

Abstract
Chronic inflammation increases the risk of a number of cancers, including gastric, colon, and hepatic cancers. Conversely, tumors, similar to tissue injury, trigger an inflammatory response coordinated by the innate immune system. Cellular and molecular mediators of inflammation modulate tumor growth both directly and by influencing the adaptive immune response. Depending on the balance of immune cell types and signals within the tumor microenvironment, inflammation can support or restrain the tumor. Adding to the complexity, research from the past two decades has revealed that innate immune cells are highly heterogeneous and plastic, with variable phenotypes depending on tumor type, stage, and treatment.
The field is now on the cusp of being able to harness this wealth of data to a) classify tumors based on their immune makeup, with implications for prognosis, treatment choice, and clinical outcome; and b) design therapeutic strategies that activate anti-tumor immune responses by targeting innate immune cells.
Keywords: inflammation, innate immunity, immuno-oncology, cancer immunology, tumor microenvironment, plasticity
Introduction
Tumor development elicits a host response that resembles inflammation and is similarly coordinated by the innate immune system. Cellular and extracellular components engaged in this process shape tumor growth and progression by modifying the abundance and functions of one another, interacting with cancer cells, and modulating the adaptive immune response. Inflammation is a plastic process and ultimately, whether inflammation promotes or inhibits cancer depends on the balance of a complex and still incompletely understood cellular and molecular circuit [Fig. 1].
Fig. 1. Plasticity of the innate immune system.
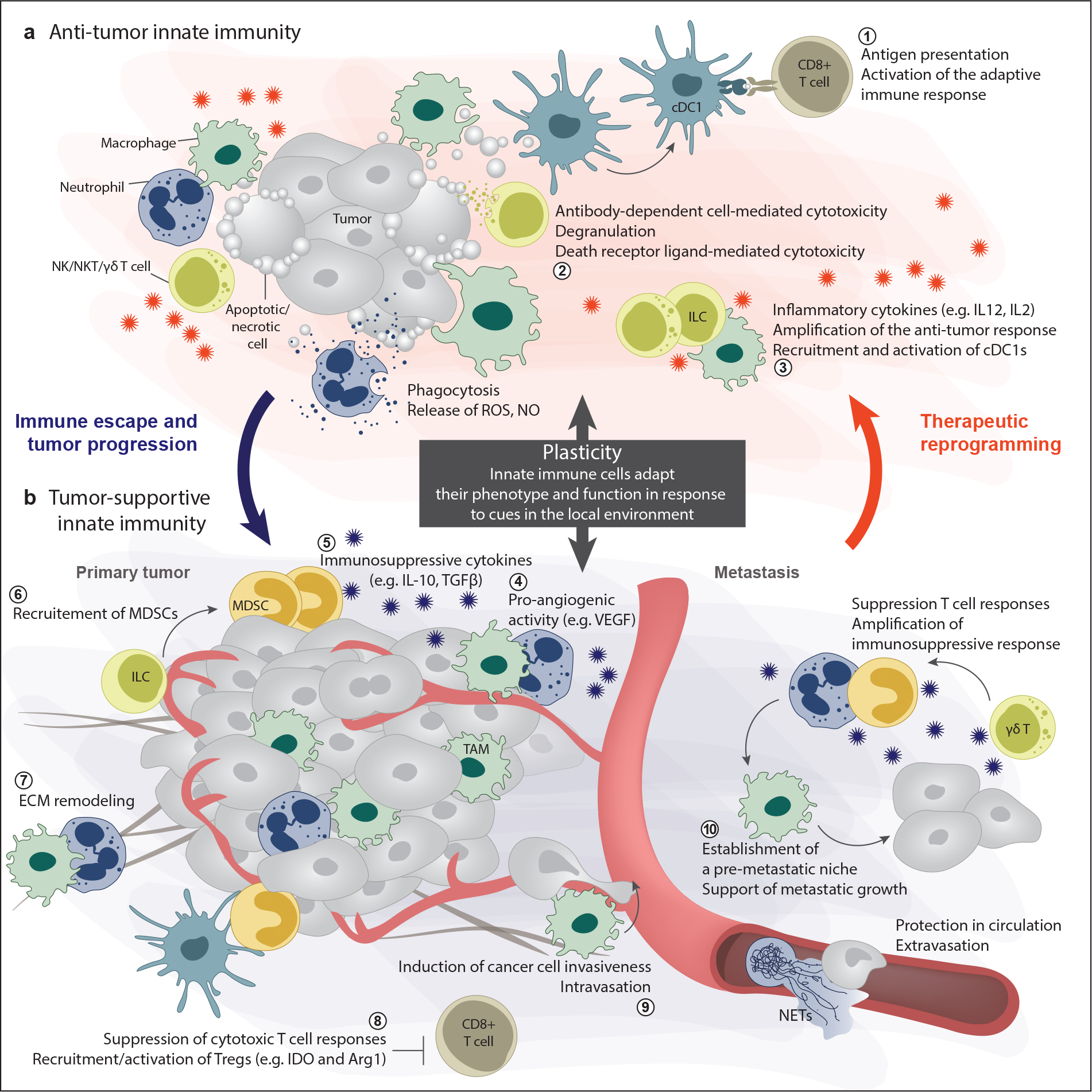
The components of the innate immune system are not inherently tumor-supportive or tumor-opposing. Rather, cells of the innate immune system are highly plastic and their phenotype and activity depend on the balance of signals within the tumor. (a) The anti-tumorigenic functions of the innate immune system include 1) antigen presentation and activation of the adaptive response, 2) direct killing of cancer cells, and 3) amplification of the anti-tumor immune response through cytokine secretion. (b) During tumor progression signals from tumor cells and other cells in the microenvironment can polarize innate immune cells towards supporting the tumor, e.g., through 4) angiogenesis, 7) ECM remodeling, 5, 6, 8) immunosuppression, and 9, 10) pro-metastatic activities. Thus, because of plasticity, the innate immune system has tumor-promoting potential. However, plasticity also affords us the opportunity to therapeutically reprogram the innate immune system to fight the tumor.
Abbreviations: DC, dendritic cell; ECM, extracellular matrix; IDO, indoleamine 2,3-dioxygenase; IL, interleukin; ILC, innate lymphoid cell; MDSC, myeloid-derived suppressor cell; NET, neutrophil extracellular trap; NK, natural killer; NKT, natural killer T; NO, nitric oxide; ROS, reactive oxygen species; TAM, tumor-associated macrophage; TGF, transforming growth factor; VEGF, vascular endothelial growth factor.
One of the earliest indications of a connection between immunity and cancer was the detection of immune cells in histological sections of tumors (1). Another clue came from clinical practice. In the 19th century, the German physicians Wilhelm Busch and Friedrich Fehleisen independently recognized that patients could experience cancer regression after contracting post-operative infections (2). This observation was further expanded upon by William B. Coley, an American surgeon, who, in the 1890s, began inoculating patients with bacteria, with the idea that the response to the pathogen could clear both the infection and the cancer. Eventually, he settled on “Coley’s toxins”, a formulation comprising heat-killed gram-positive Streptococcus pyogenes and gram-negative Serratia marcescens bacteria. The therapy had severe side effects, required daily inoculations, and beneficial responses were mostly limited to sarcoma, a rare cancer. However, Coley’s was the first concerted and broadly recognized effort to elicit an immune response against cancer (3, 4).
Early research focused mostly on inducing inflammation to elicit anti-cancer immunity; yet, the possibility that inflammation could also drive cancer was already suggested in 1828 by Jean-Nicolas Marjolin, a French surgeon, who reported the occurrence of squamous cell carcinoma in chronically inflamed wounds (5). In 1863, the German pathologist Rudolf Virchow proposed that cancer originates at sites of chronic inflammation (1). Starting in the 1990s, seminal work using genetically engineered mouse models demonstrated that innate immune cells such as neutrophils, macrophages, and mast cells contribute to cancer progression (6–13).
During the past decade, the clinical success of cancer immunotherapy has sparked renewed interest in the role of innate immunity in cancer. Although most approved strategies are aimed at boosting tumor-specific cytotoxic T cell responses, strategies to engage the innate immune system in anti-cancer responses are being developed. Moreover, sophisticated techniques to resolve gene expression and proteomic profiles at the single-cell level have allowed researchers to classify cancers based on their immune makeup: the type of immune cells present in the tumor, their phenotypes, and their location within the microenvironment. These studies have also highlighted the plasticity of innate immune cells, i.e., how their phenotype and function change in response to the microenvironment. Along with its genotype, the cancer immune makeup is refining our capacity to predict clinical outcome. Here, we review the contrasting roles of innate immunity in cancer and discuss the clinical application of these findings.
1. Inflammation and cancer are highly interconnected
Inflammation is a biological reaction the body mounts in response to infections, wounds, and chemical exposure, to restore homeostasis and prevent loss of tissue function (14, 15). The cells and molecules responsible for triggering and coordinating inflammation make up the innate immune system. Tissue-resident macrophages and mast cells are the first to recognize the insult. They secrete a variety of soluble mediators, cytokines and chemokines, to recruit other innate immune cells to the infection (or injury) site. Neutrophils are the first cells to respond to such signals, becoming activated and killing invading bacteria (16, 17). If this acute inflammatory response is not able to eliminate the insult, macrophages and T cells are next attracted to and activated by increased expression of chemokines, growth factors, and cytokines. This process is followed by a resolution and repair phase in which the local release of signals, e.g., resolvins, protectins, and transforming growth factor β (TGF-β), inhibits further neutrophil recruitment (18). Instead, monocytes are attracted to the site, where they differentiate into macrophages, remove dead cells, and initiate tissue repair mechanisms. This orchestrated immune response mediates neutralization of the offending agent. However, when the body is unable to resolve this acute inflammatory response, the result is chronic inflammation (19). In 1986, Howard Dvorak published an essay where he drew a parallel between tumors and “wounds that do not heal” (20). He argued that tumors invoke an inflammatory wound healing response similar to the one described above, creating favorable conditions for survival and growth [Fig. 2].
Fig. 2. Aberrant wound healing response, fibrosis, and cancer.
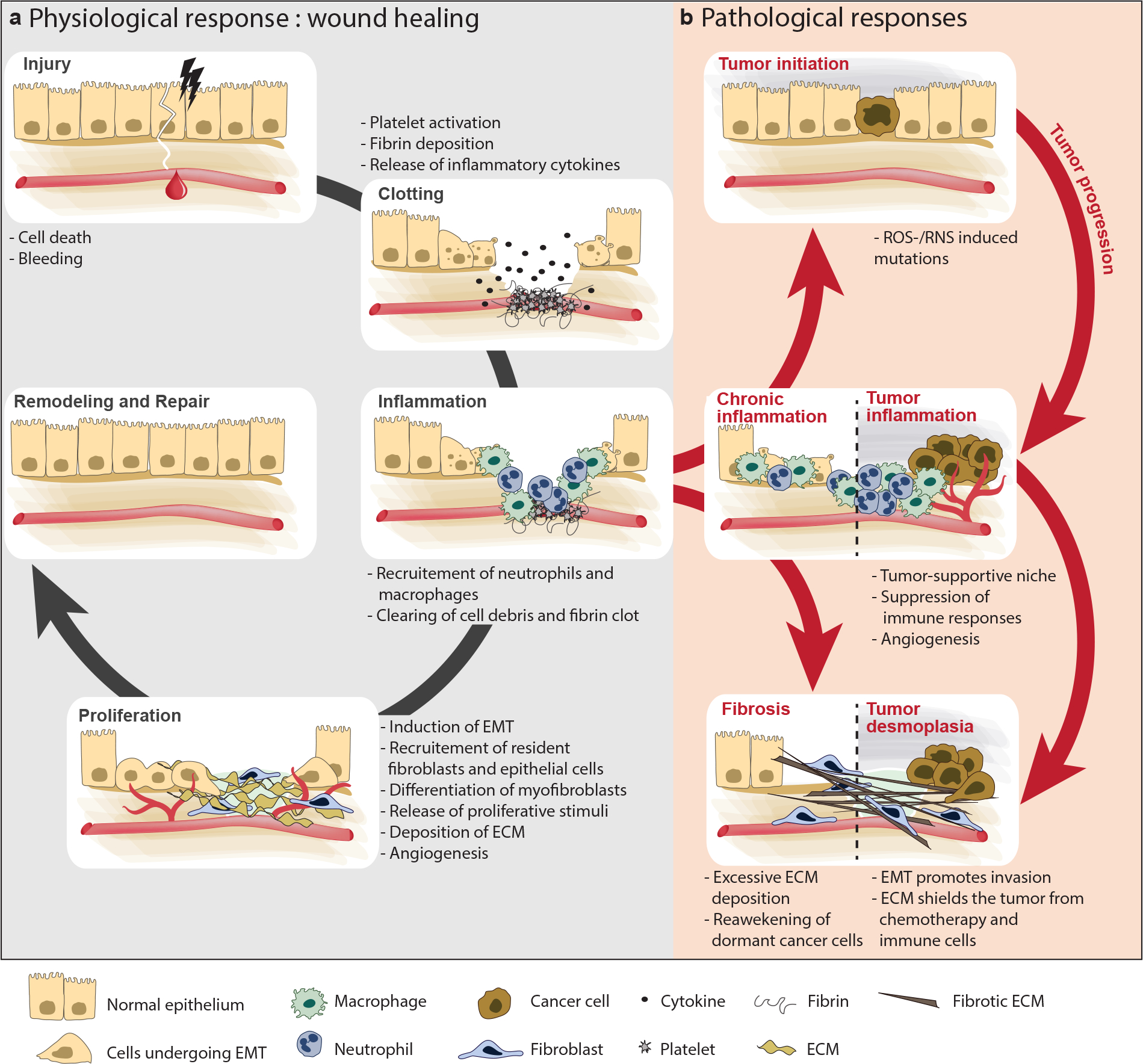
Partly owing to the activation of the innate immune system, features of the tumor microenvironment resemble an aberrant wound healing response. Wound healing consists of overlapping phases (left). Injury of adult tissue results in local hemorrhage, immediately followed by clotting. A temporary matrix of fibrin is deposited locally, which serves as a scaffold for migrating immune cells, epithelial cells, fibroblasts, and endothelial cells. During wound healing, neutrophils and macrophages kill bacteria, degrade the fibrin clot, and remove cellular debris. Neutrophils also secrete mediators such as TNF-α, IL-1β, and IL-6, amplifying the innate response. Macrophages produce VEGF and other growth factors, such as TGF-β, that stimulate the next phase: migration and proliferation of cells within the wound. In this phase blood supply is restored, new connective tissue is produced, and the wound re-epithelializes. Lastly, during the repair phase, the extracellular matrix is remodeled and new blood vessels are culled. Failure to clear the rich inflammatory infiltrate results in chronic inflammation (right). The persistence of inflammatory cells results in the accumulation of toxic compounds such as reactive oxygen and nitrogen species, as well as cytokines, which support tumor initiation and progression and sustain myofibroblast activation and fibrosis. Inflammation, fibrosis, and cancer are tightly linked in a vicious cycle, in which they can each trigger and aggravate the other.
Abbreviations: ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; IL, interleukin; RNS, reactive nitrogen species; ROS, reactive oxygen species; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Epidemiological data support the notion that chronic inflammation drives tumor development. Prospective studies have shown that patients displaying elevated levels of circulating inflammatory markers (e.g., C-reactive protein) at routine checkups have more than twice the risk of developing cancer within one year than those with normal levels (21). A 2018 study estimated that 42% of adult cancers in the United States are caused by modifiable risk factors (22), all of which cause either local or systemic inflammation. For example, cigarette smoking accounts for 19% of cancers, obesity is linked to 7.8%, alcohol intake explains 5.6% of cases, and chronic infections are the cause of 3.3% of cancers in the United States and 13% of cases worldwide (23).
Local inflammation can promote cancer development and progression within the same tissue or organ site (24, 25). Several infections can result in cancer: i) Helicobacter pylori-induced gastritis can progress to gastric cancers, ii) chronic hepatitis B or C virus infections can lead to liver cancers, and iii) unresolved infection with human papillomavirus can result in cervical cancers. Besides direct carcinogenic mechanisms associated with the infectious agents, the persistent inflammatory environment resulting from the failure to clear the infection contributes to the development of these cancers (26). Chronic inflammatory diseases in the absence of infections can also forge a local microenvironment that is primed for tumor development. For instance, inflammatory bowel diseases, such as Crohn’s disease and ulcerative colitis, increase the risk of developing colorectal cancer (27). Similarly, chronic pancreatitis carries an elevated risk of developing pancreatic cancer (28). Lastly, environmental factors can predispose patients to and promote cancer by causing local inflammation. Most notably, exposure to particulate or tobacco smoke has a well-defined relationship with chronic obstructive pulmonary disease development, which increases the risk of lung cancer (29).
In addition to changing the local inflammatory microenvironment, some insults such as tobacco smoke and obesity drive systemic inflammation. As a result, levels of circulating pro-inflammatory mediators are chronically elevated, leading to an increased risk of developing cancer in several organs (30). For instance, in a diet-induced mouse model of obesity, high levels of serum interleukin (IL)-5 and granulocyte-macrophage colony-stimulating factor (GM-CSF) cause lung inflammation and subsequent metastasis to this site (31). Consistent with inflammation’s pro-tumorigenic role, long-term treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with lower cancer incidence (32), including a notable decline in the incidence of lung cancer in chronic smokers (33). Moreover, a phase III trial (CANTOS; NCT01327846) of an inhibitory antibody targeting the pro-inflammatory molecule IL-1β for atherosclerosis also found that it significantly reduced lung cancer incidence (34), while a subsequent trial testing the anti-IL-1β antibody together with chemotherapy in established lung cancer found no effect (35). However, not all chronic inflammatory diseases increase the risk of cancer. For instance, psoriasis and rheumatoid arthritis do not appear to promote cancer, although it is possible that the drugs used to keep these inflammatory diseases in check also modify cancer risk.
Cancers not directly associated with inflammation still recruit innate immune cells, release cytokines, and exhibit angiogenesis and tissue remodeling—essentially driving the establishment of “tumor-intrinsic” inflammation (36) [Fig. 3a]. NSAIDs can also reduce mortality from some of these cancer types, e.g., prostate and brain cancers (32, 37).
Fig. 3. Interactions between cancer and the innate immune system.
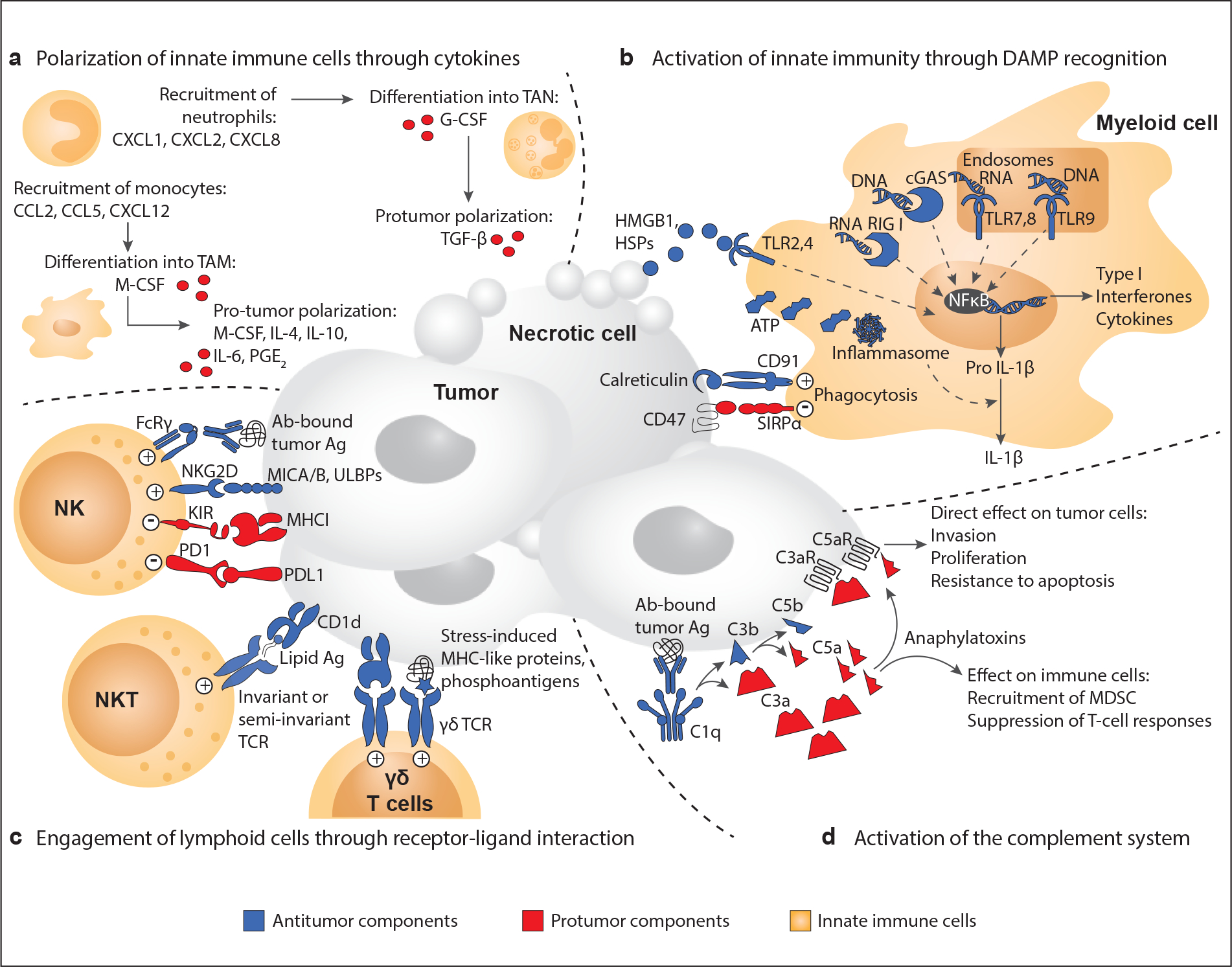
Cellular and molecular components of innate immunity interact with cancer cells through a variety of mechanisms that can support or restrain tumor growth. (a) By virtue of the high degree of plasticity of innate immune cells, cytokines secreted in the tumor microenvironment can polarize innate immune cells toward tumor-supportive phenotypes. (b) Necrotic cells release DAMPs, endogenous “danger signals” recognized by pattern recognition receptors (PRRs), e.g., Toll-like receptors (TLRs), on innate immune cells. DAMP sensing increases phagocytosis of the necrotic debris and amplifies the inflammatory response, leading to activation of the adaptive immune response. Opposite mechanisms also exist. For example, the binding of CD47 to SIRPα helps cancer cells escape phagocytosis, by transmitting a “don’t eat me” signal. (c) Innate lymphoid cells (ILCs and NK cells) and unconventional T lymphocytes (γδ T cells and NKT cells) can directly eliminate tumor cells by releasing cytotoxic granules or engaging death receptors. In NK cells, this cytotoxic activity is regulated by cancer cell ligands that bind either activating or inhibitory surface receptors. Unconventional T lymphocytes recognize cancer cells through their TCR. The γδ TCR allows for non-MHC-restricted recognition of e.g., phosphoantigens. The invariant or semi-invariant TCR on NKT cells binds lipid antigens presented on the non-polymorphic MHC-I-like molecule CD1d. (d) The complement system can mediate tumor cell lysis and phagocytosis by immune cells, e.g., by binding anti-cancer antibodies on the surface of cancer cells. By contrast, cleavage products of complement activation (C3a and C5a) can support tumor growth, either by directly affecting cancer cells or by recruiting immunosuppressive cells.
Abbreviations: DAMP, danger-associated molecular pattern; G-CSF, granulocyte colony–stimulating factor; HSP, heat shock protein; IL, interleukin; ILC, innate lymphoid cell; M-MDSC, monocytic myeloid-derived suppressor cell; M-CSF, macrophage colony stimulating factor; MHC, major histocompatibility complex; NK, natural killer; NKT, natural killer T; PGE2, prostaglandin E2; PMN-MDSC, polymorphonuclear myeloid-derived suppressor cell; PRR, pattern recognition receptor; SIRPα, signal-regulatory protein α; TAM, tumor-associated macrophage; TAN, tumor-associated neutrophil; TCR, T cell receptor; TLR, Toll-like receptor.
Drivers of tumor-intrinsic inflammation include genetic and epigenetic alterations in tumor-suppressor genes and oncogenes. The tumor-suppressor TP53 is a good example. In several cancer models and in clinical studies, immune cells are attracted to the primary tumor in response to P53 loss. For instance, in prostate cancer, loss of P53 triggers C-X-C chemokine motif ligand 17 (CXCL17) upregulation and the subsequent recruitment of immunosuppressive innate immune cells in the tumor microenvironment (TME) (38). Beyond this local effect, P53 loss in breast cancer stimulates tumor-associated macrophages (TAMs) to release high levels of IL-1β, driving systemic inflammation and ultimately supporting metastasis (39).
Another important cause of tumor-intrinsic inflammation is necrotic cell death, occurring when, e.g., rapidly growing tumors outpace the blood supply, resulting in hypoxia and necrosis. Necrotic cells release potent danger-associated molecular patterns (DAMPs), endogenous “danger signals” recognized by innate immune system cells via germline-encoded pattern recognition receptors (PRRs), e.g., Toll-like receptors (TLRs) (40). DAMP sensing increases phagocytosis of the necrotic debris and amplifies the inflammatory response so antigen-presenting cells (e.g., dendritic cells [DCs] and macrophages) can activate the adaptive immune response. High mobility group box 1 (HMGB1) is a nuclear non-histone-binding protein that serves as a DAMP by signaling through TLR3, -4, and -9, as well as the scavenger receptor RAGE (receptor for advanced glycation end products) [Fig. 3a]. HMGB1 binding to its receptors ultimately leads to inflammatory cell recruitment and induces the release of pro-inflammatory cytokines (41, 42). DAMPs are also important in eliciting the antigen-specific adaptive immune response evoked by immunogenic cell death of tumor cells. This is a unique form of cell death defined by its ability to elicit protective immunity and mainly caused by cytotoxic agents like anthracyclines and radiotherapy. Following immunogenic cell death, the chronic exposure of DCs to DAMPs, particularly calreticulin, ATP, and HMGB1, favors DC maturation and priming of protective T cell responses that can kill tumor cells and establish anti-tumor immunological memory (43). Supporting the importance of this mechanism for anti-tumor immunity, germline mutations in PRRs affect cancer risk and response to therapy. For example, patients with breast cancer who carry a TLR4 allele displaying reduced binding to HMGB1 relapse faster after chemotherapy (41) [Fig. 3b].
In summary, inflammation is a hallmark of cancer, whether it originated before or is driven by the cancer.
2. Innate immune cells in the tumor are heterogeneous and plastic
The innate immune system comprises immune cells of either myeloid or lymphoid lineage, and several classes of proteins, including cytokines, chemokines, receptors, and proteins of the complement system. These components are not inherently tumor-supportive or tumor-opposing. Rather, their activity depends on the relative abundance of each cell type in the specific tissue, the balance of signals within the TME, and the tumor progression stage [Fig. 1]. For instance, lung cancer cells engineered to express a strong antigen are rejected by cytotoxic T cells in the lung, but expression of the same antigen and a very similar genetic makeup in pancreatic cancer cells instead exacerbates the disease, as this site has fewer DCs capable of activating T cells (44). In addition, the phenotype and function of innate immune cells are plastic and change as their local environment changes. We present key findings that illustrate this context-dependence of innate immune cell types and how it affects cancer.
2.1. Myeloid cells modulate tumor-associated inflammation
Myeloid cells comprise heterogeneous cell populations derived from a common myeloid progenitor in the bone marrow. These cells are recruited to the tumor and can regulate the tumorigenic process, from initiation to invasion and metastasis. The most well-studied myeloid cells are macrophages/monocytes, neutrophils, myeloid-derived suppressor cells (MDSCs), and DCs.
2.1.1. Macrophages and monocytes
Macrophages are large phagocytic cells critical for host defense, especially against bacteria, but are also necessary for tissue homeostasis, e.g., by clearing cell debris and dysfunctional cells. Tissue-resident macrophages originate from cells seeded to tissues during embryogenesis, but macrophages can also expand from bone marrow-derived blood monocytes, which are recruited in large numbers in response to injury or infection (45).
Macrophages and their monocytic precursors constitute the largest fraction of leukocytes in most solid tumors and are critical drivers of cancer-associated inflammation (46, 47). Activated inflammatory macrophages produce potentially mutagenic reactive nitrogen species (RNS) and reactive oxygen species (ROS) and secrete cytokines, including tumor necrosis factor α (TNF-α) and interleukins (e.g., IL-1β, IL-6, IL-12), providing fertile soil for initiation and progression of chronic inflammation-associated cancers. In a model of colitis-associated cancer, constitutive genetic inactivation of the canonical nuclear factor κB (NF-κB) pathway, a master regulatory pathway of inflammation, specifically in myeloid cells (macrophages and neutrophils), results in downregulated inflammatory cytokine secretion and reduced tumor incidence (12). Furthermore, when stimulated by interferon-γ (IFN-γ) and TLR ligands, macrophages can directly kill tumor cells by generating nitric oxide (NO) (48). However, as the tumor progresses, cues in the microenvironment drive TAMs to become tumor-supportive. Early findings showed that this phenotypic shift could occur in response to IL-4/IL-13 derived from e.g., T lymphocytes (49). Yet, other cytokines (e.g., IL-10 and TGF-β), tumor cell-derived metabolic products, hypoxia, and immune complexes can also induce pro-tumorigenic polarization of TAMs (50–52). Much of the early literature on macrophages categorized anti-tumorigenic macrophages as classically activated/M1 and pro-tumorigenic macrophages as alternatively activated/M2. This dichotomy reconciled the antithetical roles that macrophages display in cancer, but it is now clear that a spectrum of phenotypes exists beyond the two extremes. Accordingly, the field has moved away from the binary nomenclature, favoring a more precise definition of populations based on how the cells are isolated and which markers define them (53). In 2018, high-dimensional profiling techniques like single-cell RNA sequencing and mass cytometry have allowed us to granularly characterize the heterogeneous macrophage populations in tumors and map their plastic evolution during disease progression or treatment (54).
TAMs’ ability to directly sustain tumor progression has been widely documented and reviewed [e.g., (47)]. In brief, TAMs are key players driving the acquisition of a tumor vasculature, the so-called “angiogenetic switch”. Tie2+ monocyte-derived macrophages are an essential source of vascular endothelial growth factor (VEGF) and support angiogenesis in several mouse models (13). In addition, intravital imaging of mammary tumors showed that perivascular TAMs aid cancer cells in entering blood vessels (55). But TAMs can also promote invasion by remodeling the extracellular matrix (ECM) through expression of proteases such as matrix metalloproteinases (MMPs) and by promoting the epithelial-mesenchymal transition of cancer cells (9, 56). At the metastatic site, macrophages can aid cancer cells to adapt to the new environment. For instance, in the lungs, α4 integrin on macrophages can serve as a receptor for vascular cell adhesion molecule 1 (VCAM-1) expressed on breast cancer cells and can activate pro-survival signaling (57). In addition, both monocytes (or monocytic MDSCs, see below) and macrophages reinforce the immunosuppressive TME by secreting cytokines, e.g., IL-10 and TGF-β, which inhibit the anti-tumor immune response (58).
A clinically relevant aspect of macrophage biology is its profound effect on treatment outcome. Depending on the treatment and their phenotype, TAMs can either contribute to or interfere with the therapeutic mechanism (59). For instance, in mice, macrophages capture therapeutic anti-programmed cell death protein 1 (PD-1) monoclonal antibodies (mAbs) from the surface of T cells, the intended target, thus blunting therapeutic efficacy (60). However, PD-1 is also expressed on TAMs, and PD-1 blockade in NOD scid gamma mice, which lack T, B, and natural killer (NK) cells, leads to a TAM-dependent decrease in tumor burden, suggesting that TAMs can be pharmacologically re-educated (61) and again highlighting the plasticity of these innate immune cells. Indeed, a colony stimulating factor 1 receptor (CSF-1R) inhibitor causes tumor regression by re-polarizing macrophages rather than simply depleting them in a mouse model of glioma (62). This inherent plasticity of macrophages represents both an obstacle in trying to untangle their complex biology and an opportunity for therapeutic targeting.
2.1.2. Neutrophils
Neutrophils are some of the first immune cells to be recruited to damaged tissues. They can eliminate pathogens by phagocytosis, by the release of antibacterial proteins and proteases, and by the formation of neutrophil extracellular traps (NETs) (63). Granulocyte-colony stimulating factor (G-CSF) and other cytokines that promote the differentiation and release of neutrophils from the bone marrow are often elevated locally in the tumor and systemically in patients with cancer, leading to the mobilization of high numbers of both mature and immature neutrophils (39). High levels of tumor-associated neutrophils and neutrophils in blood (neutrophilia) and high neutrophil-to-lymphocyte ratios are associated with poor prognosis in cancer (46, 64, 65). Like macrophages, neutrophils also have pro- or anti-tumor activities (66), depending on context. Yet, the stimuli leading to these opposite activities are much less defined than they are for macrophages.
Tumor-associated neutrophils can support cancer cell proliferation, angiogenesis, and immunosuppression in the TME (67). However, by harnessing their plasticity, they can become anti-tumor through TGF-β blockade (68). Similarly, IFN-β suppresses genes encoding homing and angiogenic factors in neutrophils, delaying tumor growth in melanoma and fibrosarcoma mouse models (69).
Early research showed that neutrophils from tumor-bearing animals could increase the invasive potential of cancer cells (70). For example, UV-damaged keratinocytes release HMGB1, resulting in a neutrophilic skin inflammatory response. In turn, neutrophil-derived TNF-α increases melanoma cells’ migration along blood vessels and consequently lung metastasis (71). Besides aiding cancer cells to escape the primary tumor, neutrophils can assist cancer cells in leaving blood vessels, e.g., by tethering cancer cells to liver sinusoids (72). During early stages of tumor progression, neutrophils are mobilized and accumulate at metastatic sites, where they help establish a premetastatic niche before cancer cells infiltrate (73–75). In the lungs of mouse mammary tumor virus–polyoma middle T antigen (MMTV-PyMT) tumor-bearing mice, neutrophil-derived leukotrienes support the preferential expansion of a highly metastatic subpopulation of cancer cells, and targeting this mechanism is sufficient to decrease metastasis (76). Lastly, at both the primary and metastatic sites, neutrophils (or the possible overlapping cell population sometimes referred to as polymorphonuclear MDSCs, see below) can drive immunosuppression (77). Contrasting these findings, in experimental metastasis models, neutrophils activated by inflammatory stimuli inhibit liver metastasis by releasing cytotoxic NO, and thrombospondin-1 released by neutrophils in the lung establishes a metastasis-resistant niche (78, 79).
In the last decade, neutrophil-expelled NETs were found to promote metastasis. NETs are extracellular networks comprising chromatin and granule-derived antimicrobial peptides and proteases, such as neutrophil elastase, cathepsin G, myeloperoxidase, and MMP9. NETs were first described as contributors to the innate immune response, with the ability to immobilize and eliminate large pathogens that could not be engulfed by phagocytosis (80). However, since 2013, NETs in the TME have been found to be associated with tumor progression both in animal models of cancer and in patients with cancer, including breast, ovarian, colorectal, or lung cancer (81–83). NETs can promote metastasis through multiple means. Our group and others reported that NETs can be chemotactic for metastatic cancer cells (82, 84). NET-associated DNA was later shown to serve as a chemotactic factor for cancer cells expressing the transmembrane protein CCDC25, which upon sensing DNA, enhances cell motility and facilitates metastasis to the liver (85). Besides increasing cancer cell migration/invasion, NETs can also trap cancer cells in the vasculature and facilitate extravasation (81). The presence of an early-stage ovarian tumor in the abdominal cavity induces neutrophils to accumulate in the omentum and release NETs in response to cell-derived factors (IL-8, G-CSF, CXCL1, and CXCL2), promoting the formation of a favorable premetastatic niche (83). Furthermore, we have shown that in the lungs, NET-associated proteases can induce cell proliferation through ECM remodeling (86). Specifically, neutrophil elastase and MMP9 cleave the basement membrane protein laminin, producing a cryptic epitope that activates integrin signaling and re-awakens dormant cancer cells. Lastly, NETs may physically shield cancer cells from cytotoxic immune cells (87). Based on these diverse pro-metastatic mechanisms, inhibiting NETs is thus a potential therapeutic strategy. Notably, targeting NETs in vivo with DNase I particles or a PAD4 inhibitor reduces metastatic burden in breast and ovarian cancers and inhibits the NET-activated awakening of dormant cancer cells (83, 84, 86).
2.1.3. Immature myeloid cells and myeloid-derived suppressor cells (MDSCs)
MDSCs are a heterogeneous population of immature myeloid cells that greatly expand during pathological conditions such as cancer. They are usually divided into two groups based on surface marker expression: monocytic MDSCs (M-MDSCs) and granulocytic or polymorphonuclear MDSCs (PMN-MDSCs). Morphologically and phenotypically, M-MDSCs and PMN-MDSCs are difficult to distinguish from monocytes and neutrophils, respectively, but they are functionally defined by their ability to restrain T cell activities. MDSCs expand in response to stem cell factors and cytokines like GM-CSF, G-CSF, M-CSF, IL-6, and VEGF. However, MDSCs’ immunosuppressive activity also requires activation by IL-4, IL-13, or TGF-β (88).
Unlike classical monocytes and neutrophils, MDSCs express high levels of molecules that inhibit T cell responses, including L-arginase, inducible nitric oxide synthase (iNOS), TGF-β, IL-10, cyclooxygenase-2 (COX2), and indoleamine 2, 3-dioxygenase (IDO). L-arginase and IDO are important suppressing factors that catabolize essential metabolites and/or produce toxic metabolites that accumulate in the TME, inhibiting T cell proliferation (89). NO production suppresses T cell function by inhibiting major histocompatibility complex (MHC) class II expression or inducing T cell apoptosis (90). Moreover, MDSCs have a dormant metabolic phenotype characterized by repressed glycolysis, which they can “pass on” to T cells by transferring the metabolite methylglyoxal, ultimately causing metabolic and functional paralysis of activated CD8+ T cells (91). A preclinical study found that depleting MDSCs after primary tumor resection delayed lung metastasis and extended survival, implicating them in metastatic progression as well. In this setting, low-dose adjuvant epigenetic therapy (5-azacytidine and entinostat) decreases MDSC trafficking, reducing metastasis (92). Consistent with the ability of MDSCs to repress T cell function, MDSC level is negatively associated with patient responses to immunotherapy, including to cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1 inhibition (93, 94). Targeting MDSCs may therefore represent an attractive therapeutic opportunity, but with the caveat that there is currently no clear method to distinguish MDSCs from neutrophils and monocytes—cell types with many complex functions.
2.1.4. Dendritic cells (DCs)
DCs are innate immune cells with a crucial role in bridging innate and adaptive immunity. DCs constantly scan their surroundings and detect antigens, danger signals, and invading pathogens. Once they take up antigens, they migrate to lymphoid tissues (thymus, spleen, and lymph node), where they present antigens via MHC class I and II complexes to stimulate antigen-reactive effector immune cells, primarily T cells and B cells (95). Traditionally, DCs have been divided into two major populations: conventional (or myeloid) DCs (cDCs) and plasmacytoid DCs (pDCs) (96).
cDCs are professional antigen-presenting cells, further classified as cDC1s and cDC2s, priming CD8+ or CD4+ T cells, respectively. They have the ability to “cross-present”, i.e., present tumor-derived MHC-I antigens to CD8+ T cells. In humans, cDC1s express CD141 and BDCA3. In mouse, the development of these cross-presenting DCs depends on the transcription factors Batf3 and Irf8, and tumor growth is increased both in Batf3−/− and Irf8−/− mice (97, 98). In lymphoid tissues, cDC1s are CD8α+, while in the periphery, they are identified by the expression of integrin αE, also known as CD103. Though they are a relatively rare population (often <1% of tumor-infiltrating immune cells), CD103+ DCs play a prominent role in recruiting and activating cytotoxic T cells. As antigen-presenting cells, cDC1s influence the breadth of the immune response, i.e., how many antigens are targeted. Adoptive cellular therapy with T cells expressing FMS-like tyrosine kinase 3 ligand (FLT3L), a DC growth factor, leads to cDC1-dependent expansion of the anti-tumor T cell repertoire (99). In a model of melanoma, CD103+ DCs were shown to be necessary for recruiting effector T cells intratumorally through CXCL9/10 expression (100). Accordingly, ablating CD103+ DCs thwarts tumor rejection after adoptive transfer of activated tumor-specific T cells in combination with immune checkpoint blockade (98, 101). cDC1s also influence chemotherapy response. For example, in the MMTV-PyMT breast cancer model, macrophages inhibit the secretion of IL-12 by CD103+ DCs and ablating macrophages restores DCs’ IL-12 production, increases T cell influx, and improves outcome (58). Exposure to chronic stress can inhibit the immunostimulatory activity of DCs, compromising the response to lung and colon cancer in mice. Stress causes elevated glucocorticoid levels, which repress the response to the important pro-inflammatory type I IFN cytokines in tumor-infiltrating DCs and curtail the immune response (102).
In contrast to cDCs, pDCs have limited antigen-presentating ability but are immunomodulatory via e.g., the production of type I IFNs (103). pDCs are largely immunoinhibitory in cancer, and pDC recruitment is associated with poor prognosis in several tumors, including ovarian and breast cancers (104, 105). However, OX40+ pDCs from patients with head and neck cancer can stimulate tumor-specific T cell responses (106). Dysfunction of pDCs or their acquisition of immunosuppressive properties are a result of e.g., tumor-derived IL-10, TGF-β, and TNF-α, which directly suppress pDCs’ IFN-α production (107, 108). In addition, by expressing IDO and inducible T cell costimulator ligand (ICOSL), pDCs support tolerogenic regulatory T cell expansion (109, 110).
DCs can tune the response between immune control or immune tolerance, whereby the tumor is recognized but not attacked. For instance, in inflammatory conditions, cDC2s activate anti-tumor CD4+ T cells, whereas in non-inflamed lymph nodes, they induce ineffective priming of CD4+ cells and a tolerogenic response (111). Single cell RNA sequencing (scRNA-Seq) of murine and human lung cancers revealed that upon antigen uptake, cDCs in the TME activate a transcriptional program that limits their immunostimulatory function and T cell priming. IL-4 signaling drives this program and IL-4 blockade abolishes its effects, supporting the notion that the cytokine milieu serves as a rheostat for DC activation and determines the outcome of the immune response (112).
2.2. Lymphoid cells and lymphocytes straddle immunomodulation and effector functions
Innate lymphoid cells and unconventional T lymphocytes [γδ T cells and natural killer T [NKT] cells) share attributes of innate and adaptive immunity. Besides contributing to cytokine secretion in the TME, they display effector functions, such as cytotoxic killing of cancer cells.
2.2.1. Innate lymphoid cells (ILCs)
ILCs have a lymphoid progenitor in common with T cells. However, they lack antigen-specific receptors, and their cytotoxic activity is instead regulated by soluble ligands that bind either activating or inhibitory surface receptors [Fig. 3]. Based on their cytokine-production pattern and the transcription factors required for their development, ILCs are categorized into five major groups: natural killer (NK) cells, group 1 ILCs (ILC1s), group 2 ILCs (ILC2s), group 3 ILCs (ILC3s), and lymphoid tissue-inducer cells (113, 114).
Like other immune cells, tissue type and cytokine milieu strongly influence whether ILCs are pro- or anti-tumor. For instance, administering IL-33 to 4T1 breast cancer-bearing mice results in accelerated tumor progression mediated by the accumulation of MDSCs and IL-13 producing ILC2 (115). In contrast, in pancreatic adenocarcinoma, IL-33-dependent expansion of ILC2s leads to therapeutic tumor immunity by recruiting CD103+ DCs and activating CD8+ T cells (116). IL-33 also triggers ILC1-dependent anti-tumor activity. In a mouse model of metastatic melanoma, IL-33 triggers ILC1 expansion and IL-5 upregulation in the lung, which in turn suppress lung metastasis via a mechanism that depends on eosinophil recruitment (117). ILC1s can also exert immune surveillance of early-stage tumors. In the MMTV-PyMT model of breast cancer, tumor initiation triggers an IL-15-dependent expansion of tissue-resident ILC1-like cells with cytotoxic activity against cancer (118). However, an ILC1-like phenotype can also curb anti-tumor immunity. For example, owing to the plasticity of NK cells, TGF-β can convert them into ILC1s, resulting in a decreased ability to restrain tumor growth (119, 120). Contrasting roles have been reported for ILC3s as well. In a murine model of melanoma, expression of C-C motif chemokine ligand 21 (CCL21) recruits C-C chemokine receptor 7+ (CCR7+) ILC3s with lymphoid tissue-inducing ability, which promotes lymphoid stroma formation and the establishment of an immunosuppressive, pro-tumorigenic milieu (121). In contrast, ILC3s from human lung cancer specimens can instead be polarized toward an inflammatory phenotype, which correlates with better clinical outcome. Interaction with tumor cells or tumor-associated fibroblasts triggers these ILC3s to produce inflammatory (TNF-α, IL-22) and chemotactic (IL-8, IL-2) cytokines, causing activation of the endothelium and potentially recruiting anti-tumor leukocytes (122).
2.2.1.1. NK cells
The role of NK cells in cancer was recognized decades before that of the other ILCs, when in 1980, Talmadge et al. showed that tumor cells transplanted to beige mice, deficient in NK cell activity, grew faster than in control mice (123). In humans, the presence of NK cell infiltrate or overexpression of NK-activating ligands by cancer cells correlates with better prognosis (124). In contrast, NK cell dysfunction, measured by flow cytometry analysis of cell-surface receptor expression and functional assays, predicts metastatic progression (125, 126).
NK cells can directly eliminate tumor cells by releasing cytotoxic granules or engaging death receptors (127). MHC-I molecules, expressed by most cells, serve as ligands for inhibitory receptors on NK cells, protecting healthy cells from being targeted. Cancer cells often lose MHC-I, especially when subjected to selective pressure by cytotoxic T cells, which rely on MHC-I to recognize their target. In such cases, the ability of NK cells to kill MHC-I-negative cells is an important fail-safe mechanism against immune escape (128). However, MHC-I downregulation is not the only signal that targets cancer cells for NK killing. Cancer cells also present ligands for NK cell-activating receptors (129). For instance, activating the DNA damage response in transformed cells leads to increased expression of ligands for the NK cell-activating receptor natural killer group 2D (NKG2D) (130). Genetic models lacking NK cell-activating receptors display impaired tumor immunosurveillance, supporting their important role in making cancer cells susceptible to NK killing. For instance, crossing NKG2D-deficient mice with a transgenic model of prostate cancer or a transgenic model of B cell lymphoma leads to the development of highly malignant, early arising cancer (131). Similarly, genetically ablating the natural cytotoxicity receptor NKp46 increases lung metastasis following transplantation of B16F10.9 melanoma or Lewis lung carcinoma cells (132). Under selective pressure, cancer cells can lose NK-activating ligand expression, leading to immune escape [e.g., (131, 133)].
Besides directly killing tumor cells, NK cells also amplify the anti-tumor immune response by secreting FLT3L, CCL5, and X-C motif chemokine ligand 1 (XCL1/2), which recruit cDC1s to the TME (134, 135). Activating cDC1s, in turn, can potentiate the NK cell response to the tumor. For instance, in the lungs of tumor-bearing mice, cDC1-derived IL-12 suppresses metastasis via an NK cell- and IFN-γ-dependent mechanism (136).
Similar to cytotoxic T cells, activated NK cells are also susceptible to so-called functional exhaustion, a state of decreased effector function. The activating cytokine IL-15 induces expression of the intracellular inhibitory molecule cytokine-inducible SH2-containing protein (CIS), which ultimately renders NK cells unresponsive to IL-15 in a negative feedback loop (137). In preclinical mouse models of metastasis, lung NK cells are activated by IL-12 but are also induced to upregulate checkpoint inhibitory receptors, e.g., PD-1, lymphocyte activating 3 (Lag-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT) (138). TIGIT expression was detected on tumor-infiltrating NK cells from patients with colon cancer and in several mouse models of cancer and was associated with dysfunction and reduced anti-tumor potential of these cells (139). In addition, NK cell activity is also restricted by modulated interleukin signaling. For instance, IL-1R8 negatively regulates ILR and TLR downstream signaling. IL-1R8-deficient mice display enhanced NK cell maturation and effector functions and are protected from liver cancer development and metastasis in the liver and lungs (140).
2.2.2. Unconventional T cells
2.2.2.1. γδ T cells
γδ T cells comprise 0.5–5% of all circulating T cells in healthy individuals and are most abundant in the gut mucosa. Instead of the classical T cell receptor (TCR) with α and β chains, these cells have a distinct TCR consisting of γ and δ chains. This TCR confers non-MHC-restricted antigen recognition [Fig. 3c]. The presence of intra-tumoral γδ T cells is associated with a favorable prognosis across many cancer types (46). Accordingly, in vitro studies have shown that γδ T cells are able to kill cancer cells via a) the granzyme-perforin pathway (141); b) expression of the death receptor ligand TNF-related apoptosis-inducing ligand (TRAIL) (142); and c) TNF-α and IFN-γ secretion (143). γδ T cells also induce enhanced cytotoxic activation of NK cells by engaging the co-stimulatory molecule 4–1BB (144). However, like other innate immune cells, γδ T cells are plastic and can acquire a pro-tumorigenic phenotype in response to soluble cues. Stimulation with IL-1β, IL-6, and IL-23 induces γδ T cell enrichment, producing the major immunosuppressive cytokine IL-17, with effects ranging from angiogenesis to immune escape (77, 145, 146). In pancreatic cancer, intratumoral γδ T cells express high levels of exhaustion-inducing ligands, leading to a curtailed adaptive response (146). In a mouse model of breast cancer, γδ T cell-secreted IL-17 and IL-1β stimulate neutrophil expansion and polarization (77). These neutrophils suppress CD8+ T cell function, which in turn facilitates the establishment of metastasis.
2.2.2.2. Natural Killer T (NKT) cells
NKT cells are a small subset of T lymphocytes that recognize lipid antigens presented on the non-polymorphic MHC-I-like molecule CD1d. This family is subdivided into Type I NKT cells or iNKT cells, presenting a semi-invariant TCR, and Type II NKT cells, with a variable TCR [Fig. 3c]. Most studies have found that type I and type II NKT cells play contrasting roles in cancer immunity, with the former promoting and the latter suppressing anti-tumor responses.
Much like NK cells, iNKT cells can directly kill cancer cells through non-antigen-specific cytotoxic mechanisms (147). iNKT cells can also target other cells expressing CD1d in the TME, including TAMs (148). In addition, iNKT cells support the efficient cross-priming of cytotoxic T cells by inducing DC maturation. For instance, iNKT stimulation with α-galactosylceramide, a high-affinity ligand for CD1d, leads to the upregulation of co-stimulatory molecules on DCs via CD40-CD40L interaction (149). DCs cross-primed by iNKT cells uniquely recruit T cells via the CCL17-CCR4 axis, potentiating anti-tumor immunity (150). Lastly, iNKT cell activation directly boosts cytotoxic cells, as exhausted NK cells and T cells can be rescued via iNKT cell-dependent production of IL-21, IL-12, and IL-2 (151). Conversely, an elegant genetic approach comparing CD1d-deficient mice (lacking all NKT cells) to Jα18−/− mice (deficient in iNKT cells only) showed that type II NKT cells suppress anti-tumor immunity in several mouse models (152). In fact, type I and type II NKT cells can be mutually antagonistic: when stimulated by specific ligands, type II NKT cells suppress the proliferation and cytokine production of type I NKT cells (153).
2.3. The complement system bridges innate and adaptive immunity
The complement system is a central part of the humoral arm of innate immunity. It comprises more than 50 plasma proteins, regulators, and receptors that serve as a first defense against pathogens and unwanted host molecules, besides mediating the effects of antibodies involved in a variety of activities, from regulating cytotoxicity to adaptive immunity and tissue homeostasis (154, 155). The complement system is activated by three major pathways (the classical, alternative, and lectin pathways) that converge into the cleavage of C3 into C3b. C3b binds to the surface of cells and marks them for phagocytosis by macrophages or neutrophils. After C3 is activated, C5 is cleaved and initiates assembly of the membrane attack complex, which accumulates on cell membranes and induces cell lysis. Furthermore, the cleavage products of C3 and C5—C3a and C5a—are chemokines with important inflammatory and chemoattractant functions [Fig. 3d].
The complement system’s activation can induce tumor cell lysis and phagocytosis by immune cells. In fact, the complement system mediates tumor cytolysis induced by rituximab, a chimeric anti-CD20 mAb developed to treat B cell lymphomas (156). Chemotherapy-induced immunogenic cell death (in breast cancer) activates signaling through the complement system, leading to a switch in B cell phenotype, which ultimately boosts anti-tumor immunity by increasing the ratio of effector T cells to regulatory T cells (157). However, in some settings, components of the complement system impair anti-tumor immunity instead. C5a generation suppresses CD8+ T cell responses by recruiting regulatory T cells and MDSCs and producing immunosuppressive molecules, e.g., L-arginase, IL-10, IL-6, CTLA-4, and PD-L1 (158, 159). Furthermore, C3a and/or C5a change the TME by recruiting TAMs, decreasing NK cell infiltration, and promoting NET formation (160–162). Another complement factor, iC3b, has been shown to promote IL-10 and TGF-β2 expression and MDSC generation (163, 164) to induce immunosuppression. Lastly, genetically or pharmacologically ablating the long pentraxin 3 (PTX3), which restrains complement activation, results in increased susceptibility to mesenchymal or epithelial carcinogenesis due to unleashing macrophage-mediated inflammation (165).
3. The TME modulates innate immunity
Besides cancer cells and immune cells in the TME, innate immune cell plasticity can be triggered by other cellular and non-cellular components, including the ECM, fibroblasts, and the microbiome.
3.1. Extracellular matrix (ECM)
The ECM is a three-dimensional scaffold of extracellular macromolecules, proteins, and polysaccharides that provides structural and biochemical support to cells. Several cell types, including fibroblasts, immune cells, and cancer cells, cooperate to produce, assemble, and modify the ECM.
Tumor progression is often accompanied by the deposition of a tumor-specific ECM, a typically stiffer and more fibrotic matrix characterized by higher levels of remodeled and cross-linked proteins (166) than regular ECM. The tumor-specific ECM can augment many hallmarks of cancer, such as resistance to cell death (167), induction of angiogenesis (168), and metastasis (169). In addition, the ECM modulates immune cell activation, polarization, and survival (170). A first layer of regulation comes from the physical and mechanical properties of the ECM. In human breast cancers, more aggressive subtypes display stiffer ECM and higher TGF-β, coinciding with higher macrophage infiltration, especially at the invasive front (171). Moreover, ECM stiffness and density regulate several immunoregulatory genes in macrophages. For example, when macrophage-like RAW 264.7 cells are cultured on high-density collagen matrix, their ability to inhibit T cell proliferation increases, while their ability to attract cytotoxic CD8+ T cells decreases (172). As a second layer of regulation, ECM components can be functional ligands of receptors on innate immune cells. As do many human tumors, Lewis lung carcinoma cells overexpress the matrix proteoglycan versican, which signals through TLR2 and stimulates macrophages to produce TNF-α, ultimately sustaining metastatic spread in mice (173). Collagen activates the immune-inhibitory receptor leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1), and overexpression of collagen by tumor cells impairs NK cell cytotoxic activity through LAIR-1 signaling (174).
Matrix remodeling is another layer of the ECM-mediated regulation of innate immune cells. Enzymes such as MMPs, members of a disintegrin and metalloproteinase (ADAM), and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) families cleave ECM proteins, producing peptide fragments, called matrikines, with immunomodulatory activity. For example, versikine, derived from versican proteolysis, enhances the differentiation of cross-presenting cDC1s from the bone marrow-derived precursor, thus promoting T cell anti-tumor immunity (175).
3.2. Fibroblasts
Within the TME, fibroblasts are a major producer of ECM and soluble factors. Normal fibroblasts can suppress tumor initiation and progression via direct cell-cell contact, secreting soluble factors and maintaining ECM integrity. However, with tumor progression, fibroblasts’ tumor-suppressive functions are lost. When normal fibroblasts turn into cancer-associated fibroblasts (CAFs) it triggers a range of tumor-supporting signals. As reviewed elsewhere, CAFs promote tumor growth by multiple mechanisms (176). One of these is driving an immunosuppressive microenvironment by secreting cytokines and chemokines that regulate the recruitment and functional differentiation of tumor-infiltrating immune cells. For instance, CAF-produced IL-6 and GM-CSF induce monocyte differentiation toward alternatively activated macrophage (M2)-like TAMs and promote metastasis in a colon cancer mouse model (177). Human colorectal cancer-derived CAFs can attract monocytes by secreting IL-8 and subsequently enhance the pro-tumor polarization of macrophages, and these macrophages in turn suppress NK cell cytotoxicity and activation (178). Tumor-derived CSF-1 can repress CAFs’ expression of chemokines that attract PMN-MDSCs. Therefore, an unwanted effect of CSF-1R inhibitors, used to block recruitment of macrophages into tumors, is the expression of PMN-MDSC-recruiting cytokines by CAFs, resulting in PMN-MDSC accumulation, and thus reducing the therapeutic benefit of CSF-1R inhibitors (179). Furthermore, CAFs induce infiltration of IDO-producing DCs, PDL1+ neutrophils, and regulatory T cells, while decreasing the infiltration and abrogating the functions of NK cells (180–183). In pancreatic adenocarcinoma, besides inflammatory fibroblasts (αSMAlowIL-6high) with a secretory phenotype and myofibroblasts (FAP+αSMAhigh) that secrete ECM, a novel population of CAFs was shown to express MHC-II and CD74, adding a potential immunomodulatory role as antigen-presenting cells (184). FAP+ fibroblasts inhibit immunological control of tumors: CXCL12 derived from FAP+ CAFs can be captured on the surface of cancer cells and mediates the exclusion of cytotoxic lymphocytes from the tumor bed (185, 186). Inhibiting CXCR4, the CXCL12 receptor, sensitizes the tumors to checkpoint inhibition and in patients with cancer produces an integrated response, recruiting adaptive and innate immune cells (185, 187).
3.3. Microbiome
As exemplified by Coley’s toxin, bacterial components can activate immunity and restrict cancer development. Microbes within a human organism interact with the host at numerous sites, like skin and mucosal surfaces. These microbes are not inert, but regulate innate and adaptive immunity, thus exerting a major influence on health and disease (188). Dysbiosis—a disruption in the homeostasis of microbial communities—has been linked to carcinogenesis. Indeed, large case-control studies have demonstrated that prolonged antibiotic use is an independent risk factor for cancer occurrence (189). Besides antibiotics, eating a high-fat diet also induces changes in the intestinal microbiome, favoring tumor progression in gastrointestinal cancer (190).
Microbiota can promote distinct inflammatory responses, thereby indirectly supporting tumor development. For instance, in a mouse model of colorectal cancer, erosion of the intestinal epithelial barrier favors entry of microbial products into the TME of early-stage lesions. This entry leads to the activation of IL-23-producing myeloid cells and enrichment of tumor-promoting cytokines, including IL-17 and IL-6 (191). Immunoregulatory effects of the intestinal microbiota, however, extend beyond the local environment, since other organs, e.g., the liver, can be exposed to the gut microbiome and its metabolites through the circulation. In mouse models of liver cancer, bile acids metabolized by gram-positive bacteria in the gut circulate back to the liver and downregulate expression of CXCL16 on endothelial cells, inhibiting the CXCL16-mediated recruitment of NKT cells and tumor control (192).
Besides the gut, bacteria and even yeast can reside within the TME of other tissues, e.g., lung and pancreas (145, 193, 194). In a genetically engineered mouse model, lung cancer development is decreased in germ-free or antibiotic-treated mice vs. specific pathogen-free mice (145). At the molecular level, the lung microbiota activates myeloid cells to release IL-1β and IL-23, thereby inducing IL-17 production from γδ T cells and ultimately inflammation and tumor cell proliferation (145). Analogously, the pancreas microbiota generates a tolerogenic environment by inducing an immunosuppressive phenotype in macrophages and monocytes in a TLR signaling-dependent fashion (193).
Aside from contributing to cancer development, pioneering studies in mice revealed that response to immunotherapy in mouse models of melanoma depends on the composition of the intestinal microbiota. Modifying the microbiome, e.g., by administering specific bacteria or performing fecal transplant, re-sensitizes non-responders, leading to increased DC function and enhanced CD8+ T cell priming (195, 196). These findings spurred clinical trials in which patients with previously immunotherapy-refractory melanoma experienced partial and complete tumor regression when the treatment was repeated after fecal transplant from responsive patients (197, 198). Further studies will need to define the distinct mechanisms responsible for the microbiota’s effect on anti-tumor immunity, as well as define more scalable treatment options to modulate the microbiome.
4. Multidimensional approaches to studying innate immune cells in the TME
While the first decades of TME research, including on innate immune cells, relied heavily on mouse genetics and cell biology to dissect the role of single cell types or pathways, recent advances have also been made using system-level approaches. The field has benefitted from -omics analyses, which produce large-scale biological datasets capturing e.g., the entire complexity of a sample’s mRNAs, proteins, or lipids. Initially focused on cancer cells, these approaches have now been extended to study the complexity of the immune infiltrate: the relative abundance of cell populations, diversity of phenotypes, and activation status. A crucial advance was the development of deconvolution techniques capable of estimating the abundance of different immune cell types in a sample from bulk transcriptomics data (199). An early large-scale analysis applied one such computational tool, CIBERSORT, to data from >27,000 patients across 25 cancers and revealed that intratumoral neutrophils are the leukocyte population most significantly associated with an adverse prognosis (46). Another group used RNA-Seq and flow cytometry to identify immune subtypes of triple negative breast cancer (TNBC) from mouse models and clinical datasets. Focusing on two myeloid compartments, they discovered that tumors with a macrophage-enriched microenvironment respond to checkpoint blockade, especially upon macrophage depletion. In contrast, the absence of immune infiltrate or the local and systemic accumulation of neutrophils correlates with a lack of immunotherapy response (200).
Additional improvements in high-throughput techniques have led to characterizing the TME at single cell resolution: capturing the heterogeneity within one cell type, including innate immune cells. Compared to scRNA-Seq, which informs on thousands of genes per cell, single cell proteomics protocols are not yet quite as powerful. Immune cells have historically been cataloged by extracellular markers; however, techniques like mass cytometry, which can probe tens of protein markers, including intracellular markers, have proven particularly informative in the field of cancer immunology. Integrating these approaches in multi-omics studies that yield single cell-level measurements for both proteins and transcripts promises to further amplify our ability to characterize the TME. Paired scRNA-Seq and mass cytometry of the immune compartment in early-stage lung tumor specimens revealed that cross-presenting CD141+ DCs (cDC1s) and NK cells are already depleted in early lesions compared to normal lungs. Stressing the importance of the single cell resolution, macrophages were as abundant as in normal tissue, but had a more pro-tumorigenic phenotype in tumors, with high expressions of IL-6 and the immune suppression-associated transcription factor peroxisome proliferator-activated receptor γ (PPARγ) and low expression of the co-stimulatory molecule CD86 (201). Later, the same group used scRNA-Seq to discover a cluster of DCs that had gone undetected by bulk methods, named “mature DCs enriched in immunoregulatory molecules” (mregDCs). The transcriptional program associated with mregDCs is activated upon antigen uptake and, depending on IL-4 signaling, can either enhance or inhibit DC immunostimulatory activity, making it an attractive candidate for clinical intervention (112).
Immunophenotyping tumors is further aided by topological techniques that collect complex data on tumor tissue sections, thus preserving spatial information. A major advantage of this analysis is that it can be applied to archival tissues, such that immune phenotype can be retrospectively correlated with outcome. For instance, a highly multiplexed version of traditional immunohistochemistry techniques was optimized to probe 12-antibody biomarker panels for lymphoid and myeloid cells on specimens from patients with pancreatic ductal adenocarcinoma. Analyzing the leukocyte infiltrates in intratumoral regions revealed that therapeutic response to neoadjuvant GVAX therapy correlates with a rich myeloid infiltrate, but not with lymphoid infiltrate (202). Methods that further increase multiplexing or dimensionality are also available. A good example is Multiplexed Ion Beam Imaging by Time of Flight (MIBI-TOF), a method that leverages mass spectrometry to image at subcellular resolution large samples (up to 1 mm2) labeled with isotope-conjugated antibodies. By integrating this technique with sophisticated digital segmentation of the images, one group was able to spatially locate 36 proteins and reconstruct the microenvironmental architecture of TNBC samples. One important takeaway from the study is that tumors with equally abundant immune infiltrates can vary dramatically in the compartmentalization between tumor cells and immune cells (203). A similar conclusion was reached in another TNBC study, where researchers identified four immune subtypes based on the spatial distribution of CD8+ T cells and gene expression profiles from laser capture microdissection of stroma and epithelium. The subtype associated with the poorest outcome had unexpectedly high infiltration of CD8+ T cells, but these were restricted to the stroma and accompanied by elevated levels of IL-17-producing cells and neutrophils (204).
As the field moves forward, we must integrate immune phenotyping obtained from multi-dimensional approaches with the other parameters that guide decision-making in the clinic to improve outcomes. Initiatives such as The Human Tumor Atlas Network, which will link multiparametric analysis of tumor samples with patients’ clinical information, promise to greatly accelerate progress (205).
5. Innate immune cells can be targeted for cancer therapy
Immunotherapies boost the immune system’s ability to fight diseases like cancer. They include antibody-based approaches, adoptive cellular therapies (including engineered immune cells), cancer vaccines, cytokine therapy, and small molecules targeting signaling pathways in immune cells. Although the approach of activating the immune system traces back to Coley’s work more than 100 years ago, it took off in earnest with the FDA approval of immune checkpoint inhibitors targeting CTLA-4 in 2011 (206). The majority of approved immunotherapies activate adaptive immune cells, i.e., B cells and CD4+ and CD8+ T cells, which are excellent targets because of their “memory” function. Nonetheless, innate immune cells can greatly influence adaptive immune responses. This potential is demonstrated by Bacillus Calmette-Guérin (BCG), a vaccine containing an attenuated Mycobacterium strain that has been used to treat non-muscle invasive bladder cancer for over 40 years. Intravesical administration of BCG activates innate immune cells through PRR signaling and subsequently leads to potent cytotoxic responses against the tumor (207). Approaches to reprogram, deplete, or reduce the recruitment of innate immune cells with immunosuppressive functions are therefore areas of very active investigation.
5.1. Macrophages: removed or reprogrammed
One strategy to target macrophages is to block their recruitment to tumors. CCL2 and the cytokine CSF-1 promote tumor infiltration of monocytes and their maturation to TAMs. Genetically ablating CCL2 or CSF-1 in mouse models limits tumor progression (11, 208). Several antibodies (carlumab, RG7155, AMG 820) and small molecules (PF-04136309, PLX3397) targeting CCL2 and CSF-1, or their receptors CCR2 and CSF-1R, have been studied in clinical trials. CCL2 inhibition correlates with reduced tumor progression and metastasis in breast cancer mouse models (209). A phase II study of carlumab (CNTO 888; a humanized antibody that binds CCL2) in metastatic castration-resistant prostate cancer did not show anti-tumor activity as a single agent (210). In contrast, the CCR2 small inhibitor PF-04136309 combined with the chemotherapeutic regime FOLFIRINOX resulted in an objective tumor response in 16 of 33 patients with pancreatic cancer (211). However, results of randomized, double-blinded trials targeting CCR2 have not yet been reported. PLX3397, a CSF-1R inhibitor, was tested in phase I and II trials in advanced tenosynovial giant cell tumors, where it was well tolerated, and 12 of 23 patients showed an anti-tumor response after treatment (212). Yet, when PLX3397 was tested in a phase II study in patients with recurrent glioblastoma, it showed no efficacy (213). The antibody RG7155 (emactuzumab) blocks CSF-1R dimerization and showed promising results in a phase I trial: 86% of the 28 patients achieved an objective response and 7% achieved a complete response (214). AMG 820, an anti-CSF-1R antibody, was recently tested in combination with pembrolizumab in patients with colorectal, pancreatic, and non-small cell lung cancer. The combination therapy showed on-target pharmacodynamic effects, such as CSF-1 accumulation in the serum, and had an acceptable safety profile, but no efficient anti-tumor responses were observed (215).
A recent study took advantage of myeloid cells’ propensity to home to metastatic niches and genetically manipulated myeloid cells to deliver IL-12. Upon homing to the metastatic niches, these IL-12-genetically engineered myeloid cells (IL-12-GEMys) elicited a strong anti-tumor immune response by activating endogenous T and NK cells and modulating the metastatic TME, resulting in greatly reduced lung and liver metastasis in mice (215a).
Strategies exploiting the plasticity of macrophages—attempting to reprogram or re-polarize them from pro-tumor to anti-tumor—are also being explored (45). Re-polarizing macrophages theoretically has the advantage of leading to the activation of cytotoxic effector immune cells (e.g., NK and T cells) by producing cytokines (including TNF-α, IL-6, and IL-12). The most straightforward method to re-polarize macrophages toward an anti-tumor phenotype is by activating PRRs, like TLRs. Synthetic ligands for diverse PRRs, many initially developed as vaccine adjuvants, are now being tested as cancer immunotherapies (216). The topical administration of the TLR7 ligand imiquimod has anti-tumor activity in superficial basal cell carcinoma and breast cancer skin metastasis (217, 218). The TLR9 ligand IMO-2055 combined with targeted and anti-angiogenic therapy in patients with non-small cell lung cancer showed good tolerability and possible anti-tumor activity (219). Motolimod, a small molecule targeting TLR8, is in clinical trials for advanced cancers (220). We are exploring how the combination of monophosphoryl lipid A (MPLA, a TLR4 agonist) with IFN-γ can reprogram TAMs to upregulate TNF-α, IL-12, and iNOS expression; decrease CD206 expression; and activate T cells in breast and ovarian cancer mouse models. The result is significantly reduced primary tumor growth and metastasis (48).
COX2 inhibitors, histone deacetylase (HDAC) inhibitors, phosphoinositide 3-kinase γ (PI3Kγ) inhibitors, and stimulator of interferon genes (STING) agonists are also used to reprogram TAMs. COX2 inhibition increases TNF-α, IL-12, and iNOS expression, shifting the macrophages to an anti-tumor phenotype (221, 222). In a phase I clinical trial for the treatment of docetaxel-resistant prostate cancer, patients treated with a COX2 inhibitor (celecoxib) and an epidermal growth factor receptor (EGFR) inhibitor (gefitinib) showed reduced tumor growth and invasion (223). HDAC inhibitors modify the epigenetic profile of monocytes and macrophages, resulting in altered gene expression and polarization toward an anti-tumor phenotype. In a mouse model of breast cancer, the HDAC inhibitor TMP195 induced the recruitment and differentiation of monocytes/macrophages into highly phagocytic and immunostimulatory cells, resulting in reduced tumor burden and metastasis and increased response to chemotherapy and immunotherapy with anti-PD1 antibodies (224). In mouse models of head and neck squamous cell carcinoma and breast cancer, selective inactivation of PI3Kγ in macrophages promoted an immunostimulatory transcriptional program in TAMs and in turn restored CD8+ T cell activation and cytotoxicity, leading to increased survival (225). A PI3Kγ inhibitor IPI-549 (eganelisib) is currently being tested in multiple phase I/II clinical trials (NCT03719326, NCT03980041, NCT03961698, NCT03795610). Finally, targeting STING induced TAM re-polarization in vitro; increased IFN-γ, iNOS, and IL-12 production; and led to promising results in mouse models (226). Different STING ligands are currently in clinical trials as sensitizers for multiple immunotherapies (227).
5.2. DCs: boosting presentation of tumor antigens
DC-based therapies utilize patient-derived DCs, generally produced by isolating circulating DCs or monocytes from the patient’s blood. Monocytes are then differentiated into monocyte-derived DCs by culturing them with GM-CSF and IL-4, and these DCs are further matured with a cocktail of substances, e.g., IL-6, TNF-α, IL-1β, prostaglandin E2 (PGE2), and polyinosinic:polycytidylic acid [poly(I:C)] (228). Maturation—involving enhanced expression of MHC-I, MHC-II, and co-stimulatory molecules and increased cytokine production—is essential, as incompletely matured DCs can induce tolerance rather than immunity (229). DCs have also been engineered using gene-editing technologies, like RNA interference, viral transduction, and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9, to promote their maturation (230, 231). Sipuleucel-T, a DC-based therapy approved in 2010 for castration-resistant prostate cancer (232), consists of matured and tumor antigen-loaded patient-derived DCs. Patients with melanoma have also been treated with ex vivo activated cDCs loaded with the tumor-associated antigens of tyrosinase and glycoprotein 100 (gp100) (233).
Various approaches have also been used to boost cDC mobilization to tumors, e.g., administering FLT3L (234) or modifying DC activities within the TME. The latter approaches include using TLR ligands, such as intra-tumoral injection of TriMix mRNA (three mRNA molecules encoding for CD70, CD40L, and constitutively active TLR4) (235), oncolytic viruses (236), self-replicating IL-12 RNA encapsulated in oncolytic nanoparticles (237), CD40-TLR4 agonists (238), or STING agonists (239). Such approaches have resulted in lasting systemic antigen-specific T cell immunity and tumor regression in many preclinical models, including of melanoma, breast carcinoma, colon cancer, and lung cancer (235, 237–239).
5.3. NK cells: a new frontier of cytotoxic immunotherapy
Different approaches have been investigated to engage NK cell function to treat cancer, including immunomodulatory cytokines, mAbs, and adoptive transfer of engineered NK cells. The most prominent cytokines used in NK cell activation are IL-2 and IL-15, both known to induce the expansion and increased cytotoxicity of NK cells (240). IL-2 treatment, the first approved immunotherapy, was approved to treat patients with metastatic renal cell carcinoma and melanoma (241). Moreover, the adoptive transfer of autologous or allogeneic NK cells is also used to improve NK cell tumor surveillance. mAbs have been designed to either block the interaction between the inhibitory NK cell receptor and its corresponding ligand on the cancer cells or engage activating receptors on NK cells. For example, lirilumab targets the inhibitory killer cell immunoglobulin-like (KIR) receptor and is in clinical trials for treating acute myeloid leukemia, multiple myeloma, lymphoma, chronic lymphocytic leukemia, and Hodgkin lymphoma (242). Another inhibitory receptor on NK cells, NKG2A, can be targeted by monalizumab, resulting in both enhanced NK cell activation and T cell function (243, 244). Recombinant reagents that increase the specificity and efficacy of NK cell activation—named bispecific and trispecific killer cell engagers (BiKEs and TriKEs, respectively)—are also in development. BiKEs bind to a surface tumor antigen, e.g., the highly expressed CD30 on Hodgkin lymphoma, and to an NK cell receptor, e.g., CD16, to trigger NK cell-mediated toxicity of the cancer cells (245). A TriKE has been designed to target CD33-positive hematological malignancies: it engages CD33 on the cancer cells; contains a CD16-heavy chain to activate NK cells; and also contains an IL-15 molecule to drive NK cell priming, expansion, and survival. This TriKE is currently in phase I clinical trials (NCT03214666) (246). Recently, NK cells have also been engineered to contain chimeric antigen receptors (CARs), which enables them to be directed against specific targets (247), much like similarly engineered CAR-T cells. Adaptive transfer with CAR-NK cells has shown promising results in preclinical models of B cell lymphoma and is currently being evaluated in a phase I/II study (248).
5.4. Neutrophils: an underrated target
There are currently no approved therapeutics to inhibit neutrophil activity in cancer. However, strategies to limit neutrophils’ pro-tumorigenic functions are being tested in early stage trials. These strategies include blocking CXC chemokine receptor 2 (CXCR2), which is critical for neutrophil recruitment (249). The CXCR2 antagonist AZD5069 markedly reduced neutrophils in a phase II clinical trial in patients with asthma and is now being evaluated in patients with advanced tumors (NCT02583477, NCT02499328, NCT03177187). In addition, the small molecule inhibitor reparixin, a non-competitive CXCR1/CXCR2 antagonist, was safe and tolerable in a phase Ib trial for HER-2-negative metastatic breast cancer (NCT02001974) (250). Neutrophils and granulocytic MDSCs can suppress anti-cancer immune responses, in part via arginase-1 activity (251). The combination of arginase-1 inhibitors with various chemotherapies or anti-PD-1 is in phase I and II clinical trials (NCT02903914, NCT03361228, NCT03314935).
6. Conclusion
Inflammation is an integral part of cancer pathology. Inflammation can favor cancer development, and cancer, in turn, elicits an inflammatory response. In fact, even cancers that are considered immunologically cold for lack of an adaptive immune response are often populated by innate immune cells. As described above, targeting the innate immune system therefore offers opportunities to a) prevent cancer development, b) stratify patients for specific treatments, and c) elicit an anti-tumor immune response.
Some chronic inflammatory conditions are well known to favor cancer development, but more research is needed to identify opportunities to interrupt the signaling that supports tumorigenesis and prevent tumor progression. In addition, we are just starting to appreciate how genetic factors [e.g., polymorphisms in immune-related genes (41)] and environmental factors [e.g., diet (31, 190), stress (102)] influence the immune system and contribute to tipping the scale towards tumorigenesis.
Technological advances now allow us to profile the composition and phenotype of the tumor and systemic immune microenvironment. Soon, we should be able to harness this knowledge to better identify patients at high risk of recurrence and to assign patients to the therapy they are most likely to benefit from (39, 200, 202, 204).
From a therapeutic standpoint, targeting inflammatory cells provides an opportunity to influence cancer growth and improve adaptive immune responses. The outcome of the inflammatory response is highly context-dependent. In fact, almost universally, components of the immune system display both pro-tumor and anti-tumor functions. This duality implies that the innate immune system has an inherent degree of plasticity that can be leveraged. More work needs to be done to define which molecular players reprogram inflammatory cells to an anti-tumor phenotype. Moreover, as innate immune cells influence both one another and adaptive immune cells, understanding the hierarchy of signals that can switch an immunosuppressive microenvironment to an inflammatory one can allow us to act on the upstream components.
Acknowledgements
M.E. is supported by grants from the Department of Defense (W81XWH-20-1-0753) and the National Cancer Institute (1R01CA237413); L.M. is supported by a James S. McDonnell Foundation postdoctoral fellowship; J.D.-P. is supported by funds from Deutsche Forschungsgemeinschaft; and L.S. is supported by METAvivor Research and Support, Inc.
Footnotes
Disclosure Statement
M.E. is a member of the research advisory board for brensocatib for Insmed, a member of the scientific advisory board for Vividion Therapeutics, a consultant for Protalix, and holds shares in Agios. The other authors are not aware of any other affiliations, memberships, funding, or financial holdings that might affect the objectivity of this review.
Literature Cited
- 1.Virchow R 1989. (1858). Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--Atheromatous affection of arteries. 1858. Nutr. Rev. 47:23–25 [DOI] [PubMed] [Google Scholar]
- 2.Oelschlaeger TA. 2010. Bacteria as tumor therapeutics? Bioeng Bugs 1: 146–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coley WB. 1991. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res: 3–11 [PubMed] [Google Scholar]
- 4.Starnes CO. 1992. Coley’s toxins in perspective. Nature 357: 11–2 [DOI] [PubMed] [Google Scholar]
- 5.Balkwill F, Mantovani A. 2001. Inflammation and cancer: back to Virchow? Lancet 357: 539–45 [DOI] [PubMed] [Google Scholar]
- 6.Starkey JR, Crowle PK, Taubenberger S. 1988. Mast-cell-deficient W/Wv mice exhibit a decreased rate of tumor angiogenesis. Int J Cancer 42: 48–52 [DOI] [PubMed] [Google Scholar]
- 7.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. 1999. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13: 1382–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. 2000. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology 2: 737–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coussens LM, Tinkle CL, Hanahan D, Werb Z. 2000. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103: 481–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin EY, Nguyen AV, Russell RG, Pollard JW. 2001. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193: 727–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. 2006. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66: 11238–46 [DOI] [PubMed] [Google Scholar]
- 12.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. 2004. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–96 [DOI] [PubMed] [Google Scholar]
- 13.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. 2005. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8: 211–26 [DOI] [PubMed] [Google Scholar]
- 14.Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, Golenbock D, Gresnigt MS, Heneka MT, Hoffman HM, Hotchkiss R, Joosten LAB, Kastner DL, Korte M, Latz E, Libby P, Mandrup-Poulsen T, Mantovani A, Mills KHG, Nowak KL, O’Neill LA, Pickkers P, van der Poll T, Ridker PM, Schalkwijk J, Schwartz DA, Siegmund B, Steer CJ, Tilg H, van der Meer JWM, van de Veerdonk FL, Dinarello CA. 2017. A guiding map for inflammation. Nat Immunol 18: 826–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hossain M, Kubes P. 2019. Innate immune cells orchestrate the repair of sterile injury in the liver and beyond. Eur J Immunol 49: 831–41 [DOI] [PubMed] [Google Scholar]
- 16.Wang J 2018. Neutrophils in tissue injury and repair. Cell Tissue Res 371: 531–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castanheira FVS, Kubes P. 2019. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 133: 2178–85 [DOI] [PubMed] [Google Scholar]
- 18.Headland SE, Norling LV. 2015. The resolution of inflammation: Principles and challenges. Semin Immunol 27: 149–60 [DOI] [PubMed] [Google Scholar]
- 19.Nathan C, Ding A. 2010. Nonresolving inflammation. Cell 140: 871–82 [DOI] [PubMed] [Google Scholar]
- 20.Dvorak HF. 1986. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315: 1650–9 [DOI] [PubMed] [Google Scholar]
- 21.Watson J, Salisbury C, Banks J, Whiting P, Hamilton W. 2019. Predictive value of inflammatory markers for cancer diagnosis in primary care: a prospective cohort study using electronic health records. Br J Cancer 120: 1045–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, McCullough ML, Patel AV, Ma J, Soerjomataram I, Flanders WD, Brawley OW, Gapstur SM, Jemal A. 2018. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin 68: 31–54 [DOI] [PubMed] [Google Scholar]
- 23.de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. 2020. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health 8: e180–e90 [DOI] [PubMed] [Google Scholar]
- 24.Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140: 883–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454: 436–44 [DOI] [PubMed] [Google Scholar]
- 26.Moore PS, Chang Y. 2010. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer 10: 878–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kappelman MD, Farkas DK, Long MD, Erichsen R, Sandler RS, Sorensen HT, Baron JA. 2014. Risk of cancer in patients with inflammatory bowel diseases: a nationwide population-based cohort study with 30 years of follow-up evaluation. Clin Gastroenterol Hepatol 12: 265–73 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vujasinovic M, Dugic A, Maisonneuve P, Aljic A, Berggren R, Panic N, Valente R, Pozzi Mucelli R, Waldthaler A, Ghorbani P, Kordes M, Hagstrom H, Lohr JM. 2020. Risk of Developing Pancreatic Cancer in Patients with Chronic Pancreatitis. J Clin Med 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouronte-Roibas C, Leiro-Fernandez V, Fernandez-Villar A, Botana-Rial M, Ramos-Hernandez C, Ruano-Ravina A. 2016. COPD, emphysema and the onset of lung cancer. A systematic review. Cancer Lett 382: 240–44 [DOI] [PubMed] [Google Scholar]
- 30.Iyengar NM, Hudis CA, Dannenberg AJ. 2015. Obesity and cancer: local and systemic mechanisms. Annu Rev Med 66: 297–309 [DOI] [PubMed] [Google Scholar]
- 31.Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, Chen IC, Wendel N, Ben-Chetrit N, Walker J, Holt PR, Dannenberg AJ, Joyce JA. 2017. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol 19: 974–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. 2011. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377: 31–41 [DOI] [PubMed] [Google Scholar]
- 33.McCormack VA, Hung RJ, Brenner DR, Bickeboller H, Rosenberger A, Muscat JE, Lazarus P, Tjonneland A, Friis S, Christiani DC, Chun EM, Le Marchand L, Rennert G, Rennert HS, Andrew AS, Orlow I, Park B, Boffetta P, Duell EJ. 2011. Aspirin and NSAID use and lung cancer risk: a pooled analysis in the International Lung Cancer Consortium (ILCCO). Cancer Causes Control 22: 1709–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, Group CT. 2017. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390: 1833–42 [DOI] [PubMed] [Google Scholar]
- 35.Novartis. Media Release. March 09 2021. https://www.novartis.com/news/media-releases/novartis-provides-update-phase-iii-study-evaluating-canakinumab-acz885-second-or-third-line-treatment-combination-chemotherapy-non-small-cell-lung-cancer
- 36.Wellenstein MD, de Visser KE. 2018. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 48: 399–416 [DOI] [PubMed] [Google Scholar]
- 37.Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, Eccles D, Evans DG, Maher ER, Bertario L, Bisgaard ML, Dunlop MG, Ho JW, Hodgson SV, Lindblom A, Lubinski J, Morrison PJ, Murday V, Ramesar R, Side L, Scott RJ, Thomas HJ, Vasen HF, Barker G, Crawford G, Elliott F, Movahedi M, Pylvanainen K, Wijnen JT, Fodde R, Lynch HT, Mathers JC, Bishop DT, Investigators C. 2011. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378: 2081–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, Mitchell C, Ng C, Katon J, Lunardi A, Signoretti S, Clohessy JG, Zhang J, Pandolfi PP. 2018. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med 24: 165–75 [DOI] [PubMed] [Google Scholar]
- 39.Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, Henneman L, Kas SM, Prekovic S, Hau CS, Vrijland K, Drenth AP, de Korte-Grimmerink R, Schut E, van der Heijden I, Zwart W, Wessels LFA, Schumacher TN, Jonkers J, de Visser KE. 2019. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 572: 538–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernandez C, Huebener P, Schwabe RF. 2016. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35: 5931–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. 2007. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13: 1050–9 [DOI] [PubMed] [Google Scholar]
- 42.He SJ, Cheng J, Feng X, Yu Y, Tian L, Huang Q. 2017. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 8: 64534–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitt JM, Kroemer G, Zitvogel L. 2017. Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. Adv Exp Med Biol 1036: 65–79 [DOI] [PubMed] [Google Scholar]
- 44.Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, Hogg GD, Tang JP, Baer JM, Mpoy C, Lee KB, Alexander KA, Rogers BE, Murphy KM, Hawkins WG, Fields RC, DeSelm CJ, Schwarz JK, DeNardo DG. 2020. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 37: 289–307 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496: 445–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, Alizadeh AA. 2015. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21: 938–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noy R, Pollard JW. 2014. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41: 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun L, Kees T, Almeida AS, Liu B, He X-Y, et al. 2021. Activating a collaborative innate-adaptive immune response to control metastasis. Cancer Cell. 39:1361–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. 2009. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16: 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R. 2014. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, Korets L, Lam J, Tawfik D, DeNardo DG, Naldini L, de Visser KE, De Palma M, Coussens LM. 2010. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 17: 121–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, Hammond R, Gimotty PA, Keith B, Simon MC. 2010. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120: 2699–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, Medrano RFV, Fronick C, Fehlings M, Newell EW, Fulton RS, Sheehan KCF, Oh ST, Schreiber RD, Artyomov MN. 2018. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 175: 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. 2007. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 67: 2649–56 [DOI] [PubMed] [Google Scholar]
- 56.Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, Zhang J, Cui X, Zheng F, Li H, Yao H, Su F, Song E. 2014. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 25: 605–20 [DOI] [PubMed] [Google Scholar]
- 57.Chen Q, Zhang XH, Massague J. 2011. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 20: 538–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. 2014. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26: 623–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mantovani A, Allavena P. 2015. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med 212: 435–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, Weissleder R, Pittet MJ. 2017. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, Ring AM, Connolly AJ, Weissman IL. 2017. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545: 495–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA. 2013. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19: 1264–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liew PX, Kubes P. 2019. The Neutrophil’s Role During Health and Disease. Physiol Rev 99: 1223–48 [DOI] [PubMed] [Google Scholar]
- 64.Keizman D, Ish-Shalom M, Huang P, Eisenberger MA, Pili R, Hammers H, Carducci MA. 2012. The association of pre-treatment neutrophil to lymphocyte ratio with response rate, progression free survival and overall survival of patients treated with sunitinib for metastatic renal cell carcinoma. European Journal of Cancer 48: 202–08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Donskov F 2013. Immunomonitoring and prognostic relevance of neutrophils in clinical trials. Seminars in Cancer Biology 23: 200–07 [DOI] [PubMed] [Google Scholar]
- 66.Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. 2010. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. The Journal of Clinical Investigation 120: 1151–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coffelt SB, Wellenstein MD, de Visser KE. 2016. Neutrophils in cancer: neutral no more. Nat Rev Cancer 16: 431–46 [DOI] [PubMed] [Google Scholar]
- 68.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. 2009. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 16: 183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. 2010. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest 120: 1151–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welch DR, Schissel DJ, Howrey RP, Aeed PA. 1989. Tumor-elicited polymorphonuclear cells, in contrast to “normal” circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proc Natl Acad Sci U S A 86: 5859–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn-Konijnenberg D, Homig-Holzel C, Reuten R, Schadow B, Weighardt H, Wenzel D, Helfrich I, Schadendorf D, Bloch W, Bianchi ME, Lugassy C, Barnhill RL, Koch M, Fleischmann BK, Forster I, Kastenmuller W, Kolanus W, Holzel M, Gaffal E, Tuting T. 2014. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 507: 109–13 [DOI] [PubMed] [Google Scholar]
- 72.Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, Ferri LE. 2012. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res 72: 3919–27 [DOI] [PubMed] [Google Scholar]
- 73.Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, Geng Y, Gray JW, Moses HL, Yang L. 2010. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res 70: 6139–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, Yu L, Ross J, Korsisaari N, Cao T, Bou-Reslan H, Kallop D, Weimer R, Ludlam MJ, Kaminker JS, Modrusan Z, van Bruggen N, Peale FV, Carano R, Meng YG, Ferrara N. 2010. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A 107: 21248–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, Lin C, Alicea-Torres K, Sehgal M, Nefedova Y, Zhou J, Languino LR, Clendenin C, Vonderheide RH, Mulligan C, Nam B, Hockstein N, Masters G, Guarino M, Schug ZT, Altieri DC, Gabrilovich DI. 2018. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol 19: 1236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wculek SK, Malanchi I. 2015. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528: 413–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels L, Jonkers J, de Visser KE. 2015. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522: 345–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Catena R, Bhattacharya N, El Rayes T, Wang S, Choi H, Gao D, Ryu S, Joshi N, Bielenberg D, Lee SB, Haukaas SA, Gravdal K, Halvorsen OJ, Akslen LA, Watnick RS, Mittal V. 2013. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov 3: 578–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, Wauters E, Walmsley S, Prenen H, Granot Z, Casazza A, Mazzone M. 2015. MET is required for the recruitment of anti-tumoural neutrophils. Nature 522: 349–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303: 1532–5 [DOI] [PubMed] [Google Scholar]
- 81.Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, Ferri L. 2013. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. The Journal of Clinical Investigation 123: 3446–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, Wang Y, Simmons RL, Huang H, Tsung A. 2016. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Research 76: 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. 2019. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med 216: 176–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, Nakasone ES, Hearn SA, Küttner V, Qiu J, Almeida AS, Perurena N, Kessenbrock K, Goldberg MS, Egeblad M. 2016. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Science Translational Medicine 8: 361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, Liu J, Xing Y, Chen X, Su S, Song E. 2020. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 583: 133–38 [DOI] [PubMed] [Google Scholar]
- 86.Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V, Bruzas E, Maiorino L, Bautista C, Carmona EM, Gimotty PA, Fearon DT, Chang K, Lyons SK, Pinkerton KE, Trotman LC, Goldberg MS, Yeh JT, Egeblad M. 2018. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, Andueza MP, Nieto CP, Resano L, Azpilikueta A, Allegretti M, de Pizzol M, Ponz-Sarvise M, Rouzaut A, Sanmamed MF, Schalper K, Carleton M, Mellado M, Rodriguez-Ruiz ME, Berraondo P, Perez-Gracia JL, Melero I. 2020. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 52: 856–71 e8 [DOI] [PubMed] [Google Scholar]
- 88.Veglia F, Perego M, Gabrilovich D. 2018. Myeloid-derived suppressor cells coming of age. Nat Immunol 19: 108–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, Zamboni N, Sallusto F, Lanzavecchia A. 2016. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167: 829–42 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harari O, Liao JK. 2004. Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr Pharm Des 10: 893–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baumann T, Dunkel A, Schmid C, Schmitt S, Hiltensperger M, Lohr K, Laketa V, Donakonda S, Ahting U, Lorenz-Depiereux B, Heil JE, Schredelseker J, Simeoni L, Fecher C, Korber N, Bauer T, Huser N, Hartmann D, Laschinger M, Eyerich K, Eyerich S, Anton M, Streeter M, Wang T, Schraven B, Spiegel D, Assaad F, Misgeld T, Zischka H, Murray PJ, Heine A, Heikenwalder M, Korn T, Dawid C, Hofmann T, Knolle PA, Hochst B. 2020. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol 21: 555–66 [DOI] [PubMed] [Google Scholar]
- 92.Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, Jiao X, Xie W, Kong X, Vaz M, Li H, Cai Y, Xia L, Huang P, Rodgers K, Lee B, Riemer JB, Day CP, Yen RC, Cui Y, Wang Y, Wang Y, Zhang W, Easwaran H, Hulbert A, Kim K, Juergens RA, Yang SC, Battafarano RJ, Bush EL, Broderick SR, Cattaneo SM, Brahmer JR, Rudin CM, Wrangle J, Mei Y, Kim YJ, Zhang B, Wang KK, Forde PM, Margolick JB, Nelkin BD, Zahnow CA, Pardoll DM, Housseau F, Baylin SB, Shen L, Brock MV. 2020. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 579: 284–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. 2014. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunology, Immunotherapy 63: 247–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. 2014. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Science Translational Medicine 6: 237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steinman RM. 1991. The Dendritic Cell System and its Role in Immunogenicity. Annual Review of Immunology 9: 271–96 [DOI] [PubMed] [Google Scholar]
- 96.Villadangos JA, Schnorrer P. 2007. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nature Reviews Immunology 7: 543–55 [DOI] [PubMed] [Google Scholar]
- 97.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. 2008. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322: 1097–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, Krummel MF. 2014. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26: 638–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, Chen AXY, Todd KL, Petley EV, Chan JD, Carrington EM, Lew AM, Solomon BJ, Trapani JA, Kedzierska K, Evrard M, Vervoort SJ, Waithman J, Darcy PK, Beavis PA. 2020. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol 21: 914–26 [DOI] [PubMed] [Google Scholar]
- 100.Spranger S, Dai D, Horton B, Gajewski TF. 2017. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31: 711–23 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka Anna K, Brown Brian D, Brody J, Ginhoux F, Merad M. 2016. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44: 924–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang H, Xia L, Chen J, Zhang S, Martin V, Li Q, Lin S, Chen J, Calmette J, Lu M, Fu L, Yang J, Pan Z, Yu K, He J, Morand E, Schlecht-Louf G, Krzysiek R, Zitvogel L, Kang B, Zhang Z, Leader A, Zhou P, Lanfumey L, Shi M, Kroemer G, Ma Y. 2019. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med 25: 1428–41 [DOI] [PubMed] [Google Scholar]
- 103.Swiecki M, Colonna M. 2015. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15: 471–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, Bremond A, Goddard S, Pin JJ, Barthelemy-Dubois C, Lebecque S. 2004. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res 10: 7466–74 [DOI] [PubMed] [Google Scholar]
- 105.Labidi-Galy SI, Treilleux I, Goddard-Leon S, Combes JD, Blay JY, Ray-Coquard I, Caux C, Bendriss-Vermare N. 2012. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. Oncoimmunology 1: 380–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Poropatich K, Dominguez D, Chan WC, Andrade J, Zha Y, Wray B, Miska J, Qin L, Cole L, Coates S, Patel U, Samant S, Zhang B. 2020. OX40+ plasmacytoid dendritic cells in the tumor microenvironment promote antitumor immunity. J Clin Invest 130: 3528–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, Renaudineau S, Poyet G, Labidi-Galy SI, Goddard-Leon S, Durand I, Le Mercier I, Bajard A, Bachelot T, Puisieux A, Puisieux I, Blay J-Y, Ménétrier-Caux C, Caux C, Bendriss-Vermare N. 2012. Impaired IFN-α Production by Plasmacytoid Dendritic Cells Favors Regulatory T-cell Expansion That May Contribute to Breast Cancer Progression. Cancer Research 72: 5188. [DOI] [PubMed] [Google Scholar]
- 108.Terra M, Oberkampf M, Fayolle C, Rosenbaum P, Guillerey C, Dadaglio G, Leclerc C. 2018. Tumor-Derived TGFbeta Alters the Ability of Plasmacytoid Dendritic Cells to Respond to Innate Immune Signaling. Cancer Res 78: 3014–26 [DOI] [PubMed] [Google Scholar]
- 109.Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. 2008. The Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. The Journal of Immunology 181: 5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, Anderson S, Atkinson N, Ramirez PT, Liu YJ, Freedman R, Gilliet M. 2012. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res 72: 5240–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, Spasic M, Giurintano JP, Chan V, Daud AI, Ha P, Ye CJ, Roberts EW, Krummel MF. 2019. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 177: 556–71 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, Merad M. 2020. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580: 257–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu T, Ramakrishnan R, Altiok S, Youn J-I, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, Gabrilovich D. 2011. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. The Journal of Clinical Investigation 121: 4015–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H. 2018. Innate Lymphoid Cells: 10 Years On. Cell 174: 1054–66 [DOI] [PubMed] [Google Scholar]
- 115.Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Lukic ML. 2014. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer 134: 1669–82 [DOI] [PubMed] [Google Scholar]
- 116.Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, Ramnarain A, Gasmi B, Gururajan M, Redmond D, Askan G, Bhanot U, Elyada E, Park Y, Tuveson DA, Gonen M, Leach SD, Wolchok JD, DeMatteo RP, Merghoub T, Balachandran VP. 2020. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 579: 130–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, Kouro T, Itakura A, Nagai Y, Takaki S, Takatsu K. 2012. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol 188: 703–13 [DOI] [PubMed] [Google Scholar]
- 118.Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, Li MO. 2016. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 164: 365–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, Rautela J, Straube J, Waddell N, Blake SJ, Yan J, Bartholin L, Lee JS, Vivier E, Takeda K, Messaoudene M, Zitvogel L, Teng MWL, Belz GT, Engwerda CR, Huntington ND, Nakamura K, Holzel M, Smyth MJ. 2017. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol 18: 1004–15 [DOI] [PubMed] [Google Scholar]
- 120.Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, Colonna M. 2017. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat Immunol 18: 995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. 2010. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 328: 749–52 [DOI] [PubMed] [Google Scholar]
- 122.Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, Benelli R, Spaggiari GM, Cantoni C, Campana S, Bonaccorsi I, Morandi B, Truini M, Mingari MC, Moretta L, Ferlazzo G. 2015. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun 6: 8280. [DOI] [PubMed] [Google Scholar]
- 123.Talmadge JE, Meyers KM, Prieur DJ, Starkey JR. 1980. Role of NK cells in tumour growth and metastasis in beige mice. Nature 284: 622–4 [DOI] [PubMed] [Google Scholar]
- 124.Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, Martos JA, Moreno M. 1997. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 79: 2320–8 [DOI] [PubMed] [Google Scholar]
- 125.Mastaglio S, Wong E, Perera T, Ripley J, Blombery P, Smyth MJ, Koldej R, Ritchie D. 2018. Natural killer receptor ligand expression on acute myeloid leukemia impacts survival and relapse after chemotherapy. Blood Adv 2: 335–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stringaris K, Sekine T, Khoder A, Alsuliman A, Razzaghi B, Sargeant R, Pavlu J, Brisley G, de Lavallade H, Sarvaria A, Marin D, Mielke S, Apperley JF, Shpall EJ, Barrett AJ, Rezvani K. 2014. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 99: 836–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.van den Broek MF, Kagi D, Zinkernagel RM, Hengartner H. 1995. Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol 25: 3514–6 [DOI] [PubMed] [Google Scholar]
- 128.Karre K, Ljunggren HG, Piontek G, Kiessling R. 1986. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319: 675–8 [DOI] [PubMed] [Google Scholar]
- 129.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. 2001. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413: 165–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gasser S, Orsulic S, Brown EJ, Raulet DH. 2005. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436: 1186–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH. 2008. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28: 571–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, Mandelboim O. 2012. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol 188: 2509–15 [DOI] [PubMed] [Google Scholar]
- 133.Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, Gastaut JA, Pende D, Olive D, Moretta A. 2002. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 99: 3661–7 [DOI] [PubMed] [Google Scholar]
- 134.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, Alvarado MD, Wolf DM, Bogunovic D, Bhardwaj N, Daud AI, Ha PK, Ryan WR, Pollack JL, Samad B, Asthana S, Chan V, Krummel MF. 2018. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med 24: 1178–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis e Sousa C. 2018. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172: 1022–37 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mittal D, Vijayan D, Putz EM, Aguilera AR, Markey KA, Straube J, Kazakoff S, Nutt SL, Takeda K, Hill GR, Waddell N, Smyth MJ. 2017. Interleukin-12 from CD103(+) Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol Res 5: 1098–108 [DOI] [PubMed] [Google Scholar]
- 137.Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang JG, Teh C, Firth M, Ushiki T, Andoniou CE, Degli-Esposti MA, Sharp PP, Sanvitale CE, Infusini G, Liau NP, Linossi EM, Burns CJ, Carotta S, Gray DH, Seillet C, Hutchinson DS, Belz GT, Webb AI, Alexander WS, Li SS, Bullock AN, Babon JJ, Smyth MJ, Nicholson SE, Huntington ND. 2016. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol 17: 816–24 [DOI] [PubMed] [Google Scholar]
- 138.Ohs I, Ducimetiere L, Marinho J, Kulig P, Becher B, Tugues S. 2017. Restoration of Natural Killer Cell Antimetastatic Activity by IL12 and Checkpoint Blockade. Cancer Res 77: 7059–71 [DOI] [PubMed] [Google Scholar]
- 139.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, Sun R, Tian Z. 2018. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 19: 723–32 [DOI] [PubMed] [Google Scholar]
- 140.Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, Popovic B, Bernardini G, Magrini E, Gianni F, Zelenay S, Jonjic S, Santoni A, Garlanda C, Mantovani A. 2017. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 551: 110–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Niu C, Jin H, Li M, Xu J, Xu D, Hu J, He H, Li W, Cui J. 2015. In vitro analysis of the proliferative capacity and cytotoxic effects of ex vivo induced natural killer cells, cytokine-induced killer cells, and gamma-delta T cells. BMC Immunol 16: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dokouhaki P, Schuh NW, Joe B, Allen CA, Der SD, Tsao MS, Zhang L. 2013. NKG2D regulates production of soluble TRAIL by ex vivo expanded human gammadelta T cells. Eur J Immunol 43: 3175–82 [DOI] [PubMed] [Google Scholar]
- 143.Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, Craft J, Yin Z. 2003. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med 198: 433–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Maniar A, Zhang X, Lin W, Gastman BR, Pauza CD, Strome SE, Chapoval AI. 2010. Human gammadelta T lymphocytes induce robust NK cell-mediated antitumor cytotoxicity through CD137 engagement. Blood 116: 1726–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, Whary MT, Meyerson M, Germain R, Blainey PC, Fox JG, Jacks T. 2019. Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell 176: 998–1013 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, Barilla R, Torres-Hernandez A, Hundeyin M, Mani VRK, Avanzi A, Tippens D, Narayanan R, Jang JE, Newman E, Pillarisetty VG, Dustin ML, Bar-Sagi D, Hajdu C, Miller G. 2016. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell 166: 1485–99 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, Taniguchi M. 1997. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 278: 1623–6 [DOI] [PubMed] [Google Scholar]
- 148.De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, Salio M, Middleton M, Cerundolo V. 2010. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol 11: 1039–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. 2003. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol 171: 5140–7 [DOI] [PubMed] [Google Scholar]
- 150.Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, Rossjohn J, Perlmutter P, Cao J, Godfrey DI, Savage PB, Knolle PA, Kolanus W, Forster I, Kurts C. 2010. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol 11: 313–20 [DOI] [PubMed] [Google Scholar]
- 151.Bae EA, Seo H, Kim BS, Choi J, Jeon I, Shin KS, Koh CH, Song B, Kim IK, Min BS, Han YD, Shin SJ, Kang CY. 2018. Activation of NKT Cells in an Anti-PD-1-Resistant Tumor Model Enhances Antitumor Immunity by Reinvigorating Exhausted CD8 T Cells. Cancer Res 78: 5315–26 [DOI] [PubMed] [Google Scholar]
- 152.Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, Berzofsky JA. 2005. A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med 202: 1627–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, Yamamura T, Kumar V, Berzofsky JA. 2007. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol 179: 5126–36 [DOI] [PubMed] [Google Scholar]
- 154.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. 2015. Complement System Part I – Molecular Mechanisms of Activation and Regulation. Frontiers in Immunology 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. 2015. Complement System Part II: Role in Immunity. Frontiers in Immunology 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, Densmore JJ, Williams ME, Taylor RP. 2004. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol 172: 3280–8 [DOI] [PubMed] [Google Scholar]
- 157.Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, Li Y, Li J, Zhou B, Ye Y, Di C, Yu S, Zeng Y, Su S. 2020. Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell 180: 1081–97 e24 [DOI] [PubMed] [Google Scholar]
- 158.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, Rouzaut A, Pajares MJ, Montuenga LM, Pio R. 2012. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol 189: 4674–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.An LL, Gorman JV, Stephens G, Swerdlow B, Warrener P, Bonnell J, Mustelin T, Fung M, Kolbeck R. 2016. Complement C5a induces PD-L1 expression and acts in synergy with LPS through Erk1/2 and JNK signaling pathways. Sci Rep 6: 33346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Janelle V, Langlois MP, Tarrab E, Lapierre P, Poliquin L, Lamarre A. 2014. Transient complement inhibition promotes a tumor-specific immune response through the implication of natural killer cells. Cancer Immunol Res 2: 200–6 [DOI] [PubMed] [Google Scholar]
- 161.Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, Leyshock P, Leitenberger JJ, Kulesz-Martin M, Margolin AA, Werb Z, Coussens LM. 2018. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 34: 561–78 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, Bazolli B, Lu B, Penna G, Rescigno M. 2016. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun 7: 11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sohn J-H, Bora PS, Suk H-J, Molina H, Kaplan HJ, Bora NS. 2003. Tolerance is dependent on complement C3 fragment iC3b binding to antigen-presenting cells. Nature Medicine 9: 206–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hsieh C-C, Chou H-S, Yang H-R, Lin F, Bhatt S, Qin J, Wang L, Fung JJ, Qian S, Lu L. 2013. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood 121: 1760–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, Greco C, Feruglio F, Molgora M, Laface I, Tartari S, Doni A, Pasqualini F, Barbati E, Basso G, Galdiero MR, Nebuloni M, Roncalli M, Colombo P, Laghi L, Lambris JD, Jaillon S, Garlanda C, Mantovani A. 2015. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 160: 700–14 [DOI] [PubMed] [Google Scholar]
- 166.Pickup MW, Mouw JK, Weaver VM. 2014. The extracellular matrix modulates the hallmarks of cancer. EMBO reports 15: 1243–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. 2003. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nature Cell Biology 5: 733–40 [DOI] [PubMed] [Google Scholar]
- 168.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LEH, Ingber DE. 2009. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457: 1103–08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. 2012. Matrix rigidity regulates a switch between TGF-β1–induced apoptosis and epithelial–mesenchymal transition. Molecular Biology of the Cell 23: 781–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sorokin L 2010. The impact of the extracellular matrix on inflammation. Nat Rev Immunol 10: 712–23 [DOI] [PubMed] [Google Scholar]
- 171.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. 2015. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative Biology 7: 1120–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Larsen AMH, Kuczek DE, Kalvisa A, Siersbæk MS, Thorseth M-L, et al. 2020. Collagen density modulates the immunosuppressive functions of macrophages. J. Immunol. 205:1461–72. 10.4049/jimmunol.1900789 [DOI] [PubMed] [Google Scholar]
- 173.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M. 2009. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457: 102–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rygiel TP, Stolte EH, de Ruiter T, van de Weijer ML, Meyaard L. 2011. Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol Immunol 49: 402–6 [DOI] [PubMed] [Google Scholar]
- 175.Hope C, Emmerich PB, Papadas A, Pagenkopf A, Matkowskyj KA, Van De Hey DR, Payne SN, Clipson L, Callander NS, Hematti P, Miyamoto S, Johnson MG, Deming DA, Asimakopoulos F. 2017. Versican-Derived Matrikines Regulate Batf3-Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J Immunol 199: 1933–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kalluri R 2016. The biology and function of fibroblasts in cancer. Nature Reviews Cancer 16: 582–98 [DOI] [PubMed] [Google Scholar]
- 177.Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, Soh J, Kim HS, Lee H, Kim J, Min JJ, Jung DW, Williams DR. 2018. Cancer-Stimulated CAFs Enhance Monocyte Differentiation and Protumoral TAM Activation via IL6 and GM-CSF Secretion. Clin Cancer Res 24: 5407–21 [DOI] [PubMed] [Google Scholar]
- 178.Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, Yuan L, Feng Y. 2019. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis 10: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, Hashimoto A, Vonteddu P, Behera R, Goins MA, Mulligan C, Nam B, Hockstein N, Denstman F, Shakamuri S, Speicher DW, Weeraratna AT, Chao T, Vonderheide RH, Languino LR, Ordentlich P, Liu Q, Xu X, Lo A, Pure E, Zhang C, Loboda A, Sepulveda MA, Snyder LA, Gabrilovich DI. 2017. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 32: 654–68 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, Queirolo P, Vermi W, Facchetti F, Moretta A, Moretta L, Mingari MC, Vitale M. 2009. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A 106: 20847–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, Liu W, Tai Y, Peng YW, Zhang Q. 2016. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 5: e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, Liu W, Zhang Q, Yang Y. 2018. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis 9: 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, Komoto S, Katsube R, Ninomiya T, Tazawa H, Shirakawa Y, Fujiwara T. 2018. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin Cancer Res 24: 4820–33 [DOI] [PubMed] [Google Scholar]
- 184.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, Sivajothi S, Armstrong TD, Engle DD, Yu KH, Hao Y, Wolfgang CL, Park Y, Preall J, Jaffee EM, Califano A, Robson P, Tuveson DA. 2019. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9: 1102–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA, Fearon DT. 2013. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences 110: 20212–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wang Z, Yan R, Li J, Gao Y, Moresco P, Yao M, Hechtman JF, Weiss MJ, Janowitz T, Fearon DT. 2020. Pancreatic cancer cells assemble a CXCL12-keratin 19 coating to resist immunotherapy. bioRxiv 776419; doi: 10.1101/776419 [DOI] [Google Scholar]
- 187.Biasci D, Smoragiewicz M, Connell CM, Wang Z, Gao Y, Thaventhiran JED, Basu B, Magiera L, Johnson TI, Bax L, Gopinathan A, Isherwood C, Gallagher FA, Pawula M, Hudecova I, Gale D, Rosenfeld N, Barmpounakis P, Popa EC, Brais R, Godfrey E, Mir F, Richards FM, Fearon DT, Janowitz T, Jodrell DI. 2020. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proceedings of the National Academy of Sciences 117: 28960–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Honda K, Littman DR. 2016. The microbiota in adaptive immune homeostasis and disease. Nature 535: 75–84 [DOI] [PubMed] [Google Scholar]
- 189.Petrelli F, Ghidini M, Ghidini A, Perego G, Cabiddu M, Khakoo S, Oggionni E, Abeni C, Hahne JC, Tomasello G, Zaniboni A. 2019. Use of Antibiotics and Risk of Cancer: A Systematic Review and Meta-Analysis of Observational Studies. Cancers (Basel) 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B, Ziegler PK, Varga J, Reindl W, Pommerenke C, Salinas-Riester G, Bock A, Alpert C, Blaut M, Polson SC, Brandl L, Kirchner T, Greten FR, Polson SW, Arkan MC. 2014. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514: 508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M. 2012. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491: 254–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, Korangy F, Wang XW, Trinchieri G, Greten TF. 2018. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, Werba G, Zhang K, Guo Y, Li Q, Akkad N, Lall S, Wadowski B, Gutierrez J, Kochen Rossi JA, Herzog JW, Diskin B, Torres-Hernandez A, Leinwand J, Wang W, Taunk PS, Savadkar S, Janal M, Saxena A, Li X, Cohen D, Sartor RB, Saxena D, Miller G. 2018. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov 8: 403–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, Guo Y, Saxena A, Vardhan M, Diskin B, Wang W, Leinwand J, Kurz E, Kochen Rossi JA, Hundeyin M, Zambrinis C, Li X, Saxena D, Miller G. 2019. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574: 264–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF. 2015. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350: 1084–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Berard M, Nigou J, Opolon P, Eggermont A, Woerther PL, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L. 2015. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350: 1079–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, Rotin D, Anafi L, Avivi C, Melnichenko J, Steinberg-Silman Y, Mamtani R, Harati H, Asher N, Shapira-Frommer R, Brosh-Nissimov T, Eshet Y, Ben-Simon S, Ziv O, Khan MAW, Amit M, Ajami NJ, Barshack I, Schachter J, Wargo JA, Koren O, Markel G, Boursi B. 2021. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371: 602–09 [DOI] [PubMed] [Google Scholar]
- 198.Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, Zidi B, Zhang S, Badger JH, Vetizou M, Cole AM, Fernandes MR, Prescott S, Costa RGF, Balaji AK, Morgun A, Vujkovic-Cvijin I, Wang H, Borhani AA, Schwartz MB, Dubner HM, Ernst SJ, Rose A, Najjar YG, Belkaid Y, Kirkwood JM, Trinchieri G, Zarour HM. 2021. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371: 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Finotello F, Trajanoski Z. 2018. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother 67: 1031–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, Bado I, Lo HC, Toneff MJ, Nguyen T, Bu W, Jiang W, Arnold J, Gu F, He J, Jebakumar D, Walker K, Li Y, Mo Q, Westbrook TF, Zong C, Rao A, Sreekumar A, Rosen JM, Zhang XH. 2019. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol 21: 1113–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, Meinhof K, Chow A, Kim-Shulze S, Wolf A, Medaglia C, Li H, Rytlewski JA, Emerson RO, Solovyov A, Greenbaum BD, Sanders C, Vignali M, Beasley MB, Flores R, Gnjatic S, Pe’er D, Rahman A, Amit I, Merad M. 2017. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 169: 750–65 e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, El Rassi E, Clayburgh DR, Kulesz-Martin MF, Lutz ER, Zheng L, Jaffee EM, Leyshock P, Margolin AA, Mori M, Gray JW, Flint PW, Coussens LM. 2017. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep 19: 203–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R, Bendall SC, Angelo M. 2018. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174: 1373–87 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, Saleh SMI, Zhao H, Souleimanova M, Johnson RM, Monette A, Ramos VM, Hallett MT, Stagg J, Lapointe R, Omeroglu A, Meterissian S, Buisseret L, Van den Eynden G, Salgado R, Guiot MC, Haibe-Kains B, Park M. 2019. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest 129: 1785–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, Ashenberg O, Cerami E, Coffey RJ, Demir E, Ding L, Esplin ED, Ford JM, Goecks J, Ghosh S, Gray JW, Guinney J, Hanlon SE, Hughes SK, Hwang ES, Iacobuzio-Donahue CA, Jane-Valbuena J, Johnson BE, Lau KS, Lively T, Mazzilli SA, Pe’er D, Santagata S, Shalek AK, Schapiro D, Snyder MP, Sorger PK, Spira AE, Srivastava S, Tan K, West RB, Williams EH, Human Tumor Atlas N. 2020. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181: 236–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.van Puffelen JH, Keating ST, Oosterwijk E, van der Heijden AG, Netea MG, Joosten LAB, Vermeulen SH. 2020. Trained immunity as a molecular mechanism for BCG immunotherapy in bladder cancer. Nature Reviews Urology 17: 513–25 [DOI] [PubMed] [Google Scholar]
- 208.Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, Saya H, Suda T. 2009. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med 206: 1089–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Li M, Knight DA, L AS, Smyth MJ, Stewart TJ. 2013. A role for CCL2 in both tumor progression and immunosurveillance. Oncoimmunology 2: e25474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F, de Bono JS. 2013. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs 31: 760–8 [DOI] [PubMed] [Google Scholar]
- 211.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, Fowler KJ, Lockhart AC, Suresh R, Tan BR, Lim KH, Fields RC, Strasberg SM, Hawkins WG, DeNardo DG, Goedegebuure SP, Linehan DC. 2016. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17: 651–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, Staddon AP, Cohn AL, Shapiro GI, Keedy VL, Singh AS, Puzanov I, Kwak EL, Wagner AJ, Von Hoff DD, Weiss GJ, Ramanathan RK, Zhang J, Habets G, Zhang Y, Burton EA, Visor G, Sanftner L, Severson P, Nguyen H, Kim MJ, Marimuthu A, Tsang G, Shellooe R, Gee C, West BL, Hirth P, Nolop K, van de Rijn M, Hsu HH, Peterfy C, Lin PS, Tong-Starksen S, Bollag G. 2015. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med 373: 428–37 [DOI] [PubMed] [Google Scholar]
- 213.Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, Huse J, Phillips J, West BL, Nolop KB, Hsu HH, Ligon KL, Molinaro AM, Prados M. 2016. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol 18: 557–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA, Ries C, Brillouet A, Muller C, Jegg AM, Broske AM, Dembowski M, Bray-French K, Freilinger C, Meneses-Lorente G, Baehner M, Harding R, Ratnayake J, Abiraj K, Gass N, Noh K, Christen RD, Ukarma L, Bompas E, Delord JP, Blay JY, Ruttinger D. 2015. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol 16: 949–56 [DOI] [PubMed] [Google Scholar]
- 215.Razak AR, Cleary JM, Moreno V, Boyer M, Calvo Aller E, Edenfield W, Tie J, Harvey RD, Rutten A, Shah MA, Olszanski AJ, Jager D, Lakhani N, Ryan DP, Rasmussen E, Juan G, Wong H, Soman N, Smit MD, Nagorsen D, Papadopoulos KP. 2020. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cancer 8 [DOI] [PMC free article] [PubMed] [Google Scholar]; 215a. Kaczanowska S, Beury DW, Gopalan V, et al. , Genetically engineered myeloid cells rebalance the core immune-suppression program in metastasis. Cell. March 24, 2021. DOI: 10.1016/j.cell.2021.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Aleynick M, Svensson-Arvelund J, Flowers CR, Marabelle A, Brody JD. 2019. Pathogen Molecular Pattern Receptor Agonists: Treating Cancer by Mimicking Infection. Clin Cancer Res 25: 6283–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Smith M, Garcia-Martinez E, Pitter MR, Fucikova J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L. 2018. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 7: e1526250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, Hochman T, Shuman N, Axelrod D, Speyer J, Novik Y, Tiersten A, Goldberg JD, Formenti SC, Bhardwaj N, Unutmaz D, Demaria S. 2012. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res 18: 6748–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Smith DA, Conkling P, Richards DA, Nemunaitis JJ, Boyd TE, Mita AC, de La Bourdonnaye G, Wages D, Bexon AS. 2014. Antitumor activity and safety of combination therapy with the Toll-like receptor 9 agonist IMO-2055, erlotinib, and bevacizumab in advanced or metastatic non-small cell lung cancer patients who have progressed following chemotherapy. Cancer Immunol Immunother 63: 787–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dietsch GN. 2016. Motolimod effectively drives immune activation in advanced cancer patients. Oncoimmunology 5: e1126037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Na YR, Yoon YN, Son D, Jung D, Gu GJ, Seok SH. 2015. Consistent inhibition of cyclooxygenase drives macrophages towards the inflammatory phenotype. PLoS One 10: e0118203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K, Ueo T, Komekado H, Kawada M, Minami M, Chiba T. 2011. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis 32: 1333–9 [DOI] [PubMed] [Google Scholar]
- 223.Lin JZ, Hameed I, Xu Z, Yu Y, Ren ZY, Zhu JG. 2018. Efficacy of gefitinibcelecoxib combination therapy in docetaxelresistant prostate cancer. Oncol Rep 40: 2242–50 [DOI] [PubMed] [Google Scholar]
- 224.Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo S, Bronson RT, Davis SP, Lobera M, Nolan MA, Letai A. 2017. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543: 428–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE, Varner JA. 2016. PI3Kgamma is a molecular switch that controls immune suppression. Nature 539: 437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Downey CM, Aghaei M, Schwendener RA, Jirik FR. 2014. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2’3’-cGAMP, induces M2 macrophage repolarization. PLoS One 9: e99988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Li A, Yi M, Qin S, Song Y, Chu Q, Wu K. 2019. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J Hematol Oncol 12: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Anguille S, Smits EL, Bryant C, Van Acker HH, Goossens H, Lion E, Fromm PD, Hart DN, Van Tendeloo VF, Berneman ZN. 2015. Dendritic Cells as Pharmacological Tools for Cancer Immunotherapy. Pharmacol Rev 67: 731–53 [DOI] [PubMed] [Google Scholar]
- 229.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. 2001. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med 193: 233–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kim JH, Kang TH, Noh KH, Kim SH, Lee YH, Kim KW, Bae HC, Ahn YH, Choi EY, Kim JS, Lee KM, Kim TW. 2010. Enhancement of DC vaccine potency by activating the PI3K/AKT pathway with a small interfering RNA targeting PTEN. Immunol Lett 134: 47–54 [DOI] [PubMed] [Google Scholar]
- 231.Theisen DJ, Davidson JTt, Briseno CG, Gargaro M, Lauron EJ, Wang Q, Desai P, Durai V, Bagadia P, Brickner JR, Beatty WL, Virgin HW, Gillanders WE, Mosammaparast N, Diamond MS, Sibley LD, Yokoyama W, Schreiber RD, Murphy TL, Murphy KM. 2018. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362: 694–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Handy CE, Antonarakis ES. 2018. Sipuleucel-T for the treatment of prostate cancer: novel insights and future directions. Future Oncol 14: 907–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Schreibelt G, Bol KF, Westdorp H, Wimmers F, Aarntzen EH, Duiveman-de Boer T, van de Rakt MW, Scharenborg NM, de Boer AJ, Pots JM, Olde Nordkamp MA, van Oorschot TG, Tel J, Winkels G, Petry K, Blokx WA, van Rossum MM, Welzen ME, Mus RD, Croockewit SA, Koornstra RH, Jacobs JF, Kelderman S, Blank CU, Gerritsen WR, Punt CJ, Figdor CG, de Vries IJ. 2016. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin Cancer Res 22: 2155–66 [DOI] [PubMed] [Google Scholar]
- 234.Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, Zhan Y, Ostrowski D, Yellin M, Marsh H, Salazar AM, Rahman AH, Brown BD, Merad M, Brody JD. 2019. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med 25: 814–24 [DOI] [PubMed] [Google Scholar]
- 235.Van Lint S, Renmans D, Broos K, Goethals L, Maenhout S, Benteyn D, Goyvaerts C, Du Four S, Van der Jeught K, Bialkowski L, Flamand V, Heirman C, Thielemans K, Breckpot K. 2016. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol Res 4: 146–56 [DOI] [PubMed] [Google Scholar]
- 236.Ylosmaki E, Cerullo V. 2020. Design and application of oncolytic viruses for cancer immunotherapy. Curr Opin Biotechnol 65: 25–36 [DOI] [PubMed] [Google Scholar]
- 237.Li Y, Su Z, Zhao W, Zhang X, Momin N, Zhang C, Wittrup KD, Dong Y, Irvine DJ, Weiss R. 2020. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nature Cancer 1: 882–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Khalil DN, Suek N, Campesato LF, Budhu S, Redmond D, Samstein RM, Krishna C, Panageas KS, Capanu M, Houghton S, Hirschhorn D, Zappasodi R, Giese R, Gasmi B, Schneider M, Gupta A, Harding JJ, Moral JA, Balachandran VP, Wolchok JD, Merghoub T. 2019. In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J Clin Invest 129: 3435–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW Jr., Gajewski TF. 2015. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11: 1018–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, Lopez-Diaz de Cerio A, Cabo M, Lopez-Botet M, Melero I. 2017. Targeting NK-cell checkpoints for cancer immunotherapy. Curr Opin Immunol 45: 73–81 [DOI] [PubMed] [Google Scholar]
- 241.Amin A, White RL Jr. 2013. High-dose interleukin-2: is it still indicated for melanoma and RCC in an era of targeted therapies? Oncology (Williston Park) 27: 680–91 [PubMed] [Google Scholar]
- 242.Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, Capanni M, Ruggeri L, Benson DM Jr., Blaser BW, Della Chiesa M, Moretta A, Vivier E, Caligiuri MA, Velardi A, Wagtmann N. 2009. Preclinical characterization of 1–7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 114: 2667–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, Goncalves A, Andre P, Romagne F, Thibault G, Viens P, Birnbaum D, Bertucci F, Moretta A, Olive D. 2011. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest 121: 3609–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.van Hall T, Andre P, Horowitz A, Ruan DF, Borst L, Zerbib R, Narni-Mancinelli E, van der Burg SH, Vivier E. 2019. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J Immunother Cancer 7: 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, Dietlein M, Kuhnert G, Kessler J, Buerkle C, Ravic M, Knackmuss S, Marschner JP, Pogge von Strandmann E, Borchmann P, Engert A. 2015. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 125: 4024–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, Zhang B, Lenvik AJ, Panoskaltsis-Mortari A, Verneris MR, Tolar J, Cooley S, Weisdorf DJ, Blazar BR, Miller JS. 2016. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin Cancer Res 22: 3440–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lin C, Zhang J. 2018. Reformation in chimeric antigen receptor based cancer immunotherapy: Redirecting natural killer cell. Biochim Biophys Acta Rev Cancer 1869: 200–15 [DOI] [PubMed] [Google Scholar]
- 248.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K. 2020. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 382: 545–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. 2003. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19: 583–93 [DOI] [PubMed] [Google Scholar]
- 250.Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, Perez RP, Kato G, Wicha M. 2017. Phase Ib Pilot Study to Evaluate Reparixin in Combination with Weekly Paclitaxel in Patients with HER-2-Negative Metastatic Breast Cancer. Clin Cancer Res 23: 5358–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, Li W, MacKinnon AL, Makkouk A, Marguier G, Murray PJ, Neou S, Pan A, Parlati F, Rodriguez MLM, Van de Velde LA, Wang T, Works M, Zhang J, Zhang W, Gross MI. 2017. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer 5: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]