

Abstract
Objectives:
Patients with established Parkinson’s disease (PD) display differences in peripheral blood markers of immune function, including leukocyte differential counts, compared with controls. These differences may be useful biomarkers to predict PD and may shed light on pathogenesis. We sought to identify whether peripheral immune dysregulation was associated with increased risk of subsequent PD diagnosis.
Methods:
We examined the relationship between incident PD, baseline differential leukocyte count and other blood markers of acute inflammation in UK Biobank (UKB), a longitudinal cohort with ~500,000 participants. We used a range of sensitivity analyses and Mendelian randomization (MR) to further explore the nature of associations.
Results:
After excluding individuals with comorbidities which could influence biomarkers of inflammation, 465 incident PD cases and 312,125 controls remained. Lower lymphocyte count was associated with increased risk of subsequent PD diagnosis (per 1-SD decrease in lymphocyte count odds ratio [OR] = 1.18, 95% confidence interval [CI] = 1.07–1.32, padjusted = 0.01). There was some evidence that reductions in eosinophil counts, monocyte counts and C-reactive protein (CRP) were associated with increased PD risk, and that higher neutrophil count was also associated. Only the association between lower lymphocyte count and increased PD risk remained robust to sensitivity analyses. MR suggested that the effect of lower lymphocyte count on PD risk may be causal (per 1-SD decrease in lymphocyte count; ORMR = 1.09, 95% CI = 1.01–1.18, p = 0.02).
Interpretation:
We provide converging evidence from observational analyses in UKB and MR that lower lymphocyte count is associated with an increased risk of subsequent PD.
Parkinson’s disease (PD) affects 2% of the population over 65 years of age.1 The diagnosis is made once motor signs appear, however, by this stage ~50% of nigrostriatal neurons have been lost.2 There is an urgent unmet clinical need for earlier identification of PD and development of therapies that could slow, prevent, or reverse the progression of the disease.
Immune dysregulation may play a role in the pathogenesis of PD. The white blood cell (WBC) differential is a crude marker of immune function but is simple to measure in large-scale observational studies. Previous studies have found lower lymphocyte counts in patients with PD compared with controls, driven by reductions in helper-CD4+ T cell and B-cell counts.3–7 Case-control studies have also identified higher neutrophil and lower lymphocyte counts in patients with established PD compared with controls.8
Converging evidence from genetic, epidemiological, and cytokine profiling studies has added further weight to the view that immune dysregulation may play a role in the pathogenesis of PD.9 Human leukocyte antigen genes (HLA; DRB1 / DRB5) have been identified as risk loci for PD in genomewide association studies (GWAS).10,11 In vitro, alpha-synuclein-derived peptides are preferentially displayed on major histocompatibility (MHC) molecules associated with PD risk, suggesting that PD-associated HLA haplotypes could drive an adaptive immune response targeted toward alpha-synuclein epitopes.12,13 Variants in the leucine-rich repeat kinase 2 (LRRK2) gene, a target for proinflammatory signals, confer effects in the same direction on risk of PD and Crohn’s disease.14 Observational studies have reported reduced risk of PD and reduced penetrance of LRRK2 variants with use of immunosuppressants and nonsteroidal anti-inflammatory drugs.15,16 The prospective ICICLE-PD cohort study found that a baseline “pro-inflammatory” serum cytokine profile in patients with PD was associated with faster motor deterioration than an “anti-inflammatory” profile.17
It is however unclear whether immune dysregulation is detectable in the years prior to PD diagnosis. To answer this, we studied the relationship between baseline blood tests (differential leukocyte count and markers of acute inflammation) and incident PD in UK Biohank (UKB), a large longitudinal cohort with ~500,000 participants. We used a range of sensitivity analyses to mitigate the likelihood that observed associations were driven by confounding or reverse causation. Given the long latency between the biological onset and clinical presentation of PD, it is plausible that cases identified as clinically incident were actually biologically “prevalent” at the time of recruitment. Observed associations between immune changes and incident PD could either be a determinant of PD or a consequence of preclinical PD, limiting our ability to make causal inferences about the direction of effect.
Methods
Population
UKB recruited ~500,000 individuals aged 40 to 69 years between 2006 and 2010. Prospective follow-up data, including census data, blood tests, and healthcare records, are regularly obtained.18 Institutional review board approval was not required as all data provided by UKB is de-identified.
PD cases were defined as individuals with an International Classification of Disease (ICD)-10 diagnosis of PD (code G20) derived from Hospital Episode Statistics or a self-reported diagnosis of PD. The date at PD diagnosis was determined using the UKB data field “Date of Parkinson’s Disease report.” Age at diagnosis was derived using this field, age at recruitment, and birth year. Cases were defined as “incident” if their age at diagnosis was greater than at recruitment. “Prevalent” PD cases (ie, with a diagnosis of PD at baseline), were excluded from analyses. “Controls” were defined as all other individuals in the dataset after applying these exclusions. Individuals without data available for the baseline visit blood tests were excluded. A flowchart of participant numbers at each stage of inclusions and exclusions is provided in Figure 1. Demographic details of excluded participants are also shown in Table 1.
FIGURE 1:
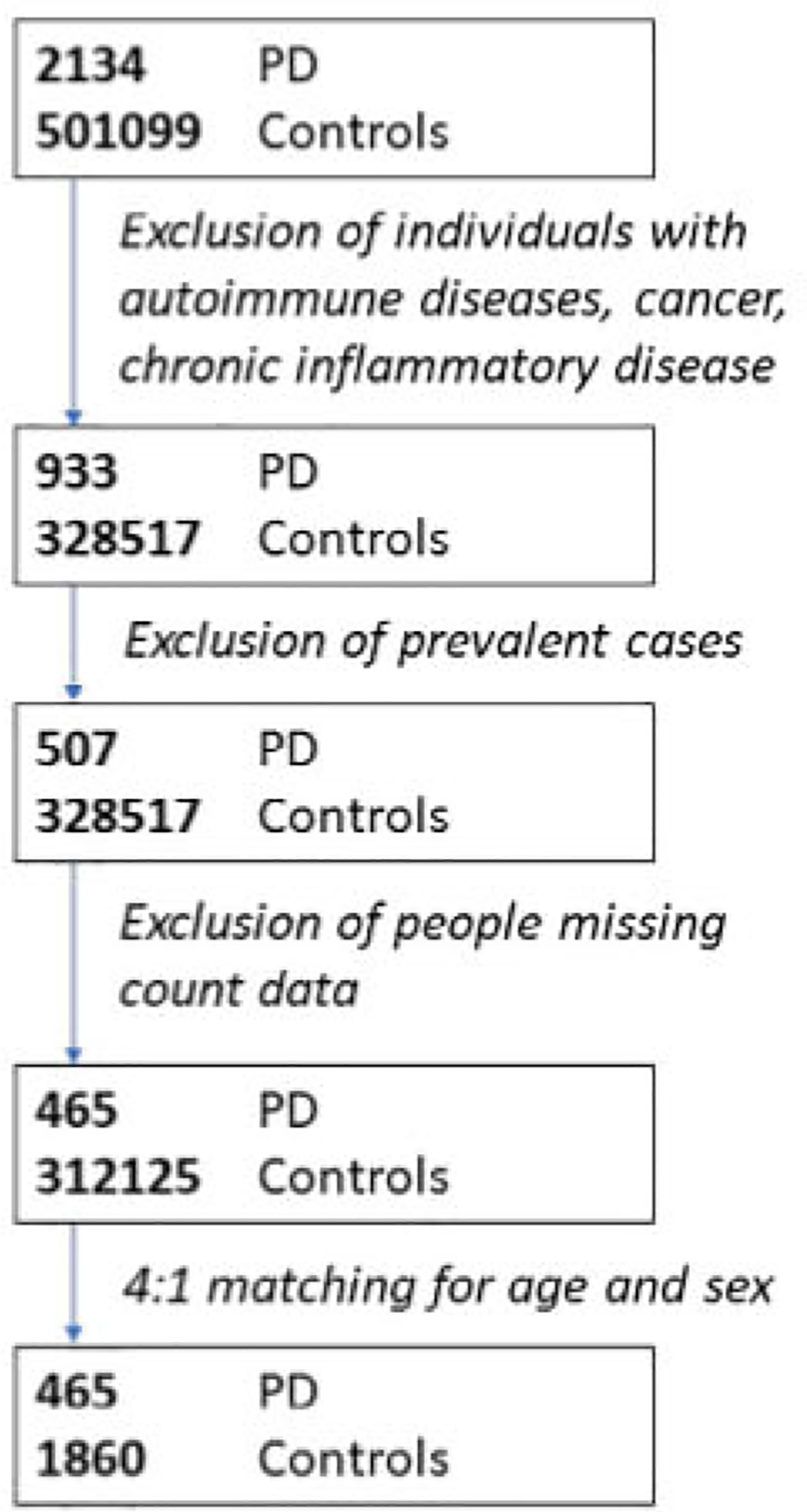
Flowchart of participants showing the number of individuals included at each stage.
TABLE 1.
Demographic Information for Incident PD Cases and Controls After Exclusions
| Excluded individuals | Matched analysis | Unmatched analysis | ||||
|---|---|---|---|---|---|---|
| Controls (n = 172,582) | Cases (n = 1,201) | Controls (n = 1,860) | Cases (n = 465) | Controls (n = 312,125) | Cases (n = 465) | |
| Age at PD report | NA | 69.65 (5.7) | NA | 68.52 (6.82) | NA | 68.52 (6.82) |
| Age at recruitment | 58.99 (7.46) | 63.58 (5.18) | 62.4 (5.81) | 62.4 (5.82) | 55.21 (8.1) | 62.4 (5.82) |
| Townsend score | −1.2 (3.15) | −1.16 (3.21) | −1.52 (3.05) | −1.51 (2.94) | −1.36 (3.06) | −1.51 (2.94) |
| Sex | ||||||
| F | 95,510 (55.35%) | 317 (41.22%) | 636 (34.19%) | 159 (34.19%) | 167,951 (53.81%) | 159 (34.19%) |
| M | 77,062 (44.65%) | 452 (58.78%) | 1,224 (65.81%) | 306 (65.81%) | 144,174 (46.19%) | 306 (65.81%) |
| Self-reported ethnic background | ||||||
| Non-While | 7,067 (4.12%) | 31 (4.05%) | 72 (3.89%) | 12 (2.61%) | 18,406 (5.93%) | 12 (2.61%) |
| White | 164,518 (95.88%) | 735 (95.95%) | 1,778 (96.11%) | 448 (97.39%) | 292,189 (94.07%) | 448 (97.39%) |
| Country of birth | ||||||
| Do not know | 47 (0.03%) | 0 (0%) | 0 (0%) | 1 (0.22%) | 90 (0.03%) | 1 (0.22%) |
| Elsewhere | 11186 (6.49%) | 47 (6.14%) | 124 (6.67%) | 24 (5.18%) | 26936 (8.64%) | 24 (5.18%) |
| England | 137,274 (79.69%) | 640 (83.55%) | 1,438 (77.4%) | 362 (78.19%) | 239,966 (76.96%) | 362 (78.19%) |
| Northern Ireland | 1,061 (0.62%) | 8 (1.04%) | 14 (0.75%) | 6 (1.3%) | 1,920 (0.62%) | 6 (1.3%) |
| Prefer not to answer | 204 (0.12%) | 0 (0%) | 1 (0.05%) | 3 (0.65%) | 475 (0.15%) | 3 (0.65%) |
| Republic of Ireland | 2,051 (1.19%) | 10 (1.31%) | 19 (1.02%) | 11 (2.38%) | 2,737 (0.88%) | 11 (2.38%) |
| Scotland | 12,742 (7.4%) | 39 (5.09%) | 174 (9.36%) | 41 (8.86%) | 25,952 (8.32%) | 41 (8.86%) |
| Wales | 7,703 (4.47%) | 22 (2.87%) | 88 (4.74%) | 15 (3.24%) | 13,724 (4.4%) | 15 (3.24%) |
“Unmatched analysis” refers to the entire cohort after exclusion of individuals with potentially confounding comorbidities. “Matched analysis” refers to 1:4 matched controls (matched to incident PD cases by age at recruitment and sex). Demographic data for excluded individuals (excluded due to potentially confounding comorbidities) are also shown. Categorical variables are presented as n (%), and continuous variables as mean (SD).
NA = not applicable; PD = Parkinson’s disease.
Various pre-existing health conditions can influence leukocyte differential counts and biomarkers of inflammation. To minimize bias from unbalanced comorbidities among cases and controls, we excluded individuals with ICD-10 diagnoses of malignant neoplasms, disease of the blood and blood-forming organs, autoimmune disease, thyrotoxicosis, demyelinating disease of the central nervous system, inflammatory respiratory conditions (asthma and bronchiectasis), noninfective enteritis, inflammatory dermatological conditions (atopic dermatitis and psoriasis), inflammatory polyarthropathies, spondylopathies, and eating disorders (Supplementary Table S1).
Blood Cell Markers
Blood cell markers (absolute and relative counts) and other markers of inflammation (CRP and albumin) were obtained from baseline blood tests of UKB participants taken at the initial assessment visit (https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=100080). Details of data processing can be found on the UKB website (http://biobank.ndph.ox.ac.uk/showcase/showcase/docs/haematology.pdf and https://biobank.ndph.ox.ac.uk/showcase/showcase/docs/serum_biochemistry.pdf).
Statistical Analysis
We determined associations of blood cell and inflammatory markers with incident risk of PD using logistic regression. As our primary analysis, we conducted multivariable logistic regression, modeling incident PD diagnosis as the outcome, and adjusting for age, sex, Townsend deprivation score, and ethnicity (dichotomized as “White” background vs all other ethnicities). Models were of the form: Incident PD ~ age + sex + deprivation + ethnicity + blood cell marker. Prior to model fitting, all exposures were transformed to Z scores to allow for comparison of effect sizes between exposures (Z = (value – mean)/SD). For clarity, we examined the effect of a decrease in each exposure on PD risk. The regression coefficient for each trait therefore represents the predicted effect of a 1-standard deviation (SD) decrease in the trait. Raw, untransformed counts and distributions for each of the blood test traits are shown in Supplementary Table S2. The strength of association was determined using the likelihood ratio test, comparing the full model to a null model consisting of the confounding covariates only (incident PD ~ age + sex + deprivation + ethnicity). The p values were adjusted to control the false discovery rate at 5% using the Benjamini-Hochberg procedure (adjusted p values are labeled as “Q” values).
We then undertook a variety of sensitivity analyses. First, we included additional covariates in the models: body mass index (BMI) at recruitment, smoking status (“ever” vs “never”), and alcohol consumption (“ever” vs “never”). Second, we excluded individuals within serial time windows of PD diagnosis (<1, <2, <3 years from diagnosis, etc.) to determine whether the effects from the primary analysis were restricted to individuals who would go on to develop PD sooner (i.e. who were more likely to already have PD and simply have not had their diagnosis coded in healthcare records yet). Third, we repeated the analysis using a matched case:control approach, individually matching controls by age and sex to PD cases with a 4:1 ratio. Fourth, we repeated the analyses with more liberal inclusion criteria (ie, including all participants in the dataset regardless of co-existent comorbidities).
To determine associations between blood markers and time until PD diagnosis, we constructed linear models for the time to PD diagnosis on age, sex, Townsend score, ethnicity, and blood cell marker. Model fit was quantified using the likelihood ratio test. Time to diagnosis/report was normalised prior to model-fitting using the rank-based inverse-normal transformation.
Mendelian Randomization
We used 2-sample Mendelian randomization (MR) to further examine robust associations from the observational study for evidence of causal influence on PD risk. MR is a type of instrumental variable (IV) analysis, which exploits the association between genetic variants and an exposure to predict the effect of that exposure on an outcome.19,20 As genetic variants are randomly allocated at birth and do not change throughout the lifetime, MR can be used in some situations to estimate the effect of an exposure while reducing the effects of confounding and reverse causation. The validity of the MR estimate depends on 3 core assumptions: that the IV is associated with the exposure of interest, that the IV is not associated with confounders of the exposure-outcome association, and that the IV is only associated with the outcome via its effect on the exposure of interest. If these assumptions are satisfied, MR can be used to approximate the causal effect of the exposure on the outcome.21
We performed 2-sample MR using the TwoSampleMR R package.22,23 For the exposure instrument, we used summary statistics from the largest published GWAS on blood cell traits.23 This GWAS of 408,112 European UKB participants used as its outcome measure the absolute lymphocyte count which was adjusted for covariates (age, sex, principal components, and study-specific factors) and rank inverse-normalized. Thus, a one unit increase in the beta coefficient represents a 1-SD increase in adjusted and normalized lymphocyte count per additional effect allele.
We applied the following steps to develop a genetic instrument for lymphocyte count:
Removed single nucleotide polymorphisms (SNPs) not typed / imputed in the outcome GWAS;
Restricted to biallelic single nucleotide variants;
Removed SNPs within the super-extended MHC region due to the complex pleiotropy and linkage disequilibrium (LD) of this region (hg19, chr6: 25,000,000–35,000,000);
Restricted to SNPs strongly associated with standardized lymphocyte count (p < 5e-08);
Clumped SNPs using stringent default parameters (LD window = 10,000 kb, r2 = 0.001);
Removed SNPs explaining more variance in the outcome than the exposure through Steiger filtering.22
Outcome data for PD were taken from the most recent and largest case-control GWAS of PD published by the IPDGC and 23andMe.25 Overlap in the controls in the exposure and outcome data can result in bias, so we used summary statistics from the PD GWAS that excluded participants from the UKB.
We harmonized exposure and outcome SNPs to ensure that effect estimates were aligned for the same effect allele. As our primary analysis, we used the inverse-variance weighted (IVW) MR estimate, which provides an estimate of the effect of the exposure on the outcome when MR assumptions are valid.26 As a secondary sensitivity analysis, we applied the Mixture of Experts approach, which applies different MR estimators and methods for SNP instrument selection (heterogeneity and directionality filtering), and predicts which method has the highest probability of estimating the effect of the exposure on the outcome based on the data characteristics.22 As a further sensitivity analysis, we used MR-PRESSO (Pleiotropy Residual Sum and Outlier) to automatically remove outliers with evidence of substantial pleiotropy. MR-PRESSO quantifies the degree of distortion of the IVW estimate contributed by each variant; more pleiotropic variants are expected to deviate more strongly from the overall IVW regression line.27 For each variant, MR-PRESSO calculates the IVW estimate without that variant, quantifies the difference between the expected and observed effect on the outcome of that variant, and compares that residual to a null distribution to generate an empirical p value.
Data and Code Availability
UK Biobank data are available via application (https://www.ukbiobank.ac.uk/). Code is available at https://github.com/Wolfson-PNU-QMUL/PD_FBC. PD GWAS summary statistics which exclude UKB data can be obtained from https://pdgenetics.org/resources and an application to 23andMe https://research.23andme.com/dataset-access/.25 Blood cell trait GWAS summary statistics have been made publicly available by the authors at ftp://ftp.sanger.ac.uk/pub/project/humgen/summary_statistics/UKBB_blood_cell_traits/.
Results
Demographics
After applying the exclusion criteria, 465 incident PD cases and 312,125 controls remained in the main (unmatched) analysis (see Fig 1). Participant demographic data are shown in Table 1.
Association of Blood Cell and Inflammatory Traits with Incident PD
Each 1-SD reduction in lymphocyte count was associated with an 18% increase in the odds of incident PD (odds ratio [OR] = 1.18, 95% confidence interval [CI] = 1.07–1.32, padjusted = 0.01; Table 2, Fig 2). There was some evidence that reductions in CRP, monocyte count, and eosinophil count were also associated with an increased risk of subsequent PD (see Table 2). There was also some evidence that a higher neutrophil count was associated with PD (see Table 2). We obtained similar results with a slight loss of precision in a matched case:control subset of the wider cohort (Supplementary Table S3).
TABLE 2.
Association of Blood Cell Traits and Inflammatory Markers with Incident PD
| Trait | OR per 1-SD reduction in exposure | p | Adjusted p value (Q, FDR 5%) |
|---|---|---|---|
| Lymphocyte count | 1.18 (95% CI = 1.07–1.32) | 0.001 | 0.011 |
| Eosinophil count | 1.16 (95% CI = 1.04–1.3) | 0.006 | 0.027 |
| CRP | 1.13 (95% CI = 1–1.29) | 0.031 | 0.073 |
| Monocyte count | 1.12 (95% CI = 1.01–1.26) | 0.032 | 0.073 |
| Neutrophil count | 0.91 (95% CI = 0.84–1) | 0.051 | 0.093 |
| Albumin | 1.04 (95% CI = 0.93–1.15) | 0.504 | 0.756 |
| Platelet count | 1.02 (95% CI = 0.93–1.13) | 0.645 | 0.829 |
| Total white cell count | 1 (95% CI = 0.91–1.1) | 0.945 | 0.979 |
| Basophil count | 1 (95% CI = 0.91–1.11) | 0.979 | 0.979 |
The table shows the odds ratios, confidence intervals, p values from likelihood ratio tests, and FDR Q values for the output of multivariable logistic regression models, modeling incident PD on age + sex + deprivation + ethnicity + trait. Odds ratios represent the predicted effect of a 1 standard deviation (SD) decrease in the trait (ie, a 1 unit decrease in Z score) on the odds of incident PD. For instance, for each 1-SD decrease in lymphocyte count, the odds of PD are predicted to increase by 18%.
CI = confidence interval; CRP = C-reactive protein; FDR = False Discovery Rate; OR = odds ratio; PD = Parkinson’s disease; SE = standard error.
FIGURE 2:

Association of blood cell traits and inflammatory markers with incident Parkinson’s disease (PD) in the UK Biobank (UKB). Betas and 95% confidence intervals (CIs) are shown from multivariable logistic regression models of the form PD ~ age + sex + ethnicity + Townsend deprivation score + blood marker. Estimates shown here are for Z-scores and are orientated such that an increase of “1” on the x axis corresponds to the effect of a 1 standard deviation (SD) decrease in the blood marker.
To determine whether these associations could be driven by confounding, we constructed models in which we also controlled for variables that can impact both PD risk and blood cell indices: BMI, smoking, and alcohol consumption. The effect estimates from these models were less precise but of a similar magnitude to the primary analysis (Supplementary Table S4). In these models, only lymphocyte count remained strongly associated with incident PD at a false discovery rate of 5%.
To examine the possibility that reverse causation could be driving our findings (ie, that early PD could be driving lower lymphocyte counts) we excluded individuals who underwent blood draw within serial time windows of diagnosis (within 1, 2, 3, 4, 5, 6, 7, and 8 years of diagnosis). There was a consistent signal in all groups (Supplementary Table S5). Exclusion of extreme lymphocyte counts (mean ± >3 SD) did not substantially alter the observed association (OR for PD per 1-SD decrease in lymphocyte count = 1.17, 95% CI = 1.06–1.29, p = 0.002). Lymphopenia – defined as a binary trait (ie, absolute lymphocyte count <1 × 109 cells/L) – was associated with an increased risk of PD (OR = 1.93, 95% CI = 1.26–2.97, p = 0.006). Repeating these analyses using the entire UKB cohort with available data (ie, including those with potentially confounding comorbidities that were excluded for the primary analysis) yielded very similar findings (Supplementary Table S6). There was no strong evidence that any blood markers were associated with time until PD diagnosis at a false discovery rate of <5% (Supplementary Table S7).
Mendelian Randomization
We used MR to determine whether the observational association between lower lymphocyte count and increased risk of incident PD might be a causal relationship. The genetic instrument for lymphocyte count consisted of 510 independent non-MHC autosomal SNPs (Supplementary Table S8). Collectively, these SNPs explained 5.37% of the variance in lymphocyte count in this sample and could be considered a strong instrument (F statistic = 45.3).
In the primary MR analysis (IVW), for each genetically estimated 1-SD decrease in lymphocyte count, the odds of PD were increased by 9% (OR = 1.09, 95% CI = 1.01–1.18, p = 0.02). The confidence interval for the IVW estimate overlapped those of the observational estimate. There was no evidence that unbalanced horizontal pleiotropy (whereby variants influence the outcome via pathways other than through the exposure) was biasing the IVW result (MR-Egger intercept = −0.0006, p = 0.76). There was evidence of substantial heterogeneity in the IVW estimate (Cochran’s Q = 642, p = 5.29 × 10−5), however, heterogeneity filtering did not alter the magnitude of the effect, and in fact increased the precision of the estimate (Supplementary Table S9). Standard sensitivity analyses and the “mixture of experts” approach yielded similar effect estimates with varying degrees of precision, collectively providing evidence that genetically lowered lymphocyte count may be a risk factor for PD (Figs 3 and 4, Supplementary Table S9). MR-PRESSO yielded an estimate of similar magnitude and precision to the primary analysis (OR = 1.09, 95% CI = 1.01–1.17, p = 0.03).
FIGURE 3:

Mendelian randomization (MR) estimates from various methods using the “Mixture of Experts (MOE) approach.” MR estimates are orientated such that they express the predicted effect on PD risk of each 1 standard deviation (SD) reduction in lymphocyte count. The y axis shows different MR methods and different approaches for filtering single nucleotide polymorphisms (SNPs) to be included in the genetic instrument (heterogeneity filtering [HF]). Note that we employed Steiger / directional filtering (DF) for the primary analysis and the MOE analysis. Estimates are colored and ordered by the “MOE” statistic, which is similar to an area under the curve statistic in that it quantifies that ability of a given MR method to distinguish a true effect from the absence of a true effect. MOE statistics closer to 1 indicate a higher likelihood that the given MR method will give an accurate estimate for the given dataset.
FIGURE 4:

Mendelian randomization (MR) analysis of the effect of lymphocyte count on Parkinson’s disease (PD) risk. Scatter plot showing per-effect-allele single nucleotide polymorphism (SNP) associations with lymphocyte count and the per-allele log odds ratio [OR] for PD. Note that to orient the MR effects in the same direction as the observational estimate, we reversed the effect directions for SNP associations with lymphocyte count such that a 1 unit increase on the x axis reflects a genetically-predicted 1 standard deviation (SD) reduction in lymphocyte count for each copy of the effect allele. The model fit lines indicate MR estimates from different MR methods.
Discussion
We used data from >300,000 individuals enrolled in the UKB to examine the association between blood markers of immune system function (full blood count, CRP, and albumin) and the risk of subsequent PD diagnosis. We found evidence that lower lymphocyte count was associated with increased risk of incident PD. This finding was robust in a range of sensitivity analyses and 2-sample MR, suggesting it was unlikely to be driven by observed confounding factors or reverse causation.
Several studies have assessed changes in leukocyte populations in PD after diagnosis. Established PD is associated with lower lymphocyte counts, driven by absolute reductions in CD4+ T-helper cells, CD19+ B-cells, and Treg cells4–6,28 In a study of 123 newly diagnosed patients with PD, the percentage of neutrophils and lymphocytes had positive and negative correlations, respectively, with Unified Parkinson Disease Rating Scale (UPDRS) motor scores.29 Flow cytometric analysis has demonstrated that leukocyte apoptosis is higher in patients with PD than in controls and is associated with dopaminergic deficits on single photon emission computed tomography (SPECT).30 However, these studies have examined immune markers in established PD and have not determined whether lower lymphocyte count influences PD risk or represents a consequence of accumulating PD pathology. Moreover, most individuals with PD in these studies are receiving dopaminergic medication, which could confound immunophenotypic patient/control differences.31–33 In contrast to previous studies relating blood counts to prevalent PD, the availability of baseline blood tests and longitudinal follow-up of PD diagnosis in this study enabled us to examine the association between leukocyte subsets and risk of incident PD.
Only one other study has explored the link between leukocyte subsets and risk of incident PD. In the Swedish Apolipoprotein-Related Mortality Risk cohort, higher lymphocyte count was associated with a lower risk of incident PD (HR = 0.74, 95% CI = 0.59–0.94).34 However, pre-existing health conditions that can influence blood counts were not excluded or adjusted for so the possibility of residual confounding remained. We took several steps to address bias and confounding in the observational study: we excluded 100,000 individuals with comorbidities, such as autoimmune disease and cancer, we corrected for potential confounders in the primary analysis (age, sex, ethnicity, and deprivation indices), and undertook sensitivity analyses (including a broader list of confounders, matching controls to cases in a 4:1 ratio for age and sex, and excluding extreme values more than 3 SDs from the mean, and including the entire cohort of individuals). Taken together, the association between lower lymphocyte count and higher risk of subsequent PD diagnosis does not appear to be driven by measured confounding factors. In addition, we show the association persisted after excluding individuals who underwent blood draw within 8 years of diagnosis, reducing the possibility of reverse causation.
We then used MR to further explore the nature of the association between lower lymphocyte count and higher risk of PD. We used the latest large-scale GWAS efforts to construct a strong genetic instrument explaining ~5% of the variation in lymphocyte count. The IVW MR estimate was commensurate with the observational effect size we observed (ORMR = 1.09, 95% CI = 1.01–1.18; ORObservational =1.18, 95% CI = 1.07–1.32). The MR-Egger regression intercept, which quantifies the degree to which net unbalanced horizontal pleiotropy may bias the IVW estimate, was close to null, suggesting that the main IVW estimate provided an unbiased estimate of the effect of lower lymphocyte count on risk of PD. Sensitivity analyses using a variety of MR estimators, heterogeneity filtering, and MR-PRESSO supported the magnitude of the IVW estimate, albeit with varying precision.
PD is known to have a long latency between biological onset and clinical presentation. As such, a proportion of the incident PD cases included may have been clinically incident hut biologically prevalent at the time of recruitment. We cannot be certain that lymphopenia precedes biological disease onset and it could therefore be a marker of preclinical PD. Alternatively, the association might arise as a result of a shared, potentially genetic, cause. Hence, we are limited in our ability to conclude that lower lymphocyte count mechanistically drives PD pathogenesis from this study. Nonetheless, there is evidence to support a mechanistic link between lower lymphocyte count and PD. Studies in patients with PD have demonstrated blood–brain barrier compromise in the vicinity of the midbrain and increased T-cells infiltrating affected brain regions.35 CD4+ T-cell counts in the amygdala correlate with activated microglia and alpha-synuclein pathology, suggesting a causative role for infiltrating T-cells in propagating neurodegeneration.36 Thus, reductions in peripheral lymphocyte counts in incident PD may reflect migration of T-cells into the central nervous system (CNS).
Strengths of our study include the large sample size derived from the UKB, the triangulation of various different sensitivity analyses, and the triangulation of the observational and MR results. The comprehensive phenotyping of individuals in the cohort allowed us to correct for multiple potential confounding factors.
Limitations of our study include the possibility of collider bias. UKB is a highly selected cohort and we analysed only ~65% of the recruited sample (due to exclusion of participants with missing covariates, prevalent PD cases, and those with comorbidities that can influence blood counts). However, despite selection leading to more favorable risk factor profiles, exposure-outcome associations in the UKB have been shown to be generalizable.37 Survival bias may distort observational study results. However, the cohort of participants we analyzed in the UKB were relatively young at study inclusion (see Table 1), which, in turn, is likely to minimize the impact of survival bias on effect estimates.38 We did not adjust for medication use, which could impact leukocyte subsets, although exclusion for comorbidities associated with use of immunosuppressant and immunomodulatory medication will have captured a significant proportion of this confounding. Due to lack of flow cytometric data in the UKB, we could not establish whether the observed association was driven by reductions in T-cells and/or B-cells. MR analysis precludes the identification of nonlinear exposure-outcome associations, although nonlinear mechanisms are not seen in other conditions in which lymphopenia influences outcome.39,40 Furthermore we used the age at PD report to determine approximate age at diagnosis - this is a proxy measure at best and reflects the first coded diagnosis of PD in linked healthcare records or by self-report (where both were available, the earlier date was used). This is not the same as the true date of diagnosis, and may be several years later. Although we tried to mitigate this concern by excluding individuals whose blood test date and date of PD report were close, we cannot exclude the possibility that for some individuals, the true date of PD diagnosis could differ considerably from the date of PD report.
In conclusion, we report that lower lymphocyte count is associated with higher risk of subsequent diagnosis of PD in a large UK cohort. The association remained robust to a range of sensitivity analyses. MR analyses suggested that this relationship may be causal and is not purely driven by confounding or reverse causation. Lower lymphocyte count, although lacking specificity as a biomarker in isolation, may enhance efforts to identify patients who are at the earliest (ie, preclinical) stages of PD. We cannot exclude the possibility that lower lymphocyte count may be a consequence of preclinical PD or that the association arises through a common aetiological factor. Further work is required to replicate these findings in other cohorts and to address the mechanisms underpinning this relationship.
Supplementary Material
Acknowledgments
This research has been conducted using the UK Biohank Resource. We thank the participants of the UK Biobank. We would also like to acknowledge the 23andMe Research Team (especially Dr Karl Heilbron) and the 23andMe research participants. The Preventive Neurology Units is funded by the Barts Charity. Melanie Jensen received support from the Isaac Schapera Trust and Benjamin Jacobs is an National Institute for Health Research (NIHR) Academic Clinical Fellow. The funding bodies had no role design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Potential Conflicts of Interest
The authors have no potential conflicts of interest to report.
Additional supporting information can be found in the online version of this article.
References
- 1.de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic diseases in the elderly research group. Neurology 2000;54:S21–S23. [PubMed] [Google Scholar]
- 2.Cheng H-C, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 2010;67:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gruden MA, Sewell RDE, Yanamandra K, et al. Immunoprotection against toxic biomarkers is retained during Parkinson’s disease progression. J Neuroimmunol 2011;233:221–227. [DOI] [PubMed] [Google Scholar]
- 4.Bas J, Calopa M, Mestre M, et al. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J Neuroimmunol 2001;113:146–152. [DOI] [PubMed] [Google Scholar]
- 5.Baba Y, Kuroiwa A, Uitti RJ, et al. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord 2005;11:493–498. [DOI] [PubMed] [Google Scholar]
- 6.Stevens CH, Rowe D, Morel-Kopp M-C, et al. Reduced T helper and B lymphocytes in Parkinson’s disease. J Neuroimmunol 2012;252:95–99. [DOI] [PubMed] [Google Scholar]
- 7.Niwa F, Kuriyama N, Nakagawa M, Imanishi J. Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson’s disease. Geriatr Gerontol Int 2011;12:102–107. [DOI] [PubMed] [Google Scholar]
- 8.Akil E, Bulut A, Kaplan I, et al. The increase of carcinoembryonic antigen (CEA), high-sensitivity C-reactive protein, and neutrophil/lymphocyte ratio in Parkinson’s disease. Neurol Sci 2015;36:423–428. [DOI] [PubMed] [Google Scholar]
- 9.Tan E-K, Chao Y-X, West A, et al. Parkinson disease and the immune system— associations, mechanisms and therapeutics. Nat Rev Neurol 2020;16:303–318. [DOI] [PubMed] [Google Scholar]
- 10.Saiki M, Baker A, Williams-Gray CH, et al. Association of the human leucocyte antigen region with susceptibility to Parkinson’s disease. J Neurol Neurosurg Psychiatry 2010;81:890–891. [DOI] [PubMed] [Google Scholar]
- 11.Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 2011;377:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017;546:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandres-Ciga S, Cookson MR. Alpha-synuclein triggers T-cell response. Is Parkinson’s disease an autoimmune disorder? Mov Disord 2017. Sep;32:1327. [DOI] [PubMed] [Google Scholar]
- 14.Hui KY, Femandez-Hemandez H, Hu J, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med 2018;10:eaai7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noyce AJ, Bestwick JP, Silveira-Moriyama L, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol 2012;72:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.San Luciano M, Tanner CM, Meng C, et al. Nonsteroidal anti-inflammatory use and LRRK2 Parkinson’s disease penetrance. Mov Disord 2020;35:1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams-Gray CH, Wijeyekoon R, Yamall AJ, et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov Disord 2016;31:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bycroft C, Freeman C, Petkova D, et al. The UKbiobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014;23:R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bandres-Ciga S, Noyce AJ, Traynor BJ. Mendelian randomization-A journey from obscurity to center stage with a few potholes along the way. JAMA Neurol 2019;77:7–8. [DOI] [PubMed] [Google Scholar]
- 21.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 2017;13:e1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vuckovic D, Bao EL, Akbari P, et al. The polygenic and monogenic basis of blood traits and diseases. Cell 2020;182:1214–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol 2013;178:1177–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charlett A, Dobbs RJ, Dobbs SM, et al. Blood profile holds clues to role of infection in a premonitory state for idiopathic parkinsonism and of gastrointestinal infection in established disease. Gut Pathog 2009;1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umehara T, Oka H, Nakahara A, et al. Differential leukocyte count is associated with clinical phenotype in Parkinson’s disease. J Neurol Sci 2020;409:116638. [DOI] [PubMed] [Google Scholar]
- 30.Lin W-C, Tsai N-W, Huang Y-C, et al. Peripheral leukocyte apoptosis in patients with parkinsonism: correlation with clinical characteristics and neuroimaging findings. Biomed Res Int 2014;2014:635923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna F, McLaughlin PJ, Lewis BJ, et al. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol 2002;132:34–40. [DOI] [PubMed] [Google Scholar]
- 32.Besser MJ, Ganor Y, Levite M. Dopamine by itself activates either D2, D3 or D1/D5 dopaminergic receptors in normal human T-cells and triggers the selective secretion of either IL-10, TNFα or both. J Neuroimmunol 2005;169:161–171. [DOI] [PubMed] [Google Scholar]
- 33.Carr L, Tucker A, Femandez-Botran R. In vivo administration of L-dopa or dopamine decreases the number of splenic IFN gamma-producing cells. J Neuroimmunol 2003;137:87–93. [DOI] [PubMed] [Google Scholar]
- 34.Yazdani S, Mariosa D, Hammar N, et al. Peripheral immune biomarkers and neurodegenerative diseases: a prospective cohort study with 20 years of follow-up. Ann Neurol 2019;86:913–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garretti F, Agalliu D, Lindestam Arlehamn CS, et al. Autoimmunity in Parkinson’s disease: the role of α-Synuclein-specific T cells. Front Immunol 2019;10:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kouli A, Camacho M, Allinson K, Williams-Gray CH. Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun 2020;8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batty GD, Gale CR, Kivimaki M, Deary IJ, Bell S. Comparison of risk factor associations in UK Biobank against representative, general population based studies with conventional response rates: prospective cohort study and individual participant meta-analysis. BMJ. 2020;368:m131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smit RAJ, Trompet S, Dekkers OM, et al. Survival bias in Mendelian randomization studies: a threat to causal inference. Epidemiology 2019;30:813–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Britanova OV, Putintseva EV, Shugay M, et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol 2014;192:2689–2698. [DOI] [PubMed] [Google Scholar]
- 40.Zidar DA, Al-Kindi SG, Liu Y, et al. Association of Lymphopenia with Risk of mortality among adults in the US general population. JAMA Network Open 2019;2:e1916526–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
UK Biobank data are available via application (https://www.ukbiobank.ac.uk/). Code is available at https://github.com/Wolfson-PNU-QMUL/PD_FBC. PD GWAS summary statistics which exclude UKB data can be obtained from https://pdgenetics.org/resources and an application to 23andMe https://research.23andme.com/dataset-access/.25 Blood cell trait GWAS summary statistics have been made publicly available by the authors at ftp://ftp.sanger.ac.uk/pub/project/humgen/summary_statistics/UKBB_blood_cell_traits/.