

Abstract
Chronic thromboembolic pulmonary hypertension (CTEPH) is a rare and underdiagnosed complication of acute pulmonary embolism (APE). CTEPH is a common cause of pulmonary hypertension (PH) with distinct management strategy including pulmonary endarterectomy, balloon pulmonary angioplasty, long-term anticoagulation and PH drugs targeting endothelial cell dysfunction. Initially, PH in chronic thromboembolic pulmonary disease (CTEPD) was thought to be due exclusively to the intravascular obstruction of pulmonary arteries by unresolved fibrotic clots. However, it is now well accepted that pulmonary vascular remodelling can include significant pulmonary microvasculopathy, which plays a role in the development of CTEPH. The histological description and clinical consequences of CTEPH microvasculopathy are now better understood. These lesions may involve not only small muscular pulmonary arteries <500 µm, but also pulmonary capillaries and veins. In addition, enlargement and proliferation of systemic bronchial arteries as well as anastomoses between the systemic and pulmonary circulations contribute to the development of microvasculopathy. In this review, we discuss the recent advances in the understanding of the pathophysiology of CTEPH.
Keywords: Chronic thromboembolic pulmonary disease (CTEPD), chronic thromboembolic pulmonary hypertension (CTEPH), pathophysiology, microvasculopathy, pulmonary hypertension (PH)
Introduction
Chronic thromboembolic pulmonary disease (CTEPD), is a general term proposed in the recent ERS Statement on Chronic Thromboembolic Pulmonary Hypertension (CTEPH) (1) to characterize symptomatic patients who present mismatched perfusion defects on ventilation/perfusion (V/Q) lung scintigraphy and specific signs of chronic organized clots on computed tomography pulmonary angiography (CTPA), conventional pulmonary angiography (CPA) or magnetic resonance imaging (MRI) after at least three months of therapeutic anticoagulation. Some of these patients have no pulmonary hypertension (PH) at rest, while the majority presents with PH at rest, corresponding to the definition of CTEPH (group 4 of the updated clinical classification of PH) (2).
It is generally accepted that CTEPH can occur as a complication of acute pulmonary embolism (APE) (3). However, it remains unclear why in some patients, fresh embolic clots cannot be resolved, despite therapeutic anticoagulation. Furthermore, the precise incidence of CTEPH after a documented and correctly treated APE is still unknown.
The natural history of CTEPH is complex (Figure 1). Initially it was considered that PH in CTEPD was exclusively due to the intravascular obstruction of pulmonary arteries by unresolved fibrotic clots. However, it is now well accepted that pulmonary vascular remodelling can lead to significant pulmonary microvasculopathy, which plays a role in the development of CTEPH. These lesions may involve not only small muscular pulmonary arteries <500 µm (4-6), but also pulmonary capillaries and veins (4,6,7). In addition, enlargement and proliferation of systemic bronchial arteries as well as anastomoses between the systemic and pulmonary circulations contribute to the development of microvasculopathy (7-9).
Figure 1.
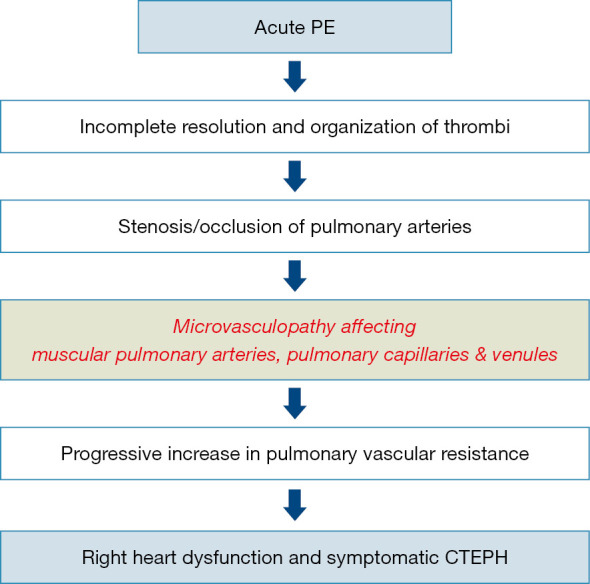
Natural history of CTEPH. Natural history of CTEPH is complex. Initially it was considered that PH in CTEPD was exclusively due to the intravascular obstruction of pulmonary arteries by unresolved fibrotic clots. Today, it is well accepted that pulmonary vascular remodelling can lead to significant pulmonary microvasculopathy, which plays a role in the development of CTEPH. These lesions may involve not only small muscular pulmonary arteries <500 µm, but also pulmonary capillaries and veins. CTEPH, chronic thromboembolic pulmonary hypertension; CTEPD, chronic thromboembolic pulmonary disease; PE, pulmonary embolism.
From APE to CTEPH
Incidence of CTEPH after an acute PE
CTEPH is generally considered to be a rare and late complication of APE, not resolved despite >three months of therapeutic anticoagulation. APE is a frequent disease and in a large prospective international CTEPH registry, 75% of patients had a confirmed or suspected history of APE (10). CTEPH can develop several months or years after an APE, despite continuing anticoagulation, and in the absence of any new acute thromboembolic events (3). The cumulative incidence of CTEPH after an APE has not yet been clearly established (11). In published prospective studies with a diagnosis of PH confirmed by a right heart catheterization, the incidence of CTEPH after a symptomatic APE is reported to range from 0.4% to 6.2% (12), giving a pooled incidence of 3.4%. Considering that around 30,000 APE cases are diagnosed each year in France (13), a CTEPH incidence of 3.4% would lead to 1,000 new CTEPH cases a year, far more than is actually observed in the French registry (around 200–300 new cases a year). One possible explanation is that many patients with CTEPH are not followed in the French PH network and thus not included in the French PH registry (14). Another reason for this apparent overestimation is that many CTEPH cases may not be identified in the community, due to poor awareness. This is supported by a French study indicating that patients recently diagnosed with CTEPH had pre-existing undiagnosed CTEPH at the time of the index APE (15). In this study, 146 patients with APE were treated with curative anticoagulation, during a median 26 months follow-up. Eight out of 146 patients had suspected CTEPH because of persistent dyspnea and abnormal echocardiographic findings suggestive of PH, and CTEPH was confirmed by right heart catheterization in seven patients. However, at the time of the index APE, five of the seven patients presented with markedly elevated systolic pulmonary artery pressure (PAP) ranging from 62 to 102 mmHg, a level not compatible with a first episode of APE because a non-adapted right ventricle cannot generate such high pressures. It is therefore likely that CTEPH was already present at the time of the index APE. This hypothesis was confirmed by review of initial CTPA by a senior radiologist, confirming that these five patients presented with several signs of CTEPH at the time of index APE. Therefore, the true cumulative incidence of APE was not 4.8%, but 1.5% (i.e., two out of 146).
Conditions contributing to a non-resolution of acute PE in CTEPH
In most patients with an APE treated with curative anticoagulation, significant resolution of the embolus occurs within a few weeks, with subsequent restoration of blood flow and normalisation of pulmonary haemodynamics, thanks to the activation of endogenous fibrinolysis (16). However, in a small subset of patients, a residual fibrotic clot remains attached to pulmonary vessel walls, leading to the development of CTEPH. The reason that only a minority of patients fail to resolve fresh thrombi and in turn develop CTEPH after an APE remains poorly understood. The main factors increasing the risk of developing CTEPH after an APE are: history of previous APE, presence of large perfusion defects and idiopathic APE at the time of presentation (17). However, these factors cannot explain the development of CTEPH in most patients and other mechanisms must be involved. Many clinical conditions (Table 1) have been suspected to increase the risk of CTEPH (18), including ventriculo-atrial shunt or infected pace-maker, splenectomy, thyroid hormone replacement, malignancy, non-O blood group and more recently implantable chambers for chemotherapy (19).
Table 1. Risk factors predisposing to CTEPH.
| Clinical conditions predisposing to CTEPH |
| Large pulmonary emboli |
| Recurrent pulmonary emboli |
| Insufficient anticoagulation |
| Underlying autoimmune and haematological disorders |
| Ventriculo-atrial shunts |
| Infected pacemaker leads |
| Implantable chambers for intravenous chemotherapy (especially when infected) |
| Totally implantable central venous access systems |
| Splenectomy |
| Cancer and chronic myeloproliferative disorders |
| Thyroid replacement therapy |
| Biological risk factors predisposing to CTEPH |
| Non-O blood group |
| Presence of lupus anticoagulant/antiphospholipid antibodies* |
*, interestingly, the classical hereditary thrombotic risk factors, e.g., protein C, protein S and antithrombin deficiencies and mutations of factor V and II are not more frequent in patients with CTEPH, compared with healthy controls and patients with pulmonary arterial hypertension. CTEPH, chronic thromboembolic pulmonary hypertension.
It has been suspected that patients with thrombus non-resolution could be in a hypercoagulable state due to biological abnormalities. Interestingly the classical hereditary thrombotic risk factors (e.g., mutations of factors V and II as well as deficiencies in protein C, protein S and antithrombin) are no more frequent in patients with CTEPH than in healthy controls. In this prospective study, only the frequency of antiphospholipid antibodies and lupus anticoagulant was higher in patients with CTEPH compared to healthy controls and patients with idiopathic PAH (20).
The lung is a rich source of endogenous procoagulants, anticoagulants and fibrinolytic agents. However, it remains unclear whether fibrinolytic system abnormalities are molecular ‘drivers’ in CTEPH.
Pulmonary vascular lesions observed in CTEPH
Anatomically, two different types of pulmonary vascular lesions contribute to the increase in pulmonary vascular resistance (PVR) in CTEPH: (I) an intra-vascular obstruction of pulmonary arteries by unresolved organized fibrotic clots, which constitutes the initial trigger for developing CTEPH, and (II) a secondary microvasculopathy characterized by a non-thrombotic obstructive remodelling involving not only the wall of muscular pulmonary arteries, but also pulmonary capillaries and veins (5).
Obstruction of pulmonary arteries by organized fibrotic clots
The intra-vascular obstruction of pulmonary arteries by unresolved fibrotic clots may involve main, lobar, segmental and subsegmental pulmonary arteries, reaching down to distal pulmonary arteries at the intra-acinar level. These histologic lesions correspond to what is called slits, webs, stenosis or pouching on vascular imaging. Distally, some obstructive chronic thromboembolic lesions appear as occluded vessels with recanalization and multiple secondary lumina called colander-like lesions (1).
Macroscopically, these chronic fibrotic clots are yellow, highly adherent to the wall of pulmonary vessels and they contain collagen, elastin, inflammatory cells and sometimes calcifications. Only a true thromboendarterectomy can remove theses organized clots. These pathological specimens are completely different from fresh clots observed in APE, which are of reddish color and mainly consist of erythrocytes and platelets in a fibrin mesh (Figure 2). These fresh clots can be easily detached from the pulmonary vessels. In CTEPH, like in pulmonary arterial hypertension (PAH), large, elastic-type pulmonary arteries that are exposed to a long-standing increase in PAP cause moderate atheromatous lesions leading to an increased wall stiffness (1).
Figure 2.

Fresh clots from a patient with acute pulmonary embolism and chronic fibrotic specimen from a CTEPH patient. (A) Fresh clots with a typical reddish color easily removed by thrombectomy from the pulmonary arteries of a patient with acute pulmonary embolism. (B) Chronic organized clots highly adherent to the wall of pulmonary arteries and removed by a true thromboendarterectomy. CTEPH, chronic thromboembolic pulmonary hypertension; PE, pulmonary embolism.
Secondary microvasculopathy (Figure 3)
Figure 3.

Typical histologic lesions encountered in lungs from patients with CTEPH [Reprinted with permission of the American Thoracic Society. Copyright © 2021 American Thoracic Society. All rights reserved. Lang IM, Dorfmüller P, Vonk Noordegraaf A. The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann Am Thorac Soc. 2016 Jul;13 Suppl 3:S215-21. doi:10.1513/AnnalsATS.201509-620AS (21). PMID: 27571003. Annals of the American Thoracic Society is an official journal of the American Thoracic Society]. (A) Pulmonary arteries; (B) bronchial arteries and pulmonary veins in patients with CTEPH (a,c) and in a piglet model (b); (C) microvessels. (A) Lung histology (HES staining) from a transplanted CTEPH patient. (a) Pulmonary artery (segmental level) with occluding organized thromboembolus. Note the angiogenesis within the occlusion and a systemic vessel within the adventitia that appears connected to the neovessels. (b) Another example of an occluded elastic-type artery. The hilar bronchial arteries are injected with blue dye right after lung transplantation; the dye can be perceived within the vasa vasorum (arrow) and within multiple neovessels and the muscular-type pulmonary artery, suggesting a systemic origin of the latter. (c) Eccentric intimal fibrosis of a smaller, muscular-type pulmonary artery, typically seen in CTEPH (top artery). (B) Lung histology (HES staining) from transplanted patients (a,c) and a CTEPH model (b). (a) Central section with a bronchial division (asterisks); hypertrophic bronchial arteries (arrows) and hyperplasia of bronchial microvessels (arrowheads) can be observed, mostly in occluded territories. (b) Central section in a piglet model of CTEPH. Blue ink has been injected into the bronchial circulation via the aortic ostia after sacrifice; note the injected bronchial arteries (left, arrowheads) which appear to discharge into the equally injected and strongly remodeled pulmonary veins running within the septa (right, arrows). (c) In human CTEPH lungs, septal veins frequently present intimal, collagen-rich, near-occlusive fibrosis. (C) Lung histology (HES staining) from transplanted CTEPH patients. (a) Microvascular disease with important remodelling of pulmonary vessels <100 microns, either arterioles or venules (arrows). (b) After injection of green dye into the hilar veins. Both septal veins (arrowhead, right) and remodelled small venules (arrow, left) are highlighted, suggesting involvement of post-capillary vessels in microvascular disease. (c) Areas with alveolar wall thickening are frequently present in occluded territories. Alveolar walls comprise multiple layers of capillaries (circle) as seen in capillary hemangiomatosis or pulmonary veno-occlusive disease; note edema and numerous macrophages within the alveoli (arrows). HES, hematoxylin-eosin-saffron; CTEPH, chronic thromboembolic pulmonary hypertension.
In CTEPH, pulmonary microvasculopathy was first described by Kenneth Moser and Colin Bloor (3) in lung tissues obtained by biopsy or at autopsy. These lesions are characterized by a major remodelling of the wall of muscular pulmonary arteries (50–500 µm), with a full range of PH lesions similar to those observed in idiopathic PAH, including eccentric intimal fibrosis and intimal fibromuscular proliferation. In CTEPH, Moser and Bloor described plexiform lesions which are classically observed in severe PAH, while other authors have not observed such lesions in CTEPH lungs (5). Interestingly, recent reports suggest that plexiform lesions in PAH might represent broncho-pulmonary shunts (22). In humans and animal models of CTEPH, these PAH-like lesions predominate in lung regions vascularized by non-obstructed proximal vessels (3,4,6). Therefore, it was proposed that in the presence of an obstruction of proximal pulmonary arteries in some territories, the pulmonary blood flow is redistributed to non-obstructed areas leading to local high flow, pressure and in consequence, shear stress, like in the case of a left-to-right cardiac shunt (23). However, in his initial report, Moser recognized that this hypothesis could not explain the presence of a microvasculopathy that was also present in lung areas downstream to completely or partially obstructed proximal vessels (3).
More recently, another form of microvasculopathy has been described in CTEPH, characterized by a remodelling of small pre-capillary vessels, capillaries (with pulmonary hemangiomatosis-like remodelling) and pulmonary venules (pulmonary veno-occlusive disease like). These lesions have been reported to be a hallmark of lung territories distal to completely or partially obstructed proximal pulmonary arteries by organised clots (4-6). In terms of pathophysiological mechanism, development of anastomoses between hypertrophic systemic bronchial arteries and the pulmonary circulation downstream to pulmonary stenosis or occlusion could play a major role (Figures 4,5). Importantly, these anastomoses are essential to keep open the capillary bed, precluding lung ischaemia in these areas. Owing to individual susceptibility, anastomoses between the high-pressure systemic circulation and both pre-capillary arterioles/capillaries and pulmonary venules will induce shear stress and vascular remodelling in some patients. These anastomoses, as well as pulmonary vascular remodelling of pre-capillary vessels, capillaries and veins, have been observed both in humans and in animal models of CTEPH (4,5).
Figure 4.

Appearance of bronchial artery hypertrophy and bronchopulmonary shunts 16 months after an acute pulmonary embolism associated with the development of CTEPD [Reprinted from Imaging as a biomarker in Behçet disease: prognostic and therapeutic implications of serial pulmonary CT angiography, Tsai HY, Chin CS, Tsai IC, The American Journal of Roentgenology 2011 Jan;196:W105-6. doi: 10.2214/AJR.10.5147 (7), Copyright© 2011]. Acute PE (A,B), evolution towards CTEPH 16 months later (C,D). (A) CTPA showing a complete obstruction of the right central pulmonary artery due to acute pulmonary embolism (asterisk). Concave filling defect at the level of obstruction (arrowhead) is characteristic of acute pulmonary embolism. (B) At the same time, slab volume-rendering image showing normal size of the bronchial artery (arrows). Right distal lobar pulmonary artery (white arrowhead) distally from proximal acute pulmonary embolism is small in caliber. No obvious connection between the right bronchial artery and distal pulmonary arteries. (C) Follow-up CTPA performed 16 months later showing typical aspect of CTEPD with convex pouching aspect (asterisk and arrowhead). (D) At the same time, slab volume-rendering image showing hypertrophy of the right bronchial artery (arrows) which connects with the vasa vasora (black arrowhead) to supply the distal pulmonary artery (white arrowhead). Note anastomoses between the bronchial artery, vasa vasora, and the right pulmonary artery are now larger. CTEPH, chronic thromboembolic pulmonary hypertension; CTEPD, chronic thromboembolic pulmonary disease; CTPA, computed tomography pulmonary angiography; AAO, ascending aorta; MPA, main pulmonary artery; DAO, descending aorta.
Figure 5.

Bronchopulmonary anastomosis detected on the late phase acquisition of a multidetector CTPA in a patient with CTEPH. Computed tomography pulmonary angiography performed with two acquisitions. (A) At early phase acquisition, typical aspect of CTEPD of the right lower lobe pulmonary artery totally obstructed by a chronic organized clot. (B) At late phase acquisition (at this time pulmonary veins, left ventricle and aorta are well opacified), we can see dilated right bronchial arteries supplying distal right lobar pulmonary artery now opacified. CTPA, computed tomography pulmonary angiography; CTEPH, chronic thromboembolic pulmonary hypertension; CTEPD, chronic thromboembolic pulmonary disease.
In a piglet model, ligation of the left main pulmonary artery associated with progressive embolization of the right lower lobe leads to hypertrophy of bronchial arteries in the territories obstructed, but not in the right upper lobe where the pulmonary artery remains open (4,5). This connection between high-pressure systemic blood flow and the low-pressure pulmonary circulation could lead to muscular and fibrotic remodelling of pre-capillary arterioles, capillaries and venules in the obstructed pulmonary vascular bed. In this piglet model of CTEPH, six weeks after surgical reperfusion of the left main pulmonary artery, the lesions concerning arterioles, capillaries and venules regressed.
In the non-obstructed right upper lobe, the pulmonary overflow due to the obstruction of other territories is considered the cause of a microvasculopathy concerning mainly muscular pulmonary arteries; these lesions also regressed after surgical reperfusion of the left main pulmonary artery, leading to normalization of the flow in the right upper lobe.
Molecular mechanisms underlying the microvasculopathy
The lack of a clear molecular basis of the processes underlying the microvasculopathy characteristic of CTEPH is a major obstacle for successful treatment of this disease. However, even as more precise identification of the multiple determinants underlying the remodelling of small vessels in CTEPH is still under investigation, there is accumulating evidence supporting the role of dysregulated pathways associated with pulmonary endothelial dysfunction. The clinical and haemodynamic improvements observed with the soluble guanylate cyclase (sGC) stimulator riociguat in patients with distal inoperable CTEPH or with persistent or recurrent PH after pulmonary thromboendarterectomy (PEA) suggest an important role of the nitric oxide (NO)-sGC-cGMP pathway in the pathophysiology of this microvasculopathy (24,25). Moreover, the phase II randomized-controlled trial MERIT-1 performed in inoperable CTEPH showed that the endothelin (ET)-1 receptor antagonist macitentan improved six-minute walk distance and PVR, as compared to placebo (26). Interestingly, background therapies with phosphodiesterase type 5 inhibitors or oral prostanoids, two effective drug classes for improving endothelial dysfunction in clinical studies, were permitted at inclusion. Lastly, subcutaneous treprostinil, a long-acting prostanoid, improved six-minute walk distance in a phase III randomized-controlled trial performed in functional class III/IV patients with inoperable CTEPH or persistent PH after PEA (27). Taken together, these data support the notion that drugs targeting the NO, ET-1 and prostacyclin (PGI2) pathways are effective in inoperable CTEPH, as previously shown in PAH (28).
Damage to the pulmonary endothelium is not only known to upset the balance between vasoconstriction and vasodilation, but can also initiate a number of events/processes that are responsible for rapid structural and functional changes of the pulmonary vasculature (29,30) (Figure 6). At the interface between the bloodstream and the vessel wall, the pulmonary endothelium indeed plays an integral role in the maintenance and preservation of vessel integrity and function. It acts as an active and dynamic receptor-effector tissue responding to the different chemical, physical or mechanical stimuli by secreting different factors via which it optimizes gas exchange and barrier function, maintains the vasomotor and haemostatic balance, controls the recruitment and retention of inflammatory cells and assures a functional angiogenic response (31,32). Among the different substances secreted by the pulmonary endothelium, the NO and its second messenger (cyclic guanosine monophosphate) not only act as vasorelaxants, but also attenuate leukocyte recruitment and activation (33), platelet adherence and aggregation (34,35) and vascular smooth muscle cell (SMC) proliferation and migration (36-40). In addition to NO synthesis, the endothelium also generates PGI2, which is another key vasodilator of the pulmonary vascular bed, and one of the most potent endogenous inhibitor of platelet aggregation. PGI2 also has anti-proliferative and immunomodulatory properties (41,42). Transgenic mice deficient in PGI2 receptor (IP-/-) are indeed more susceptible to thrombosis and develop intimal hyperplasia as compared to wild-type mice (43,44). Consistently, PGI2 is commonly found to be decreased in patients with different forms of PAH (45). It is also well established that pulmonary endothelial cells are also an important source of ET-1, which is known as a potent vasoconstrictor peptide and smooth-muscle mitogen (46). Tissue and blood levels of big ET-1 and ET-1 are elevated in patients with CTEPH (47-49) and in animal models of this condition (50,51). In addition, it has been reported that ET-1 serum levels correlate with disease severity in CTEPH patients (47).
Figure 6.

Proposed model illustrating the role of endothelial dysfunction in microvasculopathy associated with CTEPH. Dysfunction of the pulmonary endothelium leads to an altered balance of endothelium-derived vasoactive mediators including NO, endothelin-1 and prostacyclin. Other mediators such as cytokines, chemokines and growth factors may also contribute to pulmonary microvasculopathy. CTEPH, chronic thromboembolic pulmonary hypertension.
Current evidence therefore strongly suggests a central role for dysfunctions of pulmonary endothelium in the microvasculopathy of CTEPH and may explain why drugs that improve endothelial function or restore the altered balance of endothelium-derived vasoactive mediators have favorable benefit-risk profiles in CTEPH patients. Therefore, a greater understanding of the role of the pulmonary endothelium in the microvasculopathy of CTEPH and the potential molecular mechanisms will facilitate the identification of effective and powerful drugs to improve patient outcome and treat CTEPH.
Clinical consequences of the microvasculopathy and bronchial artery proliferation/anastomoses
The progressive development of a microvasculopathy downstream to non-occluded proximal pulmonary arteries may explain why some patients deteriorate even in the absence of recurrent APE (3). The extent and severity of this microvasculopathy, mainly in muscular pulmonary arteries, is highly variable between individual patients. The presence of an extensive microvasculopathy explains, at least in part, why the major elevation of PVR observed in some cases increases the risk of persistent post-operative PH and poor outcome after PEA. In contrast, the presence of a microvasculopathy downstream to obstructed pulmonary arteries probably plays a minor role in the increase of PVR before surgery. However, these lesions that mainly involve precapillary arterioles, capillaries and venules may suggest that the persistence of PH after surgery and the occurrence of pulmonary edema after PEA are possibly due to the recovery of normal blood flow in lung regions with microvascular changes, similar to those described in pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis.
Regarding systemic circulation, the anastomoses between dilated bronchial arteries and pulmonary circulation downstream to obstructed pulmonary arteries are essential in supplying the pulmonary circulation and preventing local lung ischemia. However, these bronchial artery proliferation/anastomoses may cause some deleterious effects. In addition to the development of capillary and post-capillary microvasculopathy observed in some patients (5), hypertrophy of bronchial arteries may also be involved in the occurrence of spontaneous massive hemoptysis requiring bronchial artery embolization. Hemoptysis could also occur after PEA (Figure 7) and may challenge pulmonary bypass weaning by the severity of bleeding. As demonstrated in an experimental study (50), bronchial and more systemic circulation are hypertrophied in cases of chronic pulmonary arterial obstruction, leading to large subepithelial bronchial arteries which can rupture after any local damage (Figure 8). Lastly, from our personal experience, in cases of predominantly proximal unilateral pulmonary artery obstruction inducing a modest elevation of PAP, the development of large broncho-pulmonary anastomoses may induce competitive flow after PEA, leading to a rapid post-operative re-thrombosis of the proximal pulmonary artery. Hence, we are routinely performing immediate post-endarterectomy bronchial embolization in cases of unilateral endarterectomy, with the goal of preventing re-thrombosis; in rare cases, severe hemoptysis may occur before a planned PEA and need urgent bronchial arteries embolization just before surgery (Figure 9).
Figure 7.

CTEPH patients experiencing post-endarterectomy hemoptysis treated by bronchial embolization. Hypertrophied systemic circulation to the lung in CTEPH patients. (A) Operative view of the right pleural cavity of a CTEPH patient showing vascular pulmonary adhesions to the chest wall. (B) Angiogram demonstrating the hypertrophied bronchial circulation which re-perfuses the distal pulmonary circulation. Pre- (C) and post- (D) bronchial embolization in a CTEPH patient experiencing post-endarterectomy hemoptysis. CTEPH, chronic thromboembolic pulmonary hypertension.
Figure 8.
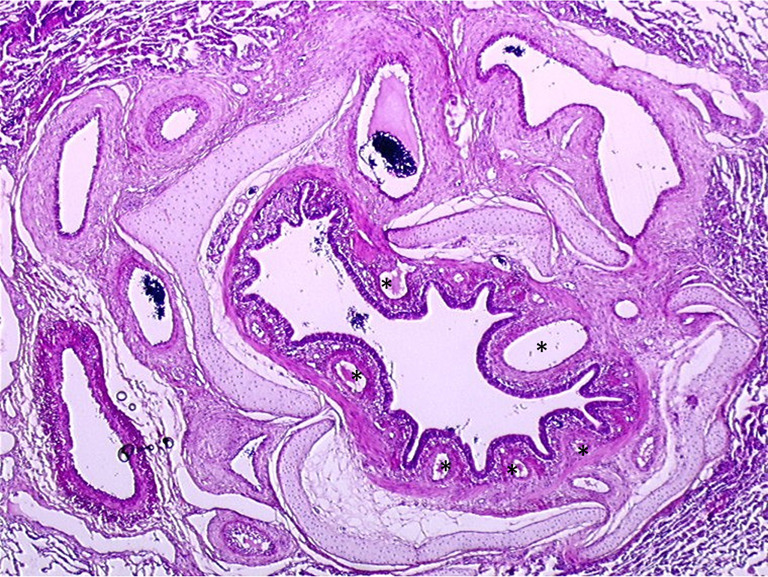
Hypertrophy of bronchial arteries in a CTEPH piglet model. Left lung bronchus after five weeks of pulmonary artery ligation from the piglet CTEPH model showing hypertrophied subepithelial bronchial arteries (black stars) explaining the potential occurrence of hemoptysis (HES staining). CTEPH, chronic thromboembolic pulmonary hypertension; HES, hematoxylin-eosin-saffron. Magnification ×20.
Figure 9.

Pulmonary endarterectomy specimen of a CTEPH patient who experienced massive preoperative hemoptysis requiring emergent embolization of bronchial arteries: 500 micron beads (white arrow) were found within the pulmonary circulation demonstrating communication between the two circulations in obstructed vascular territories. CTEPH, chronic thromboembolic pulmonary hypertension.
Respective roles of obstructive clots and microvasculopathy in the severity of CTEPH
In the initial hemodynamic evaluation of CTEPH patients, it is important to assess the respective roles of obstructive fibrotic clots and pulmonary microvasculopathy by appreciating PH severity. Indeed, these two types of lesions may benefit from different types of treatment-surgical (PEA) for proximal fibrotic lesions, interventional (balloon pulmonary angioplasty) for more distal fibrotic lesions, and medical therapy targeting microvasculopathy similar to that observed in PAH (Figure 6). The fibrotic organized clots (proximal and more distal) can be identified on high-quality CTPA and/or CPA and is associated with multiple perfusion defects on V/Q lung scintigraphy. In contrast, like in PAH patients, microvasculopathy cannot not be detected with current imaging techniques. However, the presence of a severe microvasculopathy involving muscular pulmonary arteries should be suspected when the extent of mechanical obstruction assessed by scintigraphy and angiography (only few perfusion defects) does not correlate with the hemodynamic severity suggested by high PVR (52). The hemodynamic impact of the microvasculopathy in CTEPH may also be evaluated with the pulmonary occlusion technique used to partition PVR into upstream resistance (due to organized proximal fibrotic clots) and downstream resistance (due to microvasculopathy). A lower pre-operative upstream resistance, suggesting extensive microvasculopathy, has been associated with a worse outcome after PEA (53).
Conclusions
The current understanding of the pathophysiology of CTEPH has recently moved beyond a pulmonary vascular disease caused by an intravascular mechanical obstruction due to chronic organized fibrotic material from unresolved clots, to a much more complex condition comprising proximal and more distal obstruction of pulmonary arteries associated with remodelling of muscular pulmonary arteries (mimicking PAH lesions), capillaries and venules. The processes by which residual organized clots persist after an APE are not fully understood, even if several risk factors have been identified. The mechanisms leading to the development of a microvasculopathy beyond the presence of fibrotic organized clots remains to be completely elucidated.
Video.

Chronic thromboembolic pulmonary hypertension: the magic of pathophysiology.
Acknowledgments
Funding: M. Humbert is supported by the Investissement d’Avenir programme managed by the French National Research Agency under the grant contract ANR-18-RHUS-0006 (DESTINATION 2024).
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Conflicts of Interest: GS has received consultancy and speaker fees from Actelion, Jansen, Bayer and MSD. MH has received consultancy and speaker fees from Acceleron, Bayer Janssen, MSD. PD, CG and OM have no conflicts of interest to declare.
References
- 1.Delcroix M, Torbicki A, Gopalan D, et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur Respir J 2021;57:2002828. 10.1183/13993003.02828-2020 [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913. 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moser KM, Bloor CM. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest 1993;103:685-92. 10.1378/chest.103.3.685 [DOI] [PubMed] [Google Scholar]
- 4.Boulate D, Perros F, Dorfmuller P, et al. Pulmonary microvascular lesions regress in reperfused chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant 2015;34:457-67. 10.1016/j.healun.2014.07.005 [DOI] [PubMed] [Google Scholar]
- 5.Dorfmüller P, Günther S, Ghigna MR, et al. Microvascular disease in chronic thromboembolic pulmonary hypertension: a role for pulmonary veins and systemic vasculature. Eur Respir J 2014;44:1275-88. 10.1183/09031936.00169113 [DOI] [PubMed] [Google Scholar]
- 6.Gerges C, Gerges M, Friewald R, et al. Microvascular Disease in Chronic Thromboembolic Pulmonary Hypertension: Hemodynamic Phenotyping and Histomorphometric Assessment. Circulation 2020;141:376-86. 10.1161/CIRCULATIONAHA.119.041515 [DOI] [PubMed] [Google Scholar]
- 7.Tsai HY, Chin CS, Tsai IC. Imaging as a biomarker in Behçet disease: prognostic and therapeutic implications of serial pulmonary CT angiography. AJR Am J Roentgenol 2011;196:W105-6. 10.2214/AJR.10.5147 [DOI] [PubMed] [Google Scholar]
- 8.Perino MG, Moldobaeva A, Jenkins J, et al. Chemokine localization in bronchial angiogenesis. PLoS One 2013;8:e66432. 10.1371/journal.pone.0066432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mercier O, Fadel E. Chronic thromboembolic pulmonary hypertension: animal models. Eur Respir J 2013;41:1200-6. 10.1183/09031936.00101612 [DOI] [PubMed] [Google Scholar]
- 10.Pepke-Zaba J, Delcroix M, Lang I, et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation 2011;124:1973-81. 10.1161/CIRCULATIONAHA.110.015008 [DOI] [PubMed] [Google Scholar]
- 11.Simonneau G, Hoeper MM. Evaluation of the incidence of rare diseases: difficulties and uncertainties, the example of chronic thromboembolic pulmonary hypertension. Eur Respir J 2017;49:1602522. 10.1183/13993003.02522-2016 [DOI] [PubMed] [Google Scholar]
- 12.Ende-Verhaar YM, Cannegieter SC, Vonk Noordegraaf A, et al. Incidence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: a contemporary view of the published literature. Eur Respir J 2017;49:1601792. 10.1183/13993003.01792-2016 [DOI] [PubMed] [Google Scholar]
- 13.Oger E. Incidence of venous thromboembolism: a community-based study in Western France. EPI-GETBP Study Group. Groupe d'Etude de la Thrombose de Bretagne Occidentale. Thromb Haemost 2000;83:657-60. 10.1055/s-0037-1613887 [DOI] [PubMed] [Google Scholar]
- 14.Cottin V, Avot D, Lévy-Bachelot L, et al. Identifying chronic thromboembolic pulmonary hypertension through the French national hospital discharge database. PLoS One 2019;14:e0214649. 10.1371/journal.pone.0214649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guérin L, Couturaud F, Parent F, et al. Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thromb Haemost 2014;112:598-605. 10.1160/TH13-07-0538 [DOI] [PubMed] [Google Scholar]
- 16.Banks DA, Pretorius GV, Kerr KM, et al. Pulmonary endarterectomy: part I. Pathophysiology, clinical manifestations, and diagnostic evaluation of chronic thromboembolic pulmonary hypertension. Semin Cardiothorac Vasc Anesth 2014;18:319-30. 10.1177/1089253214536621 [DOI] [PubMed] [Google Scholar]
- 17.Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004;350:2257-64. 10.1056/NEJMoa032274 [DOI] [PubMed] [Google Scholar]
- 18.Bonderman D, Jakowitsch J, Adlbrecht C, et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb Haemost 2005;93:512-6. 10.1160/TH04-10-0657 [DOI] [PubMed] [Google Scholar]
- 19.Jevnikar M, Montani D, Savale L, et al. Chronic thromboembolic pulmonary hypertension and totally implantable central venous access systems. Eur Respir J 2021;57:2002208. 10.1183/13993003.02208-2020 [DOI] [PubMed] [Google Scholar]
- 20.Wolf M, Boyer-Neumann C, Parent F, et al. Thrombotic risk factors in pulmonary hypertension. Eur Respir J 2000;15:395-9. 10.1034/j.1399-3003.2000.15b28.x [DOI] [PubMed] [Google Scholar]
- 21.Lang IM, Dorfmüller P, Vonk Noordegraaf A. The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann Am Thorac Soc 2016. Jul;13 Suppl 3:S215-21. 10.1513/AnnalsATS.201509-620AS [DOI] [PubMed] [Google Scholar]
- 22.Galambos C, Sims-Lucas S, Abman SH, et al. Intrapulmonary Bronchopulmonary Anastomoses and Plexiform Lesions in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 2016;193:574-6. 10.1164/rccm.201507-1508LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simonneau G, Torbicki A, Dorfmüller P, et al. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur Respir Rev 2017;26:160112. 10.1183/16000617.0112-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonneau G, D'Armini AM, Ghofrani HA, et al. Predictors of long-term outcomes in patients treated with riociguat for chronic thromboembolic pulmonary hypertension: data from the CHEST-2 open-label, randomised, long-term extension trial. Lancet Respir Med 2016;4:372-80. 10.1016/S2213-2600(16)30022-4 [DOI] [PubMed] [Google Scholar]
- 25.Ghofrani HA, D'Armini AM, Grimminger F, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369:319-29. 10.1056/NEJMoa1209657 [DOI] [PubMed] [Google Scholar]
- 26.Ghofrani HA, Simonneau G, D'Armini AM, et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med 2017;5:785-94. 10.1016/S2213-2600(17)30305-3 [DOI] [PubMed] [Google Scholar]
- 27.Sadushi-Kolici R, Jansa P, Kopec G, et al. Subcutaneous treprostinil for the treatment of severe non-operable chronic thromboembolic pulmonary hypertension (CTREPH): a double-blind, phase 3, randomised controlled trial. Lancet Respir Med 2019;7:239-48. 10.1016/S2213-2600(18)30367-9 [DOI] [PubMed] [Google Scholar]
- 28.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004;351:1425-36. 10.1056/NEJMra040291 [DOI] [PubMed] [Google Scholar]
- 29.Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019;53:1801887. 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guignabert C, Tu L, Girerd B, et al. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest 2015;147:529-37. 10.1378/chest.14-0862 [DOI] [PubMed] [Google Scholar]
- 31.Ricard N, Bailly S, Guignabert C, et al. The quiescent endothelium: signalling pathways regulating organ-specific endothelial normalcy. Nat Rev Cardiol 2021;18:565-80. 10.1038/s41569-021-00517-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huertas A, Guignabert C, Barberà JA, et al. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur Respir J 2018;51:1700745. 10.1183/13993003.00745-2017 [DOI] [PubMed] [Google Scholar]
- 33.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A 1991;88:4651-5. 10.1073/pnas.88.11.4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res 2001;88:756-62. 10.1161/hh0801.089861 [DOI] [PubMed] [Google Scholar]
- 35.Tziros C, Freedman JE. The many antithrombotic actions of nitric oxide. Curr Drug Targets 2006;7:1243-51. 10.2174/138945006778559111 [DOI] [PubMed] [Google Scholar]
- 36.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 1989;83:1774-7. 10.1172/JCI114081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kariya K, Kawahara Y, Araki S, et al. Antiproliferative action of cyclic GMP-elevating vasodilators in cultured rabbit aortic smooth muscle cells. Atherosclerosis 1989;80:143-7. 10.1016/0021-9150(89)90022-1 [DOI] [PubMed] [Google Scholar]
- 38.Nakaki T, Nakayama M, Kato R. Inhibition by nitric oxide and nitric oxide-producing vasodilators of DNA synthesis in vascular smooth muscle cells. Eur J Pharmacol 1990;189:347-53. 10.1016/0922-4106(90)90031-R [DOI] [PubMed] [Google Scholar]
- 39.Dubey RK, Jackson EK, Lüscher TF. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J Clin Invest 1995;96:141-9. 10.1172/JCI118014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkar R, Meinberg EG, Stanley JC, et al. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res 1996;78:225-30. 10.1161/01.RES.78.2.225 [DOI] [PubMed] [Google Scholar]
- 41.Kothapalli D, Stewart SA, Smyth EM, et al. Prostacylin receptor activation inhibits proliferation of aortic smooth muscle cells by regulating cAMP response element-binding protein- and pocket protein-dependent cyclin a gene expression. Mol Pharmacol 2003;64:249-58. 10.1124/mol.64.2.249 [DOI] [PubMed] [Google Scholar]
- 42.Li RC, Cindrova-Davies T, Skepper JN, et al. Prostacyclin induces apoptosis of vascular smooth muscle cells by a cAMP-mediated inhibition of extracellular signal-regulated kinase activity and can counteract the mitogenic activity of endothelin-1 or basic fibroblast growth factor. Circ Res 2004;94:759-67. 10.1161/01.RES.0000121568.40692.97 [DOI] [PubMed] [Google Scholar]
- 43.Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 1997;388:678-82. 10.1038/41780 [DOI] [PubMed] [Google Scholar]
- 44.Cheng Y, Austin SC, Rocca B, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002;296:539-41. 10.1126/science.1068711 [DOI] [PubMed] [Google Scholar]
- 45.Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999;159:1925-32. 10.1164/ajrccm.159.6.9804054 [DOI] [PubMed] [Google Scholar]
- 46.Chaumais MC, Guignabert C, Savale L, et al. Clinical pharmacology of endothelin receptor antagonists used in the treatment of pulmonary arterial hypertension. Am J Cardiovasc Drugs 2015;15:13-26. 10.1007/s40256-014-0095-y [DOI] [PubMed] [Google Scholar]
- 47.Reesink HJ, Meijer RC, Lutter R, et al. Hemodynamic and clinical correlates of endothelin-1 in chronic thromboembolic pulmonary hypertension. Circ J 2006;70:1058-63. 10.1253/circj.70.1058 [DOI] [PubMed] [Google Scholar]
- 48.Stewart DJ, Levy RD, Cernacek P, et al. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med 1991;114:464-9. 10.7326/0003-4819-114-6-464 [DOI] [PubMed] [Google Scholar]
- 49.Bauer M, Wilkens H, Langer F, et al. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation 2002;105:1034-6. 10.1161/hc0902.105719 [DOI] [PubMed] [Google Scholar]
- 50.Mercier O, Tivane A, Dorfmüller P, et al. Piglet model of chronic pulmonary hypertension. Pulm Circ 2013;3:908-15. 10.1086/674757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rondelet B, Kerbaul F, Motte S, et al. Bosentan for the prevention of overcirculation-induced experimental pulmonary arterial hypertension. Circulation 2003;107:1329-35. 10.1161/01.CIR.0000053443.27512.33 [DOI] [PubMed] [Google Scholar]
- 52.Azarian R, Wartski M, Collignon MA, et al. Lung perfusion scans and hemodynamics in acute and chronic pulmonary embolism. J Nucl Med 1997;38:980-3. [PubMed] [Google Scholar]
- 53.Kim NH, Fesler P, Channick RN, et al. Preoperative partitioning of pulmonary vascular resistance correlates with early outcome after thromboendarterectomy for chronic thromboembolic pulmonary hypertension. Circulation 2004;109:18-22. 10.1161/01.CIR.0000111841.28126.D4 [DOI] [PubMed] [Google Scholar]