

Abstract
Cancers adapt to increasingly potent targeted therapies by reprogramming their phenotype. Here we investigated such a phenomenon in prostate cancer, in which tumours can escape epithelial lineage confinement and transition to a high-plasticity state as an adaptive response to potent androgen receptor (AR) antagonism. We found that AR activity can be maintained as tumours adopt alternative lineage identities, with changes in chromatin architecture guiding AR transcriptional rerouting. The epigenetic regulator enhancer of zeste homologue 2 (EZH2) co-occupies the reprogrammed AR cistrome to transcriptionally modulate stem cell and neuronal gene networks—granting privileges associated with both fates. This function of EZH2 was associated with T350 phosphorylation and establishment of a non-canonical polycomb subcomplex. Our study provides mechanistic insights into the plasticity of the lineage-infidelity state governed by AR reprogramming that enabled us to redirect cell fate by modulating EZH2 and AR, highlighting the clinical potential of reversing resistance phenotypes.
Increasingly potent molecular targeted therapies have altered the archetypical course of cancers, with lineage plasticity emerging as a mechanism of therapeutic resistance1,2. A fascinating example is the conversion of prostate adenocarcinomas to a neuroendocrine state, which confers resistance to currently approved targeted therapies3,4. An unintended consequence of potent AR-pathway inhibitors (ARPIs), such as enzalutamide (ENZ), has been the stereotypic expansion of tumour heterogeneity highlighted by the clinical emergence of diverse phenotypic states to bypass ARPIs5,6, including cells with heightened plasticity and divergent differentiation. Increased plasticity is a highly reproducible feature of prostate cancer tumours following ARPI therapy and is greater in tumours in which RB1 and/or TP53 are inactivated7,8. Up to 20% of advanced ARPI-refractory prostate cancers exhibit a loss of epithelial lineage identity and shortened survival5,9–13. These tumours are characterized by loss of canonical AR signalling, expression of neuroendocrine lineage markers, and activation of stem cell transcriptional programmes9,14,15. While end-state small cell prostate cancer lacks AR, a high prevalence (>50%) of treatment-resistant tumours with molecular neuroendocrine features retain nuclear AR without activation of canonical AR signalling5,6,16,17. This plasticity can be controlled epigenetically18 with widespread changes in chromatin landscape following ARPIs enhancing tumour heterogeneity by unlocking alternative lineage programmes19. Upregulation of epigenetic reprogramming factors, including EZH2, create a stem cell-like epigenetic environment permissive for lineage plasticity7,13,20. EZH2 can function non-canonically as a co-activator of the AR through a polycomb-independent mechanism21,22. Here we establish a cooperative role for AR and EZH2 in driving the emergence of a drug-resistant, high-plasticity cell state in response to AR antagonism as a mechanism of treatment resistance.
Results
A high-plasticity state emerges following AR antagonism.
A sizeable proportion of patient tumours that relapse following treatment with ARPIs exhibit a loss of luminal identity with activation of stem cell and neuronal programmes1. The AR remains expressed in more than 50% of these patient tumours5,6,17,23, suggesting that it may continue to have a functional role in this setting. To explore this premise, we used an in vivo model of ENZ resistance24 that recapitulates clinically reported treatment-refractory phenotypes. Castration-resistant prostate cancer (CRPC) tumours (16DCRPC) treated with ENZ relapsed with AR expression but diverged with respect to canonical AR-pathway activity as measured by prostate-specific antigen (PSA, encoded by the KLK3 gene) expression (Extended Data Fig. 1a–c). ENZ-resistant AR+PSA+ cells (49FENZR cells) harbour the AR F876L mutation25 (Extended Data Fig. 1d) and exhibit activated canonical AR signalling (Extended Data Fig. 1e,f).
Conversely, ENZ-resistant AR+PSA− cells (42DENZR and 42FENZR cells) exhibited loss of canonical AR signalling with enrichment in transcriptional programmes associated with lineage plasticity (Extended Data Fig. 1e,f). This included a core set of genes that are upregulated in embryonic stem cells (Extended Data Fig. 1g) and a signature of adult epithelial stem cells (adult stem cell (ASC) score; Extended Data Fig. 1h) enriched in neuroendocrine prostate cancer (NEPC)15. Neuronal networks were activated (Extended Data Fig. 1f), reflecting the clinical manifestation of lineage plasticity often associated with neuroendocrine-like differentiation. A signature of RB1 and TP53 loss26 was also enriched (Extended Data Fig. 1i) and correlated with inactivation of RB1 by hyperphosphorylation (Extended Data Fig. 1j). These cells exhibit strong transcriptional similarity to treatment-refractory AR+ patient tumours with neuroendocrine molecular features in two clinical cohorts5,6 (Fig. 1a and Extended Data Fig. 1k). This underscores the clinical applicability of our model, and suggests that persistent AR expression may have a functional role in treatment-resistant tumours exhibiting lineage plasticity. 42D and 42F cells formed spheroids in three-dimensional (3D) culture (Extended Data Fig. 1l), co-expressed the stem-like cell marker CD44 and the neuronal lineage marker NCAM1, and exhibited elevated aldehyde dehydrogenase (ALDH) activity, a functional marker of stem and progenitor cells27 (Extended Data Fig. 1m). These data suggest that these cells represent a high-plasticity cell state with enrichment of neuroendocrine-lineage characteristics.
Fig. 1 |. The ENZ-resistant high-plasticity state possesses a distinct AR cistrome and chromatin accessibility landscape.
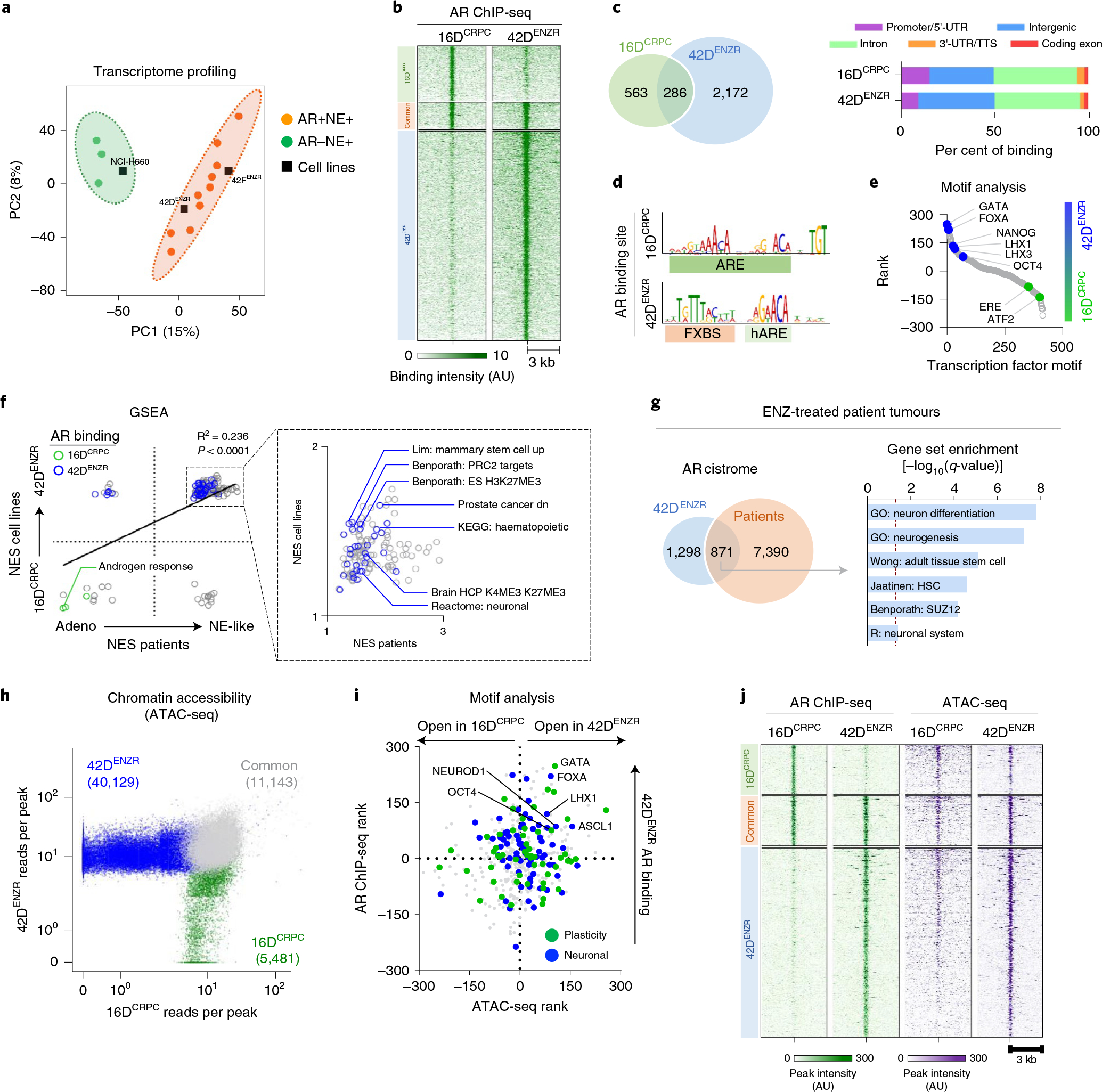
a, Partial least-squares discriminant analysis (PLS-DA) of the global transcriptome separates AR+neuroendocrine (NE)+ and AR−NE+ patient tumours from ref. 5. RNA-seq data from prostate cancer cell lines were projected onto the PLS-DA plot. Probability ellipsoid indicates the 95% confidence interval to group the samples. b, AR ChIP-seq signal intensity. Cell lines were grown in 5% FBS, with 42DENZR cells supplemented with 10 μM ENZ. The shade of green reflects the binding intensity. AU, arbitrary units. c, Overlapping AR peaks in 16DCRPC and 42DENZR cells (left) and annotations plotted as a percentage of all peaks (right). d, The most significant AR DNA-binding motif in 16DCRPC and 42DENZR cells identified by MEME-ChIP. ARE, androgen response element; hARE, half-ARE; FXBS, FOXA binding site. e, Transcription factor binding motifs within a 250-bp window surrounding AR ChIP-seq peaks in 42DENZR and 16DCRPC cells plotted by ranks generated from their associated differential P values. f, Gene set enrichment ananlysis (GSEA) pathways enriched in 42DEZNR cells (versus 16DCRPC) and treatment-induced neuroendocrine-like patient tumours (versus non-neuroendocrine tumours from ref. 5). Pathways with genes associated with AR binding (Fisher’s exact test, P < 0.05, fasle discovery rate (FDR) < 0.25) based on ChIP-seq profiles in 16DCRPC and 42DENZR cells are highlighted. NES, normalized enrichment score. g, Overlap of AR ChIP-seq peaks in 42DENZR cells and prostate tumours following three months of ENZ therapy from the DARANA trial, along with gene ontology of the shared AR-bound genes. The AR cistrome from patients represents the common AR binding sites shared between patients (n = 3). h, Scatter plot of ATAC-seq counts per peak in 16DCRPC and 42DENZR cell lines. Each dot represents an accessible region. i, Transcription factor binding motifs surrounding AR binding sites (AR ChIP-seq) and accessible chromatin (ATAC-seq) in 42DENZR versus 16DCRPC cells were ranked on the basis of differential P value. Each dot represents a motif identified by HOMER and annotated on the basis of association with plasticity (Benporath ES 1 and Wong adult tissue stem module) or neuronal (GO neurogenesis) transcription factors in MSigDB. j, Heat map indicating AR ChIP-seq and ATAC-seq signal intensity in 16DCRPC and 42DENZR cell lines. The shade of green (AR) and purple (ATACseq) reflects peak intensity. Each horizontal line represents a 6-kb locus.
The AR cistrome in treatment-resistant tumours is unique.
To decipher a possible role for AR reprogramming, we profiled the AR cistrome across CRPC (16DCRPC), ENZ-resistant AR+PSA+ (49FENZR) and ENZ-resistant AR+PSA− (42DENZR) phenotypes. Principal component analysis (PCA) revealed the AR binding pattern across the three phenotypic states to be more correlated with metastatic CRPC patient tumours than normal prostate or primary tumours28 (Extended Data Fig. 2a). The 16DCRPC and 49FENZR cells clustered together, reflecting a similar AR binding profile. By contrast, redistribution of the AR cistrome was evident between 16DCRPC and 42DENZR cells; specifically, 2,172 unique AR binding sites were detected in 42DENZR cells (Fig. 1b and Supplementary Table 1). Analysis of the AR binding profile revealed a slight bias toward intergenic regions in 42DENZR cells (Fig. 1c) and increased enrichment at a hybrid binding site comprising a hARE adjacent to a FXBS (Fig. 1d). Transcription factor motif analysis identified enrichment of stem cell (OCT4 and NANOG) and neuronal (LHX family) transcription factor-associated motifs surrounding AR peaks (Fig. 1e). The AR binding profile revealed a shift from canonical AR target genes toward neuronal-lineage transcriptional programmes (Extended Data Fig. 2b). This distinct AR cistrome remained exclusive to the high-plasticity ENZ-resistant cells when compared against AR chromatin immunoprecipitation with sequencing (ChIP-seq) datasets from prostate adenocarcinomas (Extended Data Fig. 2c).
Integrating our AR cistrome data with RNA-sequencing analysis (RNA-seq) from matched cell lines revealed that the AR is redirected from the canonical ‘AR-driven’ transcriptional programme in CRPC to positively regulate pathways implicated in stem cell plasticity and neuronal processes in 42DENZR cells (Fig. 1f). Attesting to human relevance, these same pathways were upregulated in treatment-induced neuroendocrine-like clinical specimens compared with adenocarcinomas (Fig. 1f). AR ChIP-seq performed in patient tumours following three-month ENZ therapy revealed that approximately 40% of the reprogrammed AR binding in ENZ-resistant cells overlapped with the AR cistrome in ENZ-treated patient tumours, despite the inherent heterogeneity between cell lines and patient tumours. Analysis of the shared AR-bound genes uncovered an enrichment in neuronal and plasticity-associated networks (Fig. 1g). These findings suggest that redistribution of the AR cistrome following potent AR-pathway inhibition may function to unlock alternative lineage programmes.
To delineate how the AR transcriptional programme is rewired to support different cellular phenotypes, genome-wide chromatin accessibility by assay for transposase-accessible chromatin using sequencing (ATAC-seq) was performed. Comparative analysis showed distinct chromatin accessibilities of 42DENZR compared with 16DCRPC cells (Fig. 1h), indicating large-scale chromatin remodelling occuring during the emergence of the lineage-plastic phenotype, as previously suggested29. A similar phenomenon was observed in a genetically engineered mouse model (GEMM) (PB-Cre4:Ptenf/f;Rb1f/f) that evolves from adenocarcinoma to a neuroendocrine-like state (Extended Data Fig. 3a). ATAC-seq peaks in both cell lines and GEMMs captured motifs associated with stem cell plasticity and neuronal differentiation (Fig. 1i and Extended Data Fig. 3b,c). AR binding was enriched surrounding these newly exposed transcription factor motifs (Fig. 1l). Notably, the reprogrammed AR-bound regions in ENZ-resistant cells were hypo-accessible in CRPC (Fig. 1j).
AR functions with EZH2 to regulate lineage plasticity.
To explore the mechanism underlying AR cistrome reprogramming, we surveyed the AR interactome using rapid immunoprecipitation–mass spectrometry of endogenous proteins (RIME) in 16DCRPC and 42DENZR cells. The most abundant peptides corresponded to AR and its co-factor FOXA1 (Fig. 2a and Supplementary Table 2). A strong and reproducible interaction between the AR and the core polycomb repressive complex 2 (PRC2) subunit SUZ12 was specific to the ENZ-resistant state (Fig. 2a), while the polycomb complex protein EED was pulled down at lower frequency. The AR–SUZ12 interaction was confirmed by immunoprecipitation (Fig. 2b), and the AR–EZH2 nuclear interaction was visualized by in situ proximity ligation assay (PLA) (Fig. 2c).
Fig. 2 |. The AR functions in a non-canonical polycomb complex with EZH2.
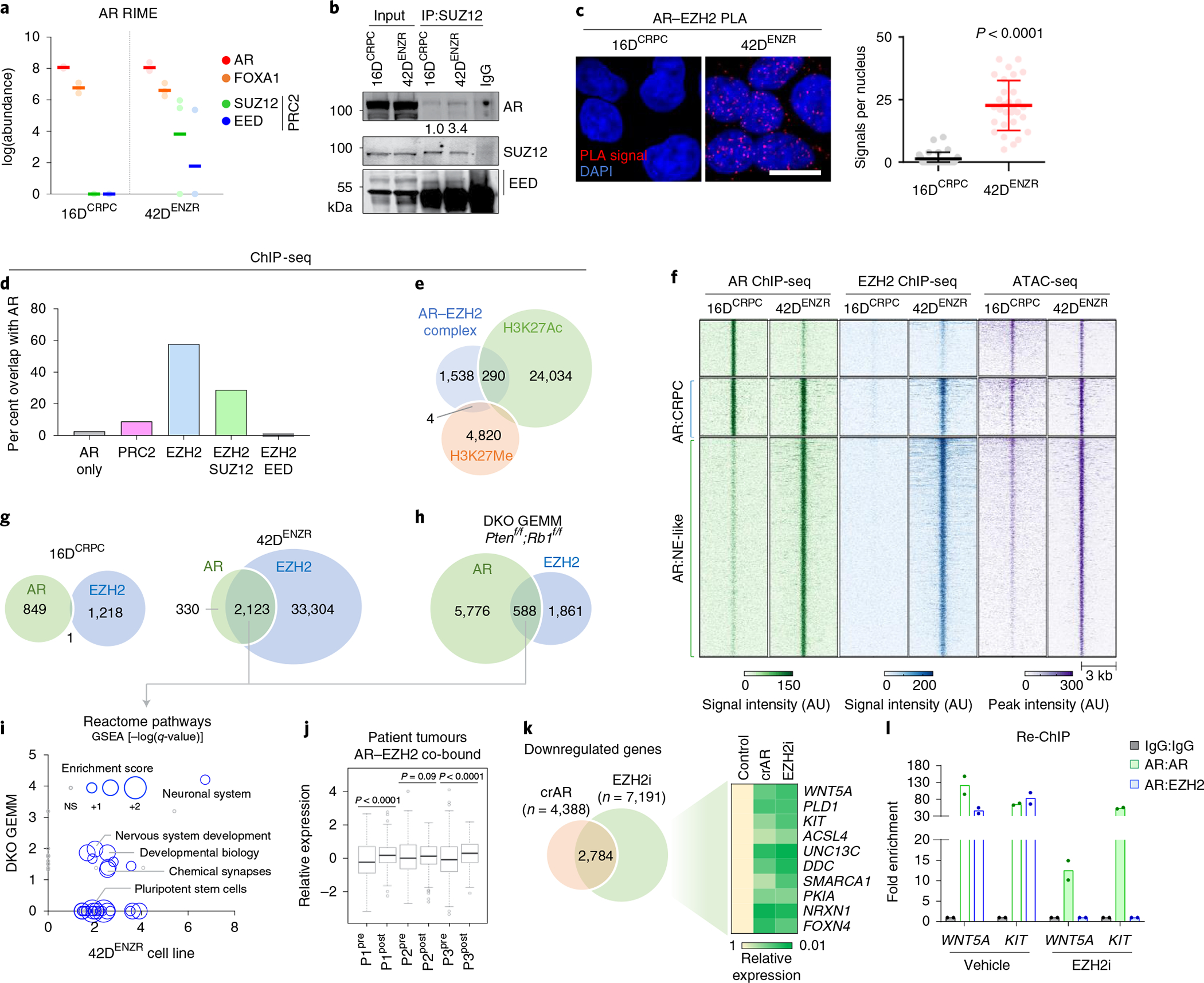
a, Abundance of AR, FOXA1, SUZ12 and EED peptides detected using RIME with AR antibodies as bait. Each dot represents an independent replicate, with a solid line denoting the mean. b, SUZ12 immunoprecipitation (IP) followed by immunoblotting for AR and PRC2 subunits. The relative abundance of AR was normalized to SUZ12 pulldown. c, AR–EZH2 PLA and quantification of nuclear PLA signals (red dots) from a single plane (mean ±s.d.; P < 0.0001, two-tailed unpaired t-test; n = 3). Each dot represents the number of PLA signals in a single nucleus. Scale bar, 10 μm. d, Frequency of AR-bound genes with EZH2, SUZ12 and/or EED co-occupancy based on ChIP-seq peak annotation (±50 kb from the nearest TSS) in 42DENZR cells. e, Overlap of genomic regions co-occupied by AR and EZH2 ChIP-seq peaks (AR–EZH2 complex) with ChIP-seq peaks for the H3K27Me3 and H3K27Ac in 42DENZR cells. f, Heat map of AR and EZH2 ChIP-seq signal intensity in 16DCRPC and 42DENZR cells, with corresponding ATAC-seq peak intensity. g, Overlap of AR and EZH2 ChIP-seq peaks in 16DCRPC and 42DENZR cell lines. h, Overlap of AR and EZH2 ChIP-seq peaks in the Ptenf/f;Rb1f/f (DKO) GEMM. i, Enriched reactome pathways with genes co-occupied by AR–EZH2 in 42DENZR cells and the Ptenf/f/Rb1f/f GEMM. The size of each circular data point reflects the degree to which genes in the pathway are enriched based on RNA-seq from 42DENZR compared with 16DCRPC cells. NS, not significant. j, Expression of AR–EZH2 co-bound genes in matched prostate tumours (P1–P3) pre- and post-ENZ therapy (n = 3) from the DARANA trial. Box plot shows mean and interquartile range. Statistical analysis was performed using a paired t-test. k, Venn diagram of overlap in genes downregulated (log2FC < 1) in 42DENZR cells following depletion of AR using CRISPR (crAR) or EZH2 inhibition (10 μm GSK126; 96 h). The heat map depicts relative expression of select AR–EZH2 co-bound genes, reported relative to parental cells. l, Sequential ChIP (Re-ChIP) for selected binding sites in 42DENZR cells treated with vehicle or EZH2 inhibitor (10 μm GSK126, 96 h). Cells were first analysed by chromatin immunoprecipitation with AR antibody and then immunoprecipitated again with an AR or EZH2 antibody, as indicated. Results are reported relative to IgG control (mean ± s.d., n = 2).
To establish how AR and the core PRC2 subunits are assembled on chromatin and the resultant effect on chromatin architecture, we performed ChIP-seq for each subunit of the PRC2 holoenzyme (EZH2, SUZ12 and EED) and histone H3 lysine 27 (H3K27) modifications. The EZH2 cistrome was expanded in 42DENZR compared with 16DCRPC (approximately 36,500 versus 1,200 binding sites, respectively), with the majority of EZH2 (around 40%) shouldered by active H3K27 acetylation (H3K27Ac) (Extended Data Fig. 4a–c). Motif analysis surrounding EZH2 peaks in the ENZ-resistant cells uncovered motifs implicated in AR signalling (for example, FOXA1, ETV1 and ARE) (Supplementary Table 3). Overlaying AR ChIP-seq peaks with those of the PRC2 subunits revealed AR-bound genes to be predominantly co-occupied with EZH2 alone or in complex with SUZ12, but less frequently with EED (Fig. 2d), suggesting that AR functions in one or more non-canonical PRC2 subcomplexes containing EZH2. Sites co-occupied by AR–EZH2 were shouldered by the active histone mark H3K27Ac (around 14% overlap) relative to the repressive H3K27 tri-methylation (H3K27Me3) mark (less than 1% overlap) (Fig. 2e and Extended Data Fig. 4d), consistent with findings of transcriptional activation by polycomb subcomplexes lacking EED30. ATAC signals were enriched surrounding AR–EZH2 binding sites, further confirming the association between AR–EZH2 co-occupancy and open, transcriptionally active chromatin (Fig. 2f). The interaction between AR and EZH2 was largely confined to the ENZ-resistant state; no overlap was observed in 16DCRPC cells (Fig. 2g). These findings were validated in a neuroendocrine-like GEMM (PB-Cre4:Ptenf/f;Rb1f/f) in which overlap between AR and EZH2 cistromes was also observed (Fig. 2h). Genes with co-bound AR–EZH2 in cell lines and GEMM tumours converged on a shared set of transcriptional programmes governing stem cell plasticity and neuronal differentiation. RNA-seq data confirmed positive regulation of these pathways (Fig. 2j). Attesting to human relevance, genes with co-bound AR–EZH2 were upregulated in patient tumours following ENZ treatment (Fig. 2j). These data across multiple models highlight that AR and EZH2 co-operate to positively regulate lineage-plastic and neuroendocrine-associated genes.
To determine the relative contribution of AR and EZH2 at lineage-plastic genes, AR was deleted using clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) and EZH2 was pharmacologically inhibited using GSK136 (hereafter referred to as EZH2i). Disruption of either AR or EZH2 yielded downregulation of genes co-bound to AR–EZH2 (Fig. 2k), including those associated with stemness such as WNT5A, KIT and PLD1. EZH2i displaced EZH2, but not AR (Fig. 2l). Almost no EZH2 was detected at AR–EZH2 co-bound regions in AR-null prostate cancer cell lines (Extended Data Fig. 4e), suggesting that AR may be required to recruit EZH2.
EZH2 is required to establish the lineage-infidelity state.
To interrogate the requirement of EZH2 for cells to enter the lineage-infidelity state and gain neuroendocrine features, EZH2 was deleted in 16DCRPC cells using CRISPR–Cas9 (resulting in CRPCcrEZH2 cells). Relative to control CRPC, CRPCcrEZH2 cells were markedly diminished for plasticity and neuroendocrine-associated genes following ENZ treatment (Fig. 3a). Tumour growth was restrained in CRPCcrEZH2 xenografts treated with ENZ (Fig. 3b) and plasticity and neuroendocrine-associated genes were not upregulated (Fig. 3c). These data support a functional role for EZH2 in establishing the lineage-infidelity state following AR inhibition.
Fig. 3 |. EZH2 is required to establish the lineage-infidelity state.
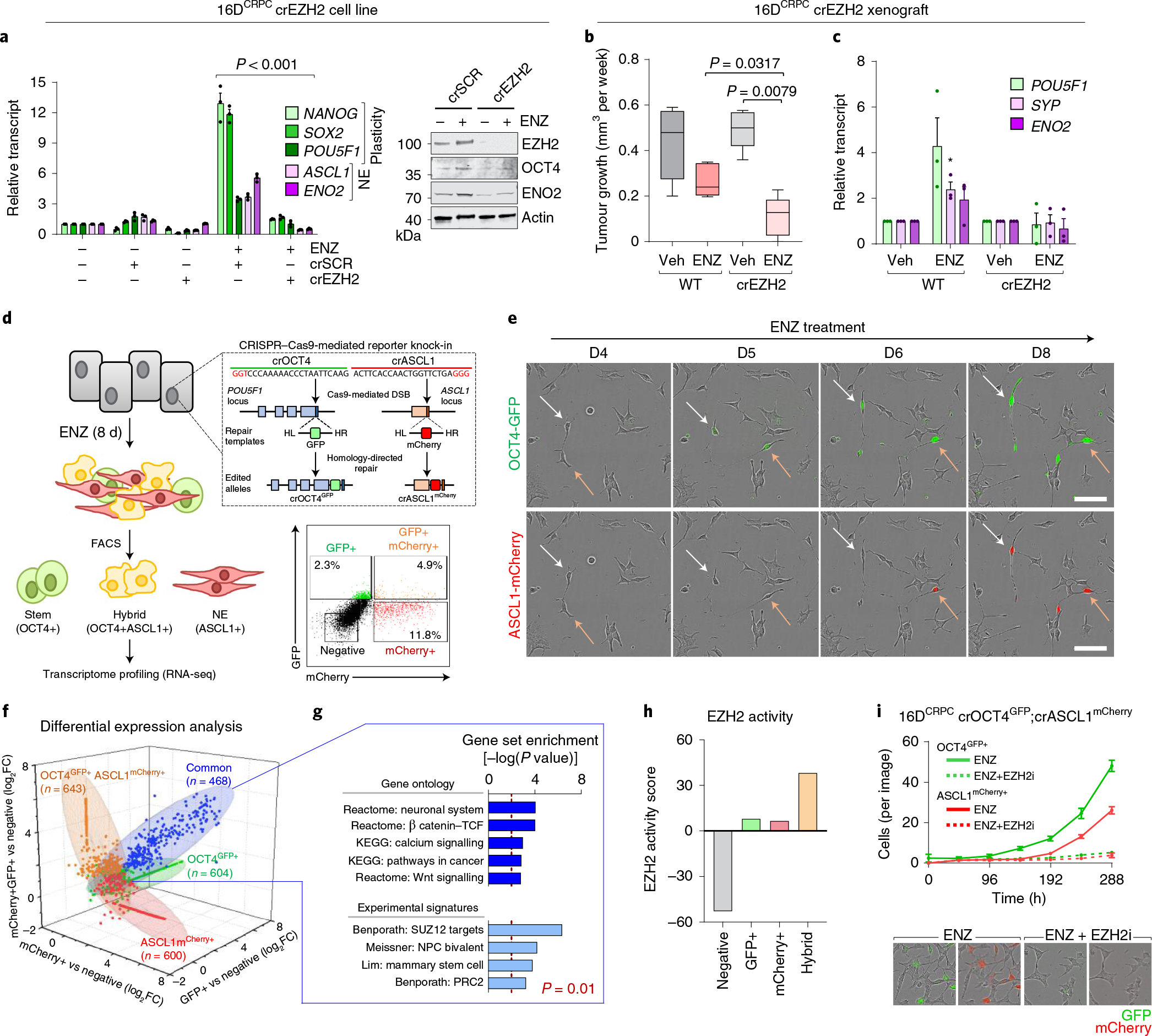
a, Expression of plasticity and neuroendocrine markers by real-time PCR (rtPCR) and Western blot in 16DCRPC cells with CRISPR-mediated EZH2 knockout (16DCRPC crEZH2) following 7 d ENZ treatment. Cells transfected with a non-silencing scrambled guide RNA (crSCR) served as a control. Data are reported relative to non-transfected cells (mean ± s.d., n = 3). Two-tailed unpaired t-test. b, Tumour growth velocity of CRPC cells with CRISPR-mediated EZH2 knockout transplanted subcutaneously into nude mice, followed by treatment with vehicle (veh) or ENZ (n = 5 mice per group). Box plots show mean and interquartile range. Mann–Whitney test. c, Gene expression analysis (by rtPCR) in 16DCRPC control and crEZH2 xenograft tumours at the experimental end point. Data are reported relative to vehicle-treated mice (mean ± s.d.; *P = 0.05, two-tailed unpaired t-test; n = 3 mice per treatment group). d, Strategy used to establish the 16Dreporter cell line carrying GFP and mCherry fluorescent reporters in the endogenous OCT4 and ASCL1 loci, respectively. Fluorescence-activated cell sorting (FACS) plot shows gating used to isolate the individual cell populations. HL, left homology arm; HR, right homology arm. e, Immunofluorescence images for OCT4-GFP (green) and ASCL1-mCherry (red) in CRPCreporter cells at the indicated time points after ENZ treatment. Single cells were tracked and are denoted with arrows. Scale bar, 100 μm. f, Fold change in transcript abundance of genes unique and common to the OCT4+, ASCL1+ and hybrid (OCT4+ASCL1+) FACS-isolated CRPCreporter cell populations relative to the negative population (log2FC cut-off of 1.5), by RNA-seq. g, MSigDB pathways enriched for common genes (n = 468) upregulated (defined as log2FC > 1.5) across OCT4+, ASCL1+ and hybrid (OCT4+ASCL1+) CRPCreporter populations relative to the negative population. Statistical analysis was performed using a hypergeometric test. h, EZH2 activity score, calculated on the basis of z-score-transformed expression of genes in the ‘Kondo EZH2 targets’ signature from MSigDB, in negative, OCT4+, ASCL1+ and hybrid (OCT4+ASCL1+) CRPCreporter FACS-isolated cell populations. i, Quantification of GFP+ and ASCL1+ fluorescent CRPCreporter cells following treatment with ENZ (10 μM) alone or in combination with EZH2 inhibitor (10 μM GSK126) using the IncuCyte fluorescent object counting algorithm (mean ± s.d., n = 2). Representative images at 8 d after treatment are shown. Scale bar, 50 μm.
To explore the importance of EZH2 during the emergence of stem cell and/or neuronal phenotype following ENZ therapy, a dual-reporter system was engineered in 16DCRPC cells by knocking-in GFP and mCherry reporter cassettes before the POU5F1 (which encodes OCT4, a pluripotency reprogramming factor31) and ASCL1 (which encodes a neuronal lineage-guiding factor32) stop codon sequences using CRISPR–Cas9 (referred to as CRPCreporter) (Fig. 3d). Tracking single CRPCreporter cells by live-cell imaging following ENZ revealed the emergence of OCT4-GFP-expressing cells, which subsequently co-expressed ASCL1-mCherry and acquired a neuronal-like morphology (Fig. 3e and Supplementary Video 1). The individual phenotypic groups (negative, OCT4+, ASCL1+ and hybrid OCT4+ASCL1+) were isolated and profiled by RNA-seq. A total of 468 genes were upregulated (log2(fold change (FC)) ≥ 1.5) across the OCT4+, ASCL1+ and hybrid cell populations relative to the negative phenotypic group (Fig. 3f and Supplementary Table 4). These shared genes were associated with development, cell plasticity and epigenetic regulation by EZH2 (Fig. 3g). EZH2 activity was increased in OCT4+ and ASCL1+ cells compared with the negative group and enriched in the hybrid cell population (Fig. 3h). Treatment of CRPCreporter cells with the EZH2i precluded ENZ-mediated emergence of both OCT4+ and ASCL1+ cell populations (Fig. 3i).
EZH2 is reprogrammed by phosphorylation at T350.
To gain insight into EZH2 function, post-translational modification analysis of EZH2 in 16DCRPC and 42DENZR cells was performed by immunoprecipitation–mass spectrometry. No differential phosphorylation at the T311 and T487 residues, reported to regulate EZH2 activity33,34, was detected (Extended Data Fig. 5a,b). The EZH2 S21 phosphorylation, known to activate AR signalling in CRPC22, was not enriched in ENZ-resistant cells or neuroendocrine patient tumours (Extended Data Fig. 5b–d). Expression of non-phosphorylatable (S21A) or phosphomimetic (S21D) EZH2 S21 mutants in CRPCcrEZH2 cells did not alter neuroendocrine lineage markers (Extended Data Fig. 5e). By contrast, EZH2 T350 phosphorylation was highly enriched across ENZ-resistant, neuroendocrine-like cell lines relative to CRPC (Fig. 4a). Active CDK1 (pCDK1-T161) was elevated, and its inhibition abrogated EZH2 T350 phosphorylation (Fig. 4b), consistent with CDK1 as the kinase for EZH2 phosphorylation35,36 at T350. In concordance, pEZH2-T350 and pCDK1-T161 were upregulated in neuroendocrine patient tumours compared with adenocarcinomas (Fig. 4c).
Fig. 4 |. EZH2 is reprogrammed by T350 phosphorylation to drive lineage infidelity and plasticity.
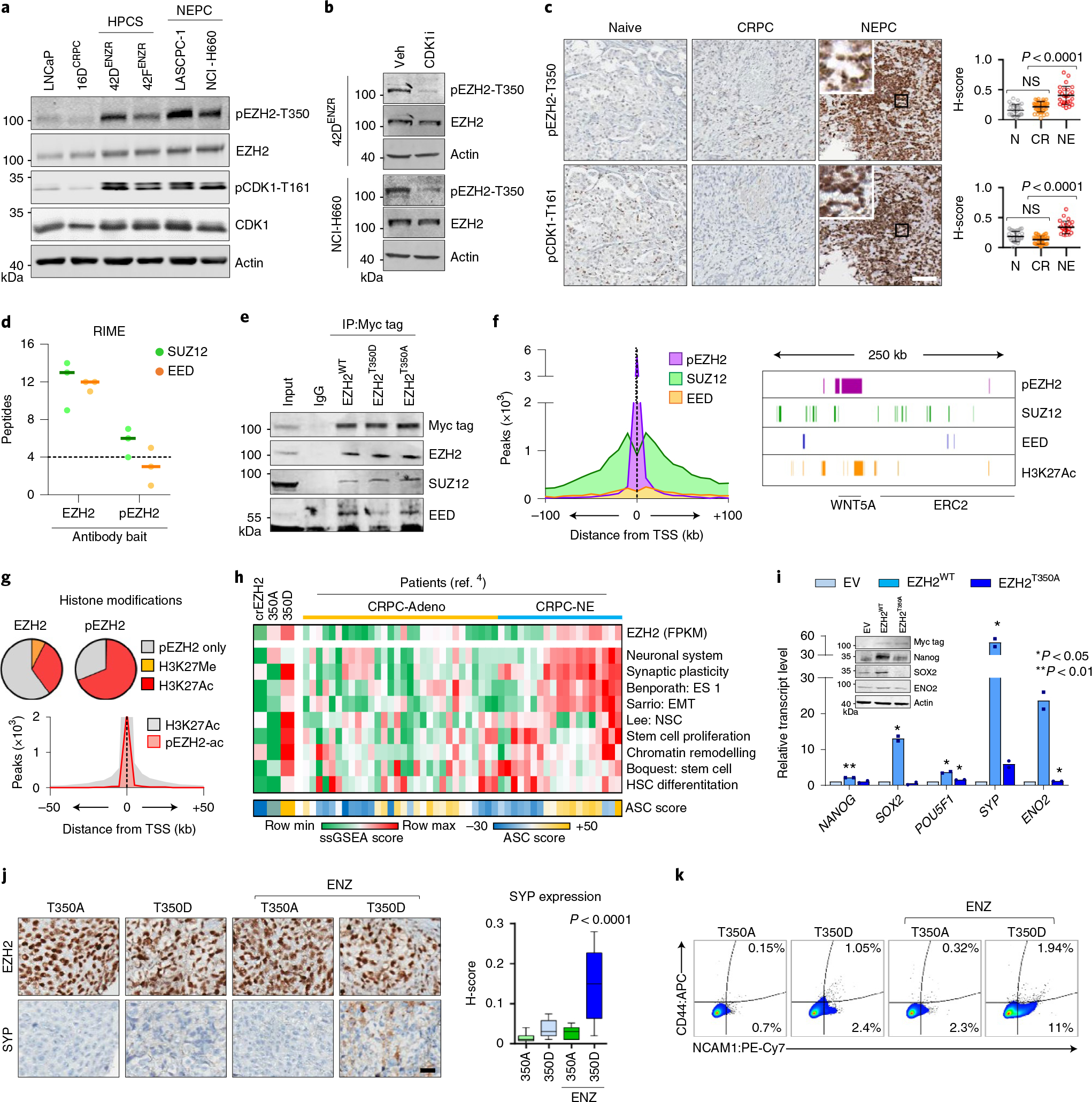
a, Immunoblot of total and phosphorylated EZH2 and CDK1 in the indicated prostate cancer cell lines. HPCS, high-plasticity cell state. b, Immunoblot of EZH2 and pEZH2-T350 following CDK1 inhibition (5 μM RO-3306, 6 h). c, Immunohistochemical staining for pEZH2-T350 and pCDK1-T161 in serial sections from treatment-naive (N, n = 30), CRPC (CR, n = 40) and NEPC (NE, n = 26) clinical samples. Scale bar, 100 μm. Staining intensity was quantified (mean ± s.d.; two-tailed unpaired t-test). d, SUZ12 and EED peptides detected by RIME using EZH2 and pEZH2-T350 antibodies as bait in 42ENZR cells. Each dot represents an independent replicate, with a solid line denoting mean. Significance was defined as ≥4 peptides. e, Myc-tagged wild-type EZH2 (EZH2WT) and T350 phospho-mimicking (EZH2T350D) and phospho-dead (EZH2T350A) mutants were transiently transfected into 16DCRPC cells with endogenous EZH2 deletion for 72 h. Immunoprecipitation was performed using a Myc tag antibody. f, Distribution of pEZH2-T350, SUZ12 and EED ChIP-seq peaks in relation to the nearest TSS. The density of polycomb subunits and H3K27Ac are shown surrounding the WNT5A locus. g, Proportion of EZH2 and pEZH2-T350 ChIP-seq peaks overlapping with H3K27Me3 and H3K27Ac ChIP-seq peaks in 42DENZR cells. The distribution of H3K27Ac alone and co-occupied with pEZH2-T350 (pEZH2-ac) in relation to the TSS is shown. h, Single-sample GSEA (ssGSEA) score of MSigDB pathways in CRPCcrEZH2 cells expressing EZH2T350A or EZH2T350D mutant, and adenocarcinoma (CRPC-Adeno) and NEPC (CRPC-NE) patient specimens from the Beltran 2016 cohort4. The ASC score is shown below each cell line or individual patient. i, rtPCR and immunoblot in 42DENZR cells with EZH2 knockdown, stably expressing siRNA-resistant Myc-tagged EZH2WT or EZH2T350A mutant for 72 h. Data are reported relative to cells transfected with empty vector (EV) (mean ± s.d.; two-tailed unpaired t-test, n = 2). j, Immunohistochemical staining for EZH2 and SYP in serial sections from CRPCcrEZH2 EZH2T350A and EZH2T350D mutant xenografts treated with vehicle or ENZ. Scale bar, 100 μm. SYP staining intensity was quantified; box plots show mean and interquartile range. k, Flow cytometry plots of CD44 and NCAM1 cell surface expression in dissociated tumour cells from EZH2T350A and EZH2T350D mutant xenografts.
To evaluate the unique function of pEZH2-T350, we assessed its binding partners by RIME. While EZH2 pulled down the core PRC2 subunits SUZ12 and EED at comparable frequencies, pEZH2-T350 was strongly associated with SUZ12 (Fig. 4d and Supplementary Table 2), mirroring our AR results. Immunoprecipitation of exogenously expressed Myc-tagged EZH2T350D mutant confirmed a bias toward binding SUZ12 compared with EED (Fig. 4e). We next evaluated the pEZH2-T350 cistrome in ENZ-resistant cells and its relationship with PRC2; about 20% of total EZH2 ChIP-seq peaks were shared with pEZH2-T350. Notably, pEZH2-T350 peaks were largely shouldered by SUZ12 and the active H3K27Ac histone mark (Fig. 4f), with nearly 70% of all pEZH2-T350 peaks overlapping with H3K27Ac, whereas less than 0.01% of peaks overlapped with H3K27Me3 (Fig. 4g). These analyses indicate that pEZH2-T350 favours the formation of a non-canonical polycomb complex.
Next, we stably introduced phosphomimetic (T350D) and non-phosphorylatable (T350A) EZH2 T350 mutants into CRPC cells with endogenous EZH2 deletion (CRPCcrEZH2) to generate isogeneic cell lines expressing EZH2T350A or EZH2T350D mutants. These cell lines exhibited no difference in proliferation (Extended Data Fig. 5f), while genes related to cellular plasticity, chromatin remodelling and neuronal differentiation were specifically enriched in EZH2T350D-expressing cells (Fig. 4h). These pathways were upregulated in CRPC-neuroendocrine patient tumours with high EZH2 expression and ASC score (Fig. 4h). Accordingly, replacing endogenous EZH2 in 42DENZR cells with the EZH2T350A mutant led to downregulation of plasticity and neuroendocrine lineage markers (Fig. 4i), establishing the importance of pEZH2-T350 for maintaining the lineage-infidelity state.
To address whether EZH2 T350 phosphorylation is a prerequisite for ENZ-induced cell plasticity, EZH2T350D- and EZH2T350A-expressing CRPC xenografts were established—synaptophysin (SYP)-positive neuroendocrine foci were observed only in EZH2T350D xenografts following ENZ treatment (Fig. 4j). Analysis of the resultant tumours revealed that a greater proportion of cells in EZH2T350D-driven tumours exhibit NCAM1 and CD44 positivity in response to ENZ (Fig. 4k). A similar dependency on EZH2 T350 phosphorylation was found in VCaP and C4–2 cells in vitro (Extended Data Fig. 5g). Together, these studies highlight a role for pEZH2-T350 in both promoting and maintaining lineage infidelity and neuroendocrine features.
EZH2 T350 phosphorylation is elevated in ENZ resistance.
A 50-gene pEZH2 signature was generated by integrating RNA-seq data with pEZH2-T350 ChIP-seq data to identify genes specifically regulated by active EZH2T350D (log2FC ≥ 1) that had pEZH2-T350 bound within proximity of the transcription start site (TSS) (Fig. 5a and Supplementary Table 5). Applying this signature to a GEMM of PTEN and TP53 inactivation (NPp53)13, we found that ARPI-treated CRPC ‘exceptional non-responders’ exhibited upregulation of pEZH2 signature genes with elevated neuroendocrine marker expression (Fig. 5a). Within human prostate cancer cohorts6,37, the pEZH2-T350 signature was strongly associated with lineage-plastic tumours with a high ASC and/or epithelial–mesenchymal transition (EMT) transcriptional signatures (Extended Data Fig. 6a). This signature was elevated in clinical NEPC tumours, however, the degree of activity varied (Fig. 5b). We measured a strong positive correlation between pEZH2 and ASC scores (R2 = 0.71, P < 0.0001) (Fig. 5c), supporting the relationship between pEZH2-T350 and enhanced plasticity.
Fig. 5 |. EZH2 T350 phosphorylation correlates with RB1 loss in neuroendocrine-like tumours.
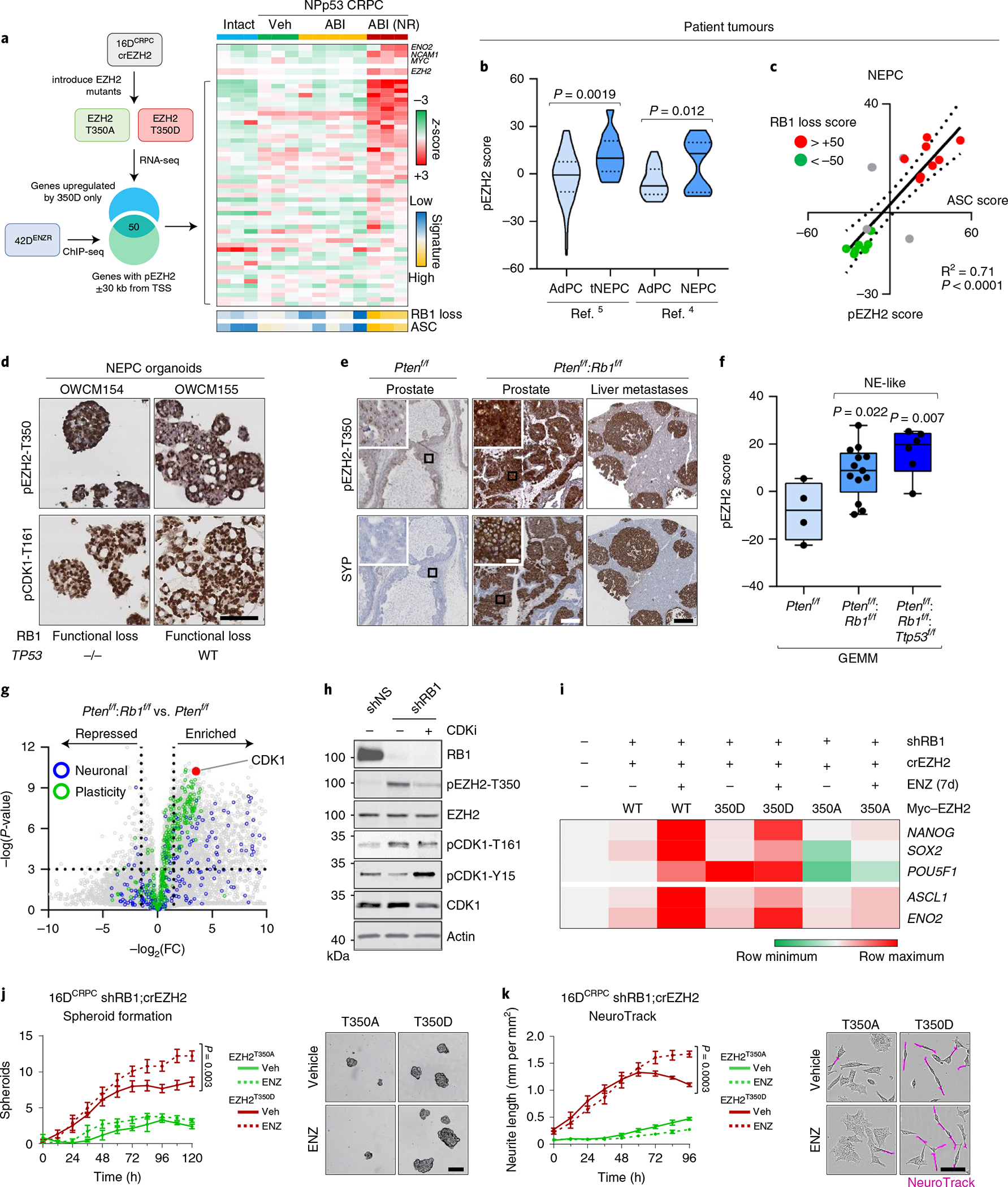
a, Heat map depicting normalized z-score for genes in the pEZH2-T350 signature in NPp53 GEMMs (Gene Expression Omnibus (GEO) accession: GSE92721). ABI, abiraterone acetate; NR, exceptional non-responder. b, pEZH2-T350 signature score in patient tumours from Aggarwal (ref. 5) (P = 0.0019) and Beltran (ref. 4) (P = 0.012). Violin plots show mean and interquartile range, with significance assessed using a two-tailed unpaired t-test. AdPC, prostate adenocarcinoma; tNEPC, treatment-induced neuroendocrine prostate cancer. c, Correlation between pEZH2-T350 and ASC scores in NEPC tumours from ref. 4. Each dot represents a patient tumour showing RB1 loss-of-function score. d, Immunohistochemical staining in patient-derived NEPC organoids. Scale bar, 100 μm. e, Immunohistochemical staining in PB-Cre4:Ptenf/f and PB-Cre4:Ptenf/f;Rb1f/f GEMMs. Scale bars: 200 μm (prostate), 1 mm (liver), 20 μm (insets). f, pEZH2-T350 score in PTEN-null GEMMs with RB1 deletion alone (P = 0.022) or concomitant with TP53 loss (P = 0074; GEO accession: GSE90891). Dots represent individual tumours. Box plot shows mean and interquartile range, with significance assessed using a two-tailed unpaired t-test. g, Volcano plot of RNA-seq from PB-Cre4:Ptenf/f and PB-Cre4:Ptenf/f;Rb1f/f GEMMs (GEO accession: GSE90891). Each dot represents a gene, with those in the ‘Reactome neuronal system’ (neuronal) and ‘Wong embryonic stem cell core’ (plasticity) MSigDB pathways highlighted. h, Immunoblot in 16DCRPC cells with stable RB1 knockdown treated with CDK1 inhibitor (5 μM RO-3306, 6 h). shNS, non-silencing control. i, Heat map of gene expression (by rtPCR) in CRPCcrEZH2 cells with shRB1 expressing EZH2WT, EZH2T350A or EZH2T350D mutant constructs following treatment with DMSO or ENZ (10 μM) for 7 d. Expression is reported relative to parental cells (n = 3). j, Left: quantification of spheroids in CRPCcrEZH2 cells with shRB1 expressing EZH2T350A or EZH2T350D treated with DMSO or ENZ (10 μM). Data reported as mean ± s.d., with significance evaluated at the end point (P = 0.003, two-tailed unpaired t-test; n = 3). Right: images at 5 d after ENZ treatment. Scale bar, 100 μm. k, Left: neurite length measured using IncuCyte in CRPCcrEZH2 cells with shRB1 expressing EZH2T350A or EZH2T350D and treated with DMSO or ENZ (10 μM). Right: phase-contrast images at 96 h after ENZ treatment, with neurite-like extensions highlighted by NeuroTrack. Data reported as mean ± s.d. with significance evaluated at end-point (P = 0.0003, two-tailed unpaired t-test; n = 3). Scale bar, 100 μm.
RB1 loss is a feature of patient tumours with higher pEZH2 signature scores (Fig. 5c and Extended Data Fig. 6b). We found that pEZH2-T350 was elevated in patient-derived NEPC organoids with functional RB1 loss (Fig. 5d) and in SYP-positive neuroendocrine foci in Ptenf/f;Rb1f/f GEMM tumours (Fig. 5e,f). Transcriptomic profiling of the GEMM tumours revealed upregulation of CDK1 (approximately 12-fold versus Ptenf/f tumours) (Fig. 5g and Extended Data Fig. 6c), which was mirrored in patients with NEPC and in patient-derived xenograft models, compared with adenocarcinomas (Extended Data Fig. 6d,e). Inhibition of CDK1 repressed pEZH2-T350 and its target genes as well as neuroendocrine lineage markers (Extended Data Fig. 6f). Silencing RB1 using short hairpin RNA (shRB1) in 16DCRPC cells enhanced CDK1 activation and EZH2 T350 phosphorylation, which was blocked by CDK1 inhibition (Fig. 5h). While repression of RB1 was sufficient to upregulate core pluripotency circuitry and neuroendocrine lineage markers in CRPCcrEZH2 cells expressing wild-type EZH2 (EZH2WT) and EZH2T350D, this was impeded by EZH2T350A, even after ENZ treatment (Fig. 5i). In contrast to EZH2T350D-expressing CRPCcrEZH2 cells treated with shRB1, EZH2T350A mutant expression precluded emergent 3D spheroids (Fig. 5j) and neuronal-like morphology (Fig. 5k). Collectively, and in support of previous studies7,8, these data convey that RB1 loss enhances cell plasticity and neuroendocrine features, in part through CDK1-mediated phosphorylation of EZH2 at T350.
AR and pEZH2-T350 co-operate to promote lineage plasticity.
Next, we investigated whether AR and pEZH2-T350 co-operate to regulate lineage plasticity. Integration of cistromes revealed that AR co-occupies both EZH2 and pEZH2-T350 sites on chromatin (Fig. 6a,b). Both AR–EZH2 co-bound sites and AR–pEZH2 co-bound sites were found largely within introns and intergenic regions (Fig. 6c). Direct interaction between AR and pEZH2-T350 was confirmed by PLA (Fig. 6d). On chromatin, AR and pEZH2-T350 were found to be in proximity to SUZ12 much more frequently than with EED (Fig. 6e). Transcriptomic analysis confirmed AR–EZH2 co-binding to be associated with active gene transcription in ENZ-resistant cell lines (Fig. 6f) and ENZ-treated patient tumours (Fig. 6g). The complex was enriched at genes associated with stem cell programmes (Fig. 6h), highlighting a role for AR–pEZH2 in positively regulating lineage plasticity. The cooperativity between AR and pEZH2-T350 in supporting a lineage-infidelity state was analysed in early-stage prostate tumours following neoadjuvant androgen deprivation and taxane therapy (ADT/TAX). AR and pEZH2-T350 were found to colocalize in SYP-positive neuroendocrine foci, and tumours with higher pEZH2-T350 expression exhibited a more lineage-plastic gene signature (Fig. 6i). Collectively, these data reveal that AR and pEZH2-T350 function to enhance lineage infidelity, with this drug-resistant, high-plasticity state arising early in tumour evolution following therapy.
Fig. 6 |. AR and pEZH2-T350 co-operate to activate lineage-plastic transcriptional programmes.
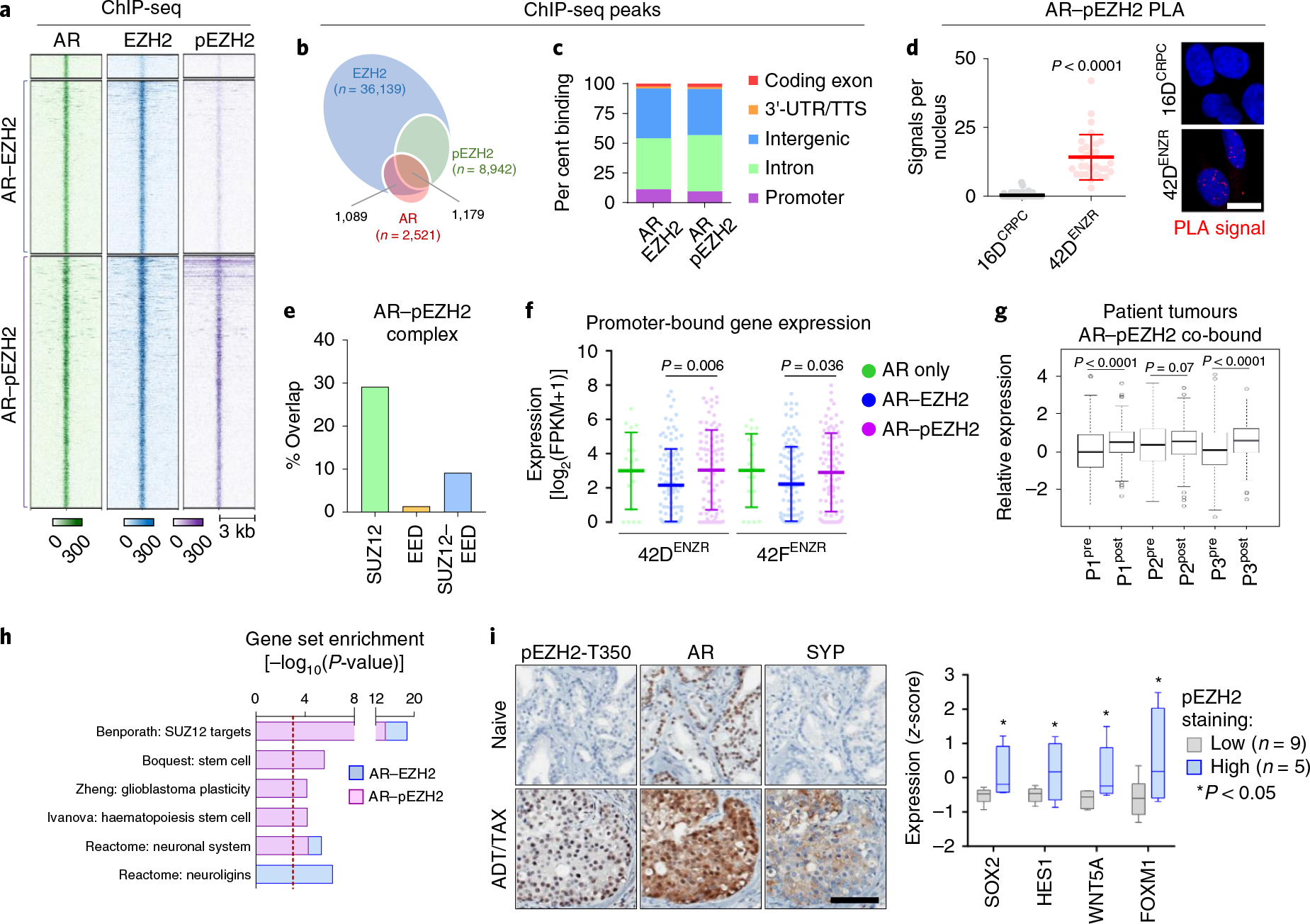
a, Heat map of AR, EZH2 and pEZH2-T350 ChIP-seq binding intensity in 42DENZR cells. Each horizontal line represents a 6-kb locus. b, Frequency of AR ChIP-seq peaks overlapping with EZH2 and pEZH2-T350 ChIP-seq peaks in 42DENZR cells. c, Distribution of AR–EZH2 and AR–pEZH2 co-bound peaks in relation to the TSS. Peaks were mapped into 5-kb bins. d, PLA analysis of the interaction between AR and pEZH2-T350, and quantification of nuclear PLA signals (red dots) from a single plane (mean ± s.d.; P = 3.8 × 10−10, two-tailed unpaired t-test; n = 3). Each dot represents the number of PLA signals in a single nucleus. Scale bar, 10 μm. e, Overlap of genes co-bound to AR–EZH2 occupied by SUZ12- and/or EED, based on ChIP-seq peak annotation in 42DENZR cells. Gene annotation was restricted to ±50 kb from TSS. f, Expression of genes with promoter-bound (defined as ±3 kb from TSS) AR alone or co-occupancy with EZH2 or pEZH2-T350 in 42DENZR and 42FENZR cell lines. Data are mean expression ± s.d., with significance assessed using a two-tailed unpaired t-test. g, Expression of AR–pEZH2 co-bound genes in matched individual patient tumours pre- and post-ENZ therapy from the DARANA trial (n = 3). Box plots show mean and interquartile range. Statistical analysis was performed using a paired t-test. h, Gene ontology signatures from MSigDB enriched for AR–EZH2 and AR–pEZH2 co-bound genes in 42DENZR cells. Statistical analysis was performed using a hypergeometric test. i, Immunohistochemical staining for AR, pEZH2-T350 and SYP (neuroendocrine marker) in serial sections from non-treated (naive) and neoadjuvant ADT/TAX-treated (4.5 months) prostate tumours from the CALGB 90203 clinical trial. Treated tumours were binned on the basis of pEZH2-T350 staining intensity, and matched NanoString-based sequencing was used to assess the expression of plasticity factors in pEZH2-low (n = 8) and pEZH2-high (n = 4) tumours. Box plots show mean and interquartile range of z-score-transformed expression values with significance assessed using a two-tailed unpaired t-test. Scale bar, 100 μm.
The AR+ lineage-infidelity state is a plastic intermediate.
Persistent AR expression in the lineage-infidelity state raises potential therapeutic implications for restoring canonical AR activity. Treatment of 42DENZR cells with EZH2i (Extended Data Fig. 7a) led to a change in the transcriptome similar to those observed in AR-driven cells (Fig. 7a), with a decrease in ASC score similar to adenocarcinoma patient tumours (Fig. 7b), while activating canonical AR signalling (Fig. 7c). By contrast, deletion of AR using CRISPR–Cas9 yielded an increased terminal NEPC score (Fig. 7b) and neuronal gene networks (Fig. 7c). Loss of AR in CRPC-adeno cells did not lead to the acquisition of neuroendocrine characteristics (Extended Data Fig. 7b) as previously reported23, suggesting that the lineage-plastic state may represent a continuum of differentiation towards terminal NEPC.
Fig. 7 |. The lineage-infidelity state exhibits dynamic plasticity.

a, PCA of global transcriptome in the indicated cell lines. 42DENZR cells with AR knockout (AR KO) and inhibited EZH2 activity (10 μM GSK126, 96 h) are shown. b, ASC and NEPC scores in patient tumours from ref. 5 (adenocarcinoma cluster 5, n = 28; AR+NE+, n = 10; AR−NE+, n = 3) and the indicated cell lines. c, GSEA signatures enriched (Fisher’s exact test, P < 0.05) in 42DENZR cells following AR knockout or EZH2 inhibition (10 μM GSK126, 96 h). d, Volcano plot of peptides detected by RIME using EZH2 antibodies as bait in 42DENZR cells treated with DMSO or EZH2 inhibitor (10 μM GSK126, 96 h). Statistical analysis was performed using a two-tailed unpaired t-test (n = 3). e, Immunoprecipitation of EZH2 in 42DENZR cells treated with 10 μM GSK126 for 96 h followed by immunoblotting. f, Immunoblot of SUZ12 in nuclear soluble and chromatin-bound fractions in 42DENZR cells treated with 10 μM GSK126 for 96 h. g, PLA analysis of AR–EZH2 in 42DENZR cells following EZH2 inhibition (10 μM GSK126, 96 h). Nuclear PLA signals from a single plane were quantified (mean ± s.d.; P = 3.1 × 10−16, two-tailed unpaired t-test; n = 3). Scale bar, 10 μm. h, Chromatin immunoprecipitation–PCR (ChIP–PCR) for AR at the AREs within the KLK3 enhancer in 42DENZR cells following treatment with EZH2 inhibitor (10 μM GSK126, 96 h). Results reported relative to IgG control (mean ± s.d.; P = 0.018, two-tailed unpaired t-test; n = 4). F, forward; R, reverse. i, rtPCR in 42DENZR cells treated with EZH2 inhibitor (10 μM GSK126 or GSK343, 96 h) or EED inhibitor (1 μM A-395, 96 h). Data reported relative to vehicle-treated cells (mean ± s.d., two-tailed unpaired t-test; n = 3). Western blot confirmed PRC2 inhibition. j, Confluency measured using IncuCyte (mean ± s.d., n = 2). At 48 h after seeding, cells were treated with EZH2 inhibitor (10 mM GSK126). k, Proliferation of 42DENZR cells treated with ENZ (10 μM) and EZH2 inhibitor (2 μM GSK126) alone or in combination, measured using IncuCyte. EZH2 inhibitor was removed (washout) at 96 h. Data plotted are mean ± s.d. (n = 3), with significance evaluated using a two-tailed unpaired t-test at the end point.
As EZH2 inhibitors can reverse ENZ-resistant cells to an AR-driven state, we queried how these inhibitors function in the context of the AR–EZH2 complex. EZH2i disrupted EZH2 binding to SUZ12 (as previously reported38) as well as the AR (Fig. 7d,e). Moreover, EZH2i displaced SUZ12 from the chromatin (Fig. 7f) and led to a loss of the AR–EZH2 interaction (Fig. 7g). This yielded a lineage reversal to a luminal state, measured by increased AR binding at the KLK3 enhancer (Fig. 7h), upregulation of PSA (Fig. 7i) and decreases in plasticity and neuroendocrine markers (Fig. 7i). This was phenocopied by small interfering RNA (siRNA)-mediated EZH2 silencing (Extended Data Fig. 8a,b), and manifested as a reduction in spheroid formation and ALDH activity (Extended Data Fig. 8c,d). Notably, washout of EZH2i reversed these cells back to the lineage-infidelity state, highlighting the bidirectional plasticity of this lineage conversion (Extended Data Fig. 8e). Inhibition of the canonical PRC2 complex using an EED inhibitor did not drive lineage reversion, suggesting that this process is polycomb-independent (Fig. 7i). Notably, ENZ sensitivity was restored by EZH2i (Fig. 7j). When primed with EZH2i alone or in combination with ENZ for 4 d followed by EZH2i washout, the combinatorial treatment diminished cell growth in a manner that was sustained (Fig. 7k). Together, our data support the reversibility of ENZ resistance using epigenetic inhibitors, at least from the lineage-plastic state.
Discussion
Current therapeutic strategies focused on inhibiting oncogenic pathways that drive aggressive phenotypes almost inevitably lead to treatment-resistant cancers. It is increasingly appreciated that tumours usurp alternative developmental transcriptional programmes to undergo lineage switching and acquire aggressive clinical characteristics15,39,40. Using diverse prostate cancer models and patient samples we uncovered a lineage-infidelity phenotype, mediated by cooperation between AR and EZH2, that emerges following AR-targeted therapy.
With increased clinical sampling, we are beginning to appreciate the diversity of treatment-resistant prostate cancer phenotypes, including AR+NE+ and AR−NE− tumours, which may represent transition states5,6. Given that lineage plasticity is a dynamic process, it is not surprising that heterogeneity is observed with multiple ‘conduits’ to end-state NEPC. The current understanding and working model posits that loss of AR expression is a late manifestation in histological small cell NEPC. Knockout of AR in prostate adenocarcinoma before hormone therapy precludes neuroendocrine differentiation23. Our studies suggest that the AR remains functional, albeit reprogrammed, to support lineage infidelity and plasticity during emergence of treatment-resistant tumours with molecular features of neuroendocrine differentiation. This work builds on observations of continued AR expression in lineage-plastic, neuroendocrine-like GEMM tumours7 and cell lines8 with RB1 and TP53 deletion. Moreover, AR expression persists in prostate cancer models of ENZ-induced lineage plasticity in the context of genomic CHD1 loss19, including a cluster defined by BRN2 expression that we have previously shown to drive a neuroendocrine phenotype24.
We report here that reprogramming of the AR cistrome in the lineage-infidelity state is coupled to EZH2. Other groups have reported that EZH2 T350 is not altered in CRPC characterized by high canonical AR transcriptional activity22, further nominating T350 phosphorylation as a unique feature of the lineage-plastic state. High levels of CDK1 have been reported in androgen-independent prostate cancer41, which may contribute to EZH2 phosphorylation and, in turn, acquisition of a lineage-plastic phenotype. Interestingly, CDK1 phosphorylates and stabilizes the AR41 and has been associated with pluripotency in hESCs42, and thus may represent a molecular circuitry contributing to lineage plasticity.
Previous studies have shown that T350 phosphorylation controls the recruitment of EZH2 on chromatin to regulate pro-oncogenic properties, such as proliferation and migration36,43. However, in contrast to a study suggesting that EZH2 T350 maintains a repressive chromatin state in androgen-sensitive prostate cancer36, we found it to be associated with active, H3K27ac-marked chromatin and positive regulation of gene transcription in the lineage-plastic state. This discrepancy may be attributed to the complexity of EZH2 post-translational modifications. Notably, we found EZH2 was also phosphorylated at S21 in lineage-plastic tumours, and this phosphorylation site has been shown to switch EZH2 into a transcriptional co-activator22. The balance between multiple phosphorylation events may therefore dictate the EZH2 cistrome and its activity at target sites.
Potent AR-targeted therapies are now being deployed earlier in the neoadjuvant and non-metastatic CRPC settings for patients with aggressive localized prostate cancer44,45. Our findings demonstrate that EZH2 inhibition can reverse the lineage-infidelity state and resensitize tumours to AR antagonists, mirroring observations from neuroendocrine-like PTEN- and RB1-deleted GEMM tumours7 as well as ENZ-resistant CRPC cell lines46,47. However, late-stage AR-negative NEPC organoids do not undergo lineage reversion48. This suggests that a therapeutic window exists before AR is lost, during which tumours retain the flexibility to transition to alternative lineages. Further studies are warranted to address when to best integrate epigenetic therapies into the clinic to capitalize on this plasticity.
In closing, emergence of treatment-resistance phenotypes, such as AR+NE+ tumours, has catalysed a deeper need for the molecular understanding of neuroendocrine prostate cancer. Our work provides insight into an intermediate drug-resistant, high-plasticity cell state that can emerge following potent AR-targeted therapy, and proposes co-targeting AR and EZH2 in patients with AR-positive, neuroendocrine-like tumours. As the mechanisms responsible for tumour cell plasticity continue to be uncovered, the ability to reprogramme aggressive tumours to a targetable state might soon become a clinical reality.
Methods
Human prostate cancer specimens.
Prostate tumours were obtained during routinely scheduled biopsies. The tissue collection protocol was approved by the Clinical Research Ethics Board at the University of British Columbia (H09–01628) and all patients signed a written informed consent. Tissue microarrays were obtained from the Vancouver Prostate Centre biobank. All remaining patient samples and data used in the present study had been collected with patient consent obtained by the respective studies.
Cell lines and tissue culture.
Generation and molecular characterization of the CRPC (16DCRPC), ENZ-resistant AR+ NE-like (42DENZR and 42FENZR) and ENZ-resistant AR-driven (49CENZR and 49FENZR) tumours and cell lines: in brief, hormone-sensitive LNCaP cells serve as the backbone of the ENZ-resistance model, which was established through serial passaging of CRPC xenografts treated with ENZ24,49 (10 mg kg−1 d−1). These cell lines were cultured in RPMI-1640 containing 5% fetal bovine serum (FBS; Gibco A3160701), with the ENZ-resistant cell lines supplemented with 10 μmol l−1 ENZ for all experiments, unless otherwise noted. The LNCaP (ATCC 1740), C4–2 (ATCC 3314), VCaP (ATCC 2876) and NCI-H660 (ATCC 5813) cell lines were acquired from ATCC, LASCPC-1 cells were obtained from O. Witte (UCLA), and SKO (Ptenf/f), DKO (Ptenf/f/Rb1f/f) and TKO (Ptenf/f/Rb1f/f/Tp53f/f) GEMM cell lines were obtained from D. Goodrich (Roswell Park). The LASCPC-1 and NCI-H660 cell lines were cultured in HITES medium and GEMM cell lines were cultured in PrE medium. When indicated, cells were treated with the following inhibitors: GSK126 (Millipore 500580), GSK343 (Sigma SML0766), A-395 (Sigma SML1923) or RO-3306 (Sigma SML0569). Cultures were assessed for mycoplasma monthly, and all cell lines have been authenticated by short tandem repeat profiling.
Mouse xenograft generation.
All animal studies were performed in accordance with protocols approved by the Animal Care Committee at the University of British Columbia (A16–0246). Mice were maintained in ventilated cages (4 mice per cage), with constant humidity (25–47%) and temperature (21–22 °C), under a 12 h:12 h light:dark cycle, and had ad libitum access to rodent chow diet and drinking water.
Immunocompromised mice (Envigo; strain: NU-Foxn1nu; sex: male; age: 6–8 weeks old) were subcutaneously injected with 1 × 106 cells in a Matrigel matrix (1:1) and when tumour volume reached ~150 mm3, mice were randomly assigned to treatment groups (10 mice in each group): vehicle (0.5% methyl cellulose) or ENZ (10 mg kg−1) administered by oral gavage 3 times per week. Tumour volumes were measured twice weekly in a blinded fashion, and calculated with the formula volume = (π × (length × width × height))/6. When tumour volume reached ≥2,000 mm3, tumour weight reached 10% of initial body weight, or body weight loss exceeded 15%, the mice were euthanized and tumours were collected for downstream analysis.
Plasmids.
The shRB1 (#25641), pcDNA3-EZH2S21A (#42663), pcDNA3-EZH2S21D (#42664) and control (#8453) plasmids were available from Addgene. The pcDNA3-EZH2WT and pcDNA3-EZH2T350A plasmids were a gift from H. Huang (Mayo Clinic). To generate the pcDNA3-EZH2T350D plasmid, we inserted a ACC>GAC mutation into pcDNA3-EZH2WT using the Agilent Site-Directed Mutagenesis Kit (Agilent) with the primers, F: 5′-ACCGCTGAGCGGATAAAGGACCCACCAAAACGTCC and R: 5′-GGACGTTTTGGTGGGTCCTTTATCCGCTCAGCGGT. The reaction was carried out using 50 ng wild-type EZH2 plasmid with the following PCR conditions: 95 °C for 30 s for 1 cycle, then 95 °C for 30 s, 55 °C for 1 min and 68 °C for 8 min repeated for 16 cycles. Plasmids used for CRISPR–Cas9-mediated genomic editing were constructed using GeneArt CRISPR Nuclease Vectors (Thermo Fisher). Double-stranded oligonucleotides encoding a target-specific crRNA were generated and cloned into the guide RNA (gRNA) expression cassette: ASCL1 (F: 5′-ACTTCACCAACTGGTTCTGAGTTTT; R: 5′-TCAGAACCAGTTGGTGAAGTCGGTG), POU5F1 (F: 5′- GAACTTAATCCCAAAAACCCGTTTT; R: 5′-GGGTTTTTGGGATTAAGTTCCGGTG), EZH2 (F: 5′- TGAGCTCATTGCGCGGGACTGTTTT; R: 5′- AGTCCCGCGCAATGAGCTCACGGTG). All plasmids were analysed for correct insertion by Sanger sequencing before use.
Stable cell line generation and siRNA transfection.
For generation of stable cell lines, cells were transfected with 2.5 μg plasmid using TransIT-2020 (Mirus) in Opti-MEM media for 24 h. Cells were maintained under antibiotic selection with G418 (for EZH2 stable cell lines; 1,200 μg ml−1 for 3 week selection, 500 μg ml−1 for maintenance) or puromycin (for shRB1 stable cell lines; 10 μg ml−1).
For siRNA knockout experiments, cells were transfected with 20 nM siRNA targeting full-length AR (5′-UCAAGGAACUCGAUCGUAUUU; Dharmacon) or EZH2 (Hs_EZH2_2; Qiagen) in Opti-MEM using Lipofectamine RNAiMAX (Thermo). Scrambled sequences (5′-AUCAAACUGUUGUCAGCGCUG, Dharmacon) were used as a control to rule out phenotypic or genotypic changes due to nonspecific targeting. For EZH2 rescue experiments, 5 μg siRNA-resistant Myc-tagged pcDNA3-EZH2WT, pcDNA3-EZH2350A or pcDNA3-EZH2350D plasmid was co-transfected with 20 nM EZH2 siRNA (5′-GAAUGGAAACAGCGAAGGA) using TransIT-2020 (Mirus) in Opti-MEM media. Cells were incubated for 18 h with siRNA, followed by a 4 h recovery in complete medium before re-transfection for 4 h.
CRISPR–Cas9-based gene editing.
The CRPCreporter cell line was generated by two rounds of co-transfection of GeneArt Cas9/gRNA targeting plasmid (POU5F1 (GAACTTAATCCCAAAAACCCTGG) followed by ASCL1 (ACTTCACCAACTGGTTCTGAGGG)) with closed circular dsDNA donor vector (GFP or mCherry cassette flanked by 1-kb homology arms targeted to the 3′ untranslated region sequence adjacent to the OCT4 or ASCL1 stop codon, respectively) using Lipofecamine 3000 (3 μg targeting, 7 μg donor). After transfection, cells were recovered in 5% charcoal-stripped serum (Thermo A3382101) for 7 d, and those expressing GFP and mCherry were isolated by FACS. Individual clones were expanded and screened for correct integration of the reporter allele by PCR-based genotyping using primers spanning the homology arms: ASCL1 (F: 5′-AGCGCAGCCTTAGTAGGAGAGG; R: 5′-CTGGTGGCCTCTTGATCTCACC), OCT4 (F: 5′-GAGCTGGAGGTAGGAATACAGG; R: 5′-CTTGGATCTCAGGGTCACAAGG). For detailed information and validation see Supplementary Data.
For generation of CRISPR knockout cell lines, cells were transfected with 3 μg GeneArt CRISPR OFP Nuclease Vector (Thermo) containing an EZH2-specific gRNA (TGAGCTCATTGCGCGGGACT) using Lipofectamine 3000 or infected with TLCV2 vector (Addgene, #87360) containing an AR-specific guide RNA (CCGCCGTCCAAGACCTACCG). A non-targeting gRNA (GTATTACTGATATTGGTGGG) was used as a control. At 5 days post-transfection, cells expressing Cas9/gRNA plasmid were isolated by FACS, seeded at single-cell density, and expanded. PCR of genomic DNA from individual clones was performed with primers flanking the gRNA target site and assayed by Sanger sequencing. Loss of AR and EZH2 was confirmed by immunoblotting.
RNA-seq and data analysis.
Cell lines were grown in recommended media, with the ENZ-resistant 42DENZR and 42FENZR lines supplemented with 10 μM ENZ. Total RNA was isolated from cell lines using the PureLink RNA Mini Kit (Thermo). Library constructions were performed using the NEBnext Ultra ii Stranded RNA Library Prep Kit, and sequencing was performed on an Illumina NextSeq 500 (42 × 42 bp paired-end reads). For tissue samples, RNA from formalin-fixed, paraffin embedded (FFPE) material was isolated from 2–10 sections of 10 μm using the AllPrep DNA/RNA FFPE kit (Qiagen). Strand-specific libraries were generated with the TruSeq RNA Exome kit (Illumina) and sequenced on a HiSeq 2500 (65 bp single-end reads).
Data were de-multiplexed using bcl2fastq2 Conversion Software (version 2.20) and the resultant read sequences were aligned to the hg19 human reference genome using STAR aligner50. Assembly and differential expression was estimated using Cufflinks software (version 2.2.1)51 available through the Illumina BaseSpace Sequence Hub. For patient tumours, sequencing data was aligned to hg38 using TopHat and number of reads per gene were measured with HTSeq count. Gene counts (FPKM) were normalized using DESeq252 and subsequently log-transformed. For visualization purposes, the data were z-transformed per gene. PCA plots were generated using ClustVis. Significance of expression level differences between pre- and post-treatment samples from the DARANA clinical trial was determined using a paired t-test. DARANA trial (NCT03297385) (https://clinicaltrials.gov/ct2/show/NCT03297385) is a prospective single-arm analysis to study the effects of ENZ on surgical margin in patients over 18 yr of age with localized prostate cancer in neo-adjuvant setting.The trial was approved by the IRB in the Netherlands Cancer Institute. Informed consent, was signed by all participants. Biopsy (pre-treatment) and prostatectomy specimens were examined pathologically for tumour cell content, and only those with a tumour cell percentage of 50% were used for ChIP-seq analysis.
ChIP and ChIP-seq.
Cell lines were grown in media supplemented with 5% FBS (Gibco #A3160701), with 42DENZR cells grown in the presence of 10 μM ENZ. Cells were processed for ChIP using the Manga ChIP Kit (Millipore 17–10085) according to the manufacturer’s instructions with the following antibodies: AR (5 μg, Millipore 06–680, lot no. GR3352799–1), EZH2 (5 μg, Active Motif 39933, lot no. 296200006), H3K27Me3 (5 μg; Millipore 07–449, lot no. 3317006 and 3170806), H3K27Ac (Clone MABI 0309; 5 μg; Active Motif 39685, lot no. 06420021), SUZ12 (5 μg, Cell Signaling 3737, lot no. 8), EED (5 μg, Millipore 17–10034, lot no. 3577475) or pEZH2-T350 (10 μg, generated in this study). For ChIP–PCR, fold enrichment relative to input was evaluated by SYBR Green-based quantitative PCR using the following primers: AR (F: 5′- GCCTGGATCTGAGAGAGATATCATC, R: 5′-ACACCTTTTTTTTTCTGGATTGTTG), KIT (F: 5′- TATTGGAGTTGGAACGCGGC, R: 5′-TGCCTTGGAACCCATTTGGA), WNT5A (F: 5′- ACACAGACTCACCAGCATGA, R: 5′- AAACCCGAGTGCCGGTTATT). For ChIP-seq, sequencing libraries (100 ng DNA per sample) were constructed using the KAPA HyperPrep Kit with Illumina TruSeq indexes (Roche). Libraries were assessed for quality using gel electrophoresis, and libraries passing quality control (for example, no primer dimers) were quantified using the KAPA Library Quantification Kit for Illumina (Roche). Libraries were sequenced using the Illumina NextSeq 500 (75 bp single-end reads; cell lines) or Illumina HiSeq 2500 (65 bp single-end reads; patient tissue specimens).
Re-ChIP.
Re-ChIP was performed in one million cells per immunoprecipitation using the Re-ChIP-IT Kit (Active Motif) with anti-AR (5 μg; Abcam 74272, lot no. GR3352799–1) and anti-EZH2 (5 μg, Active Motif 39933, lot no. 296200006) antibodies. IgG was used as a negative control. Quantitative PCR was performed using 1.6 μl of ChIPed DNA.
ATAC-seq.
ATAC-seq experiments were performed as described53. In brief, cells were collected by incubating in trypsin for 5 min at room temperature and subsequent centrifugation at 592g for 5 min at 4 °C. Fifty-thousand cells were used for tagmentation by incubating in 50 μl of 1× THS-seq buffer (25 μl 2× THS buffer (66 mM Tris acetate, pH 7.8, 132 mM potassium acetate, 20 mM magnesium acetate and 32% dimethylformamide), 5 μl 10× Digitonin, 2 μl Illumina-TDE1) for 20 minutes at 37 °C. To stop the tagmentation reaction, an equal volume of 2× Tagmentation Stop Buffer (10 mM Tris-HCl (pH 8.0), 20 mM EDTA (pH 8.0)) was added to the reaction and incubated for 10 min on ice. For cell lysis, an equal volume of 2× lysis buffer (100 mM Tris-HCl (pH 8.0), 100 mM NaCl, 40 μg ml−1 proteinase K, 0.4% SDS) was added to the tagmentation mix and incubated at 65 °C for 15 min. The tagmented DNA library was purified in 20 μl buffer EB using Qiaquick PCR purification kit (Qiagen). Number of amplification cycles and library quantification was done as described53. Paired-end sequencing was performed on an Illumina NextSeq 500.
Real-time PCR.
Total RNA was extracted and reverse transcribed using SuperScript IV Reverse Transcriptase with random hexamers (Thermo). cDNA (60 ng μl−1) was combined with FastStart TaqMan Probe Master and used as a template for TaqMan-based rtPCR, using the following probes: AR (Hs00171172_m1), ASCL1 (Hs00269932_m1), ENO2 (Hs00157360_m1), GAPDH (Hs02786624_g1), KLK3/PSA (Hs00426859_g1), NANOG (Hs02387400_g1), POU5F1/OCT4 (Hs04260367_gH), SOX2 (Hs01053049_s1) and SYP (Hs00300531_m1). Samples were run on a ABI ViiA7 rtPCR system. GAPDH was used for normalization. Fold changes in mRNA expression levels were calculated using the comparative Ct method.
Co-immunoprecipitation and immunoblotting.
For immunoprecipitation, cells were lysed in IP Lysis Buffer (Thermo) and incubated overnight at 4 °C with 20 μl magnetic protein A/G beads (Millipore) plus one of SUZ12 (5 μg, Cell Signalling 3737 S, lot no. 8) or EZH2 (5 μg, Active Motif 39933, lot no. 2962006) antibody. As a control, protein A/G beads were incubated with lysate only. Chromatin fractionation was performed using the Subcellular Protein Fractionation Kit for Cultured Cells (Thermo). Western blotting was performed as expected using RIPA buffer. Membranes were imaged using the LI-COR Odyssey Imaging System with Li-COR Image Studio (version 4.2) software.
The following antibodies were used for immunoblotting: AR (clone D6F11; 1:1,000; Cell Signaling 5153, lot no. 9), CDK1 (clone POH1; 1:1,000; Cell Signaling 9116, lot no. 7), pCDK1-T161 (1:500; Cell Signaling 9114, lot no. 4), pCDK1-Y15 (1:1,000, Cell Signaling 9111, lot no. 9), EED (1:1,000; Millipore 17–10034, lot no. 3577475), EZH2 (1:2,000; Active Motif 39933, lot no. 2962006), pEZH2-T350 (1:1,000; generated in this study), pEZH2-S21 (1:500; Bethyl, lot no. IHC-00388–6), H3K27Me3 (1:2,000; Millipore 07–449, lot no. 3317006–3170806), Myc tag (1:1,000; Abcam ab9106, lot no. GR130480–22), NANOG (1:500; Abcam ab80892, lot no. CR3280357–1), NSE/ENO2 (clone BBS/NC/VI-H14; 1:1,000; Agilent M0873), OCT4 (clone D7O5Z; Cell Signalling 75463 S, lot no. 1), PSA (clone D6B1; 1:5,000; Cell Signaling 5365, lot no. 4), RB1 (Clone IF9, 1:500; Santa Cruz sc-73598, lot no. C0619), pRB1-S780 (1:1,000; Cell Signaling 9307), SOX2 (clone D6D9; 1:1,000; Cell Signaling 3579 S, lot no. QL230817), SUZ12 (clone D39F6; 1:1,000; Cell Signaling 3737 S, lot no. 8). β-actin (clone AC-74; 1:25,000; Sigma A2228, lot no. 00959896) and vinculin (Clone hvin-1; 1:1,000; Cell Signaling 4650, lot no. 118M4777V) were used for loading controls.
pEZH2-T350 antibody generation.
A polyclonal antibody against phosphorylated EZH2 at the T350 residue was raised by immunizing rabbits with the phosphorylated human EZH2 peptide (C-AERIK(pT)PPKRP-amide), using the Thermo Fisher custom antibody service (2 Rabbit 90 Day Protocol). Crude antibody sera (50 ml) from the terminal bleed were purified using a 2-step depletion method; the serum was first passed through a phospho-peptide coupled resin followed by a control peptide resin to deplete any antibody that may cross-react with the unmodified protein. The specificity of the pEZH2-T350 antibody was confirmed by ELISA. The antibody was validated by western blot using blocking peptide and by inhibiting pEZH2 using CDK1 inhibitor. The antibody was further validated by immunohistochemistry using CRPC xenograft tumours lacking endogenous EZH2 (CRISPR-mediated deletion) that express the EZH2T350A mutant.
RIME.
RIME was carried out essentially as described19. Nuclear lysate was subjected to immunoprecipitation with AR (5 μg, Millipore 06–680), EZH2 (5 μg, Active Motif 39933, lot no. 296200006) or pEZH2-T350 (10 μg, generated in this study) antibodies. Analysis of peptides was carried out on an Orbitrap Lumos Mass Spectrometer at the Vancouver Prostate Centre. Data files were processed with Protein Discoverer 2.2.0.388 (Thermo). Spectrum files were recalibrated and features extracted with Minora. Searches were carried out with Sequest HT with SwissProt TaxID=9606 (v2017–10-25) with precursor mass tolerance 10 ppm and fragment mass tolerance 0.01. Decoy database strict and relaxed FDR targets were 0.01 and 0.05 based on q-value with filtering for high confidence peptides. Precursor quantification was intensity based with unique and razor peptides used without normalizing and peptide group modification site probability set to 75. For analysis, only co-precipitating proteins occurring in all biological replicates were considered, and further filtering was achieved by excluding proteins that appeared in matching IgG controls.
Flow cytometry and FACS.
Single-cell suspensions were pelleted at 300g and re-suspended in flow cytometry buffer (2.5 mM EDTA, 1% FBS, 0.1% NaN3 in 1× PBS) with the following fluorophore-conjugated antibodies for 45 min at 4 °C: CD56/NCAM1, PE-Cyanine7-conjugated (Clone CMSSB; 1:40, Thermo 25–0567-42, lot no. 2100317) and CD44, APC-conjugated (Clone IM7; 1:40, Thermo 17–0441-82, lot no. 2023951). Cells were washed 2 times with flow cytometry buffer, incubated with 7-AAD (Thermo A1310) for 10 min at 4 °C (to exclude dead cells), and acquired on a FACS Canto II with Diva software (minimum 30,000 events). Data were analysed using FlowJo software (version 10.4.2). A representative gating strategy is shown in Extended Data Fig. 9.
For FACS, cells were re-suspended in FACS buffer (1 mM EDTA, 1% FBS in 1× PBS) and filtered through 40 μm nylon strainers to achieve a single-cell suspension. Forward and side scatters were used to gate for singlets. Cells were sorted into FACS collection buffer (2% FBS, 1% penicillin-streptomycin in 1× PBS) using a BD FACSAria Fusion flow cytometer.
Spheroid assay.
Cells (500 per well) were seeded into 96-well ultra-low attachment cell culture plates (Sigma CLS3474) containing supplemented NeuroCult NS-A Basal Media (STEMCELL), and quantified using the IncuCyte Basic Analyzer (version 2018B) with a minimum size filter of 2,000 μm2.
Proliferation and NeuroTrack.
Cells (1,000 per well) were seeded in 96-well cell culture plates, allowed to attach overnight, treated with drug (where indicated), and imaged using the IncuCyte S3. Cell confluence was assessed using the IncuCyte Basic Analyzer with a minimum size filter of 400 μm2. Neuronal-like morphology was measured using the IncuCyte NeuroTrack software module (version 2018B) with the following settings: segmentation mode: brightness; segmentation adjustment: 0.5; minimum cell width: 15 μm; neurite filtering: better; neurite sensitivity: 0.4; neurite width: 4 μm. A minimum of four technical replicates and three biological replicates were performed for each cell line or treatment.
Proximity ligation assay.
Cells were cultured on 8-well chamber slides at a density of 5.0 × 103 cells per well, and PLA was carried out using the Duolink Proximity Ligation Assay Kit (Sigma) with AR (clone 441; Santa Cruz sc-7305) and one of EZH2 (Active Motif 39933) or pEZH2-T350 antibody (generated in this study) at a dilution of 1:200 overnight at 4 °C.
ALDH activity assay.
ALDH activity was assayed using the Aldefluor Kit (Stemcell) following the manufacturer’s instructions.
Immunohistochemistry.
Immunohistochemical staining was performed on deparaffinized FFPE sections using a Ventana Discovery XT automated immunostainer using the following antibodies: AR (clone N-20; 1:50; Santa Cruz sc-816, lot no. G1916), pCDK1-T161 (1:400; Abcam ab47329, lot no. r3260427–5), EZH2 (Clone DR69; 1:50; Cell Signaling 5246S, lot no. 9), pEZH2-T350 (1:75; generated in this study), pEZH2-S21 (1:250; Bethyl IHC-00388, lot no. 6), and SYP (1:500; Abcam ab32127, lot no. GR223336–15). Chromogenic detection was achieved using the DAB Map Detection Kit. Tumour images were annotated by a pathologist (L.F.) to select the relevant areas, and automated digital image analysis was run for each biomarker using the Aperio ImageScope Positive Pixel Count algorithm.
ChIP-seq and ATAC-seq data analysis.
Raw reads were aligned to the hg38 (human) or mm10 (mouse) reference genome using BWA-MEM software (version 0.7.17) with default parameters54. Alignments with mapping quality less than 60 were filtered out (leaving only uniquely mapped reads) using Samtools software (version 1.9)55. Peaks were called using MACS2 (version 2.1.2)56 with FDR q-value of 0.05 using the narrow peak caller for AR, EZH2 and pEZH2-T350, while the broad calling option was used for SUZ12, EED, H3K27Me3 and H3K27Ac. ATAC peaks were called from reads with a template length between 40 and 120 bp. Peaks were annotated using ChIPseeker (release 3.8)57. To compute the intersections between peak annotation files, we developed an in-house algorithm. Two peaks were considered overlapping if (1) they belong to the same gene; and (2) the length of the overlapping region is greater than 50% of the length of either peak. For visualization, bigWig files were generated using deepTools software (version 3.1.3)58 bamCoverage command with bin size of 25. Heat maps were generated using computeMatrix with reference-point mode and plotHeatmap commands of deepTools, and scatterplots with Matplotlib (version 3.1.0).
For clinical specimens, raw reads were filtered based on MAPQ quality (quality ≥ 20) and duplicate reads were removed. Peak calling over input control was performed using MACS2 (v2.1) and Dfilter (v1.6) peak callers56,59. MACS2 was run with option nomodel; DFilter was run with bs = 50, ks = 20, refine, nonzero. Only peaks that were shared by both peak callers were considered.
Motif analysis.
Motif analysis surrounding ChIP-seq peaks (250-bp window) was conducted using HOMER (version 4.10). Motifs were ranked by log P value, and the difference in rank was plotted on a waterfall plot to identify motifs significantly enriched under a given condition.
PCA and PLSR (partial least-squares regression) analysis.
PCA was performed using the prcomp() function in R. The log2-transformed transcripts per million (TPM) values were used as input. Supervised PLS-DA was carried out to identify similarity between samples from multiple independent clinical cohorts using a multivariate integrative method, MINT, as part of the mixOmics R package. The z-scaled combined expression matrix was used as the x variable and a combined vector indicating the biological phenotype of each sample was used as the y variable.
Gene ontology and pathway analysis.
Pathway analysis using GSEA software from the Broad Institute (Massachusetts Institute of Technology) was used to identify functions of differentially expressed genes within the Molecular Signatures Database (MSigDB, version 7.1). The tool was run in classic mode to identify significantly enriched biological pathways. Pathways enriched with a nominal P value < 0.05 and FDR < 0.25 were considered to be significant. ssGSEA was carried out using the ssGSEAProjection module (version 9.1.1) in GenePattern.
Generation of signature scores.
To generate the pEZH2-T350 signature score, we identified genes upregulated (≥2-fold change) following stable EZH2T350D mutant expression in 16DCRPC cells with endogenous EZH2 knockout (crEZH2). Genes regulated by EZH2 irrespective of T350 phosphorylation status were removed by filtering out genes also upregulated by stable expression of the non-phosphorylatable EZH2T350A mutant (defined as ≥10% change in gene expression). To refine the signature, only genes with pEZH2-T350 binding within ±30 kb of the TSS from 42DENZR pEZH2-T350 ChIP-seq were included. To remove cell cycle and proliferation-associated genes, we removed genes common with one of the following pathways from the Molecular Signatures Database: Benporath cycling genes, Benporath proliferation, and Positive regulation of cell proliferation. The pEZH2 score was calculated by summation of the expression z-scores of the 50 genes in the pEZH2 signature.
Previously described ASC15, EMT60, NEPC4 and RB1 loss61 scores were computed by the sum z-score-transformed expression levels across each score’s gene list.
Statistics and reproducibility.
Statistical analysis was performed using Microsoft Excel and GraphPad Prism (version 7 and 8). All experiments were independently repeated at least two times with similar results obtained. In bar graphs, unpaired, two-tailed, Student’s t-tests were performed to analyse statistical significance between groups. For longitudinal profiling experiments, a two-tailed Student’s t-test was performed to determine the statistical difference at the final time point. P < 0.05 was considered significant. For immunohistchemistry scoring, a pathologist (L.F.) was blinded to clinical characteristics (for human tissue microarrays) and mouse genotypes (for GEMMs). All statistical analyses and visualization were performed using GraphPad Prism 7 or 8, unless otherwise specified.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Characterization of the enzalutamide resistance model.
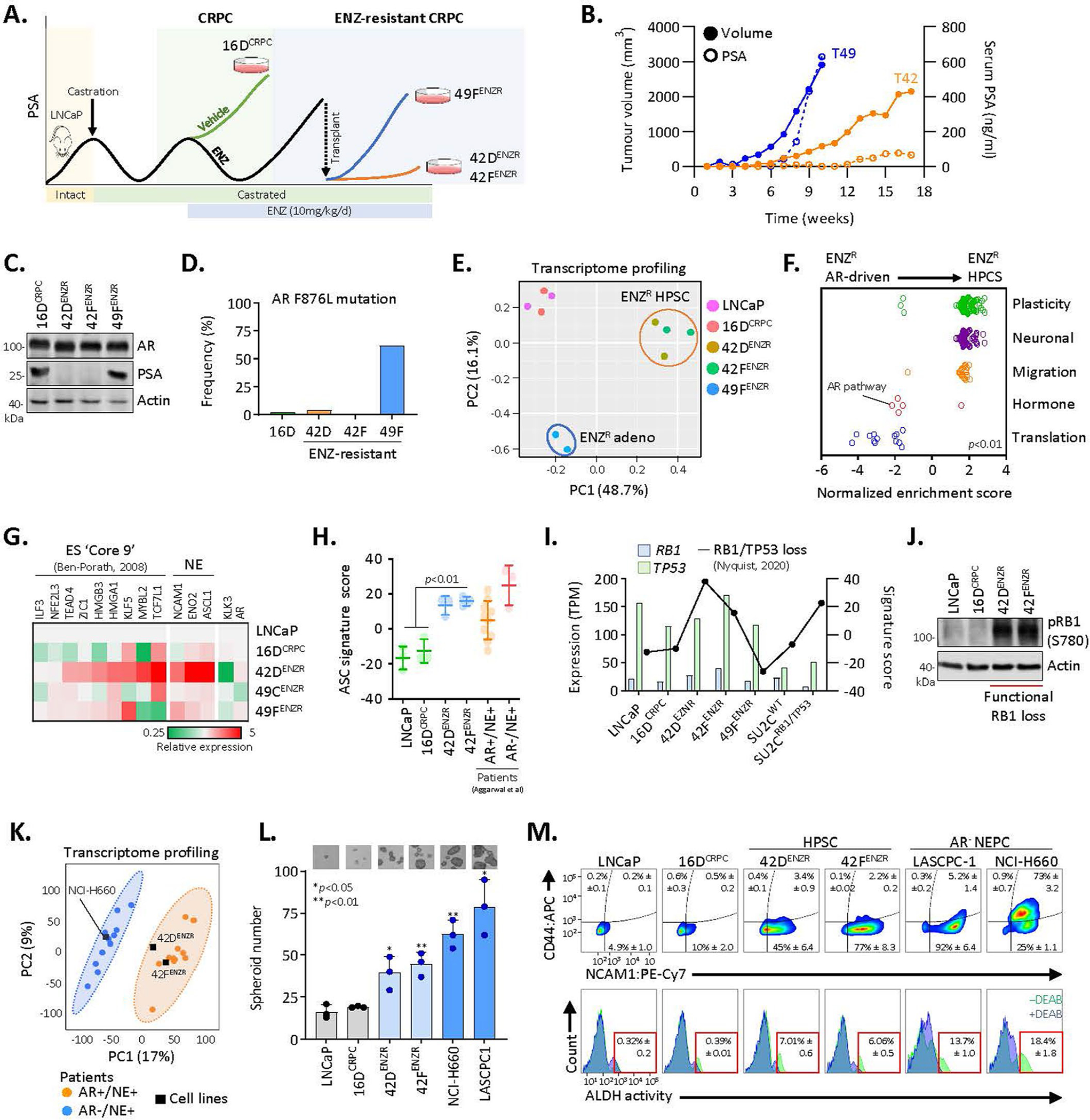
(a) Schematic depicting generation of the ENZ-driven resistance model. (b) Tumour volume and serum PSA of PSA+ (T49) and PSA− (T42) ENZ-resistant tumours at time following ENZ treatment. (c) Immunoblot of AR and PSA in cell lines derived from CRPC (16DCRPC) and ENZ-resistant AR-driven (49FENZR) and lineage plastic (42DENZR, 42FENZR) tumours. (d) Frequency of activating AR F876L mutation in CRPC and ENZ-resistant cell lines. (e) PCA of the global transcriptome in the indicated cell lines. (f) Significantly enriched (p < 0.05) gene ontology pathways in CRPC and ENZ-resistant cell lines ranked by normalized enrichment score. The following keywords were used to define functional categories: Plasticity (morphogenesis, plasticity, differentiation, mesenchymal); Neuronal (cerebral, axon, synap, neuro); Migration (chemotaxis, migration); Hormone (androgen, hormone); Translation. (g) Expression of ‘Core 9’ embryonic stem cell genes and neuronal lineage markers in the indicated cell lines, reported relative to LNCaP. (h) ASC scores in the indicated prostate cancer cell lines (n = 3) and AR+/NE+ and AR−/NE+ patient tumours from Aggarwal et al. Statistical analysis was performed using a two-tailed unpaired t-test. Error bars represent mean ± SD. (i) Transcript expression of RB1 and TP53 in cell lines and SU2C patient samples with wild-type RB1/TP53 (SU2CWT) or biallelic RB1 and TP53 deletion (SU2CRB1/TP53). An RB1/TP53 signature score was applied to cell lines and tumours (higher score indicative of functional RB1/TP53 loss). (j) Immunoblot of pRB1-S780 in the indicated cell lines. (k) Partial least squares discriminant analysis (PLS-DA) of global transcriptome separates AR+/NE+ and AR−/NE+ patient tumours from the Labrecque et al cohort (AR+/NE+, n = 11; AR−/NE+, n = 11; GEO: GSE126078). RNA-seq data from cell lines were projected on the PLS-DA plot. Probability ellipse=95% confidence. (l) Spheroid formation quantified at 8 days following seeding of single cells from the indicated cell lines (mean ± SD; two-tailed unpaired t-test, n = 3). Phase contrast images are shown. Scale bar, 100 μm. (m) Flow cytometry plots of CD44 and NCAM1 cell surface expression (top) and ALDH activity (bottom) in the indicated cell lines (mean ± SD). Diethylaminobenzaldehyde (DEAB) is as a control for background fluorescence.
Extended Data Fig. 2 |. ENZ-resistant lineage plastic tumours exhibit a distinct AR cistrome.
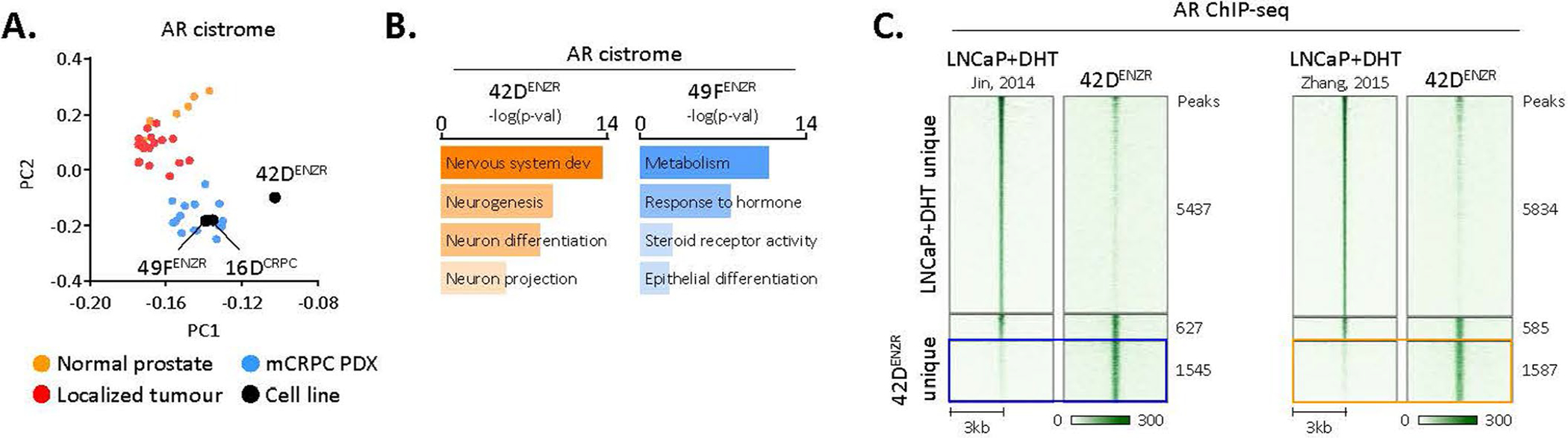
(a) Principal component analysis (PCA) reveals distinct AR binding patterns across prostate states. Each dot represents the genome-wide AR cistrome in an individual clinical specimen (6 normal prostate epithelial, 18 primary prostate cancer tumours, 15 PDX tumours derived from patient mCRPC; GEO:GSE130408) or indicated cell line. (b) Gene ontology (GO) pathways enriched surrounding AR binding sites in ENZ-resistant AR-driven (49FENZR) and lineage plastic (42DENZR) cells. The closest 2000 peaks in proximity to a transcriptional start site were used for pathway analysis. Statistical significance was determined using a hypergeometric test. Representative AR ChIP-seq tracks surrounding the KLK3/PSA locus are shown. (c) Heatmap indicating AR ChIP-seq signal intensity in 42DENZR cells and DHT-stimulated LNCaP cells from Jin et al and Zhang et al. The shade of green reflects binding intensity. Each horizontal line represents a 6-kb locus.
Extended Data Fig. 3 |. Lineage plastic NE-like tumours exhibit a unique chromatin accessibility profile.
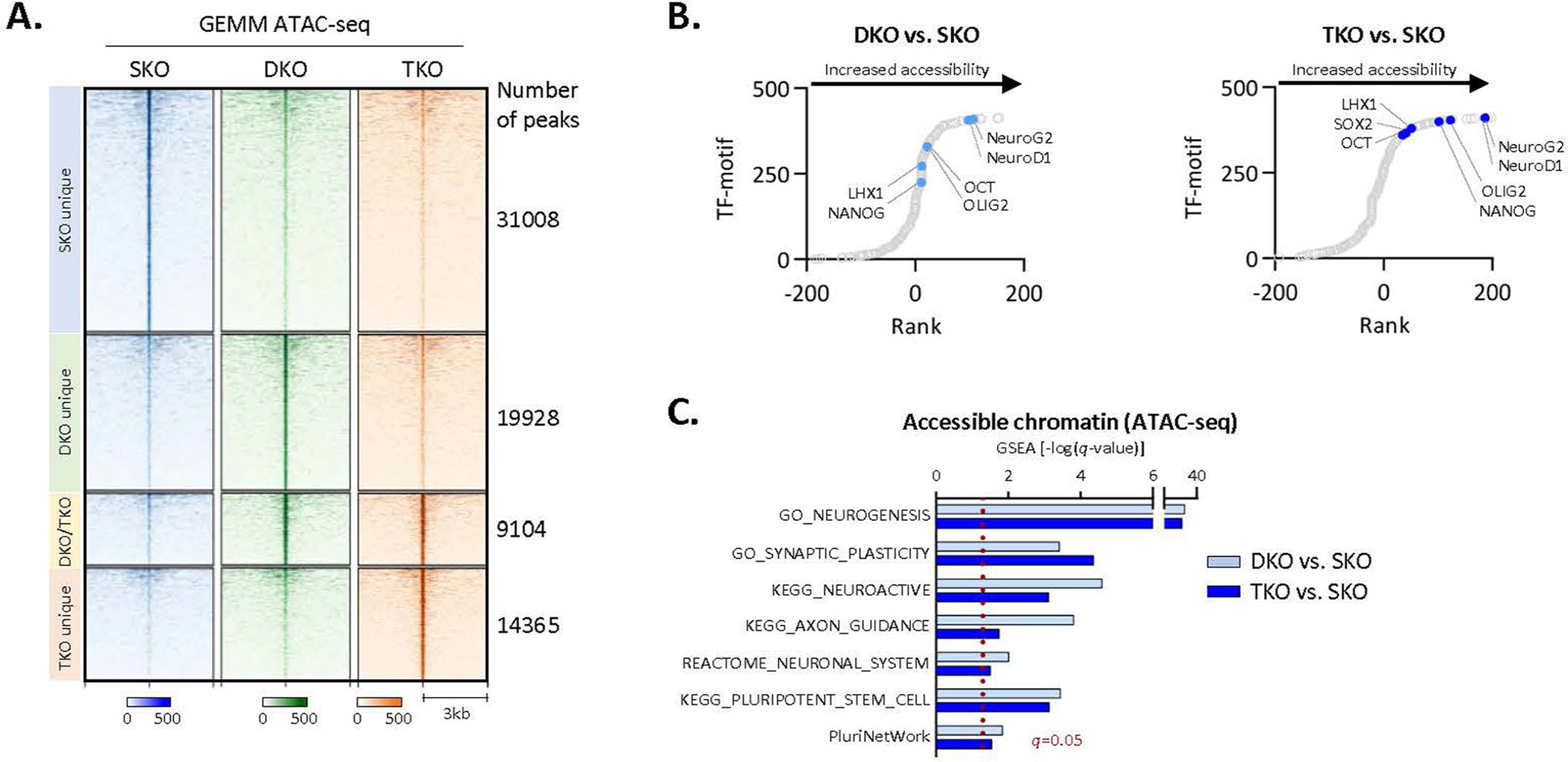
(a) Heatmap of ATAC-seq signal intensity in a GEMM of prostate adenocarcinoma (SKO, Ptenf/f) evolution to a plastic, NE-like state (DKO, Ptenf/f/Rb1f/f; TKO, Ptenf/f/Rb1f/f/Tp53f/f). Each horizontal line represents a 6-kb locus. (b) Motif analysis surrounding ATAC-seq peaks (250-bp) in NE-like DKO and TKO GEMMs compared to SKO. Transcription factor motifs identified by HOMER were plotted by ranks generated from their associated differential p values. (c) Significantly enriched pathways (gene set enrichment analysis) in accessible chromatin regions specific to NE-like DKO and TKO GEMMs compared to SKO.
Extended Data Fig. 4 |. The EZH2 cistrome is expanded in the lineage plastic state.
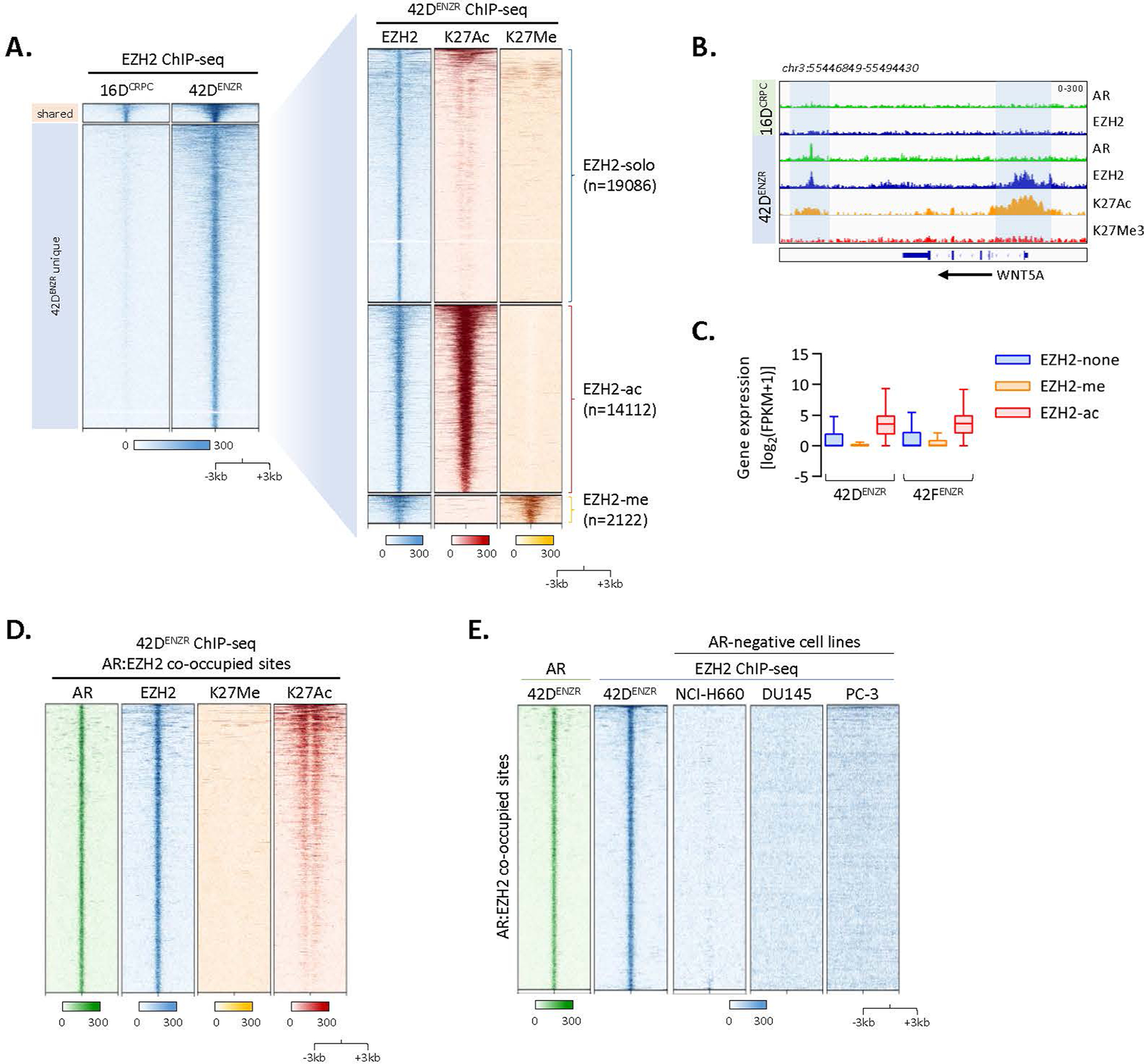
(a) Heatmap of EZH2 ChIP-seq signal intensity in CRPC 16DCRPC and 42DENZR cell lines (left), with overlaid H3K27Ac and H3K27Me3 histone mark ChIP-seq (right). Each horizontal line represents a 6-kb locus. (b) Representative ChIP-seq tracks surrounding the WNT5A locus in 16DCRPC and 42ENZR cells. Regions of EZH2 co-occupancy with the active H3K27Ac histone mark are highlighted. (c) Relative expression of genes bound by EZH2 alone (EZH2-none) or co-operatively with H3K27Me3 (EZH2-me) and H3K27Ac (EZH2-ac) histone marks in 42DENZR and 42FENZR cell lines. Box plot shows mean and interquartile range. (d) Heatmap of H3K27Me3 and K3K27Ac ChIP-seq signal intensity surrounding AR:EZH2 co-occupied regions in 42DENZR cells. (e) Heatmap indicating AR and EZH2 ChIP-seq signal intensity at AR:EZH2 co-occupied sites (n = 2155) in 42DENZR cells, and EZH2 signal intensity at the corresponding sites in AR-negative cell lines: NCI-H660, DU145 (GEO: GSE135623), and PC-3 (GEO: GSE123204). The shade of green (AR) or blue (EZH2) reflects binding intensity. Each horizontal line represents a 6-kb locus.
Extended Data Fig. 5 |. Characterization of EZH2 phosphorylation.
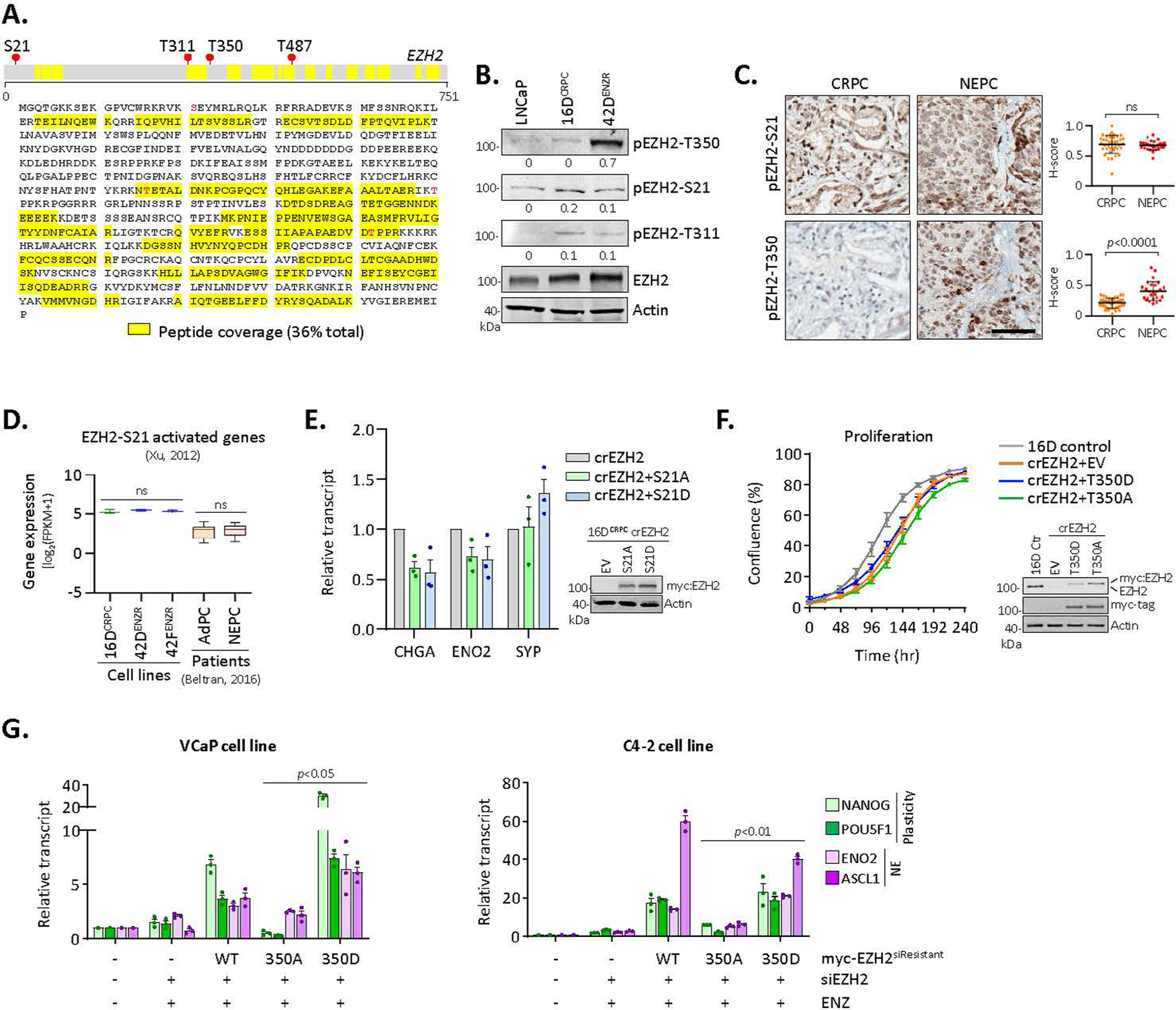
(a) EZH2 was immunoprecipitated in 42ENZR cells, trypsin digested, and analyzed by mass spectrometry. Peptides covering 36% of EZH2 were recovered and analyzed for post-translational modifications. (n = 4 independent replicates). (b) Expression of total and phosphorylated (T350, S21, and T311 residues) EZH2 in the indicated cell lines. Protein abundance was assessed by densitometry and is reported relative to total EZH2. (c) IHC staining of pEZH2-S21 and pEZH2-T350 in serial sections from representative CRPC (n = 39) and NEPC (n = 26) patient tumours (Scale bar, 100 μm). Staining area and intensity was quantified and reported (mean ± SD; two-tailed unpaired t-test). (d) Expression of genes positively regulated by EZH2 when phosphorylated at S21 [defined by Xu et al.] in the indicated cell lines and patient tumours from the Beltran 2016 cohort. Statistical analysis was performed using a two-tailed unpaired t-test. Box plots show mean and interquartile range. ns, not significant. (e) qRT-PCR of NE lineage markers in CRPCcrEZH2 cells expressing myc-tagged EZH2S21A or EZH2S21D mutants, reported relative to empty vector transfected cells. (mean ± SD; two-tailed unpaired t-test, n = 3). Immunoblotting confirmed transgene expression. (f) Proliferation of parental 16DCRPC (control) and CRPCcrEZH2 cells stably expressing EZH2T350A and EZH2T350D phospho-mutants assessed by IncuCyte (mean ± SD, n = 3 replicates). Immunoblotting confirmed transgene expression. (g) qRT-PCR of plasticity and NE markers in VCaP and C4–2 cell lines co-transfected with EZH2 siRNA and siRNA-resistant myc-tagged EZH2WT, EZH2T350A, or EZH2T350D plasmid following treatment with ENZ (10 μM) for 7 days (mean ± SD; two-tailed unpaired t-test, n = 3).
Extended Data Fig. 6 |. pEZH2-T350 is associated with lineage plasticity.
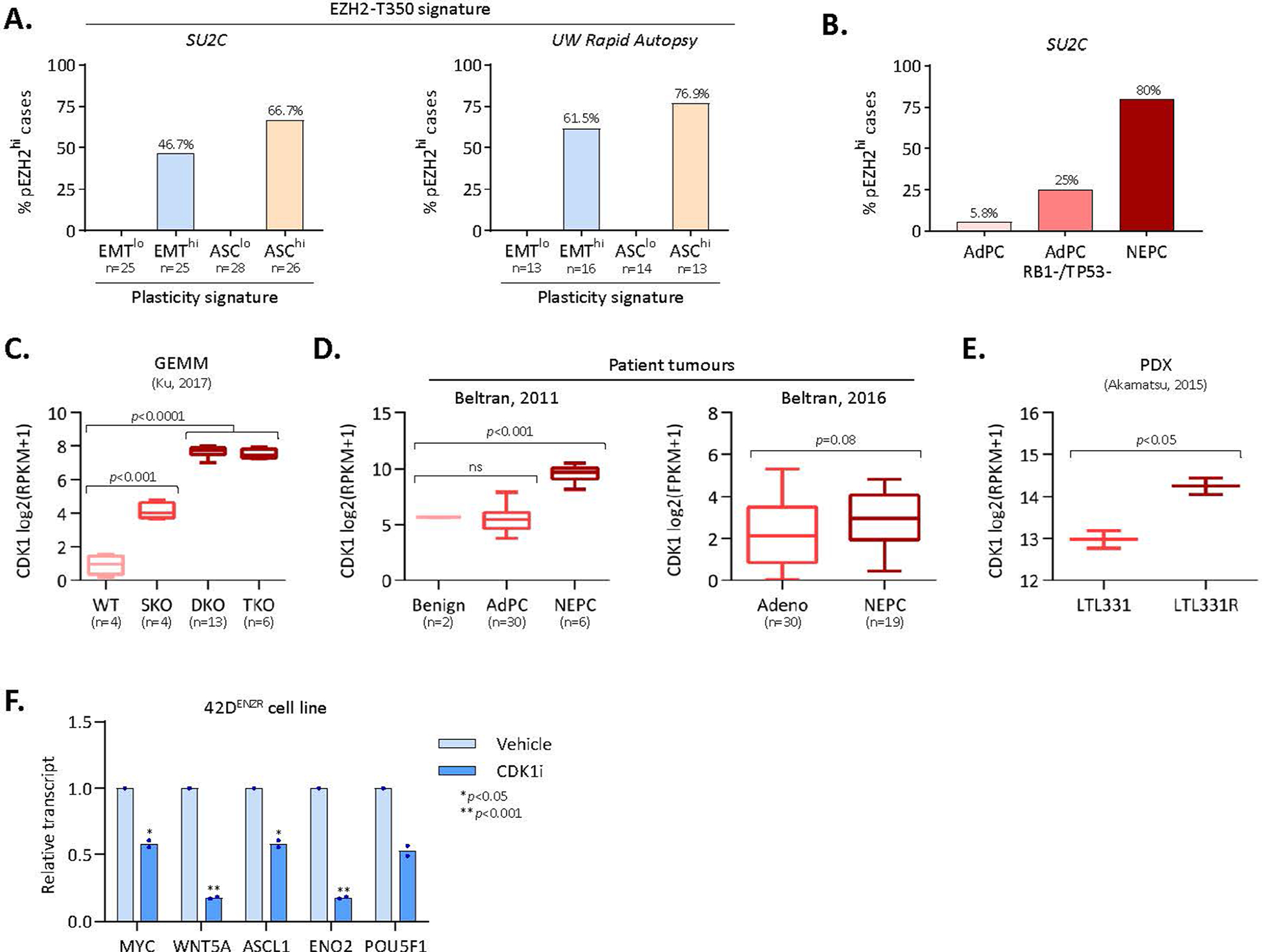
(a) Frequency of patients with low and high EMT and ASC signature scores in the SU2C37 and Labrecque et al clinical cohorts that exhibit a high pEZH2-T350 score (indicative of pEZH2-T350 phosphorylation). Patients were defined as “lo” or “hi” for each signature based on +/− 1 standard deviation from the mean signature score in the respective cohort. (b) Frequency of patients with high pEZH2-T350 score (defined as ≥1 standard deviation from cohort mean) in adenocarcinoma, adenocarcinoma with genomic RB1/TP53 loss, and NEPC patient tumours from the SU2C clinical cohort. (c) CDK1 transcript abundance in a GEMM model of prostate adenocarcinoma to NE-like tumour evolution (GEO: GSE90891). DKO and TKO tumours mimic NE-like tumours. SKO, PBCre4:Ptenf/f; DKO, PBCre4:Ptenf/f;Rb1f/f; TKO, PBCre4:Ptenf/f;Rb1f/f;Trp53f/f. Statistical analysis was performed using a two-tailed unpaired t-test. Box plots show mean and interquartile range. (d) CDK1 transcript abundance in benign prostate, adenocarcinoma (AdPC), and NEPC patient specimens from the 2011 Beltran cohort and 2016 Beltran cohort. Statistical analysis was performed using a two-tailed unpaired t-test. Box plots show mean and interquartile range. (e) CDK1 transcript abundance in a patient-derived xenograft (PDX) model of adenocarcinoma (LTL331) to NEPC (LTL331R) lineage conversion following androgen deprivation (castration) (ENA: PRJEB9660). Statistical analysis was performed using a two-tailed unpaired t-test. Mean with min/max range is reported. (f) Expression (qRT-PCR) of genes in 42DENZR cells following treatment with CDK1 inhibitor (5 μM RO-3306) for 24 hours. Data are reported relative to vehicle treated cells (mean ± SD; two-tailed unpaired t-test, n = 2).
Extended Data Fig. 7 |. Assessment of neuroendocrine differentiation following AR silencing.
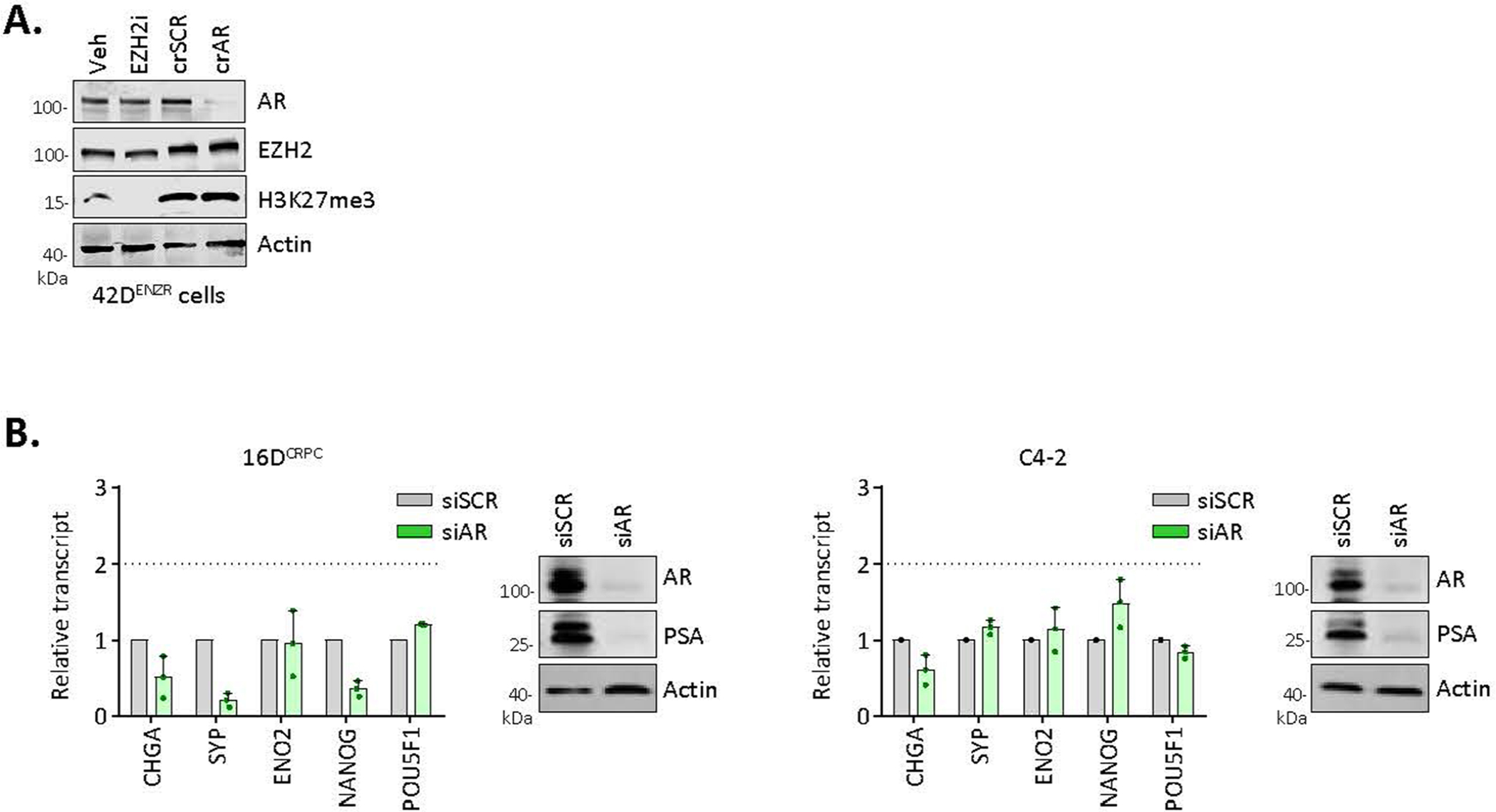
(a) Immunoblot AR, EZH2, and H3K27Me3 (a surrogate marker of EZH2 activity) in 42DENZR cells following CRISPR-mediated AR deletion (crAR) or EZH2 inhibition (10 μM GSK126, 96 hrs). (b) Relative expression (qRT-PCR) of neuroendocrine lineage markers in 16DCRPC and C4–2 cell lines following siRNA-mediated AR silencing for 96 hours. Data are reported relative to cells transfected with a non-silencing scrambled control (mean ± SD, n = 3). A fold change >2 is considered significant. Immunoblotting confirmed AR knockdown.
Extended Data Fig. 8 |. Silencing EZH2 expression and/or activity reverts the lineage-infidelity phenotype.

(a-b) qRT-PCR in 42DENZR (a) and 42FENZR (b) cells following siRNA-mediated EZH2 silencing (siEZH2) for the indicated time, reported relative to non-transfected control cells at day 0 (mean ± SD; two-tailed unpaired t-test, n = 3). NTC, non-targeting control. (c-d) Spheroid formation and ALDH activity in 42DENZR (c) and 42FENZR (d) cells following siRNA-mediated EZH2 silencing (siEZH2; left) or treatment with increasing dose of EZH2 inhibitor (GSK126; right) for 8 days (mean ± SD; two-tailed unpaired t-test, n = 2). (e) qRT-PCR in 42DENZR cells treated with EZH2 inhibitor (10 μM GSK126) for 7 days, followed by removal (washout) for 14 days. Expression is reported relative to cells at day 0 (mean ± SD; two-tailed unpaired t-test, n = 3). Immunoblotting confirmed on-target effect.
Extended Data Fig. 9 |. Flow Cytometry Gating.
Flow cytometry gating strategy used in Fig. 4k and Extended Data Fig. 1m.
Supplementary Material
Acknowledgements
This research was supported by funding from the Terry Fox Research Institute New Frontiers Program (F15-05505 to A.Z.), the Prostate Cancer Foundation (to A.Z. and H.B.), the Canadian Institutes of Health Research (399802 to A.D. and A.Z.), Dutch Cancer Society/Alpe d’HuZes (10084 to W.Z.), and a Prostate Cancer Foundation Young Investigator Award (to A.D.). We thank members of the Zoubeidi laboratory for valuable inputs in designing this research, the Molecular Pathology Core (Vancouver Prostate Centre) for assisting with tissue processing and immunohistochemistry, the Flow Cytometry Core Facility (BC Cancer Research Centre) for cell sorting, the Genomic Analysis Core (Vancouver Prostate Centre) for ChIP-seq and ATAC-seq processing, the Genomics Core Facility (Netherlands Cancer Institute) for ChIP-seq analyses on prostate tumour specimens, the Core Facility for Molecular Pathology and Biobanking (Netherlands Cancer Institute) for tissue processing and haematoxylin and eosin analyses using Slide Score, the Biomedical Research Centre Sequencing Core (University of British Columbia) for RNA-seq, and O. Witte and J. Lee (UCLA) for providing LASCPC-1 cells. L.S. is supported by a Principal Cancer Research Fellowships awarded by Cancer Council’s Beat Cancer project on behalf of its donors, the State Government through the Department of Health and the Australian Government through the Medical Research Future Fund.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41556-021-00743-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41556-021-00743-5.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41556-021-00743-5.
Data availability
RNA-seq, ChIP-seq and ATAC-seq data have been deposited in the GEO under the accession GSE138460. Previously published sequencing data were re-analysed here: publicly available data from the SU2C/PCF-West Coast Dream Team cohort was obtained from ref. 5, the SU2C cohort from GitHub (https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_2019)37, the University of Washington Rapid Autopsy cohort6 from GEO accession GSE126078, the Beltran 2016 cohort4, and the CALGB 90203 cohort60. The SKO/DKO/TKO prostate cancer GEMM dataset7 was downloaded from GEO accession GSE90891 and the NPp53 GEMM13 was downloaded from GEO accession GSE92721. Source data are provided with this paper.
References
- 1.Davies AH, Beltran H & Zoubeidi A Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 15, 271–286 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Le Magnen C, Shen MM & Abate-Shen C Lineage plasticity in cancer progression and treatment. Annu. Rev. Cancer Biol. 2, 271–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boumahdi S & de Sauvage FJ The great escape: tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 19, 39–56 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aggarwal R et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 36, 2492–2503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labrecque MP et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Invest. 129, 4492–4505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ku SY et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mu P et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beltran H et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 1, 487–495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aparicio AM et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 19, 3621–3630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzelepi V et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer Res. 18, 666–677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aparicio AM et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin. Cancer Res. 22, 1520–1530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou M et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 7, 736–749 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith BA et al. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl Acad. Sci. USA 112, E6544–E6552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith BA et al. A human adult stem cell signature marks aggressive variants across epithelial cancers. Cell Rep. 24, 3353–3366 e3355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams SG et al. Immune molecular profiling of a multiresistant primary prostate cancer with a neuroendocrine-like phenotype: a case report. BMC Urol. 20, 171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alumkal JJ et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl Acad. Sci. USA 117, 12315–12323 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge R et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 31, 470–479 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell 37, 584–598 e511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clermont PL et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenetics 7, 40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820 e2804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu K et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bluemn EG et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 32, 474–489(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bishop JL et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 7, 54–71 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Korpal M et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 3, 1030–1043 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Nyquist MD et al. Combined TP53 and RB1 loss promotes prostate cancer resistance to a spectrum of therapeutics and confers vulnerability to replication stress. Cell Rep. 31, 107669 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burger PE et al. High aldehyde dehydrogenase activity: a novel functional marker of murine prostate stem/progenitor cells. Stem Cells 27, 2220–2228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pomerantz MM et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 52, 790–799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park JW et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 362, 91–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol. Cell 57, 304–316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu P, Chen M, Liu Y, Qi LS & Ding S CRISPR-based chromatin remodeling of the endogenous Oct4 or Sox2 locus enables reprogramming to pluripotency. Cell Stem Cell 22, 252–261 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Park NI et al. ASCL1 reorganizes chromatin to direct neuronal fate and suppress tumorigenicity of glioblastoma stem cells. Cell Stem Cell 21, 209–224 e207 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Wan L et al. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol. Cell 69, 279–291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei Y et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 13, 87–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaneko S et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 24, 2615–2620 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell Biol. 12, 1108–1114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abida W et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl Acad. Sci. USA 116, 11428–11436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim KH et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 21, 1491–1496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong DJ et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2, 333–344 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Porath I et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499–507 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen S, Xu Y, Yuan X, Bubley GJ & Balk SP Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc. Natl Acad. Sci. USA 103, 15969–15974 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang XQ et al. CDK1–PDK1–PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ. 24, 38–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng X, Chen S & Huang H Phosphorylation of EZH2 by CDK1 and CDK2: a possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle 10, 579–583 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKay RR et al. Evaluation of intense androgen deprivation before prostatectomy: A randomized phase II trial of enzalutamide and leuprolide with or without abiraterone. J. Clin. Oncol. 37, 923–931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hussain M et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 378, 2465–2474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bai Y et al. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J. Biol. Chem. 294, 9911–9923 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao L et al. Epigenetic reprogramming with antisense oligonucleotides enhances the effectiveness of androgen receptor inhibition in castration-resistant prostate cancer. Cancer Res. 78, 5731–5740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puca L et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 9, 2404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuruma H et al. A novel antiandrogen, compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol. Cancer Ther. 12, 567–576 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buenrostro JD et al. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu G, Wang LG & He QY ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar V et al. Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 31, 615–622 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Beltran H et al. Impact of therapy on genomics and transcriptomics in high-risk prostate cancer treated with neoadjuvant docetaxel and androgen deprivation therapy. Clin. Cancer Res. 23, 6802–6811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ertel A et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 9, 4153–4163 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ChIP-seq and ATAC-seq data have been deposited in the GEO under the accession GSE138460. Previously published sequencing data were re-analysed here: publicly available data from the SU2C/PCF-West Coast Dream Team cohort was obtained from ref. 5, the SU2C cohort from GitHub (https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_2019)37, the University of Washington Rapid Autopsy cohort6 from GEO accession GSE126078, the Beltran 2016 cohort4, and the CALGB 90203 cohort60. The SKO/DKO/TKO prostate cancer GEMM dataset7 was downloaded from GEO accession GSE90891 and the NPp53 GEMM13 was downloaded from GEO accession GSE92721. Source data are provided with this paper.