

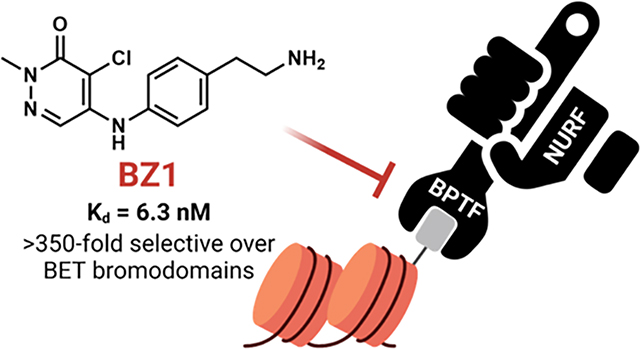
Abstract
The Nucleosome Remodeling Factor (NURF) alters chromatin accessibility through interactions with its largest subunit the Bromodomain PHD Finger Transcription Factor, BPTF. BPTF is overexpressed in several cancers and is an emerging anticancer target. Targeting the BPTF bromodomain presents a potential strategy for inhibition and evaluation of its functional significance; however, inhibitor development for BPTF has lagged behind other bromodomains. Here we describe the development of pyridazinone-based BPTF inhibitors. The lead compound, BZ1 possesses high potency (Kd = 6.3 nM) and >350-fold selectivity over BET bromodomains. We identify an acidic triad in the binding pocket to guide future designs. We show that our inhibitors sensitize 4T1 breast cancer cells to doxorubicin, but not BPTF knockdown cells, suggesting specificity to BPTF. Given the high potency and good physicochemical properties of these inhibitors, we anticipate that they will be useful starting points for chemical tool development to explore the biological roles of BPTF.
Keywords: epigenetics, NURF, bromodomains, BPTF
Graphical Abstract
INTRODUCTION
Epigenetic processes involve heritable changes in gene expression without altering the underlying DNA sequence.1 Gene accessibility leading to these changes occurs through mechanisms such as DNA methylation, covalent modifications of histones, chromatin remodeling, and exchange of histones.2 In the case of chromatin remodeling, ATP-dependent processes are catalyzed by multidomain protein complexes which include SWI/SNF, ISWI, CHD and INO80.3 Of these, SWI/SNF has been extensively studied and is implicated in ~20% of human cancers.4 The mammalian SWI/SNF complexes, BAF and PBAF, have emerged as attractive epigenetic therapeutic targets, for which chemical inhibitors and catalytically degrading molecules of complex members BRD7 and BRD9 have been developed.5,6 In contrast, the ISWI family is less well-studied for its potential role as a therapeutic target. Nucleosome Remodeling Factor (NURF) is one member of the ISWI family, consisting of an ATPase domain SNF2L, a WD-repeat protein RbAP46/48, and a chromatin-binding protein, BPTF (Figure 1A).7 Chemical probe development for these complex members remains at an early stage.8
Figure 1.

A) BPTF interacts with chromatin through the bromodomain (BRD) and PHD domain, directing the chromatin remodelling complex NURF to genes, leading to downstream phenotypic effects such as Myc regulation, MAPK signaling and resistance to chemotherapeutics. B) Part of the bromodomain phylogenetic tree, showing class I and class II (BET) bromodomains (adapted with permission from Pomerantz et al.11). C) Reported BPTF bromodomain inhibitors with in vitro affinities.
BPTF (Bromodomain PHD Finger Transcription Factor) is the largest subunit of NURF and is considered essential for its function.9 BPTF contains a bromodomain, two PHD fingers, a DNA-association domain, three nuclear receptor binding motifs, and a glutamine-rich domain.10 Both the bromodomain and C-terminal PHD domain are structurally well-characterized and are responsible for binding to acetylated and methylated histones respectively.12 While BPTF is known to be essential in normal cellular processes such as embryonic development,13 T-cell homeostasis14 and differentiation of mammary epithelial cells,15 the oncogenic effects of BPTF have been recently well-documented. BPTF is overexpressed in melanoma, where it impacts MAPK signaling, and is regulated by the melanocyte-inducing transcription factor, MITF.16,17 High BPTF levels correlate with c-Myc expression in various cancers, regulation of Myc signaling, and Myc protein-protein intractions.18,19 Additional oncogenic roles for BPTF have been found in breast cancer,15 non-small-cell lung cancer,20 colorectal cancer,21 and high-grade gliomas.22,23
BPTF also confers chemoresistance to cancer cells; overexpression of BPTF promotes resistance to BRAF inhibitors in melanoma16 and knockdown of BPTF sensitizes hepatocellular carcinoma cells to chemotherapeutic drugs.24 The implication of BPTF in cancer and its key role as a NURF subunit makes it a potential new therapeutic target for small-molecule inhibitor development. One attractive targeting element is the bromodomain, which is computationally predicted to be highly druggable.25 However, the role of the bromodomain in many of these disease states needs to be established.
While inhibitor development for class II bromodomain and extraterminal domain (BET) family proteins (Figure 1B) have resulted in translation of numerous inhibitors into the clinic,26 non-BET class I bromodomains such as BPTF have received less attention.27 To address this need, we reported AU1 as the first small-molecule inhibitor of the BPTF bromodomain (Kd = 2.8 μM, Figure 1C).28 Importantly this molecule was selective over the BET protein BRD4, given the strong phenotype of BRD4 in regulating cell cycle, proliferation, and inflammatory pathways.29 AU1 has since been used in mouse mammary epithelial cells showing decreased proliferation, cell cycle arrest, and reduced c-Myc-DNA occupancy;15 however in other cell lines, off-target activity was identified.30 Most recently, AU1 showed enhancement of anti-cancer activity when used in combination with the chemotherapeutic drug doxorubicin in vitro and in vivo in 4T1 breast cancer models.31 Mechanistic studies showed these processes to be autophagy-dependent and AU1 effects on topo2-isomerase-DNA crosslinks and DNA damage recapitulated the effects from BPTF knockdown experiments. However, the off-target kinase activity of AU1, its poor physicochemical properties, and low ligand efficiency, posed significant challenges to inhibitor development and highlighted the need for new and more potent BPTF inhibitors. Recently, several inhibitors were disclosed online by the structural genomics consortium; TP-238, a dual CECR2/BPTF chemical probe (12-fold higher affinity for CECR2 over BPTF)32 and NVS-BPTF-1, a potent BPTF inhibitor in vitro but with poor solubility and ADME properties (Figure 1C).33,34 Encouragingly, TP-238 administration to cells was shown to reduce BPTF chromatin binding, supporting the importance of bromodomain inhibition.32 However, detailed reports and their characterization have yet to be described in the primary literature. Given the emerging role of BPTF in cancer, there is a significant need for improved potent and selective inhibitors.
Here we describe the development of BPTF bromodomain inhibitor BZ1 with nanomolar affinity (Kd = 6.3 nM) and > 350-fold selectivity over BET bromodomains. This inhibitor and analogues are obtained via a facile synthesis route with no more than 2–3 synthetic steps. The high affinity, aqueous solubility, and physicochemical properties of these inhibitors enabled us to obtain several cocrystal structures with BPTF for rationalizing our structure-activity-relationship data and to identify an acidic triad as a targetable feature of the binding site for future inhibitor development. Finally, we use the 4T1 breast cancer cell chemotherapeutic synergy model previously validated for BPTF on-target engagement,31 to show that our scaffolds are both generally well-tolerated by cells, and enhance doxorubicin cytotoxic effects to wild type breast cancer cells but not identical cells with BPTF knockdown, demonstrating specificity in their biological activity. The improved biochemical properties and cellular activity of BZ1 makes it a lead scaffold to further efforts in selective BPTF inhibitor discovery for developing new BPTF-specific chemical tools. Future work will focus on engineering selectivity for BPTF over other class I bromodomains and the BRD7/9 off-targets of this scaffold.
RESULTS AND DISCUSSION
As a first step towards BPTF inhibitor development, we recently cross-validated several biophysical assays for BPTF ligand screening including a competitive inhibition AlphaScreen assay using an acetylated histone peptide and SPR binding experiments. We used several compounds reported in the literature or online, including TP-238 and GSK4027, a PCAF/GCN5L2 inhibitor35 with off-target affinity for BPTF (Kd = 1.7 μM, Figure 1C). Using these inhibitors and new fragment compounds from our lab, we reported a number of small-molecule cocrystal structures with the BPTF bromodomain.36 From these studies, we chose GSK4027 for further analysis to establish design rules for inhibitor development.
From our cocrystal structure of GSK4027 with BPTF bromodomain (Figure 2), the carbonyl group acted as the acetyl lysine histone mimic, forming a hydrogen bond with N3007, and the bromine atom pointed into the binding pocket. The pyridazinone core formed π-stacking interactions with the gatekeeper residue F3013 (not shown here, see Figure 3). The WPF shelf was engaged by hydrogen bonding to the P2951 backbone and W2950 by an edge-to-face interaction with the pendant phenyl ring, as previously shown with PCAF/GCN5L2.35 In addition, we identified the acidic patch residues D2957 and D2960 as key targets for inhibitor design (Figure 2 inset). We also showed that TP-238 could engage these side chains supporting this approach.36 Among class I bromodomains, BPTF is the only member with two acidic groups at this site so we hypothesized that interactions with these side chains could improve both affinity and potentially selectivity for BPTF. We anticipated that these interactions would provide multiple sites to fine-tune the potency and selectivity of our inhibitors.
Figure 2.

Cocrystal structure of GSK4027 (cyan) with BPTF bromodomain (gray, PDB: 7K6R). Four conserved structured waters are shown as red spheres. Hydrogen bonds are shown as yellow dashed lines and aromatic interaction as orange dashed line. The distances (Å) between key residues are indicated. Inset: Residues in other class I bromodomains (PCAF, GCN5 and CECR2) corresponding to D2957 and D2960 in BPTF.
Figure 3.

BPTF bromodomain (gray) cocrystal structures with A,B) 10 (magenta, PDB: 7RWP, 1.73 Å resolution), C) 12 (yellow, PDB: 7RWQ, 1.90 Å resolution), D) 13 (orange, PDB: 7RWO, 1.58 Å resolution) and E) 19 (blue, PDB ID: 7M2E, 1.75 Å resolution). Hydrogen bonds are shown as yellow dashed lines. The distances (Å) between key residues are indicated. Three of the conserved structured waters are excluded for clarity.
We first tested 4,5-dichloropyridazinone, as a parent fragment of GSK4027 representing the acetylated lysine pharmacophore for the BPTF bromodomain. We used protein-observed fluorine (PrOF) NMR as a sensitive biophysical assay to quantify weak interactions with BPTF, using a fluorine-labeled tryptophan at W2950.37 In this experiment, the protein resonance showed a significant dose-dependent shift and broadening below 100 μM of the compound (Figure S1). A dose-dependent chemical shift perturbation at low concentrations was consistent with significant affinity of this pharmacophore for BPTF. Encouraged by the apparent potency of the starting fragment and the relatively facile synthesis towards elaborated compounds (Scheme 1), we generated a library of pyridazinone-containing aliphatic amines similar to GSK4027 (Table 1) and tested them with BPTF using PrOF NMR and a competitive inhibition AlphaScreen assay (See SI for AlphaScreen binding isotherms).36
Scheme 1.

Nucleophilic aromatic substitution with 4,5-dichloropyridazinone and primary amines.
Table 1:
SAR with aliphatic pyridazinones and BPTF
| |||
|---|---|---|---|
| R | BPTF AlphaScreen IC50 (μM) |
L.E. | |
|
| |||
|
GSK
4027 |
|
1.5 ± 0.236 | 0.35 |
|
| |||
| 1 |
|
10 ± 236 | 0.45 |
|
| |||
| 2 |
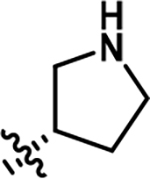
|
3136 | 0.41 |
|
| |||
| 3 |
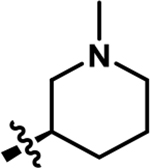
|
1936 | 0.38 |
|
| |||
| 4 |
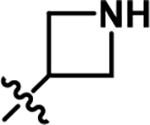
|
8.7 | 0.49 |
|
| |||
| 5 |
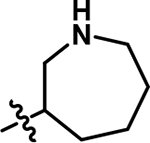
|
7.7 | 0.41 |
AlphaScreen values were an average of two technical replicates with N = 1, except for GSK4027 and 1 which were averages of six and three experimental replicates, respectively. Binding isotherms shown in Figure S2.
Aliphatic pyridazinone series.
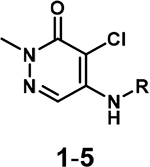
The initial synthesis started with a nucleophilic aromatic substitution reaction with various aliphatic amines, generating the desired compounds 1–5 as the major isomers (Table 1). Our initial characterization of the affinity of 1-3 was recently reported.36 The IC50 values ranged from 7.7–31 μM. Although we were unable to significantly increase the affinity of these analogues, we were encouraged by their ligand efficiency (0.38–0.49), which were a significant improvement over AU1 (0.22) and in agreement with the benchmarked value of ~0.3 for suitable drug candidates.38 To gain structural insight, we acquired cocrystal structures of compounds 1-4 with BPTF (Figure S6 and S7). These structures supported the importance of the exocyclic amine in maintaining the hydrogen bonding interaction with the backbone carbonyl of P2951, similar to GSK4027. However, the ring size and position of the endocyclic amine group did not significantly impact the affinity of the compounds and accessibility to the acidic D2957 and D2960 side chains. Interestingly, these crystal structure revealed a water-mediated hydrogen bond with E2954, an interaction not previously explored in BPTF inhibitor design.
Aromatic amine substituted pyridazinones.
Based on our hypothesis that the N-H interaction with P2951 was critical for the affinity of our pyridazinone inhibitors, we proposed that the more acidic aniline N-H could be a stronger H-bond donor compared to aliphatic amines.39 Therefore, in our second series of inhibitors, we investigated aromatic amine-substituted pyridazinones (Table 2). The aniline-substituted compound 6 (previously reported as a PCAF and BRD9 inhibitor with affinities of 10 μM and 2.5 μM respectivley35) demonstrated a 10-fold improvement in affinity and higher ligand efficiency (L.E.) compared to our previous aliphatic amine analogues.
Table 2:
Aniline-substituted pyridazinones and substituent effects for binding to BPTF
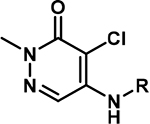
| |||
|---|---|---|---|
| R | BPTF AlphaScreen IC50 (μM) |
L.E. | |
|
| |||
| 6 |
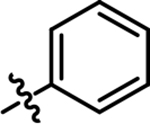
|
0.95 | 0.51 |
|
| |||
| 7 |
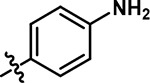
|
3.2 | 0.44 |
|
| |||
| 8 |
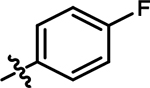
|
0.70 | 0.49 |
|
| |||
| 9 |
|
11 | 0.40 |
AlphaScreen values were an average of two technical replicates, N = 1. Binding isotherms shown in Figure S2.
We compared the effects of electron-donating and -withdrawing substituents on the aromatic ring and found that the para-fluoro group (8) led to an improved affinity compared to a para-amino group (7). This observation was consistent with the importance of the hydrogen-bonding interaction with the P2951 backbone, which would be assisted by an electron-withdrawing group on the ring and the more acidic character of the conjugate acid of the anilinic NH. In agreement with this data, compound 9, containing a benzylic amine group attached to the pyridazinone core, was also a weaker binder of BPTF compared to 6 and 7. Interestingly, an analogue of 9 was also recently identified as the starting fragment for BRD9 inhibitors, with pIC50 = 5.7 and 6-fold selectivity over PCAF.40
Aromatic pyridazinones: Effect of basic group substitution.
In a further round of SAR, based on our acidic patch hypothesis, we investigated different amine substitutions on the aromatic ring for engaging D2957 and D2960 (Table 3). Encouragingly, extending the NH2 group by just one methylene (from compound 7 to 10) resulted in a ~10-fold improvement in potency, with IC50 values of 0.29 μM and 0.31 μM, and ligand efficiencies of 0.50 and 0.44 respectively for compounds 10 and 11. We attributed this gain in affinity to a potential electrostatic interaction between the amine group and the aspartate side chains of BPTF. Such an interaction was also consistent with the loss in affinity observed when the amine was removed (14) or the positive charge neutralized via acetylation (17).
Table 3:
SAR with aromatic pyridazinones containing different basic groups substitutions for binding to BPTF
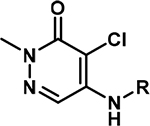
| |||
|---|---|---|---|
| R | BPTF AlphaScreen IC50 (μM) |
L.E. | |
|
| |||
| 10 |
|
0.29 ± 0.08 | 0.50 |
|
| |||
| 11 |
|
0.31 | 0.44 |
|
| |||
| 12 |
|
0.25 | 0.45 |
|
| |||
| 13 |
|
0.37 | 0.44 |
|
| |||
| 14 |
|
3.9 | 0.37 |
|
| |||
| 15 |
|
0.80 | 0.46 |
|
| |||
| 16 |
|
0.22 | 0.50 |
|
| |||
| 17 |
|
0.97 | 0.39 |
AlphaScreen values were an average of two technical replicates with N = 1, except for 10 where it was an average of three experimental replicates. Binding isotherms shown in Figure S3.
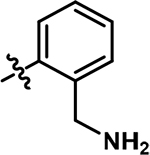
We further looked at the effect of the position of the amine group, expecting to see significant differences based on which orientation of the group was more favorable for engaging D2957 and D2960. Surprisingly, the regioisomers 12 and 13 displayed similar affinities, which were comparable to 10. In this series, 15, where the amine was no longer restricted in a ring, was a weaker binder compared to 13. While compound 16 showed a high affinity, it was obtained in the lowest synthetic yield and was also previously reported to have affinity for an off-target bromodomain, BRD9.35
We turned to X-ray crystallography to obtain structural information that could account for the similar affinities of our amine analogues 10-13 (Figures 3, S8 and S9). Similar to our aliphatic amines, all cocrystal structures displayed the canonical hydrogen bonding with N3007 and water-mediated hydrogen bonding with Y2964 and a key hydrogen bond with P2951. The phenyl groups were 3.8–5.0 Å from W2950, which could contribute to the higher potency of our aromatic series over the aliphatics, forming a CH-π interaction. The amine group on compound 10 was 2.9 Å away from D2960, which could explain the improved affinity over compounds 7, 14, and 17. While compound 12 retained these interactions, to our surprise, the different orientation of the basic group in compound 13 led to an interaction with E2954. Therefore, we attribute the improved affinities of our aromatic pyridazinone series compared to the aliphatics, to an additional aromatic interaction with the WPF shelf, strengthened H-bonding interactions with P2951, and differential engagement of side chains in a potential acidic triad (D2960, D2957 and E2954), depending upon the relative orientations of the amine moieties.
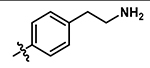
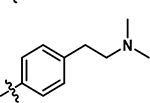
Based on this structural analysis, we proposed that extending the amine group could lead to a further improvement in potency as D2957 was 5.1–6.8 Å away from the basic group on our compounds. In support of this, compound 18, with just an additional methylene, showed a 4-fold improvement in affinity over 10, with an AlphaScreen IC50 of 67 nM and ligand efficiency 0.51 (Table 4). The N,N-dimethyl analogue, 19, was slightly less potent, but may improve cellular permeability due to fewer hydrogen bond donors.41 To test the importance of the H-bond with P2951, we also isolated the 4-position regioisomer 20. Supporting this interaction, we measured a significantly reduced potency (IC50 = 10 μM). For future cellular studies, 20 can serve as an important negative control compound (140-fold weaker affinity than 18).
Table 4:
Aromatic pyridazinones with extended basic group

|
||||
|---|---|---|---|---|
| R | R’ | BPTF Alpha Screen IC50 (μM) |
L.E. | |
|
| ||||
|
18
(BZ1) |
|
Cl | 0.067 ± 0.01 | 0.51 |
|
| ||||
| 19 |
|
Cl | 0.17 | 0.44 |
|
| ||||
| 20 |
|
Cl | 10 | 0.32 |
|
| ||||
| 21 |
|
Br | 0.036 ± 0.008 | 0.53 |
|
| ||||
| 22 |
|
Cl | 0.056 ± 0.01 | 0.45 |
AlphaScreen values were an average of two technical replicates, with N = 1 except for 18 (BZ1) which was an average of seven experimental replicates and 21–22 which were averages of three experimental replicates. Binding isotherms shown in Figure S4.
To validate our designs, we obtained a cocrystal structure of BPTF with 19, a close analogue of our lead inhibitor 18 (Figure 3E). In this case, the amino group was now within 5 Å of D2957 and D2960, supporting the enhanced affinity for engaging either acidic group via electrostatic interactions. An overlay of the apo structure with 19 indicated very little movement of acidic residues (Figure S10).
Selectivity Profile of compound 18 (BZ1) with bromodomain families.
We next conducted a preliminary assessment of the selectivity of compound 18, referred to as BZ1 here onwards, using our PrOF NMR assay (Figure 4B–D). The tryptophan residue in the WPF shelf of three class I bromodomains, BPTF, PCAF, CECR2 and one class II bromodomain, BRD4(1) were fluorine-labelled (Figure 4A) and the chemical shift perturbation on titrating in BZ1 was observed. For both BPTF and PCAF, a slow exchange regime stoichiometric titration was observed, with the bound and unbound resonances resolved at sub-stoichiometric concentrations of BZ1. CECR2 showed intermediate chemical exchange, indicating that BZ1 was a weaker binder for CECR2 compared to BPTF and PCAF in this assay. Importantly, BRD4(1) demonstrated fast-intermediate exchange, showing qualitatively that BZ1 was the weakest inhibitor for BET bromodomains under study here. We further used our AlphaScreen assay to quantify the affinity for BRD4(1) as a representative member of the BET family (Figure 4E). In this experiment, BZ1 was found to be 400-fold selective for BPTF over BRD4(1), consistent with the PrOF NMR results. Selectivity over the BET family is important for non-BET chemical probes because BET inhibition shows a strong cellular phenotype which can mask any BPTF-dependent effects. In both BPTF and PCAF, an acidic residue is present in the acidic dyad, whereas in CECR2 and BRD4(1) the 3D equivalent is a tyrosine or leucine, respectively, and may account for some of the apparent selectivity differences (Figure 2 and 5C). Moreover, our PrOF NMR data also demonstrated that BZ1 with a clogP = 1.6 can be titrated at high micromolar concentrations at 1% DMSO and shows dose dependence, indicating good solubility. The solubilities of BZ1, 19, and 20 were further confirmed up to 100 μM at 0.1% DMSO using UV-Vis spectroscopy (Figure S11).
Figure 4.

A) The tryptophan residues in the binding sites of BPTF (PDB ID: 7JT4), PCAF, CECR2 and BRD4(1) were fluorine-labeled to act as reporters for PrOF NMR. BZ1 was titrated with 50 μM of 5-fluorotryptophan (5FW)-labeled proteins. Slow chemical exchange regimes were observed with B) 5FW-BPTF and C) 5FW-PCAF, indicating the high affinity of BZ1 for these proteins. Intermediate exchange with D) 5FW-CECR2 and E) 5FW-BRD4(1) indicated BZ1 was a weaker binder. F) Affinity values of BZ1 for BPTF (blue) and BRD4(1) (red) were quantified using AlphaScreen competition experiments
Figure 5.

A) Single-point measurement of 140 nM BZ1 against a representative panel of 32 bromodomains via BROMOscan. Percent inhibition ranges are shown by: circles 95–100%, triangles 90–95% and squares 65–90%. (Adapted with permission from Pomerantz et al.)11. B) Kd values for BZ1 with BPTF and off-target class I (PCAF, GCN5L2, CECR2,) and class IV (BRD7, BRD9) bromodomains and BRD4(1) as the highest off-target from the BET family and Kd values for compound 21, 22 and 24 with BPTF, PCAF and BRD9. Values are averages of two technical replicates, N = 1, except BZ1 with BPTF and BRD9, which are averages of two experimental replicates. C) Sequence alignment of selected bromodomains highlighting WPF shelf motif (cyan), 3D equivalents of acidic triad (yellow), Kac mimetic H-bonding groups (magenta), and the gatekeeper residue (green).
Based on the preliminary assessment of BPTF selectivity and affinity of BZ1 by PrOF NMR and AlphaScreen competition assays, we next characterized our ligand using a commercial BROMOscan assay. Using this assay, the Kd of BZ1 for BPTF was determined to be 6.3 nM (Figure 5B). Given the low concentration of ligand and protein used, AlphaScreen can be used to estimate Ki values as was previously the case for characterizing BRD4-ligand interactions,42 however our assay for BPTF may slightly underestimate the affinity. Given the high affinity of BZ1, we measured its selectivity against a panel of 32 representative bromodomains with a one-point measurement in the same assay format (Table S1). We screened at 140 nM, approximately twenty times above the Kd of BZ1 for BPTF (Figure 5A). Encouragingly, consistent with our PrOF NMR and AlphaScreen results, the BET family proteins, were weakly inhibited with the highest estimated affinity for BRD4(1) (71% inhibition). For Class I, bromodomains, BPTF and PCAF were significantly inhibited as expected (100% inhibition), with lower levels of inhibition for CECR2 and GCN5L2. Although, BRD7 and BRD9 lack acidic residues corresponding to the acidic triad (Figure 5C), they were also strongly inhibited (99%). Recently reported pyridazinone-based inhibitors also bind to these proteins40 and BRD9 was reported as the closest off-target for TP-238.32 These studies supported good on target-BPTF inhibition, and identified several off-targets bromodomains for a more quantitative selectivity analysis.
Given that these measurements were only estimates of affinity, we carried out a full titration for five additional bromodomains (Figures 5B and S12). In this case, we obtained a 350-fold selectivity over BRD4(1). However, our selectivity over class I bromodomains, PCAF, CECR2, and GCN5L2 was reduced. Surprisingly, the affinity for BRD7 and 9 was stronger than expected (Kd = 0.76 and 0.47 nM respectively) and represents an important off-targets for future inhibitor designs. During the course of preparing this manuscript, a new BPTF inhibitor was reported with the highest affinity of 428 nM.43 However selectivity studies against BRD9 and class I bromodomains were not conducted in this study to allow comparisons. Currently the ability to potently inhibit both the SWI/SNF and NURF nucleosome remodeling complexes have yet to be explored and may provide a novel mechanism for therapeutic applications.
As an initial evaluation of two additional analogs to improve activity, we synthesized and tested 21 and 22. 21 is an analogue of BZ1 which replaces the chloro group with a bromine atom, analogous to GSK4027. 22 is an analog of 19 which extends the amino group by one additional methylene to further engage D2957. In the case of 21, there was a small but significant improvement in affinity by AlphaScreen relative to BZ1 and a 3-fold increase in potency of 22 relative to 19 (Table 4). We further measured their affinity and selectivity by BROMOscan (Figures S13, S14). While the Kd of 22 was weaker (Kd = 70 nM), both BRD9 affinity and PCAF affinity were weakened more significantly and now result in a modest selectivity over BRD9 and further selectivity over PCAF. These results support our design strategy for targeting the two acidic residues of BPTF to enhance the selectivity of our inhibitor series.
Exploring the SAR at the pyridazinone N-CH3.
As a second attempt to improve selectivity and/or affinity, in our final SAR series, we investigated the N-CH3 position on the pyridazinone core. Using the cocrystal structure reported for NVS-BPTF-1,33 we hypothesized that the cyclopropyl-substituted pyrazole ring may contribute to the affinity and selectivity for BPTF. In our scaffold, the analogous position would be the R’ substituent in Table 5. We observed that small alkyl groups and a propargyl group were tolerated at that position, albeit with no improvement in affinity. However, all the analogues retained their selectivity over BRD4(1).
Table 5:
Aromatic pyridazinones with 2-position N-alkyl substituents
| |||
|---|---|---|---|
| R’’ | BPTF AlphaScreen IC50 (μM) |
BRD4(1) AlphaScreen IC50 (μM) |
|
|
| |||
| 10 | CH3 | 0.29 | 42 |
|
| |||
| 23 |

|
0.55 | 35 |
|
| |||
| 24 |
|
0.38 | 71 |
|
| |||
| 25 |
|
0.79 | NB |
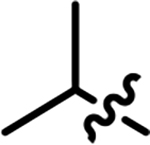
We further characterized the affinity of 24 with BPTF and PCAF using BROMOscan, obtaining Kd values of 200 nM and 230 nM respectively (Figure S15). Although we were not able to improve selectivity over the closely-related bromodomain of PCAF, the alkyne group can serve as a useful click-chemistry handle for further modifications of the pyridazinone scaffold.
Enhancing toxicity of chemotherapeutics in a model breast cancer cell line.
With potent inhibitors in hand, we sought to conduct an initial assessment of cellular activity prior to further selectivity optimization. BPTF has been implicated in resistance to chemotherapeutics for treating hepatocellular carcinoma,24 and BRAF inhibitors for melanoma therapy.16 We recently identified BPTF suppression of Topoisomerase 2 poisons, including doxorubicin and etoposide, whose cytotoxic activity was enhanced with BPTF knockdown or bromodomain inhibition with AU1.31 While knockdown of BPTF in 4T1 mouse breast cancer cells44 does not exhibit toxicity on its own,45 AU1 treatment exhibited toxicity at higher concentrations consistent with an off-target effect. We first tested several of our pyridazinones and found them to be well-tolerated by the 4T1 cells up to mid-micromolar concentrations, with the exception of BZ1 which started to exhibit some toxicity at 8 μM (% survival = 56 and 89% in two separate experiments) (Figure 6A and S12). BZ1, 19 and a regioisomer control, 20,46 were further used for combination treatment with doxorubicin at concentrations lacking significant toxicity with inhibitor alone (Figure 6A and B). Encouragingly, BZ1 and 19 sensitized 4T1 cells to doxorubicin, exhibiting sensitization similar to BPTF shRNA knockdown levels, while 20 did not. A separate dose dependence experiment showed BZ1 maintained strong biological effects down to 2.5 μM while 19 was 2–4-fold less effective (Figure S17). This result is consistent with the weaker affinity of 19 towards the BPTF bromodomain. It remains unclear if the lack of an effect at concentrations closer to the inhibitors’ biochemical potencies are due to a lack of cellular uptake, or if alternate mechanism are also important such as engagement of additional BPTF domains with chromatin. As a control for off-target effects no further toxicity was observed when BPTF knockdown cells were treated with BPTF pyridazinone inhibitors and doxorubicin at these concentrations despite the high BRD9 affinity (Figures 6C and D). Additional toxicity was observed for AU1 at the highest concentrations tested. Together, these results are consistent with an on-target BPTF bromodomain inhibition effect of a new inhibitor class.
Figure 6.

AU1, 19 and BZ1 synergize with chemotherapy drug doxorubicin in 4T1 breast cancer cells. Compound 20 was used as a negative control. 4T1 cells were tested A) without doxorubicin B) in the presence of 50 nM doxorubicin. As a control for off-target effects, shRNA-mediated BPTF knockdown (KD) cells were treated with BPTF inhibitors with and without doxorubicin in C) and D) respectively. Fraction survival values are averages of three experimental replicates, except DMSO controls which are averages of nine experimental replicates.
Pyridazinones effect on BPTF target genes.
As a final evaluation of BPTF-dependent cellular effects, we tested the effects of 19 on several potential BPTF target genes. 19 was chosen due to its low level of toxicity in 4T1 cells, and its regioisomer control 20. We also tested AU1 as a second control for BPTF inhibition. Given the lack of BPTF inhibitors, few-BPTF dependent genes have been validated for bromodomain inhibition and prior work has shown BPTF bromodomain inhibitors do not replicate all genes affected by BPTF depletion.34
We previously showed BPTF inhibition was associated with alteration to lineage commitment and stem cell maintenance. Loss of BPTF expression in a mixed population of Krt5-expressing mammary stem cells induced differentiation, a process that was accompanied by changes to chromatin accessibility and altered gene expression activation.15 We thus decided to investigate the effects of our BPTF inhibitors in mammary luminal cells. We used the murine Eph4 cell line, an immortalized, normal-like system previously shown to activate molecular process of luminal cell differentiation,47,48 and were responsive to AU1 treatment.15 Here, Eph4 cells were treated with AU1 (5 μM), 19 (5 μM), and its regioisomer control 20 (5 μM), followed by either apoptosis analysis or RNA extraction (Figure S18). The mRNA levels of the three genes were analyzed via RT-qPCR based on our prior analysis of BPTF knockout studies in mammary epithelial luminal cells which included two highly upregulated genes, Stratifin (Sfn), and Small proline rich protein 1A (Sprr1a). We also analyzed Myc levels given prior reports on BPTF regulation,22 although our prior knockout data did not show a statistically significant effect (Table S2).
We found that compound 19 treatment led to minimal toxicity against Eph4 cells (Figure S18) and induced a statistically significant increase in Sfn which was not significantly affected by 20 (Figure 7A). AU1 upregulated Sfn but did not reach a high enough level of statistical significance. Conversely, Sprr1a was not significantly affected by any treatment (Figure 7B). This result suggests potential differential effects between bromodomain inhibition and whole protein knockout. Myc levels were also unaffected relative to DMSO treatment (Figure 7C). Unaffected Myc levels are consistent with a lack of caspase activation by 19 (Figure S19). From this preliminary analysis, we show that compound 19 treatment can induce cellular effects in at least one gene associated with BPTF knockout studies, and warrants further investigation. A limitation of the analysis is the comparison to BPTF knockout cells from a mixed population, and a need for analysis with more selective bromodomain inhibitors which is the focus of future work.
Figure 7: RT-qPCR Analysis of BPTF Regulated Genes.

A) Sfn: DMSO vs AU1: p= 0.5994, 19 vs 20: p= 0.0263*, DMSO vs 19: p=0.0388*, DMSO vs 20: p= 0.9798 B) Sprr1a: DMSO vs AU1: p= 0.515 NS, 19 vs 20: p= 0.3264 NS, DMSO vs 19: p= 0.8727 NS, DMS0 vs 20: p=0.9037 NS C) Myc: DMSO vs AU1: p>0.9999 NS, 19 vs 20: p= 0.0568, DMSO vs 19: p= 0.3265 NS, DMSO vs 20: p=0.9557 NS.
CONCLUSIONS
We describe the development of new BPTF inhibitors based on a pyridazinone scaffold with our lead molecule BZ1 having a high affinity for BPTF (Kd = 6.3 nM) and >350-fold selectivity over the BET family, making it the most potent inhibitor for the BPTF bromodomain in the published literature. We use the cocrystal structures of our analogues to establish a framework of structure-based design that can aid future efforts in rational development of chemical probes and to engineer selectivity over off-target bromodomains (Figure 8). Molecule 22 is one such example for reducing affinity towards BRD7/9. As not all bromodomain inhibitors exhibit cellular effects, here we use breast cancer cell lines to show that our inhibitors have on-target activity for BPTF and sensitize to the chemotherapy drug doxorubicin. Their activity is significantly improved relative to AU1, which is less effective with a sharp toxicity profile starting above 16 μM. The high potency, solubility and ligand efficiency (0.51) of BZ1 makes it a suitable lead for further medicinal chemistry optimization and the development of new chemical biology tools.
Figure 8.

Design rules established from pyridazinone SAR studies.
EXPERIMENTAL SECTION
Materials and Methods.
All commercially available reagents were used without further purification. Flash column chromatography was performed on a Teledyne-Isco Rf-plus CombiFlash instrument with RediSep columns. NMR spectra were collected on a Bruker Avance III AX-400 or a Bruker Avance III HD-500 equipped with a Prodigy TCI cryoprobe. Chemical shifts (δ) were reported in parts per million (ppm) and referenced to residual solvent signals for Chloroform-d (1H 7.26 ppm), Dimethyl Sulfoxide-d6 (1H 2.50 ppm, 13C 39.5 ppm) and Methanol-d4 (1H 3.31 ppm, 13C 49.0 ppm). Coupling constants (J) are in Hz. Splitting patterns were reported as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). High resolution ESI-MS spectra were recorded on a Thermo Fischer Orbitrap Velos equipped with an autosampler. Where stated, compounds were purified by reverse-phase high-performance liquid chromatography (RP-HPLC) on a C-18 column using 0.1% TFA water and CH3CN as solvents and TFA salts were quantified using the procedure described by Carlson et. al.49
Purity Analysis
All compounds tested in cells were ≥95% pure by RP-HPLC. Compounds 18–20 were run on a RP-HPLC with a C-18 column over a gradient of 0–10% ACN in 0.1% TFA H2O over 60 min. Spectral traces are shown in Figure S21.
General procedure A for the synthesis of compounds 1-16, 18-25.
Step 1: The nucleophilic aromatic substitution procedure was adapted from Humphreys et al.35 4,5-dichloro-2-methylpyridazin-3(2H)-one (1.0 eq.) was stirred in DMSO (1 mL) at room temperature, followed by addition of the primary amine (1.2 eq) and N,N-Diisopropylethylamine (2.0 eq.). The reaction mixture was heated in a sealed tube at 120 °C for 18 h. Following completion of the reaction, the reaction mixture was extracted into ethyl acetate, washed with saturated sodium bicarbonate solution (3×20 mL) and finally with brine (20 mL). The organic layer was dried over magnesium sulfate, filtered, concentrated in vacuo and purified by flash column chromatography (CombiFlash Rf system: 4 g silica, hexanes/ethyl acetate, 0–100% ethyl acetate, 30 minutes unless stated otherwise). The 4- and 5-positional isomers were obtained, with the 5-positional isomer as the more polar fraction. Step 2: The product from Step 1 was stirred in DCM (1 mL) at RT, followed by addition of trifluoroacetic acid (5.0 eq.) and stirred at RT for an additional 2 h. Step 3: The DCM was removed under vacuum and the product was isolated either as a TFA salt or a free base compound. To obtain the TFA salt, cold diethyl ether was added dropwise to precipitate out the product and the diethyl ether was removed in vacuo. For the free amine compounds, the mixture from Step 2 was extracted into DCM and treated with 1 M NaOH to attain a pH > 10. The DCM layer was dried with magnesium sulfate, filtered and the DCM was removed in vacuo to obtain the product.
General procedure B for the synthesis of compounds 26-28.
4,5-dichloropyridazin-3(2H)-one (1.0 eq.) was stirred in DMF (5 mL) followed by addition of sodium hydride (1.1 eq) and the alkyl bromide (1.4 eq.). The reaction mixture was stirred at room temperature for 12 h. Following completion of the reaction, the reaction mixture was extracted into ethyl acetate, washed with distilled water and finally with brine. The organic layer was dried over magnesium sulfate, filtered, concentrated in vacuo and purified by flash column chromatography (CombiFlash Rf system: 24 g silica, hexanes/ethyl acetate, 0–100% ethyl acetate, 20 minutes).
GSK4027 was purchased from Cayman Chemicals. The synthesis and characterization of compounds 1-3 were described previously.36
5-(azetidin-3-ylamino)-4-chloro-2-methylpyridazin-3(2H)-one (4).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (150 mg, 0.838 mmol, 1.0 eq.), tert-butyl 3-aminoazetidine-1-carboxylate (173 mg, 1.01 mmol, 1.2 eq.), N,N-Diisopropylethylamine (292 μL, 1.68 mmol, 2.0 eq.)), product 4 was obtained as a brown solid (211 mg, 77% yield over two steps). 1H NMR (400 MHz, DMSO-d6) δ 8.78 (d, J = 68.6 Hz, 2H), 7.76 (s, 1H), 7.16 (d, J = 7.3 Hz, 1H), 4.76 (h, J = 7.5 Hz, 1H), 4.28 – 4.19 (m, 2H), 4.18 – 4.08 (m, 2H), 3.60 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 158.5 (q, J = 34.9 Hz), 156.8, 143.2, 126.6, 116.1 (q, J = 293.4 Hz), 106.4, 52.6, 44.1 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C8H12ClN4O+ [M+H]+: 215.0694, observed 215.0686.
5-(azepan-3-ylamino)-4-chloro-2-methylpyridazin-3(2H)-one (5).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (114 mg, 0.636 mmol, 1.0 eq.), tert-butyl 3-aminoazepane-1-carboxylate (150 mg, 0.699 mmol, 1.1 eq.), N,N-Diisopropylethylamine (222 μL, 1.27 mmol, 2.0 eq.)), product 5 was obtained as a brown oil (49 mg, 21% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 9.05 (s, 2H), 7.93 (s, 1H), 6.35 (d, J = 9.2 Hz, 1H), 4.27 – 4.14 (m, 1H), 3.60 (s, 3H), 3.27 – 3.09 (m, 4H), 2.06 – 1.94 (m, 1H), 1.91 – 1.83 (m, 1H), 1.82 – 1.71 (m, 3H), 1.62 – 1.47 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 158.5, 156.8, 143.2, 126.5, 105.7, 49.4, 49.4, 46.4, 33.2, 24.8, 22.1 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C11H18ClN4O+ [M+H]+: 257.1164, observed 257.1154.
4-chloro-2-methyl-5-(phenylamino)pyridazin-3(2H)-one (6).
Following step 1 of the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (634 mg, 3.54 mmol, 1.1 eq.), aniline (300 mg, 3.22 mmol, 1.0 eq.), N,N-Diisopropylethylamine (1.12 mL, 6.44 mmol, 2.0 eq.)), product 6 was obtained as a yellow solid (73 mg, 10% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.73 (s, 1H), 7.64 (d, J = 1.7 Hz, 1H), 7.39 (d, J = 7.7 Hz, 2H), 7.25 (d, J = 7.9 Hz, 2H), 7.20 (t, J = 7.4 Hz, 1H), 3.61 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.5, 142.8, 139.0, 129.9, 128.1, 125.4, 124.0, 109.0 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C11H11ClN3O+ [M+H]+: 236.0585, observed 236.0575.
5-((4-aminophenyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (7).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (300 mg, 1.68 mmol, 1.0 eq.), tert-butyl (4-aminophenyl)carbamate (419 mg, 2.01 mmol, 1.2 eq.), N,N-Diisopropylethylamine (584 μL, 3.35 mmol, 2.0 eq.)), product 7 was obtained as a brown solid (49 mg, 8% yield over two steps). 1H NMR (500 MHz, Methanol-d4) δ 7.49 (s, 1H), 7.00 (d, J = 8.6 Hz, 2H), 6.77 (d, J = 8.6 Hz, 2H), 4.59 (s, 1H), 3.70 (s, 3H). 13C NMR (126 MHz, Methanol-d4) δ 160.1, 148.3, 146.2, 129.2, 128.8, 128.3, 117.0, 107.4, 40.5. HRMS (ESI-TOF) calculated for C11H12ClN4O+ [M+H]+: 251.0694, observed 251.0683.
4-chloro-5-((4-fluorophenyl)amino)-2-methylpyridazin-3(2H)-one (8).
Following step 1 of the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (532 mg, 2.97 mmol, 1.1 eq.), 4-fluoroaniline (300 mg, 2.70 mmol, 1.0 eq.), N,N-Diisopropylethylamine (940 μL, 5.40 mmol, 2.0 eq.)), product 8 was obtained as a white solid (52 mg, 8% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.57 (s, 1H), 7.34 – 7.18 (m, 4H), 3.60 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 13C NMR (126 MHz, DMSO-d6) δ 159.5 (d, J = 242.0 Hz), 157.0, 142.6, 134.8, 134.7, 127.4, 126.2 (d, J = 8.5 Hz), 116.1 (d, J = 22.6 Hz), 108.0 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C11H10ClFN3O+ [M+H]+: 254.0491, observed 254.0477.
5-((3-aminobenzyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (9).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (150 mg, 0.838 mmol, 1.0 eq.), tert-butyl (3-(aminomethyl)phenyl)carbamate (224 mg, 1.01 mmol, 1.2 eq.), N,N-Diisopropylethylamine (292 μL, 1.68 mmol, 2.0 eq.)), product 9 was obtained as a brown solid (50 mg, 23% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 7.61 (s, 1H), 7.22 (t, J = 6.5 Hz, 1H), 6.96 (t, J = 7.7 Hz, 1H), 6.45 (d, J = 1.9 Hz, 1H), 6.42 (dt, J = 7.9, 1.9 Hz, 2H), 5.08 (s, 2H), 4.41 (d, J = 6.5 Hz, 2H), 3.54 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 156.8, 149.0, 144.7, 139.6, 129.1, 126.5, 113.9, 112.7, 111.6, 104.8, 45.3 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C12H14ClN4O+ [M+H]+: 265.0851, observed 265.0842.
5-((4-(aminomethyl)phenyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (10).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (250 mg, 1.39 mmol, 1.0 eq.), tert-butyl (4-aminobenzyl)carbamate (279 mg, 1.26 mmol, 0.9 eq.), N,N-Diisopropylethylamine (487 μL, 2.79 mmol, 2.0 eq.)), product 10 was obtained as a brown solid (65 mg, 14% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.13 (s, 3H), 7.63 (s, 1H), 7.46 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H), 4.03 (q, J = 5.6 Hz, 2H), 3.62 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.1, 142.1, 138.9, 130.2, 130.1, 127.8, 123.1, 109.3, 41.85 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C12H14ClN4O + [M+H]+: 265.0851, observed 265.0839.
4-chloro-5-((4-((dimethylamino)methyl)phenyl)amino)-2-methylpyridazin-3(2H)-one (11).
Following step 1 of the general procedure A, 4,5-dichloro-2-methylpyridazin-3(2H)-one (328 mg, 1.83 mmol, 1.1 eq.), 4-((dimethylamino)methyl)aniline (245 μL, 1.66 mmol, 1.0 eq.), N,N-Diisopropylethylamine (579 μL, 3.32 mmol, 2.0 eq.) and purification by flash column chromatography (CombiFlash Rf system: 4 g silica, DCM/methanol, 0–20% methanol, 20 minutes), product 11 was obtained as a brown solid (46 mg, 9% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.66 (s, 1H), 7.36 (d, J = 7.9 Hz, 2H), 7.15 (d, J = 7.9 Hz, 2H), 6.39 (s, 1H), 3.76 (s, 3H), 3.43 (s, 2H), 2.26 (s, 6H) (NH not observed). 13C NMR (126 MHz, DMSO-d6) δ 157.0, 142.4, 137.2, 135.7, 129.7, 127.6, 123.5, 108.2, 62.8, 44.9. HRMS (ESI-TOF) calculated for C14H18ClN4O+ [M+H]+: 293.1164, observed 293.1150.
4-chloro-2-methyl-5-((1,2,3,4-tetrahydroisoquinolin-6-yl)amino)pyridazin-3(2H)-one (12).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (198 mg, 1.11 mmol, 1.1 eq.), tert-butyl 6-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate (250 mg, 1.01 mmol, 1.0 eq.), N,N-Diisopropylethylamine (351 μL, 2.01 mmol, 2.0 eq.)), product 12 was obtained as a yellow solid (51 mg, 17% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.60 (s, 1H), 7.05 (d, J = 8.2 Hz, 1H), 6.99 (dd, J = 8.3, 2.2 Hz, 1H), 6.96 (s, 1H), 3.85 (s, 2H), 3.60 (s, 3H), 2.96 (t, J = 5.7 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 157.0, 142.6, 136.0 (two resonances partially overlapping), 132.8, 127.5, 127.1, 124.2, 121.4, 107.8, 47.1, 42.8, 28.4. HRMS (ESI-TOF) calculated for C14H16ClN4O+ [M+H]+: 291.1007, observed 291.0996.
4-chloro-2-methyl-5-((1,2,3,4-tetrahydroisoquinolin-7-yl)amino)pyridazin-3(2H)-one (13).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (198 mg, 1.11 mmol, 1.1 eq.), tert-butyl 7-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate (250 mg, 1.01 mmol, 1.0 eq.), N,N-Diisopropylethylamine (351 μL, 2.01 mmol, 2.0 eq.)), product 13 was obtained as a yellow solid (29 mg, 10% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.61 (s, 1H), 7.59 (s, 1H), 7.09 (d, J = 8.1 Hz, 1H), 6.99 (dd, J = 8.1, 2.2 Hz, 1H), 6.90 (d, J = 2.3 Hz, 1H), 3.83 (s, 2H), 3.60 (s, 3H), 2.95 (t, J = 5.9 Hz, 2H), 2.67 (t, J = 5.9 Hz, 2H) (NH not observed). 13C NMR (126 MHz, DMSO-d6) δ 157.0, 142.6, 136.9, 135.7, 131.9, 129.9, 127.5, 121.7, 121.5, 107.8, 47.3, 43.0, 27.9. HRMS (ESI-TOF) calculated for C14H16ClN4O+ [M+H]+: 291.1007, observed 291.0995.
4-chloro-2-methyl-5-((5,6,7,8-tetrahydronaphthalen-2-yl)amino)pyridazin-3(2H)-one (14).
Following step 1 of the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (334 mg, 1.87 mmol, 1.1 eq.), 5,6,7,8-tetrahydronaphthalen-2-amine (250 mg, 1.70 mmol, 1.0 eq.), N,N-Diisopropylethylamine (592 μL, 3.40 mmol, 2.0 eq.)), product 14 was obtained as a yellow solid (175 mg, 36% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.57 (s, 1H), 7.59 (s, 1H), 7.06 (d, J = 8.1 Hz, 1H), 6.97 – 6.92 (m, 2H), 3.59 (s, 3H), 2.70 (t, J = 4.8, 2.4 Hz, 4H), 1.72 (t, J = 3.3 Hz, 4H). 13C NMR (126 MHz, DMSO) δ 157.5, 143.1, 138.2, 136.1, 134.2, 130.2, 127.9, 124.7, 121.8, 108.1, 29.2, 28.8, 23.2, 23.0 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C15H17ClN3O+ [M+H]+: 290.1055, observed 290.1038.
5-((3-(aminomethyl)phenyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (15).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (250 mg, 1.39 mmol, 1.0 eq.), tert-butyl (3-aminobenzyl)carbamate (279 mg, 1.26 mmol, 0.9 eq.), N,N-Diisopropylethylamine (487 μL, 2.79 mmol, 2.0 eq.)), a portion of the crude product (71 mg, crude 15% yield) was then purified by reverse-phase HPLC (5–40% CH3CN gradient over 30 minutes) to obtain product 15 as a brown solid. 1H NMR (500 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.14 (s, 3H), 7.75 (s, 1H), 7.44 (m, 1H), 7.31 (m, 1H), 7.25 (m, 2H), 4.03 (q, J = 5.7 Hz, 2H), 3.63 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.0, 142.0, 138.9, 135.2, 129.8, 127.8, 125.1, 123.4, 123.0, 109.3, 42.1 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C12H14ClN4O + [M+H]+: 265.0851, observed 265.0842.
5-((2-(aminomethyl)phenyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (16).
Compound previously characterized in literature.35 Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (250 mg, 1.39 mmol, 1.0 eq.), tert-butyl (2-aminobenzyl)carbamate (279 mg, 1.26 mmol, 0.9 eq.), N,N-Diisopropylethylamine (487 μL, 2.79 mmol, 2.0 eq.)), a portion of the crude product (12 mg, 3% yield) was then purified by reverse-phase HPLC (5–40% CH3CN gradient over 30 minutes) to obtain product 16 as a brown solid. 1H NMR (500 MHz, DMSO-d6) δ 8.34 (s, 1H), 8.12 (s, 3H), 7.50 (m, 1H), 7.36 (m, 2H), 7.22 (m, 1H), 7.26 (s, 1H), 4.06 (q, J = 5.2 Hz, 2H), 3.61(s, 3H). HRMS (ESI-TOF) calculated for C12H14ClN4O + [M+H]+: 265.0851, observed 265.0840.
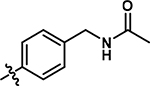
N-(4-((5-chloro-1-methyl-6-oxo-1,6-dihydropyridazin-4-yl)amino)benzyl)acetamide (17).
5-((4-(aminomethyl)phenyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (10) (20 mg, 0.05 mmol, 1.0 eq.) was stirred in dichloromethane (0.5 mL) followed by addition of acetic anhydride (6.4 mg, 0.06 mmol, 1.2 eq) and triethylamine (26 mg, 0.26 mmol, 5 eq.). The reaction mixture was stirred at room temperature for 0.5 h. Following completion of reaction, the reaction mixture was washed with diethyl ether. The solid was concentrated in vacuo and purified by flash column chromatography (CombiFlash Rf system: 12 g silica, dichloromethane/methanol, 0–10% methanol, 20 minutes). Product 17 was obtained as a white solid (9 mg, 56% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.66 (s, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 4.46 (d, J = 8.3 Hz, 2H), 3.77 (s, 3H), 2.08 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 170.3, 157.9, 142.1, 136.8, 136.7, 129.4, 126.7, 124.2, 110.2, 43.2, 40.4, 23.4. HRMS (ESI-TOF) calculated for C14H16ClN4O2+ [M+H]+: 307.0956, observed 307.0949.
5-((4-(2-aminoethyl)phenyl)amino)-4-chloro-2-methylpyridazin-3(2H)-one (18/BZ1).
Following the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (250 mg, 1.39 mmol, 1.0 eq.), tert-butyl (2-(4-amino-phenyl)-ethyl)carbamate (307 mg, 1.30 mmol, 0.9 eq.), N,N-Diisopropylethylamine (487 μL, 2.79 mmol, 2.0 eq.)), product 18 was obtained as a brown solid (140 mg, 21% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.69 (s, 1 H), 7.61 (s, 1 H), 7.27 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 7.9 Hz, 2H), 3.61 (s, 3 H), 2.93 (t, J = 7.5 Hz, 2 H), 2.76 (t, J = 7.6 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 157.1, 142.6, 136.7, 135.6, 129.7, 127.6, 123.9, 108.1, 41.5, 35.4 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C13H16ClN4O+ [M+H]+: 279.1007, observed 279.1002.
4-chloro-5-((4-(2-(dimethylamino)ethyl)phenyl)amino)-2-methylpyridazin-3(2H)-one (19).
Following step 1 of the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (250 mg, 1.39 mmol, 1.0 eq.), 4-(2-dimethylamino-ethyl)aniline (214 mg, 1.26 mmol, 0.9 eq.), N,N-Diisopropylethylamine (487 μL, 2.79 mmol, 2.0 eq.)) and purification by flash column chromatography (CombiFlash Rf system: 4 g silica, DCM/methanol, 0–20% methanol, 20 minutes), product 19 (more polar fraction) was obtained as a yellow solid (42 mg, 11% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.65 (s, 1H), 7.59 (s, 1H), 7.25 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 3.61 (s, 3 H), 2.70 (t, J = 7.6 Hz, 2H), 2.45 (t, J = 7.7 Hz, 2H), 2.18 (s, 6H). 13C NMR (126 MHz, Chloroform-d) δ 158.0, 142.4, 139.1, 135.4, 130.3, 126.8, 124.4, 109.7, 61.4, 45.6, 40.4, 33.9. HRMS (ESI-TOF) calculated for C15H20ClN4O+ [M+H]+: 307.1320, observed 307.1314.
5-chloro-4-((4-(2-(dimethylamino)ethyl)phenyl)amino)-2-methylpyridazin-3(2H)-one (20).
Following step 1 of the general procedure A, (4,5-dichloro-2-methylpyridazin-3(2H)-one (250 mg, 1.39 mmol, 1.0 eq.), 4-(2-dimethylamino-ethyl)aniline (214 mg, 1.26 mmol, 0.9 eq.), N,N-Diisopropylethylamine (487 μL, 2.79 mmol, 2.0 eq.)) and purification by flash column chromatography (CombiFlash Rf system: 4 g silica, DCM/methanol, 0–20% methanol, 20 minutes), product 20 (less polar fraction) was obtained as a yellow solid (37 mg, 10% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.75 (s, 1H), 7.11 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.3 Hz, 2H), 3.67 (s, 3H), 2.66 (t, J = 7.7 Hz, 2H), 2.44 (t, J = 7.8 Hz, 2H), 2.18 (s, 6H). 13C NMR (126 MHz, Chloroform-d) δ 158.0, 142.4, 139.1, 135.4, 130.3, 126.8, 124.4, 109.7, 61.4, 45.6, 40.4, 33.9. HRMS (ESI-TOF) calculated for C15H20ClN4O+ [M+H]+: 307.1320, observed 307.1310.
5-((4-(2-aminoethyl)phenyl)amino)-4-bromo-2-methylpyridazin-3(2H)-one (21).
Following the general procedure A, (4,5-dibromo-2-methylpyridazin-3(2H)-one (150 mg, 0.56 mmol, 1.0 eq.), tert-butyl (2-(4-amino-phenyl)-ethyl)carbamate (146 mg, 0.62 mmol, 1.1 eq.), N,N-Diisopropylethylamine (195 μL, 1.12 mmol, 2.0 eq.)), product 21 was obtained as a yellow solid (14 mg, 7% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 7.48 (s, 1H), 7.24 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 3.61 (s, 3H), 2.82 (t, J = 7.3 Hz, 2H), 2.68 (d, J = 7.3 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 157.3, 144.6, 136.8, 136.5, 129.6, 127.4, 124.1, 100.1, 42.7, 37.8 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C13H16BrN4O + [M+H]+: 323.0502, observed 323.0488.
4-chloro-5-((4-(3-(dimethylamino)propyl)phenyl)amino)-2-methylpyridazin-3(2H)-one (22).
Following step 1 of the general procedure A, 4,5-dichloro-2-methylpyridazin-3(2H)-one (552mg, 3.08 mmol, 1.1 eq.), 4-(3-(dimethylamino)propyl)aniline (500mg, 2.80 mmol, 1.0 eq.) and N,N-Diisopropylethylamine (975 μL, 5.60 mmol, 2.0 eq.) and purification by flash column chromatography (CombiFlash Rf system: 4 g silica, DCM/methanol, 0–20% methanol, 20 minutes). A portion of the product (91 mg, 10% crude yield) was further purified by reverse-phase HPLC (5–45% CH3CN gradient over 30 minutes) to obtain product 22 as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.75 (s, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.3 Hz, 2H), 3.67 (s, 3H), 2.57 – 2.52 (m, 2H), 2.23 (t, J = 7.2 Hz, 2H), 2.14 (s, 6H), 1.68 (t, J = 7.4 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 156.1, 138.2, 137.2, 136.6, 136.3, 127.6, 122.7, 111.0, 58.2, 44.9, 32.1, 28.6 (one resonance obscured by solvent). HRMS (ESI-TOF) calculated for C16H22ClN4O+ [M+H]+: 321.1477, observed 321.1463.
5-((4-(aminomethyl)phenyl)amino)-4-chloro-2-ethylpyridazin-3(2H)-one (23).
Following the general procedure A, (compound 26 (479 mg, 2.48 mmol, 1.0 eq.), tert-butyl (4-aminobenzyl)carbamate (607 mg, 2.73 mmol, 1.1 eq.), N,N-Diisopropylethylamine (864 μL, 4.96 mmol, 2.0 eq.)), product 23 was obtained as a yellow solid (62 mg, 9% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 7.61 (s, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 4.03 (q, J = 7.2 Hz, 2H), 3.73 (s, 2H), 1.22 (t, J = 7.1 Hz, 3H) (NH not observed). 13C NMR (126 MHz, DMSO-d6) δ 156.5, 142.4, 140.9, 136.5, 128.0, 127.6, 123.8, 107.9, 46.2, 45.0, 13.5. HRMS (ESI-TOF) calculated for C13H16ClN4O+ [M+H]+: 279.1007, observed 279.0995.
5-((4-(aminomethyl)phenyl)amino)-4-chloro-2-(prop-2-yn-1-yl)pyridazin-3(2H)-one (24).
Following the general procedure A, (compound 27 (185 mg, 0.911 mmol, 1.0 eq.), tert-butyl (4-aminobenzyl)carbamate (223 mg, 1.00 mmol, 1.1 eq.), N,N-Diisopropylethylamine (317 μL, 1.82 mmol, 2.0 eq.)), a portion of the crude product was then purified by reverse-phase HPLC (5–45% CH3CN gradient over 30 minutes) to obtain product 24 as a white solid (20 mg, 8% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 8.95 (s, 1H), 8.20 (s, 3H), 7.66 (s, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 4.94 (d, J = 10.3 Hz, 1H), 4.04 (s, 2H), 2.18 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 156.8, 142.3, 138.6, 130.4, 130.1, 128.2, 125.5, 123.5, 108.7, 60.7, 41.8, 27.2. HRMS (ESI-TOF) calculated for C14H14ClN4O+ [M+H]+: 289.0851, observed 298.0840.
5-((4-(aminomethyl)phenyl)amino)-4-chloro-2-isopropylpyridazin-3(2H)-one (25).
Following the general procedure A, (compound 28 (270 mg, 1.31 mmol, 1.0 eq.), tert-butyl (4-aminobenzyl)carbamate (320 mg, 1.44 mmol, 1.1 eq.), N,N-Diisopropylethylamine (456 μL, 2.62 mmol, 2.0 eq.)), product 25 was obtained as a brown solid (65 mg, 17% yield over two steps). 1H NMR (500 MHz, DMSO-d6) δ 7.66 (s, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 5.09 (hept, 6.6 Hz, 1H), 3.74 (s, 2H), 1.23 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 156.4, 141.9, 140.2, 136.6, 128.1, 127.4, 123.7, 107.8, 48.8, 44.8, 20.8 (NH2 resonance not observed). HRMS (ESI-TOF) calculated for C14H18ClN4O+ [M+H]+: 293.1164, observed 293.1153.
4,5-dichloro-2-ethylpyridazin-3(2H)-one (26).
Following the general procedure B, (4,5-dichloropyridazin-3(2H)-one (1 g, 6.06 mmol, 1.0 eq.), ethyl bromide (680 μL, 9.09 mmol, 1.5 eq.), sodium hydride (160 mg, 6.67 mmol, 1.1 eq.)), product 26 was obtained as a white solid (525 mg, 45% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.21 (s, 1H), 4.12 (d, J = 7.3 Hz, 2H), 1.27 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 155.5, 136.6, 135.8, 132.8, 47.4, 13.1. HRMS (ESI-TOF) calculated for C6H7Cl2N2O+ [M+H]+: 192.9930, observed 192.9925.
4,5-dichloro-2-(prop-2-yn-1-yl)pyridazin-3(2H)-one (27).
Following the general procedure B, (4,5-dichloropyridazin-3(2H)-one (2 g, 12.1 mmol, 1.0 eq.), propargyl bromide 80 wt. % in toluene (2.02 mL, 18.2 mmol, 1.5 eq.), sodium hydride (320 mg, 13.3 mmol, 1.1 eq.)), product 27 was obtained as a white solid (493 mg, 20% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.25 (s, 1H), 4.90 (d, J = 2.6 Hz, 2H), 3.42 (t, J = 2.5 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 155.1, 136.5, 136.4, 133.1, 77.5, 76.2, 41.9. HRMS (ESI-TOF) calculated for C7H5Cl2N2O+ [M+H]+: 202.9773, observed 202.9769.
4,5-dichloro-2-isopropylpyridazin-3(2H)-one (28).
Following the general procedure B, (4,5-dichloropyridazin-3(2H)-one (1.5 g, 9.1 mmol, 1.0 eq.), isopropyl bromide (1.2 mL, 13.6 mmol, 1.5 eq.), sodium hydride (240 mg, 10.0 mmol, 1.1 eq.)), product 28 was obtained as a white solid (294 mg, 16% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.25 (s, 1H), 5.15 – 5.00 (m, 1H), 1.29 (d, J = 6.7 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 155.4, 136.5, 135.6, 132.5, 50.6, 20.6.
Protein-Observed Fluorine (PrOF) NMR.
Fluorinated BPTF, PCAF, CECR2 and BRD4 D1 were expressed and purified as described previously.28 40–50 μM of protein in 50 mM TRIS, 100 mM NaCl, and pH 7.4 was diluted by adding 25 μL of D2O and 2 μL of 0.1% TFA for NMR locking and referencing purposes, respectively. Two spectra were taken of the control protein sample in the presence of 5 μL of DMSO (1% final concentration) at an O1P = −75 ppm, NS = 16, d1 = 1 s, AQ = 0.5 s (samples were referenced to trifluoroacetate at −75.25 ppm) and an O1P = −125 ppm, NS = 500–750, d1 = 0.7 s, AQ = 0.05 s (protein resonances). Ligands were titrated and the change in chemical shift relative to the control sample was plotted as a function of ligand concentration to generate binding isotherms. The data was processed in Mestrenova and isotherms were fit using GraphPad Prism with the equation below. Δδobs is the change in chemical shift, [L] is the total ligand concentration, and [P] is the total protein concentration:
General procedure for AlphaScreen assay.36
Unlabeled His9-tagged BPTF and BRD4 D1 were expressed and purified as described previously.36 The AlphaScreen assay procedures for BPTF and BRD4 bromodomains were adapted from the manufacturers protocol (PerkinElmer, USA). Nickel chelate (Ni-NTA) acceptor beads and streptavidin donor beads were purchased from PerkinElmer (Cat. #: 6760619M). The biotinylated Histone H4 KAc5,8,12,16 peptide was purchased from EpiCypher, with the sequence:
Ac-SGRGK(Ac)GGK(Ac)GLGK(Ac)GGAK(Ac)RHRKVLR-Peg(Biot).
All reagents were diluted in the assay buffer (50 mM HEPES-Na+ (ChemImpex), 100 mM NaCl (SigmaAldrich), 0.05% CHAPS (RPI), 0.1% BSA (SigmaAldrich), pH 7.4). The final assay concentrations (after the addition of all assay components) of 30 nM for His9-tagged BPTF bromodomain and 50 nM for the biotinylated peptide were used. For BRD4 D1, 7.5 nM His9-BRD4 and 25 nM of the peptide were used. 3-fold serial dilutions were prepared with varying concentrations of the compounds and a fixed protein concentration, keeping the final DMSO concentration at either 0.25% or 0.5% v/v, depending upon the solubility of the compounds. 5 μL of these solutions were added to a 384-well plate (ProxiPlate-384, PerkinElmer). The plate was sealed and kept at room temperature for 30 min, followed by the addition of 5 μL of the biotinylated peptide. 5 μL of nickel chelate acceptor beads was added to each well under low light conditions (<100 lux), to a final concentration of 20 μg/mL, and the plate was incubated at room temperature in the dark for 30 minutes. This was followed by the addition of 5 μL (20 μg/mL final concentration) of streptavidin donor beads in low light conditions. After incubation for 30 min in the dark, the plate was read in AlphaScreen mode using a PerkinElmer EnSpire plate reader. Each compound was run in two technical replicates and the data was normalized against 0 μM inhibitor signal to obtain the % normalized AlphaScreen signal. IC50 values were calculated in GraphPad Prism 5 using sigmoidal 4-parameter logistic (4PL) curve fit.
Cell culture methods.
4T1 cells were grown to a confluency of 50–60% using media containing DMEM with 10% fetal bovine serum (FBS), 2 mM glutamine and penicillin-streptomycin. 4T1 cells with shRNA-mediated BPTF knockdown (KD) were prepared as described previously.45 Figure S15 shows the western blot indicating BPTF KD. For NURF inhibitor toxicity study, 4000 cells/well were seeded in a 96-well plate and allowed to adhere overnight. The next day, 10 different dilutions of inhibitors were prepared starting with a highest concentration of 1.0 mM and further serially diluted 10 times to get the lowest concentration of 1.95 μM. Cells were treated with the inhibitors in complete media for 4 days. Thereafter, the MTS reagent was prepared using the CellTitre 96 aqueous MTS reagent (Promega, Cat #G1111) and phenazine methosulphate (Sigma, Cat #P9625). The MTS assay was performed as per manufacturer’s protocol and the absorbance was recorded at a wavelength of 490 nm. Fraction cell survival was calculated using untreated control cells to indicate complete survival (1.0) and blank solutions as 0.0 survival. The data was derived from three independent experiments (N=3) and fraction survival was plotted as mean fraction survival ± SEM using GraphPad Prism software. For checking the toxicity on wildtype and BPTF knockdown cells, three doses were selected for each inhibitor based on their toxicity curves and treated for 4 days alone or in combination with 50 nM doxorubicin. Fraction survival was measured and calculated by MTS assay as mentioned above.
Cytotoxicity experiments with Eph4 cells.
Eph4 cells were treated with either DMSO, AU1, 19 or 20 for 72 hours. Media containing each condition were changed every 12 hours. Cells were then incubated with Magic Red Caspase 3/7 (ImmunoChemistry Technologies, #936) to manufacturers specifications. Cells were also stained with Live/Dead Violet (Thermo Scientific, #L34964) in accordance to manufacturers specifications. All flow was performed on a Macsquant 10 (Miltenyi Biotec) and analyzed on FlowJo (TreeStar/BD). Statistically significant differences for cell line treatment groups were considered with a t-test p-value lower than 0.05 (p<0.05).
qPCR methods.
Eph4 cells were treated for 24 h and harvested in trizol. RNA extraction was carried out via chloroform extractions. cDNA creation was completed via SuperScript III cDNA creation kit (Invitrogen, #12574026). All qpcrs are normalized to EPH4 DMSO and the house keeping gene beta actin. Bars represent 2 biological replicates and 3 technical replicates. All statistical analysis are student’s t-test carried out on GraphPad. Reactions were carried out on the Quantstudio 6 platform using Sybr Green PCR Master Mix (Applied Biosystems, #4309155). Statistically significant differences for cell line treatment groups were considered with a one-way Anova p-value lower than 0.05 (p<0.05).
UV-Vis Methods
Compounds were diluted in DMSO at a top concentration of 100 mM. 2-fold serial dilutions in DMSO where performed followed by 1000-fold dilution into phosphate saline buffer (PBS) to get a final top concentration of 100 μM in 0.1% DMSO for each compound. UV-Vis measurements at 254 nm were taken on a Biomate 3S Spectrophotometer.
X-ray crystallography conditions and data collection methods.
BPTF bromodomain purification and crystallography for compounds 1-4:
Protein purification was performed at 4 °C by FPLC using columns and chromatography resins from GE Healthcare. Cell pellets were re-suspended in 50 mM Na/K Phosphate buffer (pH 7.4) containing 100 mM NaCl, 20 mM imidazole, 0.01% w/v lysozyme, 0.01% v/v Triton X-100 and 1mM DTT. Cells were lysed using a homogenizer, the lysate was clarified by centrifugation and subjected to purification on immobilized Ni2+-affinity chromatography (Qiagen) using a linear gradient of 20 – 500 mM imidazole. Fractions containing BPTF were pooled and incubated overnight with TEV protease at 4 °C. Cleaved BPTF was subjected to a second Ni2+-affinity chromatography run to remove His-TEV and the cleaved His-tag. The flow-through containing BPTF was concentrated and purified to homogeneity by size exclusion chromatography using a Superdex 26/60 column. Protein was eluted using 50 mM Tris/HCl (pH 8.0) containing 100 mM NaCl and 1 mM DTT. Peak fractions were combined, concentrated to 5 mg/mL, flash frozen in liquid N2 and stored at −80 °C. Crystallization was performed at 18 °C with precipitant solutions from Hampton Research using a Mosquito liquid handler (TTP Labtech). Robust crystallization conditions were established using 25% PEG 3,350, 0.2 M lithium sulfate monohydrate, 0.1 M Bis-Tris pH 6.5 mixed with an equal volume of protein in vapor diffusion hanging droplets. Compounds were cocrystallized with BPTF at 1 mM final concentration. Crystals were cryoprotected by addition of 20% ethylene glycol in the precipitant, flash frozen and stored in liquid N2. During data collection, crystals were maintained under a constant stream of N2 gas. X-ray diffraction data were recorded at beamlines 22-BM hosted by Ser-Cat and 23-ID-D hosted by GM/CA of Argonne National Laboratory. Data were indexed and scaled with XDS.50 Phasing and refinement was performed using PHENIX51 and model building with Coot.52 PDB entry 7K6R served as the search model for molecular replacement. Initial models for small molecule ligands were generated through MarvinSketch (ChemAxon, Cambridge, MA) and ligands restraints through eLBOW of the PHENIX suite. All structures have been validated by MolProbity. Figures were prepared using PyMOL (Schrödinger, LLC). Data processing and refinement statistics are given in Table S2.
Crystallography methods for compounds 10-13:
Unlabeled BPTF was expressed and purified as described previously.36 200–300 μM BPTF (in 50 mM TRIS, 100 mM NaCl, 10% (v/v) ethylene glycol, pH 7.4) was crystallized with 700 μM of compounds 10-13 using the hanging drop method at 4 °C. Crystals grew to harvestable size in 3–4 days. 10 was crystallized using 200 mM potassium acetate and 20% (v/v) PEG 3350. 11 and 13 were crystallized using 200 mM manganese acetate and 20% (v/v) PEG 3350. 12 was crystallized with 200 mM magnesium chloride and 10% (v/v) PEG 3350. Crystals were harvested, cryoprotected with ethylene glycol and flash frozen. Data was acquired at the Advanced Photon Source with the NECAT 24-IDE beamline. The structures were solved using molecular replacement with Phaser-MR and the PDB structure 3UV2. PHENIX51 and Coot52 were used for structure refinement and model building. Data processing and refinement statistics are given in Table S3.
Crystallography method for compound 19:
Unlabeled BPTF was expressed and purified as described previously.36 BPTF was concentrated to 16 mg/mL and previously reported crystallization conditions53 were chosen for optimization using a Dragonfly liquid handler (TTP Labtech). Drops consisting of 150 nL reservoir solution and 150 nL protein solution were set up in 96-well hanging drop plates using a mosquito crystallization robot (TTP Labtech). Thin needles formed and grew over 14–16 days in 0.2M NaCl and 23% PEG 3350 at 277 K. Larger needle crystals were grown in 24-well VDX hanging drop plate using micro-seeding. These crystals were soaked in solutions containing 1 mM of compound 19 for 1 hour, cryoprotected using the well solution supplemented with additional 10% glycerol, flash frozen and X-ray diffraction data were collected at 100 K on beam line SER-CAT 22ID at the Advanced Photon Source. Diffraction images were indexed, integrated, and scaled using HKL2000 suite. Phases were obtained by rigid body refinement using 3UV2 as the initial model. Residues were renumbered using 7K6R as a template. Model building was carried out using Coot. The final model was refined using PHENIX, and torsion-angle molecular dynamics with a slow-cooling simulated annealing. Data processing and refinement statistics are given in Table S4.
Supplementary Material
ACKNOWLEDGMENT
This work was conducted at the NE-CAT, GM/CA and SER-CAT beamlines at the Advanced Photon Source, which are supported by the NIH (P30-GM124165, Y1-CO-1020 and Y1-GM-1104). The Pilatus 6M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10-RR029205). We thank staff at the NE-CAT, GM/CA and SER-CAT beamlines for assistance in data collection. We thank the Moffitt Chemical Biology Core for use of the crystallization and crystallography facility (National Cancer Institute grant P30-CA076292). The authors thank Jose Valentin Lopez for contributing to the synthesis of related compounds published here.
Funding Sources
This work was supported by the National Institute of General Medical Sciences R01GM121414-04 and R35 GM140837-01, University of Minnesota IEM Engineering in Medicine Doctoral Fellowship 2020 (H.Z), National Institutes of Health Chemistry-Biology Interface Training Grant 5T32GM132029-02 (C.R.B), Department of Defense Grant W81XWH1910489 (J.W.L), R35-GM118047 (H.A). M.F is supported by the American Lebanese Syrian Associated Charities (ALSAC) and R35GN142772-01. C.O.D.S. is financially supported by the CSHL and Northwell Health affiliation, the NIH/NCI grant R01CA248158-01, and the NIH/NIA grant R01 AG069727-01.
ABBREVIATIONS
- ADME
absorption distribution metabolism and excretion
- BAF
BRG1/BRM-associated factor
- BET
bromodomain and extratermina
- BPTF
bromodomain PHD-finger transcription factor
- CECR2
cat eye syndrome chromosome region candidate 2
- ISWI
imitation switch
- NURF
nucleosome remodeling factor
- PBAF
polybromo-associated BAF
- PCAF
p300/CBP-associated factor
- PHD
plant homeodomain
- PrOF NMR
protein-observed fluorine nuclear magnetic resonance
- 5FW
5-fluorotryptophan
Footnotes
W.C.K.P, H.Z. and C.B. at the University of Minnesota have filed a provisional patent application on the inhibitors disclosed in this report.
ASSOCIATED CONTENT
Supporting Information. Characterization data of small molecules, biophysical and cellular assays data and X-ray crystallography statistics. The Supporting Information is available free of charge at http://pubs.acs.org.
Molecular-string files for the final compounds (csv)
Figures were created with BioRender and PyMOL.
Figure 1B and 5A adapted by permission from Springer Nature, Applied Biophysics for Bromodomain Drug Discovery by William C. K. Pomerantz, Jorden A. Johnson, Peter D. Ycas Copyright 2019.
PDB IDs:
7JT4
7K6R
7M2E
7LPK
7LP0
7LRO
7LRK
7RWN
7RWO
7RWP
7RWQ
Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- (1).Goldberg AD; Allis CD; Bernstein E Epigenetics: A Landscape Takes Shape. Cell 2007, 128 (4), 635–638. [DOI] [PubMed] [Google Scholar]
- (2).Allis CD; Jenuwein T The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17 (8), 487–500. [DOI] [PubMed] [Google Scholar]
- (3).Clapier CR; Iwasa J; Cairns BR; Peterson CL Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18 (7), 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hohmann AF; Vakoc CR A Rationale to Target the SWI/SNF Complex for Cancer Therapy. Trends Genet. 2014, 30 (8), 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).St. Pierre R; Kadoch C Mammalian SWI/SNF Complexes in Cancer: Emerging Therapeutic Opportunities. Curr. Opin. Genet. Dev. 2017, 42, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zoppi V; Hughes SJ; Maniaci C; Testa A; Gmaschitz T; Wieshofer C; Koegl M; Riching KM; Daniels DL; Spallarossa A; Ciulli A Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62 (2), 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Barak O; Lazzaro MA; Lane WS; Speicher DW; Picketts DJ; Shiekhattar R Isolation of Human NURF: A Regulator of Engrailed Gene Expression. EMBO J. 2003, 22 (22), 6089–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zahid H; Olson NM; Pomerantz WCK Opportunity Knocks for Uncovering the New Function of an Understudied Nucleosome Remodeling Complex Member, the Bromodomain PHD Finger Transcription Factor, BPTF. Curr. Opin. Chem. Biol. 2021, 63, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Xiao H; Sandaltzopoulos R; Wang H-M; Hamiche A; Ranallo R; Lee K-M; Fu D; Wu C Dual Functions of Largest NURF Subunit NURF301 in Nucleosome Sliding and Transcription Factor Interactions. Mol. Cell 2001, 8 (3), 531–543. [DOI] [PubMed] [Google Scholar]
- (10).Jones MH; Hamana N; Shimane M Identification and Characterization of BPTF, a Novel Bromodomain Transcription Factor. Genomics 2000, 63 (1), 35–39. [DOI] [PubMed] [Google Scholar]
- (11).Pomerantz WCK; Johnson JA; Ycas PD Applied Biophysics for Bromodomain Drug Discovery; 2019; pp 287–337. [Google Scholar]
- (12).Ruthenburg AJ; Li H; Milne TA; Dewell S; McGinty RK; Yuen M; Ueberheide B; Dou Y; Muir TW; Patel DJ; Allis CD Recognition of a Mononucleosomal Histone Modification Pattern by BPTF via Multivalent Interactions. Cell 2011, 145 (5), 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Landry J; Sharov AA; Piao Y; Sharova LV; Xiao H; Southon E; Matta J; Tessarollo L; Zhang YE; Ko MSH; Kuehn MR; Yamaguchi TP; Wu C Essential Role of Chromatin Remodeling Protein Bptf in Early Mouse Embryos and Embryonic Stem Cells. PLoS Genet. 2008, 4 (10), e1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wu B; Wang Y; Wang C; Wang GG; Wu J; Wan YY BPTF Is Essential for T Cell Homeostasis and Function. J. Immunol. 2016, 197 (11), 4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Frey WD; Chaudhry A; Slepicka PF; Ouellette AM; Kirberger SE; Pomerantz WCK; Hannon GJ; dos Santos CO BPTF Maintains Chromatin Accessibility and the Self-Renewal Capacity of Mammary Gland Stem Cells. Stem Cell Reports 2017, 9 (1), 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dar AA; Nosrati M; Bezrookove V; de Semir D; Majid S; Thummala S; Sun V; Tong S; Leong SPL; Minor D; Billings PR; Soroceanu L; Debs R; Miller JR III; Sagebiel RW; Kashani-Sabet M The Role of BPTF in Melanoma Progression and in Response to BRAF-Targeted Therapy. JNCI J. Natl. Cancer Inst. 2015, 107 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Koludrovic D; Laurette P; Strub T; Keime C; Le Coz M; Coassolo S; Mengus G; Larue L; Davidson I Chromatin-Remodelling Complex NURF Is Essential for Differentiation of Adult Melanocyte Stem Cells. PLOS Genet. 2015, 11 (10), e1005555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Richart L; Carrillo-de Santa Pau E; Río-Machín A; de Andrés MP; Cigudosa JC; Lobo VJSS-A; Real FX BPTF Is Required for C-MYC Transcriptional Activity and in Vivo Tumorigenesis. Nat. Commun. 2016, 7 (1), 10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Richart L; Real FX; Sanchez-Arevalo Lobo VJ C-MYC Partners with BPTF in Human Cancer. Mol. Cell. Oncol. 2016, 3 (3), e1152346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dai M; Lu J-J; Guo W; Yu W; Wang Q; Tang R; Tang Z; Xiao Y; Li Z; Sun W; Sun X; Qin Y; Huang W; Deng W; Wu T BPTF Promotes Tumor Growth and Predicts Poor Prognosis in Lung Adenocarcinomas. Oncotarget 2015, 6 (32), 33878–33892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lee JH; Kim MS; Yoo NJ; Lee SH BPTF, a Chromatin Remodeling-Related Gene, Exhibits Frameshift Mutations in Gastric and Colorectal Cancers. Apmis 2016, 124 (5), 425–427. [DOI] [PubMed] [Google Scholar]
- (22).Green AL; DeSisto J; Flannery P; Lemma R; Knox A; Lemieux M; Sanford B; O’Rourke R; Ramkissoon S; Jones K; Perry J; Hui X; Moroze E; Balakrishnan I; O’Neill AF; Dunn K; DeRyckere D; Danis E; Safadi A; Gilani A; Hubbell-Engler B; Nuss Z; Levy JMM; Serkova N; Venkataraman S; Graham DK; Foreman N; Ligon K; Jones K; Kung AL; Vibhakar R BPTF Regulates Growth of Adult and Pediatric High-Grade Glioma through the MYC Pathway. Oncogene 2020, 39 (11), 2305–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pan Y; Yuan F; Li Y; Wang G; Lin Z; Chen L Bromodomain PHD-Finger Transcription Factor Promotes Glioma Progression and Indicates Poor Prognosis. Oncol. Rep. 2019, 41 (1), 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhao X; Zheng F; Li Y; Hao J; Tang Z; Tian C; Yang Q; Zhu T; Diao C; Zhang C; Chen M; Hu S; Guo P; Zhang L; Liao Y; Yu W; Chen M; Zou L; Guo W; Deng W BPTF Promotes Hepatocellular Carcinoma Growth by Modulating HTERT Signaling and Cancer Stem Cell Traits. Redox Biol. 2019, 20 (October 2018), 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (25).Vidler LR; Brown N; Knapp S; Hoelder S Druggability Analysis and Structural Classification of Bromodomain Acetyl-Lysine Binding Sites. J. Med. Chem. 2012, 55 (17), 7346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Petretich M; Demont EH; Grandi P Domain-Selective Targeting of BET Proteins in Cancer and Immunological Diseases. Curr. Opin. Chem. Biol. 2020, 57, 184–193. [DOI] [PubMed] [Google Scholar]
- (27).Clegg MA; Tomkinson NCO; Prinjha RK; Humphreys PG Advancements in the Development of Non-BET Bromodomain Chemical Probes. ChemMedChem 2019, 14 (4), 362–385. [DOI] [PubMed] [Google Scholar]
- (28).Urick AK; Hawk LML; Cassel MK; Mishra NK; Liu S; Adhikari N; Zhang W; Dos Santos CO; Hall JL; Pomerantz WCK. Dual Screening of BPTF and Brd4 Using Protein-Observed Fluorine NMR Uncovers New Bromodomain Probe Molecules. ACS Chem. Biol. 2015, 10 (10), 2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Belkina AC; Denis GV BET Domain Co-Regulators in Obesity, Inflammation and Cancer. Nat. Rev. Cancer 2012, 12 (7), 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kirberger SE; Ycas PD; Johnson JA; Chen C; Ciccone MF; Woo RWL; Urick AK; Zahid H; Shi K; Aihara H; McAllister SD; Kashani-Sabet M; Shi J; Dickson A; dos Santos CO; Pomerantz WCK Selectivity, Ligand Deconstruction, and Cellular Activity Analysis of a BPTF Bromodomain Inhibitor. Org. Biomol. Chem. 2019, 17 (7), 2020–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tyutyunyk-Massey L; Sun Y; Dao N; Ngo H; Dammalapati M; Vaidyanathan A; Singh M; Haqqani S; Haueis J; Finnegan R; Deng X; Kirberger SE; Bos PD; Bandyopadhyay D; Pomerantz WCK; Pommier Y; Gewirtz DA; Landry JW Autophagy-Dependent Sensitization of Triple-Negative Breast Cancer Models to Topoisomerase II Poisons by Inhibition of The Nucleosome Remodeling Factor. Mol. Cancer Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Structural Genomics Consortium. TP-238 A chemical probe for CECR2/BPTF bromodomains https://www.thesgc.org/chemical-probes/TP-238.
- (33).Structural Genomics Consortium. NVS-BPTF-1 A chemical probe for BPTF.
- (34).Mélin L; Calosing C; Kharenko OA; Hansen HC; Gagnon A Synthesis of NVS-BPTF-1 and Evaluation of Its Biological Activity. Bioorg. Med. Chem. Lett. 2021, 47 (June), 128208. [DOI] [PubMed] [Google Scholar]
- (35).Humphreys PG; Bamborough P; Chung CW; Craggs PD; Gordon L; Grandi P; Hayhow TG; Hussain J; Jones KL; Lindon M; Michon AM; Renaux JF; Suckling CJ; Tough DF; Prinjha RK Discovery of a Potent, Cell Penetrant, and Selective P300/CBP-Associated Factor (PCAF)/General Control Nonderepressible 5 (GCN5) Bromodomain Chemical Probe. J. Med. Chem. 2017, 60 (2), 695–709. [DOI] [PubMed] [Google Scholar]
- (36).Ycas PD; Zahid H; Chan A; Olson NM; Johnson JA; Talluri SK; Schonbrunn E; Pomerantz WCK New Inhibitors for the BPTF Bromodomain Enabled by Structural Biology and Biophysical Assay Development. Org. Biomol. Chem. 2020, 18 (27), 5174–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mishra NK; Urick AK; Ember SWJ; Schönbrunn E; Pomerantz WC Fluorinated Aromatic Amino Acids Are Sensitive 19 F NMR Probes for Bromodomain-Ligand Interactions. ACS Chem. Biol. 2014, 9 (12), 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hopkins AL; Keserü GM; Leeson PD; Rees DC; Reynolds CH The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discov. 2014, 13 (2), 105–121. [DOI] [PubMed] [Google Scholar]
- (39).pKa Table (Evans and Ripin) https://organicchemistrydata.org/hansreich/resources/pka/#pka_water_compilation_evans.
- (40).Clegg MA; Bamborough P; Chung C; Craggs PD; Gordon L; Grandi P; Leveridge M; Lindon M; Liwicki GM; Michon A-M; Molnar J; Rioja I; Soden PE; Theodoulou NH; Werner T; Tomkinson NCO; Prinjha RK; Humphreys PG Application of Atypical Acetyl-Lysine Methyl Mimetics in the Development of Selective Inhibitors of the Bromodomain-Containing Protein 7 (BRD7)/Bromodomain-Containing Protein 9 (BRD9) Bromodomains. J. Med. Chem. 2020, 63 (11), 5816–5840. [DOI] [PubMed] [Google Scholar]
- (41).Matsson P; Doak BC; Over B; Kihlberg J Cell Permeability beyond the Rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [DOI] [PubMed] [Google Scholar]
- (42).Cui H; Divakaran A; Pandey AK; Johnson JA; Zahid H; Hoell ZJ; Ellingson MO; Shi K; Aihara H; Harki DA; Pomerantz WCK. Selective N-Terminal BET Bromodomain Inhibitors by Targeting Non-Conserved Residues and Structured Water Displacement. Angew. Chemie Int. Ed. 2021, 60 (3), 1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Xiong L; Mao X; Guo Y; Zhou Y; Chen M; Chen P; Yang S; Li L Discovery of Selective BPTF Bromodomain Inhibitors by Screening and Structure-Based Optimization. Biochem. Biophys. Res. Commun. 2021, 545, 125–131. [DOI] [PubMed] [Google Scholar]
- (44).Aslakson CJ; Miller FR Selective Events in the Metastatic Process Defined by Analysis of the Sequential Dissemination of Subpopulations of a Mouse Mammary Tumor. Cancer Res. 1992, 52 (6), 1399–1405. [PubMed] [Google Scholar]
- (45).Mayes K; Alkhatib SG; Peterson K; Alhazmi A; Song C; Chan V; Blevins T; Roberts M; Dumur CI; Wang X-Y; Landry JW BPTF Depletion Enhances T-Cell–Mediated Antitumor Immunity. Cancer Res. 2016, 76 (21), 6183–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lin A; Giuliano CJ; Palladino A; John KM; Abramowicz C; Yuan M. Lou; Sausville EL; Lukow DA; Liu L; Chait AR; Galluzzo ZC; Tucker C; Sheltzer JM Off-Target Toxicity Is a Common Mechanism of Action of Cancer Drugs Undergoing Clinical Trials. Sci. Transl. Med. 2019, 11 (509), eaaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Xu R; Nelson CM; Muschler JL; Veiseh M; Vonderhaar BK; Bissell MJ Sustained Activation of STAT5 Is Essential for Chromatin Remodeling and Maintenance of Mammary-Specific Function. J. Cell Biol. 2009, 184 (1), 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Geletu M; Hoskin V; Starova B; Niit M; Adan H; Elliot B; Gunning P; Raptis L Differentiation of Mouse Breast Epithelial HC11 and EpH4 Cells. J. Vis. Exp 2020, No. 156. [DOI] [PubMed] [Google Scholar]
- (49).Carlson AS; Cui H; Divakaran A; Johnson JA; Brunner RM; Pomerantz WCK; Topczewski JJ Systematically Mitigating the P38α Activity of Triazole-Based BET Inhibitors. ACS Med. Chem. Lett. 2019, 10 (9), 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kabsch W XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX : A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Müller S; Pawson T; Gingras AC; Arrowsmith CH; Knapp S Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149 (1), 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.