

Summary
Background
Guadecitabine is a next-generation hypomethylating agent whose active metabolite decitabine has a longer in-vivo exposure time than intravenous decitabine. More effective hypomethylating agents are needed for the treatment of myelodysplastic syndromes. In the present study, we aimed to compare the activity and safety of two doses of guadecitabine in hypomethylating agent treatment-naive or relapsed or refractory patients with intermediate-risk or high-risk myelodysplastic syndromes.
Methods
This phase 2 part of the phase 1/2, randomised, open-label study enrolled patients aged 18 years or older from 14 North American medical centres with International Prognostic Scoring System intermediate-1-risk, intermediate-2-risk, or high-risk myelodysplastic syndromes, or chronic myelomonocytic leukaemia. They were either hypomethylating agent treatment-naive or had relapsed or refractory disease after previous hypomethylating agent treatment as determined by the investigators’ judgment. Eligible patients had Eastern Cooperative Oncology Group performance status of 0–2. Patients were randomly assigned (1:1) using a computer algorithm for dynamic randomisation to subcutaneous guadecitabine 60 or 90 mg/m2 on days 1–5 of a 28-day treatment cycle. Treatment was stratified by previous treatment with hypomethylating agents and neither patients nor investigators were masked. The primary endpoint was overall response (a composite of complete response, partial response, marrow complete response, and haematological improvement) assessed in all patients who received at least one dose of study drug. This study is registered with ClinicalTrials.gov, number NCT01261312.
Findings
Between July 9, 2012, and April 7, 2014, 105 patients were enrolled: 55 (52%) were allocated to guadecitabine 60 mg/m2 (28 patients were treatment-naive and 27 had relapsed or refractory disease after previous hypomethylating agent treatment) and 50 (48%) patients to 90 mg/m2 (23 patients were treatment-naive and 27 had relapsed or refractory disease). Three (3%) patients of 105 did not receive study treatment and were excluded from analyses. Median follow-up was 3·2 years (IQR 2·8–3·5). The proportion of patients achieving an overall response did not significantly differ between dose groups (21 of 53 [40%, 95% CI 27–54] with 60 mg/m2 and 27 of 49 [55%, 95% CI 40–69] with 90 mg/m2; p=0·16). 25 of 49 (51%, 95% CI 36–66) patients who were treatment-naive and 23 of 53 (43%, 30–58) patients with relapsed or refractory disease achieved an overall response. The most common grade 3 or worse adverse events in both groups, regardless of relationship to treatment, were thrombocytopenia (22 [41%] of 53 patients in the 60 mg/m2 group and 28 [57%] of 49 in the 90 mg/m2 group), neutropaenia (21 [40%] and 25 [51%]), anaemia (25 [47%] and 24 [49%]), febrile neutropaenia (17 [32%] and 21 [43%]), and pneumonia (13 [25%] and 15 [31%]). Seven (7%) of 102 patients died due to adverse events (three with 90 mg/m2 and four with 60 mg/m2), and all except one were in the relapsed or refractory cohort. Two deaths were deemed treatment related (septic shock with 60 mg/m2; pneumonia with 90 mg/m2).
Interpretation
Guadecitabine was clinically active with acceptable tolerability in patients with intermediate-risk and high-risk myelodysplastic syndromes. Responses and overall survival in the relapsed or refractory cohort offer the potential of a new therapeutic option for patients for whom currently available hypomethylating agents are not successful. We therefore recommend guadecitabine at a dose of 60 mg/m2 on a 5-day schedule for these patients.
Funding
Astex Pharmaceuticals and Stand Up To Cancer.
Introduction
Myelodysplastic syndromes are a heterogeneous group of haemopoietic stem-cell disorders characterised clinically by peripheral blood cytopenias caused mainly by ineffective bone-marrow haemopoiesis.1 These syndromes are associated with a high symptom burden, due mainly to complications arising from cytopenias, and a high rate of progression to acute myeloid leukaemia, which occurs in around 30% of cases. The phenotypic presentation varies depending on the transforming mutations present and how these dysregulate signal transduction in the affected haemopoietic stem cells.2 Treatment of myelodysplastic syndromes is based on prognostic risk evaluated using the International Prognostic Scoring System (IPSS) or the 2012 revision (IPSS-R).3,4 The only curative treatment for high-risk myelodysplastic syndromes is stem-cell transplantation; however, because the median age for diagnosis of myelodysplastic syndromes is 70 years, many high-risk patients have age-related comorbidities that preclude consideration for stem-cell transplantation.
Frequent epigenetic alterations are found in myelodysplastic syndromes, including aberrant DNA hyper-methylation of gene promoters, which contributes to altered transcription of genes involved in cell-cycle regulation, apoptosis, cell adhesion and motility, and tumour suppression.5–7 Given that epigenetic hyper-methylation is reversible,8 hypomethylating agents are a viable treatment option for patients with myelodysplastic syndromes. These agents have become part of the standard-of-care treatment for most patients who are ineligible for stem-cell transplant, and a key component of bridge therapy for patients who are eligible for stemcell transplant.9–11 Two first-generation hypomethylating agents—decitabine and azacitidine—have been approved for the treatment of myelodysplastic syndromes after demonstrating improved outcomes in clinical trials.12–15 Azacitidine was first approved in the USA in 2004, and decitabine was approved in 2006 for higher-risk myelodysplastic syndromes and chronic myelomonocytic leukaemia. Although these are effective agents that lead to haematological improvement and enhanced health-related quality of life for many patients, they produce a complete response in only a few patients,12–16 and are limited by their short half-lives and in-vivo exposure time.
Guadecitabine (SGI-110; Astex Pharmaceuticals, Pleasanton, CA, USA), a next-generation hypomethylating agent, is a dinucleotide comprising decitabine and deoxyguanosine.17,18 Unlike decitabine, guadecitabine is relatively resistant to degradation by cytidine deaminase. Guadecitabine slowly releases its active metabolite decitabine following subcutaneous administration. This attribute results in prolonged exposure time and a reduced maximum plasma concentration of decitabine.19 The prolonged exposure time might contribute to better activity, whereas the reduced maximum plasma concentration might avoid peak-related toxicities. In a phase 1 dose escalation trial done in heavily pretreated patients with myelodysplastic syndromes and acute myeloid leukaemia, guadecitabine 60 mg/m2 administered as a small-volume subcutaneous injection for 5 consecutive days every 28 days was identified as the biologically effective dose and 90 mg/m2 on the same 5-day schedule was identified as the maximum tolerated dose in patients with myelodysplastic syndromes; the maximum tolerated dose was not reached in patients with acute myeloid leukaemia.19 We aimed to compare the activity and safety of guadecitabine 60 and 90 mg/m2 on a 5-day schedule in patients who were treatment-naive or patients with relapsed or refractory disease with intermediate-risk or high-risk myelodysplastic syndromes, chronic myelomonocytic leukaemia or acute myeloid leukaemia using a phase 2 dose-expansion study. Phase 2 results for the acute myeloid leukaemia cohorts showed promising activity.20,21 The phase 2 study results for patients with myelodysplastic syndromes, including chronic myelomonocytic leukaemia, given guadecitabine 60 or 90 mg/m2 on the 5-day schedule are presented here.
Methods
Study design and participants
This dose-expansion study was part of a phase 1/2 multicentre, open-label, randomised, dose-ranging study of guadecitabine in patients with myelodysplastic syndromes, including chronic myelomonocytic leukaemia or acute myeloid leukaemia. Phase 1 results for patients with myelodysplastic syndrome or acute myeloid leukaemia,19 and phase 2 results for patients who were treatment-naive with acute myeloid leukaemia who were not candidates for intensive chemotherapy,20 and patients with relapsed or refractory acute myeloid leukaemia,21 have been published previously. The present phase 2 study in patients with myelodysplastic syndromes or chronic myelomonocytic leukaemia who were hypo-methylating agent treatment-naive or had relapsed or refractory disease was done at 13 US centres and one Canadian centre (hospitals and specialist cancer clinics; appendix p 1).
Patients enrolled in the study were aged 18 years or older, with a confirmed diagnosis of IPSS intermediate-1, intermediate-2, or high-risk myelodysplastic syndromes, or chronic myelomonocytic leukaemia. They were either hypomethylating agent treatment-naive or had relapsed or refractory disease after previous hypomethylating agent treatment as determined by the investigators’ judgment (two or more complete full-dose cycles of a hypomethylating agent). The IPSS score in patients with relapsed or refractory myelodysplastic syndromes had been established at the time of their initial diagnosis. Eligible patients had Eastern Cooperative Oncology Group performance status of 0–2; adequate renal function (serum creatinine ≤1·5-times the upper limit of normal [ULN]) and adequate hepatic function (total bilirubin ≤2-times the ULN, and aspartate and alanine transaminases ≤2·5-times the ULN); had not undergone major surgery within 4 weeks or haemopoietic stem-cell transplantation within 8 weeks; or received chemotherapy within 2 weeks or nitrosoureas within 6 weeks of guadecitabine treatment (hydroxycarbamide was allowed during treatment cycle 1). Patients with previous allogeneic stem-cell transplants were only eligible if they had no evidence of active graft-versus-host disease and had discontinued immunosuppressive therapy 2 weeks or more before receiving the study drug.
Patients with acute promyelocytic leukaemia, previous malignancy (except for adequately treated basal cell or squamous cell skin cancer, in-situ cervical cancer, or other cancers from which the patient had been disease free for ≥3 years), life-threatening illness other than myelodysplastic syndromes or acute myeloid leukaemia, symptomatic arrhythmias, New York Heart Association class 3 or 4 heart disease, symptomatic CNS metastases, known HIV infection, or active infection with hepatitis B or C virus were excluded. Patients with grade 2 or worse toxicity (using Common Terminology Criteria for Adverse Events version 4.0) from previous therapy (except for alopecia), individuals who had been given any investigational drug within 2 weeks of randomisation, individuals who received radiotherapy for extramedullary disease within 2 weeks, and individuals with treatment concurrent with systemic corticosteroids for myelo-dysplastic syndromes were also excluded.
All patients provided written informed consent before participating in the study. The study was done in accordance with the ethical principles of the Declaration of Helsinki and in compliance with International Conference on Harmonisation Good Clinical Practice guidelines. The protocol was approved by the institutional review board or independent ethics committee at each centre.
Randomisation and masking
Randomisation was done centrally by Astex Pharmaceuticals using a stratified, dynamic randomisation process provided by the Clintrial (release 4.7) data management system to randomly assign patients (1:1) to receive subcutaneous guadecitabine at either 60 or 90 mg/m2. Treatment allocation was stratified by disease status: hypomethylating agent treatment-naive or relapsed or refractory (previous treatment with hypomethylating agent). Randomisation was dynamic (ie, the probability of assigning a patient to a dose group by the programme was dependent on the number of patients already randomised to each group in the overall trial). Treatment assignment information was communicated by Astex to the clinical sites at the time of patient randomisation. The trial was open-label and thus no masking was involved. Patients were enrolled by investigators and other site staff, who continued to be involved in their clinical care.
Procedures
Patients received subcutaneous guadecitabine 60 or 90 mg/m2 on days 1–5 of a 28-day treatment cycle. The intention was to provide the standard planned dose for four or more cycles; dose reduction could be instituted, as necessary, and the length of treatment cycles could be extended up to 42 days to allow for bone-marrow recovery in cases of severe cytopenia. Additional delay or dose reduction was allowed for recovery from toxicity in previous cycles based on the physician’s judgment.
Haematological responses were monitored by analysis of blood and bone-marrow aspiration. After the initial bone-marrow aspirate screening at baseline, the results of peripheral blood assessments determined the frequency of subsequent bone-marrow aspiration to confirm response or assess drug-related bone-marrow toxicity. Complete peripheral blood counts and white blood cell differentials were measured at least once a week, including granulocyte and platelet numbers, and haemoglobin concentration.
Safety was monitored throughout the study by physical examinations and clinical laboratory tests, including haematology, chemistry for liver and renal function, urinalysis, pregnancy tests, pharmacokinetics, epigenetics, buccal swab, and pharmacogenetic markers. Electrocardiograms were obtained at baseline screening and day 1 of each treatment cycle. Adverse events were coded to system organ class and preferred term using the Medical Dictionary for Regulatory Activities version 14.0. Severity of adverse events was categorised according to the Common Terminology Criteria for Adverse Events version 4.0.
Whole-blood samples were collected immediately before treatment, once a week during the first treatment cycle, and then on day 1 of subsequent cycles for demethylation analysis. Global DNA methylation was measured by the long interspersed nuclear element-1 (LINE-1) methylation assay and changes in methylation from baseline were assessed as previously described.22
Outcomes
The primary endpoint was overall response, which was a composite of complete response, partial response, marrow complete response, and haematological improvement, based on 2006 International Working Group response criteria in myelodysplasia.23 Overall response was assessed by local investigators. Secondary endpoints included the individual components of overall response, time to response, duration of response, overall survival, blood transfusion and platelet transfusion independence at weeks 8 and 16, and safety, including the incidence of adverse events, and all-cause early mortality at 30, 60, and 90 days. Genetic data were collected in a subset of patients from both phases 1 and 2, and will be reported separately. Although baseline cytogenetics were collected, follow-up samples were not and, thus, the proportion of patients who had a cytogenetic response are not available. Exploratory analyses were done to examine the association between baseline characteristics and overall response.
Statistical analysis
Initially, a minimum of 30 patients were to be enrolled in each treatment group (treatment-naive and relapsed or refractory myelodysplastic syndrome groups). This sample size was selected such that if no responses were observed, it could be concluded with 95% confidence that the proportion of patients with a response was less than 10% and further evaluation of that dose was not warranted. The protocol allowed a safety review committee to expand the number of participants in either or both groups to 50 if justified by promising activity and safety data. Initially, a minimum of 30 patients were enrolled at each dose so that if no overall response was observed, it could be concluded with 95% confidence that the proportion with an overall response was less than 10% and not worthy of further study. Because overall responses were seen at both doses, enrolment continued to approximately 50 patients per dose level. These criteria were pre-specified.
Activity and safety endpoints were evaluated for all patients who received at least one dose of study drug, and no patients who received treatment were excluded from the analyses. The proportion of patients who had an overall response was defined as the number of patients achieving an overall response divided by the total number of patients; its 95% CI was based on binomial distribution. Comparison of the proportion of patients who had an overall response between the guadecitabine 60 and 90 mg/m2 groups was made using Fisher’s exact test. Time to response and duration of response were summarised in days using descriptive statistics. Overall survival was analysed using the Kaplan-Meier method, with survival time censored on the last date of contact if the patient was alive or lost to follow-up. Blood and platelet transfusion independence were summarised descriptively in the subset of patients who were transfusion-dependent at baseline. Change from baseline LINE-1 methylation and maximum LINE-1 demethylation were also summarised descriptively, with maximum LINE-1 demethylation defined as the largest percent decrease from baseline in methylation values by patient between days 8 and 22 of the first treatment cycle.
Treatment-emergent adverse events, defined as events that first occurred or worsened after the first dose of study drug until 30 days after the last dose or until the start of an alternative anticancer treatment (whichever occurred first), and all-cause 30-day, 60-day, and 90-day cumulative mortality were summarised descriptively. All statistical analyses were done with SAS 9.3.
This study is registered with ClinicalTrials.gov, number NCT01261312.
Role of the funding source
The funder ofthe study had a role in the study design, data collection, data analysis (including LINE-1 assays), data interpretation, and writing of the report. Representatives of the funder (YH, HNK, and MA) had access to the raw data. All authors had full access to all study data and the corresponding author had final responsibility for the decision to submit for publication.
Results
Between July 9, 2012, and April 7, 2014, 105 patients were enrolled of whom 51 were hypomethylating agent treatment-naive and 54 had relapsed or refractory disease (figure 1; appendix p 1). Overall, 55 patients were randomly assigned to guadecitabine 60 mg/m2 and 50 patients were randomly allocated to guadecitabine 90 mg/m2. Three patients did not receive guadecitabine (one patient who was treatment-naive and one patient with relapsed or refractory disease allocated to the 60 mg/m2 group, and one patient who was treatment-naive allocated to the 90 mg/m2 group) and were not included in these analyses.
Figure 1: Trial profile.
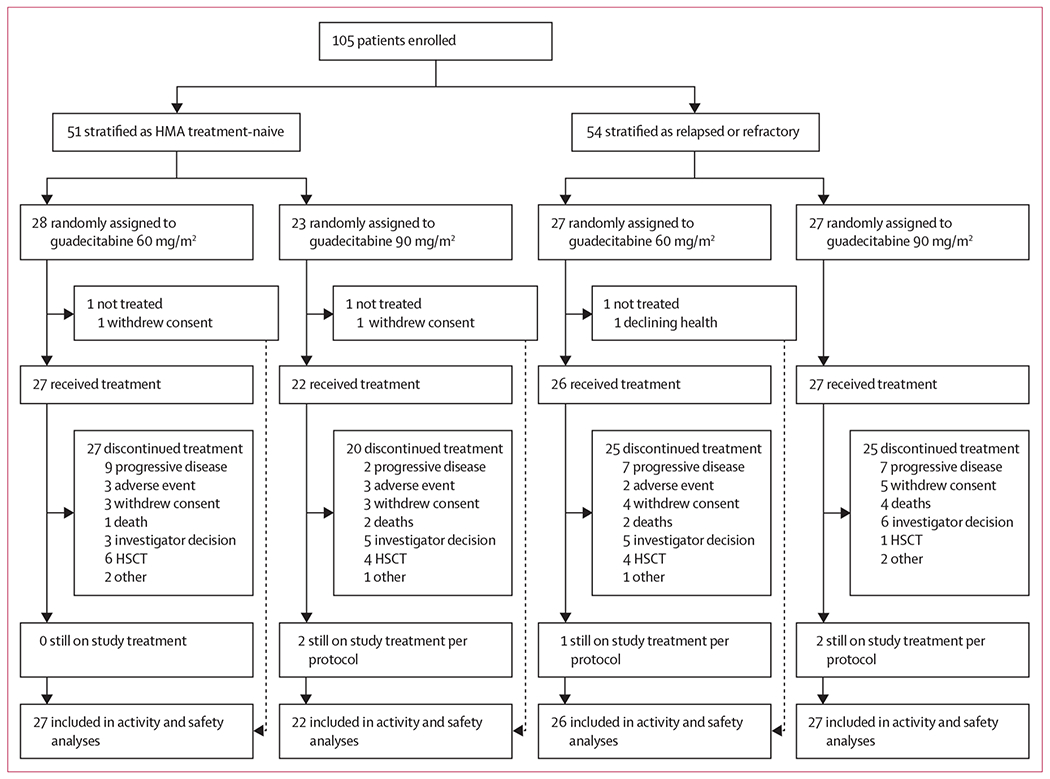
Per protocol, treatment continues until progression. HMA=hypomethylating agent. HSCT=haemopoietic stem-cell transplantation.
Median follow-up was 3·2 years (IQR 2·8–3·5) for the entire study population. Despite the long-term follow-up in this study, 22 patients were still alive at the time of the database lock on Aug 24, 2016, including five patients who continued to receive study treatment and eight patients who underwent or planned to undergo haemopoietic stem-cell transplantation.
Median patient age was 72 years (range 18–89 years), with similar demographic characteristics between guadecitabine dose groups and disease cohorts (table 1). Among patients who were hypomethylating agent treatment-naive, baseline characteristics were generally well balanced between guadecitabine dose groups, although numerically fewer patients in the 60 mg/m2 group had a bone-marrow blast percentage of more than 5% than in the 90 mg/m2 group. As expected, the relapsed or refractory cohort had a longer disease duration, and higher proportions of patients with high-risk myelodysplastic syndromes and bone-marrow blast percentages of more than 5% than the hypomethylating agent treatment-naive cohort. Among patients with relapsed or refractory disease, baseline characteristics were also generally balanced between guadecitabine groups, except that the 60 mg/m2 group had a higher proportion of patients with chronic myelomonocytic leukaemia, and lower proportions with high-risk myelodysplastic syndromes and bone-marrow blast percentages of more than 5%. 51 (96%) of 53 patients with relapsed or refractory disease had received previous hypomethylating agent treatment; 30 (57%) patients had received the treatment within the preceding 3-month period and 41 (77%) for an adequate duration of therapy of 6 months or more. In addition to hypomethylating agents, previous treatment regimens included lenalidomide, other cytotoxic agents, and supportive drugs. Overall, 58 (57%) of 102 patients were red blood cell (RBC) transfusion-dependent and 28 (27%) were platelet transfusion-dependent.
Table 1:
Demographic and baseline characteristics
| HMA treatment-naive MDS |
Relapsed or refractory MDS |
|||
|---|---|---|---|---|
| Guadecitabine 60 mg/m2 (n=27) | Guadecitabine 90 mg/m2 (n=22) | Guadecitabine 60 mg/m2 (n=26) | Guadecitabine 90 mg/m2 (n=27) | |
| Age (years) | 72 (18–85) | 71 (64–85) | 73 (55–86) | 72 (52–89) |
|
| ||||
| Sex | ||||
| Men | 21 (78%) | 14 (64%) | 16 (62%) | 16 (59%) |
| Women | 6 (22%) | 8 (36%) | 10 (38%) | 11 (41%) |
|
| ||||
| Ethnic origin* | ||||
| White | 25 (93%) | 22 (100%) | 25 (96%) | 25 (93%) |
| Asian | 1 (4%) | 0 | 1 (4%) | 1 (4%) |
| Black | 0 | 0 | 0 | 1 (4%) |
|
| ||||
| ECOG performance status | ||||
| 0 | 6 (22%) | 7 (32%) | 6 (23%) | 5 (19%) |
| 1 | 19 (70%) | 14 (64%) | 14 (54%) | 17 (63%) |
| 2 | 2 (7%) | 1 (5%) | 6 (23%) | 5 (19%) |
|
| ||||
| IPSS classification | ||||
| Intermediate-1 risk | 14 (52%) | 9 (41%) | 2 (8%) | 2 (7%) |
| Intermediate-2 risk | 1 (4%) | 4 (18%) | 6 (23%) | 7 (26%) |
| High risk | 6 (22%) | 3 (14%) | 9 (35%) | 16 (59%) |
| CMML | 6 (22%) | 6 (27%) | 9 (35%) | 1 (4%) |
| Not evaluable | 0 | 0 | 0 | 1 (4%) |
|
| ||||
| Time since diagnosis (days) | 35 (6–2257) | 34 (3–2237) | 727 (26–3090) | 466 (15–3202) |
|
| ||||
| Previous regimens (number)† | 0 (0–1) | 0 (0–1) | 1 (1–4) | 1 (1–4) |
|
| ||||
| Previous HMA (decitabine or azacytidine) | 1 (4%) | 0 | 24 (92%) | 27 (100%) |
|
| ||||
| Time since last HMA treatment (% of those with previous HMA) | ||||
| <3 months | 1/1 (100%) | NA | 16/24 (67%) | 14/27 (52%) |
| ≥3 months | 0/1 | NA | 8/24 (33%) | 13/27 (48%) |
|
| ||||
| Duration of previous HMA treatment (% of those with previous HMA) | ||||
| <6 months | 1/1 (100%) | NA | 3/24 (13%) | 7/27 (26%) |
| ≥6 months | 0/1 | NA | 21/24 (88%) | 20/27 (74%) |
|
| ||||
| Bone-marrow blasts | 2 (0–13) | 7 (0–14) | 6 (0–18) | 9 (1–19) |
| ≤5% | 20 (74%) | 10 (45%) | 13 (50%) | 6 (22%) |
| >5% | 7 (26%) | 12 (55%) | 13 (50%) | 21 (78%) |
|
| ||||
| Peripheral blood blasts | 0 (0–13) | 0 (0–6) | 0 (0–32) | 0 (0–21) |
|
| ||||
| Platelets (× 109 per L) | 54 (12–424) | 73 (9–1202) | 39 (15–328) | 35 (7–210) |
|
| ||||
| Neutrophils (× 109 per L) | 1·2 (0·2–36·9) | 2·6 (0·8–16·1) | 1·2 (0·1–13·3) | 0·5 (0·1–15·6) |
|
| ||||
| Haemoglobin (g/dL) | 9·3 (6·9–16·4) | 9·0 (7·7–12·3) | 9·3 (7·1–12·9) | 9·5 (74–13·5) |
|
| ||||
| RBC transfusion-dependent | 15 (56%) | 9 (41%) | 16 (62%) | 18 (67%) |
|
| ||||
| Platelet transfusion-dependent | 7 (26%) | 5 (23%) | 6 (23%) | 10 (37%) |
Data are median (range) or n (%) unless otherwise specified.
HMA=hypomethylating agent. MDS=myelodysplastic syndrome. ECOG=Eastern Cooperative Oncology Group. IPSS=International Prognostic Scoring System. CMML=chronic myelomonocytic leukaemia. RBC=red blood cell. NA=not applicable.
Ethnic origin data missing for one individual.
All 53 patients with relapsed or refractory disease received previous therapy for MDS; all received previous HMA treatment, except for one patient with CMML who received multiple cycles of ruxolitinib, and one with high-risk MDS who received multiple cycles of rigosertib and lenalidomide. One patient in the HMA treatment-naive cohort received one cycle of decitabine, consistent with the eligibility criteria for this cohort.
The median number of treatment cycles was 5·0 (range 1–49) in the guadecitabine 60 mg/m2 group and 4·5 (range 1–41) in the guadecitabine 90 mg/m2 group among patients who were hypomethylating agent treatment-naive. 13 (48%) patients who were treatment-naive receiving guadecitabine 60 mg/m2 and ten (45%) patients who were treatment-naive receiving guadecitabine 90 mg/m2 went on to receive six or more cycles of therapy. Likewise, nine (35%) patients with relapsed or refractory disease on the 60 mg/m2 dose and 12 (44%) patients with relapsed or refractory disease on the 90 mg/m2 dose received the drug for six or more cycles. 95 (93%) patients received at least 90% of the planned total dose in the treatment cycles received. Regardless of dose, the proportions of patients with treatment delays generally increased over time in both patients who were treatment-naive (from 14 [32%] of 44 in cycle 2 to 13 [52%] of 25 in cycle 5) and patients with relapsed or refractory disease (from 18 [38%] of 47 in cycle 2 to 15 [56%] of 27 in cycle 5). The proportions of patients with dose reductions also increased over time (from four [9%] of 44 in cycle 2 to seven [28%] of 25 in cycle 5 in the treatment-naive cohort and from seven [15%] of 47 to 15 [56%] of 27 in the relapsed or refractory cohort). The proportions of dose delays and reductions were generally similar between the two guadecitabine doses. Duration of guadecitabine exposure was longer in the hypomethylating agent treatment-naive cohort than in the relapsed or refractory cohort, which was expected because of the longer survival of patients who were treatment-naive.
The primary endpoint of overall response was achieved by 48 (47%; 95% CI 37–57) of 102 patients who received guadecitabine, including 21 (40%; 27–54) of 53 patients in the 60 mg/m2 group and 27 (55%; 40–69) of 49 patients in the 90 mg/m2 group (table 2). There was no clear relationship between overall response and IPSS group as responders were seen at all risk levels in both dose groups (post-hoc analysis). The difference in the proportions of patients who had an overall response between dose levels was not significant (p=0·16). Complete response was similar between groups, occurring in six (11%) of 53 patients in the 60 mg/m2 group and seven (14%) of 49 patients in the 90 mg/m2 group. Haematological improvement was similar between groups. More patients in the 90 mg/m2 group than the 60 mg/m2 group had a marrow complete response, possibly reflecting the greater proportion of patients in that group with baseline bone-marrow blast percentages of more than 5% that could qualify for a marrow complete response or, perhaps, more potent effects in the bone marrow seen with the larger dose. By disease cohort, overall response was achieved by 25 (51%, 95% CI 36–66) of 49 patients who were hypomethylating agent treatment-naive and by 23 (43%, 30–58) of 53 patients with relapsed or refractory disease. A complete response with guadecitabine was reached in 11 (22%) patients who were treatment-naive and two (4%) patients with relapsed or refractory disease. In an exploratory analysis, the proportions of patients who had an overall response were similar between patients with chronic myelomonocytic leukaemia (10 [45%] of 22) and myelodysplastic syndromes (38 [48%] of 80). There was no difference in overall response based on presence or absence of genetic mutations of DNA methyltransferase 3α, tet methylcytosine dioxygenase 2, or tumour protein p53 (appendix p 3).
Table 2:
Proportion of patients with an objective response or haematological improvement
| Guadecitabine dose |
Disease cohort |
All patients (n=102) | |||
|---|---|---|---|---|---|
| 60 mg/m2 (n=53) | 90 mg/m2 (n=49) | Treatment-naive (n=49) | Relapsed or refractory (n=53) | ||
| Overall response* | 21 (40%; 26·5–54·0) | 27 (55%; 40·2–69·3) | 25 (51%; 36·3–65·6) | 23 (43%; 29·8–57·7) | 48 (47%; 37·1–57·2) |
|
| |||||
| Complete response | 6 (11%) | 7 (14%) | 11 (22%) | 2 (4%) | 13 (13%) |
|
| |||||
| Partial response | 0 | 0 | 0 | 0 | 0 |
|
| |||||
| Marrow complete response | 6 (11%) | 16 (33%) | 7 (14%) | 15 (28%) | 22 (22%) |
|
| |||||
| HI | 18 (34%) | 18 (37%) | 21 (43%) | 15 (28%) | 36 (35%) |
| HI-neutrophil | 7 (13%) | 6 (12%) | 7 (14%) | 6 (11%) | 13 (13%) |
| HI-platelet | 10 (19%) | 13 (27%) | 13 (27%) | 10 (19%) | 23 (23%) |
| HI-erythroid | 10 (19%) | 8 (16%) | 13 (27%) | 5 (9%) | 18 (18%) |
| Single-lineage HI | 11 (21%) | 10 (20%) | 11 (22%) | 10 (19%) | 21 (21%) |
| Bilineage HI | 5 (9%) | 7 (14%) | 8 (16%) | 4 (8%) | 12 (12%) |
| Trilineage HI | 2 (4%) | 1 (2%) | 2 (4%) | 1 (2%) | 3 (3%) |
|
| |||||
| No response | 29 (55%) | 20 (41%) | 20 (41%) | 29 (55%) | 49 (48%) |
|
| |||||
| Not evaluable | 3 (6%) | 2 (4%) | 4 (8%) | 1 (2%) | 5 (5%) |
Data are n (%; 95% CI) or n (%). Responses are based on modified 2006 International Working Group Response Criteria in Myelodysplasia.22
HI=haematological improvement.
Defined as complete response, partial response, marrow complete response, or HI. Some patients in the HI category have also been counted in one of the other response categories (ie, complete, partial, or marrow complete response). Patients were counted only once for overall response.
The maximum extent of global DNA demethylation measured by LINE-1 methylation analysis occurred around day 8 and then returned to pretreatment levels by day 28. The mean maximum LINE-1 demethylation with guadecitabine was greater for patients receiving the 90 mg/m2 dose (28·2%, SE 1·5) than individuals receiving the 60 mg/m2 dose (23·9%, 1·4; appendix p 2). Mean maximum LINE-1 demethylation did not differ between patients who were treatment-naive (28·6%, 1·6) and patients with relapsed or refractory disease (28·9%, 1·1), nor did it differ between patients with (28·1%, 1·4) and without (29·4%, 1·3) objective treatment responses.
The median time to response in patients who achieved a complete response or marrow complete response was 85 days (range 23–512) in the entire study population. The median duration of complete response, partial response, or marrow complete response for the entire population was 203 days, and duration of response was longer for patients in the guadecitabine 60 mg/m2 group versus the 90 mg/m2 dose (295 vs 207 days).
Among the 58 patients who were dependent on RBC transfusions at baseline, 15 (26%) became RBC transfusion independent for at least 8 weeks and nine (16%) became transfusion independent for at least 16 weeks (table 3). The rates of RBC transfusion independence were similar between guadecitabine dose levels.
Table 3:
Transfusion independence
| Guadecitabine dose |
Disease cohort |
All patients | |||
|---|---|---|---|---|---|
| 60 mg/m2 | 90 mg/m2 | Treatment-naive | Relapsed or refractory | ||
| Baseline RBC dependence | 31 | 27 | 24 | 34 | 58 |
|
| |||||
| RBC independence for 8 weeks | 7 (23%) | 8 (30%) | 10 (42%) | 5 (15%) | 15 (26%) |
|
| |||||
| RBC independence for 16 weeks | 5 (16%) | 4 (15%) | 6 (25%) | 3 (9%) | 9 (16%) |
|
| |||||
| Baseline platelet dependence | 13 | 15 | 12 | 16 | 28 |
|
| |||||
| Platelet independence for 8 weeks | 3 (23%) | 8 (53%) | 6 (50%) | 5 (31%) | 11 (39%) |
|
| |||||
| Platelet independence for 16 weeks | 2 (15%) | 4 (27%) | 4 (33%) | 2 (13%) | 6 (21%) |
Data are n or n (%) of patients with baseline dependence.
RBC=red blood cell.
Median overall survival was 460 days (95% CI 384–663) for the entire study population, 611 days (408–771) for the guadecitabine 60 mg/m2 group, and 399 days (303–663) for the 90 mg/m2 group, with 67% (52–78) 12-month survival and 39% (26–52) 24-month survival for the 60 mg/m2 group versus 60% (45–72) 12-month survival and 30% (18–43) 24-month survival for the 90 mg/m2 group (figure 2). The number of deaths at database lock for the 60 mg/m2 group was 18 (67%) of 27 patients and 15 (68%) of 22 in the 90 mg/m2 group in the treatment-naive cohort, and 19 (73%) of 26 for the 60 mg/m2 group and 20 (74%) of 27 for the 90 mg/m2 group in the relapsed or refractory cohort. There was no difference in overall survival between doses (p=0·47). Median overall survival was 703 days (95% CI 458–920) in the hypomethylating agent treatment-naive cohort and 352 days (262–505) in the relapsed or refractory disease cohort, with 2-year survival rates of 44% (30–58) in the hypomethylating agent treatment-naive cohort and 25% (14–38) in the relapsed or refractory disease cohort. Survival within each disease cohort did not differ by guadecitabine dose (p=0·56 in patients who were treatment-naive; p=0·89 in patients with relapsed or refractory disease).
Figure 2:
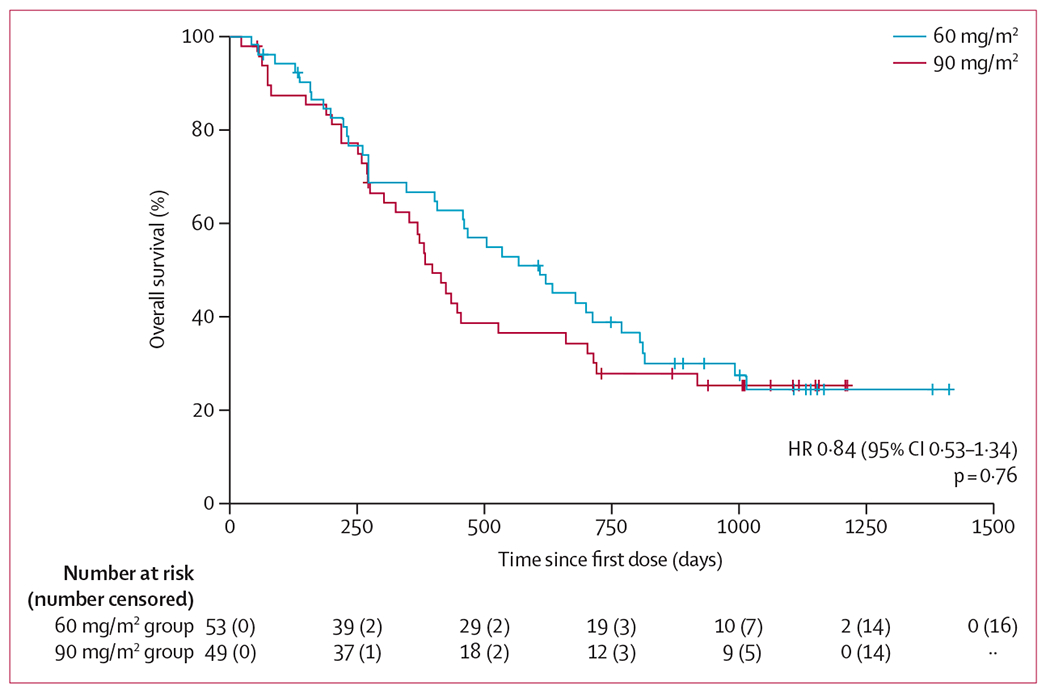
Overall survival by guadecitabine dose group
Adverse events occurring in 10% or more of patients are shown in table 4. The incidence of grade 3 or worse adverse events, regardless of relationship to treatment, tended to be lower with the guadecitabine 60 mg/m2 dose versus the 90 mg/m2 dose (44 [83%] of 53 patients vs 47 [96%] of 49 patients; p=0·054). In the 60 mg/m2 group, the most frequent grade 3 or worse adverse events were thrombocytopenia, neutropaenia, anaemia, febrile neutropaenia, and pneumonia. In the 90 mg/m2 group, the most frequent grade 3 or worse adverse events were thrombocytopenia, neutropaenia, anaemia, febrile neutropaenia, and pneumonia.
Table 4:
Adverse events
| Guadecitabine 60 mg/m2 (n=53) |
Guadecitabine 90 mg/m2 (n=49) |
|||||
|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
| Haematological events | ||||||
|
| ||||||
| Anaemia | 4 (8%) | 23 (43%) | 2 (4%) | 4 (8%) | 19 (39%) | 5 (10%) |
| Neutropaenia | 4 (8%) | 2 (4%) | 19 (36%) | 1 (2%) | 4 (8%) | 21 (43%) |
| Thrombocytopenia | 1 (2%) | 0 | 22 (41%) | 0 | 5 (10%) | 23 (47%) |
| Febrile neutropaenia | 0 | 15 (28%) | 2 (4%) | 0 | 20 (41%) | 1 (2%) |
| Leukopaenia | 0 | 2 (4%) | 5 (9%) | 0 | 2 (4%) | 6 (12%) |
|
| ||||||
| Non-haematological events | ||||||
|
| ||||||
| Injection site pain | 19 (36%) | 0 | 0 | 21 (43%) | 0 | 0 |
| Injection site haematoma | 6 (11%) | 0 | 0 | 11 (22%) | 0 | 0 |
| Injection site nodule | 9 (17%) | 0 | 0 | 8 (16%) | 0 | 0 |
| Fatigue | 21 (40%) | 4 (8%) | 0 | 13 (27%) | 6 (12%) | 0 |
| Diarrhoea | 17 (32%) | 0 | 0 | 21 (43%) | 1 (2%) | 0 |
| Nausea | 18 (34%) | 0 | 0 | 19 (39%) | 1 (2%) | 0 |
| Pneumonia | 2 (4%) | 13 (25%) | 0 | 2 (4%) | 14 (29%) | 1 (2%) |
| Constipation | 15 (28%) | 0 | 0 | 18 (37%) | 0 | 0 |
| Cough | 15 (28%) | 0 | 0 | 15 (31%) | 0 | 0 |
| Confusion | 16 (30%) | 0 | 0 | 11 (22%) | 1 (2%) | 0 |
| Decreased appetite | 12 (23%) | 0 | 0 | 15 (31%) | 0 | 0 |
| Insomnia | 11 (21%) | 0 | 0 | 14 (29%) | 0 | 0 |
| Dyspnoea | 10 (19%) | 1 (2%) | 0 | 12 (24%) | 1 (2%) | 0 |
| Stomatitis | 3 (6%) | 2 (4%) | 0 | 15 (31%) | 3 (6%) | 0 |
| Hypokalaemia | 7 (13%) | 1 (2%) | 0 | 11 (22%) | 3 (6%) | 0 |
| Vomiting | 10 (19%) | 0 | 0 | 11 (22%) | 1 (2%) | 0 |
| Dizziness | 12 (23%) | 0 | 0 | 9 (18%) | 0 | 0 |
| Hypomagnesaemia | 13 (25%) | 0 | 0 | 7 (14%) | 1 (2%) | 0 |
| Peripheral oedema | 12 (23%) | 0 | 0 | 9 (18%) | 0 | 0 |
| Epistaxis | 10 (19%) | 1 (2%) | 0 | 8 (16%) | 1 (2%) | 0 |
| Headache | 6 (11%) | 1 (2%) | 0 | 13 (27%) | 0 | 0 |
| Rash | 11 (21%) | 0 | 0 | 9 (18%) | 0 | 0 |
| Asthaenia | 9 (17%) | 1 (2%) | 0 | 9 (18%) | 0 | 0 |
| Pain in extremity | 9 (17%) | 1 (2%) | 0 | 8 (16%) | 0 | 0 |
| Petechiae | 11 (21%) | 0 | 0 | 7 (14%) | 0 | 0 |
| Cellulitis | 3 (6%) | 3 (6%) | 0 | 4 (8%) | 7 (14%) | 0 |
| Pyrexia | 3 (6%) | 1 (2%) | 0 | 8 (16%) | 3 (6%) | 0 |
| Arthralgia | 7 (13%) | 2 (4%) | 0 | 5 (10%) | 0 | 0 |
| Back pain | 6 (11%) | 2 (4%) | 0 | 5 (10%) | 1 (2%) | 0 |
| Dyspepsia | 5 (9%) | 0 | 0 | 9 (18%) | 0 | 0 |
| Myalgia | 10 (19%) | 0 | 0 | 4 (8%) | 0 | 0 |
| Upper respiratory tract infection | 5 (9%) | 0 | 0 | 8 (16%) | 1 (2%) | 0 |
| Hyponatraemia | 3 (6%) | 4 (8%) | 0 | 5 (10%) | 1 (2%) | 0 |
| Nasal congestion | 6 (11%) | 0 | 0 | 7 (14%) | 0 | 0 |
| Oropharyngeal pain | 6 (11%) | 0 | 0 | 7 (14%) | 0 | 0 |
| Hypotension | 4 (8%) | 1 (2%) | 0 | 6 (12%) | 1 (2%) | 0 |
| Night sweats | 4 (8%) | 0 | 0 | 8 (16%) | 0 | 0 |
| Dehydration | 3 (6%) | 1 (2%) | 0 | 6 (12%) | 1 (2%) | 0 |
| Muscle spasms | 8 (15%) | 0 | 0 | 3 (6%) | 0 | 0 |
| Abdominal pain | 7 (13%) | 0 | 0 | 2 (4%) | 1 (2%) | 0 |
| Rhinorrhea | 6 (11%) | 0 | 0 | 4 (8%) | 0 | 0 |
| Sepsis | 0 | 2 (4%) | 1 (2%) | 0 | 1 (2%) | 4 (8%) |
| Transfusion reaction | 4 (8%) | 1 (2%) | 0 | 4 (8%) | 1 (2%) | 0 |
| Weight decreased | 6 (11%) | 0 | 0 | 4 (8%) | 0 | 0 |
Data are n (%). Two treatment-related deaths occurred (pneumonia with 90 mg/m2 and septic shock with 60 mg/m2) Events occurring in at least 10% of patients and all grade 3 or worse events are shown.
The most common serious adverse events, regardless of relationship to study treatment, were febrile neutropaenia (16 [30%] of 53 patients receiving 60 mg/m2 and 20 [41%] of 49 patients receiving 90 mg/m2) and pneumonia (11 [21%] patients receiving 60 mg/m2 and 14 [29%] receiving 90 mg/m2). 24 (24%) of 102 patients treated had drug-related serious adverse events (11 [21%] of 53 receiving 60 mg/m2 and 13 [27%] of 49 receiving 90 mg/m2). Overall, the most common drug-related serious adverse events were febrile neutropaenia (11 [11%] of 102 patients), pneumonia (seven [7%]), anaemia (three [3%]), and thrombocytopenia (three [3%]).
Seven patients died from serious adverse events, including six patients with relapsed or refractory myelo-dysplastic syndromes (three each in the guadecitabine 60 and 90 mg/m2 groups) and one patient who was hypomethylating agent treatment-naive with myelodysplastic syndromes (60 mg/m2 group). Serious adverse events that led to death were sepsis (two patients with relapsed or refractory disease in the 90 mg/m2 group), septic shock (one patient with relapsed or refractory disease in the 60 mg/m2 group), pneumonia (one patient with relapsed or refractory disease in each group), respiratory failure (one patient with relapsed or refractory disease in the 60 mg/m2 group), and subdural haematoma (one patient who was treatment-naive in the 60 mg/m2 group). Only two serious adverse events that led to death were considered treatment related by the investigator (pneumonia with 90 mg/m2 and septic shock with 60 mg/m2).
Overall, 12 (12%) of 102 patients discontinued treatment with guadecitabine due to adverse events. The overall all-cause mortality with guadecitabine was low. 30-day all-cause mortality occurred in one (1%) of 102 patients, 60-day all-cause mortality occurred in four (4%) patients, and 90-day all-cause mortality occurred in nine (9%) patients. Most deaths occurred in the 90 mg/m2 group (appendix p 4).
Discussion
In the phase 2 part of this phase 1/2 study, guadecitabine 60 and 90 mg/m2 administered for 5 consecutive days every 28 days were clinically active in patients with intermediate-risk and high-risk myelodysplastic syndrome and chronic myelomonocytic leukaemia, with no clinically important differences in activity observed between dose levels. The proportion of patients who had an overall response was numerically but not significantly higher in the 90 mg/m2 group than in the 60 mg/m2 group, whereas duration of response and median overall survival were numerically but not significantly longer in the 60 mg/m2 group than in the 90 mg/m2 group. For secondary endpoints, the prevalence of complete response, haematological improvement, and RBC transfusion independence did not differ between doses. The incidence of marrow complete response was higher in the 90 mg/m2 group, but probably reflected a greater proportion of patients with baseline bone-marrow blast percentages of more than 5% who could qualify for a marrow complete response. Finally, both dose levels produced effective and similar DNA demethylation as measured in the LINE-1 analysis, suggesting that a plateau effect had been achieved at these doses. These findings are consistent with the phase 1 study of heavily pretreated patients with myelodysplastic syndromes and acute myeloid leukaemia19 and the phase 2 study of treatment-naive acute myeloid leukaemia, which identified 60 mg/m2 as the optimal biologically effective dose of guadecitabine.20
Both guadecitabine doses were clinically active in the hypomethylating agent treatment-naive and relapsed or refractory disease cohorts. The results observed with guadecitabine in the treatment-naive cohort compare well with those reported previously for patients who were treatment-naive receiving azacitidine or decitabine in terms of complete response, the proportion of patients who had an overall response, time to onset of response, and duration of response. The variability in reported treatment results in previous studies, however, confounds any comparison across studies.12–14,16 The relatively high proportion of patients who had an overall response (23 [43%] of 53) and long median overall survival (352 days [95% CI 262–505] or 11·7 months) in previously treated patients with myelodysplastic syndromes are of particular interest. Historically, patients with higher-risk myelodysplastic syndromes who have not had successful previous hypomethylating agent treatment have a poor prognosis, with an estimated survival of 4–6 months and no approved therapies. Further, in a 2016 randomised study,24 rigosertib compared with best supportive care had a median overall survival of 8·2 months (95% CI 6·1-10·1) versus 5·9 months (4·1–9·3). The majority of patients with relapsed or refractory myelodysplastic syndromes in the present study received at least 4–6 months of treatment with a previous hypomethylating agent—21 (88%) of 26 in the 60 mg/m2 group and 20 (74%) of 27 in the 90 mg/m2 group had 6 months or more of previous hypomethylating agent treatment.
No major differences were observed in LINE-1 demethylation between the two disease cohorts, suggesting that previous hypomethylating agent treatment did not affect the extent of demethylation by guadecitabine. Likewise, the activity of hypomethylating agents is S phase-dependent, but due to the short half-lives of the first-generation hypomethylating agents (decitabine and azacitidine), there might be insufficient drug exposure to S phase cancer cells. The conversion of guadecitabine to decitabine is efficient, and the mean half-life and exposure time of decitabine following guadecitabine administration are more than doubled compared with intravenous decitabine dosing,19 and nearly double the half-life of azacitidine after its subcutaneous administration.25 Consequently, exposure time of the S phase cells to the hypomethylating agent would be much longer after subcutaneous guadecitabine administration than that expected after decitabine or azacitidine administration. This mechanism might explain how guadecitabine could rescue patients from pharmacological resistance (although probably not biological resistance).
The safety profile of guadecitabine was consistent with the myelosuppression known to occur with hypomethylating agents and the complications typically associated with myelodysplastic syndromes. Grade 3 or worse adverse events tended to be more common overall with the 90 mg/m2 dose than with the 60 mg/m2 dose, reflecting small increases in the incidence of thrombo-cytopenia, neutropenia, febrile neutropenia, and pneumonia. In the context of this fatal disease, guadecitabine showed an acceptable safety profile.
Several study limitations should be recognised. First, the small sample size limits any definite conclusions that can be drawn from statistical comparisons; however, there were no major differences observed between the guadecitabine dose groups. Second, the study randomly assigned patients to two different guadecitabine dose levels, with no other treatment comparator, which limits the ability to compare study results with those from other studies. Lastly, potential clearance of some mutations in patients who achieved prolonged response was not available for analysis.
In summary, guadecitabine 60 mg/m2 showed similar activity, and a trend for better safety and tolerability than with 90 mg/m2 and, therefore, offers the better benefit-to-risk ratio for patients with myelodysplastic syndromes. Moreover, for patients with relapsed or refractory disease, guadecitabine showed promising activity that warrants further investigation. A phase 3 study of guadecitabine 60 mg/m2 compared with the investigators’ choice of treatment in patients with relapsed or refractory myelo-dysplastic syndrome is ongoing (NCT02907359).
Supplementary Material
Research in context.
Evidence before this study
Published data and clinical literature on the treatment of myelodysplastic syndromes were reviewed by experts in the field. Azacitidine and decitabine are first-generation hypomethylating agents that were shown to be more effective than best supportive care in phase 3 clinical trials, and were subsequently approved for use in treating myelodysplastic syndromes. The proportion of patients who have an overall response to these agents is small. Prognosis for relapsed or refractory myelodysplastic syndromes is very poor. Data from a phase 1 trial of guadecitabine, a next-generation hypomethylating agent, administered for 5 days every 28-day treatment cycle, identified 60 mg/m2 as the biologically effective dose and 90 mg/m2 as the maximum tolerated dose. Responses in patients with relapsed or refractory disease were observed in a phase 1 trial, but the dose level providing an optimal benefit-to-risk profile was not found.
Added value of this study
This phase 2 study investigated the activity and safety of guadecitabine in adult patients with International Prognostic Scoring System intermediate-1-risk, intermediate-2-risk, or high-risk myelodysplastic syndromes, including chronic myelomonocytic leukaemia, using the 60 and 90 mg/m2 doses identified in the phase 1 section of the trial. The study included both patients who were hypomethylating agent treatment-naive and individuals with relapsed or refractory disease following previous hypomethylating agent treatment. The results show that both dose levels provided similar proportions of patients who had overall response, had clinical activity in treatment-naive and relapsed or refractory patients, and showed an acceptable safety profile; however, grade 3 or higher adverse event rates tended to be increased with the 90 mg/m2 dose. To date, this is the largest study with guadecitabine in these populations.
Implications of all the available evidence
Based on previous evidence we recommend guadecitabine in patients with myelodysplastic syndrome at 60 mg/m2 on a 5-day schedule, reflecting the similar activity and better safety profile than the 90 mg/m2 dose. The proportion of patients who had a response with relapsed or refractory disease was higher than expected and suggests guadecitabine could be a potential new therapeutic option for such patients and warrants further investigation. A phase 3 trial (NCT02907359) in this population compared with the investigators’ treatment choice is ongoing.
Acknowledgments
This trial was supported by Astex Pharmaceuticals and Stand Up To Cancer Dream Team Translational Research Grant SU2C-AACR-DT0109. Stand Up To Cancer is a programme of the Entertainment Industry Foundation administered by the American Association for Cancer Research. We also acknowledge Geoff Marx, Amy Volpert, and Barry M Weichman of BioScience Communications, New York, NY, USA, for providing medical writing and editorial support (funded by Astex Pharmaceuticals).
Declaration of interests
GG-M has received honoraria and research funding from Astex/Otsuka. GR has received research funding from Astex and Cellectis; and has consulted for Astex, AbbVie, Amphivena, Argenx, Astellas, Bayer, Celgene, Celltrion, Daiichi Sankyo, Eisai, Janssen, Jazz, Novartis, MEI Pharma, Orsenix, Otsuka, Pfizer, Roche/Genentech, Sandoz, and Takeda. KW and RT have received research funding from Astex. HK has received honoraria from AbbVie, Agios, Amgen, Immunogen, Orsinex, Pfizer, and Takeda; has participated in advisory board meetings for Actinium; and has received grants from Astex, AbbVie, Agios, Amgen, Ariad, BMS, Cyclacel, Daiichi-Sankyo, Immunogen, Jazz, Novartis, and Pfizer. ER has received grants from Jazz and Pfizer; and has received personal fees from Agios, Celgene, Incyte, Novartis, Pfizer, and Toledo. CO has received grants from Stand Up To Cancer and Van Andel Research Institute; and has received personal fees from Otsuka. KY has received research funding from Astex, GlaxoSmithKline, Millennium, and MSD; and has participated in advisory board meetings for Celgene and Otsuka. WS has participated in advisory board meetings for Amgen, Jazz, Novartis, and Pfizer; and has consulted for Adaptive Biotechnology. EG has received research funding from Astex; has participated in advisory boards for Alexion, Celgene, New Link Genetics, and Otsuka; has received manuscript development support from Novartis; and has participated in educational development programs for the National Organization for Rare Diseases. NP has received research funding for his institution from Astex, Boehringer Ingelheim, Celator, CTI Biopharma, Daiichi Sankyo, Genentech, LAM Therapeutics, Pfizer, and Sunesis; has participated in advisory board meetings for Alexion, CTI Biopharma, and Pfizer; and has received grants from Celgene. JB has received grants from Astex, AbbVie, Amgen, Bluebird, BMS, Celgene, Genentech, Glenmark, Janssen, Novartis, Poseida, Sanofi, Takeda, and Teva. J-PJI has received personal fees from Astex, GlaxoSmithKline, Janssen, Otsuka, and Teva. YH, HNK, and MA are employees of Astex. MRS has received research funding from Astex, Boehringer Ingelheim, Gilead, Incyte, Millennium, Sunesis, and TG Therapeutics; has consulted for Astex, Celgene, Gilead, Incyte, Karyopharm, Millennium, Sunesis, and TG Therapeutics; and has equity in Karyopharm. PK, SL, TR, JM, and EJ have nothing to declare.
Footnotes
Data sharing
Data from this study will be posted approximately 1 year after last patient dosing on ClinicalTrials.gov (number NCT01261312) under study protocol SGI-110-01.
References
- 1.Gangat N, Patnaik MM, Tefferi A. Myelodysplastic syndromes: contemporary review and how we treat. Am J Hematol 2015; 91: 76–89. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med 2009; 361: 1872–85. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079–88. [PubMed] [Google Scholar]
- 4.Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System for myelodysplastic syndromes. Blood 2012; 120: 2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Issa JP. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am 2010; 24: 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia 2014; 28: 497–506. [DOI] [PubMed] [Google Scholar]
- 7.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal hematopoiesis to secondary leukemia. Nat Rev Cancer 2017; 17: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamazaki J, Issa JPJ. Epigenetic aspects of MDS and its molecular targeted therapy. Int J Hematol 2013; 97: 175–82. [DOI] [PubMed] [Google Scholar]
- 9.Kim DY, Lee JH, Park YH, et al. Feasibility of hypomethylating agents followed by allogeneic hematopoietic cell transplantation in patients with myelodysplastic syndrome. Bone Marrow Transplant 2012; 47: 374–79. [DOI] [PubMed] [Google Scholar]
- 10.Montalban-Bravo G, Garcia-Manero G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am J Hematol 2018; 93: 129–47 [DOI] [PubMed] [Google Scholar]
- 11.Sohn SK, Moon JH. When is the optimal timing for allogeneic transplantation in the case of MDS patients treated with hypomethylating agents? Expert Rev Hematol 2013; 6: 389–95. [DOI] [PubMed] [Google Scholar]
- 12.Fenaux P, Mufti G, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10: 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantarjian H, Issa JPJ, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes. Results of a phase III randomized study. Cancer 2006; 106: 1794–803. [DOI] [PubMed] [Google Scholar]
- 14.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the Cancer and Leukemia Group B. J Clin Oncol 2002; 20: 2429–40. [DOI] [PubMed] [Google Scholar]
- 15.Kaminskas E, Farrell A, Abraham S, et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res. 2005; 11: 3604–08. [DOI] [PubMed] [Google Scholar]
- 16.Lübbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol 2011; 29: 1987–96. [DOI] [PubMed] [Google Scholar]
- 17.Chuang JC, Warner SL, Vollmer D, et al. S110, a 5-aza-2’-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol Cancer Ther 2010; 9: 1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoo CB, Jeong S, Egger G, et al. Delivery of 5-aza-2’-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res 2007; 67: 6400–08. [DOI] [PubMed] [Google Scholar]
- 19.Issa JJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol 2015; 16: 1099–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantarjian HM, Roboz GJ, Kropf PL, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol 2017; 18: 1317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roboz GJ, Kantarjian HM, Yee KWL, et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer 2018; 124: 325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang AS, Estécio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–25. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Manero G, Fenaux P, Al-Kali A, et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol 2016; 17: 496–508. [DOI] [PubMed] [Google Scholar]
- 25.Marcucci G, Silverman L, Eller M, Lintz L, Beach CL. Bioavailability of azacitidine subcutaneous versus intravenous in patients with myelodysplastic syndromes. J Clin Pharmacol 2005; 45: 597–602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.