

Summary:
Store-operated Ca2+ entry (SOCE) is a mechanism used by many cells types including lymphocytes and other immune cells to increase intracellular Ca2+ concentrations to initiate signal transduction. Activation of immunoreceptors such as the T-cell receptor, B-cell receptor, or Fc receptors results in the release of Ca2+ ions from endoplasmic reticulum (ER) Ca2+ stores and subsequent activation of plasma membrane Ca2+ channels such as the well-characterized Ca2+ release-activated Ca2+ (CRAC) channel. Two genes have been identified that are essential for SOCE: ORAI1 as the pore-forming subunit of the CRAC channel in the plasma membrane and stromal interaction molecule-1 (STIM1) sensing the ER Ca2+ concentration and activating ORAI1-CRAC channels. Intense efforts in the past several years have focused on understanding the molecular mechanism of SOCE and the role it plays for cell functions in vitro and in vivo. A number of transgenic mouse models have been generated to investigate the role of ORAI1 and STIM1 in immunity. In addition, mutations in ORAI1 and STIM1 identified in immunodeficient patients provide valuable insight into the role of both genes and SOCE. This review focuses on the role of ORAI1 and STIM1 in vivo, discussing the phenotypes of ORAI1- and STIM1-deficient human patients and mice.
Keywords: ORAI1, STIM1, Ca2+, T cells, B cells, mast cells
Introduction
Modulation of intracellular Ca2+ levels provides a signal transduction mechanism that is used by virtually all cells types – including lymphocytes – for the control of both short-term and long-term cellular functions. In the immune system, engagement of immunoreceptors such as the T-cell receptor (TCR), B-cell receptor (BCR), or Fc receptors on mast cells, dendritic cells, and macrophages results in a robust influx of Ca2+ from the extracellular space (1, 2). This influx in many instances is due to store-operated Ca2+ entry (SOCE) mediated by SOC channels of which the Ca2+ release-activated Ca2+ (CRAC) channel is the best characterized. While other pathways for Ca2+ influx may co-exist with SOCE in cells of the immune system, SOCE and the prototypical store-operated CRAC channel play a prominent role in Ca2+ signaling. In fact, the CRAC channel current ICRAC was first described in T cells and mast cells (3, 4). SOCE, by definition, is activated by the depletion of endoplasmic reticulum (ER) Ca2+ stores that in immune cells generally occurs following antigen recognition, immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation, phospholipase C (PLC) activation, and production of inositol-1,4,5-triphosphate (IP3). Binding of IP3 to IP3 receptors (IP3Rs) in the ER membrane induces Ca2+ efflux from the ER through IP3R resulting in (i) a transient increase in intracellular Ca2+ concentrations and (ii) activation of SOC channels in the plasma membrane. Major break-throughs in the understanding of SOCE have been made in the past few years with the discovery of stromal interaction molecule 1 (STIM1) and ORAI1 (or CRACM1) as essential molecular components of this pathway (5). Their identification has resulted in a flurry of research describing both the mechanisms underlying CRAC channel activation and the role of SOCE in many tissues including the immune system. This review focuses mainly on the role of SOCE for immune function by describing insights gained from studying human patients with mutations in STIM1 and ORAI1 and mice with targeted deletion of Stim1, Stim2, and Orai1.
Mechanisms and roles of Ca2+ signaling in the immune system
Ca2+ signals contribute to the function of many cell types in the immune system, including T and B cells, natural killer (NK) cells, mast cells, dendritic cells, and macrophages, where they control to diverse cell functions ranging from differentiation, proliferation, gene expression, cell motility, to secretion of vesicles containing cytokines, cytotoxic, or proinflammatory proteins (2, 6). In immune cells, Ca2+ signals result from engagement of antigen receptors at the cell surface and therefore directly determine the strength of the immune response to antigen. In addition, Ca2+ signals can be initiated through cell surface molecules and coreceptors such as CD19, CD20, and CD81 on B cells (7–9) and G-protein-coupled chemokine receptors on a variety of immune cells including dendritic cells (10). Details of how these receptors induce Ca2+ signals and which Ca2+ channels are activated is not well understood in many cases, although CRAC channel currents were observed in neutrophils in response to interleukin-8 (IL-8) treatment (11) and dendritic cells following passive store depletion (12). Other channels that may be involved in Ca2+ influx in immune cells include TRP (transient receptor potential) channels (13, 14), the adenosine triphosphate (ATP)-responsive P2Y and P2X purinoreceptors expressed on T cells and mast cells (15–18), and – although controversial – voltage-gated Ca2+ channels (19, 20). While TRP channels have not been shown to function as Ca2+ -selective cation channels in T cells, it is of note that TRPC1 could be co-immunoprecipitated together with STIM1 and ORAI1 in HEK293 and salivary gland cells (22) and that STIM1 was shown to gate TRPC1 and other TRPC channels (21). TRPM4, by contrast, functions as a calcium-activated non-selective cation channel that regulates Ca2+ influx in T cells and dendritic cells by effecting plasma membrane depolarization (23, 24). In a similar manner, potassium channels such as the voltage-sensitive Kv1.3 and the Ca2+ sensitive IKCa1 channel do not themselves conduct Ca2+ but are essential for Ca2+ entry by maintaining a negative membrane potential and providing the electrical driving force required for Ca2+ influx. As a consequence, inhibition of K+ channel function suppresses Ca2+ entry and T-cell activation (25). The strength and duration of cytosolic Ca2+ signals is limited by re-uptake of Ca2+ into the ER or export into the extracellular space by Ca2+ pumps (26). B cells in addition use a variety of inhibitory receptors and signaling mechanisms to attenuate Ca2+ signaling through molecules such as SHP-1, CD22, Dok-3, Grb2, PD-1 and FcγRIIB in B cells (27–29).
Ca2+ signals are involved in a multitude of short- and long-term functions of immune cells (1, 2, 6). These include the regulation of T-cell motility during TCR-mediated antigen recognition on antigen-presenting cells (APCs) and formation of the immunological synapse (30–32), secretion of vesicles in mast cells and cytotoxic T cells (33), and phagocytosis in neutrophils (34) and macrophages (35–37). In mast cells, Ca2+ signals were shown to be involved in FcεRI-mediated degranulation and the release of histamine, leukotrienes, and prostaglandins (38–41). A similar process, the exocytosis of cytolytic granules by CD8+ cytotoxic T cells, also depends on Ca2+ influx (33). When CD8+ T cells recognize virus-infected or tumor cells, they form a synapse-like structure with their target cell that allows for the directional secretion of cytolytic granules containing perforin and granzyme proteases, resulting in the induction of apoptosis or necrosis in target cells (42). In the absence of Ca2+ influx, granule exocytosis and target cell apoptosis are impaired (33, 43).
Ca2+ signals are also involved in more long-term processes such as the regulation of cytokine and chemokine gene expression through Ca2+ -dependent transcription factors such as nuclear factor for activation of T cells (NFAT) (reviewed in 44–47) and certain cell fate decisions. The differentiation of naive CD4+ T cells into T-helper 1 or 2 cells was shown to depend on the strength of signals mediated by the TCR including Ca2+ signals (48), and sustained increases in intracellular Ca2+ in the presence or absence of costimulatory signals are involved in the decision whether a T cell is activated or becomes unresponsive to future TCR stimulation (reviewed in 49–51). The role of Ca2+ signals, particularly SOCE, for T-cell development is discussed in more detail further below in the context of human patients and mice lacking SOCE (52). Taken together, Ca2+ influx is critically involved in many effector functions and cell-fate decisions controlling adaptive and innate immune responses. The absence of Ca2+ influx through SOCE results in immunodeficiency and autoimmunity, as discussed in detail further below.
Molecules mediating SOCE: the STIM and ORAI protein families
STIM1 and STIM2
STIM1 is a single-pass transmembrane protein, which is localized predominantly in the membrane of the ER where it functions as a sensor of ER Ca2+ concentrations and essential activator of ORAI1⁄CRAC channels (53, 54). The N-terminus of STIM1 contains a pair of low-affinity EF hand calcium-binding domains adjacent to a sterile α motif (SAM) protein–protein interaction domain; the longer C-terminus of STIM1 features two coiled-coil domains, serine⁄proline-rich, and lysine-rich domains (68, 69). Depletion of Ca2+ from the ER results in dissociation of Ca2+ from the N-terminal EF hand domains of STIM1, unfolding of the EF-SAM domain, and multimerization of STIM1, ultimately leading to the assembly of STIM1 in large clusters in the ER membrane, which are conventionally called puncta (55, 56). STIM1 puncta formation causes aggregation of ORAI1 in the plasma membrane and was shown to coincide with localized Ca2+ influx (56–58). Multimerization of STIM1 in the ER is sufficient to activate CRAC channels, elegantly demonstrated by substituting the ER-luminal N-terminus of STIM1 with artificial inducible protein–protein interaction domains (59). Expression of the cytoplasmic C-terminus of STIM1 as a soluble protein, however, is also able to constitutively activate CRAC channels in the absence of ER store depletion (60) and to induce assembly of ORAI1 into functional tetrameric complexes (61, 62).
Several laboratories including ours have now identified a minimal CRAC channel activation domain within the C terminus of STIM1 (63–66). This domain, when expressed by itself, is sufficient to colocalize with and bind to ORAI1 and activate the CRAC channel. We found that coiled-coil domain containing fragment b9 (CCb9) encompassing amino acids 339–446 is sufficient for binding to the C-terminus of ORAI1 and activation of CRAC channels in the absence of store depletion (66). Minimal activation domains identified by other groups are very similar in extent and comprise amino acids 342–448 [CRAC channel activation domain(CAD)](64), 344–442 [STIM1 Orai activating region (SOAR)](62), and 233–450⁄474 [Orai1-activating small fragment (OASF)] (67). Further truncation of the CCb9 fragment [and CAD (64)] by 10 amino acids at its C-terminal end resulted in a non-activating fragment (CCb10). Some minimal activation domains were shown to form tetramers and cluster ORAI1, but clustering itself does not seem to be sufficient for CRAC channel activation (63, 64). The CCb9 minimal activation domain is flanked at its C-terminal end by an approximately 31 amino acid peptide (445–475) that when applied directly to the cytoplasm inhibits CRAC channel activation (66). This finding suggests that the STIM1 minimal activation domain is masked in the context of full-length protein under resting conditions (i.e., replete Ca2+ stores) by an adjacent inhibitory peptide. This inhibition is presumably released when STIM1 undergoes conformational changes following depletion of Ca2+ stores (Fig. 1). Taken together, many new details regarding the structure and functional domains of STIM1 and their interactions with ORAI1 are emerging that elucidate the mechanisms involved in CRAC channel activation.
Fig. 1. A minimal ORAI1 activation domain in STIM1 (CCb9).
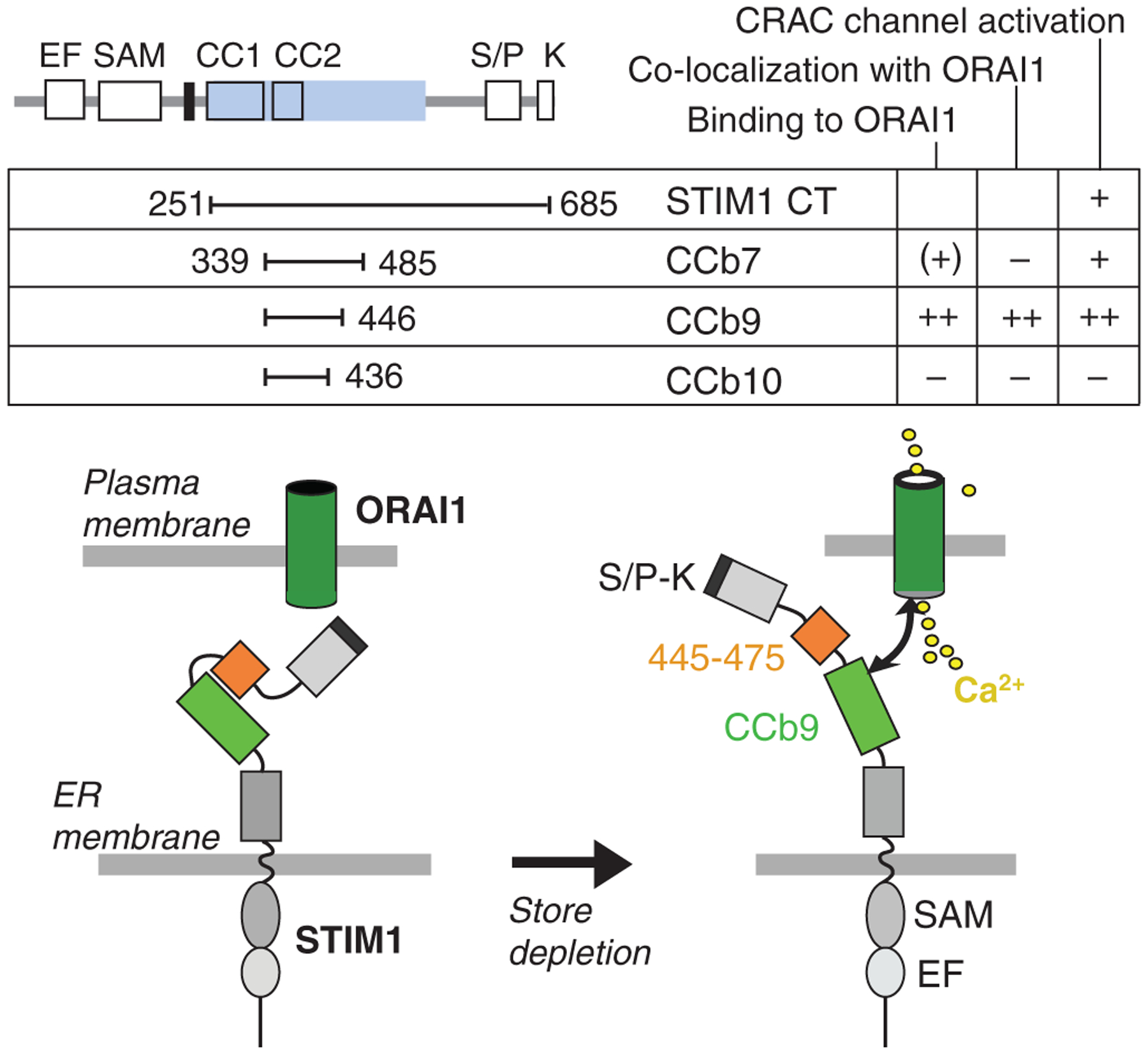
In the resting state with replete Ca2+ stores, the CCb9 ORAI1-activation domain in STIM1 is masked by an inhibitory STIM1445–475 peptide. Upon store depletion, the CCb9 domain is released, binds to and activates the ORAI1–CRAC channel. CC, coiled-coil; CT, STIM1 C-terminus; EF, EF hand; ERM, ezrin⁄radixin⁄moesin; SAM, sterile α motif; TM, transmembrane; S⁄P, serine–proline; K, lysine. Modified and reproduced with permission from (66).
STIM2 is a closely related paralogue of STIM1 that shares its overall protein domain architecture including the EF hand Ca2+ binding, SAM, and coiled-coiled domains (68). STIM2, like STIM1, is located in the ER and was shown to heterodimerize with STIM1 (68). Early experiments on the function of STIM2 using RNA interference (RNAi)-mediated knock-down of STIM2 showed either no or only moderate effects on Ca2+ influx in HeLa or HEK293 cells in contrast to knock-down of STIM1 (53, 54). While one study overexpressing STIM2 suggested that it may have a potential inhibitory effect on STIM1-mediated store-operated Ca2+ influx (70), the majority of evidence now indicates that STIM2 is a positive regulator of SOCE and CRAC channel activation. STIM2 activates Ca2+ influx upon smaller decreases in ER Ca2+ concentrations than STIM1 and was suggested to regulate basal cytosolic Ca2+ concentrations (71). These data are consistent with a lower affinity of the EF hand Ca2+ -binding domain in STIM2 compared to that of STIM1. Analyzing T cells from mice lacking STIM2 expression, we observed that Stim2-deficient T cells have almost normal Ca2+ influx and CRAC channel function in the first few minutes following T-cell activation (72). They fail, however, to sustain increased intracellular Ca2+ concentrations for prolonged periods of time after store depletion compared with control cells. As a consequence, nuclear translocation of the Ca2+-dependent transcription factor NFAT is rapidly reversed resulting in impaired NFAT-dependent cytokine gene expression in Stim2-deficient T cells despite normal expression of STIM1 (72). This delayed role of STIM2 in the regulation of store-operated Ca2+ influx relative to STIM1 is consistent with the much slower unfolding kinetics of the EF-SAM domain in STIM2 compared with STIM1 (74). These findings suggest that STIM1 and STIM2 respond with different thresholds and different activation kinetics to ER Ca2+ store depletion but also that both molecules synergize in their regulation of store-operated Ca2+ entry. This synergy is apparent in (i) the ability of STIM2 – when overexpressed – to restore SOCE in cells from human patients and mice lacking STIM1 (72, 75) and (ii) the more severe phenotype in mice with T-cell-specific deletion of both Stim1 and Stim2 compared with individual knockout mice (72).
ORAI1, ORAI2, and ORAI3
ORAI1 (or CRACM1) was identified as a regulator of Ca2+ signaling in RNAi screens and as the gene responsible for immunodeficiency in patients with a defect in CRAC channel function (76–78). The ORAI1 gene on human chromosome 12q24 encodes an evolutionarily highly conserved tetraspanning plasma membrane protein that is structurally unrelated to other proteins or ion channels. Two paralogues ORAI2 (or CRACM2) and ORAI3 (or CRACM3) (79) are encoded by genes on human chromosomes 7q22 and 16p11, respectively. Strong functional and genetic evidence indicates that ORAI1 functions as the pore-forming subunit of the CRAC channel (80–82). Two negatively charged glutamate residues in the first and third transmembrane domain of ORAI1, E106, and E190, are thought to act as Ca2+ -binding sites in the ion channel pore. Mutations in either glutamate residue do not interfere with protein expression but abolish or significantly alter CRAC channel function (80–82). The native CRAC channel is characterized by its high Ca2+ selectivity and other distinctive properties including an inwardly rectifying current–voltage (I–V) relation and very low single-channel conductance (approximately 1 pS) (83). ORAI1-CRAC channels mutated at residues E106 or E190 cease to selectively conduct Ca2+ but become permeable to Na+ and Cs+, presumably by interference of the mutations with Ca2+ binding in the selectivity filter of the CRAC channel pore. Recent studies indicate that functional CRAC channels likely consist of ORAI1 tetramers (61, 62, 84) reminiscent of voltage-gated Ca2+ channels. In this model, it is likely that each ORAI1 subunit contributes one or two glutamate residues for coordinated Ca2+ binding in the CRAC channel pore.
ORAI2 and ORAI3 share the predicted tetraspanning membrane topology with ORAI1 and show a high degree of sequence identity when compared with full-length ORAI1 (60.3% for ORAI1 and ORAI2, 63.2% for ORAI1 and ORAI3, and 66.4% for ORAI2 and ORAI3). Sequence homology is almost complete when comparing only the transmembrane domains (92.5%, 93.8%, and 93.8%, respectively). Both ORAI2 and ORAI3 form Ca2+ -permeable ion channels when overexpressed in vitro together with STIM1 (85, 86). The biophysical properties of currents recorded under these conditions are similar to native ICRAC and those observed in cells co-expressing ORAI1 and STIM1. They differ, however, from ORAI1-mediated and native CRAC channel currents in a number of respects, for instance Ca2+ -dependent inactivation, monovalent permeation, and responsiveness to 2-APB (85, 86). These findings suggest that ORAI2 and ORAI3 may play a role in CRAC channel function and store-operated Ca2+ entry in vivo. No direct evidence for endogenous ORAI2 and ORAI3 function currently exists, however, and the tissues and cell types in which the two proteins may play a role remain to be determined.
STIM and ORAI expression in cells of the immune system
SOCE and CRAC channel activity have been documented in a variety of immune and non-immune cells (reviewed in 87). Outside the immune system, these cells include (but are not limited to) vascular endothelial (88–90) and smooth muscle cells (91, 92), pancreatic acinar cells (93), hepatocytes (94), and adrenal chromaffine cells (95). In the immune system, SOCE and ICRAC have been observed in many different cell types of the lymphoid and myeloid lineage such as T cells (3), mast cells (4), B cells (96–98), dendritic cells (12), macrophages (99), NK cells (100), and neutrophils (11). It is likely that SOCE plays a role in other cell types in the immune system as well and contributes to proper innate and adaptive immune responses.
With the cloning of the STIM and ORAI gene families, expression data can be added to the picture to explain in which cells SOCE and CRAC channels are functional. mRNA for STIM1 and STIM2 is widely expressed in many tissues in both human and mouse including cells of the immune system (68, 69, 101) (Fig. 2). Human STIM1 and STIM2 are strongly expressed in lymphoid and myeloid cells such as T cells, B cells, NK cells, and monocytes (101, 102). In mice, STIM1 mRNA levels are the highest in mast cells, NK cells, and T cells, whereas STIM2 expression is fairly uniform and average in immune cells compared with other organs (101, 102) (Fig. 2). Using a transgenic expression system, Stiber et al. (103) detected STIM1–LacZ fusion protein in skeletal muscle, cerebellum, spleen, and thymus of mice heterozygous for the transgene. We observed strong STIM1 protein expression in naive and differentiated murine CD4+ T cells (72), whereas STIM2 was undetectable in naive CD4+ T cells and upregulated only upon differentiation into Th1 and Th2 cells in vitro. Expression levels of STIM2 were low, however, compared with those of STIM1 in both naive and differentiated CD4+ T cells (72).
Fig. 2. Murine Orai and Stim family members are expressed in a wide variety of cells in the immune system.
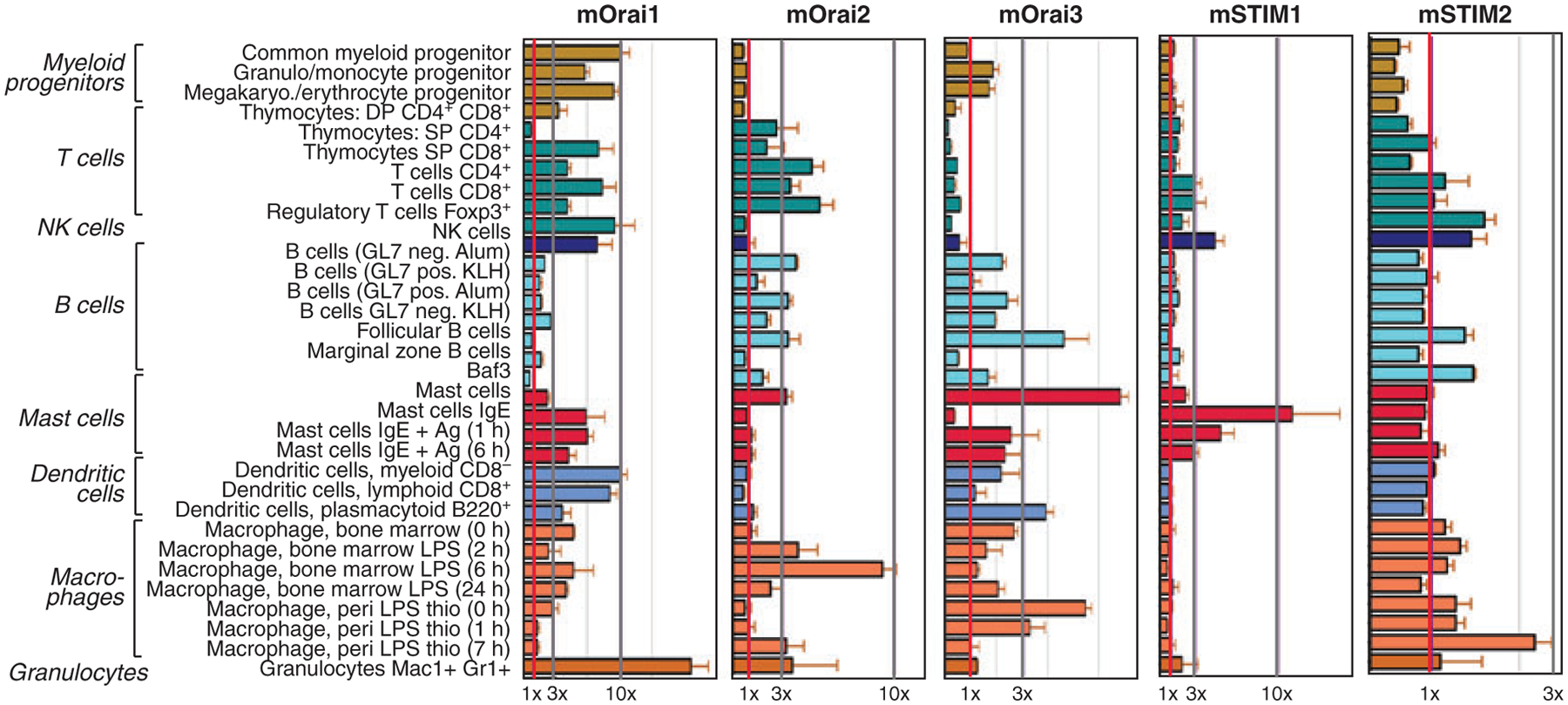
mRNA levels of mStim and mOrai based on expression data retrieved from BioGPS (http://biogps.gnf.org). Cell lines and tissues were sourced from 8–10-week-old C57Bl⁄6 mice and mRNA expression analyzed using Affymetrix MOE430_2 microarrays (102). Vertical gray lines indicate 1×, 3×, and 10× fold expression over the median of all tissues analyzed.
All three ORAI paralogues are expressed in a large variety of human and mouse tissues and cell types including lymphoid organs (101, 102, 104, Stefan Feske, unpublished data) (Fig. 2). Generally, the expression of human ORAI1 and ORAI3 mRNA is more ubiquitous than that of ORAI2, which in human is predominantly expressed in kidney, lung, and spleen (104). Murine ORAI1, ORAI2, and ORAI3 transcripts are found in most myeloid and lymphoid cells such as macrophages, dendritic cells, mast cells, T cells, and B cells (101, 102, 105) (Fig. 2). Particularly, high levels of ORAI1 mRNA were observed in mouse granulocytes, whereas ORAI2 and ORAI3 expression was strongest in lipopolysaccharide (LPS)-stimulated macrophages and mast cells, respectively (101) (Fig. 2). Interestingly, ORAI2 expression is high in naive CD4+ T cells (38, 106) and is downregulated upon differentiation into Th1 or Th2 cells (106). This led to the suggestion that ORAI2, not ORAI1, is responsible for SOCE in naive T cells (38).
Expression data on ORAI proteins is very limited due to the current lack of reliable commercial antibodies against ORAI1, ORAI2, and ORAI3. Using a custom-made ORAI1-antibody, we evaluated the distribution of ORAI1 protein expression in human and mouse tissues including primary and secondary lymphoid organs (Stefan Feske, unpublished data). In human lymphoid organs, ORAI1 was detected in a subset of cells in the thymus, spleen, and tonsils by immunohistochemistry. The highest expression was seen in cells of the periarterial lymphoid sheath (PALS) of the spleen and the paracortical zone in tonsils, consistent with ORAI1 expression in T cells. In mice, ORAI1 protein was detected fairly uniformly in all lymphocyte populations tested including CD4+ and CD8+ T cells, B cells, NK cells, and NKT cells isolated from spleen and lymph nodes (Fig. 3). No significant differences in expression were detected between T-cell subsets including effector T cells, regulatory T cells (defined as CD4+CD25+GITR+), naive and memory CD4+ and CD8+ T cells, or between resting (CD69−) and stimulated (CD69+) T cells (Fig. 3). ORAI1 protein was equally expressed at all stages of T-cell development in the thymus (Fig. 3). Similar protein expression data for ORAI2 and ORAI3 are not available. Taken together, both STIM1 and ORAI1 are expressed almost ubiquitously in human and mouse tissues and many if not all cells of the immune system.
Fig. 3. ORAI1 expression in developing and mature lymphocytes.
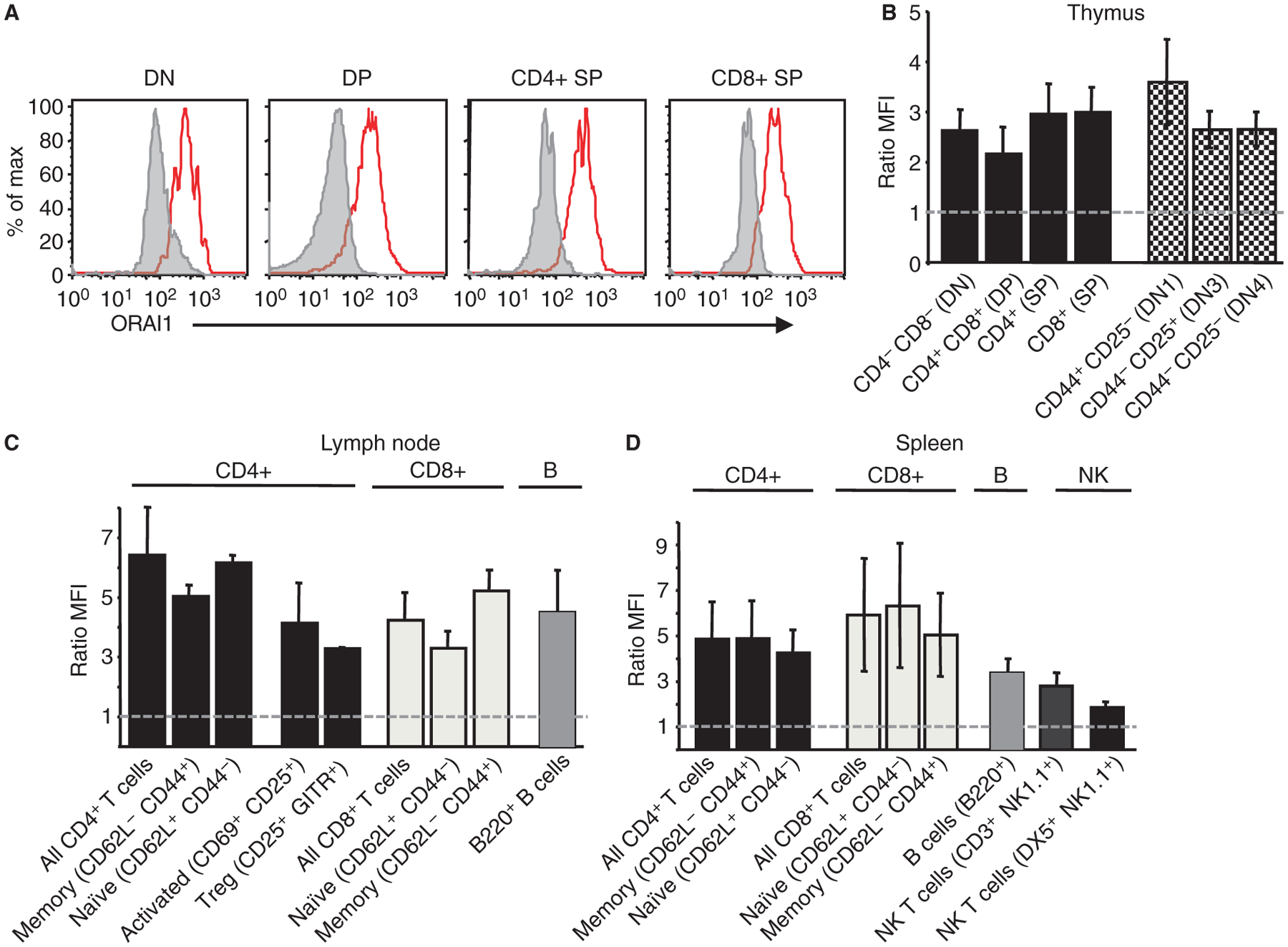
ORAI1 protein expression was analyzed in cells isolated from primary and secondary lymphoid organs of 6–8-week-old C57Bl⁄6 mice incubated with polyclonal anti-ORAI1 antibody or secondary antibody alone. (A,B) ORAI1 is expressed at all stages of T-cell development in the thymus. (C,D) ORAI1 expression in T, B, and NK cells in lymph node and spleen as indicated. ORAI1 expression was comparable in lymphocyte populations at various stages of differentiation and activation; weakest expression was found in splenic NK cells (n = 8) and highest expression in lymph node CD4+ T cells. Expression is shown as the ratio of MFI ORAI1 (red trace in A): MFI secondary antibody alone (gray trace in A). n = 2–8 mice analyzed (spleen), n = 2–19 (lymph node), n = 6–10 (thymus). MFI, mean fluorescence intensity.
CRAC-channelopathies due to mutations in ORAI1 and STIM1 in human patients
Mendelian diseases caused by mutations in single genes have provided important insights into the function of many genes based on the pathophysiology associated with absent, nonfunctional, or dominant negative mutant gene products. Early evidence for the function and crucial role of SOCE and CRAC channels in vivo came from the analysis of patients with rare but very instructive inherited immunodeficiency diseases (78, 107–109). Patients lacking store-operated Ca2+ influx and CRAC channel function show a severe defect in T-cell activation and suffer from immunodeficiency characterized by life-threatening viral, bacterial, and fungal infections. We have identified mutations in ORAI1 that interfere with CRAC channel function and STIM1 that generate a null phenotype by abolishing protein expression (75, 78). The clinical phenotypes of ORAI1 and STIM1 deficiency overlap to a large degree and are characterized by immunological and non-immunological symptoms, suggesting that the clinical syndrome associated with CRAC-channelopathy is pathway and not gene specific.
ORAI1 deficiency
Mutations and molecular characterization
ORAI1 was identified as the gene encoding the CRAC channel through the combination of linkage mapping in immunodeficient patients lacking SOCE and CRAC channel function (78, 110) and a genome-wide functional RNAi screen in Drosophila melanogaster S2 cells for genes regulating NFAT nuclear translocation and Ca2+ influx (76–78). Sequencing of the ORAI1 gene revealed that immunodeficient patients are homozygous for a missense mutation in exon 1 of ORAI1 that results in substitution of an arginine at position 91 of the protein sequence with tryptophane (R91W) (78) (Fig. 4). The single amino acid substitution abolishes CRAC channel function, SOCE, and T-cell activation but does not interfere with ORAI1 expression or its localization in the plasma membrane. R91 is located at the beginning of the first transmembrane domain of ORAI1, i.e., at the interface of cytoplasm and plasma membrane (Fig. 4). Substitution of R91 with other amino acids had variable effects on CRAC channel function with hydrophobic residues exerting the strongest inhibitory effect (111). The R91W mutation does not seem to interfere with interactions between ORAI1 and STIM1 (112, 113) and does not have a strong dominant negative effect on channel function. T cells isolated from heterozygous carriers of the R91W mutation showed partially impaired Ca2+ influx ranging from approximately 30–80% of that observed in wildtype control cells depending on the extracellular Ca2+ concentration (78). By contrast, expression of a single ORAI1-R91W mutant subunit together with three wildtype ORAI1 molecules in the context of a concatenated ORAI1 tetramer almost completely abolished CRAC channel function (114). Such a negative interfering effect of the ORAI1-R91W mutation would be significant because ORAI1 was shown to form functional homotetramers (61, 62, 84) and may form heteromultimers with ORAI2 and ORAI3 based on co-immunoprecipitation experiments (104). Whether such heteromultimers between ORAI1, ORAI2, and ORAI3 are formed in vivo, however, and whether the R91W mutation interferes with their function is unclear.
Fig. 4. Mutations in ORAI1 and STIM1 in immunodeficient patients lacking store-operated Ca2+ influx.
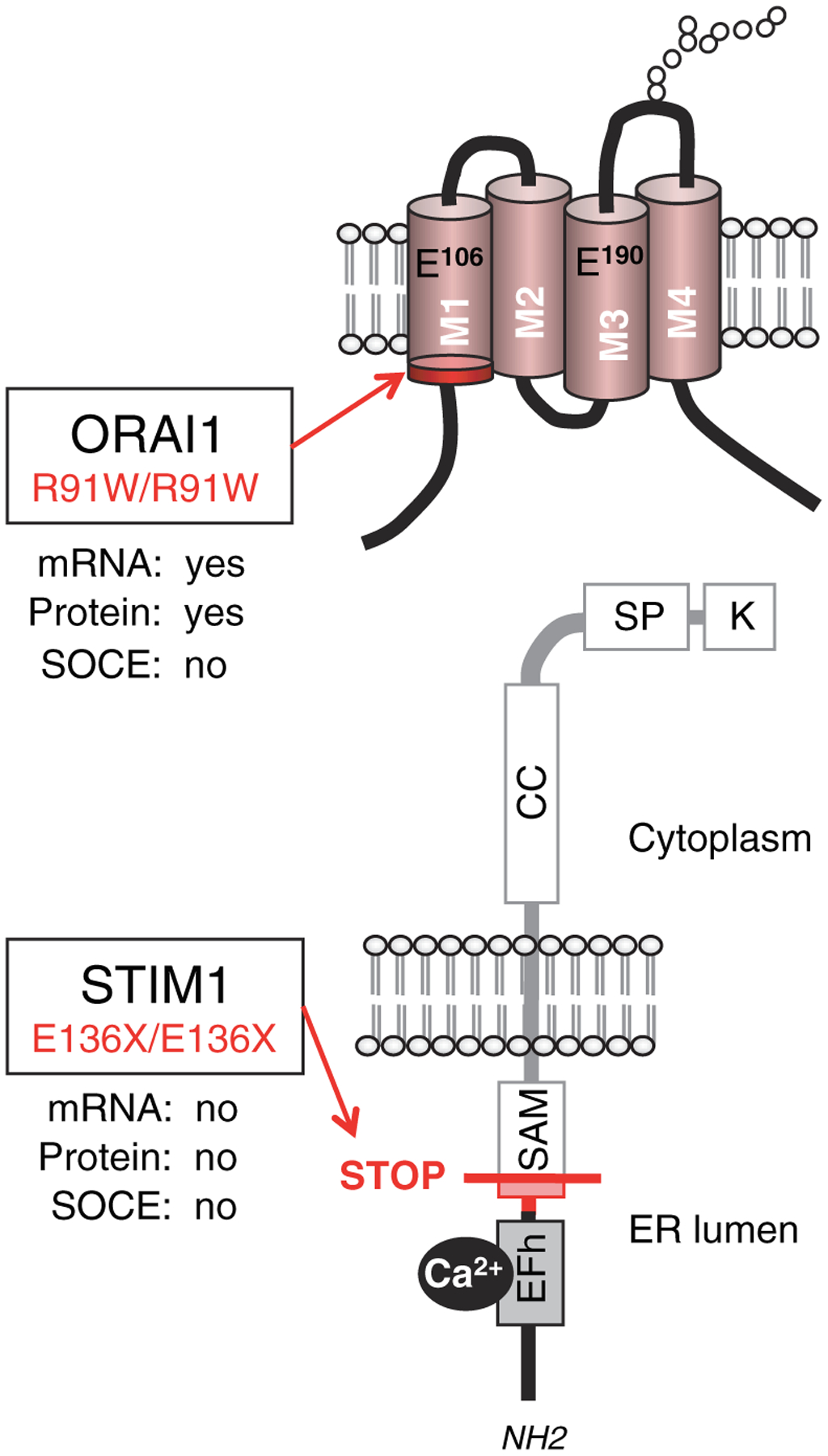
Locations of homozygous missense and nonsense mutations in ORAI1 and STIM1. An arginine to tryptophan single amino acid substitution at the cytoplasmic end of the first transmembrane domain of ORAI1 results in expression of a nonfunctional protein (ORAI1R91W) (77). An insertion mutation in codon 128 of STIM1 results in a frame shift (fs) and premature STOP (X) in codon 136; the frameshifted region of STIM1 protein is indicated in red. No STIM1 mRNA or protein is found in cells of the patient.
Clinical phenotype of ORAI1 deficiency
Lack of functional ORAI1 in human patients with ORAI1-R91W mutation is dominated clinically by immunodeficiency with severe infections early in life and, in addition, congenital myopathy and ectodermal dysplasia. Immunodeficiency is characterized by recurrent severe infections including rotavirus enteritis, BCGitis, pneumonia, meningitis, and gastrointestinal sepsis (107, 115, 116) (Table 1). Antibiotics and intravenous immunoglobulin (IVIg) only inefficiently controlled infections necessitating hematopoietic stem cell transplantation (HSCT). One patient survived after HSCT, while his older brother died before an appropriate donor could be found. Clinical symptoms of immunodeficiency in patients with mutations in ORAI1 are very similar to those observed in patients with severe combined immunodeficiency (SCID), although in contrast to the majority of SCID patients, absolute lymphocyte counts and numbers of CD4+ and CD8+ T cells and B cells were normal in ORAI1-deficient patients (Table 1). These findings suggest that lymphocyte development, positive selection of T cells in the thymus, and peripheral lymphocyte maturation are independent of ORAI1 or potentially SOCE. By contrast, activation of peripheral T cells is severely compromised, apparent in reduced or absent skin delayed-type hypersensitivity reactions in vivo and impaired proliferative responses to TCR-dependent and independent stimuli in vitro (107–109) (Table 1). While immunoglobulin levels were normal, ORAI1-deficient patients failed to mount antigen-specific antibody responses in response to vaccination and infections. Taken together, the immunodeficiency in ORAI1-deficient patients, despite its severity and life-threatening nature, is caused by the impaired function of T cells (and B cells) but not impaired lymphocyte development.
Table 1.
Clinical and immunological phenotypes of ORAI1- and STIM1-deficient patients
| ORAI1 | STIM1 | |
|---|---|---|
| Gene defect | R91W/R91W | E136X/E136X |
| Chromosome and inheritance | 12q24 homozygous (AR) | 11p15 homozygous (AR) |
| No of Patients | Two (P1, P2) | Three (P3–P5) |
| Gene expression & Ca2+ channel function | ||
| mRNA/protein | Present/present | Absent/absent |
| SOCE/ICRAC | Absent/absent | Absent/not tested |
| Cell types tested | T cells, B cells, fibroblasts | Fibroblasts |
| Immunological manifestations | ||
| Immunodeficiency & infections | P1: BCG infection, rota virus enteritis, Interstitial pneumonia, gastrointestinal sepsis P2: none; HSCT at 4 m |
P3: Pneumonia, urinary tract infections, sepsis (Escherichia coli, Streptococcus pneumoniae); cytomegalo and varizella zoster virus P4: Epstein Barr virus, viral enteritis, enteroviral encephalitis P5: sepsis; HSCT at 15 m |
| Lymphocyte counts | Normal | Normal |
| Lymphocyte subsets | Normal | CD4+ CD25+ Foxp3+ Treg ↓ |
| T cell activation (in vitro) | Proliferation ↓↓ Cytokine production ↓↓ |
Proliferation ↓-↓↓ |
| Antibody production | Normal - ↑ Ig levels (infections), no specific Ab response | Normal Ig levels, no specific Ab response |
| Autoimmunity & lymphoproliferation | No | AIHA (P3, P4), Thrombocytopenia (P3–P5), splenomegaly & lymphadenopathy (P3, P4) |
| Extraimmunological manifestations | ||
| Congenital myopathy | Yes | Yes |
| Ectodermal dysplasia | Enamel dentition defect (Hypocalcified amelogenesis imperfecta) Anhydrosis |
Enamel dentition defect |
| Other | No | P4: Nephrotic syndrome |
| Outcome | P1: Death from sepsis (11 m) P2: Survival after HSCT (now 15 y) |
P3: Death from HSCT complications (9 y) P4: Death from encephalitis (18 m) P5: Survival after HSCT (now 6 y) |
| References | 78,107,110,115,116 | 75 |
Patients lacking STIM1 and functional ORAI1 suffered from similar clinical syndromes comprising immunodeficiency early in life, congenital myopathy, and an enamel calcification defect. STIM1-deficient patients in addition presented with autoimmunity and lymphoproliferation. Ab, antibody; AIHA, autoimmune hemolytic anemia; AR, autosomal recessive; BCG, Bacille Calmette-Guérin; CMV, cytomegalovirus; ICRAC, Ca2+ release-activated Ca2+ (CRAC) channel current; HSCT, hematopoietic stem cell transplantation; m, months; P, patient; SOCE, store-operated Ca2+ entry; y, years.
In addition to the defect in immune function, ORAI1 deficiency is characterized by congenital myopathy and ectodermal dysplasia. The myopathy manifests clinically very early in life as global muscular hypotonia with decreased head control, delayed ambulation, reduced muscle strength and endurance (77, Stefan Feske, unpublished data). Chronic pulmonary disease is likely to result from impaired respiratory muscle function. The ectodermal dysplasia in ORAI1-deficient patients is characterized by impaired sweat production and a defect in dental enamel formation. Sweat provocation tests in the patient homozygous for the ORAI1-R91W mutation were abnormal, and the reduced sweat production results in dry skin and heat intolerance with recurrent fever, especially in the summer. The severely dysplastic dental enamel matrix is hypocalcified, resulting in painful exposure of the underlying yellow dentin. The phenotype is consistent with the diagnosis of ectodermal dysplasia with anhydrosis (EDA), although other symptoms of EDA such as sparse scalp hair or eyebrows were not present. The clinical phenotype of EDA suggests that SOCE mediated by ORAI1 is involved in Ca2+ transport in eccrine sweat glands and that SOCE is required for calcification of the dental enamel matrix.
Defects in SOCE and CRAC channel function were reported in additional patients from two unrelated families suffering from congenital immunodeficiency (108, 109). Patients from both families suffered from severe T-cell immunodeficiency with recurrent viral, fungal, and bacterial infections similar to the individuals with ORAI1-R91W mutation. SOCE was severely impaired in lymphocytes of all patients; in addition, ICRAC was undetectable in one of the patients. The nature of the gene defect in these patients remained unknown, because ORAI1 and STIM1 had not been identified as mediators of SOCE at the time (108, 109).
STIM1 deficiency
Mutations and molecular characterization
Lack of STIM1 resulting from RNAi in vitro or genetic ablation of Stim1 expression in mice in vivo results in a severe defect in CRAC channel activation and store-operated Ca2+ entry (53, 54, 72). We recently identified a homozygous nonsense mutation in STIM1 as the cause for immunodeficiency in three patients from one family (75). Fibroblasts from one of the patients available for analysis showed a pronounced defect in SOCE in response to thapsigargin, an inhibitor of the sarco⁄endoplasmic reticulum Ca2+ ATPase (SERCA), which induces passive depletion of intracellular Ca2+ stores. No mutations in ORAI1, ORAI2, and ORAI3 were observed in the index patient or her younger brother (no DNA of the third patient was available for analysis). Instead, both patients were homozygous for an insertion of an adenine in exon 3 of STIM1 resulting in a frameshift and premature termination at position 136 of STIM1 protein (E136X, or E128RfsX9 as the mutation leads to a frame shift beginning at codon 128 and a STOP codon nine amino acids later). We failed to detect either full-length STIM1 or the predicted N-terminal STIM1 fragment in the patients cells. Strongly reduced STIM1 mRNA transcript levels indicated that the nonsense mutation likely results in nonsense-mediated mRNA decay and a STIM1 null-phenotype. SOCE was rescued by reintroducing STIM1 into the patients’ cells, whereas expression of ORAI proteins had no effect. Ectopic expression of STIM2 was able to partially rescue SOCE, suggesting that STIM1 and STIM2 have overlapping functions; nevertheless, endogenous expression levels of STIM2 do not seem to be sufficient to compensate for the lack of STIM1 in the patients’ cells.
Clinical phenotype of STIM1 deficiency
Lack of STIM1 expression is characterized clinically by immunodeficiency, congenital myopathy, and ectodermal dysplasia reminiscent of the phenotype observed in ORAI1-deficient patients, and, in addition, autoimmune disease (75)(Table 1). The immunodeficiency in STIM1-deficient patients is marked by recurrent bacterial and viral infections. The index patient suffered from urinary tract infections, otitis media, pneumonia and multiple episodes of sepsis, caused by a spectrum of pathogens such as Streptococcus pneumoniae, Escherichia coli, cytomegalovirus, and varizella zoster virus. Her younger sister had died from encephalitis and enteroviral infection at the age of 18 months, and a younger brother suffered from sepsis during the first 2 months of his life before receiving HSCT. Total lymphocyte counts and numbers of T cells, B cells, and NK cells were normal in the patients’ blood as was the TCR repertoire in T cells of the index patient. T cells of two of the patients, however, showed a severe defect in proliferation in vitro. Serum Ig levels for all subtypes were mostly normal in two of the patients, but strongly reduced IgG levels were found in the third patient due to nephrotic syndrome. Taken together, STIM1-deficient patients – like those lacking functional ORAI1 – do not show a gross defect in lymphocyte development but are severely compromised in T-cell activation.
Immunodeficiency in all three STIM1-deficient patients was complicated by the presence of lymphoproliferative and autoimmune disease (75). Two of the patients showed lymphadenopathy and hepatosplenomegaly despite normal Fas-induced apoptosis measured in T cells of one patient. All three patients presented with thrombocytopenia and two suffered from Coombs-positive autoimmune hemolytic anemia (AIHA). The thrombocytopenia, like the anemia, is due to autoimmunity and not a defect in platelet development, because platelets were coated with autoantibodies against platelet glycoprotein Ib⁄IX and platelet numbers recovered following glucocorticoid therapy but not in response to platelet transfusions. A likely cause for the autoimmunity in the STIM1-deficient patients is the reduced number of CD4+CD25+FoxP3+ regulatory T cells (Tregs) found in the peripheral blood of the index patient (blood samples from the other patients were not available for analysis). This situation is reminiscent of mice with T-cell-specific deletion of both Stim1 and Stim2, which show greatly reduced numbers and impaired function of Treg cells (72). The mice display many signs of an autoimmune lymphoproliferative phenotype such as splenomegaly, lymphadenopathy, leukocytic organ infiltration, dermatitis, and blepharitis. In the absence of STIM1 expression in humans and that of Stim1 and Stim2 in mice, SOCE is abolished and with it the activation of the Ca2+ -regulated transcription factor NFAT (72). Binding sites for NFAT exist in the promoter of the Treg-lineage specific transcription factor Foxp3 (73), and NFAT was shown to form a complex with Foxp3 at a composite DNA-binding site in the IL-2 promoter (117). In the absence of SOCE and NFAT activation, Foxp3 expression is presumably reduced, and NFAT:Foxp3 complexes are not formed providing a potential explanation for the reduced numbers of Foxp3+ Treg cells in Stim1⁄Stim2-deficient mice and STIM1-deficient patients.
Despite reduced numbers of Tregs, STIM1-deficient patients did not present clinical features observed in X-linked immune dysregulation, polyendocrinopathy, enteropathy (IPEX) syndrome (118). The IPEX syndrome constitutes an immune disorder due to Foxp3 deficiency and is characterized by severe enteropathy and autoimmune diabetes. The absence of more severe IPEX-like symptoms in STIM1-deficient patients may be explained by the impaired antigen-specific activation of effector T cells in the absence of STIM1 and SOCE. While the reduced numbers of Treg cells are likely to be the main cause of the autoimmune phenotype in the STIM1-deficient patients, we cannot exclude that in addition lack of SOCE in developing T cells results in abnormal checkpoint control of self-reactive T cells during negative selection in the thymus. The severe defect in T-cell activation and immunodeficiency observed in STIM1-deficient patients is very similar to that seen in ORAI1-deficient patients with the notable exception of autoimmunity and reduced numbers of Treg cells.
In addition to immunodeficiency and autoimmunity, STIM1-deficient patients suffer from ectodermal dysplasia and congenital myopathy reminiscent of patients lacking functional ORAI1. Ectodermal dysplasia is characterized by a severe defect in dental enamel formation. Myopathy presented as non-progressive global muscular hypotonia and partial iris hypoplasia. A muscle biopsy and electromyography did not show abnormalities indicative of common neuropathies or myopathies. The myopathy observed in the STIM1-deficient patients correlates well with that observed in ORAI1-deficient patients, and the defect in skeletal muscle development and function found in Stim1-deficient mice (103). The myopathy is consistent with the function of ORAI1 and STIM1 in store-operated Ca2+ influx in skeletal muscle and the demonstration that STIM1 is required for myoblast differentiation (103, 119, 120).
Comparison of ORAI1 and STIM1 deficiency
The clinical phenotypes of ORAI1- and STIM1-deficient patients indicate that ORAI1 and STIM1 play important roles in CRAC channel function and SOCE during T-cell activation, skeletal muscle development and⁄or function and ectodermal derived tissues such as teeth and sweat glands (Table 2). The clinical phenotypes associated with defects in both genes overlap to a large degree, suggesting that they are not gene but pathway specific, i.e. that they result from the absence of SOCE and CRAC channel function. The severity of immunodeficiency and the spectrum of pathogens causing infections are similar in patients lacking functional ORAI1 and STIM1. Importantly, the immunodeficiency in both cases is characterized by a severe defect in T-cell activation but not a gross defect in lymphocyte development. T cells failed to proliferate in vitro and showed impaired skin delayed type hypersensitivity reactions in vivo. By contrast, total lymphocyte counts and numbers of T, B, and NK cells were normal in ORAI1- and STIM1-deficient patients, suggesting that SOCE mediated by neither gene is required for T-cell development. Although consistent with normal lymphocyte development in Stim1- and Orai1-deficient mice, this finding is surprising, because Ca2+ signals following TCR engagement are considered necessary for thymic T-cell development and selection. Potential explanations are discussed in more detail further below.
Table 2.
Summary of main clinical findings associated with CRAC channelopathy due to mutations in human STIM1 and ORAI1
| ORAI1 | STIM1 | |
|---|---|---|
| Immunodeficiency | ||
| Viral, bacterial, fungal infections | + | + |
| Autoimmunity | + | |
| Autoimmune hemolytic anemia | + | |
| Thrombocytopenia | + | |
| Lymphoadenopathy/ hepatosplenomegaly | + | |
| Congenital myopathy | + | + |
| Ectodermal dysplasia | ||
| Enamel dentition defect | + | + |
| Anhydrosis | + |
For details see Table 1.
SOCE seems required however for the development of CD4+Foxp3+ Tregs, as indicated by the reduced numbers of Tregs in the peripheral blood of one STIM1-deficient patient and mice with T-cell-specific deletion of Stim1 and Stim2 resulting in lymphoproliferative disease and autoimmunity. Autoimmunity has not been observed in ORAI1-deficient patients and mice, and Treg numbers were normal in Orai1−⁄− mice (38, 78, 106); blood samples of ORAI1-deficient patients pre-HSCT were not available for analysis of Treg cells. A possible explanation for the differential effect of ORAI1 and STIM1 deficiency on Treg development and autoimmunity may lie in the distinct residual levels of SOCE in immature T cells. In mice, lack of both STIM1 and STIM2 completely abolished SOCE in naive T cells whereas residual Ca2+ influx was observed in naive T cells from Orai1−/− mice (106). It can be speculated that small amounts of SOCE are sufficient for Treg cell development.
Comparison of CRAC channelopathy with similar immunodeficiency syndromes
ORAI1 and STIM1-deficient patients suffer from very similar non-immunological symptoms including global muscular hypotonia and ectodermal dysplasia. The combination of immunodeficiency and ectodermal dysplasia was also observed in patients with hypomorphic mutations in NFκB essential modulator (NEMO) and a hypermorphic mutation in IκBα, in both cases impairing activation of the transcription factor NF-κB (121–123). These patients suffer from severe bacterial infections early in life, despite normal numbers and subset distribution of T, B, and NK cells. One patient with mutation of Ser32 in IκBα that prevents phosphorylation and degradation of IκBα lacked γ⁄δ T cells and memory α⁄β T cells. T-cell responses following TCR stimulation were significantly impaired. Similar to ORAI1- and STIM1-deficient patients, NEMO and IκBα mutant patients also suffered from EDA, which in their case is characterized by hypodontia and conical teeth. A dental enamel calcification defect and the myopathy present in SOCE-deficient patients were not observed, suggesting that the pathophysiology of disease in both groups of patients is different (123, 124).
Role of STIM1 and ORAI1 outside the immune system
SOCE and CRAC channel function have been reported in a variety of cell types outside the immune system years before the cloning of STIM and ORAI genes (reviewed in 87). Since then, SOCE was shown to require functional STIM1 and ORAI1 in a number of different cell types and tissues such as platelets, skeletal muscle, vascular endothelium, and smooth muscle (103, 125–128). An important role of both proteins outside the immune system is emphasized by the non-immunological symptoms observed in SOCE-deficient patients (75, 78, Stefan Feske, unpublished data) and mice (38, 39, 72, 105, 125, 127, 129). The disease spectrum in both ORAI1- and STIM1-deficient patients is limited to immunodeficiency, congenital myopathy, ectodermal dysplasia, and, in the case of STIM1 deficiency, autoimmunity (75, 78, Stefan Feske, unpublished data) (Tables 2 and 3). This does not, however, exclude a role for ORAI1, STIM1, and SOCE in other cell types or organs but rather points to a non-redundant role of these genes in the affected tissues. Potential reasons for the lack of more extensive disease in the patients include that (i) ORAI1 and STIM1 have partially redundant roles in SOCE and can be functionally replaced by ORAI2, ORAI3, and STIM2 or that (ii) SOCE co-exists with non-store operated Ca2+influx in many other tissues compensating for the lack of functional ORAI1 and STIM1. As discussed earlier, in vitro studies showed that coexpression of ORAI2 and ORAI3 together with STIM1 results in large Ca2+ currents (85, 86), suggesting that both genes can form functional calcium channels. While in vivo evidence for a physiological role of endogenous ORAI2 and ORAI3 is currently missing, it is likely that both genes may play a role in SOCE in some cell types inside or outside the immune system.
Table 3.
Phenotypes of transgenic mice lacking Stim1, Stim2, and Orai1 gene expression
| Orai1 | Stim1 | Stim2 | Stim1/Stim2 | |
|---|---|---|---|---|
| Gene-targeting method | Deletion (38,106,127); knock-in (129) | Deletion (all) | Deletion | Deletion |
| gt(38, 127); hr (106,129) | gt (35,103,126,136); hr (39,72) | hr | hr | |
| Conventional (all) | Conventional (35,103,126,136) loxP: CMV-Cre (39,72); CD4-Cre (72) | loxP: CD4-Cre, CMV-Cre | loxP: CD4-Cre, CMV-Cre | |
| Viability | Perinatal death & runted (all) | Perinatal death & runted: conventional (35,103,126,136), CMV-Cre (39,72) Viable: CD4-Cre (72) |
Death 4–5 w p.p.: CMV-Cre Viable: CD4-Cre |
Viable: CD4-Cre |
| Ca2+ influx | Mast cells: ↓↓ (38) T cells: normal(38), ↓↓ (106) B cells: ↓↓ (106) Platelets: ↓↓ (127,129) |
Mast cells: ↓↓ (39) T cells: ↓↓ (72,136) Macrophages: ↓↓ (35) Platelets: ↓↓ (126) Skeletal myotubes: ↓↓ (103) |
T cells: peak normal, sustained [Ca2+]i ↓ | T cells: ↓↓ Foxp3+ Treg: ↓↓ MEF: ↓↓ |
| Immunological phenotypes | ||||
| In vitro | Mast cells: degranulation ↓↓: LTC4 synthesis ↓↓: cytokines ↓ (38) T cells: cytokines ↓ (38) to ↓↓ (106) B cells: proliferation ↓ (106) |
Mast cells: degranulation ↓, cytokines ↓ (39) T cells: cytokines ↓↓ (72) Macrophage function ↓ (35) |
T cells: cytokines ↓ | T cells: cytokines ↓↓ |
| In vivo | Anaphylaxis in vivo ↓ (38) | Anaphylaxis in vivo ↓ (39) T-depend. B cell reponse normal (136) Ability of T cells to induce GvHD normal to ↓ (136) Protection from autoimm. hemolytic anemia and thrombocytopenia (35) |
Autoimmunity (lympho-myeloproliferation) | |
| Development | Normal: T cells (38,106), Foxp3+ Treg (106), B cells (106), Mast cells (38) |
Normal: T cells (72,136), Foxp3+ Treg (72,136), Mast cells (39) |
Normal: T cells | Normal: T cells, B cells Treg (Foxp3+) ↓↓ |
| Non-immunological Phenotypes | ||||
| Myopathy (38,106) Hair loss (106) Platelet activation ↓↓ (127,129), protection from thromboischemia (127) |
Myopathy (103) Platelet activation ↓↓ (126), protection from thromboischemia (126) |
|||
| References | (38, 106, 127, 129) | (35, 39, 72, 103, 126, 136) | (72) | (72) |
This table provides a summary of mouse lines generated by homologous recombination (hr) using conventional or conditional (loxP⁄Cre) gene-targeting and insertional mutagenesis (gt, gene-trapping), respectively. All Orai1- and Stim1-deficient mice displayed early postnatal lethality and a runted phenotype. CMV, cytomegalovirus; GvHD, graft-versus-host disease; LTC4, leukotriene C4; MEF, mouse embryonic fibroblasts; p.p., post partum; w, week.
In skeletal muscle, SOCE was described and both STIM1 and ORAI1 are expressed at high levels (38, 68, 106, 130). In addition, STIM1 and ORAI1 mediate SOCE in primary cultures of murine skeletal myotubes (103, 119) and are required for differentiation of human myoblasts from isolated satellite cells, the stem cells of adult skeletal muscle (120). Myotubes from Stim1-deficient mice have a contractile defect that is associated with rapid fatigue following repeated stimulation. Importantly, the majority of mice lacking functional STIM1 expression die early postnatally; surviving mice are runted and show a myopathy that was suggested to cause or contribute to the mortality in these mice (103). The myopathy observed in Stim1-deficient animals is consistent with the congenital myopathy in human patients lacking functional ORAI1 or STIM1(75, 78), and together these observations strongly suggest that SOCE mediated by STIM1 and ORAI1 plays a somewhat unexpected but important role in skeletal muscle function and development.
SOCE is required for the development and⁄or function of ectodermal derived tissues such as sweat glands and teeth given the EDA phenotype in patients with mutations in STIM1 and ORAI1 (75, 78, Stefan Feske, unpublished data). EDA is characterized mainly by hypocalcification of the dental enamel matrix and a defect in sweat production. Dental enamel is the most highly calcified tissue in the human body, but the mechanisms used by ameloblasts, enamel epithelial cells, to transport Ca2+ to the enamel matrix are poorly understood. Several ameloblast Ca2+ transport mechanisms have been proposed, including paracellular and transcellular routes; the latter may occur via the cytoplasm of amenoblasts or via the ER facilitated by SOCE (131). Direct experimental evidence for the ER route is missing, but it seems plausible given the defect in enamel calcification in STIM1- and ORAI1-deficient patients. The secretory activity of eccrine sweat glands is known to depend on Ca2+ influx (132, 133). SOCE has been reported in a variety of epithelia including equine epithelial sweat gland cells where it occurs at the basolateral membrane and is required for anion secretion (134). Given the expression of ORAI1 in human eccrine sweat glands and the defect in sweat production in ORAI1-deficient patients (78, Stefan Feske, unpublished data), SOCE seems to be required for sweat gland function. Roles for SOCE in other tissues and cell types mediated by STIM and ORAI proteins undoubtedly exist but cannot be discussed in this review.
Mouse models of ORAI1 and STIM1 function
Mice lacking Orai1, Stim1, and Stim2 expression have been generated by a number of laboratories, and their phenotypes will be discussed here with an emphasis on the similarities and differences to human patients (Table 3). In contrast to ORAI1- and STIM1-deficient patients (75, 78), mice lacking expression of functional Orai1 and Stim1 genes die early postnatally with certain variations due to the method and genetic background used for gene-targeting. The precise cause of death is unclear, but morphological abnormalities in skeletal muscle of Stim1-deficient (103) and Orai1 knock in mice (Stefan Feske, unpublished data) as well as functional defects in myoblasts of Stim1−⁄− mice (102) are likely to cause or contribute to their severely runted phenotype (38, 103, 106, 126, 127, Stefan Feske, unpublished data). The skeletal muscle defect in mice matches the congenital myopathy observed in human patients lacking functional ORAI1 and STIM1 (75, 78). In contrast to mice, the myopathy in humans is not life-threatening, and survival of ORAI1- and STIM1-deficient patients is limited by immunodeficiency and infections.
Role of ORAI1 for SOCE and immune function in mice
Lack of Orai1 expression in mice strongly reduces the amplitude of CRAC currents in mast cells (38) and T cells (106), with residual currents observed in both cells types. As a consequence, SOCE is impaired in mast cells (38), B cells, and T cells (106) of Orai1−⁄− mice, although normal SOCE was observed in CD4+ T cells in one study (38). A likely explanation for this discrepancy is that the SOCE defect was more pronounced in CD4+ and CD8+ T cells differentiated in vitro into Th1, Th2 cells, and cytotoxic T cells, respectively, compared with freshly isolated naive CD4+ and CD8+ T cells (38, 106, Stefan Feske, unpublished data). These findings suggest that regulation of SOCE changes during T-cell differentiation. Indeed, Orai2 mRNA expression was observed in naive T cells from wildtype mice (38, 106) but decreased substantially upon differentiation into Th1 and Th2 cells (106). In naive mouse T cells, ORAI2 may therefore contribute to SOCE, whereas ORAI1 is the predominant ORAI family member in differentiated T cells (106). In human T cells, by contrast, ORAI1 is required for CRAC channel function given the complete absence of ICRAC and SOCE in T cells from immunodeficient patients lacking functional ORAI1 (78). B cells from Orai1-deficient mice showed severely impaired SOCE (106, Stefan Feske, unpublished data). The defect was however more pronounced in response to passive store depletion with thapsigargin compared with BCR stimulation by anti-IgM crosslinking (106). This finding is consistent with the recently described ORAI1- and ORAI2-independent Ca2+ influx in response to BCR stimulation and suggests the presence of a non-store operated Ca2+ influx pathway in B cells (135).
In the absence of Orai1 gene expression in mice, the function of mast cells, T cells, and B cells is compromised. Mast cell degranulation and cytokine secretion in vitro and passive cutaneous anaphylaxis in vivo are impaired in Orai1−⁄− mice (38) (Table 3). Furthermore, B cells proliferated poorly in response to BCR but not LPS stimulation in the absence of ORAI1, suggesting that Ca2+ signals are essential for antigen-dependent expansion of mouse B cells (106). Variable effects on T-cell function were observed in Orai1-deficient mice. Cytokine expression in Orai1−⁄− mice was substantially reduced for IL-2, IFN-γ, IL-4, and IL-10 in Th1 and Th2 cells, respectively (106) (Table 3). This finding is consistent with the greatly diminished cytokine gene expression found in T cells from human ORAI1-deficient patients (116). By contrast, no defect in T-cell proliferation or IL-2 and IFN-γ production was seen in another study (38). The cause of this discrepancy is not immediately clear but may have to do with the different methods used for gene targeting, genetic backgrounds of mice, or, most likely, the differentiation stage of T cells at the time of analysis.
Remarkably, the development of lymphoid and myeloid cells such as T, B, and mast cells in the thymus and bone marrow appears to be unperturbed in the absence of ORAI1 in mice. T-cell development in the thymus and B-cell development in the bone marrow and spleen appeared normal in Orai1−⁄− mice (106). These findings are consistent with normal lymphocyte numbers and subsets observed in human ORAI1-deficient patients (38, 107) (Tables 1 and 3) as well as Stim1- and Stim2- deficient mice (72), suggesting that SOCE may be dispensable for lymphocyte development.
Role of STIM1 and STIM2 for SOCE and immune function in mice
Stim1-deficient mice, like those lacking functional Orai1 expression, die early postnatally, and most studies have therefore been conducted using fetal liver chimeras (126, 136) or conditional T-cell-specific deletion of Stim1 and Stim2 using the Cre-loxP system (72). Lack of Stim1 expression abolishes CRAC channel function and SOCE in mast cells, macrophages, and CD4+ and CD8+ T cells, both naive and in vitro differentiated (35, 39, 72). By contrast, deletion of Stim2 in T cells had only a very moderate effect on SOCE and CRAC channel function, when tested in the first 5–10 min following T-cell stimulation but resulted in a marked defect in sustained Ca2+ influx and Ca2+ -dependent translocation of the transcription factor NFAT >10 min poststimulation, as discussed earlier (72). While STIM1 appears to play a greater role for CRAC channel activation than STIM2 in the immediate response to cell stimulation, STIM1 alone is not sufficient to maintain Ca2+ levels in Stim2-deficient T cells (72). A potential explanation is that STIM2 activation - compared to that of STIM1 - is (i) delayed due to the slower unfolding kinetics of its EFh-SAM domain and subsequent oligomerization (74) but (ii) maintained longer when calcium stores are refilling, thus contributing to sustained SOCE.
STIM1 is required for activation of mast cells, T cells, and macrophages (35, 39, 72), and presumably other immune cells as well. Mast cells lacking STIM1 were impaired in FscεRI-induced degranulation and cytokine production in vitro and showed an impaired passive cutaneous anaphylactic reaction in vivo (39), similar to observations made in Orai1-deficient mice (38). Stim1-deficient T cells also showed greatly reduced production of cytokines such as IL-2, IFN-γ, and IL-4, a defect that was also observed, although in milder form, in Stim2-deficient T cells, suggesting that both proteins are required for maintaining SOCE and inducing T-cell activation (72). It is likely that SOCE mediated by STIM1 is important for T-cell functions in vivo such as alloreactivity, control of viral infections, and anti-tumor responses by CD4+ and CD8+ T cells, although a recent study showed that T-cell-dependent antibody responses and graft-versus-host disease were comparable in Stim1-deficient and wildtype mice (136).
The role of Ca2+ influx for macrophage function has been controversial (36, 37, 137), but STIM1 and SOCE may play a role in FcRγ receptor-mediated activation of macrophages and phagocytosis in vivo (35). Murine macrophages lacking Stim1 expression had severely compromised FcRγII⁄III-mediated Ca2+ influx (35) and were impaired in a number of in vivo models of autoantibody and immune complex induced macrophage function. Interestingly, when Stim1-deficient mice were injected with autoantibodies against platelets or red blood cells, they were protected from thrombocytopenia and anemia due to erythrophagocytosis by Kupffer cells in the liver. By contrast, human patients lacking STIM1 expression presented with the very diseases that Stim1-deficient mice were protected from, i.e., AIHA and thrombocytopenia (75). Apparently, lack of STIM1 and SOCE did not protect the patients’ red blood cells and platelets from macrophage-mediated phagocytosis, pointing to a potential difference in the role of STIM1 for macrophage function in human and mouse.
While T-cell activation is profoundly impaired in the absence of STIM1 and ORAI1, T-cell development is unperturbed as numbers of T and B cells in the peripheral blood of ORAI1- and STIM1-deficient patients (75, 107–109), and T and B-cell development in the thymus and bone marrow of Stim1-, Stim1⁄Stim2-, and Orai1-deficient mice were normal (38, 72, 106, 136). This finding is unexpected, because TCR-induced Ca2+ signals have been considered necessary for proper T-cell development (138, 139). Mice with defective Ca2+ signaling such as Slp76, Lat, and Itk mutant mice (140–143) have blocks at various stages of T-cell development. Furthermore, positive selection of T cells in the thymus was impaired in mice lacking the regulatory B1 subunit of the Ca2+-dependent phosphatase calcineurin (144) or the transcription factor NFAT4 (145, 146, reviewed in 45). Positive selection was also shown to be associated with Ca2+ oscillations in thymocytes (30). Most of this evidence is indirect, but collectively it suggested that SOCE as the main source of Ca2+ signaling in T cells is required for T-cell development.
Using Stim1-deficient mice (Stim1fl⁄fl CMV-Cre, crossed to ICR outbred mice to ameliorate early postnatal lethality), we found normal T-cell development in the thymus despite undetectable Ca2+ influx induced by TCR crosslinking or thapsigargin treatment in double negative, double positive, and single positive thymocytes (Masatsugu Oh-hora, Anjana Rao, Stefan Feske, unpublished data). Similar observations were made in other strains of Stim1−⁄− mice (38, 136) and in Orai1−⁄− mice (106). These findings are not easily reconciled with the idea that TCR-induced SOCE is required for T-cell development, as patients and mice lacking STIM1 and ORAI1 provide the most direct model systems to test this hypothesis. It can be speculated that ORAI1 is not required for store-operated Ca2+ influx in immature T cells, because ORAI2 and ORAI3 may mediate SOCE in those cells. However, T-cell development is also unperturbed in the absence of both STIM1 and STIM2 in double deficient mice (Stim1fl⁄fl, Stim2fl⁄fl CD4-Cre) (72). If the widely accepted model that STIM1 and ORAI1 constitute the elementary unit of SOCE (5) is true, then SOCE is not required for T-cell development. In immature T cells, repeated release of Ca2+ from ER Ca2+ stores or influx through non-store operated channels such as TRP channels or P2 receptors may provide the Ca2+ signal necessary for T-cell development. Further studies are necessary to investigate the nature of Ca2+ signals in T-cell development.
In contrast to normal development of T and B cells, we observed a substantial defect in the development of CD4+ Foxp3+ regulatory T cells in human patients lacking STIM1 expression and mice with T-cell-specific deletion of both Stim1 and Stim2 (Stim1fl⁄fl⁄Stim2fl⁄fl CD4-Cre mice) (72, 75). Treg numbers in the thymus, spleen, and lymph nodes of Stim1⁄Stim2-deficient mice were approximately 10% of those found in wildtype littermates. Reduced Treg cell numbers are due to an intrinsic defect in regulatory T-cell development as Stim1⁄Stim2-deficient Treg cells also failed to develop in the presence of wildtype Treg cells in mixed bone marrow chimeras (72). As discussed further above, a potential explanation for impaired development of Treg cells is the impaired Ca2+-and NFAT-dependent induction of Foxp3 expression and the impaired formation of a NFAT:Foxp3 DNA-binding complex (73, 117). Stim1-deficient mice, in contrast to human STIM1-deficient patients, have normal Treg numbers, potentially because STIM2 is not expressed at significant levels in immature human T cells to compensate for a lack of STIM1 in contrast to mouse T cells. That said, STIM2 protein levels in naive mouse T cells were below the detection limit in our experiments, and it remains to be addressed whether STIM2 is functional in immature mouse T cells. Treg cell development in Orai1-deficient mice is normal despite impaired SOCE in T cells (106, Stefan Feske, unpublished data), and it can be speculated that residual SOCE in Orai1-deficient immature T cells in the thymus (potentially mediated by ORAI2 or ORAI3) is sufficient for Treg development.
Impaired development of functional Treg cells in Stim1⁄Stim2-deficient mice and STIM1-deficient patients results in an autoimmune, lympho-myeloproliferative phenotype characterized by hepatosplenomegaly and lymphadenopathy (72, 75). In mice, this phenotype can be prevented by transfer of wildtype Treg cells into 2-week-old Stim1⁄Stim2-deficient mice, indicating that the lympho-myeloproliferative disease is mainly caused by the regulatory T-cell defect. In addition, ORAI1-deficient human patients and mice that have normal numbers of Treg cells lacked overt signs of autoimmunity and lymphoproliferation (78, 106). Disease manifestations due to Treg cell deficiency vary between human patients and mice. In STIM1-deficient patients, autoimmunity presents as autoimmune hemolytic anemia and thrombocytopenia that was not observed in mice. Conversely, leukocytic infiltration of liver and lung, dermatitis, blepharitis, and colitis were not detected in the patients (72). Taken together, the mouse models for ORAI1 and STIM1⁄STIM2 function in combination with the characterization of ORAI1- and STIM1-deficient patients have greatly enhanced our understanding of Ca2+ signaling and SOCE in immune function in vivo and yielded some unexpected results regarding the role of SOCE in T-cell development.
The role of Ca2+ influx in immunodeficiency and autoimmune and inflammatory diseases
Immunodeficiency
The importance of SOCE for adaptive immune responses is unequivocal given the severe immunodeficiency observed in patients with mutations in STIM1 and ORAI1 (75, 78) and the functional defects in T cells from Orai1-, Stim1-, and Stim2-deficient mice (72, 106). Aside from T cells, functional defects in other cell types of the adaptive and innate immune system cannot be ruled out to contribute to immunodeficiency in the patients. This assumption is plausible as STIM1 and ORAI1 are expressed in practically all lymphoid and myeloid cells and a role of STIM1 and ORAI1 in SOCE has been demonstrated in B cells (135), macrophages (35), and mast cells (38, 39). In addition, defects in Ca2+ signaling unrelated to STIM1 and ORAI1 are likely to contribute to the pathology of other forms of immunodeficiency (reviewed in 2). Ca2+ influx was found to be impaired in T and B cells of patients suffering from X-linked agammaglobulinemia (XLA), common variable immunodeficiency (CVID), and Wiskott–Aldrich syndrome (WAS) due to mutations in Bruton’s tyrosine kinase (BTK) (147, 148), CD19 (149), and WAS protein (WASP), respectively (149, reviewed in 2). CD19 functions by amplifying BCR signaling including Ca2+ influx and loss of CD19 function in human patients and mice results in defective Ca2+ influx, Ig class switching, and reduced numbers of memory B cells (149, 151, 152). WASP, apart from being a key regulator of F-actin polymerization, has a role in signal transduction in T cells and is involved in, for instance, the regulation of transcription factors NFAT, Erk, Elk1, and c-fos, and IL-2 production (153). While WASP controls Ca2+ signaling most likely by acting upstream of store depletion, its homologue WAVE2 (WASP-family verprolin homologous protein 2) has been proposed to directly regulate CRAC channel activation (154). Thus, mutations in a number of genes impair efficient activation of Ca2+ influx in lymphocytes and contribute to the severity of immunodeficiency disease.
Autoimmune disease and inflammation
Abnormal lymphocyte Ca2+ signaling was observed in several models of autoimmune disease including those for systemic lupus erythematosus (SLE) (2). Removing negative regulators of BCR signaling, thereby modulating the strength of the BCR Ca2+ signal, resulted in the occurrence of autoimmunity in a number of transgenic mouse models. Lack of FcγRIIB (155), SH2 domain-containing phosphatase 1 (SHP1) (156), LYN (157), or CD22 (158, 159) is associated with autoantibody production or autoimmune disease in mice. Mature follicular B cells but not transitional stage 1 B cells from lyn−⁄− and cd22−⁄− mice showed substantially enhanced Ca2+ influx, suggesting that the CD22–LYN–SHP1 pathway attenuates Ca2+ responses and B-cell activation in mature B cells and thereby maintains B-cell tolerance (160). Furthermore, Duty et al. (161) observed a subset of autoantibody-producing IgD+ mature B cells that had greatly reduced Ca2+ responses, a finding that was interpreted to indicate that these cells are kept in an anergic state to prevent autoreactivity. It should be emphasized, though, that while these observations are intriguing, the evidence is mostly circumstantial, because Ca2+ influx is likely to be one of several signaling pathways modulated in the studies described above. In addition, it is difficult to prove that altered Ca2+ signaling in T and B cells in fact causes autoimmune disease. Conversely however, inhibition of Ca2+ influx has been reported to have an attenuating effect on autoimmunity and inflammation in several animal models. Pharmacological inhibition of K+ channels expressed in T cells significantly ameliorated disease outcome in animal models of multiple sclerosis (25, 162, 163). This effect is indirect, because in the absence of K+ channel function, the negative plasma membrane potential required for passive Ca2+ influx is dissolved. Inhibition of either of the two K+ channels expressed in T cells, the voltage-gated potassium channel Kv1.3 (KCNA3) and the intermediate conductance calcium-activated potassium channel IKCa1 (KCNN4), impairs SOCE. As the main effect of K+ channel inhibition is the attenuation of Ca2+ influx, it can be surmised that Ca2+ signals are necessary for the autoreactive functions of T cells.
T cells, mast cells, and macrophages are essential mediators of pathophysiological processes in a number of inflammatory diseases including asthma, atherosclerosis, and colitis. Inhibition of Ca2+ influx in these proinflammatory cells has been shown to ameliorate disease in a number of animal models. In atherosclerosis, T cells infiltrate atherosclerotic plaques, where they are activated by plasmacytoid dendritic cells resulting in activation of macrophages and killing of endothelial cells. Inhibition of IKCa1 K+ channels in T cells, macrophages, and vascular smooth muscle cells was able to suppress the development of atherosclerosis in the Apoe−⁄− mouse model (164). The non-store operated Ca2+ channel, transient receptor potential family member M2 (TRPM2) was recently shown to be involved in monocyte Ca2+ signaling, chemokine production, and activation of inflammatory neutrophils in a mouse model of colitis in which mucosal neutrophil infiltration and ulceration were attenuated in Trpm2−⁄− mice (165). Asthma is an inflammatory immune disease in which mast cells and T cells play a central role in disease pathology (reviewed in 166, 167), and it seems likely that SOCE mediated by STIM1 and ORAI1 is required for activation of both cell types during disease initiation and progression (38, 39, 72, 106). Mast cells and T cells from Stim1- and Orai1-deficient mice were severely impaired in cytokine production (38, 39, 72, 106), and mast cells in addition showed defects in degranulation in vitro upon FcεRI stimulation and anaphylactic responses in vivo (38, 39). Evidence for an involvement of Ca2+ influx in asthma also comes from Slat-deficient mice that lack Ca2+ store depletion and influx and that are impaired in their ability to mount both Th1 and Th2 inflammatory responses in their lungs (168). More roles for SOCE, STIM1, and ORAI1 in autoimmune and inflammatory diseases are likely to be discovered in the future.
Conclusion
With the molecular identification of STIM1 and ORAI1, the mechanisms and role of SOCE and CRAC channel function in cells of the immune system and other tissues are far better understood than just a few years ago. In vitro experiments showed that ORAI1 and STIM1 are sufficient to mediate SOCE through CRAC channels, and many details regarding the structure, functional domains, and interactions of STIM1 with ORAI1 have emerged. The details of how ORAI1 functions as the CRAC channel and how it is gated by STIM1 need to be elucidated. Significant insight can be expected from solving the crystal structure of both STIM1 and ORAI1. Most research has focused on ORAI1 and STIM1 both in vitro and in vivo, where several transgenic mouse models for Stim1 and Orai1 have been generated. In addition, the identification of patients with hypomorphic mutations in ORAI1 and STIM1 has illustrated that both genes are indispensable for lymphocyte function, skeletal muscle function, and dental enamel formation. The function and in vivo role of the paralogues ORAI2, ORAI3, and STIM2 remain largely unresolved, although they seem to have overlapping functions with ORAI1 and STIM1 in vitro. All five molecules are expressed in a large variety of tissues, and it needs to be determined if different combinations of STIM and ORAI proteins mediate SOCE in diverse tissues or if STIM1 and ORAI1 are the main regulators of SOCE. An exclusive role for STIM1 and ORAI1 is unlikely given the observation of SOCE in many cell types and the limited spectrum of disease in patients lacking functional ORAI1 and STIM1 comprising ‘only’ immunodeficiency, ectodermal dysplasia, and myopathy.
In the immune system, SOCE and expression of STIM1 and ORAI1 have been observed in many cell types involved in innate and adaptive immune responses. Lack of STIM1 or ORAI1 in human patients and mice strongly compromises T-cell function, but data from mouse models indicate that mast cell, phagocyte, and B-cell function are also impaired in the absence of SOCE. Nevertheless, the role of SOCE, ORAI1, and STIM1 in adaptive and innate immune responses is not well understood yet. While functional responses of immune cells are impaired their development from hematopoietic stem cells seems unperturbed, which is especially puzzling for T cells because Ca2+ signals have been implicated in T-cell development and thymic selection. Other Ca2+ channels need to be considered and the role of Ca2+ released from ER stores revisited during T-cell development. By contrast, lack of SOCE interferes with the development of regulatory T cells both in human patients and mice, although the mechanisms underlying this specific Ca2+ requirement for Treg cell differentiation remain unclear. Similarly, it is not known how SOCE is regulated in different types of T cells such as Th1, Th2, Th17, and Treg cells and whether altering SOCE in these subsets is involved in shifting the balance between normal immune responses and autoimmunity and inflammation. Given the strong attenuation of T-cell and mast cell function in the absence of STIM1 and ORAI1, it is not unreasonable to expect that inhibition of SOCE will have beneficial therapeutic effects in inflammatory and autoimmune diseases.
Acknowledgements
This work was supported by grants from the March of Dimes Foundation and National Institutes of Health (NIH). Special thanks to Jens Röther for ORAI1 expression analysis (Fig. 3). The author is a scientific co-founder of CalciMedica, a biotechnology company that seeks to develop CRAC channel inhibitors.
References
- 1.Oh-hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol 2008;20:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feske S Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 2007;7:690–702. [DOI] [PubMed] [Google Scholar]
- 3.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA 1993;90: 6295–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992;355:353–356. [DOI] [PubMed] [Google Scholar]
- 5.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature 2007;446:284–287. [DOI] [PubMed] [Google Scholar]
- 6.Gwack Y, Feske S, Srikanth S, Hogan P, Rao A. Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 2007;42:145–156. [DOI] [PubMed] [Google Scholar]
- 7.Uchida J, et al. Mouse CD20 expression and function. Int Immunol 2004;16:119–129. [DOI] [PubMed] [Google Scholar]
- 8.Fujimoto M, Poe JC, Hasegawa M, Tedder TF. CD19 amplification of B lymphocyte Ca2+ responses: a role for Lyn sequestration in extinguishing negative regulation. J Biol Chem 2001;276:44820–44827. [DOI] [PubMed] [Google Scholar]
- 9.Tsitsikov EN, Gutierrez-Ramos JC, Geha RS. Impaired CD19 expression and signaling, enhanced antibody response to type II T independent antigen and reduction of B-1 cells in CD81-deficient mice. Proc Natl Acad Sci USA 1997;94: 10844–10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Partida-Sanchez S, et al. Chemotaxis and calcium responses of phagocytes to formyl peptide receptor ligands is differentially regulated by cyclic ADP ribose. J Immunol 2004;172:1896–1906. [DOI] [PubMed] [Google Scholar]
- 11.Schorr W, Swandulla D, Zeilhofer HU. Mechanisms of IL-8-induced Ca2+ signaling in human neutrophil granulocytes. Eur J Immunol 1999;29:897–904. [DOI] [PubMed] [Google Scholar]
- 12.Hsu S, et al. Fundamental Ca2+ signaling mechanisms in mouse dendritic cells: CRAC is the major Ca2+ entry pathway. J Immunol 2001;166:6126–6133. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Lu ZH, Gabius HJ, Rohowsky-Kochan C, Ledeen RW, Wu G. Cross-linking of GM1 ganglioside by galectin-1 mediates regulatory T cell activity involving TRPC5 channel activation: possible role in suppressing experimental autoimmune encephalomyelitis. J Immunol 2009;182: 4036–4045. [DOI] [PubMed] [Google Scholar]
- 14.Philipp S, et al. TRPC3 mediates T-cell receptor-dependent calcium entry in human T-lymphocytes. J Biol Chem 2003;278:26629–26638. [DOI] [PubMed] [Google Scholar]
- 15.Freedman BD, Liu QH, Gaulton G, Kotlikoff MI, Hescheler J, Fleischmann BK. ATP-evoked Ca2+ transients and currents in murine thymocytes: possible role for P2X receptors in death by neglect. Eur J Immunol 1999;29:1635–1646. [DOI] [PubMed] [Google Scholar]
- 16.Yip L, et al. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J 2009;23:1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osipchuk Y, Cahalan MD. Cell-to-cell spread of calcium signals mediated by ATP receptors in mast cells. Nature 1992;359:241–244. [DOI] [PubMed] [Google Scholar]
- 18.Ross PE, Ehring GR, Cahalan MD. Dynamics of ATP-induced calcium signaling in single mouse thymocytes. J Cell Biol 1997;138:987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badou A, et al. Critical role for the beta regulatory subunits of Cav channels in T lymphocyte function. Proc Natl Acad Sci USA 2006;103:15529–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stokes L, Gordon J, Grafton G. Non-voltage-gated L-type Ca2+ channels in human T cells: pharmacology and molecular characterization of the major alpha pore-forming and auxiliary beta-subunits. J Biol Chem 2004;279:19566–19573. [DOI] [PubMed] [Google Scholar]
- 21.Zeng W, Yuan J, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell 2008;32: 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem 2008;283:12935–12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barbet G, et al. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol 2008;9:1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP. TRPM4 regulates calcium oscillations after T cell activation. Science 2004;306:1374–1377. [DOI] [PubMed] [Google Scholar]
- 25.Chandy GK, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 2004;25:280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bautista DM, Lewis RS. Modulation of plasma membrane calcium-ATPase activity by local calcium microdomains near CRAC channels in human T cells. J Physiol 2004;556:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engelke M, Engels N, Dittmann K, Stork B, Wienands J. Ca2+ signaling in antigen receptor-activated B lymphocytes. Immunol Rev 2007;218:235–246. [DOI] [PubMed] [Google Scholar]
- 28.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A 2001;98:13886–13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature 1994;368: 70–73. [DOI] [PubMed] [Google Scholar]
- 30.Bhakta NR, Oh DY, Lewis RS. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol 2005;6:143–151. [DOI] [PubMed] [Google Scholar]
- 31.Delon J, Bercovici N, Liblau R, Trautmann A. Imaging antigen recognition by naive CD4+ T cells: compulsory cytoskeletal alterations for the triggering of an intracellular calcium response. Eur J Immunol 1998;28:716–729. [DOI] [PubMed] [Google Scholar]
- 32.Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity 1996;4:421–430. [DOI] [PubMed] [Google Scholar]
- 33.Lyubchenko TA, Wurth GA, Zweifach A. Role of calcium influx in cytotoxic T lymphocyte lytic granule exocytosis during target cell killing. Immunity 2001;15:847–859. [DOI] [PubMed] [Google Scholar]
- 34.Lew DP, Andersson T, Hed J, Di Virgilio F, Pozzan T, Stendahl O. Ca2+-dependent and Ca2+-independent phagocytosis in human neutrophils. Nature 1985;315:509–511. [DOI] [PubMed] [Google Scholar]
- 35.Braun A, et al. STIM1 is essential for Fc receptor activation and autoimmune inflammation. Blood 2008;113:1097–1104. [DOI] [PubMed] [Google Scholar]
- 36.Hishikawa T, Cheung JY, Yelamarty RV, Knutson DW. Calcium transients during Fc receptor-mediated and nonspecific phagocytosis by murine peritoneal macrophages. J Cell Biol 1991;115:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg S, el Khoury J, di Virgilio F, Kaplan EM, Silverstein SC. Ca2+-independent F-actin assembly and disassembly during Fc receptor-mediated phagocytosis in mouse macrophages. J Cell Biol 1991;113:757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vig M, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol 2008;9:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol 2008;9:81–88. [DOI] [PubMed] [Google Scholar]
- 40.Hide M, Beaven MA. Calcium influx in a rat mast cell (RBL-2H3) line. Use of multivalent metal ions to define its characteristics and role in exocytosis. J Biol Chem 1991;266:15221–15229. [PubMed] [Google Scholar]
- 41.Penner R, Neher E. The role of calcium in stimulus-secretion coupling in excitable and non-excitable cells. J Exp Biol 1988;139:329–345. [DOI] [PubMed] [Google Scholar]
- 42.Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol 2007;19:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keefe D, et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 2005;23:249–262. [DOI] [PubMed] [Google Scholar]
- 44.Savignac M, Mellström B, Naranjo JR. Calcium-dependent transcription of cytokine genes in T lymphocytes. Pflugers Arch 2007;454:523–533. [DOI] [PubMed] [Google Scholar]
- 45.Macian F NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005;5:472–484. [DOI] [PubMed] [Google Scholar]
- 46.Hogan P Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003;17:2205–2232. [DOI] [PubMed] [Google Scholar]
- 47.Feske S, Okamura H, Hogan PG, Rao A. Ca2+⁄calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun 2003;311:1117–1132. [DOI] [PubMed] [Google Scholar]
- 48.Leitenberg D, Bottomly K. Regulation of naive T cell differentiation by varying the potency of TCR signal transduction. Semin Immunol 1999;11:283–292. [DOI] [PubMed] [Google Scholar]
- 49.Bandyopadhyay S, Soto-Nieves N, Macián F. Transcriptional regulation of T cell tolerance. Semin Immunol 2007;19:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borde M, Barrington RA, Heissmeyer V, Carroll MC, Rao A. Transcriptional basis of lymphocyte tolerance. Immunol Rev 2006;210:105–119. [DOI] [PubMed] [Google Scholar]
- 51.Heissmeyer V, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol 2004;5:255–265. [DOI] [PubMed] [Google Scholar]
- 52.Oh-Hora M Calcium signaling in the development and function of T lineage cells. Immunol Rev 2009;231:210–224. [DOI] [PubMed] [Google Scholar]
- 53.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 2005;169:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 2005;15:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008;135: 110–122. [DOI] [PubMed] [Google Scholar]
- 56.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 2007;104:9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depeltion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 2006;174:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol 2006;174:815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008;454:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang GN, et al. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat Cell Biol 2006;8:1003–1010. [DOI] [PubMed] [Google Scholar]
- 61.Penna A, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 2008;456: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ji W, et al. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA 2008;105:13668–13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan J, Zeng W, Dorwart MR, Choi Y, Worley P, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 2009;11: 337–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009;136: 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muik M, et al. A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem 2009;284:8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun 2009;385:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muik M, et al. A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J Biol Chem 2008;284: 8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams RT, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J 2001;357:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manji SS, et al. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta 2000;1481:147–155. [DOI] [PubMed] [Google Scholar]
- 70.Soboloff J, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol 2006;16:1465–1470. [DOI] [PubMed] [Google Scholar]
- 71.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007;131:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oh-Hora M, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 2008; 9:432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol 2008;9:194–202. [DOI] [PubMed] [Google Scholar]
- 74.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem 2009;284:728–732. [DOI] [PubMed] [Google Scholar]
- 75.Picard C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 2009;360: 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang SL, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA 2006;103:9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006;312:1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006;441: 179–185. [DOI] [PubMed] [Google Scholar]
- 79.Cai X Molecular evolution and structural analysis of the Ca2+ release-activated Ca2+ channel subunit, Orai. J Mol Biol 2007;368: 1284–1291. [DOI] [PubMed] [Google Scholar]
- 80.Yeromin AV, Zhang S, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006;443: 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vig M, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol 2006;16:2073–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006;443:230–233. [DOI] [PubMed] [Google Scholar]
- 83.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium 2003;33: 311–321. [DOI] [PubMed] [Google Scholar]
- 84.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol 2008; 586:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lis A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol 2007;17:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DeHaven WI, Smyth JT, Boyles RR, Putney JW. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem 2007;282:17548–17556. [DOI] [PubMed] [Google Scholar]
- 87.Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev 2005;85:757–810. [DOI] [PubMed] [Google Scholar]
- 88.Vaca L, Kunze DL. Depletion of intracellular Ca2+ stores activates a Ca2+-selective channel in vascular endothelium. Am J Physiol 1994;267:C920–C925. [DOI] [PubMed] [Google Scholar]
- 89.Fasolato C, Nilius B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embryonic kidney cell lines. Pflugers Arch 1998; 436:69–74. [DOI] [PubMed] [Google Scholar]
- 90.Freichel M, et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−⁄− mice. Nat Cell Biol 2001;3:121–127. [DOI] [PubMed] [Google Scholar]
- 91.Gibson A, McFadzean I, Wallace P, Wayman CP. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends Pharmacol Sci 1998;19:266–269. [DOI] [PubMed] [Google Scholar]
- 92.McFadzean I, Gibson A. The developing relationship between receptor-operated and store-operated calcium channels in smooth muscle. Br J Pharmacol 2002; 135:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krause E, Pfeiffer F, Schmid A, Schulz I. Depletion of intracellular calcium stores activates a calcium conducting nonselective cation current in mouse pancreatic acinar cells. J Biol Chem 1996;271: 32523–32528. [DOI] [PubMed] [Google Scholar]
- 94.Rychkov G, Brereton HM, Harland ML, Barritt GJ. Plasma membrane Ca2+ release-activated Ca2+ channels with a high selectivity for Ca2+ identified by patch-clamp recording in rat liver cells. Hepatology 2001;33:938–947. [DOI] [PubMed] [Google Scholar]
- 95.Fomina AF, Nowycky MC. A current activated on depletion of intracellular Ca2+ stores can regulate exocytosis in adrenal chromaffin cells. J Neurosci 1999;19: 3711–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peinelt C, Beck A, Monteilh-Zoller MK, Penner R, Fleig A. IP receptor subtype-dependent activation of store-operated calcium entry through ICRAC. Cell Calcium 2009;45:326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu QH, et al. Distinct calcium channels regulate responses of primary B lymphocytes to B cell receptor engagement and mechanical stimuli. J Immunol 2005;174:68–79. [DOI] [PubMed] [Google Scholar]
- 98.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol 2001;536:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Semenova SB, Kiselev KI, Mozhaeva GN. Low-conductivity calcium channels in the macrophage plasma membrane: activation by inositol-1,4,5-triphosphate. Neurosci Behav Physiol 1999;29:339–345. [DOI] [PubMed] [Google Scholar]
- 100.Hess SD, Oortgiesen M, Cahalan MD. Calcium oscillations in human T and natural killer cells depend upon membrane potential and calcium influx. J Immunol 1993;150:2620–2633. [PubMed] [Google Scholar]
- 101.BioGPS. http://biogps.gnf.org Accessed 15 May 2009.
- 102.Lattin JE, et al. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res 2008;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stiber J, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol 2008;10:688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gwack Y, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem 2007;282:16232–16243. [DOI] [PubMed] [Google Scholar]
- 105.Huang YH, Hoebe K, Sauer K. New therapeutic targets in immune disorders: ItpkB, Orai1 and UNC93B. Expert Opin Ther Targets 2008;12:391–413. [DOI] [PubMed] [Google Scholar]
- 106.Gwack Y, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol 2008;28:5209–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Feske S, et al. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur J Immunol 1996;26:2119–2126. [DOI] [PubMed] [Google Scholar]
- 108.Le Deist F, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood 1995;85:1053–1062. [PubMed] [Google Scholar]
- 109.Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem 1994;269:32327–32335. [PubMed] [Google Scholar]
- 110.Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med 2005;202:651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Derler I, et al. Increased hydrophobicity at the N-terminus⁄membrane interface impairs gating of the SCID-related ORAI1 mutant. J Biol Chem 2009;284:15903–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol 2008;586: 5383–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muik M, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem 2008;283:8014–8022. [DOI] [PubMed] [Google Scholar]
- 114.Thompson J, Mignen O, Shuttleworth T. The Orai1 SCID mutation and CRAC channel function in the heterozygous condition. J Biol Chem 2009;284:6620–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schlesier M, et al. Primary severe immunodeficiency due to impaired signal transduction in T cells. Immunodeficiency 1993;4:133–136. [PubMed] [Google Scholar]
- 116.Feske S, Draeger R, Peter HH, Eichmann K, Rao A. The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J Immunol 2000;165:297–305. [DOI] [PubMed] [Google Scholar]
- 117.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006;126:375–387. [DOI] [PubMed] [Google Scholar]
- 118.Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res 2007;38:112–121. [DOI] [PubMed] [Google Scholar]
- 119.Lyfenko A, Dirksen R. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol 2008;586:4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Darbellay B, et al. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J Biol Chem 2008;284:5370–5380. [DOI] [PubMed] [Google Scholar]
- 121.Courtois G, et al. A hypermorphic IkappaB-alpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest 2003;112:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Doffinger R, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 2001;27:277–285. [DOI] [PubMed] [Google Scholar]
- 123.Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol 2004;16:34–41. [DOI] [PubMed] [Google Scholar]
- 124.Ku CL, et al. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics 2005;115:e615–e619. [DOI] [PubMed] [Google Scholar]
- 125.Potier M, et al. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J 2009; doi: 10.1096/fj.09-131128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Varga-Szabo D, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med 2008;205:1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Braun A, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 2009;113: 2056–2063. [DOI] [PubMed] [Google Scholar]
- 128.Abdullaev I, Bisaillon J, Potier M, Gonzalez J, Motiani R, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res 2008;103: 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bergmeier W, Oh-Hora M, McCarl CA, Roden RC, Bray PF, Feske S. R93W mutation in Orai1 causes impaired calcium influx in platelets. Blood 2009;113: 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kurebayashi N, Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J Physiol 2001;533:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hubbard MJ. Calcium transport across the dental enamel epithelium. Crit Rev Oral Biol Med 2000;11:437–466. [DOI] [PubMed] [Google Scholar]
- 132.Sato K, Sato F. Relationship between quin-2 determined cytosolic [Ca2+] and sweat secretion. Am J Phys 1988;254:C310–C317. [DOI] [PubMed] [Google Scholar]
- 133.Prompt CA. Functions of calcium in sweat secretion. Nature 1978;272:171–172. [DOI] [PubMed] [Google Scholar]
- 134.Ko WH, Chan HC, Wong PY. Anion secretion induced by capacitative Ca2+ entry through apical and basolateral membranes of cultured equine sweat gland epithelium. J Physiol 1996;497 (Pt 1):19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Morita T, Tanimura A, Baba Y, Kurosaki T, Tojyo Y. A Stim1-dependent, noncapacitative Ca2+-entry pathway is activated by B-cell-receptor stimulation and depletion of Ca2+. J Cell Sci 2009;122:1220–1228. [DOI] [PubMed] [Google Scholar]
- 136.Beyersdorf N, et al. STIM1-independent T cell development and effector function in vivo. J Immunol 2009;182:3390–3397. [DOI] [PubMed] [Google Scholar]
- 137.McNeil PL, Swanson JA, Wright SD, Silverstein SC, Taylor DL. Fc-receptor-mediated phagocytosis occurs in macrophages without an increase in average [Ca2+]i. J Cell Biol 1986;102:1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Aifantis I, Gounari F, Scorrano L, Borowski C, Von Boehmer H. Constitutive pre-TCR signaling promotes differentiation through Ca2+ mobilization and activation of NF-kappaB and NFAT. Nat Immunol 2001;2:403–409. [DOI] [PubMed] [Google Scholar]
- 139.Von Boehmer H, Fehling HJ. Structure and function of the pre-T cell receptor. Annu Rev Immunol 1997;15:433–452. [DOI] [PubMed] [Google Scholar]
- 140.Zhang W, et al. Essential role of LAT in T cell development. Immunity 1999;10: 323–332. [DOI] [PubMed] [Google Scholar]
- 141.Pivniouk V, Tsitsikov EN, Swinton P, Rathbun G, Alt FW, Geha RS. Impaired viability and profound block in thymocyte development in mice lacking the adaptor protein SLP-76. Cell 1998;94: 229–238. [DOI] [PubMed] [Google Scholar]
- 142.Clements JL, et al. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science 1998;281:416–419. [DOI] [PubMed] [Google Scholar]
- 143.Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity 1995; 3:757–769. [DOI] [PubMed] [Google Scholar]
- 144.Neilson JR, Winslow MM, Hur EM, Crabtree GR. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity 2004;20:255–266. [DOI] [PubMed] [Google Scholar]
- 145.Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity 1998; 9:295–304. [DOI] [PubMed] [Google Scholar]
- 146.Cante-Barrett K, Winslow MM, Crabtree GR. Selective role of NFATc3 in positive selection of thymocytes. J Immunol 2007;179: 103–110. [DOI] [PubMed] [Google Scholar]
- 147.Fluckiger AC, et al. Btk⁄Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J 1998;17:1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Takata M, Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med 1996;184:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.van Zelm MC, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med 2006;354:1901–1912. [DOI] [PubMed] [Google Scholar]
- 150.Simon HU, Mills GB, Hashimoto S, Siminovitch KA. Evidence for defective transmembrane signaling in B cells from patients with Wiskott-Aldrich syndrome. J Clin Invest 1992;90:1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang Y, Carter RH. CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity 2005;22:749–761. [DOI] [PubMed] [Google Scholar]
- 152.Fehr T, et al. Antiviral protection and germinal center formation, but impaired B cell memory in the absence of CD19. J Exp Med 1998;188:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cianferoni A, et al. Defective nuclear translocation of nuclear factor of activated T cells and extracellular signal-regulated kinase underlies deficient IL-2 gene expression in Wiskott-Aldrich syndrome. J Allergy Clin Immunol 2005;116:1364–1371. [DOI] [PubMed] [Google Scholar]
- 154.Nolz JC, et al. The WAVE2 complex regulates actin cytoskeletal reorganization and CRAC-mediated calcium entry during T cell activation. Curr Biol 2006;16:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000;13:277–285. [DOI] [PubMed] [Google Scholar]
- 156.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol 1997;15: 302–307. [DOI] [PubMed] [Google Scholar]
- 157.Hibbs ML, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 1995;83:301–311. [DOI] [PubMed] [Google Scholar]
- 158.O’Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med 1999;189: 1307–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sato S, et al. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity 1996;5:551–562. [DOI] [PubMed] [Google Scholar]
- 160.Gross AJ, Lyandres JR, Panigrahi AK, Prak ET, DeFranco AL. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol 2009;182:5382–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Duty JA, et al. Functional anergy in a sub-population of naive B cells from healthy humans that express autoreactive immunoglobulin receptors. J Exp Med 2009;206: 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Reich EP, et al. Blocking ion channel KCNN4 alleviates the symptoms of experimental autoimmune encephalomyelitis in mice. Eur J Immunol 2005;35:1027–1036. [DOI] [PubMed] [Google Scholar]
- 163.Beeton C, et al. Selective blockade of T lymphocyte K(+) channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci USA 2001;98:13942–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Toyama K, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest 2008;118:3025–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yamamoto S, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 2008;14: 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rivera J, Fierro NA, Olivera A, Suzuki R. New insights on mast cell activation via the high affinity receptor for IgE. Adv Immunol 2008;98:85–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Meyer EH, DeKruyff RH, Umetsu DT. T cells and NKT cells in the pathogenesis of asthma. Annu Rev Med 2008;59:281–292. [DOI] [PubMed] [Google Scholar]
- 168.Bécart S, et al. SLAT regulates Th1 and Th2 inflammatory responses by controlling Ca2+⁄NFAT signaling. J Clin Invest 2007; 117:2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]