

Abstract
CD8+ T cell responses are the foundation of the recent clinical success of immunotherapy in oncologic indications. Although checkpoint inhibitors have enhanced the activity of existing CD8+ T cell responses, therapeutic approaches to generate Ag-specific CD8+ T cell responses have had limited success. Here, we demonstrate that cytosolic delivery of Ag through microfluidic squeezing enables MHC class I presentation to CD8+ T cells by diverse cell types. In murine dendritic cells (DCs), squeezed DCs were ∼1000-fold more potent at eliciting CD8+ T cell responses than DCs cross-presenting the same amount of protein Ag. The approach also enabled engineering of less conventional APCs, such as T cells, for effective priming of CD8+ T cells in vitro and in vivo. Mixtures of immune cells, such as murine splenocytes, also elicited CD8+ T cell responses in vivo when squeezed with Ag. We demonstrate that squeezing enables effective MHC class I presentation by human DCs, T cells, B cells, and PBMCs and that, in clinical scale formats, the system can squeeze up to 2 billion cells per minute. Using the human papillomavirus 16 (HPV16) murine model, TC-1, we demonstrate that squeezed B cells, T cells, and unfractionated splenocytes elicit antitumor immunity and correlate with an influx of HPV-specific CD8+ T cells such that >80% of CD8s in the tumor were HPV specific. Together, these findings demonstrate the potential of cytosolic Ag delivery to drive robust CD8+ T cell responses and illustrate the potential for an autologous cell-based vaccine with minimal turnaround time for patients.
Key Points
Cytosolic delivery of Ag enables MHC-I presentation by diverse immune cells.
Squeeze-engineered APCs prime CD8+ T cells with antitumor activity.
Introduction
CD8+ T cells are the driving force for adaptive immune responses against intracellular targets; hence, they play a key role in combating infectious diseases and cancer. The success of checkpoint inhibitors in many cancer indications has generated excitement about the prospect of immunotherapy (1–3), and their impact in cancers with high mutational burden has correlated with the presence of CD8+ T cells in the tumor (4). Although these approaches have enhanced the potential of existing CD8+ T cell responses by combating immunosuppression, the ability to prime CD8+ T cell responses in an Ag-specific manner has proved challenging (5). Preclinical and clinical studies with mRNA, peptide, DNA, viral, and bacterial vaccination methods, which are normally effective for CD4 and humoral responses, have shown some promise but have not generated adequate CD8+ T cell responses to merit widespread adoption in clinical practice (6). Although the potential impact of an effective vaccine that elicits potent tumor-specific CD8+ T cells is immense, a potent approach has remained elusive.
The physiological interaction that underpins the priming of CD8+ T cell responses is that between an APC and the T cell. A critical bottleneck for CD8+ T cell activation is routing the desired Ag into the MHC class I (MHC-I) pathway of the APC to facilitate TCR stimulation. Peptides presented on MHC-I are derived from proteins that are degraded within the cytosol, transported into the endoplasmic reticulum, where they are loaded onto MHC-I, and then trafficked to the surface for display to CD8+ T cells (7). Although the cytosolic presence of target Ag is critical to this mechanism, current vaccination approaches have by and large relied on introducing Ags into the extracellular vicinity of APCs or targeting their surface. In these contexts, APCs will endocytose the target material, thus segregating it from the cytosol in endosomal vesicles. There is, however, a process termed “cross-presentation” by which a fraction of Ags in endosomes escape into the cytosol and can be presented on MHC-I (8). Data from in vitro experimental models suggest that this transfer of Ags into the cytosol is a major rate-limiting step in cross-presentation (9). The process of cross-presentation is most often associated with dendritic cells (DCs), which has made them the favored cell type for cell-based approaches seeking to elicit CD8+ T cell responses. Commercial and clinical success with this approach has been limited (10) due to the limited number of DCs in peripheral blood and the inefficiency with which Ags are cross-presented onto MHC-I by DCs.
Although delivery of materials to primary immune cells is challenging (11), microfluidic squeezing, which can deliver biomaterials directly into the cytosol of a wide array of cell types, represents an attractive approach for engineering Ag presentation to CD8+ T cells. The squeezing process uses rapid mechanical deformation of cells to generate temporary pores that enable material delivery while minimally altering normal cellular function (12). In this work, we describe the implementation of the squeeze technology to facilitate direct cytosolic delivery of Ags. We studied its impact across various primary human and murine immune cells, including DCs, T cells, B cells, and heterogeneous populations (splenocytes or PBMCs). Our results demonstrate that direct cytosolic loading dramatically improved Ag presentation across the tested cell types. In the context of DCs, for example, direct cytosolic delivery of protein Ag was shown to be ∼1000 times more effective than cross-presentation after endocytic uptake of protein Ag. These engineered cells also showed potent abilities to stimulate naive and previously activated CD8+ T cells, both in vitro and in vivo. We also demonstrate that microfluidic squeezing can enable Ag presentation by human cells at manufacturing scale for potential clinical application. Finally, we show that immune responses elicited by squeezed cells, in combination with an adjuvant, are capable of driving antitumor effects that correlate with an influx of tumor-specific CD8+ T cells. By overcoming the fundamental barrier to effective MHC-I presentation of Ag, squeeze-engineered cells could potentially be used as the basis for a potent, rapid turnaround, cell-based vaccine that is applicable across tumor types.
Materials and Methods
Ethics statement
All experimental methods were carried out in accordance with approved guidelines. The blood collection procedure was performed in accordance with guidelines approved by the New England Independent Review Board. All donors signed an informed consent for scientific research statement. All animal work was carried out in accordance with guidelines approved by the Institutional Animal Care and Use of Laboratory Animals and the US Government Principles for Utilization and Care of Vertebrate Animals Used in Testing, Research and Training.
Cell isolation
Human PBMCs were isolated from fresh blood using Ficoll gradient centrifugation. Human T cells were isolated using the EasySep Human T Cell Enrichment Kit (19051; STEMCELL Technologies). Human B cells were isolated using the EasySep Human B Cell Enrichment Kit (19054; STEMCELL Technologies). Murine T cells were isolated directly from the spleen using the EasySep Mouse T Cell Isolation Kit (19851; STEMCELL Technologies). Murine B cells were isolated directly from the spleen using the EasySep Mouse Pan-B Cell Isolation Kit (19844; STEMCELL Technologies). To generate a murine cell composition that approximates human PBMCs, a PBMC surrogate was generated with murine splenocytes by combining B cell–depleted splenocytes and untouched splenocytes in a ratio of 4:1. Four dissociated spleens were depleted of B cells using the EasySep Mouse CD19 Positive Selection Kit II (18954; STEMCELL Technologies) and combined with an untouched dissociated spleen before ammonium-chloride-potassium buffer RBC lysis. The method generates a cell composition of 70–80% CD3+ cells and 10–20% B220+ cells, with smaller fractions of NK cells, monocytes, and neutrophils. For manufacturing scale, full Leukopaks were enriched for WBCs by density centrifugation. T cells were purified using a CliniMACS Plus (151-01; Miltenyi Biotec).
Cell squeezing and microfluidic devices
Cells were resuspended at 10–50 × 106 cells/ml in RPMI 1640 (11875-093; Life Technologies) as the delivery buffer. Cell squeezing was performed using previously established methods (13, 14) with the specifications described below. Human and murine cells were squeezed through microfluidic channels containing a single 3.5–6-µm-wide, 10–30-µm-long constriction at 45–60 pounds per square inch, depending on the cell type being squeezed. When appropriate, cargo was added to the cells and delivery buffer before squeezing. Delivery for each cell type was evaluated using 100 µg/ml 3 kDa Dextran Cascade Blue (D7132; Invitrogen). Viability was measured using the Zombie Yellow Fixable Viability Kit (423103; BioLegend). For manufacturing scale, 10 µg/ml 3 kDa Dextran Alexa Fluor 680 (D34681; Thermo Fisher Scientific) was used for squeezing.
Dendritic cell culture
Murine bone marrow-derived dendritic cells (BMDCs) were generated by culturing bone marrow cells in the presence of 55 µM 2-ME (Thermo Fisher Scientific), 20 ng/ml murine GM-CSF (R&D Systems), and 10 ng/ml murine IL-4 (R&D Systems). After 6–8 d, BMDCs were matured for 1 h with 100 international units of edotoxin/ml LPS (InvivoGen) and 100 ng/ml IFN-γ (R&D Systems) before squeezing. Human monocyte-derived DCs (moDCs) were generated from human monocytes isolated from PBMCs using the EasySep Human CD14+ Enrichment Kit without CD16 Depletion (19058; STEMCELL Technologies). Cells were cultured for 6 d in the presence of 800 U/ml GM-CSF (R&D Systems) and 1000 U/ml IL-4 (R&D Systems). Flow cytometry was performed to confirm an immature DC phenotype of CD14−/low and CD11c+.
Immunization after adoptive transfer of TCR-transgenic cells
The following female 8–10-wk-old mice obtained from The Jackson Laboratory were used: C57BL/6J (000664), B6.SJL-Ptprca Pepcb/BoyJ (002014), B6.129P2-B2mtm1Unc/J (i.e., MHC-I knockout [β2m−/−]; 002087), C57BL/6-Tg(TcraTcrb)1100Mjb/J (i.e., OT-I; 003831), and B6.Cg-Thy1a/Cy-Tg(TcraTcrb)8Rest/J (i.e., pmel; 005023). CD8+ T cells were isolated from OT-I or pmel mice using the EasySep Mouse CD8+ T Cell Isolation Kit (19853; STEMCELL Technologies) and labeled with the CellTrace CFSE Cell Proliferation Kit (C34554; Invitrogen). CFSE-labeled CD8+ T cells (2.5 × 106 per mouse) were administered i.v. on day 0 into either wild-type (WT) or β2m−/− recipients. Murine T or B cells were squeezed with 400 µg/ml EndoFit OVA (vac-pova-100; InvivoGen) as described above or incubated in the presence of OVA, and 5 × 106 cells were coinjected with 3 µg LPS i.v. on day 0 into the same WT or β2m−/− recipients. For studies using the gp100 Ag, 2.5 × 106 CFSE-labeled pmel CD8+ T cells were transferred on day 0 along with B cells squeezed or incubated with 250 µg/ml gp100 synthetic long peptide (SLP) (AVIGALLAVGALKVPRNQDWLGVSRQLRTKAWNRQ; BIOSYNTAN). Epitope pulsed B cell conditions were prepared by incubating B cells with minimal epitope (SIINFEKL or KVPRNQDWL; AS-60193 and AS-62589, respectively; Anaspec) for 1 h at 37°C, washing, and administering on day 0. After 3 d, lymph nodes were harvested, then processed into a single-cell suspension, and proliferation of OT-I cells was assessed by CFSE dilution using flow cytometry.
In vivo immunization and ex vivo restimulation
Cells of interest were cultured as described or harvested from the spleen and isolated accordingly. Cells were squeezed with OVA (vac-pova-100; InvivoGen) or E7 SLP (GQAEPDRAHYNIVTFSSKSDSTLRLSVQSTHVDIR; BIOSYNTAN). For DC immunizations, DCs were treated for 1 h with 100 international units of edotoxin/ml LPS (InvivoGen) and 100 ng/ml IFN-γ (R&D Systems) before squeezing. B cells were incubated with 1 µM CpG 1826 (vac-1826-1; InvivoGen) for 16 h at 37°C after squeezing. T cells were coinjected with 25 µg CpG. PBMC surrogate cells were incubated with 1 µM CpG for 4 h at 37°C after squeezing. After 7 d, spleens were harvested and restimulated with 1 µg/ml SIINFEKL (AS-60193) or 1 µg/ml RAHYNIVTF (AS-61022) and 2 µg/ml anti-CD28 (16-0281-86; eBioscience). After 1 h of incubation, GolgiPlug (555029; BD Biosciences) and GolgiStop (554724; BD Biosciences) were added for an additional 4 h. Cells were stained, fixed, and permeabilized according to the manufacturer’s instructions (554714; BD Biosciences) and subsequently analyzed by flow cytometry.
In vitro stimulation of human T cell responders
CMV pp65-specific CD8+ T cells that recognize the HLA-A*02 restricted peptide, NLVPMVATV, or the HLA-B*35 restricted peptide, IPSINVHHY, were purchased from Astarte Biologics. Human moDCs, T cells, B cells, or PBMCs squeezed with 50 µM SLP derived from CMV pp65 (HLA-A*02 restricted: PPWQAGILARNLVPMVATVQGQNLKYQEFFWDAND; BIOSYNTAN) were cocultured with CMV-specific CD8+ T cells in XVivo15 media (04-418Q; Lonza) supplemented with 5% Human AB Serum (H4522-100ML; Corning) with 20 U/ml recombinant human IL-2 (202-IL-010; R&D Systems) overnight. After overnight culture, supernatants were collected, and functional responses were assessed by production of IFN-γ by ELISA (430104; BioLegend). For HLA-B*35 restricted responses, B cells were squeezed with 50 µM HLA-A*02 restricted pp65 SLP, 50 µM HLA-B*35 restricted pp65 SLP (HLA-B*35 restricted: LPLKMLNIPSINVHHYPSAAERKHR; BIOSYNTAN), 50 µM of each SLP. Cryopreserved T cells were resuspended to 1 × 107 cells/ml in cryoprotectant media and frozen using a controlled rate freezer. T cells were thawed, and the buffer was exchanged before coculture with responder cells.
E.G7-OVA tumor studies
C57BL/6J mice were anesthetized using isoflurane and shaved on the right flank. E.G7-OVA cells (1 × 105, CRL-2113; American Type Culture Collection) were s.c. injected into the flank with a 25-gauge needle. Mice were subsequently monitored twice weekly for tumor volume and body weight.
TC-1 tumor studies
TC-1 cells were obtained from Dr. T.C. Wu (Johns Hopkins University). C57BL/6J mice were anesthetized using isoflurane and shaved on the right flank. TC-1 cells (5 × 104) were s.c. injected into the flank with a 25-gauge needle. Mice were subsequently monitored twice weekly for tumor volume and body weight.
Tumor-infiltrating lymphocytes
Tumors were dissected from the right rear flank of the animal and weighed. Tumors were minced using scissors, then dissociated using a gentleMACS dissociator and incubated at 37°C for 45 min with continuous rotation. The suspension was then filtered over a 70-µm cell strainer and processed for flow cytometric analysis. The E7 tetramer was from MBL International (TB-5008-1).
Flow cytometric analysis
Flow cytometry was performed using Attune NxT (Thermo Fisher Scientific). Data were examined using FlowJo (BD Biosciences). Fluorescently conjugated Abs were purchased from BioLegend.
Statistics
An unpaired two-tailed Student t test (for two-group comparisons) or a two-way ANOVA (for comparisons of more than two groups) was performed using Prism (GraphPad Software) to calculate statistical significance of the difference in mean values and p values. A p value <0.05 was considered statistically significant.
Results
Microfluidic squeezing enables Ag presentation in vivo by diverse immune cell types
Cellular approaches to priming CD8+ T cell responses have traditionally used DCs, given their ability to cross-present Ags (8). To assess whether cytosolic delivery of Ag by microfluidic squeezing was superior to cross-presentation, mouse BMDCs were loaded with increasing concentrations of fluorescent OVA by either 30-min incubation (endocytosis) or squeezing. The mean fluorescence intensity (MFI) of fluorescent OVA showed that incubation alone resulted in approximately two to four times greater internalization of OVA within the range evaluated (Fig. 1A and (1B). However, the inefficiency of escape from endosomes into the cytosol and subsequent presentation on MHC-I was demonstrated by coculture of these BMDCs with OT-I CD8+ T cells (specific for the OVA-derived epitope, SIINFEKL). Using CD69 upregulation on OT-I T cells as a measure for Ag presentation to CD8+ T cells, squeezed BMDCs induced 70% upregulation of CD69 on OT-I T cells at a concentration of 5 µg/ml (MFI 170), whereas cross-presenting BMDCs induced similar CD69 upregulation at a concentration 1,000-fold higher (5,000 µg/ml OVA; MFI 42,000) (Fig. 1C). Presentation of OVA-derived SIINFEKL across the different concentrations was also quantified using the 25-D1.16 Ab, which recognizes SIINFEKL in the context of H-2Kb (15). This reinforced what was seen with activation of OT-I cells in that there was substantially greater 25-D1.16 staining on DCs that had been squeezed with OVA than on those that had been incubated with OVA (Fig. 1D and (1E). This suggests that cytosolic entry of endocytosed Ags is a major bottleneck for MHC-I presentation of proteins and that cytosolic delivery by squeezing bypasses this inefficient process.
FIGURE 1.
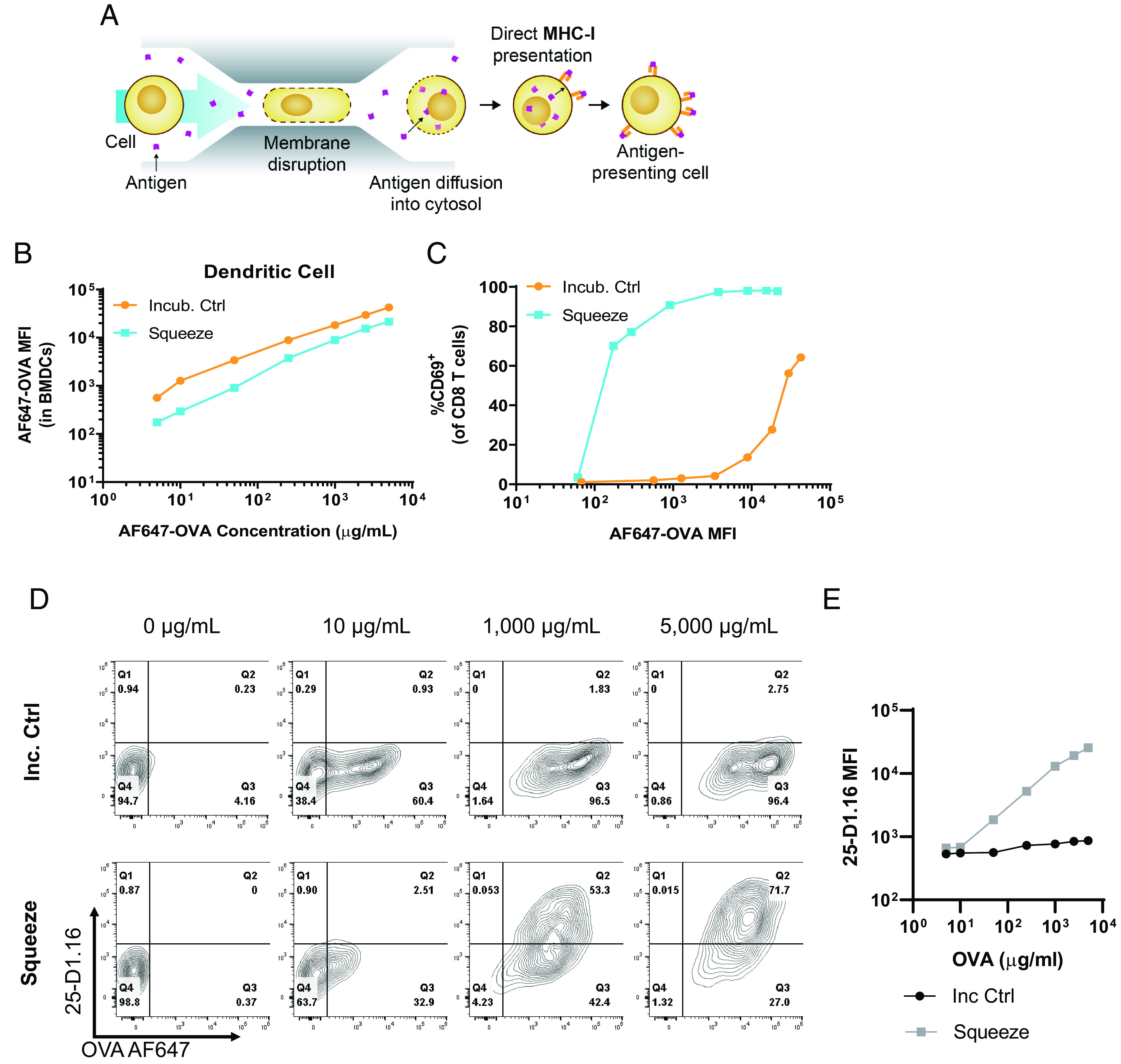
Microfluidic squeezing enhances Ag presentation by professional APCs. (A) Cells are squeezed in the presence of Ag in a microfluidic chip that creates temporary pores in the cells, allowing Ag to diffuse into the cytosol, from which it can be presented on MHC-I. (B–E) Mouse BMDCs were incubated with varying concentrations of fluorescently labeled OVA for 30 min at 37°C (Incub. Ctrl) or squeezed with the same concentrations (Squeeze). (B) OVA MFI for Incub. Ctrl and Squeeze groups at varying OVA concentrations. (C) Incub. Ctrl and Squeeze BMDCs were cocultured with OT-I CD8+ T cells for 24 h, and CD69 staining on the OT-I cells was assessed by flow cytometry. (D and E) Mouse BMDCs were incubated with varying concentrations of fluorescently labeled OVA for 30 min at 37°C (Incub. ctrl) or were squeezed with the same concentrations (Squeeze). Incub. ctrl and squeezed BMDCs were incubated for 4 h at 37°C and subsequently stained with 25-D1.16 Ab and analyzed by flow cytometry. Representative plots for indicated concentrations are shown in (D). Summary data for all concentrations are shown in (B). Data are representative of two independent experiments.
Demonstrating superior activation of CD8+ T cell responses by squeezed DCs in comparison with conventional cross-presenting DCs suggested that squeezing could enable presentation by other immune cells. B cells are attractive for use as APCs because they are abundant in the circulation and thus do not require expansion (up to 5 × 105/ml in human blood). Furthermore, B cells can migrate into lymphoid organs from the blood, which is where CD8+ T cell responses are primed. Delivery of material to B cells was demonstrated using fluorescent 3 kDa dextran (Supplemental Fig. 1A), as had been shown previously (16). To assess the kinetics of MHC-I presentation, OVA was delivered to B cells, and presentation was monitored using the 25-D1.16 Ab (15). SIINFEKL-MHC class I complexes could be found in as little as 2 h after squeeze and continued to accumulate for at least 4 h (Supplemental Fig. 1B). To evaluate the ability of squeezed B cells to prime CD8+ T cell responses, endogenous CD8+ T cell responses in naive mice were measured. B cells were squeezed with OVA and subsequently incubated with or without the TLR9 agonist, CpG, which will induce upregulation of costimulatory molecules as well as inflammatory cytokines by B cells. In this context, the importance of CpG in generating a CD8+ T cell response was apparent as B cells squeezed with OVA but incubated without the CpG failed to prime endogenous responses. Conversely, squeezed B cells incubated with CpG before injection induced robust CD8+ T cell responses as measured by an increase in IFN-γ–producing CD8+ T cells in the spleen when restimulated ex vivo with the SIINFEKL epitope (Fig. 2A).
FIGURE 2.

Microfluidic squeezing enables Ag presentation in vivo by diverse immune cell types and requires adjuvant. (A) Mice were left untreated or immunized with 1 × 106 B cells treated as indicated. Spleens were harvested 7 d later and restimulated with SIINFEKL peptide to assess IFN-γ–producing CD8+ T cells by flow cytometry. N = 5 mice per group. (B) Mice were left untreated or were immunized with 1 × 106 T cells treated as indicated. Spleens were harvested 7 d later and restimulated with SIINFEKL peptide to assess IFN-γ–producing CD8+ T cells by flow cytometry. N = 5 mice per group. (C) Mice were left untreated or were immunized with 1 × 106 murine PBMC surrogate cells treated as indicated. All groups were matured with CpG before injection. Spleens were harvested 7 d later and restimulated with SIINFEKL peptide to assess IFN-γ–producing CD8+ T cells by flow cytometry. n = 5 mice per group. (D) Mouse splenocytes were left untreated (NC), incubated at room temperature with fluorescent 3 kDa dextran (Incub. ctrl), or squeezed in the presence of fluorescently labeled 3 kDa dextran (Squeeze). Viability (left) and dextran delivery (right) of each cell type were immediately determined by flow cytometry. (E) Mouse splenocytes were squeezed without cargo (Squeeze only) or squeezed in the presence of OVA (Squeeze + OVA). At the indicated time points, cells were stained with the 25-D1.16 Ab to assess SIINFEKL presentation on H-2Kb and phenotypic markers to define cell subsets. ***p < 0.001.
When considering cell subsets that are readily available to engineer as a cell therapy, T cells are the most abundant cell type in human PBMCs (up to 1.5 × 106 cells/ml of blood) and efficiently home to compartments and regions containing other T cells. However, T cells are poorly endocytic and rarely upregulate costimulatory molecules, thus making them poor candidates for Ag presentation by traditional methods. To test whether T cells could be used as APCs for priming CD8+ T cell responses, delivery via squeeze was first optimized for murine T cells (Supplemental Fig. 1C). After OVA delivery to T cells, SIINFEKL-MHC complexes appeared on the surface within 2 h and continued to accumulate (Supplemental Fig. 1D). This was similar to the observed kinetics in B cells and DCs. To test the ability of squeezed T cells to elicit a naive CD8+ T cell response, mice were immunized using squeezed T cell APCs with and without coinjected CpG. When OVA-squeezed T cells were injected alone, OVA-specific responses failed to be generated. However, when CpG was coinjected alongside OVA-squeezed T cells, endogenous OVA-specific responses were primed (Fig. 2B), demonstrating the need for an inflammatory context for effective priming.
Given that unconventional cell types could elicit a CD8+ T cell response and that squeezing could mediate delivery to different immune cells, murine splenocytes were evaluated for their Ag-presenting capacity after squeezing. When unfractionated murine splenocytes were squeezed with fluorescent dextran, high delivery was achieved to all cell types evaluated with minimal impact on viability (Fig. 2D). Delivery to total splenocytes was >80%, with no individual cell type having <75% delivery. The ability of these cells to display delivered Ag in MHC-I was then assessed using the OVA model system. Similarly to isolated B and T cells, delivery to the mixture resulted in presentation of SIINFEKL peptide in as little as 2 h (Fig. 2E, histograms in Supplemental Fig. 1E). For evaluation in vivo, a splenocyte composition (termed “PBMC surrogate”) was crafted to more closely align with the composition of human PBMCs, which have substantially fewer B cells than the mouse spleen. This was done by combining one spleen with four B cell–depleted spleens. To evaluate the ability of squeezed splenocytes to prime CD8+ T cell responses in vivo, mice were immunized using the PBMC surrogate incubated with OVA, squeezed alone, or squeezed with OVA. Each group was incubated with CpG for 4 h before immunization. OVA-squeezed cells incubated with CpG were able to prime a CD8+ T cell response (Fig. 2C).
Mechanisms of priming by squeezed nonconventional APCs
Given that mouse splenocytes are not traditionally used as APCs for priming CD8+ T cell responses, experiments were performed to assess whether splenocytes squeezed with Ag were directly presenting to CD8+ T cells or whether squeezed splenocytes were indirectly presenting through professional APCs. To evaluate this, OT-I cells were adoptively transferred into β2m−/− or WT mice. Mice were subsequently immunized with splenocytes that had been squeezed with OVA and proliferation of transferred OT-I cells measured in spleens 3 d later. In β2m−/− mice, MHC-I presentation was restricted to the transferred splenocytes, ruling out the possibility for Ag presentation by host APCs. Proliferation of OT-I CD8+ T cells in WT and β2m−/− recipients was equivalent (Fig. 3A), highlighting that squeezed splenocytes directly present to Ag-specific T cells.
FIGURE 3.

Ag presentation by squeezed immune cells is direct and primes antitumor immunity. (A) A total of 2.5 × 106 CFSE-labeled CD8+ OT-I cells were transferred into either WT or MHC-I−/− (knockout [KO]) mice. A few hours later, 5 × 106 splenocytes squeezed in the presence of OVA and incubated for 4 h with CpG were injected i.v. After 3 d, lymph nodes were harvested, and proliferation of OT-I cells was assessed by CFSE dilution. n = 5 mice per group. (B) Mouse B cells were not in contact with OVA (NC), incubated at room temperature with OVA (Incub. ctrl), squeezed in the presence of OVA (Squeeze), or pulsed with SIINFEKL peptide for 1 h at 37°C. A total of 5 × 106 B cells were coinjected with 3 µg LPS to immunize mice that had also received 2.5 × 106 CFSE-labeled OT-I CD8+ T cells. CFSE dilution by the OT-I cells was assessed 3 d after immunization in lymph nodes. n = 5 mice per group. (C) Mouse B cells were not in contact with gp100 SLP (NC), incubated at room temperature with gp100 SLP (Incub. ctrl), squeezed in the presence of the gp100 SLP (Squeeze), or pulsed with short peptide for 1 h at 37°C (PP). A total of 5 × 106 B cells were coinjected with 3 µg LPS to immunize mice that had also received 2.5 × 106 CFSE-labeled pmel CD8+ T cells. CFSE dilution by the pmel CD8+ T cells was assessed 3 d after immunization. n = 5 mice per group. (D) Mouse splenocytes were squeezed with or without OVA. After squeezing, indicated populations of cells were magnetically separated and cocultured with OT-I TCR-transgenic cells overnight. CD69 MFI on OT-I cells was assessed by flow cytometry. Data are representative of two independent experiments. (E) Mice were left untreated or immunized on day −14 and day −7 with 1 × 106 murine PBMC surrogate cells incubated or squeezed with OVA protein and matured with CpG. On day 0, mice were s.c. implanted with E.G7-OVA and monitored for tumor growth. ***p < 0.001.
To test the applicability of squeezing across Ags, CD8+ T cell responses were measured for the highly immunogenic OVA-derived SIINFEKL and the less immunogenic self-Ag from gp100, KVPRNQDWL. For OVA responses, mice were immunized using B cells incubated with OVA protein or squeezed with OVA or cells incubated with the minimal epitope, SIINFEKL. OVA-specific responses were monitored by CFSE dilution of adoptively transferred OT-I CD8+ T cells. Three days after immunization, OT-I T cells from lymph nodes had divided an average of four times in response to the OVA-squeezed B cells compared with 0.5 times for B cells incubated with or without OVA protein (Fig. 3B). B cells pulsed with SIINFEKL peptide elicited strong CD8+ T cell responses, with OT-I cells from these mice dividing an average of six times. To evaluate responses to the less immunogenic Ag from gp100, KVPRNQDWL, B cells were squeezed with a gp100 SLP, incubated with gp100 SLP, or pulsed with the minimal epitope, KVPRNQDWL. CD8+ T cell responses were measured by adoptive transfer of CFSE-labeled pmel TCR-transgenic cells. B cells squeezed with gp100 SLP drove strong proliferation of CD8+ T cells, whereas B cells incubated with or without gp100 SLP failed to activate gp100-specific CD8+ T cells. In contrast to the SIINFEKL-pulsed B cells, KVPRNQDWL-pulsed B cells failed to drive any proliferation of pmel CD8+ T cells (Fig. 3C). This difference between the two Ags is likely due to the half-life of the bound epitope (t1/2 of ∼2 h for KVPRNQDWL and t1/2 of ∼8 h for SIINFEKL [17, 18]) and the kinetics with which Ag-presenting B cells meet their cognate T cell.
In order to assess the relative Ag-presenting capacity of cell types within the mouse PBMC surrogate, PBMC surrogate cells were squeezed with or without OVA and subsequently fractionated into constituent cell types. Ag-presenting capacity was measured using OT-I TCR-transgenic cells and CD69 MFI as a measure of activation. Although the magnitude of CD69 expression differed when cocultured with different cell types in the PBMC surrogate, T cells, B cells, NK cells, and monocytes were all capable of activating OT-I cells (Fig. 3D).
To measure the functional antitumor activity of primed CD8+ T cells, mice were immunized using the PBMC surrogate either squeezed with OVA or incubated alone. Mice were then challenged by s.c. inoculation with the OVA-expressing tumor E.G7-OVA. In untreated mice, 9 of 10 mice grew tumors, and median survival was reached in 27 d. For mice that were immunized using cells that had been incubated with OVA, 7 of 10 mice grew tumors, and median survival was reached within 31 d. In the group squeezed with OVA, 7 of 10 mice were completely protected, and median survival was significantly extended compared with the untreated mice or the incubation control (Fig. 3E). These data demonstrate that the OVA-specific T cells primed by squeezed cells are capable of antitumor activity.
Microfluidic squeezing enables Ag presentation in vitro by diverse human cell types
To assess whether microfluidic squeezing could enable Ag presentation by diverse human cell types, healthy human donor cells were squeezed or incubated with an SLP containing the HLA-A2–restricted epitope (NLVPMVATV) derived from the human CMV pp65 protein. Responses were monitored by measuring IFN-γ secretion from ex vivo expanded pp65-reactive CD8+ T cells (Fig. 4A). moDCs, B cells, T cells, and unfractionated PBMCs presented low amounts of pp65 peptide on HLA-A2 when incubated with pp65 SLP. However, when cells were squeezed with the same SLP, IFN-γ responses increased by >10-fold for all cell types compared with the incubation control (Fig. 4B–(4E). To demonstrate that multiple Ags could be delivered and presented by the same cell, an additional pp65 SLP that spanned the HLA-B35–restricted minimal epitope, IPSINVHHY, was generated. Both the HLA-A2–restricted SLP and the HLA-B35–restricted SLP were squeezed into HLA-A2+HLA-B35+ B cells, and responses were measured using corresponding ex vivo expanded Ag-specific T cells. Responses could be seen for both HLA-A2–restricted and HLA-B35–restricted SLPs when delivered individually; responses were of similar magnitude when both SLPs were delivered together (Fig. 4F). At the concentrations tested, multiple Ags can be delivered together and presented by the same cell without affecting the response to the other.
FIGURE 4.
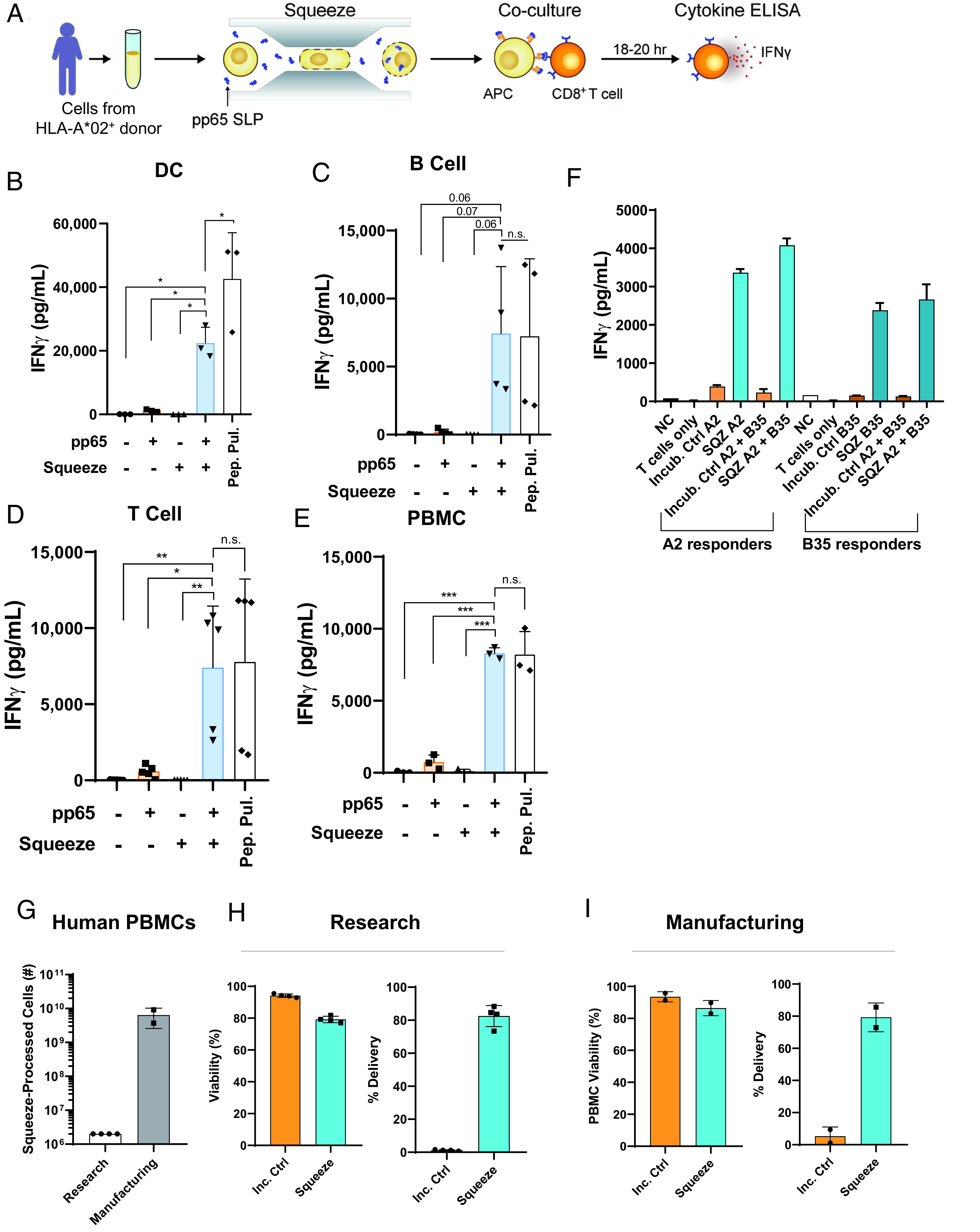
Microfluidic squeezing enables MHC-I presentation of Ags by diverse human immune cells. (A) Immune cells from healthy human donors were squeezed with an SLP containing the HLA-A2–restricted CMV pp65 epitope, NLVPMVATV, and incubated overnight with HLA-A2–restricted pp65 responder CD8+ T cells. Supernatants were collected for IFN-γ analysis by ELISA. (B–E) Human moDCs (n = 3), B cells (n = 4), T cells (n = 3), or PBMCs (n = 3) were left untreated, incubated with pp65 SLP, squeezed without Ag, squeezed with pp65 SLP, or pulsed with the minimal epitope NLVPMVATV (Pep. Pul.). IFN-γ production from HLA-A2–restricted pp65 responder CD8+ T cells after overnight incubation is shown. (F) Human B cells were squeezed or incubated with a pp65 SLP containing the HLA-A2–restricted epitope, a pp65 SLP containing an HLA-B35–restricted epitope, or squeezed with both. IFN-γ production from HLA-A2– or HLA-B35–restricted pp65 responder CD8+ T cells is shown. (G–I) For research scale, cells were isolated from human blood, and 2 × 106 cells were squeezed. For manufacturing scale, cells were isolated from apheresis products, and the isolated product was squeezed. Viability and delivery were determined by flow cytometry immediately after delivery. Each dot is representative of a single donor. Delivery was determined on the basis of cells that were not incubated with dextran. *p < 0.05, **p < 0.01, ***p < 0.001.
For in vitro assessment of Ag-presenting activity, 1 million to 2 million cells were squeezed because this was sufficient for downstream assays. However, billions of cells are isolated from human apheresis products (19) and would be needed for therapeutic applications. The number of constrictions within the microfluidic chips was therefore increased compared with “research” chips. This led to an increase in cells squeezed per second from 2 million to 3 million per second up to 100 million cells per second (data not shown). To test whether squeezing using these “manufacturing”-scale chips generated a similar viability and delivery profile, human PBMCs were compared using both chip types. When squeezing 2 million PBMCs using research chips, 75% viability and 80% delivery of 3 kDa dextran was achieved (Fig. 4G and (4H). Similar viability and delivery could be shown for PBMCs using manufacturing chips when squeezing >6 billion PBMCs (Fig. 4G and (4I). Delivery to different cell types within PBMCs was also similar across research and manufacturing-scale chips (Supplemental Fig. 1F, 1G).
Squeezed cells prime antitumor activity in a mouse model of human papillomavirus 16–positive tumors
To demonstrate that the Ag-presenting capacity of squeezed cells could elicit an antitumor CD8+ T cell response with relevance to a human tumor, T cells, B cells, or a PBMC surrogate were squeezed with an SLP derived from the human papillomavirus 16 (HPV16) E7 protein. This E7 SLP spans the immunodominant H2-Db-restricted epitope, RAHYNIVTF (20), and allows immunization of C57BL/6 mice. The antitumor activity of the ensuing E7-specific response could then be assessed using the TC-1 tumor model, which is transformed with the oncogenic HPV16 proteins E6 and E7, as well as H-RAS (21).
To determine whether squeezed T cells could elicit an E7-specific CD8+ T cell response, mouse T cells were squeezed with E7 SLP and coinjected with CpG. As was seen for OVA, squeezed T cells primed a CD8+ T cell response specific for E7 (Fig. 5A). Next, the antitumor activity of primed E7-specific cells was measured by prophylactic immunization with squeezed T cells 7 d before challenge with TC-1 tumor cells. Immunization with squeezed T cells significantly extended survival compared with untreated mice, with 8 of 10 mice surviving to day 60 when immunized with squeezed T cells, whereas all of the untreated mice reached endpoint tumor size (Fig. 5B). To evaluate the efficacy of squeezed T cell immunization in the context of an established tumor, mice were immunized 10 d after TC-1 inoculation using T cells squeezed with E7 SLP and coinjected with CpG or T cells squeezed with E7 SLP alone. Mice immunized with squeezed T cells alone had minimal antitumor activity, extending median survival from 34 d in untreated mice to 41 d, whereas median survival in mice immunized with squeezed T cells and coinjected with CpG was 59 d (Fig 5C, spider plots in Supplemental Fig. 2A). Thus, squeezed T cells may be able to present Ag to CD8+ T cells, but an adjuvant improves their ability to prime protective antitumor immunity.
FIGURE 5.

Squeezed cells prime antitumor activity in a mouse model of HPV16+ tumors. (A) Mice were left untreated (Untr.) or immunized with T cells squeezed with the E7 SLP containing the H2-Db-restricted epitope RAHYNIVTF and coinjected with CpG. Seven days later, E7-specific responses were measured in the spleen by intracellular cytokine staining (ICS). (B) Mice were prophylactically immunized with 1 × 106 T cells squeezed with E7 SLP and coinjected with CpG on day −7. On day 0, mice were challenged s.c. with TC-1. n = 10 mice per group. (C) Ten days after TC-1 inoculation, mice were immunized with 1 × 106 T cells squeezed with E7 SLP with or without coinjection of CpG. Mean tumor volume + SEM is shown. n = 10 mice per group. (D) Mice were left untreated or immunized with 5 × 106 B cells squeezed with E7 SLP and matured with CpG. Seven days later, E7-specific responses were measured by ICS. (E) Mice were immunized with 5 × 106 B cells squeezed with E7 SLP and matured with CpG overnight or left untreated. Seven days later, mice were challenged s.c. with TC-1. Survival is shown. n = 10 mice per group. (F) Mice were immunized 14 d after TC-1 implantation using 5 × 106 B cells pulsed with minimal epitope (Min. Epi.), RAHYNIVTF, or squeezed with E7 SLP and matured with CpG overnight. An additional group of mice was immunized and boosted on days 14 and 28 s.c. with 150 µg E7 SLP and 50 µg CpG. Mean tumor volume + SEM is shown. n = 10 mice per group. (G) Mice were immunized as indicated 14 d after TC-1 implantation. Seven days later, tumors were harvested for analysis by flow cytometry. Mean ± SD is shown. (H) Mice were left untreated or immunized with 1 × 106 splenocytes squeezed with E7 SLP and matured overnight with CpG. Seven days later, E7-specific responses were measured in the spleen by ICS. (I) Mice were immunized on days −14 and −7 with 1 × 106 M-SQZ-PBMC-HPV or left untreated. On day 0, TC-1 tumors were implanted, and tumor growth was monitored. Sixty days after inoculation, immunized mice were rechallenged with TC-1 on the opposite flank, and a new cohort of untreated mice was introduced. (J) Ten days after TC-1 implantation, mice were immunized with 1 × 106 splenocytes squeezed with E7 SLP that had been matured with CpG for 4 h or incubated without CpG. Tumor volume + SEM is shown. n = 10 mice per group. (K) Mice were primed with M-SQZ-PBMC-HPV at a dose of 1 × 106 or 1 × 105 cells on day 10. An additional group of mice was primed on day 10 and boosted two more times on days 17 and 24 (P/B/B). (L) Mice were immunized with 1 × 106 M-SQZ-PBMC-HPV or s.c. 150 μg E7 SLP with 50 μg CpG 16 d after TC-1 implantation. Twelve days later, tumor-infiltrating lymphocytes (TILs) were analyzed by flow cytometry. Mean percentage ± SD of indicated populations is shown. *p < 0.05, **p < 0.01, ***p < 0.001.
B cells squeezed with E7 SLP generated E7-specific CD8+ T cell responses when incubated with CpG before injection (Fig. 5D). To assess whether CD8+ T cell responses elicited by squeezed B cells were protective, mice were immunized, and TC-1 tumor cells were implanted 7 d later. In this prophylactic setting, a single immunization 7 d before TC-1 inoculation was capable of protecting 90% of mice (Fig. 5E). To compare B cell immunization with other therapeutic approaches, mice were immunized with B cells incubated with E7 minimal epitope (which binds exogenously to MHC-I) and CpG, immunized with B cells squeezed with E7 SLP and incubated with CpG, or immunized by s.c. injection with 150 µg free E7 SLP and CpG (the basis for a clinical approach to immunization) (22, 23). B cells incubated with minimal epitope and CpG failed to have any effect on tumor growth (Fig. 5F, spider plots in Supplemental Fig. 2B), despite effectively eliciting responses from E7-specific cells in vitro (Supplemental Fig. 2C). Injection s.c. with 150 µg free E7 SLP and CpG had antitumor activity but was inferior to squeezed B cells, despite administering only 0.07 µg E7 SLP, a 3000-fold difference in administered Ag (Supplemental Table 1). To assess the tumor infiltrate after immunization, mice were immunized 14 d after tumor inoculation with B cells incubated with minimal epitope and CpG, B cells squeezed with E7 SLP and incubated with CpG, or s.c. injected with free E7 SLP and CpG. Seven days after immunization, tumors were collected and analyzed by flow cytometry for the presence of various immune cells. In mice that had received B cells squeezed with E7 SLP, a significant influx of CD45+ cells was apparent, the majority of which were CD8+ T cells (Supplemental Fig. 2D, 2E). Of these infiltrating CD8+ T cells, >90% were specific for E7, which was 10-fold higher than s.c. immunization with E7 SLP and CpG (Fig. 5G). Interestingly, despite poor tumor control and low recruitment of CD8+ T cells, the group immunized with B cells pulsed with minimal epitope had a high percentage of E7-specific T cells (50%) of those few CD8+ T cells that were recruited to the tumor.
To generate a mouse surrogate for human PBMCs, a fraction of mouse splenocytes were depleted of B cells to more closely approximate the cellular composition of human PBMCs. These cells were then squeezed with E7 SLP containing the H2-Db-restricted epitope, RAHYNIVTF; incubated with CpG; then washed. We term this mouse surrogate “M-SQZ-PBMC-HPV.” When mice were immunized with M-SQZ-PBMC-HPV, E7-specific responses could be detected in the spleen 7 d later (Fig. 5H). To better understand the durability of immunity induced by M-SQZ-PBMC-HPV, mice were primed and boosted 14 and 7 d before TC-1 inoculation. All 15 mice in the M-SQZ-PBMC-HPV group were protected and showed no signs of tumor burden 60 d after challenge. On day 60, these mice were challenged on the opposite flank with TC-1 tumor. Again, all mice in the M-SQZ-PBMC-HPV group exhibited delayed tumor growth, with 11 of 15 mice clearing the tumor entirely (Fig. 5I), highlighting the durability of immune responses elicited by M-SQZ-PBMC-HPV immunization.
To test the antitumor activity of M-SQZ-PBMC-HPV against an established tumor and the importance of CpG incubation, mice were immunized 10 d after TC-1 inoculation with M-SQZ-PBMC-HPV or cells that had been squeezed with E7 SLP, but not incubated with CpG. Immunization with M-SQZ-PBMC-HPV showed significant antitumor activity, extending median survival to 56 d, whereas immunization with squeezed cells without CpG incubation had a median survival of 35 d compared with 28 d in untreated mice (Fig 5J, spider plots in Supplemental Fig. 3A). To assess the impact of priming and boosting on antitumor activity, mice were immunized with a single dose of 1 × 106 M-SQZ-PBMC-HPV, a single dose of 1 × 105 M-SQZ-PBMC-HPV, or immunized and boosted twice (1 wk apart) with 1 × 105 M-SQZ-PBMC-HPV. Although a single dose of 1 × 105 M-SQZ-PBMC-HPV had minimal impact on tumor growth, boosting an additional two times with 1 × 105 M-SQZ-PBMC-HPV showed antitumor activity and extended median survival to 49 d compared with 52 d for a single dose of 1 × 106 M-SQZ-PBMC-HPV (Fig. 5K, spider plots in Supplemental Fig. 3B).
To assess the immune cells contributing to this antitumor activity, tumor-infiltrating lymphocytes were measured 12 d after immunization with M-SQZ-PBMC-HPV or immunization with s.c. E7 SLP and CpG. Similar to what was seen with squeezed B cells, immunization with M-SQZ-PBMC-HPV led to a significant influx of CD45+ cells (Supplemental Fig. 3C), 40% of which were CD8+ T cells (Fig. 5L, cell numbers in Supplemental Fig. 3D). Of these infiltrating CD8+ T cells, >80% of them were E7-specific, whereas only 30% of CD8+ T cells were E7 specific after immunization with E7 SLP and CpG.
Discussion
By overcoming the fundamental barrier to effective MHC-I presentation of Ag, we have shown that cytosolic delivery of Ag by microfluidic squeezing enables MHC-I presentation to CD8+ T cells by diverse immune cell types and populations. Furthermore, this Ag presentation elicits activation and expansion of CD8+ T cells that have antitumor activity. In contrast to other therapeutic vaccination approaches, microfluidic squeezing bypasses the requirement for endocytosis of Ag and subsequent cross-presentation, which allows non-DCs to be used for cell-based immunization. Even in DCs, where cross-presentation is most efficient (24), a 1000-fold greater amount of protein Ag was required to activate a similar fraction of CD8+ T cells when comparing endocytic loading to squeeze-facilitated loading. These data highlight that although DCs, which are considered the most effective cross-presenting cells, may be capable of transporting endosomal Ags into the cytosol for presentation on MHC-I, cross-presentation is an inefficient process that can be compensated for using squeezed Ag.
Most approaches for priming CD8+ T cells seek to deliver Ags to DCs in situ because of their cross-presenting capacity. Ags may be delivered in the form of DNA (25), mRNA (26), or peptides (23, 27) and can be given naked or packaged into nanoparticles, bacteria, or viruses. Given the extracellular nature of administration for these approaches, the Ags delivered through these methods will inefficiently reach target DCs and especially the MHC-I pathway. For many of them, such as mRNA, DNA, and nanoparticles, they often rely on expression of the Ag cargo by non-APCs at the site of injection before uptake by an APC due to apoptosis or secretion of Ag. Ultimately, these Ags are more likely to be presented via the MHC-II pathway and prime CD4+ T cells, as has been observed in preclinical models and in human clinical studies (27–29). Infectious agents can have means of cytosolic entry but often do not target APCs and are prone to neutralization through preexisting immunity or acquired immunity (30). In contrast, the majority of squeezed cells have material in their cytosol, which can then be directly processed and presented to CD8+ T cells on MHC-I. The potency of squeezed cells is highlighted by comparisons with SLP immunization, where as much as 15,000-fold more Ag was needed to be administered to mice, and the immune responses are still weaker than those elicited by immunization with Ag-loaded squeezed cells. Additionally, squeezing Ag into cells provides a store of Ag that can be processed and presented with kinetics distinct from pulsing cells with minimal epitope. This was effectively demonstrated with gp100 SLP and E7 SLP, which generated robust Ag-specific responses when squeezed into cells but failed to elicit a response when cells were pulsed with corresponding minimal epitopes. This occurred despite the fact that, in vitro, pulsing with minimal epitope led to Ag-specific responses equivalent to those against cells squeezed with SLPs. However, in the in vitro case, cells interact with Ag-specific cells immediately, whereas in vivo, hours will pass before cells have homed to lymphoid organs and encountered Ag-specific T cells (31). Thus, induction of responses in vivo by peptide pulsing will be dependent on the half-life of peptides on MHC-I, whereas squeezed cells have an intracellular store of Ag from which the epitope can be generated, resulting in a greater duration of presentation and more robust responses in vivo.
In this work, we did not directly compare cell squeezing with other means of engineering cell-based vaccines, but we believe cell squeezing offers multiple advantages. moDCs are the most common APC used for cell-based immunization strategies, which are generated through a multistep process: monocyte isolation from PBMCs, followed by an extended culture in the presence of IL-4, GM-CSF, and additional maturation agents such as TLR ligands and/or CD40L (32). This multistep process requires days to complete, and the resulting moDCs remain phenotypically and transcriptionally distinct from naturally occurring DC populations (32). Autologous moDCs electroporated with tumor-associated Ag mRNAs and TriMix, a mixture of constitutively activated TLR4, CD40L, and CD70 mRNAs, have proved both tolerable and immunogenic in the clinic (33, 34). However, these approaches remain limited by the time-consuming process of moDC differentiation. Because cell squeezing generates APCs from unfractionated PBMCs without the need for extended ex vivo culture, it reduces the processing time from days to hours. Furthermore, moDC vaccines may be hampered by the limited ability of ex vivo generated moDCs to traffic to lymph nodes (35). By using PBMCs as APCs, we exploit the lymph node homing properties inherent to T and B cell populations.
Although the current studies focused on the impact of cytosolic delivery of Ag, future work could explore the potential of simultaneously engineering other facets of cell function. Incubation with CpG or coinjection of CpG was required for in vivo induction of Ag-specific CD8+ T cell responses by squeezed cells, but more potent or targeted approaches could be explored. For example, material that would enhance expression of costimulatory molecules, such as CD80/CD86, or secretion of cytokines could be codelivered with microfluidic squeezing. Moreover, this approach could be combined with other immune-modulating agents, such as checkpoint inhibitors, to further enhance the Ag-specific impact of the generated CD8+ T cell responses. To enhance the clinical applicability of such an approach for indications such as cancer or infectious disease, future studies could explore the potential of codelivering multiple Ags to simultaneously generate CD8+ T cell responses against more than one disease target. The relative immunogenicity of various disease Ags could be explored and their form (peptide versus protein versus mRNA) and sequence optimized to ensure efficient loading of MHC-I with the desired epitopes.
Finally, although many cell therapies have significant challenges in manufacturing and cost (36), the cell-squeezing approach enables the use of abundant blood-borne cells that can be collected in the 5 billion to 15 billion range from a single leukapheresis (19). In contrast, conventional DCs are rare in the blood (37) and therefore require 4–10 d of manufacturing (38) to differentiate sufficient DCs from monocytes for therapeutic use. Similarly, T cell–based therapies, such as chimeric Ag receptor T cells, TCRs, and tumor-infiltrating lymphocytes, can require weeks of expansion in a good manufacturing practice manufacturing environment and continue to rely on toxic preconditioning regimens (39, 40). In addition to its effective targeting of endogenous CD8+ T cells, the cell-squeezing approach’s rapid processing time, lack of need for expansion/differentiation, and expected independence from immune-ablating preconditioning regimens could have significant practical advantages. By solving a critical biological challenge through a relatively simple, robust process, squeeze-engineered APCs have the potential for high impact across oncologic and infectious disease indications.
Supplementary Material
This work was supported by Foundation for the National Institutes of Health Grant R44GM112259 (to H.B. and S.M.L.).
A. Stockmann, L. Tian, E.H., D.S., A.V., C.Y., E.I.O., O.P., C.S., K.V., K.B., M.K.J., A.M., L.P., J.T.G., E.C., H.A., S.L., S.M., and M.H. performed research; M.G.B., K.A.H., E.I.O., C.S., K.B., I.V.-S., L. Talarico, D.Y., M.M., J.C., T.A., U.H.v.A., H.K., C.T., P.U., H.B., A. Sharei, and S.M.L. designed research; S.M.L. wrote the manuscript; M.G.B., K.A.H., U.H.v.A., K.F.J., R.L., H.K., C.T., P.U., and A. Sharei reviewed and edited the manuscript.
The online version of this article contains supplemental material.
- BMDC
- bone marrow-derived dendritic cell
- DC
- dendritic cell
- β2m−/−
- MHC class I knockout
- MFI
- mean fluorescence intensity
- MHC-I
- MHC class I
- moDC
- monocyte-derived dendritic cell
- SLP
- synthetic long peptide
- WT
- wild type
Disclosures
M.G.B., K.A.H., A Stockmann, L.T., E.H., D.S., A.V., C.Y., E.I.O., O.P., C.S., K.V., K.B., I.V.-S., D.Y., M.M., A.M., J.C., L.P., L. Talarico, M.K.J., E.C., J.T.G., T.A., H.A., H.B., A. Sharei, and S.M.L. are or were employed by SQZ Biotech. H.K., C.T., and P.U. are employed by Roche, which has a partnership with SQZ Biotech. R.L., K.F.J., and U.H.v.A. consult for SQZ Biotech. The other authors have no financial conflicts of interest.
References
- 1. Schachter J., Ribas A., Long G. V., Arance A., Grob J.-J., Mortier L., Daud A., Carlino M. S., McNeil C., Lotem M., et al. 2017. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390: 1853–1862. [DOI] [PubMed] [Google Scholar]
- 2. Brahmer J. R., Tykodi S. S., Chow L. Q. M., Hwu W.-J., Topalian S. L., Hwu P., Drake C. G., Camacho L. H., Kauh J., Odunsi K., et al. 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Larkin J., Hodi F. S., Wolchok J. D.. 2015. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. [Published erratum appears in 2018 N. Engl. J. Med. 379: 2185.] N. Engl. J. Med. 373: 1270–1271. [DOI] [PubMed] [Google Scholar]
- 4. Tumeh P. C., Harview C. L., Yearley J. H., Shintaku I. P., Taylor E. J. M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., et al. 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burg, van der S. H., Arens R., Ossendorp F., van Hall T., Melief C. J. M.. 2016. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 16: 219–233. [DOI] [PubMed] [Google Scholar]
- 6. Klebanoff C. A., Acquavella N., Yu Z., Restifo N. P.. 2011. Therapeutic cancer vaccines: are we there yet? Immunol. Rev. 239: 27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vyas J. M., Van der Veen A. G., Ploegh H. L.. 2008. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 8: 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joffre O. P., Segura E., Savina A., Amigorena S.. 2012. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12: 557–569. [DOI] [PubMed] [Google Scholar]
- 9. Lu Q., Grotzke J. E., Cresswell P.. 2018. A novel probe to assess cytosolic entry of exogenous proteins. Nat. Commun. 9: 3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kong B. Y., Bolton H., Kim J. W., Silveira P. A., Fromm P. D., Clark G. J.. 2019. On the other side: manipulating the immune checkpoint landscape of dendritic cells to enhance cancer immunotherapy. Front. Oncol. 9: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stewart M. P., Sharei A., Ding X., Sahay G., Langer R., Jensen K. F.. 2016. In vitro and ex vivo strategies for intracellular delivery. Nature 538: 183–192. [DOI] [PubMed] [Google Scholar]
- 12. DiTommaso T., Cole J. M., Cassereau L., Buggé J. A., Hanson J. L. S., Bridgen D. T., Stokes B. D., Loughhead S. M., Beutel B. A., Gilbert J. B., et al. 2018. Cell engineering with microfluidic squeezing preserves functionality of primary immune cells in vivo. Proc. Natl. Acad. Sci. USA 115: E10907–E10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharei A., Cho N., Mao S., Jackson E., Poceviciute R., Adamo A., Zoldan J., Langer R., Jensen K. F.. 2013. Cell squeezing as a robust, microfluidic intracellular delivery platform. J. Vis. Exp. (81): e50980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharei A., Zoldan J., Adamo A., Sim W. Y., Cho N., Jackson E., Mao S., Schneider S., Han M.-J., Lytton-Jean A., et al. 2013. A vector-free microfluidic platform for intracellular delivery. Proc. Natl. Acad. Sci. USA 110: 2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Porgador A., Yewdell J. W., Deng Y., Bennink J. R., Germain R. N.. 1997. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 6: 715–726. [DOI] [PubMed] [Google Scholar]
- 16. Szeto G. L., Van Egeren D., Worku H., Sharei A., Alejandro B., Park C., Frew K., Brefo M., Mao S., Heimann M., et al. 2015. Microfluidic squeezing for intracellular antigen loading in polyclonal B-cells as cellular vaccines. Sci. Rep. 5: 10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Stipdonk M. J. B., Badia-Martinez D., Sluijter M., Offringa R., van Hall T., Achour A.. 2009. Design of agonistic altered peptides for the robust induction of CTL directed towards H-2Db in complex with the melanoma-associated epitope gp100. Cancer Res. 69: 7784–7792. [DOI] [PubMed] [Google Scholar]
- 18. Chefalo P. J., Harding C. V.. 2001. Processing of exogenous antigens for presentation by class I MHC molecules involves post-Golgi peptide exchange influenced by peptide-MHC complex stability and acidic pH. J. Immunol. 167: 1274–1282. [DOI] [PubMed] [Google Scholar]
- 19. Garcia A., Keinonen S., Sanchez A. M., Ferrari G., Denny T. N., Moody M. A.. 2014. Leukopak PBMC sample processing for preparing quality control material to support proficiency testing programs. J. Immunol. Methods 409: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feltkamp M. C., Smits H. L., Vierboom M. P., Minnaar R. P., Jongh B. M., Drijfhout J., Schegget J., Melief C. J., Kast M. W.. 1993. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur. J. Immunol. 23: 2242–2249. [DOI] [PubMed] [Google Scholar]
- 21. Lin K. Y., Guarnieri F. G., Staveley-O’Carroll K. F., Levitsky H. I., August J. T., Pardoll D. M., Wu T. C.. 1996. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 56: 21–26. [PubMed] [Google Scholar]
- 22. Zwaveling S., Ferreira Mota S. C., Nouta J., Johnson M., Lipford G. B., Offringa R., van der Burg S. H., Melief C. J. M.. 2002. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J. Immunol. 169: 350–358. [DOI] [PubMed] [Google Scholar]
- 23. Kenter G. G., Welters M. J. P., Valentijn A. R. P. M., Lowik M. J. G., Berends-van der Meer D. M., Vloon A. P. G., Essahsah F., Fathers L. M., Offringa R., Drijfhout J. W., et al. 2009. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 361: 1838–1847. [DOI] [PubMed] [Google Scholar]
- 24. Rodriguez A., Regnault A., Kleijmeer M., Ricciardi-Castagnoli P., Amigorena S.. 1999. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1: 362–368. [DOI] [PubMed] [Google Scholar]
- 25. Best S. R., Peng S., Juang C.-M., Hung C.-F., Hannaman D., Saunders J. R., Wu T.-C., Pai S. I.. 2009. Administration of HPV DNA vaccine via electroporation elicits the strongest CD8+ T cell immune responses compared to intramuscular injection and intradermal gene gun delivery. Vaccine 27: 5450–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sahin U., Türeci Ö.. 2018. Personalized vaccines for cancer immunotherapy. Science 359: 1355–1360. [DOI] [PubMed] [Google Scholar]
- 27. Ott P. A., Hu Z., Keskin D. B., Shukla S. A., Sun J., Bozym D. J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., et al. 2017. An immunogenic personal neoantigen vaccine for patients with melanoma. [Published erratum appears in 2018 Nature 555: 402.] Nature 547: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kreiter S., Vormehr M., van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J. C., et al. 2015. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. [Published erratum appears in 2015 Nature 523: 370.] Nature 520: 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Löwer M., Bukur V., Tadmor A. D., Luxemburger U., Schrörs B., et al. 2017. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547: 222–226. [DOI] [PubMed] [Google Scholar]
- 30. Morris S. J., Sebastian S., Spencer A. J., Gilbert S. C.. 2016. Simian adenoviruses as vaccine vectors. Future Virol. 11: 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park C., Hwang I.-Y., Sinha R. K., Kamenyeva O., Davis M. D., Kehrl J. H.. 2012. Lymph node B lymphocyte trafficking is constrained by anatomy and highly dependent upon chemoattractant desensitization. Blood 119: 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saxena M., Balan S., Roudko V., Bhardwaj N.. 2018. Towards superior dendritic-cell vaccines for cancer therapy. Nat. Biomed. Eng. 2: 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Nuffel A. M., Benteyn D., Wilgenhof S., Corthals J., Heirman C., Neyns B., Thielemans K., Bonehill A.. 2012. Intravenous and intradermal TriMix-dendritic cell therapy results in a broad T-cell response and durable tumor response in a chemorefractory stage IV-M1c melanoma patient. Cancer Immunol. Immunother. 61: 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilgenhof S., Corthals J., Heirman C., van Baren N., Lucas S., Kvistborg P., Thielemans K., Neyns B.. 2016. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 34: 1330–1338. [DOI] [PubMed] [Google Scholar]
- 35. Perez C. R., De Palma M.. 2019. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 10: 5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elverum K., Whitman M.. 2020. Delivering cellular and gene therapies to patients: solutions for realizing the potential of the next generation of medicine. Gene Ther. 27: 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Breton G., Zheng S., Valieris R., da Silva I. T., Satija R., Nussenzweig M. C.. 2016. Human dendritic cells (DCs) are derived from distinct circulating precursors that are precommitted to become CD1c+ or CD141+ DCs. J. Exp. Med. 213: 2861–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sabado R. L., Balan S., Bhardwaj N.. 2017. Dendritic cell-based immunotherapy. Cell Res. 27: 74–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X., Rivière I.. 2015. Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther. 22: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Levine B. L., Miskin J., Wonnacott K., Keir C.. 2017. Global manufacturing of CAR T cell therapy. Mol. Ther. Methods Clin. Dev. 4: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.