. Distribution of calcium‐binding proteins in the cerebellum. Cerebellum. 2003;2:242‐262. [DOI] [PubMed] [Google Scholar]
- 39. Maguire‐Zeiss KA, Li ZW, Shimodal LMN, Hamill RW. Calbindin D28k mRNA in hippocampus, superior temporal gyrus and cerebellum: comparison between control and Alzheimer disease subjects. Brain Res Mol Brain Res. 1995;30:362‐366. [DOI] [PubMed] [Google Scholar]
- 40. Kondo H, Tanaka K, Hashikawa T, Jones EG. Neurochemical gradients along monkey sensory cortical pathways: calbindin‐immunoreactive pyramidal neurons in layers II and III. Eur J Neurosci. 1999;11:4197‐4203. [DOI] [PubMed] [Google Scholar]
- 41. Miguel JC, Perez SE, Malek‐Ahmadi M, Mufson EJ. Cerebellar calcium‐binding protein and neurotrophin receptor defects in down syndrome and alzheimer's disease. Front Aging Neurosci. 2021;13:645334 10.3389/fnagi.2021.645334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Erausquin GA, Snyder H, Carrillo M, Hosseini AA, Brugha TS, Seshadri S. The chronic neuropsychiatric sequelae of COVID‐19: the need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 2021;17:1056‐1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dekosky ST, Kochanek PM, Valadka AB, et al. Blood biomarkers for detection of brain injury in COVID‐19 patients. J Neurotrauma. 2021;38:1‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ermis U, Rust MI, Bungenberg J, et al. Neurological symptoms in COVID‐19: a cross‐sectional monocentric study of hospitalized patients. Neurol Res Pract. 2021;3:17. 10.1186/s42466-021-00116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ng Kee Kwong KC, Mehta PR, Shukla G, Mehta AR. COVID‐19, SARS and MERS: a neurological perspective. J Clin Neurosci. 2020;77:13‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeng X, Blancett CD, Koistinen KA, et al. Identification and pathological characterization of persistent asymptomatic Ebola virus infection in rhesus monkeys. Nat Microbiol. 2017;2:17113. 10.1038/nmicrobiol.2017.113. [DOI] [PubMed] [Google Scholar]
- 47. Billioux BJ, Smith B, Nath A. Neurological complications of ebola virus infection. Neurotherapeutics. 2016;13:461‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cai Y, Kim DJ, Takahashi T, et al. Kynurenic acid may underlie sex‐specific immune responses to COVID‐19. Sci Signal. 2021;14. 10.1126/scisignal.abf8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thomas T, Stefanoni D, Reisz JA, et al. COVID‐19 infection alters kynurenine and fatty acid metabolism, correlating with IL‐6 levels and renal status. JCI Insight. 2020;5. 10.1172/jci.insight.140327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lawler NG, Gray N, Kimhofer T, et al. Systemic perturbations in amine and kynurenine metabolism associated with acute SARS‐CoV‐2 infection and inflammatory cytokine responses. J Proteome Res. 2021;20:2796‐2811. [DOI] [PubMed] [Google Scholar]
- 51. Oka M, Fujisaki N, Maruko‐Otake A, et al. Ca2+/calmodulin‐dependent protein kinase II promotes neurodegeneration caused by tau phosphorylated at Ser262/356 in a transgenic Drosophila model of tauopathy. J Biochem. 2017;162:335‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang J‐Z, Grundke‐Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32. 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kushnir A, Todd JJ, Witherspoon JW, et al. Intracellular calcium leak as a therapeutic target for RYR1‐related myopathies. Acta Neuropathol. 2020;139:1089‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365‐376. [DOI] [PubMed] [Google Scholar]
- 56. Shan J, Betzenhauser MJ, Kushnir A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta‐adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375‐4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bellinger AM, Reiken S, Carlson C, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Braga L, Ali H, Secco I, et al. Drugs that inhibit TMEM16 proteins block SARS‐CoV‐2 spike‐induced syncytia. Nature. 2021;594:88‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Danta CC. SARS‐CoV‐2, Hypoxia, and Calcium Signaling: the Consequences and Therapeutic Options. ACS Pharmacol Transl Sci. 2021;4:400‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimers Dement. 2021;17:115‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wehrens XHT, Lehnart SE, Reiken SR, et al. Protection from cardiac arrhythmia through ryanodine receptor‐stabilizing protein calstabin2. Science. 2004;304:292‐296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information