

SUMMARY
PI5P4Ks are a class of phosphoinositide kinases that phosphorylate PI-5-P to PI-4,5-P2. Distinct localization of phosphoinositides is fundamental for a multitude of cellular functions. Here, we identify a role for peroxisomal PI-4,5-P2 generated by the PI5P4Ks in maintaining energy balance. We demonstrate that PI-4,5-P2 regulates peroxisomal fatty acid oxidation by mediating trafficking of lipid droplets to peroxisomes, which is essential for sustaining mitochondrial metabolism. Using fluorescent-tagged lipids and metabolite tracing, we show that loss of the PI5P4Ks significantly impairs lipid uptake and b-oxidation in the mitochondria. Further, loss of PI5P4Ks results in dramatic alterations in mitochondrial structural and functional integrity, which under nutrient deprivation is further exacerbated, causing cell death. Notably, inhibition of the PI5P4Ks in cancer cells and mouse tumor models leads to decreased cell viability and tumor growth, respectively. Together, these studies reveal an unexplored role for PI5P4Ks in preserving metabolic homeostasis, which is necessary for tumorigenesis.
In brief
Ravi et al. demonstrate that PI5P4K activity is required for maintaining energy homeostasis by regulating the crosstalk between peroxisomes and mitochondria. Their findings suggest that the metabolic susceptibility induced by PI5P4K inhibition can be exploited for treating diseases such as cancer.
Graphical Abstract
INTRODUCTION
The phosphatidylinositol-5-phosphate 4-kinases (PI5P4Ks) are a family of non-canonical phosphoinositide kinases that phosphorylate the minor lipid phosphatidylinositol 5-monophosphate (PI-5-P) at the 4-position to generate distinct pools of phosphatidylinositol 4,5-bisphosphate (PI-4,5-P2) largely on intracellular membranes (Rameh et al., 1997). Localization of phosphoinositides (PIs) on cellular membranes is a very dynamic process, where they rapidly cycle between one form to another, leading to the regulation of various cellular functions (Balla, 2013). In mammals, the PI5P4K family consists of three isoforms (α, β, and γ), and the genes encoding the PI5P4Ks are called PIP4K2A, PIP4K2B, and PIP4K2C (Emerling et al., 2013; Lamia et al., 2004; Shim et al., 2016). Notably, the PI5P4K family is conserved back to Drosophila (Lecompte et al., 2008) and C. elegans (Emerling et al., 2013; Weinkove et al., 2008), each having a single catalytically active PI5P4K. The importance of this evolutionarily conserved alternative pathway for generating PI-4,5-P2 as well as the roles of the PI5P4Ks in several aspects of cellular metabolism and cancer are still under active investigation.
To date, we have shown that knocking down both PI5P4Kα and PI5P4Kβ in TP53-mutant breast cancer cells inhibits cell growth and, importantly, that deletion of the enzymes in mice suppresses tumor formation with Trp53 deletion (Emerling et al., 2013). Further, we discovered that PI5P4Ks localize to lysosomes and that they are required for autophagosome-lysosome fusion in the context of p53 loss (Lundquist et al., 2018). Recent studies have also shown that cholesterol transport from lysosomes to peroxisomes is facilitated by membrane contacts formed between the lysosomal protein synaptogamin VII (Syt7) and peroxisomal PI-4,5-P2, the specific product of PI5P4Kα (Chu et al., 2015; Hu et al., 2018). Together, these findings emphasize the importance of PI5P4Ks and their lipid product PI-4,5-P2 for key metabolic signaling nodes as well as for organelle crosstalk that are necessary to maintain metabolic homeostasis. Here, we identify a new role for peroxisomal PI-4,5-P2 generated by the PI5P4Ks in maintaining cellular bioenergetics via coordinating peroxisome- mitochondria communication. Peroxisomes are key regulators of cell metabolism, as they play an indispensable role in many biochemical pathways in mammalian cells. Yet, in order to execute their roles efficiently peroxisomes must also interact both functionally and physically with other cell organelles (Fransen et al., 2017). To this end, more specific metabolic processes like fatty acid (FA) β-oxidation and the maintenance of cellular reactive oxygen species (ROS) homeostasis depend on the concerted actions of both peroxisomes and mitochondria (Fransen et al., 2017; Wanders et al., 2015).
FA β-oxidation is a multistep process that takes place in both peroxisomes and mitochondria in mammalian cells to catalyze the chain shortening of acyl-CoA esters between carbons 2 and 3, yielding various chain-shortened products (Poirier et al., 2006). For each reaction step, peroxisomes and mitochondria contain unique sets of enzymes as well as have different substrate specificities. Very-long-chain fatty acids (VLCFAs), branched-chain fatty acids (BCFAs), and leukotrienes are β-oxidized by peroxisomes (Van Veldhoven, 2010), whereas medium-chain fatty acids (MCFAs) and long-chain fatty acids (LCFAs) are preferentially b-oxidized in mitochondria (Wanders et al., 2010). Unlike the mitochondria that can β -oxidize FAs fully to CO2 and H2O, peroxisomes are only able to chain-shorten their substrates. Therefore, peroxisomal b-oxidation products can only be completely oxidized after they have been shuttled to mitochondria (Fransen et al., 2017; Poirier et al., 2006). Further, NAD+ required by peroxisomes during FA β -oxidation is generated by the mitochondria within the electron transport chain (ETC) (Wanders et al., 2015). With these functions, peroxisomes and mitochondria are interconnected to maintain their integrity and function, and more often than not, peroxisomal disorders are associated with mitochondrial deformities and dysfunction (Lismont et al., 2015; Tanaka et al., 2019).
Metabolic adaptations to the microenvironment (Hanahan and Weinberg, 2011) and particularly alterations in lipid metabolism (Munir et al., 2019) are becoming major hallmarks of cancer. This, together with the dysregulation of redox homeostasis, which are consequential to tumor growth (Ježek et al., 2018), puts mitochondria in the center of the changes that cancer cells undergo. With mitochondrial and peroxisomes tied-in to these processes, peroxisomal functions become relevant to tumor growth and progression. Indeed, studies have shown an increase in peroxisomal lipid metabolism in prostate (Wu et al., 2014), breast (Kim et al., 2015), and various other cancers (Dahabieh et al., 2018). When nutrients are sparse, as in a tumor microenvironment, FAs can be transferred to both peroxisomes and mitochondria by lipid droplets (LDs), organelles that store FAs as neutral lipids (Chang et al., 2019; Rambold et al., 2015), to gear cancer cells toward β -oxidation (Koundouros and Poulogiannis, 2020).
Unquestionably, peroxisomal and mitochondrial metabolic activities are interconnected, and their interactions become essential under metabolic stress, yet the mechanisms that facilitate this communication are still poorly understood. Here, we demonstrate that the PI5P4Ks and their product PI-4,5-P2 are vital for trafficking VLCFAs into peroxisomes, which in the end is necessary for mitochondrial function. This in turn leads to nutrient susceptibility, leading to cell death upon loss of the kinases. This discovery uncovers a new and fundamental role of peroxisomal PI4,5-P2 in maintaining metabolic homeostasis and with a role to play in regulating autophagy, further supports that targeting the PI5P4Ks could be beneficial for targeting the metabolic vulnerabilities of nutrient stress that are commonly associated with the tumor microenvironment as well as being possible targets in the treatment of peroxisome-related diseases.
RESULTS
PI-4,5-P2 generated by PI5P4Ks at the peroxisome is important for lipid metabolism
To assess the role of PI-4,5-P2 generated by PI5P4Ks at the peroxisomes, we first confirmed the localization of the kinases as well as the phospholipid to this organelle in our system. Previous studies have shown that PI-4,5-P2 can be generated on peroxisomal membranes (Jeynov et al., 2006) and that PI5P4Kα is responsible for this pool (Hu et al., 2018). For the first time, we show the physical localization of PI5P4Kα at the peroxisome in mouse prostate tissue (Figure 1A). To enable us to study the function of the kinases, we generated PI5P4Kα and PI5P4Kβ double-knockout lines in mouse embryonic fibroblasts (αβ DKO MEFs) (Figure S1A). Indeed, losing the two most catalytically active isoforms of PI5P4Ks leads to a decrease in PI-4,5-P2 staining at the peroxisome as seen in the αβ DKO MEFs compared with the wild-type (WT) MEFs (Figure 1B). To confirm this, we also used the human epithelial line, 293A and generated a double deletion of PI5P4Kα and PI5P4Kβ using CRISPR (αβ DKO), as well as various rescue lines expressing WT PI5P4Kα (αRes), WT PI5P4Kβ (βRes), and kinase-dead PI5P4Kα D359N (αKD) (Figure S1B). We observed a similar decrease in peroxisomal PI-4,5-P2 localization in the 293A αβ DKO line as compared with 293A WT, and this phenotype was rescued only by PI5P4KαRes and PI5P4KαRes but not PI5P4KαKD (Figure S1C). RNA-seq revealed a dramatic decrease in the peroxisome gene signature in the αβ DKO MEFs as compared with the WT (Figure S1D). We observed that the loss of the kinases does not alter the peroxisome number (Figure S1E) or expression of other membrane markers as measured by qRT-PCR (Figure S1F). However, as previously reported, decrease in the phospholipid levels at the peroxisome can profoundly affect the trafficking events (Chu et al., 2015). Apart from the cholesterol trafficking into the lysosome, peroxisomes are also important for the breakdown of VLCFAs, which undergo sequential twocarbon reduction to generate MCFAs. Further examination of the RNA-seq data revealed that the major peroxisome gene signature that is altered in the αβ DKO MEFs is that of peroxisomal β-oxidation (Figure S1G). qRT-PCR confirmed that the loss of the kinases decreases the expression of various peroxisome lipid transporters (Figure S1H) as well as peroxisome β-oxidation genes (Figure S1I). To test whether loss of PI-4,5P2 at the peroxisome alters its ability to traffic FAs from LDs, which might in turn affect peroxisomal β-oxidation, we treated the cells with cumene hydroperoxide (Cu-OOH), a lipid peroxidation agent that enhances LD-peroxisome contacts, which are necessary for lipid trafficking from the LDs to the peroxisome (Chang et al., 2019). While under control conditions, there is minimal association of LDs and peroxisomes, using fluorescently tagged dye, we observe increased uptake of the FAs from LDs into the peroxisome in the WT but not the αβ DKO MEFs upon lipid peroxidation (Figure 1C). Further, expression of PI5P4KαRes in αβ DKO MEFs rescues this trafficking event into the peroxisome, whereas the PI5P4KαKD fails to do so (Figure S1J), indicating a critical role for PI-4,5-P2 on the peroxisomal membrane.
Figure 1. Lipid trafficking into the peroxisome is regulated by PI5P4Ks.

(A) Mouse prostate epithelium stained for PI5P4Kα (green) and peroxisomal membrane protein PMP70 (red) protein using fluorescent IHC staining of tissue serial sections. Nuclei counterstain with DAPI (blue). Zoomed in inset of the main figure is shown to highlight the specific region of colocalization. Scale bar, 5 μm.
(B) Immunofluorescence detection of PI-4,5-P2 (green), peroxisomal membrane protein PMP70 (red), and nuclear stain DAPI (blue) in MEFs (left) and graph showing the percent of peroxisomes that colocalize with PI-4,5-P2 (right). Imaging was done using 40× oil on Zeiss LSM710. Scale bar, 10 μm.
(C) MEF lines expressing mEmerald-SKL were pulsed with BODIPY 665/676 overnight and treated with 50 μM Cu-OOH for 30 min and stained with Hoechst 33342 (nucleus) prior to imaging (top). Live cell imaging was done 63× oil for on Zeiss LSM710. Scale bar, 10 mm. Graph showing the quantification of BODIPY/ peroxisome colocalization (bottom).
(D) Crystal violet assay was performed at 72 h in WT and ab DKO MEFs grown in control medium or medium supplemented with 100 μM of lignoceric acid (C24:0) or 100 μM hexacosanoic acid (C26:0) as indicated and cell proliferation was plotted. Statistical analysis: n = 3, mean ± SD. A two-way ANOVA was performed, and Tukey’s method was used to correct for multiple comparisons for (C and D). A paired two-tailed t test was used for (B). For imaging experiments ≥ 30 cells were examined per experiment.
The inability of peroxisomes to take up FAs could lead to organellar dysfunction, which is known to be associated with accumulation of VLCFAs as seen in multiple neurodegenerative disorders (Doria et al., 2019). To examine the possible defect in the functioning of the peroxisome, we treated cells with extracellular VLCFAs, namely lignoceric acid (C24:0) and hexacosanoic acid (C26:0). WT MEFs are able to utilize these FAs to supplement their growth, whereas the αβ DKO MEFs are unable to do so and, furthermore, experience a reduction in their growth as a result of lipotoxicity (Figure 1D). Another indication of reduced peroxisomal function is that the decrease in expression and activity of catalase, an enzyme predominantly localized to the peroxisome, is essential for the breakdown of hydrogen peroxide (H2O2) formed as a by-product of peroxisomal b-oxidation. Indeed, the ab DKO MEFs have both decreased catalase expression (Figure S1H) as well as activity (Figure S1K). Notably, catalase activity can be recovered by expression of PI5P4KαRes in the αβ DKO MEFs but not by PI5P4KαKD (Figure S1K). This, along with the decreased capacity of the peroxisome to take up and metabolize FAs further emphasizes the importance of the PI5P4Ks and their product PI-4,5-P2 for peroxisome function.
Loss of PI5P4Ks regulates mitochondrial integrity downstream of the peroxisome
The interplay between peroxisomes and mitochondria is essential for maintaining the energy homeostasis in the cell (Fransen et al., 2017). To investigate if the peroxisomal dysfunction leads to defects in the mitochondria, we used transmission electron microscopy (TEM) to look at cellular structures. We observed myelinoid figures in the mitochondria in the αβ DKO MEFs (Figure 2A), which are consistent with mitochondrial defects seen in peroxisomal disorders such as X-linked adrenoleukodystrophy (X-ALD) (López-Erauskin et al., 2013; McGuinness et al., 2003). Also, the WT MEFs show a lack of staining of the phospholipid on the organelle as compared with the peroxisome, suggesting that these defects are not due to the absence of PI-4,5-P2 on the mitochondria (Figure S2A) but due to the downstream effects of peroxisomal dysfunction. We next looked at how this affects other mitochondrial functions. The oxygen consumption rate (OCR) in the αβ DKO MEFs is lower than the WT cells (Figure 2B). Upon ectopic expression of PI5P4KαRes in the αβ DKO MEFs basal respiration can be partially restored, whereas expression of PI5P4KαKD had no effect (FigureS2B). Mitochondrial respiration oroxidative phosphorylation (OXPHOS) is the process that leads to the generation of ATP in cells (Chatterjee et al., 2011), and in αβ DKO MEFs, we observed reduced ATP levels compared with WT cells (Figure 2C). During the process of OXPHOS, the proton gradient generated across mitochondrial membranes along with an electrical potential is known as the mitochondrial membrane potential (ΔΨm) (MMP) (Zorova et al., 2018). To measure this, we used a dye, namely, tetramethylrhodamine ethyl ester (TMRE), which accumulates in regions of high MMP. Consistent with the lower OCR in αβ DKO MEFs, we observed reduced MMP, as indicated by the decreased accumulation of TMRE in the mitochondria (Figure 2D) compared with the WT MEFs. Substrates for electron transfer to generate ATP during OXPHOS are produced from the breakdown of glucose. The NADH generated during this process gets converted to NAD+, which is the sole supply of NAD+ for peroxisomal β-oxidation (Wandersetal.,2015). The NAD+ generated in αβ DKO MEFs is lower than the WTMEFs, which is rescued by expression of PI5P4KαRes but not PI5P4KαKD (Figure S2C). Taken together with the lack of PI-4,5-P2 on the mitochondrial membrane, the observed mitochondrial defects are conceivably downstream of peroxisome dysfunction.
Figure 2. Defect in mitochondrial structure and function downstream of peroxisome upon loss of the PI5P4Ks.

(A)TEM images of WT and αβ DKO MEFs showing mitochondrial morphology as indicated by the white arrows. Scale bar, 500 nm.
(B) Relative OCR as measured by the Seahorse XFe96 analyzer normalized to protein concentration in the MEFs. Data are presented as mean ± SD of n = 7–8 wells pooled from three independent experiments.
(C) ATP production measured in WT and αβ DKO MEFs.
(D) Mitochondrial membrane potential as visualized by accumulation of TMRE (red) in the mitochondria (MitoTracker Green) and Hoechst 33342 (blue; nucleus) (top). Live cell imaging was done using 63× oil on Zeiss LSM710. Scale bar, 10 μm. Graph showing the fluorescent density ratio TMRE:MitoTracker Green (bottom).
(E) Percent enrichment of glycolysis-derived (left) and TCA cycle (right) metabolites obtained from [U-13C6] glucose tracing.
(F) Cell proliferation measured at 72 h by crystal violet assay in WT and ab DKO MEFs in control medium (left) and upon glucose starvation (right).
(G) Quantification of fluorescence intensity of MitoSOX analyzed by flow cytometry. Statistical analysis: n = 3, mean ± SD. A two-way ANOVA was performed, and Tukey’s method was used to correct for multiple comparisons for (G). A paired two-tailed t test was used for (C–F). For imaging experiments ≥ 30 cells were examined per experiment.
Since glucose catabolism contributes to the generation of the NADH and FADH2 to act as electron donors, as well as other mitochondrial functions, we asked if this process itself is altered. To study this, we performed labeled isotope tracing with [U-13C6] glucose (Figure S2D) in MEFs. While there is no significant difference in glucose enrichment in glycolysis-derived metabolites between WT and ab DKO MEFs, interestingly, the TCA cycle metabolites are enriched in the αβ DKO MEFs (Figure 2E). This is consistent with the increase in expression of mitochondrial pyruvate carrier, Mpc1 (McCommis and Finck, 2015), upon loss of the kinases (Figure S2E). Metabolite analysis also revealed that there is no difference in glucose consumption upon the loss of the kinases (Figure S2F), but there is a reduction in lactate production in these cells (Figure S2G). This is consistent with the increase in TCA cycle metabolites downstream of glucose tracing (Figure 2E). We also observed no change in glutamine consumption (Figure S2H) or glutamate production (Figure S2I), indicating that the changes we see in mitochondrial respiration are glucose dependent. Further, we observe no significant differences in proliferation between WT and αβ DKO MEFs under nutrient-replete conditions, yet removal of glucose from the culture medium leads to cell death in the αβ DKO MEFs, suggesting an enhanced reliance of these cells on glucose as a source of energy (Figure 2F). To confirm that this is a result of mitochondrial dysfunction, cells were grown in glucose-depleted medium supplemented with galactose, which cannot be utilized by the cells, causing them to solely rely on mitochondrial OXPHOS for energy generation (Rossignol et al., 2004). As expected, we observe cell death in the αβ DKO MEFs (Figure S2J). In functional mitochondria, there is formation of ROS due to electron leakage to oxygen (Chatterjee et al., 2011). We next confirmed that the αβ DKO MEFs have lowered ROS production consistent with the decrease in mitochondrial functions, which drastically increases upon glucose starvation, possibly contributing to the cell death (Figure 2G). In correlation to the ROS generated in the cells, we see higher expression of superoxide dismutase (Sod2), an enzyme that is required for the clearance of ROS in the αβ DKO MEFs compared with WT cells, which is negligible upon glucose starvation (Figure S2K). These results indeed indicate that downstream of peroxisome dysfunction, mitochondrial functions are defective as seen by severely impaired OXPHOS upon loss of PI5P4Ks.
Mitochondria upregulate lipid metabolism in response to increased energy load upon loss of PI5P4Ks
Mitochondrial metabolism encompasses glucose as well as FA metabolism. Any sort of nutrient starvation triggers signaling and metabolic changes to allow the cells to cater to energy demands. One such change upon glucose starvation is that, synthesis of ATP now relies on mitochondrial FA β -oxidation, which requires uptake of FAs from LDs (Henne et al., 2018). These LDs are formed to prevent cytotoxicity from excess FAs released into the cytoplasm due to upregulated autophagy, as a result of nutrient starvation (Nguyen and Olzmann, 2017). As glucose depletion leads to a striking phenotype and an increase in ROS in the αβ DKO MEFs, we next examined how FA metabolism is affected in response. To address this, we looked at LD accumulation in the cells in response to glucose deprivation. While both WT and ab DKO MEFs have similar number of LDs, as seen by imaging of BODIPY 493/503, under nutrient-replete conditions, WT MEFs accumulate LDs when subjected to glucose starvation (Figure 3A). This phenotype could be a result of increased lipogenesis or lipolysis. Interestingly, expression of two of the enzymes involved in the LD formation and breakdown, Dgat1 and Atgl, respectively, measured by qRT-PCR are much higher in αβ DKO MEFs under both control and glucose starvation conditions (Figures S3A and S3B). Studies have shown that glucose starvation in MEFs does not lead to an increase in LD biogenesis (Nguyen et al., 2017); therefore, we hypothesized that this accumulation in the WT MEFs is not due to increased biogenesis but likely due to the decreased breakdown of the LDs and their uptake into the mitochondria.
Figure 3. Mitochondria exhibit increased lipid uptake and β-oxidation upon loss of PI5P4Ks.

(A) WT and αβ DKO MEFs were plated and then grown under control or glucose starved conditions for 16 h followed by staining (A) BODIPY FL (green; LDs), LysoTracker Deep Red (red; lysosomes) and Hoechst 33342 (blue; nucleus) (top) and graph showing number of LDs/per cell (bottom).
(B) MitoTracker Green (mitochondria), LysoTracker Deep Red (lysosomes) and Hoechst 33342 (blue; nucleus) (top) and graph showing quantification of percent of short and long fragments of mitochondria per cell (bottom).
(C) MEFs were pulsed with Red-C12 (lipid) overnight and then chased in control medium or HBSS for 16 h. Cells were stained with MitoTracker Green (mitochondria) and Hoechst 33342 (blue; nucleus) for 30 min (top) and graph showing the quantification of colocalization of LDs into the mitochondria as a measure of uptake (bottom). Live cell imaging for (A–C) was done using 63× oil on Zeiss LSM710. Scale bar, 10 μm.
(D) Tracer map showing accumulation of carbons from labeled palmitate in various metabolites (right) and percent citrate enrichment from [U-13C16] palmitate tracing in MEFs (left). Statistical analysis: n = 3, mean ± SD. A two-way ANOVA was performed, and Tukey’s method was used to correct for multiple comparisons for (A–C). # indicates p < 0.001; *** and $ indicate p < 0.01; ** compared with their respective control medium. A paired two-tailed t test was used for (D). For imaging experiments ≥ 30 cells were examined per experiment.
Rambold et al. have shown that fused mitochondria are necessary for the even distribution and uptake of FAs from LDs to ensure efficient mitochondrial β-oxidation (Rambold et al., 2015). Decreased mitochondrial fragmentation in response to glucose starvation in the αβ DKO MEFs compared with WT MEFs (Figure 3B), could provide an explanation for the lack of LD accumulation. Consistent with this observation, the uptake of the fluorescently tagged neutral lipid into the mitochondria is much higher in the ab DKO cells (Figure 3C). Not surprisingly, the mitochondrial lipid transporter, Cpt1a, is highly expressed in the αβ DKO MEFs (Figure S3C). Notably, ectopic expression of PI5P4KαRes reversed the effect of lipid uptake into the mitochondria under starvation compared with ab DKO MEFs, whereas PI5P4KαKD had no effect on the same (Figure S3D). To confirm if this enhanced lipid uptake translates to an increase in the breakdown of lipids in the mitochondria, we performed labeled isotope tracing with [U-13C16] palmitate which showed an enrichment in the citrate derived from palmitate upon the loss of the PI5P4Ks (Figure 3D). However, we observed a lower fraction of M16 palmitate derived from the isotope labeling in the αβ DKO MEFs (Figure S3E). Taken together these results suggest that loss of the kinases increases the energy load on the mitochondria, effects of which are exacerbated upon nutrient starvation. This leads to dysregulated lipid uptake and β-oxidation creating a lipotoxic environment for the mitochondria resulting in cell death.
Mitochondrial dysfunction in PI5P4K/ppk-2-deficient nematodes
C. elegans have a single isoform of the PI5P4K enzyme, namely, PPK-2 (Weinkove et al., 2008). We have previously shown that the PPK-2 is able to convert PI-5-P to PI-4,5-P2 in vitro as well as that the role of this enzyme is conserved in autophagy (Lundquist et al., 2018). To further assess other roles of PPK-2 in C. elegans metabolism, we analyzed the integrity of mitochondria in ppk-2(pk1343) mutant worms. Using MitoTracker Green we show that ppk-2(pk1343) mutant animals have reduced mitochondrial staining and fragmented mitochondria (Figure 4A). This phenotype was accompanied by a lower mitochondrial membrane potential as shown by decreased uptake of TMRE (Figure 4A) compared with the wild-type worms (N2). To confirm that the decreased mitochondrial staining was due to alterations in mitochondrial structure and not content, we used qRT-PCR to measure both mitochondrial (mtDNA) and nuclear DNA (nucDNA). This showed that there was no significant difference in the mtDNA/nucDNA ratio between the N2 and ppk-2(pk1343) deficient worms (Figure 4B). These data support a conserved role for the kinases in regulating mitochondrial structure and function. To validate the dysregulation of mitochondrial function, we next measured the OCR in C. elegans. Similar to our observations in MEFs, loss of ppk-2(pk1343) significantly decreased mitochondrial respiration as compared with N2 worms (Figure 4C). We have previously shown that ppk-2(pk1343)-deficient worms have enhanced sensitivity to metabolic stress and reduced lifespan in response to paraquat, a superoxide inducer (Lundquist et al., 2018). Therefore, we next performed lifespan assays to investigate how this mutant responds to oxidative stress caused by H2O2 (Figure 4D) as well as the mitochondrial toxin, carbonylcyanide p-trifluoromethoxy-phenylhydrazone (FCCP) (Figure 4E). Likewise, loss of ppk-2(pk1343) makes the worms susceptible to both these forms of stress. Further, RNA-seq also indicates a decreased expression of ETC genes and an increased expression of genes involved in mitochondrial β-oxidation in the ppk-2(pk1343)-mutant animals (Figure 4F). These findings further corroborate that the role of PPK-2 is evolutionarily conserved, and this important kinase is indeed a major metabolic sensor.
Figure 4. C. elegans as a model organism for evolutionarily conserved role of PI5P4K in regulating mitochondrial phenotype.

(A) Mitochondrial structure and membrane potential in C. elegans as visualized by accumulation of MitoTracker Green (mitochondria) and TMRE (red). Live cell imaging was done using 63× oil on Zeiss LSM710. Scale bar, 10 μm. Graph showing the intensity of MitoTracker Green (left) and fluorescent density ratio TMRE:MitoTracker Green (right).
(B) Ratio of mtDNA:nucDNA in ppk-2 (KO) worms normalized to the ratio in the N2 (WT) worms.
(C) Relative OCR as measured by the Seahorse XFe24 analyzer normalized to C. elegans number per well. Data are presented as mean ± SD of n = 7–8 wells for an average of 400 animals per experiment. Best of 3 experiments is shown.
(F) Animals were exposed to 15 mM H2O2 over a period of 6 h and percentage of dead animals were recorded.
(G) Lifespan of C. elegans measured in response to chronic exposure to 50 mM FCCP or vehicle (DMSO).
(H) Heatmaps of ETC and mitochondrial b-oxidation genes obtained from RNA-seq data of N2 (WT) and ppk-2 (KO) worms.
Regulation of mitochondrial function extends to multiple cancer cell types
To further evaluate whether the effect of the loss of the kinases seen in MEFs could have a cancer relevance, we generated doxycycline (dox) inducible control (shRenilla) and PI5P4Kα/PI5P4Kβ double-knockdown [shMulti (α,β)] sarcoma cell lines, KPS-142 [undifferentiated pleomorphic high-grade sarcoma (UPS)], HT1080 (human fibrosarcoma), SW872 (human liposarcoma), and OH931 (human myxofibrosarcoma) (Figures S4A–S4D). We selected sarcoma cell lines spanning multiple different subtypes, as sarcomas are very heterogeneous and encompass more than 70 different subtypes (Cancer Genome Atlas Research Network, 2017; Fletcher, 2014). Additionally, for our studies, we generated a human breast epithelial line, MCF10A, in which we deleted PIP4K2A using CRISPR followed by dox-inducible shRNA targeting PIP4K2B as well as inducible shRenilla and shMulti (a,b) triple-negative breast cancer (TNBC) lines (4T1, and MDA-MB-468) (Figures S4E–S4G).
Mitochondrial morphology and functional dynamics have been shown to be highly relevant to tumor establishment and dissemination (Porporato et al., 2018). The mitochondria have the ability to switch their functional phenotypes, which gives cancer cells metabolic plasticity as well as respond to signals from the tumor microenvironment to further support the growth of cancer cells (Roth et al., 2020). Cancers such as TNBC have increased mitochondrial fragmentation, which has been shown to be important for tumor establishment (Kashatus et al., 2015; Serasinghe et al., 2015) and metastasis (Zhao et al., 2013), respectively. Further, recent studies have shown that mitochondrial metabolism is an emerging target for treating multiple subtypes of high-grade sarcomas (Huangyang et al., 2020). To examine if the inhibition of the kinases alters mitochondrial function in the various cancer cells, we measured OCR. Basal mitochondrial respiration is decreased upon knockdown of PI5P4Ks as compared with controls in all sarcoma subtypes tested (Figure 5A) as well as in TNBC cells (Figure S4H). Consistent with the lower OCR, we also observed a concomitant decrease in MMP, as seen by the reduced uptake of TMRE in the sarcoma lines (Figure 5B) as well as in MCF10A and the TNBC lines upon knockdown of the kinases (Figures S4I and S4J). Studies have shown that the majority of cancer types have elevated ROS, which plays a role in not only tumor initiation but is linked to various phenotypes necessary for cancer progression (Storz, 2005), such as in TNBC cells which require high ROS for cancer cell survival (Sarmiento-Salinas et al., 2019). Flowcytometric analysis of all the cell lines for accumulation of MitoSox further revealed a reduction in the ROS generated upon silencing of the PI5P4Ks (Figures 5C, S4K, and S4L).
Figure 5. Silencing of PI5P4Ks affects mitochondrial function in cancer cells.

For all experiments, the shRenilla and shMulti (α,β) lines, KPS-142, HT1080, SW872, and OH931 were treated with 1 μg/mL dox for 14 days prior to the experiment.
(A) Basal respiration rate as measure by Seahorse Xfe24 analyzer and normalized to protein concentrations. Data are presented as a mean ± SD of n = 3 wells.
(B) Quantification of the fluorescent density ratio of TMRE:MitoTracker Green. Mitochondrial membrane potential in OH931 cells as visualized by accumulation of TMRE (red) in the MitoTracker Green (mitochondria) and Hoechst 33342 (nucleus). Live cell imaging was done using 63× oil on Zeiss LSM710. Scale bar, 10 mm.
(C) Quantification of fluorescence intensity of MitoSOX analyzed by flow cytometry. Cell proliferation measured at 72 h by crystal violet in control medium and upon low glucose (1%) in the (D–F) (D) KPS-142, (E) HT1080, (F) SW872, and (G) OH931 cell lines (left) and representative image of crystal violet accumulation in the OH931 cells under the different treatments. Statistical analysis: n = 4, mean ± SD. A two-way ANOVA was performed, and Tukey’s method was used to correct for multiple comparisons for (D–G). A paired two-tailed t test was used for (A–C).
Interestingly, in sarcomas, targeting mitochondrial functions allows for exploitation of metabolic vulnerabilities upon glucose starvation (Huangyang et al., 2020; Issaq et al., 2014). Upon the suppression of the PI5P4Ks, all cancer cell lines other than MCF10A cells show inhibition of cell growth even under control conditions (Figures 5D–5G and S4M–S4O). This phenotype observed in the MCF10A cells, a breast epithelial line, under basal conditions is consistent with our observation in the MEFs (Figure 2F). However, all cell lines subjected to glucose deprivation show substantial cell death upon PI5P4K inhibition (Figures 5D–5G and S4M–S4O). This provides strong evidence for the role of the kinases as key metabolic regulators and potential therapeutic targets in both sarcomas and TNBC, cancers which currently do not have effective treatment options.
Transcriptomics and lipidomics corroborate role of PI5P4Ks in peroxisome-mitochondria crosstalk in cancer
To investigate whether the kinases play a role in sarcoma, we examined the correlation of their expression in normal adipose tissue and liposarcoma and observed that liposarcomas indeed have higher levels of both PIP4K2A (Figure 6A) and PIP4K2B (Figure 6B) expression (Barretina et al., 2010). These expression levels also correlate with decreased survival in sarcoma patients as recently reported by Zhang et al. (Zhang et al., 2018) and in Figure S5A. Due to the limitation of available data for sarcoma in The Cancer Genome Atlas (TCGA), we focused on the breast cancer dataset. Previously, we have established the elevated expression of both PI5P4Kα and PI5P4Kβ in breast tumors compared with normal breast tissue (Emerling et al., 2013). Through examination of TCGA dataset for breast adenocarcinomas (TCGA-BRCA), we sought to interpret correlations between elevated PIP4K2 expression and that of regulatory genes involved in peroxisome-mitochondria interactions. Our initial assessment of normal breast tissue to primary breast tumor samples reveals marked regulatory expression differences of this axis (Figure 6C). We next analyzed whether the varying expression levels of PIP4K2A within the tumor samples have an effect on the regulation of peroxisomal genes as well as those controlling the formation and dissolution of LDs. To this end, we segregated the samples into low expression of PIP4K2A in the bottom quartile (<25%) or high expression of PIP4K2A in the top quartile (>75%) and compared the FKPM (fragments per kilobase million) values of individual genes. Comparable with the qRT-PCR data obtained in the MEFs, breast tumor tissues with high expression of PIP4K2A show correlation with increased expression of peroxisomal b-oxidation genes (Figures S5B– S5D). TCGA data also shows an increased PIP4K2A expression in a multitude of cancers including ovarian, pancreatic, and prostate. Further, we examined the effect of knockdown of PIP4K2A in prostate cancer cells, LNCaP (Figure S5E). RNA-seq revealed that in fact, siRNA knockdown of PIP4K2A alone in prostate cancer cells is sufficient to increase FA metabolism as compared with the control (Figure 6D). Mass spectrometric (MS) analysis also showed an accumulation of the VLCFA, C26:0, upon knockdown of PIP4K2A (Figure 6E) in the LNCaP cells. Interestingly, accumulation of C26:0 has been shown to correlate with the ability of this lipid to be transported to the peroxisomal membrane as well as the subsequent degradation via peroxisomal β-oxidation (Shi et al., 2016; Wiesinger et al., 2013). Altogether, these results strongly indicate a role for PI5P4Ks in regulating peroxisomal function in multiple cancers.
Figure 6. Data from cancer samples indicate a role for PI5P4Ks in regulating FA metabolism.

(A and B) Gene expression of (A) PIP4K2A and (B) PIP4K2B in de-differentiated liposarcoma samples as compared with normal adipose tissue. Statistical significance was determined by comparison of the gene between two groups (Barretina et al., 2010).
(C) Heatmap showing gene expression for TCGA-BRCA normal tissue versus primary tumor samples, exemplifying a number of select genes important for the(peroxisome-mitochondrial function). Red to green color represents high to low log2(FPKM+0.1) expression.
(D) RNA-seq of siPIP4K2A human prostate cancer cells, LNCaP, show an enrichment of FA metabolism compared with the non-targeting (NT) control.
(E) LNCaP cells transfected with siRNA targeting PIP4K2A, cultured in charcoal stripped (C/S) medium, have enriched hexacosanoic acid (C26:0) by mass spectrometry. Transfection of NT siRNAs used as control. Statistical analysis: an unpaired two-tailed t test for used for (A and B) and a paired two-tailed t test was used for (E).
PI5P4K depletion in vivo results in sarcoma tumor regression
Formerly, we have shown a role for both PI5P4Kα and PI5P4Kβ in tumor initiation as well as that the loss of the kinases reduced the prevalence of spontaneous tumor formation in Trp53−/− mice (Emerling et al., 2013). Importantly, the most frequently occurring tumor types in the Trp53−/− model are lymphomas and soft tissue sarcomas (Jacks et al., 1994). Based on such data, we examined the PI5P4Kα protein expression in human UPS tumors. Looking at full tumor sections, we see higher PI5P4Kα protein accumulation in sarcoma tumor cells and lymphocytes compared with the adjacent normal connective tissue (Figure 7A). We further explored a tissue microarray (TMA) representing 50 different soft tissue entities, covering a spectrum from benign tumors to highly aggressive, treatment-naive sarcoma subtypes (Figure 7B). We see high PI5P4Kα protein accumulation in a group of adult high-grade sarcomas with complex karyotypes as well as in some subtypes harboring distinct gene translocations. Interestingly, benign soft tissue tumors and lower grade sarcomas show significantly less or no PI5P4Kα protein accumulation.
Figure 7. In vivo sarcoma tumor models support the role of PI5P4Ks as a potential target for cancer therapy.

(A) IHC showing cytoplasmic staining for PI5P4Kα in a full tissue section of a human UPS. Scale bar, 500 μm.
(B) Representative images of TMA spots showing differential cytoplasmic accumulation of PI5P4Kα in high-grade sarcomas, (top) as compared with low-grade sarcoma and benign soft tissue neoplasms (bottom). Scale bar, 250 μm for all panels except benign soft tissue neoplasms (200 μm).
(C) The Kaplan-Meier time to tumor event curve of Ad-cre injected KrasLSL-G12D/+ Trp53flx/flx (KP) versus KrasLSL-G12D/+ Trp53flx/flx Pip4k2aflx/flx Pip4k2b−/− (KP αβ DKO) adult mice. p = 0.0166 (*) (log-rank Mantel-Cox test).
(D) Established tumor volume change over days post-orthotopic implantation of R-shRenilla and R-shMulti (A and B) KPS-142 sarcoma cells into the hindlimb. At day 7, 8, and 9, dox was administered to both the R-shRenilla and R-shMulti (A and B) KPS-142 tumors for hairpin activation. n = 4 mice per group. p = 0.0012 (**) at day 11.
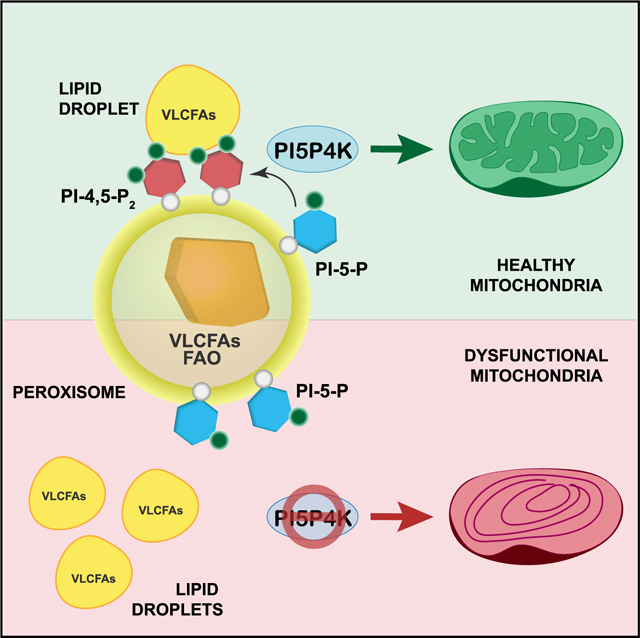
(E) Model shows the role for PI-4,5-P2 generated by PI5P4Ks at the peroxisome for trafficking VLCFAs into the organelle. Upon loss of PI5P4Ks decreased pools of PI-4,5-P2 on the peroxisome limits its capacity to sustain b-oxidation, escalating the energy-generation load on the mitochondria. This triggers mitochondrial structural deformity and functional impairment, leading to an energy crisis and, consequently, a failure to maintain metabolic homeostasis. Thus, supporting PI5P4Ks as an attractive target for cancer therapy.
To further establish a role for these enzymes in sarcoma tumor supportive mechanisms, we used the well-established KrasLSL-G12D/+ Trp53flx/flx (KP) mouse to model UPS (Kirsch et al., 2007). To first validate if the kinases are required for the formation of sarcomas, KP and KrasLSL-G12D/+Trp53flx/flx Pip4k2aflx/flx Pip4k2b−/− (KP- αβ DKO) mice were injected with adenovirus expressing cre-recombinase (Ad-cre) into the hindlimb and tumor initiation was monitored. Remarkably, deletion of the kinases restricts tumor formation to a significant degree compared with the control KP mice (Figure 7C). Further, histopathology of the tumors from the KP mice confirmed them to be high-grade serous UPS tumors (Figure S6A) as well as to express high levels of both PI5P4Kα and PI5P4Kβ compared with the normal connective tissue, analogous to the human UPS tumor (Figures S6B and 7A). Using the tumors formed in the KP mouse, we derived the tumor cell line, KPS-142. Using KPS-142, we generated inducible knockdown cell lines containing either R-shRenilla (control) or R-shMulti (α.β) to investigate the effect of kinase suppression in an established tumor (Figure S6C). The inducible lines were orthotopically injected into the hindlimb of a syngeneic recipient and tumor progression was monitored until reaching ~200 mm3. While the rate of tumor establishment was similar in both the mice cohorts, treatment with dox significantly led to the reduction in tumor size in the KP- αβ DKO mice (Figure 7D). These outcomes indicate an essential role for PI5P4Ks not only in tumor establishment but also for the first time in tumor maintenance. Further these results expose the PI5P4Ks as a metabolic vulnerability of UPS tumors and as a possible therapeutic target for this aggressive sarcoma subtype.
Collectively, all these data provide support for PI5P4Ks being regulators of metabolic homeostasis (Figure 7E). PI-4,5-P2 generated by the PI5P4Ks is indispensable for peroxisomal uptake of VLCFAs and their β-oxidation. In the absence of this, as observed in various disease conditions associated with peroxisome dysfunction, mitochondria bear the brunt of maintaining the energy homeostasis in the cells and, thus, making them highly susceptible to metabolic perturbations. With being essential for autophagy (Lundquist et al., 2018), along with the newly discovered role in mitochondrial functions, PI5P4Ks are emerging as exciting and attractive putative targets for cancer therapy.
DISCUSSION
Peroxisomes and mitochondria are small yet exceptionally dynamic organelles. They both play key roles in cellular metabolism, particularly in terms of FA metabolism and ROS production. Consequently, it is well appreciated that the exchange of contents and signals between these organelles are critical for sustaining metabolic homeostasis and any dysfunction in communication can lead to various diseases, including cancer. However, there are still gaps in our understanding of the actual mechanisms that support their cooperative and complex interactions. Here, we uncover an unexpected role for the non-canonical family of lipid kinases, the PI5P4Ks, and their lipid product PI-4,5-P2 in sustaining peroxisome-mitochondria metabolic interplay by directly regulating trafficking of LDs to the peroxisomes, resulting in proper peroxisomal function and, ultimately, mitochondrial integrity.
A key question that arises from our findings is how the loss of peroxisomal PI-4,5-P2 contributes to the peroxisome-mitochondria communication defect. Is the PI-4,5-P2 lipid required for LD-peroxisome contacts or for the FA trafficking from the LDs to peroxisomes? Recently, Chang et al. elegantly showed that M1 Spastin on LDs together with peroxisomal ABCD1 tethers LDs to peroxisomes which then recruits endosomal sorting complexes required for transport (ESCRT)-III proteins to the LD-peroxisome contact sites for FA trafficking (Chang et al., 2019). The fact that Abcd1 gene expression is increased in the αβ DKO cells compared with WT (Figure S1H) indicates that LD-peroxisome contacts can still be established upon the loss of the PI5P4Ks. This notion is further supported by the observation that upon treatment with Cu-OOH, a peroxidation agent, the LDs are found in closer proximity to the peroxisome in all the cell lines. But the uptake of lipids from these LDs by the peroxisomes occur only in the WT and PI5P4KαRes MEFs and not in the αβ DKO and PI5P4KαKD MEFs (Figures 1C and S1J). Instead, we observe coalescing of LDs around the peroxisomes in these cells, indicating that while the LD-peroxisome contacts are not affected, their uptake into the peroxisome is impaired. Interestingly, ESCRT-III complexes, which are evolutionarily conserved, are comprised of proteins that are proficient at modifying lipid bilayers, which enables FA trafficking (McCullough et al., 2015) and preferentially associate with PI-4,5-P2 containing membranes (De Franceschi et al., 2018). Therefore, while the PI4,5-P2 might not be needed for LD-peroxisome contacts, it will most likely play a significant role in FA trafficking through binding with proteins within the ESCRT-III complex. It will be interesting to further study the physical interactions with PI-4,5-P2 and the ESCRT-III protein players not only at peroxisomal membranes but other metabolically active membranes. Moreover, besides cholesterol transport and FA trafficking, PI-4,5-P2, as well as the PI5P4Ks, may regulate other functions of the peroxisome, such as autophagy and mTOR signaling. Previous studies have reported a role of the tuberous sclerosis complex (TSC) at peroxisomes where it regulates mTORC1 signaling in response to ROS (Zhang et al., 2013), and we have shown that PI5P4K deficiency results in autophagy defects and a downregulation of mTORC1 signaling (Lundquist et al., 2018). Future work is warranted to explore in detail and dissect the specific roles of the PI5P4K signaling pathway for these activities and the possibilities of these functional roles to intersect in order to maintain organelle health and prevent defects leading to disease.
Besides uncovering a fundamental role for the PI5P4Ks in sustaining peroxisome-mitochondria metabolic interplay, this study highlights the importance of these enzymes for the first time in multiple sarcoma tumor subtypes as well as for sarcoma tumor maintenance. Our previous work revealed that a subset of breast cancers express high levels of the PI5P4Ks as well as established a role for the PI5P4Ks in tumor formation in Trp53-deficient mice (Emerling et al., 2013). Here, we provide further evidence that the PI5P4Ks are metabolic drivers in cancers and a compelling rationale for targeting them as a therapeutic option. In particular, for UPS, as it is one of the most aggressive sarcoma subtypes and the efficacy of targeted therapies have been limited (Linch et al., 2014). In summary, due to the broad implications of these findings to organelle biology and cellular metabolism, PI5P4Ks, apart from cancer targets, also present as appealing targets for peroxisome-related diseases, where similar structural and metabolic dysfunctions are observed. Likewise, the prospect of the intracellular PI-4,5-P2 lipid to be used as a measurable indicator of disease severity and for the effectiveness of PI5P4K inhibitors, which are actively in clinical development, is exciting.
Limitations of study
PI5P4Ks, like all lipid kinases, are very dynamic in nature and their localization depends largely on substrate availability. Their substrate, PI-5-P, is the least abundant phosphoinositide, making it not only difficult to visualize the phospholipid but also the kinases at their site of function. With better tools being made available, we hope to be able to answer precisely, the questions regarding spatial and temporal regulation of the kinases in the cell, allowing us to better understand how the loss of PI5P4Ks can regulate cellular metabolism to such a drastic extent. Visualizing and quantifying PI-4,5-P2 on internal membranes also present a sizable challenge. First, because the majority of cellular PI-4,5-P2 is found on the plasma membrane and next, due to the rapid cycling of the phospholipids from one form to another. Recently, the technique described by Hammond et al. (2009), has allowed us to stain for PI-4,5-P2 on internal membranes, mitigating to a certain extent, one of the challenges, leaving us with the question of understanding the contribution of other kinases and phosphatases to this dynamic pool of PI-4,5-P2 in the cell.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Brooke M. Emerling (bemerling@sbpdiscovery.org).
Materials availability
All plasmids, cell lines and mouse models generated in this work can be requested from the Lead Contact. All antibodies, chemicals, and most cell lines used in this study are commercially available.
Data and code availability
The supplemental tables accompanying the published article include all datasets generated in this study.
EXPERIMENTAL MODELS
Cell lines
All cells were incubated in a 37C humidified incubator with 5% CO2. Mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco’s modified Eagle medium (Corning 10–013-CV). Media was supplemented with 10% fetal bovine serum (Life Technologies), 100 U/ ml penicillin/streptomycin (Gibco; 15140122). MEFs were isolated from E13.5 embryos and genotyped as described previously (Lundquist et al., 2018). For the generation of SV40 MEFs, the retroviral construct pBabe-Zeo-LT-ST was used (generously provided by Jorge Moscat, SBPMDI) and cells were selected post-infection in zeocin (Invivogen).
MDA-MD-468 (TNBC) was purchased from ATCC and authenticated by Short Tandem Repeat (STR) analysis. SoftGenetics GeneMarker software was used for analyzing the sequencing data. 293A, MCF10A, 4T1, HT1080 (human fibrosarcoma), SW872 (human liposarcoma) and LNCaP were also purchased from ATCC. OH931 (human myxofibrosarcoma) was a gift from Dr Julia Bridge (Krause et al., 1997).
Mouse sarcoma line, KPS-142 [undifferentiated pleomorphic high-grade sarcoma (UPS)], was generated in Emerling lab from a primary tumor originated in the KrasLSL-G12D/+ Trp53flx/flx mouse. In brief, primary tumors were extracted in a sterile environment and digested (or dissociated) with collagenase IV for four hours at 37C. Digested tumors were centrifuged at 1,0000 RPM for four minutes and supernatant was washed with sterile PBS to remove the remaining collagenase. Primary tumor cells were then maintained under standard tissue culture protocol. Additionally, cells were tested using IDEXX IMPACT III to ensure they were pathogen free and safe for subsequent animal work.
293A, human TNBC line MDA-MB-468, mouse TNBC line 4T1, human sarcoma lines HT1080, SW872 and OH931, and mouse sarcoma line KPS-142 were all grown in DMEM with 10% FBS and 100 U/ml penicillin/streptomycin. Human breast epithelial cell line MCF10A was grown in DMEM/F-12 medium (Gibco; 11330032) supplemented with 5% Horse serum (Gibco 16050122), 20ng/ml EGF (Sigma-Aldrich; E9644), 10mg/ml Insulin (Sigma-Aldrich; I1882), 100ng/ml Cholera toxin (Sigma-Aldrich; C8052), 0.5mg/ml Hydrocortisone (Sigma-Aldrich; H0888) and 100 U/ml penicillin/streptomycin. LNCaP cells were grown in RPMI 1640 (Gibco; 11875093) with 10% FBS and 100u/ml penicillin/streptomycin.
C. elegans
The following strains were used for the experiments: N2 and ppk-2 (pk1343). The details of these strains were previously described in (Lundquist et al., 2018). Animals were grown on nematode growth media with E. coli OP50. All experiments were performed on synchronized young adult animals at 20C (Stiernagle, 2006).
Mouse strains
All animal care and treatments were carried out in compliance with Institutional Animal Care and Use Committee (IACUC) guidelines. The generation of Pip4k2aflx/flx; Pip4k2b−/− (Lundquist et al., 2018) and LSL-KrasG12D/+; Trp53flx/flx (KP) (Kirsch et al., 2007) mice has been previously described. These strains were then crossbred to generate the LSL-KrasG12D/+; Trp53flx/flxPip4k2aflx/flx; Pip4k2b−/−(KP-αβDKO) mice used for the tumor-free survival study.
METHOD DETAILS
Virus production and infection
293T packaging cell line was used for retroviral and lentiviral amplification. In brief, viruses were collected 48 hours after transfection, filtered, and used for infecting cells in the presence of 10μg/ml polybrene (Santa Cruz Biotechnology; NC9840454), prior to puromycin selection or upon cell sorting for GFP. pCL-Ampho and pCL-Eco were used for retroviral packaging for human and mouse cells respectively. pVSVg and pPax were used for lentiviral packaging.
Generation of knockout and knockdown cell lines
MEFs
Pip4k2aflx/flx Pip4k2b−/− MEFs were infected at a MOI=10 of FIVCMVCre VSVG to delete Pip4k2a from the lines and generate Pip4k2a−/− Pip4k2b−/− MEFs (αβ DKO MEFs). We also generated rescue lines of the αβ DKO MEFs with expression of human HA-tagged WT PI5P4Kα (αRes) and kinase-dead PI5P4Kα D359N (αKD).
293A
PIP4K2A and PIP4K2B were deleted using CRISPR to generate 293A αβDKO cell line. sgRNA targeting PIP4K2A and PIP4K2B was cloned into pX330 (A gift from Feng Zhang) digested with BbsI. See Table S1: Targeting sgRNA oligonucleotide sequence. We also generated rescue lines of the 293A αβ DKO line with expression of HA-tagged WT PI5P4Kα (αRes), WT PI5P4Kβ (βRes) and kinase-dead PI5P4Kα D359N (αKD).
MCF10A
PIP4K2A was initially deleted using CRISPR followed by infection with dox-inducible shRNA targeting PIP4K2B and the cell were selected in 2μg/ml puromycin. To generate MCF10A lines lacking both isoforms of the kinases, cells were treated with 3μg/ml dox for 3 consecutive days before carrying out experiments in the presence of dox.
Cancer lines
Lentiviral constructs, pCF806_Reci and pCF806_shRenilla (shRen.713), were a generous gift from Dr. Fellmann. In brief, the pCF806 vector encodes a miR-E cassette (Fellmann et al., 2013) that has been optimized for dual shRNA expression, under the control of an all-in-one Tet-On system for dox regulated shRNA induction (Fellmann et al., manuscript in preparation). EGFP in these constructs were mutated to allow for fluorescent imaging. Human and mouse shRNA sequences targeting PIP4K2A and PIP4K2B were designed using the SplashRNA algorithm (Pelossof et al., 2017). Sequences for human and mouse PI5P4Kα and PI5P4Kβ were then sequentially cloned into pCF806_Reci to yield pCF806_shMulti (a,b) for human and mouse. See Table S2: Sequences for the shRNA oligonucleotides cloned into pCF806_Reci. HT1080, SW872, OH931 and MDA-MB-468 and 4T1 and KPS-142 were infected with human and mouse pCF806_shMulti (α,β), respectively and pCF806_shRenilla as control. Cells were selected in 2mg/ml puromycin. The generated control and knockdown lines were treated with 1μg/ml of dox for 2 weeks prior to all experiments, to be able to see long-term effects of the silencing of the kinases.
siRNA knockdown in LNCaP
Prostate cancer cells, LNCaP, were plated on poly-L-Lysine coated plates and transfected with pooled siRNA targeting PIP4K2A or non-targeting (NT) control oligos (ON-TARGET Plus, Dharmacon). Next day switched to charcoal stripped (C/S) medium, light RPMI + 5% C/S FBS for 48 hours.
Immunoblot analysis
Total cell lysates were prepared by washing cells with cold phosphate-buffered saline. The cells were then lysed with RIPA lysis buffer containing 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl pH 7.5, as well as Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78440). Protein was measured using the BCA assay (Thermo Scientific; 23225) and 20–30 μg of total cell lysates were run on an SDS–polyacrylamide gel electrophoresis. The proteins were transferred on to a nitrocellulose membrane and membranes were probed overnight at 4°C with the appropriate primary antibody. Antibodies used were as follows: PIP4K2α (Cell Signaling; 5527), PIP4K2β (Cell Signaling; 9694), β-actin (Santa Cruz Biotechnology; sc-517582) and a-tubulin (Sigma-Aldrich; T6199).
Microscopy
Fluorescence microscopy
For live cell imaging, cells were grown on chambered #1.5 slide (CellVis; C4–1.5H-N). For immunofluorescence, cells were grown on 10-chambered glass bottom dish (Greiner Bio-one; 07000554).
PI-4,5-P2 staining and localization
PI-4,5-P2 staining for internal membranes was done using the protocol described in (Hammond et al., 2009). In brief, cells were fixed in 2% pre-warmed PFA followed by permeabilization with 20 μM digitonin. Samples were blocked in 5% NGS in Buffer A with 50mM NH4Cl before being incubated in primary antibodies namely, anti- PI-4,5-P2 (1:100) (Echelon Biosciences; Z-P045–2), anti-TOM20 (1:100) (ABclonal Science; A6774) and anti-PMP70 (1:100) (Thermo Fisher Scientific; PA1650) as indicated. Samples were washed thrice before incubation with secondary antibodies, followed by post-fixation with 2% PFA in PBS. During the final washes DAPI was added to the wash buffer. The samples were imaged at 40x water on Zeiss LSM710 microscope. Colocalization was calculated in ImageJ using the Spot Localization plug-in plugin ComDet.
Mouse prostate tissue fluorescent immunohistochemistry (IHC)
Adult C57BL/6JRj mouse prostates were harvested and paraffin embedded. Tissue serial sections were deparaffinized in xylene, rehydrated using decreasing concentrations of ethanol (100 to 95 to 70% to H20). Heat-induced epitope retrieval used at settings of 98C and 1400rpm for 35min in Tris-EDTA buffer. Slides were washed in TBS, blocked in 5% normal goat serum at room temperature then probed with primary antibodies overnight at 4C. Proteins were detected using 1:200 PIP4K2A polyclonal antibody (Proteintech, 1249–1-AP), 1:100 Goat anti-rabbit Alexa488 secondary (Jackson ImmunoResearch, 111–545-003), and 1:200 PMP70 polyclonal antibody (Thermo Fisher Scientific; PA1650-A555). DAPI (100ng/mL) counterstain and mounting using ProLong Gold (Thermo Fisher Scientific; P36930). Images acquired with 63x (oil immersion) magnification using Leica DMI4000 B microscope, DFC360 FX B&W fluorescence camera, and LAS V3.6 software.
Peroxisome and lipid localization
For this experiment, cells expressing mEmerald-SKL (gift from Dr Jennifer Lippincott-Schwartz) to mark peroxisomes was used. To label LDs, BODIPY 665/676 (Invitrogen; B3932) was added to cells at 500 ng/ml 16 h before imaging. To induce lipid peroxidation, cells were treated with 50 μM cumene hydroperoxide (Cu-OOH) (Frontier Scientific; JK298118) for 30mins at 37°C (Chang et al., 2019). Treated cells were imaged in media without Cu-OOH. Cells were imaged at 63x oil using Zeiss LSM710 microscope. Colocalization was calculated in ImageJ using the colocalization plug-in JACoP.
Mitochondrial imaging
To label the mitochondria, cells were incubated with 100mM MitoTracker Green FM probe (Thermo Fisher Scientific; M7514) at 37°C for 30mins. Cells were imaged at 63× oil using Zeiss LSM710 microscope.
For mitochondrial fragmentation measurements, cells were incubated in control or no (0%) glucose medium for 16 h. Cells were then treated with mitochondrial dye along with 100mM LysoTracker DeepRed (ThermoFisher Scientific; L12492) and Hoechst33342 (Sigma-Aldrich; B2261). Mitochondrial fragmentation was calculated in ImageJ using a modified version of MiNA plug-in to threshold for fragment length.
For membrane potential measurements cell were treated with 100 μM TMRE (ThermoFisher Scientific, T669) along with MitoTracker Green FM and Hoechst33342 for 30mins at 37°C. TMRE:MitoTracker ratio was calculated in ImageJ using the MitoMorphology macro.
For lipid trafficking into the mitochondria, MEFs were incubated with 1μM BODIPY 558/568 C12 (Thermo Fisher Scientific; D3835) overnight and then chased in control medium or HBSS for 16 hours (Rambold et al., 2015). The cells were then stained with the mitochondrial dye and Hoechst33342 for 30mins at 37°C and imaged. Colocalization was calculated in ImageJ using the colocalization plug-in JACoP for the lipid trafficking experiment.
Lipid droplet accumulation
Cells were incubated with BODIPY 493/503 (Thermo Fisher Scientific; D3922) for 30mins at 37°C, along with LysoTracker DeepRed and Hoechst33342. Cells were imaged at 63x oil using Zeiss LSM710 microscope. Lipid droplet counting was done in ImageJ using the Spot Localization plug-in ComDet.
Human sarcoma tissue immunohistochemistry (IHC)
All work including human specimens were conducted under the ethical approval (KEK-ZH-Nr2013–0430). All patients further signed the hospital general consent to agree to use their tissue for research. 2μm thick tissue sections from archived FFPE material and the sarcoma tissue micro array were deparaffinized in xylene, rehydrated using decreasing concentrations of ethanol (100 to 95 to 70% to H20). Heat-induced epitope retrieval was done at 95°C for 30 min in Tris-EDTA buffer. The entire staining procedure was conducted with the Ventana Benchmark semi-automated staining system using Ventana reagents for the entire procedure (including iVIEW DAB detection kit). Incubation with primary antibody PIP4K2A (Proteintech, 1249–1-AP) was done at a 1:100 dilution. Images were scanned using the Ventana DP 200 slide scanner.
Mouse sarcoma tissue immunohistochemistry (IHC)
Mouse tissue sections were processed using standard histology procedures. Fixation with 4% formaldehyde, paraffin embedded, and sections cut at 2.5μm. Antibody staining was optimized for PIP4K2A (Proteintech, 1246–1-AP) with 30min Tris buffer antigen retrieval with 1:200 dilution antibody and PIP4K2B (Proteintech, 13218–1-AP) with 30min Citrate buffer antigen retrieval and 1:200 dilution. Slide staining was done using the BOND RX autostainer (Leica Biosystems) as previously described (Berezowska and Galvá n, 2017). Slide images were acquired using the Panoramic 250 Flash II slide scanner (3DHistech).
Transmission Electron Microscopy (TEM)
Cell pellets were fixed in oxygenated 4% paraformaldehyde and 2.5% glutaraldehyde (Johnson, 1987) in 0.1M sodium cacodylate buffer (pH7.4) for 5 min at 37°C, then kept at 4°C for approximately 4 hours and washed in buffer. Cells were post-fixed in buffered 1% osmium tetroxide for 1 hour at room temperature followed by water rinses and dehydration through a graded series of ethanol. Cells were infiltrated in LX112 epoxy resin (Ladd) using HPMA (2-hydroxypropyl methacrylate) as the transition fluid and polymerized at 60°C. Ultrathin sections (silver) were cut using a Leica UC6 ultramicrotome and collected on 200-mesh copper grids that were post-stained with uranyl acetate and lead citrate. Samples were analyzed at 80kV with a ThermoFisher Talos L120C transmission electron microscope and images were acquired with a CETA 16M CMOS camera.
Proliferation assay
Cells were plated in 24 or 48-well plates 24 hours before the start of the experiment. The following day, the cells were washed in PBS followed by the treatment as indicated. For MCF10A cell lines, 5μM 2-deoxyglucose (2-DG) was used. For lipotoxicity assay, Hexacosanoic (Sigma-Aldrich, H0388) acid and Lignoceric acid (Sigma-Aldrich, L6641–1G) were dissolved in warm ethanol to make a stock solution of 100mM. Fatty acid-free (FAF) BSA (Sigma-Aldrich, A8806) was used as a carrier. The lipids were dissolved in 32 mg/ml fatty acid-free (FAF) BSA solution to make a stock of 1mM lipid conjugated to BSA and the cells were treated at the indicated concentration with the control cells treated with the carrier alone. Proliferation on day 0 and the respective time points were measured using crystal violet. The cells were rinsed with PBS and fixed with 3.7% formaldehyde (RICCA chemical company; RSOF0010) at room temperature for 20mins, following which they were rinsed with distilled water and incubated with necessary volume of 0.05% crystal violet (Sigma Aldrich; C0775) solution for 20mins at room temperature. The plates were rinsed after staining, dried and imaged before the crystals were dissolved in a lysis solution (0.1M sodium citrate, 25% ethanol, pH 4.2) for half an hour at room temperature. The optical density was measured at 590nm in a plate reader (Molecular Devices).
Catalase assay
The catalase assay was performed using the Amplex Red Catalase Assay Kit (Thermo Fisher Scientific, A22180). In brief, cells were lysed in RIPA lysis buffer containing Halt protease and phosphatase inhibitor cocktail and the experiment performed according to the manufacturer’s protocol in 96-well plates. Data were determined by luminescence (Molecular Devices). Data were collected from multiple replicate wells for each experiment and were normalized to protein concentration.
ATP measurement
ATP production was measured using the ATP Determination Kit (Thermo Fisher Scientific, A22066) according to the manufacturer’s protocol. In brief, cells were lysed in RIPA lysis buffer supplemented with Halt protease and phosphatase inhibitor cocktail. Data were determined by luminescence (Molecular Devices). Data were collected from multiple replicate wells for each experiment and were normalized to protein concentration.
ROS measurement
Cells were incubated with 5mM MitoSOX reagent (Thermo Fisher Scientific, M36008) and Hoechst333422 at 37C for 30 min. The cells were then gently washed three times with warm PBS. Cells were trypsinized and resuspended to ensure a single cell suspension. Samples were analyzed by flow cytometry using Novocyte 7000. Data were collected from triplicate wells in each experiment and experiment repeated thrice.
Oxygen consumption rate (OCR) measurement by Seahorse
The mitochondrial respiratory capacity was determined using XF Cell Mito Stress Test Kit (Agilent Technologies, 103015–100). Cells were seeded in the XF96 Cell Culture Microplate at a density of 1.5–2.5 × 104 cells per well based on the cell line. 6–8 replicates were set up for each of the groups as indicated and incubated for 24 h at 37°C. The sensor cartridge was hydrated in the utility plate filled with 200 ml of Seahorse Calibrant overnight in a non-CO2 incubator at 37°C. Next day, cells were incubated with the base medium containing 2 mM L-glutamine, 1 mM sodium pyruvate, and 10 mM glucose for 1 hour prior to assay. 1.5 μM oligomycin A, 1 μM FCCP, and 0.5 μM rotenone/antimycin A were sequentially injected and the oxygen consumption rate was measure by the XFe96 flux analyzer. The OCR value is normalized to protein concentration.
NAD/NADH quantification
This assay was performed using the NAD/NADH Quantitation kit (Sigma-Aldrich, MAK037). Briefly, 200,000 cells were collected per sample and washed in cold PBS and the experiment was performed as per manufacturer’s protocol. Data were determined by luminescence (Molecular Devices). Data were collected from multiple replicate wells for each experiment and were normalized to protein concentration.
RNA Sequencing
For MEFs
RNA was extracted from cell pellets washed with PBS using Qiagen RNeasy kit. RNA yield and purity was tested using the Qubit 4 Fluorometer. Sequencing libraries were prepared using 250ng RNA and sequencing was performed using the Illumina NextSeq 500. Mouse fastq samples were processed using the following steps. First, adapter remnants of sequencing reads were removed using cutadapt v1.18 (Martin, 2011). Next, RNA-Seq sequencing reads were aligned to mouse genome version 38 and Ensembl gene annotation version 84 using STAR aligner version 2.7 (Dobin et al., 2013). RSEM v1.3.1 (Li and Dewey, 2011) was used to estimate gene expression. See Table S3: Deposited data from RNA sequencing of MEFs.
For LNCaP cells
RNA was extracted from cell pellets washed with PBS using Qiagen RNeasy kit, DNase treated (Ambion, AM1906), and quality assessed using Bioanalyzer and Qubit. Sequencing libraries were prepped using 500ng RNA and Illumina TruSeq Stranded mRNA with IDT for Illumina Truseq UD RNA indexes. FASTQ read files were analyzed as previously described (Cyrta et al., 2020) using STARv2.3.0e (Dobin et al., 2013), RSEQtools (Habegger et al., 2011), and GENCODE v19 (http://www.gencodegenes.org/releases/19.html). See Table S4: Deposited data from RNA sequencing of LNCaP cells.
For C.elegans
RNA was extracted from age synchronized young adult animals using TRZIOL digestion and followed by Directzol RNA MiniPrep (Zymo Research Corporation; 50–444-628). Raw sequenced reads were aligned to the Caenorhabditis elegans reference genome (Version WBcel235.91 from ENSEMBL) using STAR (Version 2.4.2) aligner. Aligned reads were quantified against the reference annotation GTF (WBcel235.91from ENSEMBL) to obtain FPKM (Fragments per Kilobase per million) using CuffLinks (v 2.2.1). See Table S5: Deposited data from RNA sequencing of C.elegans.
qRT-PCR
Total RNA was prepared using Directzol RNA MiniPrep (Zymo Research Corporation; 50–444-628). cDNA was synthesized using High-capacity cDNA Reverse Transcription kit (ThermoFisher Scientific; 4368814) and qRT-PCR performed utilizing PowerUp SYBR green (ThermoFisher Scientific; A25742) and the LightCycler 96 (Roche). See Table S6: Mouse qRT-PCR primers.
Prior to mRNA isolation, 150,000–200,000 cells were plated in 6-well plastic dishes. 24 hours later, the RNA in the lysates was extracted using the Directzol RNA prep protocol. The RNA was resuspended in nuclease- free H2O at a final concentration of 1 μg/mL. cDNA was transcribed using the High Capacity cDNA reverse transcription kit.
Metabolomics and lipidomics
YSI analysis
Cells were initially plated at 2.5 × 104 cells per well in a 24 well plate in triplicate. The following day, cells were washed 2x with 500uL of PBS and replaced with 500uL of DMEM culture media (10% FBS, no antibiotic). Sample media was collected at the 24-hour time point and cleared for cell debris by centrifugation at room temperature prior to storage at −80c prior to analysis. In addition, cell lysates were collected per sample for protein normalization by BCA assay. Metabolite analysis was performed by the SBP Cancer Metabolism core using the YSI 2950 metabolite analyzer.
13C Tracing
13C isotope tracing media was formulated using glucose and glutamine free DMEM 5030 supplemented 10% FBS, 20 mM [U- 13C6] glucose and 4 mM glutamine. Fatty acid oxidation experiments were conducted using glucose and glutamine free DMEM 5030 supplemented 10% delipidated FBS, 100 mM BSA-[U-13C16]palmitate, 20 mM glucose and 4 mM glutamine. Cultured cells were washed with 1 mL PBS prior to applying tracing media for 24 hours.
Isotopomer Spectral Analysis (ISA)
Isotopomer spectral analysis (ISA) was performed to estimate the percent of newly synthesized palmitate as well as the contribution of a tracer of interest to the lipogenic acetyl-CoA pool (Cordes and Metallo, 2019; Young, 2014). Parameters for contribution of 13C tracers to lipogenic acetyl-CoA (D value) and percentage of newly synthesized fatty acid (g(t) value) and their 95% confidence intervals are then calculated using best-fit model from INCA MFA software. Experimental fatty acid labeling from [U-13C6] glucose after 24 hours trace was compared to simulated labeling using a reaction network where C16:0 is condensation of 8 AcCoA. ISA data plotted as mean ± 95% CI. * indicates statistical significance by non-overlapping confidence intervals.
Metabolite Extraction and GC-MS Analysis
At the conclusion of the tracer experiment, media was aspirated. Then, cells were rinsed twice with 0.9% saline solution and lysed with 250 μL ice-cold methanol. After 1 minute, 200 μL water containing 0.5 μg/ml norvaline was added to each sample and vortexed for one minute. 500 μL chloroform was added to each sample, and all were vortexed again for 1 minute. After centrifugation at 21,130 g for 10 minutes at 4°C, 500 μL of the upper aqueous layer was collected and evaporated under vacuum at 4°C. Then, 500 μL of the upper aqueous layer was collected and evaporated under air at room temperature.
Dried polar and nonpolar metabolites were processed for gas chromatography (GC) mass spectrometry (MS) as described previously in Cordes and (Cordes and Metallo, 2019). Briefly, polar metabolites were derivatized using a Gerstel MultiPurpose Sampler (MPS 2XL). Methoxime-tBDMS derivatives were formed by addition of 15 μL 2% (w/v) methoxylamine hydrochloride (MP Biomedicals, Solon, OH) in pyridine and incubated at 45°C for 60 minutes. Samples were then silylated by addition of 15 μL of N-tert-butyldimethylsily-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylchlorosilane (tBDMS) (Regis Technologies, Morton Grove, IL) and incubated at 45°C for 30 minutes. Nonpolar metabolites were saponified and transesterified to fatty acid methyl esters (FAMEs) by adding 500 μL of 2% H2SO4 in methanol to the dried nonpolar layer and heating at 50°C for 1 hour. FAMEs were then extracted by adding 100 μL of a saturated salt solution and 500 μL hexane and vortexing for 1 minute. The hexane layer was removed, evaporated, and resuspended with 60μL hexane for injection.
Derivatized polar samples were injected into a GC-MS using a DB-35MS column (30m x 0.25mm i.d. x 0.25μm, Agilent J&W Scientific, Santa Clara, CA) installed in an Agilent 7890B GC system integrated with an Agilent 5977a MS. Samples were injected at a GC oven temperature of 100°C which was held for 1 minute before ramping to 255°C at 3.5°C/min then to 320°C at 15°C/min and held for 3 minutes. Electron impact ionization was performed with the MS scanning over the range of 100–650 m/z for polar metabolites.
Derivatized nonpolar samples were injected into a GC-MS using a Fame Select column (100m x 0.25mm i.d. x 0.25μm, Agilent J&W Scientific, Santa Clara, CA) installed in an Agilent 7890A GC system integrated with an Agilent 5977A MS. Samples were injected at a GC oven temperature of 80°C which was held for 1 minute before ramping to 170°C at 20°C/min then to 188°C at 1°C/min then to 250°C at 20°C/min and held for 10 minutes. Electron impact ionization was performed with the MS scanning over the range of 54–400 m/z for nonpolar metabolites.
Data analysis
Metabolite levels and isotopologue distributions were analyzed with an in house Matlab script which integrated the metabolite fragment ions and corrected for natural isotope abundances.
C:26 lipid detection
Lipid extraction
LNCaP cell pellets were collected by scraping, following the addition of MeOH:H2O (4:1). These extracts were evaporated and resuspended in Butanol:MeOH (1:1). Protein quantification was performed on cell pellet and samples were normalized based on the total protein content.
Targeted lipid analysis
Extracted cell lysates were analyzed by Hydrophilic Interaction Liquid Chromatography coupled to tandem mass spectrometry (HILIC - MS/MS) 3–5 in both positive and negative ionization modes using a Q-TRAP 6500 LC-MS/MS system (Sciex Technologies) (Dunn et al., 2011; Hines et al., 2017).
Quality Control (QC)
Pooled QC samples (representative of the entire sample set) were analyzed periodically (every 6 samples) throughout the overall analytical run in order to assess the quality of the data, correct the signal intensity drift (if any) and remove the peaks with poor reproducibility (CV > 30%). In addition, a series of diluted quality controls (dQC) were prepared by dilution with buthanol:methanol: 100% QC, 50%QC, 25%QC, 12.5%QC and 6.25%QC.
Data Processing
Raw LC-MS/MS data were processed using the MultiQuant Software (version 3.0.3, Sciex technologies). Relative quantification of metabolite was based on EIC (Extracted Ion Chromatogram) areas for the monitored MRM transitions. Signal intensity drift correction on peak area data were done within the LOWESS/Spline normalization program 8 (R software) followed by noise filtering (CV (QC features) > 30%) and visual inspection of linear response. See Table S7: Deposited data of lipid ion counts of LNCaP cells.
C. elegans experimental protocol
Wild type N2 Bristol and ppk-2 (pk1343) C. elegans strains were grown on nematode growth media (NGM) media seeded with E. coli OP50.
Mitochondrial DNA quantification
DNA was extracted from age synchronized young adult animals using TRZIOL digestion, following which chloroform was used for phase separation. DNA was precipitated from the colorless interphase using ethanol. It was then washed with 0.1M sodium citrate in 10% ethanol. The cleaned DNA pellet was resuspended in 8mM NaOH. qRT-PCR was performed utilizing PowerUp SYBR green (ThermoFisher Scientific; A25742) and the LightCycler 96 (Roche). The mitochondrial DNA content was calculated using the CT values obtained. See Table S8: C.elegans qRT-PCR primers.
Mitochondrial imaging
In C. elegans mitochondria were visualized using MitoTracker Green FM probe (Thermo Fisher Scientific; M7514) and TMRE (ThermoFisher Scientific, T669). Mitotracker and TMRE were diluted in OP50 suspension at 10mM and 500nM respectively. This was then plated on NGM plates and allowed to dry. Synchronized L4 worms were transferred to the experimental plates and incubated at 20°C overnight. Prior to imaging, worms were transfer to die-free OP50 plates for an 30min to minimize background. Animals were mounted on 3% agarose pads and immobilized using 0.25mM Levamisole solution (Acros Organics; AC187870100). Imaging was done using at 63x oil using Zeiss LSM710 microscope. Approximately 10 animals per grouped were imaged, and a total of 3 experiments were performed.
Oxygen consumption rate (OCR)
OCR was assayed using a Seahorse Extracellular Flux Analyzer (XFe24 Analyzer, Agilent) as previously described by Haroon and Vermulst (2019). In brief, synchronized young adult animals were picked into 24 well plates with M9 buffer to an average 50 animals/well, 7 wells/condition. Animals were allowed to equilibrate at room temperature for 30min before the experimental run. FCCP was used to a final concentration of 10μm and sodium azide to a final concentration of 40mm. The oxygen consumption values generated by the software ware normalized to the number of worms per condition, and the timepoints represent the average of all replicate wells/experiment. A total of three experiments were performed.
Oxidative stress assay
For oxidative stress assays, synchronized young adult animals were transferred to NGM plates supplemented with 15 mM Tert-Butyl Hydroperoxide (Acros Organics; AC180340050). The fraction of animals alive was recorded after seven hours of treatment. Animals that responded by movement to touch with the platinum wire were scored as alive. n=100 animals per condition.
Life span assay
For life span assay, synchronized L4 animals were transferred to NGM plates supplemented with 50mM FCCP (Sigma-Aldrich; C2920) or vehicle (DMSO) and live animals were scored daily. Animals that responded by movement to touch with the platinum wire were scored as alive. n ≥ 75 animals per condition.
Mouse Models
Survival Curve
Experimental mice aged 8–12 weeks from each group (KrasLSL-G12D/+ Trp53flx/flx and KrasLSL-G12D/+ Trp53flx/flx Pip4k2aflx/flx Pipk42b−/−). Adenovirus CMV-Cre was purchased from the University of Iowa Gene Transfer Vector Core (Iowa City, IA). Intramuscular injections of Ad-Cre into the hindlimb were performed as previously described (Kirsch et al., 2007). Mice were monitored biweekly for tumor formation by palpitation of the hindlimb. Tumor bearing mice were removed from the study after tumor volume reached ~300mm3 and necropsy was followed by tumor excision. Tumors were then processed for histological examination to confirm the formation of high grade, spindle cell neoplasms.
Multi inducible cell line generation
After establishment of the primary tumor cell line (KPS-142) as described earlier, the cells were then transduced with retrovirus expressing rtTA3 and separately with the retroviral vector RT3REVIN (Fellmann et al., 2013), expressing the miR-E shRNA. The shRNAs targeting Pip4k2a and Pip4k2b were generated using the SplashRNA algorithm (Pelossof et al., 2017). Single hairpins were then further screened using the RT3REVIN vector for knockdown efficiency and selected hairpins were chosen to generate the ‘‘multiinducible’’ shRNAs [R-shRenilla, R-shMulti (a,b)] as previously described (Fellmann et al., 2013; Pelossof et al., 2017; C. Fellmann, unpublished data) . Sequences for the shRNAs are provided in STAR Methods table.
Tumor growth experiment using multi-hairpin
Primary tumor cells (KPS-142) expressing either the R-shRenilla or R-shMulti (α,β) were used for orthotopic injection into the hindlimb of a syngeneic C57BL/6 animal. In brief, cells were centrifuged and counted using an automated cell counter to achieve 500,000 cells per 30uL injection. Mice were anesthetized using 1% isofluorane at a constant flow rate and hair was removed from the injection site using clippers. The hindlimb was wiped with 70% ethanol and cells were injected slowly with constant pressure into the caudal thigh to minimize animal distress. The syringe was moved slowly to ensure the cell suspension did not leak from the injection site. Animals were then moved to a heating pad in a clean cage for recovery. After injection, animals were monitored daily for tumor growth. Measurements were taken using a digital caliper while one user restrained the mouse, and another user took measurements for tumor length and width. Tumor volume was calculated using the following formula: V=0.5(L*W2). Final tumor volume was calculated by subtracting the daily measurement from the first day following injection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Data are expressed as means ±SD. Statistical analyses for all data, including microscopy quantification, qRT-PCR data, metabolite tracing and lipidomics, were performed by student’s two-tailed t-test or ANOVA with Tukey’s method used to correct for multiple comparisons, as indicated, using GraphPad Prism. For survival lifespan curve comparison in C. elegans, we used the log-rank (Mantel-Cox) test using GraphPad software. Statistical evaluation of tumor free survival in mouse experiments was based on the log-rank (Mantel-Cox) test for comparison of the Kaplan-Meier event-time format. Statistical significance is indicated in figures [(*p<0.05, **p<0.01, ***p<0.001, n.s., not significant (p >0.05)] unless specified otherwise.
Pathway analysis using GSEA
Gene set enrichment analysis (Subramanian et al., 2005) was performed using GSEAv4.0.3. Gene sets for KEGG:Fatty Acid Biosynthesis and Hallmarks:Peroxisome are listed in supplemental files. Normalized enrichment scores (NES) and p values were used to determine significance of findings.
Analysis of Liposarcoma dataset
Liposarcoma dataset (Barretina et al., 2010) was analyzed to compare the FKPM values of PIP4K2A and PIP4K2B in normal adipose tissue versus de-differentiated liposarcoma.
Analysis of The Cancer Genome Atlas (TCGA)
Data analysis was performed using Array Studio software (QIAGEN, Cary, NC). Expression data were extracted from OmicSoft OncoLand release TCGA_B38_20190215_v8. For tumor versus normal comparison, patient data were analyzed from the TCGA-Breast Invasive Carcinoma (TCGA-BRCA) sample set and further subset into groups containing solid tissue normal (n=158) and primary tumor (n=1113), respectively. For individual FPKM analyses, BRCA primary tumor normal samples were subset into groups based on FPKM values for PIP4K2A. This was achieved by filtering for the highest quartile (≥75%) (n=196) and lowest quartile (≤25%) (n=293) of PIP4K2A FPKM values. Individual FPKM values were then queried for select genes and statistically analyzed through the software. FKPM values for PIP4K2B for tumor (n=261) versus normal tissue (n=2) comparison for patient data from TCGA-Sarcoma (TCGASARC) were queried as well as highest (R50%) and lowest (≥50%) of the tumors expressing PIP4K2B was used for patient survival data.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| PIP4K2A (D83C1) Rabbit mAb | Cell Signaling Technology | Cat# 5527; RRID: AB_2722636 |
| Anti-PIP4K2B Antibody, Unconjugated | Cell Signaling Technology | Cat# 9694; RRID: |
| Anti-α-Tubulin antibody, Mouse monoclonal | Sigma-Aldrich | Cat# T6199; RRID: AB_477583 |
| β-Actin Antibody (2A3) | Santa Cruz Biotechnology | Cat# sc-517582 |
| Purified Anti-PtdIns (4,5)P2 IgM antibody | Echelon Biosciences | Cat# Z-P045; RRID: AB_427225 |
| TOMM20 Polyclonal Antibody | Abclonal | Cat# A6774; RRID: AB_2767357 |
| PIP4K2A antibody | Proteintech | Cat# 12469–1-AP; RRID: AB_10639518 |
| PMP70 Polyclonal Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# PA1–650-A555; RRID: AB_2662745 |
| Goat Anti-Rabbit Alexa488 | Jackson ImmunoResearch | Cat#111–545-003 |
| PIP4K2B antibody | Proteintech | Cat#13218–1-AP |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Adenovirus CMV-Cre | University of Iowa Gene Transfer Vector Core | Ad5CMV-Cre |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Horse serum | Thermo Fisher Scientific | Cat#16050122 |
| Insulin | Sigma-Aldrich | Cat# i1882 |
| EGF | Sigma-Aldrich | Cat# E9644 |
| Cholera toxin | Sigma-Aldrich | Cat# C8052 |
| Hydrocortisone | Sigma-Aldrich | Cat# H0888 |
| Polybrene | Santa Cruz Biotechnology | Cat# NC9840454 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| Puromycin | Invivogen | Cat#NC9138068 |
| Poly-L-Lysine | Fisher Scientific | Cat#343820001 |
| 16% Formaldehyde (w/v) methanol free | Thermo Fisher Scientific | Cat# 28908 |
| Digitonin | EMD Millipore | Cat#300410250MG |
| Normal Goat Serum | Thermo Fisher Scientific | Cat#016201 |
| DAPI | Biolegend | Cat#422801 |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | Cat# P36930 |
| BODIPY 665/676 (Lipid Peroxidation Sensor) | Thermo Fisher Scientific | Cat# B3932 |
| Cumene hydroperoxide, 80% | Frontier Scientific | Cat# JK298118 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M7514 |
| LysoTracker Deep Red | Thermo Fisher Scientific | Cat# L12492 |
| Hoechst 33342 | Sigma-Aldrich | Cat# B2261 |
| Tetramethylrhodamine, Ethyl Ester, Perchlorate (TMRE) | Thermo Fisher Scientific | Cat# T669 |
| BODIPY 558/568 C12 | Thermo Fisher Scientific | Cat# D3835 |
| BODIPY 493/503 | Thermo Fisher Scientific | Cat# D3922 |
| MTT Reagent | Sigma-Aldrich | Cat#M5655 |
| Halt Protease and Phosphatase Inhibitor Cocktail (100X) | Thermo Fisher Scientific | Cat# 78440 |
| MitoSOX Red Mitochondrial Superoxide Indicator, for live-cell imaging | Thermo Fisher Scientific | Cat# M36008 |
| Oligomycin A | Fisher Scientific | Cat#ICN15178605 |
| Rotenone | Sigma-Aldrich | Cat#R8875 |
| Antimycin A | Sigma-Aldrich | Cat#A8674 |
| DNA-free DNA Removal Kit | Thermo Fisher Scientific | Cat# AM1906 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat# A25742 |
| tert-Butyl hydroperoxide, 70% Solution in water, ACROS Organics | Fisher Scientific | Cat# AC180340050 |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) | Sigma-Aldrich | Cat# C2920 |
| Polybrene | Santa Cruz Biotechnology | Cat# NC9840454 |
| 37% Formaldehyde | RICCA chemical company | Cat#RSOF0010 |
| Crystal Violet | Sigma-Aldrich | Cat#C0775 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| ATP Determination Kit | Thermo Fisher Scientific | Cat# A22066 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | Cat# 103015–100 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Directzol RNA MiniPrep | Zymo Research Corporation | Cat# 50–444-628 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368814 |
| Amplex Red Catalase Assay Kit | Thermo Fisher Scientific | Cat# A22180 |
| NAD/NADH Quantitation kit | Sigma-Aldrich | Cat# MAK037 |
|
| ||
| Deposited Data | ||
|
| ||
| MEFs_RNAseq_FPKM | This Paper | Table S3: Deposited data from RNA sequencing of MEFs |
| LNCaP_RNAseq_FPKM | This Paper | Table S4: Deposited data from RNA sequencing of LNCaP cells |
| C. elegans_RNAseq_FPKM | This Paper | Table S5: Deposited data from RNA sequencing of C.elegans |
| Lipid ion counts_hexacosanoic acid | This Paper | Table S7: Deposited data of lipid ion counts of LNCaP cells |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| HEK293T | ATCC | RRID: CRL-3216 |
| Mouse Embryonic Fibroblasts (Pip4k2aflx/flxPip4k2b−/−) | This Paper | N/A |
| Mouse Embryonic Fibroblasts (WT;Pip4k2aflx/flx) | This Paper | N/A |
| MDA-MD-468 | ATCC | RRID: CVCL_0419 |
| MCF10A | ATCC | RRID: CVCL_0598 |
| 4T1 | ATCC | RRID: CVCL_0125 |
| LNCaP | ATCC | RRID: CVCL_0395 |
| KPS-142 | This Paper | N/A |
| HT1080 | ATCC | RRID: CVCL_0317 |
| SW872 | ATCC | RRID: CVCL_1730 |
| OH931 | A gift from Julia Bridge | N/A |
| 293A | ATCC | RRID: CVCL_6910 |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Pip4k2aflx/flx Pip4k2b−/− | Lundquist et al., 2018 | N/A |
| LSL-KrasG12D/+ Trp53flx/flx (KP) | This Paper | N/A |
| LSL-KrasG12D/+ Trp53flx/flx Pip4k2aflx/flx Pip4k2b−/− (KP-αβDKO) | This Paper | N/A |
| N2 | Lundquist et al., 2018 | N/A |
| ppk-2 (pk1343) | Lundquist et al., 2018 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| sgRNA targeting sequences for human PIP4K2A and PIP4K2B | This Paper | Table S1: Targeting sgRNA oligonucleotide sequence |
| A full list of shRNA oligos for PIP4K2A and PIP4K2B cloned into the pCF_806 vector | This Paper | Table S2: Sequences for the shRNA oligonucleotides cloned into pCF806_Reci |
| A full list of qPCR primers synthesized. | This Paper (Primer-BLAST) | Table S6: Mouse qRT-PCR primers |
| A full list of qPCR primers C. elegans synthesized is included in Table S8 | Rooney et al., 2015 | Table S8: C.elegans qRT-PCR primers |
|
| ||
| Recombinant DNA | ||
|
| ||
| pBabe-Zeo-LT-ST | A gift from Jorge Moscat | N/A |
| pCL-Eco | Naviaux et al., 1996 | Addgene plasmid Cat # 12371 |
| pCL-Ampho | Novus Biologicals | Cat# NBP2–29541 |
| pVSV-G | A gift from Sammie Jeffrey | N/A |
| psPax2 | Addgene | Cat# 12260 |
| pCF806_Reci | A gift from Christof Fellman | N/A |
| pCF806_shRenilla | A gift from Christof Fellman | N/A |
| pCF806_shMulti (a,b) | This paper | N/A |
| rtTA3 | Fellmann et al., 2013 | N/A |
| pBabe-3xHA PIP4K2A (WT) | This paper | N/A |
| pBabe-3xHA PIP4K2A D359N (Kinase-dead) | This paper | N/A |
| pBabe-3xHA PIP4K2B (WT) | This paper | N/A |
| RT3REVIN | Fellmann et al., 2013 | N/A |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Cong et al., 2013 | Addgene plasmid Cat # 42230 |
|
| ||
| Software and algorithms | ||
|
| ||
| SoftGenetics GeneMarker® software | N/A | |
| STARv2.3.0e | N/A | |
| GENCODE v19 | N/A | |
| STAR (Version 2.4.2) aligner | N/A | |
| MultiQuant Software (version 3.0.3, Sciex technologies) | N/A | |
| Image J | N/A | |
| GraphPad | N/A | |
Highlights.
PI-4,5-P2 generated at peroxisomes by PI5P4Ks is essential for lipid trafficking
Loss of PI5P4K activity leads to peroxisomal dysfunction and accumulation of VLCFAs
PI5P4K inhibition impairs mitochondrial function downstream of peroxisomes
Therapeutic rationale for targeting PI5P4Ks for metabolically vulnerable diseases
ACKNOWLEDGMENTS
We would like to thank the following cores at SBPMDI (NCI P30 CA030199): Cell Imaging and Histology, Flow cytometry, Viral vectors, Cancer metabolism and Genomics. We acknowledge the expert assistance of Scott Henderson, Kimberly Vanderpool, and Theresa Fassel of the Core Microscopy Facility at the Scripps Research Institute. We would like to thank Julijana Ivanisevic and Hector Gallart Ayala at the metabolomics Platform, Faculty of Biology and Medicine, University of Lausanne, Switzerland, and John gallon and Salvatore Piscuoglio at University of Basel for the prostate cancer RNA-seq data. We are grateful to Jennifer Lippincott-Schwartz for the mEmerald-SKL construct and to both Akanksha Verma and Mark Lundquist for the C. elegans RNA-seq data. We are very thankful to Scott W. Lowe for his insightful scientific input and discussion. Research was supported by DOD (W81XWH-14-10440), NCI (R01 CA237536), and ACS (RSG-20–064-01-TBE) to B.M.E., MSCA (797949) to J.T., NIGMS (R00GM118909) to C.F., SNSF (31003A_175609) to M.A.R., and Weill Cornell Medicine Englander Institute for Precision Medicine (EIPM).
Footnotes
DECLARATION OF INTERESTS
C.P. is a scientific advisor for SEngine Precision Medicine and is currently a study pathologist for the CUPISCO trial, which is sponsored by Roche. C.P. receives reimbursement for study-related travels and remuneration for her work as a study pathologist for the benefit of her employer. C.F. is a co-founder of Mirimus.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.04.019.
REFERENCES
- Balla T. (2013). Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev 93, 1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, et al. (2010). Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet 42, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezowska S, and Galvá n JA (2017). Immunohistochemical detection of the autophagy markers LC3 and p62/SQSTM1 in formalin-fixed and paraffin-embedded tissue. Methods Mol. Biol 1560, 189–194. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2017). Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171, 950–965.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Weigel AV, Ioannou MS, Pasolli HA, Xu CS, Peale DR, Shtengel G, Freeman M, Hess HF, Blackstone C, and Lippincott-Schwartz J. (2019). Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J. Cell Biol 218, 2583–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Dasgupta S, and Sidransky D. (2011). Mitochondrial subversion in cancer. Cancer Prev. Res. (Phila) 4, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL, and Song BL (2015). Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran RA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes T, and Metallo CM (2019). Quantifying intermediary metabolism and lipogenesis in cultured mammalian cells using stable isotope tracing and mass spectrometry. Methods Mol. Biol 1978, 219–241. [DOI] [PubMed] [Google Scholar]
- Cyrta J, Augspach A, de Filippo MR, Prandi D, Thienger D, Benelli M, Cooley V, Bareja R, Wilkes D, Chae S-S, et al. (2020). Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. bioRxiv 10.1101/2020.03.06.949131v1.full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahabieh MS, Di Pietro E, Jangal M, Goncalves C, Witcher M, Braverman NE, and Del Rincón SV (2018). Peroxisomes and cancer: the role of a metabolic specialist in a disease of aberrant metabolism. Biochim. Biophys. Acta Rev. Cancer 1870, 103–121. [DOI] [PubMed] [Google Scholar]
- De Franceschi N, Alqabandi M, Miguet N, Caillat C, Mangenot S, Weissenhorn W, and Bassereau P. (2018). The ESCRT protein CHMP2B acts as a diffusion barrier on reconstituted membrane necks. J. Cell Sci 132, jcs217968. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria M, Nury T, Delmas D, Moreau T, Lizard G, and Vejux A. (2019). Protective function of autophagy during VLCFA-induced cytotoxicity in a neurodegenerative cell model. Free Radic. Biol. Med 137, 46–58. [DOI] [PubMed] [Google Scholar]
- Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, Brown M, Knowles JD, Halsall A, Haselden JN, et al. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc 6, 1060–1083. [DOI] [PubMed] [Google Scholar]
- Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, Bell EL, Shim HS, Lamia KA, Rameh LE, et al. (2013). Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell 155, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Fletcher CDM (2014). The evolving classification of soft tissue tumours - an update based on the new 2013 WHO classification. Histopathology 64, 2–11. [DOI] [PubMed] [Google Scholar]
- Fransen M, Lismont C, and Walton P. (2017). The peroxisome-mitochondria connection: how and why? Int. J. Mol. Sci 18, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habegger L, Sboner A, Gianoulis TA, Rozowsky J, Agarwal A, Snyder M, and Gerstein M. (2011). RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics 27, 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GRV, Schiavo G, and Irvine RF (2009). Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5) P2. Biochem J 422, 23–35. 10.1042/BJ20090428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Haroon S, and Vermulst M. (2019). Oxygen consumption measurements in Caenorhabditis elegans using the seahorse XF24. Bio Protoc 9, e3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Reese ML, and Goodman JM (2018). The assembly of lipid droplets and their roles in challenged cells. EMBO J. 37, e98947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines KM, Herron J, and Xu L. (2017). Assessment of altered lipid homeostasis by HILIC-ion mobility-mass spectrometry-based lipidomics. J. Lipid Res 58, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A, Zhao XT, Tu H, Xiao T, Fu T, Wang Y, Liu Y, Shi XJ, Luo J, and Song BL (2018). PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P2 homeostasis. J. Lipid Res 59, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangyang P, Li F, Lee P, Nissim I, Weljie AM, Mancuso A, Li B, Keith B, Yoon SS, and Simon MC (2020). Fructose-1,6-bisphosphatase 2 inhibits sarcoma progression by restraining mitochondrial biogenesis. Cell Metab. 31, 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaq SH, Teicher BA, and Monks A. (2014). Bioenergetic properties of human sarcoma cells help define sensitivity to metabolic inhibitors. Cell Cycle 13, 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Jeynov B, Lay D, Schmidt F, Tahirovic S, and Just WW (2006). Phosphoinositide synthesis and degradation in isolated rat liver peroxisomes.FEBS Lett. 580, 5917–5924. [DOI] [PubMed] [Google Scholar]
- Ježek J, Cooper KF, and Strich R. (2018). Reactive oxygen species and mitochondrial dynamics: the yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel) 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TJ (1987). Glutaraldehyde fixation chemistry: oxygen-consuming reactions. Eur. J. Cell Biol 45, 160–169. [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, and Kashatus DF (2015). Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 57, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee Y, and Koo JS (2015). Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS One 10, e0119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, Nielsen GP, Quade BJ, Chaber CJ, Schultz CP, et al. (2007). A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat. Med 13, 992–997. [DOI] [PubMed] [Google Scholar]
- Koundouros N, and Poulogiannis G. (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause AK, Hinrichs SH, Orndal C, DeBoer J, Neff JR, and Bridge JA (1997). Characterization of a human myxoid malignant fibrous histiocytoma cell line, OH931. Cancer Genet. Cytogenet 94, 138–143. [DOI] [PubMed] [Google Scholar]
- Lamia KA, Peroni OD, Kim YB, Rameh LE, Kahn BB, and Cantley LC (2004). Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta/ mice. Mol. Cell. Biol 24, 5080–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecompte O, Poch O, and Laporte J. (2008). PtdIns5P regulation through evolution: roles in membrane trafficking? Trends Biochem. Sci 33, 453–460. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linch M, Miah AB, Thway K, Judson IR, and Benson C. (2014). Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat. Rev. Clin. Oncol 11, 187–202. [DOI] [PubMed] [Google Scholar]
- Lismont C, Nordgren M, Van Veldhoven PP, and Fransen M. (2015). Redox interplay between mitochondria and peroxisomes. Front. Cell Dev.Biol 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Erauskin J, Galino J, Ruiz M, Cuezva JM, Fabregat I, Cacabelos D, Boada J, Martínez J, Ferrer I, Pamplona R, et al. (2013). Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet 22, 3296–3305. [DOI] [PubMed] [Google Scholar]
- Lundquist MR, Goncalves MD, Loughran RM, Possik E, Vijayaraghavan T, Yang A, Pauli C, Ravi A, Verma A, Yang Z, et al. (2018). Phosphatidylinositol-5-phosphate 4-kinases regulate cellular lipid metabolism by facilitating autophagy. Mol. Cell 70, 531–544.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnetjournal 17. [Google Scholar]
- McCommis KS, and Finck BN (2015). Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem. J 466, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Clippinger AK, Talledge N, Skowyra ML, Saunders MG, Naismith TV, Colf LA, Afonine P, Arthur C, Sundquist WI, et al. (2015). Structure and membrane remodeling activity of ESCRT-III helical polymers.Science 350, 1548–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness MC, Lu JF, Zhang HP, Dong GX, Heinzer AK, Watkins PA, Powers J, and Smith KD (2003). Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol. Cell. Biol 23, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munir R, Lisec J, Swinnen JV, and Zaidi N. (2019). Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 120, 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, and Verma IM (1996). The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, and Olzmann JA (2017). DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev.Cell 42, 9–21.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, and Olzmann JA (2017). Lipid droplets and lipotoxicity during autophagy. Autophagy 13, 2002–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelossof R, Fairchild L, Huang C-H, Widmer C, Sreedharan VT, Sinha N, Lai D-Y, Guan Y, Premsrirut PK, Tschaharganeh DF, et al. (2017). Prediction of potent shRNAs with a sequential classification algorithm. Nat. Biotechnol 35, 350–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier Y, Antonenkov VD, Glumoff T, and Hiltunen JK (2006). Peroxisomal beta-oxidation–a metabolic pathway with multiple functions. Biochim. Biophys. Acta 1763, 1413–1426. [DOI] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, and Galluzzi L. (2018). Mitochondrial metabolism and cancer. Cell Res. 28, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, and Lippincott-Schwartz J. (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameh LE, Tolias KF, Duckworth BC, and Cantley LC (1997). A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature 390, 192–196. [DOI] [PubMed] [Google Scholar]
- Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre J.t., and Meyer JN, J N. (2015). PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol 1241, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, and Capaldi RA (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 64, 985–993. [DOI] [PubMed] [Google Scholar]
- Roth KG, Mambetsariev I, Kulkarni P, and Salgia R. (2020). The mitochondrion as an emerging therapeutic target in cancer. Trends Mol. Med 26, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiento-Salinas FL, Delgado-Magallón A, Montes-Alvarado JB, Ramírez-Ramírez D, Flores-Alonso JC, Cortés-Hernández P, ReyesLeyva J, Herrera-Camacho I, Anaya-Ruiz M, Pelayo R, et al. (2019). Breast cancer subtypes present a differential production of reactive oxygen species (ROS) and susceptibility to antioxidant treatment. Front. Oncol 9, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, and Chipuk JE (2015). Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 57, 521–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sun X, Sun Y, Hou L, Yao M, Lian K, Li J, Lu X, and Jiang L. (2016). Elevation of cortical C26:0 due to the decline of peroxisomal beta-oxidation potentiates amyloid beta generation and spatial memory deficits via oxidative stress in diabetic rats. Neuroscience 315, 125–135. [DOI] [PubMed] [Google Scholar]
- Shim H, Wu C, Ramsamooj S, Bosch KN, Chen Z, Emerling BM, Yun J, Liu H, Choo-Wing R, Yang Z, et al. (2016). Deletion of the gene Pip4k2c, a novel phosphatidylinositol kinase, results in hyperactivation of the immune system. Proc. Natl. Acad. Sci. U S A 113, 7596–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. (2006). Maintenance of C. elegans In WormBook, pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P. (2005). Reactive oxygen species in tumor progression. Front. Biosci 10, 1881–1896. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Okazaki T, Aoyama S, Yokota M, Koike M, Okada Y, Fujiki Y, and Gotoh Y. (2019). Peroxisomes control mitochondrial dynamics and the mitochondrion-dependent apoptosis pathway. J. Cell Sci 132, jcs224766. [DOI] [PubMed] [Google Scholar]
- Van Veldhoven PP (2010). Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res 51, 2863–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJ, Waterham HR, and Ferdinandusse S. (2015). Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol 3, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJ, Ruiter JP, IJLst L, Waterham HR, and Houten SM (2010). The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis 33, 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinkove D, Bastiani M, Chessa TA, Joshi D, Hauth L, Cooke FT, Divecha N, and Schuske K. (2008). Overexpression of PPK-1, the Caenorhabditis elegans Type I PIP kinase, inhibits growth cone collapse in the developing nervous system and causes axonal degeneration in adults. Dev. Biol 313, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesinger C, Kunze M, Regelsberger G, Forss-Petter S, and Berger J. (2013). Impaired very long-chain acyl-CoA beta-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J. Biol. Chem 288, 19269–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Daniels G, Lee P, and Monaco ME (2014). Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol 2, 111–120. [PMC free article] [PubMed] [Google Scholar]
- Young JD (2014). Inca: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, Han JM, et al. (2013). A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol 15, 1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Li Z, Yan X, Bao L, Deng Y, Zeng F, Wang P, Zhu J, Yin D, Liao F, et al. (2018). Regulatory network and prognostic effect investigation of PIP4K2A in leukemia and solid cancers. Front. Genet 9, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, and Tu Y. (2013). Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, et al. (2018). Mitochondrial membrane potential. Anal. Biochem 552, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The supplemental tables accompanying the published article include all datasets generated in this study.