

Abstract
Introduction
Neurological complications among hospitalized COVID‐19 patients may be associated with elevated neurodegenerative biomarkers.
Methods
Among hospitalized COVID‐19 patients without a history of dementia (N = 251), we compared serum total tau (t‐tau), phosphorylated tau‐181 (p‐tau181), glial fibrillary acidic protein (GFAP), neurofilament light chain (NfL), ubiquitin carboxy‐terminal hydrolase L1 (UCHL1), and amyloid beta (Aβ40,42) between patients with or without encephalopathy, in‐hospital death versus survival, and discharge home versus other dispositions. COVID‐19 patient biomarker levels were also compared to non‐COVID cognitively normal, mild cognitive impairment (MCI), and Alzheimer's disease (AD) dementia controls (N = 161).
Results
Admission t‐tau, p‐tau181, GFAP, and NfL were significantly elevated in patients with encephalopathy and in those who died in‐hospital, while t‐tau, GFAP, and NfL were significantly lower in those discharged home. These markers correlated with severity of COVID illness. NfL, GFAP, and UCHL1 were higher in COVID patients than in non‐COVID controls with MCI or AD.
Discussion
Neurodegenerative biomarkers were elevated to levels observed in AD dementia and associated with encephalopathy and worse outcomes among hospitalized COVID‐19 patients.
Keywords: Alzheimer's disease, biomarker, COVID‐19, glial fibrillary acidic protein, mortality, neurodegeneration, neurofilament light chain, SARS‐CoV‐2, tau
1. INTRODUCTION
Neurological complications, particularly encephalopathy, are common among hospitalized COVID‐19 patients, 1 , 2 , 3 and long‐term cognitive abnormalities persist in nearly 50% of hospital survivors. 4 However, the mechanisms underpinning cognitive dysfunction in acute and post‐acute COVID‐19 patients are not well understood. One possibility is that protracted hypoxia and the hyperinflammatory state encountered in acute, severe COVID‐19 may lead to neuronal and glial cell injury, which could be measured in the blood using sensitive digital enzyme‐linked immunosorbent assay or single molecule array technology (SIMOA).
In this study, we hypothesized that blood biomarkers of neuronal and glial injury would be elevated in hospitalized COVID‐19 patients with clinical evidence of new onset of cognitive dysfunction (specifically toxic‐metabolic encephalopathy [TME]), and that elevated neurodegenerative biomarkers would be associated with a higher risk of in‐hospital death and reduced rates of discharge home. We further aimed to compare neurodegenerative biomarker levels in hospitalized COVID patients to non‐COVID controls with varying degrees of cognitive impairment (normal, mild cognitive impairment [MCI], or Alzheimer's disease [AD]) to gauge their degree of brain injury.
We chose to assess neurofilament light chain (NfL), a cytoskeletal intermediate filament protein integral to axons in the central and peripheral nervous system, which has been noted to be elevated in patients with COVID‐19, 5 , 6 , 7 , 8 , 9 , 10 as well as glial fibrillary acidic protein (GFAP), which is a specific indicator of glial/astrocyte injury, and ubiquitin carboxy‐terminal hydrolase L1 (UCHL1) and total tau (t‐tau), which are a neuron‐specific proteins. Plasma t‐tau, NfL, and GFAP have also been reported to be elevated in AD. 11 , 12 , 13 Additionally, we evaluated phosphorylated tau‐181 (p‐tau181), and amyloid beta (Aβ)40 and 42, which are more specific biomarkers for AD‐type pathology. 11 , 14 , 15 , 16 Notably, UCHL1, 17 , 18 GFAP, 19 tau, 19 and NfL 20 , 21 are also elevated after blood–brain barrier (BBB) disruption, which has been documented in neuropathological studies of COVID‐19 decedents 22 as well as in AD.
2. METHODS
2.1. Study design and patient cohort
1. HIGHLIGHTS
Neurodegenerative biomarkers were elevated in hospitalized COVID‐19 patients.
Biomarker levels increased with older age and severity of COVID illness.
Biomarkers were higher in those with encephalopathy and in‐hospital death.
Lower biomarker levels predicted discharge home (vs. other dispositions).
Neurofilament light chain, glial fibrillary acidic protein, and ubiquitin carboxy‐terminal hydrolase L1 levels were as high as levels in non‐COVID Alzheimer's disease controls.
RESEARCH IN CONTEXT
Systematic review: Literature published on PubMed and preprint servers was reviewed. Cognitive effects of SARS‐CoV‐2 have emerged as an area of active research, yet the underpinning mechanisms are not well understood. While some COVID‐19 case series have described elevated neurodegenerative biomarkers, we did not identify any studies that explored a full range of neuronal, glial, axonal, and amyloid beta markers in relation to in‐hospital neurological complications or outcomes, nor did we identify any studies that compared levels to those in non‐COVID mild cognitive impairment or Alzheimer's disease patients.
Interpretation: Our findings suggest that significant neurodegenerative injury may be occurring in hospitalized COVID‐19 patients, as is associated with encephalopathy and poor discharge disposition.
Future directions: Studies tracking trajectories of neurodegenerative biomarkers over time, correlations with neuroimaging evidence of neurodegenerative disease, and the association of biomarker levels with long‐term cognitive outcomes may provide insights into underlying mechanisms of cognitive dysfunction among COVID‐19 survivors.
We conducted a retrospective analysis of COVID‐19 patients who were prospectively enrolled in the Study of Neurologic and Psychiatric Events in Acute COVID‐19 (SNaP Acute COVID) study, 3 and had serum biospecimens collected and banked during their index hospitalization for COVID‐19. Briefly, SNaP Acute COVID is a prospective study of consecutive COVID‐19 patients hospitalized at four New York City–area hospitals within the same hospital system between March 10, 2020, and May 20, 2020. Patients were prospectively evaluated by a team of neurologists for development of new neurological disorders during acute COVID‐19 hospitalization. A total of 4491 patients were included in SNaP Acute COVID (N = 606 with new neurological events and N = 3885 without new neurological events). 3 All COVID‐19 patients capable of consent were approached for blood banking at admission. For our current study, inclusion criteria were hospital admission, reverse‐transcriptase polymerase‐chain‐reaction (RT‐PCR) positive SARS‐CoV‐2 infection from nasopharyngeal sampling, and consent to store blood biospecimens in the New York University (NYU) Center for Biospecimen Research and Development biorepository for use in experimental analyses. Exclusion criteria were negative or missing SARS‐CoV‐2 RT‐PCR test, evaluation in an outpatient or emergency department setting only, history of dementia or cognitive impairment (including but not limited to: pre‐existing diagnoses of MCI, AD, vascular dementia, Lewy body/Parkinson's‐related dementia, progressive supranuclear palsy, multiple system atrophy, corticobasal degeneration, frontotemporal dementia, normal pressure hydrocephalus or Creutzfeld‐Jakob disease), and inadequate biospecimens available for analyses.
Control populations of non‐COVID‐19 subjects with blood samples banked prior to January 1, 2020 (prior to the first reported cases of SARS‐CoV‐2 infection in New York City) were selected from the NYU Alzheimer's Disease Research Center (ADRC) Clinical Core cohort. Three control populations were included: cognitively normal (defined by normal Uniform Data Set version 3 [UDS‐3] psychometric testing and Clinical Dementia Rating [CDR] score of 0), MCI (defined by abnormal UDS‐3 psychometric testing and CDR = 0.5) and dementia due to AD (defined as abnormal UDS‐3 psychometric testing, CDR ≥ 1, and clinical phenotype/biomarker suggestive of primary AD and not other dementia subtypes). 23 , 24
2.2. Blood banking process
After obtaining consent, leftover blood samples drawn on hospital day 0 during index COVID‐19 hospitalization were banked for future research. Serum samples were collected from COVID‐19 patients in gold‐ or red‐top tubes and kept at room temperature for 30 to 45 minutes after blood draw to assess for clot formation prior to centrifugation at 4°C 2000 X g for 10 minutes.
Non‐COVID control plasma biospecimens were collected at the NYU ADRC in ethylene diamine tetraacetic acid tubes and kept on wet ice until centrifugation at 4°C 2000X g for 10 minutes. Both serum and plasma samples were aliquoted 0.200 mL into 1 mL polypropylene tubes and stored in –80°C freezers equipped with 24‐hour alarm systems for detecting temperature excursions. Because only plasma control specimens were available, we evaluated only NfL, GFAP, and UCHL1 levels for comparison to the COVID‐19 samples. These three biomarkers been shown to have equivalent levels in serum and plasma samples, whereas significant differences have been identified in t‐tau and Aβ levels between serum and plasma. 25 , 26
2.3. Neurodegenerative biomarker analyses
Serum and plasma biomarker assays were conducted by the Biomarker Core of the NYU ADR Center using the SIMOA SR‐X Analyzer (Quanterix Corporation). T‐tau, NfL, GFAP, and UCHL1 were measured using the Simoa Neurology 4‐plex A kit. The Simoa pTau‐181 Advantage Kit was used to measure p‐tau181. Aβ40 and Aβ42 were measured using the Simoa Neurology 3‐plex A kit. Following the manufacturer's recommendation for handling and analyzing serum samples, each Simoa kit's run included: an 8‐point calibration curve for each marker and two internal controls.
To avoid batching effects, experiments were predesigned including a similar number of individuals from all study groups once sufficient samples were collected. Investigators running the experiments were blinded to study group assignments. The samples were thawed once, in ice, before each run, and centrifuged 5 minutes at 10,000 g before being manually diluted 1:4 in the 96‐well plates with the appropriate buffer included in Simoa kits. Each sample was run in duplicate and the average value of both runs was used for analyses.
2.4. Other laboratory data
Blood inflammatory markers collected as part of clinical care during the hospital encounter were abstracted from the medical record including interleukin‐6 (IL‐6), C‐reactive protein (CRP), D‐dimer, and ferritin levels. The values obtained at hospital day 0 were used for analyses.
2.5. Neurological diagnoses and severity of illness scales
Neurological diagnoses made during hospitalization (including TME, hypoxic–ischemic encephalopathy, stroke [ischemic or hemorrhagic], seizure, neuropathy, myopathy, movement disorder, encephalitis/meningitis, myelopathy, myelitis) followed established criteria and were coded for COVID‐19 patients found to have a new neurological complication (excluding recrudescence or worsening of old neurological deficits) as diagnosed by in‐hospital neurology teams. TME was coded for patients with new changes in mental status in the absence of focal neurological deficits (except in cases of hypo/hyperglycemia), clinical or electrographic seizures, or primary structural brain disease. For patients who had received sedating medications, an adequate washout (4–5 half‐lives) was required for mental status assessment prior to diagnosis with TME. Etiologies of TME included: septic encephalopathy, uremia, hypoxia, hypercapnia, liver failure, electrolyte disturbances, hyper/hypothermia, thyroid dysfunction, nutritional deficiencies, environmental exposures, and acid/base abnormalities, among others. 1 Hypoxic–ischemic encephalopathy (HIE), a subcategory of TME, was defined as a global cerebral insult due to oxygen deprivation to the brain or lack of perfusion to the brain caused by systemic hypoxemia, hypotension, or cardiac arrest. 27 HIE was diagnosed among patients with new neurologic deficits and/or characteristic radiographic findings on head computed tomography or magnetic resonance imaging (MRI) scans. Patients with altered mental status due to another acute neurological diagnosis that could account for the observed exam findings (e.g., stroke, seizure, traumatic brain injury) 28 or abnormal mental status due to sedative medications were excluded from the TME diagnostic category. Subsidiary review of all neurological diagnoses was performed by relevant neurological subspecialists on the study team (e.g., stroke, neurocritical care, epilepsy subspecialists). Patients could be coded for more than one neurological complication.
Demographic data, past medical history, clinical course, and hospital outcomes (mortality rates, discharge disposition, ventilator days, and hospital length of stay) were collected. Severity of illness during hospitalization was assessed using the worst recorded Sequential Organ Failure Assessment (SOFA) score. Past neurological history included: history of ischemic or hemorrhagic stroke, hydrocephalus, brain tumor, headache, seizure, traumatic brain injury, neuropathy, myasthenia gravis, multiple sclerosis, or movement disorder.
2.6. Study outcomes
The primary outcomes were serum levels of t‐tau, p‐tau181, NfL, GFAP, UCHL1, Aβ40, Aβ42, the ratio of Aβ42/Aβ40, and the ratio of p‐tau181/Aβ42 compared between hospitalized COVID‐19 patients who (1) developed TME versus those who did not, (2) died in‐hospital or were discharged to hospice versus those who survived to discharge, and (3) were discharged home versus other discharge dispositions (in‐hospital death/hospice, discharge to a nursing home, long‐term acute care facility, acute or subacute rehabilitation facility). Secondary outcomes included the comparison of serum biomarker levels among COVID‐19 patients to plasma biomarker levels of NfL, GFAP, and UCHL1 in non‐COVID controls with normal cognition, MCI, or AD.
2.7. Standard protocol approvals and patient consents
This study was approved by the NYU Grossman School of Medicine Institutional Review Board. All patients or their surrogates provided consent for participation in blood banking.
2.8. Statistical analyses
Based on the number of subjects in our sample for whom we were able to measure the biomarkers, we had approximately 80% power to detect (with a two‐sided, 0.05 level t test) the following effect sizes (noted in Cohen d values, which reflect the difference between the two groups measured in standard deviations) for the biomarkers between those with TME compared to those without TME: d = 0.39 for NfL, UCHL1, GFAP (with 75 with TME and 176 without TME), d = 0.39 for total tau (73 with TME and 168 without TME), d = 0.50 for Aβ40 (47 with TME and 99 without TME), and d = 0.55 for Aβ42 (39 with TME and 81 without). The actual effect sizes in our data were comparable to these for NfL and UCHL1, but were lower for the other markers, indicating that we had lower power to detect differences in t‐tau, p‐tau181, Aβ40, and Aβ42.
Neurodegenerative and inflammatory laboratory values were reported as median and interquartile ranges (IQR). Specialized nonparametric U‐statistics were used to compare biomarker levels between patients with and without TME, death, or discharge home. This statistic calculates pairwise rankings for comparable pairs, and accounts for the time needed to have the opportunity to be diagnosed with a specific outcome prior to the occurrence of a competing event. This statistic reduces to the Mann Whitney U‐statistic when there is no adjustment for relative timing of diagnosis and length of hospital stay. We used the simple bootstrap (500 repetitions) to calculate the variances of the U‐statistics. Biomarker levels were compared between COVID‐19 patients, and non‐COVID cognitively normal, MCI and AD patients using univariate Mann‐Whitney U non‐parametric tests. We then fit multiple linear regression models to predict neurodegenerative biomarker levels in COVID patients versus COVID negative control groups (cognitively normal, MCI, AD) adjusting for age (tertiles) and sex. Linearity was assessed by partial regression plots and plots of studentized residuals against predicted values. Independence of residuals was confirmed by Durbin‐Watson statistics (≈2 considered acceptable) and homoscedasticity was evaluated by visual inspection of plots of studentized residuals versus unstandardized predicted values. Absence of multicollinearity was confirmed by tolerance values > 0.1. For all models, studentized deleted residuals were confirmed to be < 3 standard deviations, leverage values were < 0.2, and Cook distances were < 1. The assumption of normality was required for all models as assessed by Q‐Q plots. When necessary, non‐normally distributed dependent variables were transformed (e.g., log10 transformation).
Correlations between biomarker levels and demographics, severity of illness measures, and inflammatory laboratory values were assessed using two‐tailed Spearman's rank correlation coefficients.
Using cause‐specific multivariable Cox proportional hazard regression models, we fit the event times of (1) diagnosis of TME (2) in‐hospital death/hospice discharge, and (3) discharge to home. For the outcome of TME, in‐hospital death or hospital discharge (home, skilled nursing facility, acute or subacute rehab, or long‐term acute care hospital [LTACH]) were treated as censoring events; for the outcome of in‐hospital death, any discharge disposition other than death or discharge to hospice was treated as a censoring event; and for the outcome of discharge home, we treated death or discharge to hospice, a skilled nursing facility, acute or subacute rehab or LTACH as censoring events. No patients remained under observation in the hospital at the end of follow‐up for this study. All models were adjusted for confounders, including age, sex, race, history of neurological disease, admission SOFA score, and admission oxygen saturation. Covariates were selected based on known predictors of in‐hospital death, poor discharge disposition, biological plausibility, and bivariate associations within our own data. 1 , 3 , 4 Analyses were conducted using IBM SPSS Statistics for Mac V25 (IBM Corp.) and R studio V1.1.456.
3. RESULTS
A total of 302 patients from the SNaP Acute COVID cohort had banked serum specimens available for analysis. After excluding 51 patients with a history of dementia or cognitive impairment, 251 COVID‐19 patients were included in the analysis (Figure 1). Due to limited sample availability, the number of patients tested for each biomarker varied: NfL, GFAP, and UCHL1 were assayed in N = 246, t‐tau in N = 241, p‐tau181 in N = 157, Aβ40 in N = 146, and Aβ42 in N = 120. A total of 161 controls underwent neurodegenerative biomarker testing (N = 54 cognitively normal, N = 54 MCI, N = 53 AD).
FIGURE 1.
Flowchart of patient inclusion. Aß, amyloid beta; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; pTau, phosphorylated tau; SNaP Acute COVID, Study of Neurologic and Psychiatric Events in Acute COVID‐19; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1
The median age of COVID‐19 patients was 71 years (IQR 60–83) and 63% were male, compared to 71 years (IQR 65–76) and 35% male among non‐COVID cognitively normal controls (P = .997 for age; P = .001 for sex), 77 years (range 70–86) and 20% male among non‐COVID MCI controls (P = .002 for age; P < .001 for sex), and 82 years (range 72–88) and 40% male among non‐COVID AD patients (P < .001 for age; P = .002 for sex; Table 1).
TABLE 1.
Demographics of COVID and control patients
| COVID patients | No cognitive impairment | MCI | Alzheimer's disease | |
|---|---|---|---|---|
| N | 251 | 54 | 54 | 53 |
| Age, (median, IQR) | 71 (60–83) | 71 (65–76) | 77 (70–86) | 82 (72–88) |
| Male sex, (N, %) | 158 (63%) | 19 (35%) | 11 (20%) | 21 (40%) |
| Median age of males, (IQR) | 66 (58–81) | 72 (63–78) | 69 (60–74) | 84 (73–89) |
| Median age of females, (IQR) | 77 (66–87) | 70 (66–76) | 79 (74–86) | 82 (70–88) |
| Sample type | Serum | Plasma | Plasma | Plasma |
Abbreviations: IQR, interquartile range; MCI, mild cognitive impairment.
Among COVID‐19 patients, 31% required mechanical ventilation, 25% died in‐hospital, and 53% were discharged home (Table 2). New neurological events during hospitalization occurred in 120/251 (48%) of patients with the most common diagnoses being TME in 75/120 (63%) and hypoxic/ischemic brain injury in 55/120 (46%). Among patients diagnosed with TME during their hospital stay, the median time from admission to diagnosis of TME was 0 days (IQR 0–3 days), the median time to death among those who died in‐hospital was 11 days (IQR 6–22 days), and the median hospital length of stay was 10 days (IQR 5–20 days).
TABLE 2.
Characteristics of hospitalized COVID‐19 patients (N = 251)
| Characteristic | N (%) or Median (IQR) |
|---|---|
| Hospital course | |
| Intensive care unit vs. non‐ICU unit no./total no. (%) | 78/251 (31%) |
| Intubation no./total no. (%) | 78/251 (31%) |
| Worst SOFA score median (IQR) | 4(3–7) |
| Lowest oxygen saturation (%), median (IQR) | 85% (76‐90%) |
| Lowest mean arterial pressure (mmHg), median (IQR) | 64(52‐72) |
| Acute renal failure no./total no. (%) | 34/251 (14%) |
| New neurological events during hospitalization | |
| Any new neurological event | 120/251 (48%) |
| Toxic metabolic encephalopathy no./total no. (%) | 75/251 (30%) |
| Hypoxic/ischemic brain injury no./total no. (%) | 55/251 (22%) |
| Stroke (any type) no./total no. (%) | 16/251 (6%) |
| Ischemic/TIA | 13/251 (5%) |
| Intracerebral/intraventricular hemorrhage | 2/251 (1%) |
| Spontaneous subarachnoid hemorrhage | 1/251 (0.4%) |
| Seizure (clinical or electrographic) no./total no. (%) | 10/251 (4%) |
| Movement disorder no./total no. (%) | 3/251 (1%) |
| Neuropathy no./total no. (%) | 9/251 (4%) |
| Myopathy no./total no. (%) | 6/251 (2%) |
| Guillain–Barre syndrome no./total no. (%) | 0 |
| Encephalitis/meningiti‐ no./total no. (%) | 0 |
| Myelopathy/myelitis no./total no. (%) | 0 |
| Admission neurodegenerative biomarkers | |
| Serum tau, pg/mL (N = 241), median (IQR) | 0.45 (0.19–0.99) |
| Serum p‐tau181, pg/mL (N = 157), median (IQR) | 1.3 (0.60–2.54) |
| Serum NfL, pg/mL (N = 246), median (IQR) | 73.2 (30.2–180.5) |
| Serum GFAP, pg/mL (N = 246), median (IQR) | 443.5 (191.9–813.9) |
| Serum UCHL1, pg/mL (N = 246), median (IQR) | 43.0 (24.9–77.3) |
| Serum Aβ40, pg/mL (N = 146) median (IQR) | 13.8 (3.5–60.8) |
| Serum Aβ 42, pg/mL (N = 120) median (IQR) | 2.8 (1.0–6.4) |
| Aβ40/Aβ40, (N = 110) median (IQR) | 0.12 (0.08–0.25) |
| p‐tau181/Aβ 42, (N = 83) median (IQR) | 0.46 (0.25–1.18) |
| Inflammatory biomarkers | |
| Highest serum IL‐6, pg/mL, median (IQR) | 24(7‐82) |
| Highest serum C‐reactive protein, mg/L, median (IQR) | 8464 (5352–9758) |
| Highest serum D‐dimer, ng/mL, median (IQR) | 855 (594–979) |
| Highest serum Ferritin, ng/mL, median (IQR) | 922 (650–1631) |
| Hospital outcomes | |
| Died in‐hospital no./total no. (%) | 64/251 (25%) |
| Home no./total no. (%) | 124/234 (53%) |
| Acute rehabilitation facility no./total no. (%) | 6/235 (2%) |
| Nursing home no./total no. (%) | 40/235 (16%) |
| Length of stay median (IQR) | 9.7 (5.4–20.0) |
| Ventilator days median (IQR) | 11.6 (4.4–18.6) |
Abbreviations: Aβ, amyloid beta; ICU, intensive care unit; IL‐6, interleukin‐6; IQR, interquartile range; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; p‐tau, phosphorylated tau; SOFA, Sequential Organ Failure Assessment; TIA, transient ischemic attack; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1.
Elevations in neurodegenerative biomarkers in COVID‐19 patients were correlated with older age, and increased severity of illness (requirement of mechanical ventilation, worse SOFA scores, lower O2 saturations, lower mean arterial blood pressures; Table 3). Levels of t‐tau, p‐tau181, and NfL correlated most strongly with severity of COVID illness (green color on heat map indicates increasing Spearman rho correlation coefficients). Significant correlations were also identified between p‐tau181, NfL, GFAP, and elevated admission D‐dimer levels, though correlations were not observed with other inflammatory markers sampled at admission (IL‐6, CRP, or ferritin; Table 3).
TABLE 3.
Heat map of Spearman's correlation coefficients (95% confidence intervals) among neurodegenerative biomarkers and demographics, severity of illness, and inflammatory markers among hospitalized COVID‐19 patients
| Tau N = 241 | p‐tau181 N = 157 | NfL N = 246 | GFAP N = 246 | UCHL1 N = 246 | Aβ 40 N = 146 | Aβ42 N = 120 | |
|---|---|---|---|---|---|---|---|
| Demographics | |||||||
| Age |
0.213 (0.09–0.33) |
0.367 (0.22–0.50) |
0.273 (0.15–0.39) |
0.435 (0.32–0.54) |
0.084 (–0.04–0.21) |
0.294 (0.13–0.44) |
0.14 (–0.04–0.31) |
| Male sex |
0.024 (–0.10–0.15) |
0.068 (–0.09–0.22) |
0.031 (–0.09–0.16) |
0.058 (–0.07–0.18) |
0.124 (0.00–0.25) |
0.044 (–0.12–0.21) |
0.079 (–0.10–0.25) |
| Race (White vs. other) |
0.149 (0.02–0.27) |
0.147 (–0.01–0.30) |
0.058 (–0.07–0.18) |
0.129 (0.00–0.25) |
0.061 (–0.06–0.18) |
0.096 (–0.07–0.25) |
0.104 (–0.08–0.28) |
| Severity of COVID‐19 illness | |||||||
| Intubation |
0.232 (0.11–0.35) |
0.054 (–0.10–0.21) |
0.276 (0.15–0.39) |
0.108 (–0.02–0.23) |
0.186 (0.06–0.30) |
0.005 (–0.16–0.17) |
0.021 (–0.16–0.20) |
| Worse SOFA score |
0.345 (0.23–0.45) |
0.261 (0.11–0.40) |
0.461 (0.35–0.56) |
0.25 (0.13–0.37) |
0.313 (0.19–0.42) |
0.13 (–0.03–0.29) |
0.138 (–0.04–0.31) |
| Lowest O2 saturation |
0.138 (0.01–0.26) |
0.039 (–0.12–0.19) |
0.176 (0.056‐0.30) |
0.117 (–0.01–0.24) |
0.126 (0.00–0.25) |
0.079 (–0.08–0.24) |
0.033 (–0.15–0.21) |
| Lowest mean arterial blood pressure |
0.312 (0.19–0.42) |
0.256 (0.10—0.40) |
0.385 (0.27–0.49) |
0.178 (0.05–0.30) |
0.271 (0.15–0.39) |
0.075 (–0.09–0.23) |
0.065 (–0.12–0.24) |
| Hypoxic ischemic brain injury |
0.177 (0.05‐0.30) |
0.264 (0.11‐0.41) |
0.206 (0.08‐0.32) |
0.133 (0.01‐0.25) |
0.125 (0.00‐0.25) |
0.044 (‐0.12‐0.21) |
0.034 (‐0.15‐0.21) |
| Ventilator days |
0.215 (0.09–0.33) |
0.279 (0.12–0.42) |
0.085 (–0.04–0.21) |
0.141 (0.02–0.26) |
0.099 (–0.03–0.22) |
0.586 (0.46–0.69) |
0.352 (0.18–0.50) |
| LOS |
0.135 (0.01–0.26) |
0.046 (–0.11–0.20) |
0.291 (0.17–0.40) |
0.101 (–0.02–0.22) |
0.193 (0.07–0.31) |
0.046 (–0.12–0.21) |
0.009 (–0.17–0.19) |
| Inflammatory Markers | |||||||
| Admission IL‐6 |
–0.031 (–0.16–0.10) |
0.026 (–0.13–0.18) |
0.069 (–0.06–0.19) |
0.003 (–0.12–0.13) |
0.038 (–0.09–0.16) |
0.144 (–0.02–0.30) |
0.022 (–0.16–0.20) |
| Admission CRP |
0.006 (–0.12–0.13) |
–0.017 (–0.17–0.14) |
0.044 (–0.08–0.17) |
–0.059 (–0.18–0.07) |
0.003 (–0.12–0.13) |
–0.096 (–0.25–0.07) |
0.056 (–0.12–0.23) |
| Admission ferritin |
–0.015 (–0.14–0.11) |
0.026 (–0.13–0.18) |
0.023 (–0.10–0.15) |
–0.005 (–0.13–0.12) |
0.002 (–0.12–0.13) |
0.113 (–0.05–0.27) |
0.02 (–0.16–0.20) |
| Admission D‐dimer |
–0.022 (–0.15–0.10) |
0.188 (0.03–0.34) |
0.167 (0.04–0.29) |
0.139 (0.01–0.26) |
0.035 (–0.09–0.16) |
–0.026 (–0.19–0.14) |
–0.074 (–0.25–0.11) |
Notes: Green color signifies stronger correlation and red signifies weaker. Bold indicates P < .05.
Abbreviations: Aβ, amyloid beta; CRP, C‐reactive protein; GFAP, glial fibrillary acidic protein; IL‐6, interleukin‐6; LOS, length of stay; Max, maximum recorded during hospitalization; NfL, neurofilament light chain; O2, oxygen; p‐tau, phosphorylated tau; SOFA, Sequential Organ Failure Assessment; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1.
Compared to COVID‐19 patients without any new neurological events, those with new neurological events during hospitalization had elevations in t‐tau (median 0.56 [95% confidence interval (CI) 0.45–0.79] vs. 0.34 pg/mL [95% CI 0.26–0.45], P = .009), p‐tau181 (median 1.58 [95% CI 1.08–2.03] vs. 1.11 [95% CI 0.86–1.43] pg/mL, P = .042), NfL (median 100.7 [95% CI 73.7–136.3] vs. 61.8 [95% CI 47.9–73.2] pg/mL, P = .006), and UCHL1 (median 47.9 [95% CI 43.2–64.7] vs. 36.8 [95% CI 29.9–43.9] pg/mL, P = .002) levels. Neurodegenerative biomarker levels for t‐tau, p‐tau181, GFAP, and NfL were even higher among COVID‐19 patients with TME compared to those without TME (Figure 2). Similarly, patients who died in‐hospital had significant elevations in these biomarkers compared to those who survived, and patients who were discharged home had significantly lower levels than patients with other discharge dispositions. Aβ40 and Aβ42 did not have clear associations with TME, in‐hospital death, or discharge home, though the ratio of p‐tau181/Aβ42 was significantly associated with TME and in‐hospital death, and Aβ42/40 was associated with discharge home.
FIGURE 2.
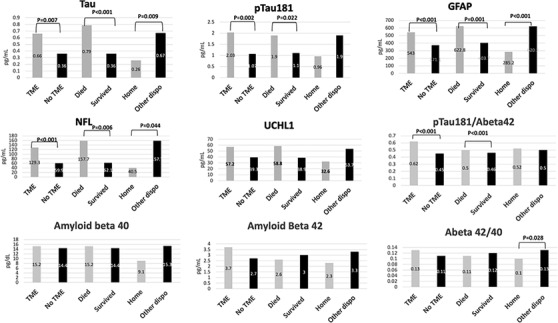
Serum neurodegenerative biomarkers in hospitalized COVID‐19 patients (N = 251) with and without toxic metabolic encephalopathy (TME), in‐hospital death versus survival, and discharge home versus other discharge dispositions. GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; pTau, phosphorylated tau; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1
In multivariable Cox regression analysis adjusting for age, sex, race, history of neurological disease, admission SOFA score, and admission oxygen saturation, TME was significantly associated with increased admission p‐tau181 (hazard ratio [HR] per 10 pg/mL increase 1.86, 95% CI 1.18–2.94, P = .007) and UCHL1 (HR 1.01, 95% CI 1.00–1.02, P = .037). In‐hospital death was associated with elevated GFAP (HR 1.004, 95% CI 1.00–1.01), P = .027) and elevated p‐tau181/Aβ42 (HR 1.05, 95% CI 1.01–1.08, P = .045). Higher levels of t‐tau (HR 0.02, 95% 0.001–0.37, P = .008), NFL (HR 0.94, 95% CI 0.91–0.97, P < .001), and GFAP (HR 0.99, 95% CI 0.99–1.000, P = .012) were associated with lower rates of discharge home, while higher serum levels of Aβ40 (HR 1.03, 95% CI 1.00–1.05, P = .028) were associated with increased rates of discharge home (Table 4).
TABLE 4.
Multivariable Cox proportional hazard ratios among COVID‐19 patients for each measured biomarker and the outcomes of Neurological events, toxic metabolic encephalopathy (TME), in‐hospital death, and discharge home
| Biomarker | N (%) and hazard ratio of any new neuro complicationper 10 pg/mL change in biomarker*HR (95% OR) P | N (%) and hazard ratio of TMEper 10 pg/mL change in biomarker*HR (95% OR) P | N (%) and hazard ratio of deathper 10 pg/mL change in biomarker*HR (95% OR) P | N (%) and hazard ratio of discharge home per 10 pg/mL change in biomarker*HR (95% OR) P |
|---|---|---|---|---|
|
Tau (N = 241) |
115/241 (48%) 0.98 (0.91–1.06) P = .631 |
73/241 (30%) 0.96 (0.81–1.14) P = .671 |
62/241 (26%) 1.02 (0.99–1.04) P = .166 |
116/222 (52%) 0.02 (0.001–0.37) P = .008 |
|
p‐tau181 (N = 157) |
74/157 (47%) 1.63 (0.99–2.67) P = .056 |
49/157 (31%) 1.86 (1.18–2.94) P = .007 |
46/157 (29%) 1.02 (0.50–2.08) P = .953 |
69/146 (47%) 0.49 (0.22–1.09) P = .080 |
|
NfL (N = 246) |
117/246 (48%) 1.002 (1.000–1.01) P = .418 |
73/246 (30%) 1.004 (1.000–1.01) P = .067 |
62/246 (25%) 1.001 (0.99–1.01) P = .789 |
121/227 (53%) 0.94 (0.91–0.97) P < .001 |
|
GFAP (N = 246) |
118/246 (48%) 1.003 (1.000–1.01) P = .061 |
73/246 (30%) 1.001 (1.000–1.01) P = .755 |
61/246 (25%) 1.004 (1.000–1.01) P = .027 |
121/227 (53%) 0.99 (0.99–1.00) P = .012 |
|
UCHL1 (N = 246) |
117/246 (48%) 1.01 (1.00–1.02) P = .118 |
73/246 (30%) 1.01 (1.00–1.02) P = .037 |
62/246 (25%) 1.01 (1.00–1.02) P = .140 |
121/227 (53%) 0.98 (0.96–1.01) P = .159 |
|
Aβ40 (N = 146) |
69/146 (47%) 1.001 (0.96–1.03) P = .811 |
47/146 (32%) 0.97 (0.91–1.02) P = .242 |
37/146 (25%) 0.97 (0.90–1.05) P = .873 |
62/134 (46%) 1.03 (1.00–1.05) P = .028 |
|
Aβ42 (N = 120) |
53/120 (44%) 0.83 (0.52–1.32) P = .423 |
39/120 (33%) 0.69 (0.38–1.27) P = .236 |
32/120 (27%) 1.03 (0.71–1.49) P = .984 |
51/109 (47%) 0.95 (0.73–1.24) P = .736 |
|
Aβ42/Aβ40* (N = 110) |
49/110 (45%) 0.43 (0.18–1.57) P = .200 |
35/110 (32%) 0.20 (0.02–1.87) P = .157 |
31/110 (28%) 0.90 (0.29–2.78) P = .860 |
42/99 (42%) 0.75 (0.42–1.33) P = .482 |
|
p‐tau181/Aβ42* (N = 83) |
32/83 (39%) 1.12 (0.95‐1.32) P = .171 |
26/83 (31%) 1.03 (0.99–1.056) P = .086 |
22/83 (27%) 1.05 (1.01–1.08) P = .045 |
36/76 (47%) 0.87 (0.71–1.05) P = .144 |
Notes: Adjusted for age, sex, race, past neurological disease, admission oxygen saturation, admission SOFA score.
For Aβ42/Aβ40 and Ptau181/Aβ42 hazard ratios represent a 1 unit change in ratio.
Abbreviations: Aβ, amyloid beta; GFAP, glial fibrillary acidic protein; HR, hazard ratio; NfL, neurofilament light chain; OR, odds ratio; p‐tau, phosphorylated tau; SOFA, Sequential Organ Failure Assessment; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1.
Compared to non‐COVID controls, after adjusting for age and sex in multivariable linear regression models, COVID‐19 patients had significantly higher NfL and GFAP levels than non‐COVID AD, MCI, and normal controls, while UCHL1 levels were significantly higher in COVID patients compared to MCI and normal subjects, and were similar to levels observed in AD subjects (Figure 3, Table S1 in supporting information).
FIGURE 3.
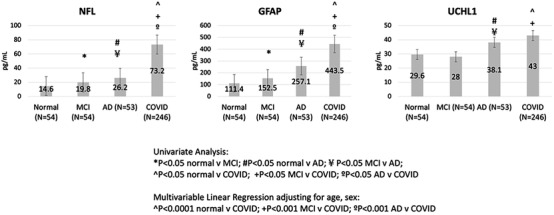
Plasma neurodegenerative biomarkers in controls (N = 161 no cognitive impairment, mild cognitive impairment, and AD dementia patients) and serum biomarker levels in hospitalized COVID patients (N = 251). NfL, GFAP, and UCHL1 levels were significantly higher in COVID patients compared to no cognitive impairment, and MCI patients; and NfL and GFAP were additionally significantly higher than AD patients, after adjusting for age and sex differences between groups. Abeta, amyloid beta; AD, Alzheimer's disease; GFAP, glial fibrillary acidic protein; MCI, mild cognitive impairment; NfL, neurofilament light chain; pTau, phosphorylated tau; UCHL1, ubiquitin carboxyl‐terminal hydrolase isozyme L1
4. DISCUSSION
In this study, we identified significant elevations in blood biomarkers of neuronal and glial degeneration among hospitalized COVID‐19 patients with clinical signs of neurological injury, specifically TME. Indeed, higher admission levels of neuronal degeneration markers p‐tau181 and UCHL1 were significantly associated with TME, even after adjusting for age, sex, race, prior neurological disease, and severity of COVID‐19 illness. Similarly, in multivariable analyses, elevations in total tau, NfL, and GFAP, in particular, were associated with reduced likelihood of discharge home. Furthermore, we found that levels of NfL, GFAP, and UCHL1 were as high as, or significantly higher than, those observed in non‐COVID patients with AD, indicating a profound neurological insult in these patients. Strengths of this study include the prospective ascertainment of new acute neurological disorders among COVID‐19 patients, inclusion of a variety of both neural and glial degenerative markers and AD‐specific p‐tau181, use of blood rather than cerebrospinal fluid (CSF) biomarkers (which makes this study feasible across a larger number of patients), and comparison to well‐characterized COVID‐negative control groups with discrete levels of cognitive impairment.
We found significant correlations between neurodegenerative biomarkers and the inflammatory marker D‐dimer, which may provide some insight into mechanisms of acute brain injury after SARS‐CoV‐2 infection. Hypoxia and hyperinflammation, both hallmarks of acute COVID‐19, have been linked to the development of AD‐type pathology in non‐COVID populations via upregulation of enzymes in the amyloidogenic pathway and downregulation of proteins that break down Aβ. 29 Hypoxia‐induced tau phosphorylation with corresponding memory deficits has been documented in animal models exposed to hypoxia for 6 hours/day for 1 to 8 weeks 30 and elevations of inflammatory cytokines such as IL‐6 and IL‐1 correlate with cognitive dysfunction and the promotion of amyloid plaque and neurofibrillary tangle pathology in animal models. 31 We have previously found that COVID‐19–related TME is significantly associated with elevations in inflammatory markers, including IL‐6, D‐dimer, CRP, and ferritin. 3 IL‐6, in particular, is known to promote endothelial dysfunction and vascular permeability and may play a role in BBB dysfunction after SARS‐CoV‐2 infection. 32 Neuropathological data among COVID‐19 decedents have revealed evidence of hypoxic injury as well as endothelial inflammation, and BBB disruption 22 , 33 , 34 , 35 , 36 that may be mediated, in part, by a COVID‐related hyperinflammatory state. BBB disruption has also been implicated in AD‐type pathology among non‐COVID patients. 37 Finally, another important association between COVID‐19 and AD is the linkage to the apolipoprotein E ε4 genotype, which is both a marker for increased COVID‐19 severity, 38 , 39 and the most impactful genetic risk factor for late‐onset AD. 40 We did not, however, identify correlates with blood Aβ40 or Aβ42 levels, which may reflect the fact that we were underpowered to detect differences in amyloid biomarkers, or be related to the use of serum (rather than plasma) for these measurements, which is known to increase variability. 25
Elevations in blood and CSF NfL, GFAP, and t‐tau among COVID‐19 patients have been described by others, and in some cases, compared to normal control groups. 5 , 6 , 7 , 9 , 10 , 41 , 42 , 43 , 44 , 45 However, none of these studies explicitly excluded COVID‐19 patients with a baseline history of dementia or cognitive decline, which likely would confound results. Additionally, NfL is not specific to the central nervous system (CNS) and can be elevated in the context of peripheral neuropathy, 46 , 47 , 48 including COVID‐related critical illness neuropathy/myopathy 8 and Guillain‐Barre Syndrome. 48 Similarly, elevated GFAP has been reported in COVID‐19 patients with critical illness neuropathy/myopathy and levels correlate with nerve amplitudes. 8 More AD‐specific biomarkers, such as p‐tau181 have not previously been explored in COVID‐19 patients. In neuropathological studies of plasma p‐tau181 and NfL, both biomarkers accurately distinguish pathology‐confirmed AD from healthy controls, but only p‐tau181 distinguished AD from non‐AD dementia cases and showed specificity for neuritic plaque pathology and Braak stage. 15 , 49 Similarly, p‐tau181 levels escalate progressively with worsening CDR scores and correlate with multiple cognitive domains, while NfL, t‐tau, and Aβ levels do not perform as well. 16 , 50 In non‐COVID patients, increases in CSF and plasma biomarkers of t‐tau, p‐tau, and NfL (comparing pre‐operative to post‐operative levels) have been found to correlate with the incidence and severity of delirium in patients undergoing surgery. 51 , 52 , 53 , 54 Furthermore, elevations in NfL have been associated with worse cognitive and functional outcomes in post‐operative patients and patients with septic encephalopathy. 54 , 55 A large meta‐analysis of non‐COVID survivors of critical illness has also identified associations of delirium with Aβ40, IL‐6, and IL‐1R antagonist. 56 Our study is unique in that we identified elevations across a spectrum of CNS specific markers, including neuronal (t‐tau, UCHL1), and astrocytic/glial markers (GFAP), as well as AD‐related markers (p‐tau181).
There are limitations to this study. First, though we excluded patients with a history of dementia or cognitive decline, it is possible that some COVID‐19 patients may have had preclinical or undiagnosed cognitive impairment. Second, biomarkers were only measured at one time point and we do not have data on trajectories of these markers. Some studies have identified persistent elevations in blood NfL levels for 30 to 50 days in small COVID‐19 cohorts, 9 , 42 while GFAP levels may initially spike and then decline after the acute phase of infection. 42 The association of these biomarkers with formal cognitive testing after the acute phase of COVID‐19 has not been demonstrated and is an active area of research needed to unravel the long‐term cognitive implications of elevated neurodegenerative biomarkers. Third, we did not have inflammatory marker data (e.g., IL‐6, CRP, D‐dimer, fibrinogen) in control patients. Fourth, due to limited sample availability, fewer specimens were available for p‐tau181, Aβ40, or Aβ42 analysis, which may have limited our ability to detect important differences among these biomarkers. Last, we compared serum biomarkers in COVID‐19 patients to plasma levels in non‐COVID controls, because pre‐COVID banked serum specimens were not available in controls. Though NfL, GFAP, and UCHL1 levels are equivalent in serum and plasma, 25 we were unable to compare t‐tau, p‐tau181, or Aβ levels to controls due to differences in specimen type. 25 , 26 Additionally, we did not have cerebrospinal fluid specimens available for biomarker analysis, which may have provided a more accurate assessment of the cerebral milieu.
5. CONCLUSIONS
Serum neuronal, glial, and axonal neurodegenerative biomarkers, including t‐tau, p‐tau181, UCHL1, GFAP, and NfL were significantly elevated in patients with encephalopathy and worse discharge disposition after hospitalization for COVID‐19. These markers correlated with the severity of COVID illness. Furthermore, levels of NfL, GFAP, and UCHL1 in hospitalized COVID patients were similar to, or higher than, levels observed in non‐COVID AD dementia patients. Additional studies tracking trajectories of these biomarkers over time and their association with long‐term cognitive outcomes among COVID‐19 survivors are warranted.
CONFLICTS OF INTEREST
Drs. J.A. Frontera, L. Balcer, and T. Wisniewski received support from NIH/NIA (PI: Wisniewski) COVID‐19 administrative supplement 3P30AG066512‐01, which supported the work of this manuscript. Drs. T. Wisniewski, A. Masurkar, R. Betensky, and A. Vedvyas received funding from P30AG066512‐01, which also supported the work of this manuscript. Dr. Y. Ge also received support from NIH grants RF1 NS11041, R01 NS108491, and R13 AG067684, as well as a grant from the Alzheimer's Association (AARG‐17‐533484) that supported his effort on this manuscript. Support outside the submitted work includes Dr. Frontera served on the advisory boards of NIH/NINDS SHINE and DSMB. Dr. Frontera is a member of these societies: Neurocritical Care Society and the American Neurological Association. Dr. A Boutajangout has received support from the Amylon Therapeutic Company and a grant from the Saudi Arabia Cultural Mission. Dr. A. Masurkar received support from NIH/NIA grants RF1AG072507, R21AG070880, P30AG066512, as well as BrightFocus Foundation A2019602S and AACF‐17‐524288. Dr. Masurkar has given a seminar at Rockefeller University. Dr. Masurkar also serves on the steering committee of the Alzheimer's Disease Cooperative Study (unpaid), the Council of the Alzheimer's Association International Research Grants Program (for which he received free ISTAART membership 2020 & 2021), the editorial advisory board of the Journal of Neuro‐Ophthalmology (unpaid). Dr. R. Betensky has received support from NIH grants 5R01NS094610‐05, 1R25AG067931‐01, 5P30DK040561‐24, 2P01AG036694‐11, and R01DA054990‐01. Dr. Betensky received payment for expert testimony from the attorneys for plaintiffs in Bard vena cava litigation, from Teva for a patent case, from Amarin for a patent case, and from Amazon. Dr. Betensky also is an advisor for the National Cancer Institute Board of Epidemiology and Clinical Sciences (and received consulting fees). Dr. Y. Ge has received support from NIH grants RF1NS11041, R01 NS108491, and R13AG067684, as well as from the Alzheimer's Association grant AARG‐17‐533484. Mr. A. Vedvyas has received support from NIH Grant P30AG066512. Mr. L. Debure has nothing to disclose. A. Moreira has served on the advisory board of the Olympus Corporation and is a member of the Binford Dammin Society of Infectious Disease Pathologists. A. Lewis received support from the ABPN. A. Lewis is a deputy editor of Seminars in Neurology (for which he receives payment) and is a member of Neurodiem Ology. A. Lewis has unpaid positions as Ethics committee chair, in the Neurocritical Care Society Steering committee, the World brain death project, and as a deputy editor of Neurology. Drs. J. Huang, and S. Thawani have nothing to disclose. Dr. S. Galetta has been a consultant for Biogen and Genentech. Other relationships/interests/activities include Dr. T. Wisniewski is chief editor of Frontiers in Aging Neuroscience. Dr. T. Wisniewski was a board member of the NYC chapter of the Alzheimer's Association. Drs. T. Wisniewski and A. Boutajangout hold patents unrelated to this manuscript. Dr. R. Betensky services on safety monitoring boards of Apotex, Reata, Biogen, PTI, and Alexion, as well as being an advisor to Alexion, Apotex, Reata, Biogen, and PTI. Dr. Betensky has given seminar at the University of Vigo.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank the patients and families who participated in this study. This study was funded by a grant from the NIH/NIA (PI: Wisniewski) COVID‐19 administrative supplement 3P30AG066512‐01.
Frontera JA, Boutajangout A, Masurkar AV, et al. Comparison of serum neurodegenerative biomarkers among hospitalized COVID‐19 patients versus non‐COVID subjects with normal cognition, mild cognitive impairment, or Alzheimer's dementia. Alzheimer's Dement. 2022;18:899–910. 10.1002/alz.12556
REFERENCES
- 1. Frontera JA, Melmed K, Fang T, et al. Toxic metabolic encephalopathy in hospitalized patients with COVID‐19. Neurocrit Care. 2021;1‐14. 10.1007/s12028-021-01220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kleineberg NN, Knauss S, Gulke E, et al. Neurological symptoms and complications in predominantly hospitalized COVID‐19 patients: Results of the European multinational LEOSS registry. Eur J Neurol. 2021;28:3925‐3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frontera JA, Sabadia S, Lalchan R, et al. A prospective study of neurologic disorders in hospitalized patients with COVID‐19 in New York City. Neurology. 2021;96(4):e575‐e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frontera JA, Yang D, Lewis A, et al. A prospective study of long‐term outcomes among hospitalized COVID‐19 patients with and without neurological complications. J Neurol Sci. 2021;426:117486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aamodt AH, Hogestol EA, Popperud TH, et al. Blood neurofilament light concentration at admittance: a potential prognostic marker in COVID‐19. J Neurol. 2021;268:3574‐3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ameres M, Brandstetter S, Toncheva AA, et al. Association of neuronal injury blood marker neurofilament light chain with mild‐to‐moderate COVID‐19. J Neurol. 2020;267(12):3476‐3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Lorenzo R, Lore NI, Finardi A, et al. Blood neurofilament light chain and total tau levels at admission predict death in COVID‐19 patients. J Neurol. 2021;268:4436‐4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frithiof R, Rostami E, Kumlien E, et al. Critical illness polyneuropathy, myopathy and neuronal biomarkers in COVID‐19 patients: a prospective study. Clin Neurophysiol. 2021;132(7):1733‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prudencio M, Erben Y, Marquez CP, et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID‐19. Sci Transl Med. 2021;13(602). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Virhammar J, Naas A, Fallmar D, et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID‐19 and associated with neurological symptoms and disease severity. Eur J Neurol. 2021;28:3324‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated Tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78(4):396‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shekhar S, Kumar R, Rai N, et al. Estimation of Tau and phosphorylated Tau181 in serum of Alzheimer's disease and mild cognitive impairment patients. PLoS One. 2016;11(7):e0159099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐beta but not tau pathology in Alzheimer's disease. Brain. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19(5):422‐433. [DOI] [PubMed] [Google Scholar]
- 15. Thijssen EH, La Joie R, Strom A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer's disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20(9):739‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao Z, Wu X, Wu W, et al. Plasma biomarker profiles and the correlation with cognitive function across the clinical spectrum of Alzheimer's disease. Alzheimers Res Ther. 2021;13(1):123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blyth BJ, Farahvar A, He H, et al. Elevated serum ubiquitin carboxy‐terminal hydrolase L1 is associated with abnormal blood‐brain barrier function after traumatic brain injury. J Neurotrauma. 2011;28(12):2453‐2462. [DOI] [PubMed] [Google Scholar]
- 18. Larsen CN, Krantz BA, Wilkinson KD. Substrate specificity of deubiquitinating enzymes: ubiquitin C‐terminal hydrolases. Biochemistry. 1998;37(10):3358‐3368. [DOI] [PubMed] [Google Scholar]
- 19. Bogoslovsky T, Wilson D, Chen Y, et al. Increases of plasma levels of glial fibrillary acidic protein, Tau, and amyloid beta up to 90 days after traumatic brain injury. J Neurotrauma. 2017;34(1):66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernick C, Zetterberg H, Shan G, Banks S, Blennow K. Longitudinal performance of plasma neurofilament light and Tau in professional fighters: the Professional Fighters Brain Health Study. J Neurotrauma. 2018;35(20):2351‐2356. [DOI] [PubMed] [Google Scholar]
- 21. Gao W, Zhang Z, Lv X, et al. Neurofilament light chain level in traumatic brain injury: a system review and meta‐analysis. Medicine (Baltimore). 2020;99(38):e22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee MH, Perl DP, Nair G, et al. Microvascular injury in the brains of patients with Covid‐19. N Engl J Med. 2021;384(5):481‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quanterix; Pages https://www.quanterix.com/wp‐content/uploads/2020/12/N4PA‐Data‐Sheet‐SR‐X.pdf [Accessed August 24, 2021].
- 26. O'Connell GC, Alder ML, Webel AR, Moore SM. Neuro biomarker levels measured with high‐sensitivity digital ELISA differ between serum and plasma. Bioanalysis. 2019;11(22):2087‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Busl KM, Greer DM. Hypoxic‐ischemic brain injury: pathophysiology, neuropathology and mechanisms. NeuroRehabilitation. 2010;26(1):5‐13. [DOI] [PubMed] [Google Scholar]
- 28. Frontera JA. Metabolic encephalopathies in the critical care unit. Continuum (Minneap Minn). 2012;18(3):611‐639. [DOI] [PubMed] [Google Scholar]
- 29. Lall R, Mohammed R, Ojha U. What are the links between hypoxia and Alzheimer's disease?. Neuropsychiatr Dis Treat. 2019;15:1343‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang CE, Yang X, Li L, Sui X, Tian Q, Wei W, et al. Hypoxia‐induced tau phosphorylation and memory deficit in rats. Neurodegener Dis. 2014;14(3):107‐116. [DOI] [PubMed] [Google Scholar]
- 31. Shen XN, Niu LD, Wang YJ, et al. Inflammatory markers in Alzheimer's disease and mild cognitive impairment: a meta‐analysis and systematic review of 170 studies. J Neurol Neurosurg Psychiatry. 2019;90(5):590‐598. [DOI] [PubMed] [Google Scholar]
- 32. Pons S, Fodil S, Azoulay E, Zafrani L. The vascular endothelium: the cornerstone of organ dysfunction in severe SARS‐CoV‐2 infection. Crit Care. 2020;24(1):353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirschenbaum D, Imbach LL, Rushing EJ, et al. Intracerebral endotheliitis and microbleeds are neuropathological features of COVID‐19. Neuropathol Appl Neurobiol. 2021;47(3):454‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanley B, Naresh KN, Roufosse C, et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID‐19: a post‐mortem study. Lancet Microbe. 2020;1(6):e245‐e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pajo AT, Espiritu AI, Apor A, Jamora RDG. Neuropathologic findings of patients with COVID‐19: a systematic review. Neurol Sci. 2021;42(4):1255‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thakur KT, Miller EH, Glendinning MD, et al. COVID‐19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y). 2018;4:575‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuo CL, Pilling LC, Atkins JL, et al. APOE e4 genotype predicts severe COVID‐19 in the UK Biobank community cohort. J Gerontol A Biol Sci Med Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Del Ser T, Fernandez‐Blazquez MA, Valenti M, et al. Residence, clinical features, and genetic risk factors associated with symptoms of COVID‐19 in a cohort of older people in Madrid. Gerontology. 2021:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wisniewski T, Drummond E. Future horizons in Alzheimer's disease research. Prog Mol Biol Transl Sci. 2019;168:223‐241. [DOI] [PubMed] [Google Scholar]
- 41. Eden A, Kanberg N, Gostner J, et al. CSF biomarkers in patients with COVID‐19 and neurologic symptoms: a case series. Neurology. 2021;96(2):e294‐e300. [DOI] [PubMed] [Google Scholar]
- 42. Kanberg N, Ashton NJ, Andersson LM, et al. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID‐19. Neurology. 2020;95(12):e1754‐e9. [DOI] [PubMed] [Google Scholar]
- 43. Ameres M, Brandstetter S, Toncheva AA, et al. Association of neuronal injury blood marker neurofilament light chain with mild‐to‐moderate COVID‐19. J Neurol. 2020;267:3476‐3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pilotto A, Masciocchi S, Volonghi I, et al. SARS‐CoV‐2 encephalitis is a cytokine release syndrome: evidences from cerebrospinal fluid analyses. Clin Infect Dis. 2021;73:e3019‐e3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prudencio M, Erben Y, Marquez C, et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID‐19. Sci Transl Med. 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hayashi T, Nukui T, Piao JL, et al. Serum neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. Brain Behav. 2021;11(5):e02084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Millere E, Rots D, Simren J, et al. Plasma neurofilament light chain as a potential biomarker in Charcot‐Marie‐Tooth disease. Eur J Neurol. 2021;28(3):974‐981. [DOI] [PubMed] [Google Scholar]
- 48. Martin‐Aguilar L, Camps‐Renom P, Lleixa C, et al. Serum neurofilament light chain predicts long‐term prognosis in Guillain‐Barre syndrome patients. J Neurol Neurosurg Psychiatry. 2020;jnnp‐2020‐323899. 10.1136/jnnp-2020-323899 [DOI] [PubMed] [Google Scholar]
- 49. Grothe MJ, Moscoso A, Ashton NJ, et al. Alzheimer's Disease Neuroimaging Initiative . Associations of Fully Automated CSF and Novel Plasma Biomarkers With Alzheimer Disease Neuropathology at Autopsy. Neurology. 2021;97(12):e1229‐e12242. 10.1212/WNL.0000000000012513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen SD, Huang YY, Shen XN, et al. Longitudinal plasma phosphorylated tau 181 tracks disease progression in Alzheimer's disease. Transl Psychiatry. 2021;11(1):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parker M, White M, Casey C, et al. Cohort Analysis of the Association of Delirium Severity with Cerebrospinal Fluid Amyloid‐Tau‐Neurodegeneration Pathologies. J Gerontol A Biol Sci Med Sci. 2021;glab203. 10.1093/gerona/glab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ballweg T, White M, Parker M, et al. Association between plasma tau and postoperative delirium incidence and severity: a prospective observational study. Br J Anaesth. 2021;126(2):458‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saller T, Petzold A, Zetterberg H, et al. A case series on the value of tau and neurofilament protein levels to predict and detect delirium in cardiac surgery patients. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2019;163(3):241‐246. [DOI] [PubMed] [Google Scholar]
- 54. Fong TG, Vasunilashorn SM, Ngo L, et al. Association of Plasma Neurofilament Light with Postoperative Delirium. Ann Neurol. 2020;88(5):984‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ehler J, Petzold A, Wittstock M, et al. The prognostic value of neurofilament levels in patients with sepsis‐associated encephalopathy ‐ A prospective, pilot observational study. PLoS One. 2019;14(1):e0211184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chan CK, Song Y, Greene R, et al. Meta‐analysis of ICU Delirium Biomarkers and Their Alignment With the NIA‐AA Research Framework. Am J Crit Care. 2021;30(4):312‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information