

Abstract
The emergence of SARS‐CoV‐2 variants is a significant concern in developing effective therapeutics and vaccines in the middle of the ongoing COVID‐19 pandemic. Here, we have identified a novel small molecule that inhibited the interactions between SARS‐CoV‐2 spike RBDs and ACE2 by modulating ACE2 without impairing its enzymatic activity necessary for normal physiological functions. Furthermore, the identified compounds suppressed viral infection in cultured cells by inhibiting the entry of ancestral and variant SARS‐CoV‐2. Our study suggests that targeting ACE2 could be a novel therapeutic strategy to inhibit SARS‐CoV‐2 entry into host cells and prevent the development of COVID‐19.
Keywords: ACE2 Inhibition, Allostery, COVID-19, PPI Inhibition, Virus Entry
Targeting the receptor‐binding domain (RBD) of the SARS‐CoV‐2 spike protein could be limited in inhibiting viral entry via the RBD–ACE2 interaction due to the emerging spike mutations. Instead, targeting ACE2 with a small molecule could suppress the entry of ancestral and variant SARS‐CoV‐2 into the host cells, without impairing the enzymatic activity of ACE2, which is necessary for the physiological function of the host cells.
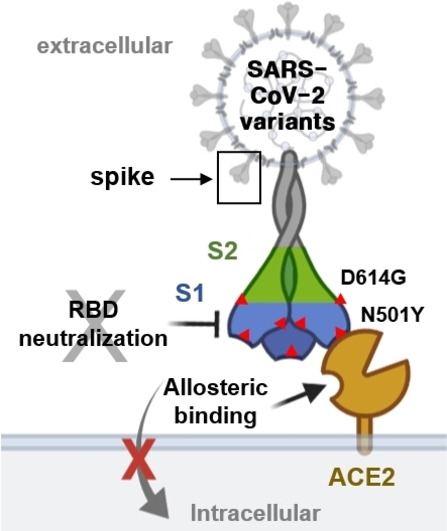
The coronavirus disease 2019 (COVID‐19), caused by the severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2), was declared a pandemic in March 2020 by World Health Organization. Accelerated research into the treatment of COVID‐19 resulted in the rapid development of prophylactic measures and therapeutics, including vaccines. [1] However, the proactive development of therapeutics targeting this virus is urgently needed, especially considering the current (and future) SARS‐CoV‐2 mutation rate. [2]
The prevention of viral entry is an efficient approach to interfere with viral transmission at the initial step of viral infection. [3] SARS‐CoV‐2 relies on the spike, a surface‐located trimeric glycoprotein, to enter host cells. The viral infection starts with the interaction between the receptor‐binding domain (RBD) of the S1 subunit of the spike and the host receptor angiotensin‐converting enzyme‐2 (ACE2), followed by spike cleavage at the S2′ site by host proteases, such as transmembrane serine protease 2 (TMPRSS2). [4] This step primes the fusion between the virus and host cell membrane through the S2 subunit of the spike. Therefore, antibodies, peptides, and small molecules that could inhibit interactions or functions of proteins, such as the spike, ACE2, and TMPRSS2, would be promising therapeutics for treating COVID‐19 (Figure 1).
Figure 1.
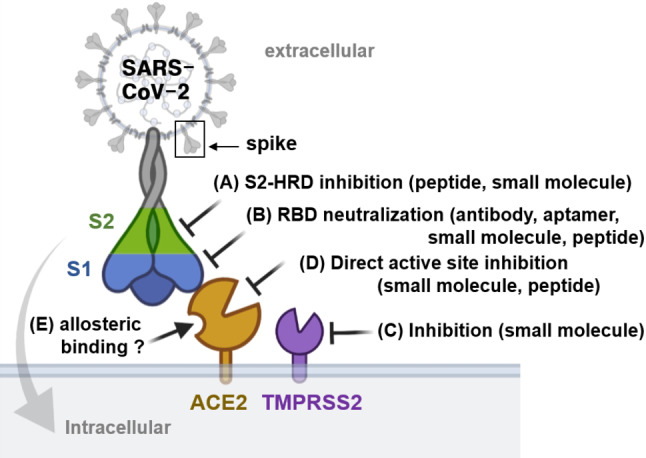
Identification of the main therapeutic targets that would inhibit SARS‐CoV‐2 entry. Image was created using BioRender.com.
Molecular binders for the heptad repeat domain (HRD) of the S2 subunit [5] and the RBD of the S1 subunit [6] within the spike protein have been shown to inhibit SARS‐CoV‐2 infection effectively. However, the emerging spike mutations would require extended verification and optimization of binder efficacy (Figure 1A and B). TMPRSS2 inhibitors can protect cells expressing TMPRSS2 and ACE2 from SARS‐CoV‐2 infection (Figure 1C).[ 4 , 7 ] The approach of targeting ACE2 can be broadly applicable to various species of coronavirus that share the exact viral entry mechanism involving ACE2 as their host receptor (Figure 1D). More importantly, targeting ACE2 would allow us to avoid problems caused by spike mutations.[ 4 , 8 ] However, the inhibition of the ACE2 active site has not been considered a viable therapeutic modality because ACE2 plays a vital role as an endogenous cardiovascular system regulator suppressing hypertension and heart failure, [9] and protects lung injury caused by the acute respiratory distress syndrome. [10] ACE2 has non‐enzymatic functions, such as regulating renal amino acid transport mediated by Collectrin (Tmem27), a homolog of ACE2 sharing 47.8 % sequence homology with ACE2, suggesting that the selective modulation of ACE2 without inhibiting its physiological functions would be critical to prevent possible side effects. On the other hand, biochemical [11] and structural analyses [12] suggested an allosteric binding site in ACE2. Therefore, the inhibition of the ACE2 interaction with the RBD subunit was proposed as a novel strategy for COVID‐19 therapeutics (Figure 1E). [13]
In this study, we explored whether small molecules can inhibit the interaction between ACE2 and the RBD of spike protein. The RBD–ACE2 binding event induces the coronavirus attachment and the subsequent invasion to host cells, which is reported to have a high affinity (K d value 4.7 nM) with a large contact area that mainly consists of polar interactions, suggesting that the RBD–ACE2 interface would not be efficiently affected by orthosteric small‐molecule binders. [14] On the other hand, allosteric small‐molecule binders can disrupt interactions that involve large polar interfaces. We were intrigued by increasing evidence demonstrating the allosteric regulation of RBD [15] and ACE2. [13a] A small‐molecule screen may identify specific binders, which could be used as effective therapeutics for COVID‐19. Here, using the ELISA‐based screening, we discovered a novel heterocyclic skeleton inhibiting the protein–protein interaction (PPI) between RBD and ACE2. Furthermore, it suppressed SARS‐CoV‐2 infection in cultured cells by inhibiting viral entry via the modulation of ACE2, suggesting a future potential as a broad‐spectrum antiviral agent against coronaviruses.
To identify the PPI inhibitors of the RBD–ACE2 interaction, we performed a sandwich ELISA‐based screening using the plate‐bound RBD (Arg319‐Phe541) and extracellular domain of human ACE2 (Gln18‐Ser740), incubated in the presence of our in‐house drug‐like polyheterocyclic compound library, generated using a privileged substructure‐based diversity‐oriented synthesis (pDOS) strategy (Figure S1). [16] We identified a number of hits including tricyclic compound 1 (SB27001) with a pyrimidodiazepine core structure and consistent inhibition (up to ≈50 %) of the RBD–ACE2 interaction (Figure S1). To improve the potency of the initial hit compound 1, we synthesized a series of analogues based on a structure–activity relationship (SAR) analysis (Figure 2A). Compound 6 (SB27012) containing cyclopropyl amide at the R2 position showed improved activity with a half‐maximal inhibitory concentration (IC50) value of 7.7 μM based on an ELISA assay (Figures 2B–D). Interestingly, compound 2 (SB27016), although it contained the identical tricyclic pyrimidodiazepine core structure without R3 substituents, did not show any inhibitory activity and, therefore, was used as a partial negative control in further investigations (Figures 2C and D).
Figure 2.

Small‐molecule inhibitors of RBD–ACE2 interaction bind ACE2 without affecting ACE2 enzymatic activity. A) Compounds synthesized for the structure–activity relationship study. B) RBD–ACE2 sandwich ELISA assay data presented as % inhibition using compounds shown in (A) at 20 μM, n=2. C) Chemical structures of active compound 6 (SB27012) and partial negative compound 2 (SB27016). D) RBD–ACE2 ELISA assay, dose‐response experiments for SB27012 and SB271016; IC50 for SB27012 is 7.7±0.5 μM. E) Surface plasmon resonance (SPR) analysis of immobilized ACE2 and RBD, representative SPR data, SB27012 (0.156–5 μM) and SB27016 (0.625–5 μM), n=3. Kinetic constants for SB27012 with immobilized ACE2: for association (k a) (7.5±4.8)×105 M−1 s−1, for dissociation (k d) (9.9±4.1)×10−2 s−1, for binding affinity (K d) (2.1±0.6)×10−7 M. F) ACE2 enzymatic assay data presented as % activity, n=3.
To determine the direct target of SB27012, we performed a biophysical study using surface plasmon resonance (SPR) analyses with recombinant RBD (Arg319‐Phe541) and hACE2 (Gln18‐Ser740) proteins, identical to the proteins used for the ELISA assay. SB27012 showed direct binding to hACE2, but not to RBD, with a binding affinity with a K d value of ≈210 nM (Figure 2E). In contrast, partial negative SB27016 did not bind to hACE2 or RBD, confirming that only SB27012 could bind hACE2 with a high affinity to disrupt the RBD–ACE2 interaction.
Since the enzymatic activity of ACE2 is necessary for the processing of angiotensin I and II,[ 9a , 10 ] we evaluated the effect of SB27012 on ACE2 protease activity. DX‐600, a potent peptide‐based ACE2 inhibitor (K i value 2.8 nM), [17] inhibited ACE2 activity at 1 μM, while SB27012 did not have any adverse effect on ACE2 function even at high concentrations (Figure 2F). These results suggested that SB27012 did not bind to the active site of ACE2. We can exclude the possibility of orthosteric inhibition of the RBD–ACE2 interaction by SB27012, since the hydrophilic nature of the RBD–ACE2 interface thermodynamically disfavors the association with hydrophobic SB27012. This hypothesis was supported by the docking study measuring the potential interaction of SB27012 with the RBD‐binding surface of ACE2 (PDB: 6LZG, RBD bound ACE2) and the ACE2 active site (PBD: 1R4L, MLN‐4760 bound ACE2). Still, we did not observe any successful docking modes for both interactions (data not shown). Our SPR results clearly showed that SB27012 binds to ACE2 but not RBD (Figure 2E). Therefore, our data collectively indicated that SB27012 inhibits the ACE2–spike interaction by inducing allosteric changes in ACE2, although we cannot completely exclude the possibility of an orthosteric PPI inhibition by SB27012.
Next, we examined whether SB27012 can block the SARS‐CoV‐2 infection of cultured cells expressing endogenous ACE2. Monkey kidney epithelial Vero cells were infected with SARS‐CoV‐2 for 24 h in the presence of SB27012 or SB27016; remdesivir (RDV), the inhibitor of the RNA‐dependent RNA polymerase (RdRp) that has antiviral effects on a broad range of RNA viruses in vitro and in vivo, was used as a positive control. [18] Figures 3A and S2 show that RDV blocked the SARS‐CoV‐2 spread with IC50 6.0 μM. Although SB27012 did not fully inhibit the viral infection, it showed a more potent inhibitory effect at the lower doses (3.13 μM) than RDV, while not affecting cell viability (Figures 3D and S3). In contrast, SB27016 (partial negative control) did not suppress virus spreading at any concentration. To test whether SB27012 blocked the entry stage of SARS‐CoV‐2 infection, we performed a time‐of‐addition study, which is a well‐accepted protocol to examine the effectiveness and working mechanism of the compound in suppressing the viral infection (Figure 3B). Lopinavir (LPV), an HIV‐1 protease inhibitor previously shown to inhibit SARS‐CoV‐2 replication, [19] was used as a positive control. LPV inhibited SARS‐CoV‐2 at all tested time points, while SB27012 reduced infection predominantly at early time points (−1 and 0 hpi), suggesting that SB27012 blocked the entry step of viral infection (Figures 3C, E, and S4).
Figure 3.
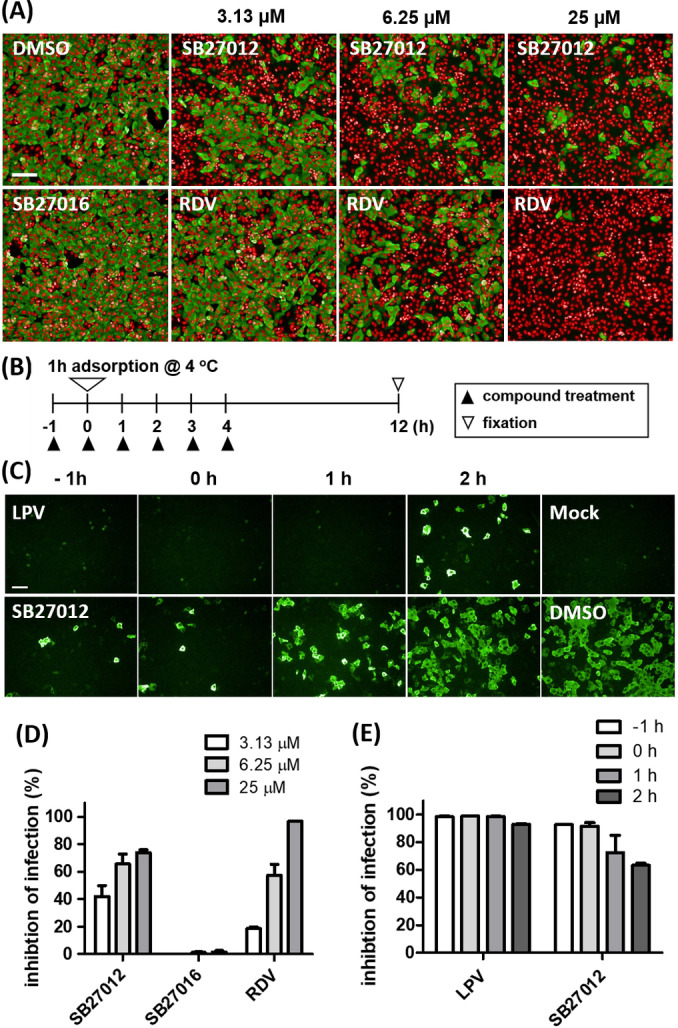
SB27012 suppressed SARS‐CoV‐2 infection by inhibiting viral entry. A) Immunofluorescence images of Vero cells infected with SARS‐CoV‐2 for 24 h and treated with active SB27012, remdesivir (RDV, positive control), and SB27016 (6.25 μM, negative control). Cell nuclei were stained red and SARS‐CoV‐2 nucleocapsid (N) protein was stained green. B) The timeline for the time‐of‐addition study. C) Immunofluorescence data from the time‐of‐addition study, SARS‐CoV‐2 N protein staining (green). Vero E6 cells were treated with compounds (25 μM) at the indicated time points. D, E) Quantification of (A) and (D) presented as % inhibition. Complete data for this study are presented in Figure S4. Scale bar=100 μm.
We hypothesized that SB27012 inhibited SARS‐CoV‐2 entry by allosterically modulating ACE2, thus suppressing infection by SARS‐CoV‐2 variants. Therefore, it is necessary to investigate whether our hit compounds can be also effective on variants since neutralizing antibodies and vaccines for COVID‐19 had been developed based on the ancestral SARS‐CoV‐2 spike protein sequence, and emerging spike mutations could impact their effectiveness. [20] In this study, SARS‐CoV‐2 variants including lineages B.1.1.7 (identified from the United Kingdom, alpha), B.1.351 (identified from South Africa, beta), and B.1.617.2. (identified from India, delta) were used to test our hypothesis. These variants contain specific mutations such as N501Y on RBD for the former two variants and D614G near the S1, S2 conjunction for all three variants. [20b] A decreased antiviral activity of SB27012 against these variants, compared to the ancestral SARS‐CoV‐2, was observed in Vero cells (Figure 4A). The negative control compound SB27016 did not show any inhibitory effect against the infection by these variants.
Figure 4.
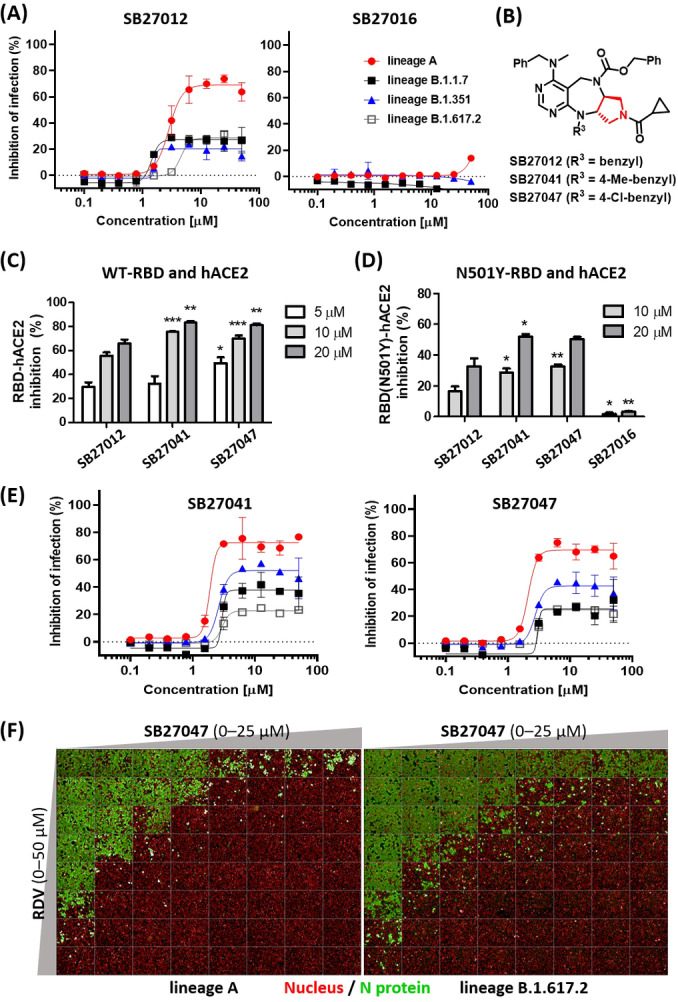
Vero cell infection by SARS‐CoV‐2 variants suppressed by SB27041 and SB27047. A, E) Dose–response curve analysis of the tested compounds in Vero cells infected for 24 h with lineage A (red), lineage B.1.1.7 (black), lineage B.1.351 (blue), and B.1.617.2 (white) SARS‐CoV‐2, respectively. B) Structure of the improved compounds from the second SAR study. C, D) RBD–ACE2 and RBD (N501Y)–ACE2 sandwich ELISA data presented as % inhibition, n≥4. Statistical differences between SB27041‐, SB27047‐, and SB27016‐treated groups compared to the same concentration of SB27012‐treated group was assessed using the one‐way ANOVA test with Dunnett's post hoc analysis (P>0.05, * P≤0.05, ** P≤0.01, *** P≤0.001). F) Immunofluorescence data from the drug combination study with remdesivir (RDV) and SB27047 in Vero cells infected with ancestral (left image) or delta variant (right image) of SARS‐CoV‐2. Data for SB27041 and synergistic scores are shown in Figures S6–S10.
To improve the potency of SB27012, we performed a second SAR study by changing the R1 and R3 positions (Figure 4B, Table S1). We evaluated the efficacy of modified compounds with ELISA assay using ancestral SARS‐CoV‐2 RBD and hACE2, leading to SB27041 and SB27047 with improved efficacy (Figure 4C). These compounds are only different from SB27012 by containing para‐chloro and para‐methyl substituents on the R3 benzyl ring, respectively, that may promote better interactions to the binding site in ACE2 with their hydrophobic nature and moderate bulkiness. To test their inhibitory effect on PPI between variant SARS‐CoV‐2 RBD and ACE2, the ELISA assay was performed using N501Y‐RBD, which was demonstrated to be the major challenger of vaccine efficacy and, therefore, necessary to evaluate during drug development. [20] Both SB27041 and SB27047 showed improved inhibition of N501Y‐RBD and ACE2 interaction compared to SB27012 (Figure 4D). SB27041 and SB27047 also showed enhanced inhibition of variant SARS‐CoV‐2 infection in Vero cells compared to SB27012, without affecting cell viability and ACE2 activity (Figures 4E, S3, and S5). These observations indicated that the physiological ACE2 binders, SB27041 or SB27047, could modulate the interaction of ACE2 to either the ancestral or variant RBDs, presumably via an allosteric manner.
The selected compounds did not elicit a complete inhibition of viral infection by SARS‐CoV‐2 in Vero cells, due to insufficient potency or the innate limitation of ACE2 allosteric regulation. These compounds might require combination treatments with other drugs that have a different mode of action to fully inhibit the viral infection. To investigate the synergistic effect of our viral entry inhibitor in combination with other drugs, especially remdesivir (RDV), an FDA‐approved drug with orthogonal mechanism (viral replication/ transcription inhibition), [18] we pursued the drug combination study as shown in Figures 4F and S10. RDV and SB27047 were used to co‐treat Vero cells infected with SARS‐CoV‐2 lineage A (ancestral) or lineage B.1.617.2 (delta variant) by systematically varying their concentrations. Intriguingly, the antiviral activities of both compounds were significantly enhanced in combined treatment, indicating that both compounds showed clear synergy in antiviral activities via different mechanisms of action (Figures 4F, S6–S10). It is worth noting that in the case of infection with delta variant, even 25–50 μM RDV could not cause the complete inhibition of the viral infection. However, in combination with 1.5–3.5 μM of SB27047 treatment, 3.12 μM of RDV ensures full inhibitory effect with >90 synergy scores, suggesting the physiological value of the ACE2 inhibitor as a viral entry suppressor for the treatment of COVID‐19.
To confirm the specific interaction of our selected compounds with ACE2, we re‐analyzed the ELISA screening data to verify other molecular skeletons that contained the pyrimidodiazepine moiety (Figure S11). All other pyrimidodiazepine‐based skeletons did not have any inhibitory effects on the RBD–ACE2 interaction. Next, we validated the compound specificity to ACE2 by designing and synthesizing SB27051, SB27053, and SB27052, the enantiomers of SB27012, SB27041, and SB27047, respectively (Figure S12). The newly synthesized enantiomers showed residual activities due to their structural similarities (Figure S13). SB27051, an enantiomer of the least active SB27012 among three compounds, exhibited the least activity difference to SB27012 for the suppression of the RBD‐ACE2 interaction and viral infection in Vero cells. However, all three compounds SB27051, SB27052, and SB27053 that bear similar polarities to their enantiomers but with different R‐groups showed significantly decreased activities on the viral infection inhibition in Vero cells. This suggests that SB27012, SB27041, and SB27047 inhibited the PPI via specific interaction with ACE2, and not a result of their hydrophobic properties (Figure S12, Tables S2 and S3).
The evolution of SARS‐CoV‐2 within the human population introduced a number of mutations on the spike protein that affected virus characteristics, including transmissibility and antigenicity, possibly due to changing immune profiles during human‐to‐human transmission. [2] For example, D614G mutation induces the open conformation of the spike trimer, perhaps leading to increased infectivity and transmissibility. Over time, this mutation became predominant in local populations where it was found. [21] Mutations in the N‐terminal domain (NTD) of the spike S1 region have been reported to escape neutralizing antibodies. [22] N501Y is located at the RBD–ACE2 interface and enhances the affinity of RBD to ACE2, raising concerns regarding vaccine efficacy. [20] Therefore, COVID‐19 therapeutics should overcome the fast‐evolving spike protein mutations, including mutations in RBD, one of the most challenging locations.
Here, we proposed a new therapeutic strategy targeting the ACE2 receptor instead of RBD to inhibit the RBD–ACE2 interaction, which would allow us to bypass spike protein mutations. ACE2 plays critical physiological roles as a transmembrane protein and has not been considered a therapeutic target in COVID‐19 due to potentially serious side effects.[ 9a , 10 ] But, growing evidence indicates that ACE2 may be subjected to conformational changes due to the dynamic interactions of its subunits or, in other words, allosteric changes, [13] and the catalytic function of ACE2 is not dependent on spike binding. [23] In fact, a potent ACE2 enzymatic inhibitor, MLN‐4760, does not inhibit the interaction between ACE2 and spike proteins of SARS‐CoV‐1, [11b] even though the allosteric connection between two sites was suggested. [13] There have been no specific studies of ACE2 allostery or identification of ACE2 allosteric compounds to modulate the ACE2–RBD interaction. Here, we demonstrated for the first time that pyrimidodiazepine‐based tricyclic compounds from our pDOS library showed specific binding to the ACE2 receptor but not to RBD, disrupting the RBD–ACE2 interaction without affecting the ACE2 enzymatic function with some evidence of allosteric modulation of ACE2. Our findings provide evidence that the regulation of ACE2 leads to the suppression of SARS‐CoV‐2 cell entry by inhibiting the RBD–ACE2 interaction. The identification of putative allosteric sites of ACE2, which still needs to be confirmed computationally and experimentally, will provide a new therapeutic strategy to confront rapidly evolving SARS‐CoV‐2.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The pathogen resources (NCCP43326, NCCP43381, NCCP43382, and NCCP43390) for this study were provided by the National Culture Collection for Pathogens. This work was supported by the National Creative Research Initiative Grant (2014R1A3A2030423 to S.B.P) and the Bio & Medical Technology Development Program (2012M3A9C4048780 to S.B.P, and 2017M3A9G6068245 and 2020M3A9I2108520 to S.K) through the National Research Foundation of Korea (NRF) funded by the Korean Government (Ministry of Science & ICT).
Y.-H. Shin, K. Jeong, J. Lee, H. J. Lee, J. Yim, J. Kim, S. Kim, S. B. Park, Angew. Chem. Int. Ed. 2022, 61, e202115695; Angew. Chem. 2022, 134, e202115695.
Contributor Information
Dr. Seungtaek Kim, Email: seungtaek.kim@ip-korea.org.
Prof. Dr. Seung Bum Park, Email: sbpark@snu.ac.kr.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Li Y. Z., Tenchov R., Smoot J., Liu C., Watkins S., Zhou Q. Q., ACS Cent. Sci. 2021, 7, 512–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. van Dorp L., Acman M., Richard D., Shaw L. P., Ford C. E., Ormond L., Owen C. J., Pang J., Tan C. C. S., Boshier F. A. T. et al., Infect. Genet. Evol. 2020, 83, 104351; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. McCormick K. D., Jacobs J. L., Mellors J. W., Science 2021, 371, 1306–1308. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Wang Q. H., Zhang Y. F., Wu L. L., Niu S., Song C. L., Zhang Z. Y., Lu G. W., Qiao C. P., Hu Y., Yuen K. Y. et al., Cell 2020, 181, 894–904; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Xiu S. Y., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X. Y., J. Med. Chem. 2020, 63, 12256–12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A. et al., Cell 2020, 181, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Outlaw V. K., Bovier F. T., Mears M. C., Cajimat M. N., Zhu Y., Lin M. J., Addetia A., Lieberman N. A. P., Peddu V., Xie X. P. et al., mBio 2020, 11, e01935-20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. de Vries R. D., Schmitz K. S., Bovier F. T., Predella C., Khao J., Noack D., Haagmans B. L., Herfst S., Stearns K. N., Drew-Bear J. et al., Science 2021, 371, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Cao L. X., Goreshnik I., Coventry B., Case J. B., Miller L., Kozodoy L., Chen R. E., Carter L., Walls A. C., Park Y. J. et al., Science 2020, 370, 426–431; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Huo J. D., Le Bas A., Ruza R. R., Duyvesteyn H. M. E., Mikolajek H., Malinauskas T., Tan T. K., Rijal P., Dumoux M., Ward P. N. et al., Nat. Struct. Mol. Biol. 2020, 27, 846–854. [DOI] [PubMed] [Google Scholar]
- 7.M. D. L. C. Muus, G. Eraslan, A. Waghray, G. Heimberg, L. Sikkema, Y. Kobayashi, E. D. Vaishnav, A. Subramanian, C. Smilie et al., bioRxiv 2020, 10.1101/2020.04.19.049254. [DOI]
- 8.
- 8a. Li W. H., Moore M. J., Vasilieva N., Sui J. H., Wong S. K., Berne M. A., Somasundaran M., Sullivan J. L., Luzuriaga K., Greenough T. C. et al., Nature 2003, 426, 450–454; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pohlmann S., Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R. et al., Circ. Res. 2000, 87, E1–E9; [DOI] [PubMed] [Google Scholar]
- 9b. Santos R. A. S., Sampaio W. O., Alzamora A. C., Motta-Santos D., Alenina N., Bader M., Campagnole-Santos M. J., Physiol. Rev. 2018, 98, 505–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H. et al., Nature 2005, 436, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Guy J. L., Jackson R. M., Acharya K. R., Sturrock E. D., Hooper N. M., Turner A. J., Biochemistry 2003, 42, 13185–13192; [DOI] [PubMed] [Google Scholar]
- 11b. Li W. H., Zhang C. S., Sui J. H., Kuhn J. H., Moore M. J., Luo S. W., Wong S. K., Huang I. C. et al., EMBO J. 2005, 24, 1634–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Towler P., Staker B., Prasad S. G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N. A. et al., J. Biol. Chem. 2004, 279, 17996–18007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Huentelman M. J., Zubcevic J., Prada J. A. H., Xiao X. D., Dimitrov D. S., Raizada M. K., Ostrov D. A., Hypertension 2004, 44, 903–906; [DOI] [PubMed] [Google Scholar]
- 12c. Yan R. H., Zhang Y. Y., Li Y. N., Xia L., Guo Y. Y., Zhou Q., Science 2020, 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Gross L. Z. F., Sacerdoti M., Piiper A., Zeuzem S., Leroux A. E., Biondi R. M., ChemMedChem 2020, 15, 1682–1690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Williams-Noonan B. J., Todorova N., Kulkarni K., Aguilar M. I., Yarovsky I., J. Phys. Chem. B. 2021, 125, 2533–2550. [DOI] [PubMed] [Google Scholar]
- 14. Lan J., Ge J. W., Yu J. F., Shan S. S., Zhou H., Fan S. L., Zhang Q., Shi X. L., Wang Q. S., Zhang L. Q., Wang X. Q., Nature 2020, 581, 215–220. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Olotu F. A., Omolabi K. F., Soliman M. E. S., Inf. Med. Unlocked 2020, 21, 100451; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Raghuvamsi P. V., Tulsian N. K., Samsudin F., Qian X. L., Purushotorman K., Yue G., Kozma M. M., Hwa W. Y., Lescar J., Bond P. J. et al., eLife 2021, 10, e63646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Kim J., Kim H., Park S. B., J. Am. Chem. Soc. 2014, 136, 14629–14638; [DOI] [PubMed] [Google Scholar]
- 16b. Kim J., Jung J., Koo J., Cho W., Lee W. S., Kim C., Park W., Park S. B., Nat. Commun. 2016, 7, 13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang L. L., Sexton D. J., Skogerson K., Devlin M., Smith R., Sanyal I., Parry T., Kent R., Enright J., Wu Q. L. et al., J. Biol. Chem. 2003, 278, 15532–15540. [DOI] [PubMed] [Google Scholar]
- 18. Wang M. L., Cao R. Y., Zhang L. K., Yang X. L., Liu J., Xu M. Y., Shi Z. L., Hu Z. H., Zhong W., Xiao G. F., Cell Res. 2020, 30, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choy K. T., Wong A. Y., Kaewpreedee P., Sia S. F., Chen D., Hui K. P. Y., Chu D. K. W., Chan M. C. W., Cheung P. P., Huang X., Peiris M., Yen H. L., Antiviral Res. 2020, 178, 104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Li Q. Q., Wu J. J., Nie J. H., Zhang L., Hao H., Liu S., Zhao C. Y., Zhang Q., Liu H., Nie L. L. et al., Cell 2020, 182, 1284–1294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Wang P. F., Nair M. S., Liu L. H., Iketani S., Luo Y., Guo Y. C., Wang M., Yu J., Zhang B. S., Kwong P. D. et al., Nature 2021, 593, 130–135. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Korber B., Fischer W. M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E. E., Bhattacharya T., Foley B. et al., Cell 2020, 182, 812–827; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Yurkovetskiy L., Wang X., Pascal K. E., Tomkins-Tinch C., Nyalile T., Wang Y. T., Baum A., Diehl W. E., Dauphin A., Carbone C. et al., Cell 2020, 183, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang P., Casner R. G., Nair M. S., Wang M., Yu J., Cerutti G., Liu L., Kwong P. D., Huang Y., Shapiro L., Ho D. D., Cell Host Microbe 2021, 29, 747–751 e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moore M. J., Dorfman T., Li W. H., Wong S. K., Li Y. H., Kuhn J. H., Coderre J., Vasilieva N., Han Z. C. et al., J. Virol. 2004, 78, 10628–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.