

Abstract
Objective
To identify the class of evidence for aducanumab use for the treatment of Alzheimer disease and present clinical considerations regarding use.
Methods
The author panel systematically reviewed available clinical trial data detailing aducanumab use in individuals with early symptomatic Alzheimer disease. Level of evidence statements were assigned in accordance with the American Academy of Neurology's 2017 therapeutic classification of evidence scheme. Safety information, regulatory decisions, and clinical context were also reviewed.
Results
Data were identified from 4 clinical trials, 1 rated Class I and 3 rated Class II. The Class I study showed that single doses of aducanumab up to 30 mg/kg were safe and well tolerated. All 3 Class II studies provided evidence that aducanumab (3–10 mg/kg) decreased amyloid deposition on brain PET at 1 year vs placebo. Efficacy data in the Class II studies varied by dose and outcome, but aducanumab either had no effect on mean change on the Clinical Dementia Rating Sum of Boxes scores or resulted in less worsening (vs placebo) that was of uncertain clinical importance. Adverse amyloid-related imaging abnormalities occurred in approximately 40% of individuals treated with aducanumab vs 10% receiving placebo.
Clinical Context
Administration of aducanumab will require expanded clinical infrastructure. Evidence-based guidance is needed to address key questions (e.g., safety in populations not enrolled in phase 3 studies, expected benefits on daily function, treatment duration) and critical issues relating to access to aducanumab (e.g., coverage, costs, burden of monthly infusions) that will inform shared decision making between patients and providers.
Alzheimer disease (AD) is a relentlessly progressive neurodegenerative disorder and the leading cause of mild cognitive impairment (MCI) and dementia, affecting an estimated 6.2 million Americans aged 65 years and older, and as many as 200,000 younger individuals.1,2 Although the primary initiators of AD continue to be debated,3 confluent lines of evidence implicate dysregulation of β-amyloid (Aβ) metabolism in early AD pathogenesis, leading to the formation of Aβ plaques, tau-containing neurofibrillary tangles, synaptic dysfunction, neurodegeneration, and ultimately cognitive impairment.4,5 Several large-scale clinical trials have been conducted to evaluate the safety and efficacy of investigational agents that modulate Aβ production, aggregation, and clearance in patients with symptomatic AD.6-9 These trials include amyloid PET measurements as a surrogate outcome (in addition to clinical outcome measures), asserting that a reduction in amyloid plaques will convey clinical benefit, although convincing evidence of this is lacking.10
Aducanumab is a recombinant human immunoglobulin G1 (IgG1) monoclonal antibody designed to promote the clearance of cerebral amyloid aggregates and insoluble forms of Aβ.11-13 On June 7, 2021, the Food and Drug Administration (FDA) granted provisional approval of aducanumab (Aduhelm) for the treatment of patients with symptomatic AD.12 Approval of this first-in-class antiamyloid therapy was met with considerable controversy, centered upon the interpretation and application of the evidence supporting the safety and efficacy of aducanumab,13-16 and the decision to approve this medication “based on reduction in Aβ plaques observed in patients treated with Aduhelm” rather than compelling evidence of a change in clinical measures.12 In response, the American Academy of Neurology (AAN) created a number of resources,18 published a viewpoint regarding the ethics of the approval process,19 and undertook a systematic review of available evidence. This Evidence in Focus represents the evidence review process. The accompanying discussion is intended to aid the practicing neurologist, other medical professionals, patients, and families in interpreting published data concerning aducanumab to inform clinical decision making.
Description of the Analytic Process
Evidence in Focus articles advance the Guidelines Subcommittee mission (eAppendix 1, links.lww.com/WNL/B831) by providing timely evidence-based discussions of focused topics of high relevance for neurologists and patients. This type of systematic review uses an abbreviated version of the AAN guideline methodology20 to highlight the strength of evidence underlying new therapies and facilitate discussion concerning appropriate use. This process does not generate specific recommendations for care.
In June 2021, the AAN initiated this Evidence in Focus document and appointed a guidelines subcommittee member with content expertise (N.S.), a former guidelines subcommittee member with content expertise (G.S.D.), a patient representative (A.M.), a care partner representative (B.W.), a solo/small practice neurologist also representing the AAN Coding and Payment Policy Subcommittee (K.C.), a guideline dissemination manager (S.R.W.), a guideline methodologist (R.D.), and a former guideline methodologist with expertise with the Evidence in Focus product (M.J.A.) to develop this document. Guidelines Subcommittee leadership and an AAN staff person reviewed potential authors' conflict of interest forms and their curricula vitae and approved panel composition. Panel members did not have relevant financial or intellectual conflicts of interest.
A pragmatic PubMed literature search for clinical trials using aducanumab was conducted on July 8, 2021, using the term “aducanumab” in the Clinical Queries PubMed section,21 and the validated therapy (filter menu) and narrow (scope menu) options.22 The search was repeated on November 29, 2021. Clinicaltrials.gov was also searched for “aducanumab” and “BIIB037” (July 8, 2021); data from the FDA were included. Conference abstracts were not included in the formal evidence review. As this process focused on aducanumab, publications involving other antiamyloid therapies were not included. Two panel members independently reviewed titles and abstracts for inclusion. Articles and study data were classified by 2 independent raters following the 2017 AAN therapeutic classification scheme.20 Use of surrogate outcomes (e.g., amyloid PET) is discouraged in the AAN guideline development process, but this does not affect the classification of evidence, which is based on study design and conduct.20 Level of evidence statements were developed according to the process used for Neurology® level of evidence reviews. This document does not involve a critique of statistical analyses performed as part of the FDA review or the FDA process for aducanumab approval and confirmatory trial requirements. This article was reviewed and approved by the AAN Guidelines Subcommittee (eAppendix 2, links.lww.com/WNL/B831) and Quality Committee before submission to Neurology.
Evidence Summary
The initial PubMed search yielded 4 articles (eAppendix 3, links.lww.com/WNL/B831), 2 of which were deemed relevant. One additional article was identified within the references of PubMed articles and included. The clinicaltrials.gov search yielded 9 trials. The updated PubMed search yielded 2 new articles (eAppendix 3); 1 was deemed relevant.
Level of Evidence
Table 1 summarizes studies investigating aducanumab and level of evidence statements. The first study (Single Ascending Dose Study of BIIB037 in Participants With Alzheimer's Disease; Unique Identifier: NCT01397539) was a Class I, phase 1 randomized, double-blind, placebo-controlled single ascending-dose study in individuals with mild to moderate AD dementia.23 Primary outcomes were safety and tolerability. Eight participants were planned for enrollment into each of 7 sequential cohorts randomized 6:2 to receive a single dose of aducanumab (0.3, 1, 3, 10, 20, 30, or 60 mg/kg) or placebo. Enrollment within the 60 mg/kg cohort was stopped early after all 3 individuals developed symptomatic amyloid-related imaging abnormalities (ARIA) with edema/effusion (ARIA-E) or ARIA-E with microhemorrhage/hemosiderosis (ARIA-H) (n = 1). Imaging abnormalities resolved 8–15 weeks following treatment cessation in all participants. The 13-item Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS-Cog 13) was used as an exploratory efficacy outcome measure. Mean change in ADAS-Cog 13 scores 3 and 24 weeks after administration of aducanumab did not significantly differ among groups (n = 39 on treatment, n = 14 on placebo). Doses of aducanumab up to 30 mg/kg were tolerated without serious adverse events (AEs).23
Table 1.
Clinical Trials of Aducanumab in Patients With Alzheimer Disease Listed on Clinicaltrials.gova

The second study (PRIME [Multiple Dose Study of Aducanumab (BIIB037) (Recombinant, Fully Human Anti-Aβ IgG1 mAb) in Participants With Prodromal or Mild Alzheimer's Disease]; Unique Identifier: NCT01677572; 221AD103) was a Class II (due to <80% completion), phase 1 randomized, double-blind, placebo-controlled study of multiple doses of aducanumab (BIIB037) in participants with prodromal (very mild) or mild AD. Results were published in 2 articles,24,25 one of which was a secondary analysis of amyloid PET data.25 PRIME randomized 165 individuals with prodromal or mild symptomatic AD and a visually positive amyloid PET to monthly infusions of aducanumab (1, 3, 6, or 10 mg/kg) or placebo for 1 year. Forty (24.2%) participants discontinued treatment: 20 due to AEs (9 from the 10 mg/kg group; 11 spread across other groups), 14 withdrew consent, and 6 for other reasons.24 The primary outcome was the number of participants with AEs. Secondary outcomes were change in amyloid PET imaging, pharmacokinetic serum concentrations, and development of antibodies against aducanumab. Amyloid PET standardized uptake value ratio (SUVR) values at 1 year decreased more in the 3 mg/kg (mean ± SE −0.135 ± 0.022), 6 mg/kg (−0.210 ± 0.024), and 10 mg/kg (−0.268 ± 0.025) groups vs placebo (0.003 ± 0.021; all p < 0.001), but not in the 1 mg/kg group (−0.055 ± 0.024; p > 0.05). Subsequent analyses confirmed a dose-dependent reduction in amyloid (p < 0.0001). This study was not powered to detect clinical change; thus, analyses considering the effect of aducanumab on clinical outcome measures were exploratory. The mean (±SE) change in Clinical Dementia Rating Sum of Boxes (CDR-SB) scores between baseline and week 54 were significantly different only between the 10 mg/kg group (0.63 ± 0.47) and placebo (1.87 ± 0.41; p < 0.05). There were no significant differences between placebo and the 1 mg/kg (1.72 ± 0.46), 3 mg/kg (1.37 ± 0.43), or 6 mg/kg (1.11 ± 0.44) groups.24 For reference, a 1- to 2-point change in the CDR-SB may indicate a clinically important change.26 Change in the Mini-Mental State Examination (MMSE) score between baseline and week 52 was significantly different for the 3 mg/kg (−0.70 ± 0.75) and 10 mg/kg groups (−0.56 ± 0.76) vs placebo (−2.81 ± 0.67; both p < 0.05), but not the 1 mg/kg (−2.18 ± 0.75) or 6 mg/kg (−1.96 ± 0.75) groups. The p value for the dose–response analyses was <0.05 for both clinical outcomes.24
Two identically designed Class II phase 3 randomized double-blind placebo-controlled studies evaluated the efficacy and safety of aducanumab in patients with prodromal or mild symptomatic AD: ENGAGE (221AD301 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer's Disease; Unique Identifier: NCT02477800; 221AD301) and EMERGE (221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer's Disease; Unique Identifier: NCT02484547; 221AD302).27 At the time of the original review, data for these trials were available only through FDA documentation.27 In the updated literature search, ARIA-related results were published ahead of print.28 Participants were randomized 1:1:1 to aducanumab low dose (3 mg/kg for APOE ε4 carriers and 6 mg/kg for noncarriers), aducanumab high dose (initially 6 mg/kg for APOE ε4 carriers and 10 mg/kg for noncarriers, later increasing to 10 mg/kg for both groups), or placebo. Participants were followed for 78 weeks (20 doses), which included 8–24 weeks of titration, and for an additional 18 weeks after the final dose for safety.
Although the studies were blinded, the occurrence of ARIA resulted in functional unblinding and necessitated dose modification following a standard protocol. The primary efficacy endpoint was the change in the CDR-SB between baseline and week 78. Secondary clinical endpoints included the MMSE, ADAS-Cog 13, and the Alzheimer's Disease Cooperative Study–Activities of Daily Living scale adapted for Mild Cognitive Impairment (ADCS-ADL-MCI).12,13,27 An interim futility analysis was planned after half of participants completed the week 78 visit, with futility presumed if conditional power for the primary efficacy analysis was <20% in treatment groups in both studies (i.e., if there was a low probability that additional trials would result in conclusive evidence). An independent, unblinded statistician conducted the futility analysis in spring 2019, after which Biogen made the decision to terminate ongoing studies of aducanumab.27 FDA documents included additional analyses informing primary and secondary study outcomes, which were reviewed and incorporated within this document.27 Post hoc analyses limited to subgroups (e.g., participants with rapidly progressive courses and participants exposed to the target 10 mg/kg dose for varying lengths of time) were outside of the scope of this Evidence in Focus document.
ENGAGE (Class II due to <80% completion) randomized 1,647 participants (547 to low-dose aducanumab, 555 to high-dose aducanumab, and 545 to placebo). Of these, 720 (43.7%) participants discontinued treatment, most due to study termination after the interim analysis. There was no significant difference in change on the CDR-SB between baseline and week 78 in either group (difference between low-dose aducanumab and placebo was −0.18; 95% CI, −0.469 to 0.110; difference between high-dose aducanumab and placebo was 0.03; 95% CI, −0.262 to 0.326). There was no significant difference in mean change on the MMSE between baseline and week 78 in the low-dose (0.2; 95% CI, −0.35 to 0.74) or high-dose groups (−0.1; 95% CI, −0.62 to 0.49) vs placebo. There was no significant difference in the mean change on the ADAS-Cog 13 between baseline and week 78 in the low-dose (−0.58; 95% CI, −1.58 to 0.42) or high-dose groups (−0.59; 95% CI, −1.61 to 0.43) vs placebo. There was also no significant difference in the mean change on the ADCS-ADL-MCI between baseline and week 78 in the low-dose (0.7; 95% CI, −0.19 to 1.64) or high-dose groups (0.7; 95% CI, −0.25 to 1.61) vs placebo. Mean change from baseline brain amyloid signal to week 78 was measured using SUVR. The adjusted mean change vs placebo (124 participants) was −0.167 for the low-dose group (138 participants) and −0.232 for the high-dose group (112 participants; SE not explicitly reported), indicating a reduction in cerebral amyloid in treated individuals (p < 0.0001). There was no correlation between change from baseline SUVR at week 78 and change from baseline CDR-SB at week 78 (numbers not provided).27
EMERGE (Class II due to <80% completion) randomized 1,643 participants (547 to low-dose aducanumab, 547 to high-dose aducanumab, and 549 to placebo). Of these, 783 participants (47.7%) discontinued treatment, most due to study termination after the interim analysis. There was no difference in change in mean CDR-SB between baseline and week 78 in the low-dose group vs placebo (−0.26; 95% CI, −0.569 to 0.041). The high-dose group experienced less worsening than the placebo group (−0.39; 95% CI, −0.694 to −0.086), which was statistically significant but less than the suggested clinically important change on the CDR-SB (1–2 points).26 There was no difference in mean change on the MMSE between baseline and week 78 in the low-dose (−0.1; 95% CI, −0.65 to 0.48) or high-dose groups (0.6; 95% CI, 0.00–1.13) vs placebo. In the low-dose group vs placebo, there was no difference in the mean change on the ADAS-Cog 13 between baseline and week 78 (−0.70; 95% CI, −1.76 to 0.36) or the ADCS-ADL-MCI (0.7; 95% CI, −0.27 to 1.73). The high-dose group showed less worsening vs placebo on the same measures (ADAS-Cog 13: −1.40; 95% CI, −2.46 to −0.34; ADCS-ADL-MCI: 1.7; 95% CI, 0.75–2.74). Mean change from baseline brain amyloid signal to week 78 was measured by the SUVR. The adjusted mean change vs placebo (93 participants) was −0.179 for the low-dose group (100 participants) and −0.278 for the high-dose group (109 participants; SE not explicitly reported), indicating a reduction in cerebral amyloid in treated individuals (p < 0.0001). In a subanalysis of 329 participants, there was a weak relationship between change in the amyloid PET composite SUVR and change in CDR-SB between baseline and week 78 (Spearman correlation 0.19; 95% CI, 0.048–0.327).27
An inverse variance meta-analysis was performed using a random effects model that incorporated data from EMERGE and ENGAGE primary outcomes (CDR-SB). Although the study designs were identical, a random effects model was chosen to account for potential differences between study cohorts. According to this model, low-dose aducanumab resulted in less worsening on the CDR-SB at 78 weeks (−0.22; 95% CI, −0.43 to −0.01; I2 0%) compared with placebo. The effect of high-dose aducanumab was not statistically significant (−0.18; 95% CI, −0.59 to 0.23; I2 74%), with substantial heterogeneity in the results. None of the measured effects surpassed thresholds for CDR-SB change associated with clinically important effects.26
Post hoc FDA analyses performed on the subset of patients with amyloid PET scans showed a decrease in amyloid burden (EMERGE: placebo = no change [n = 93]; low-dose aducanumab = 0.179 reduction in mean SUVR [n = 100]; high-dose aducanumab = 0.278 reduction in mean SUVR [n = 109]).27 Tau PET was added to the EMERGE and ENGAGE studies late in their course and pooled analysis was performed. Early study termination and the low number of enrolled participants precluded meaningful analysis.27
Safety
Aducanumab, like most antiamyloid agents, can cause ARIA (Figure 1 and Table 2).29 In this context, ARIA refers to imaging findings attributable to vasogenic edema (ARIA-E), including patients with mild asymptomatic parenchymal edema and those with sulcal effacement and localizing signs or blood-degradation products (ARIA-H).30 In PRIME, ARIA-E occurred in 1 (3%), 2 (6%), 11 (37%), and 13 (41%) participants receiving aducanumab 1, 3, 6, and 10 mg/kg, respectively (no occurrences in the placebo group). ARIA typically occurred early during treatment and resolved within 4–12 weeks. Approximately half (15/27, 56%) of participants experiencing ARIA continued treatment. There were no hospitalizations or deaths reported in the initial publication.24 One subsequent fatal intracranial hemorrhage was assessed as treatment-related.27 The other most common AEs were headache, urinary tract infection, and upper respiratory tract infection.24
Figure 1. Severe Amyloid-Related Imaging Abnormalities.
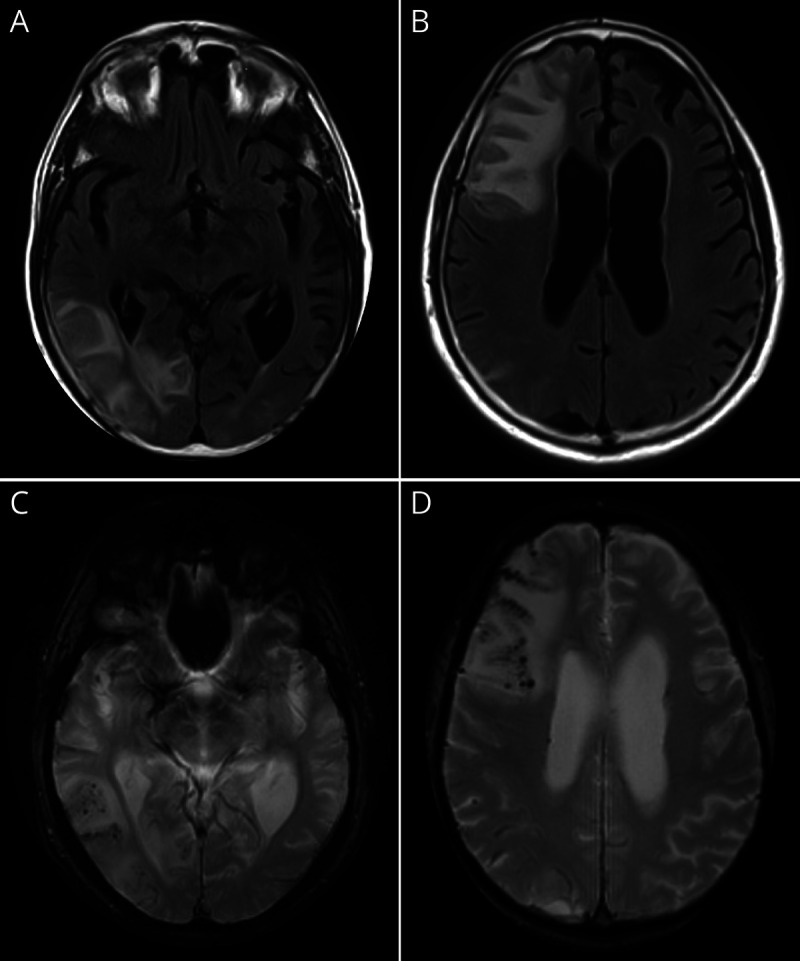
Magnetic resonance (3T) neuroimaging performed in a 75-year-old woman with early symptomatic Alzheimer disease who presented with increasing headache, confusion, and left-sided weakness. Symptoms developed 4 months following randomization within a phase 3 study of aducanumab. (A, B) T2 fluid-attenuated inversion recovery sequences depicting right frontal and temporoparietal edema with localized mass effect amyloid-related imaging abnormalities with edema/effusion (ARIA-E). (C, D) Susceptibility-weighted images depicting multiple areas of signal change within areas of edema, corresponding to microhemorrhage (amyloid-related imaging abnormalities with edema/effusion with concurrent microhemorrhage/hemosiderosis [ARIA-H]). Images displayed in radiologic convention. Images courtesy of Dr. T.L.S. Benzinger (Washington University in St. Louis, Missouri).
Table 2.
ARIA Radiographic (Brain MRI) Classification Used in Clinical Studies of Aducanumab11

In ENGAGE and EMERGE, ARIA occurred more frequently in participants treated with aducanumab vs placebo (Table 3), particularly in APOE ε4 carriers. This effect was most pronounced in the 10 mg/kg dose group (APOE e4 carriers: 43.0%; noncarriers: 20.3%).27,28 Although ARIA can occur at any time following treatment with aducanumab, most events occurred within the first 8 doses (7 months of initiation). Aducanumab-related ARIA-H usually occurred within the setting of coexisting ARIA-E. Almost all (98%) ARIA-E cases in the high-dose aducanumab group resolved (i.e., no residual brain edema) during the study, with 69% resolving within 3 months and 83% within 4 months.28 In the 10 mg/kg group, ARIA-H microhemorrhages stabilized within 2 months in 69% of participants with ARIA-H and 93% with superficial siderosis.28
Table 3.
Proportions of Participants With ARIA-E and ARIA-H in the EMERGE and ENGAGE Phase 3 Studies
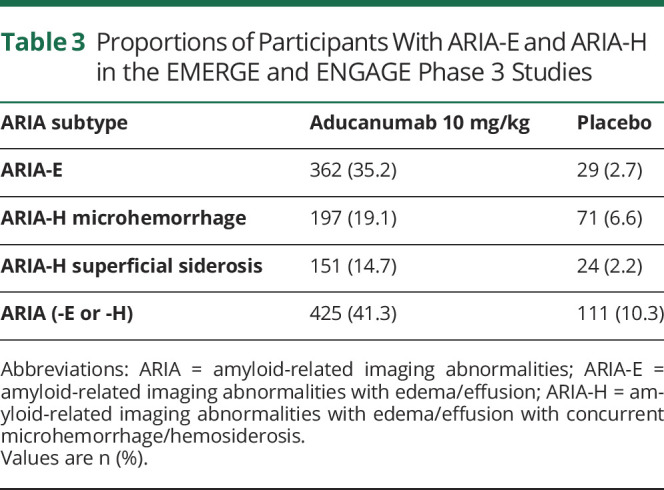
Of the 425 patients receiving 10 mg/kg who experienced ARIA during the placebo-controlled period, 103 (24.2%) had associated symptoms, including headache (47%), confusion (15%), dizziness (11%), and nausea (8%). In the 10 mg/kg group, ARIA-related symptoms resolved in a median 5 weeks (interquartile range, 1–13 weeks).28
Approximately 9% of individuals enrolled in ENGAGE/EMERGE who received the recommended 10 mg/kg aducanumab dose discontinued due to AEs (vs ∼4% of patients receiving placebo). ARIA was the most common reason for drug discontinuation.27 There was no overall imbalance in mortality in aducanumab- vs placebo-treated participants.27 A November 2021 publication31 mentioned the death of a participant receiving aducanumab through an extension trial, but formal determination of whether this death was potentially related to aducanumab is pending. Other severe AEs in ENGAGE/EMERGE were rare, with angioedema and urticaria (hypersensitivity reaction) reported in a single patient during aducanumab infusion.12,13
Regulatory Decisions
The FDA provided the following approval on June 7, 2021:
Aduhelm is an Aβ-directed antibody indicated for the treatment of AD. This indication is approved under accelerated approval based on reduction in Aβ plaques observed in patients treated with Aduhelm. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s).12
Prescribing indications were modified on July 8, 2021, to stipulate that aducanumab “should be initiated in patients with MCI or mild dementia stage of the disease, the population in which treatment was initiated in clinical trials.”13 Other international drug regulatory agencies have refused marketing authorization or not approved aducanumab.32 Aducanumab is administered every 4 weeks via a weight-based IV infusion, titrated to 10 mg/kg over a 6-month period.
Clinical Context
The approval of aducanumab marks a shift in AD therapeutics from medications targeting symptoms (e.g., acetylcholinesterase inhibitors) to those targeting neuropathology associated with AD. Whether aducanumab will result in a clinically meaningful slowing of AD symptoms remains to be determined, as does the safety of aducanumab in clinical populations.
Populations for Use
The phase 3 aducanumab trials (ENGAGE and EMERGE) enrolled patients aged 50–85 years with MCI or mild dementia due to AD, corresponding to a global Clinical Dementia Rating score of 0.5 and an MMSE score between 24 and 30, and abnormal cerebral amyloid deposition determined via amyloid PET. Whereas initial FDA indications did not specify AD stage,12 prescribing recommendations were updated in July 2021 to clarify that aducanumab is indicated for use in patients with early symptomatic AD and acknowledge that “there are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease.“13 Contrary to the clinical trials, neither the original nor revised recommendations require objective demonstration of amyloid deposition prior to use of aducanumab.12,13 A substantial proportion of patients with suspected early symptomatic AD evaluated in clinical and research settings (20%–40%) do not have abnormal amyloid on testing.33-35 Logically, therapies approved for the reduction of cerebral amyloid deposits should be reserved for patients with elevated cerebral amyloid. Amyloid PET and CSF measures of AD biomarkers (e.g., Aβ42, Aβ40, total tau, and phosphorylated tau181) are validated for this purpose, with comparable diagnostic accuracy in patients with early symptomatic AD36 and well-defined appropriate use criteria.37,38
The FDA did not list contraindications to administration of aducanumab.12,13 However, phase 3 studies of aducanumab excluded individuals with concurrent medical, neurologic, or psychiatric conditions that might contribute to cognitive impairment (including suspected dementia with Lewy bodies, vascular cognitive impairment, or clinically significant unstable psychiatric illness). Patients with a history of bleeding disorders, prior brain hemorrhage, or cerebrovascular abnormalities, including stroke, and those taking antiplatelets other than daily aspirin (≤325 mg/d) were also excluded from participation.27 Strict extrapolation of clinical trial criteria to real-world populations would dramatically limit the number of eligible patients. A recent analysis of 2,870,023 Medicare beneficiaries with symptomatic AD and related disorders identified that 91% of patients diagnosed with AD dementia and 86% of patients with MCI met at least 1 exclusion criterion for the aducanumab trials (e.g., cardiovascular disease, anticoagulation, chronic kidney disease, age >85 years). Most of these individuals met multiple exclusion criteria (77% with AD dementia, 64% with MCI).39 In lieu of clear directions from the FDA, expert consensus suggests avoiding aducanumab use in patients at the highest risk of serious AEs, including individuals taking anticoagulants and those with a history of symptomatic hemorrhage.40
Participants from traditionally underrepresented minority groups were not well represented in phase 3 studies of aducanumab.12,13,27,41 ENGAGE (study 301, n = 1,647) enrolled only 8 Black or African American individuals (5 assigned to placebo, 1 assigned to low-dose aducanumab, 2 assigned to high-dose aducanumab) and 37 Hispanic or Latino individuals (13 assigned to placebo, 11 assigned to low-dose aducanumab, and 13 assigned to high-dose aducanumab). EMERGE (study 302, n = 1,638) enrolled only 11 Black or African American individuals (1 assigned to placebo, 6 assigned to low-dose aducanumab, and 4 assigned to high-dose aducanumab) and 67 Hispanic or Latino individuals (22 assigned to placebo, 22 assigned to low-dose aducanumab, and 23 assigned to high-dose aducanumab).27 In the recently published secondary analysis of ARIA data, only race (not ethnicity) was reported in the baseline characteristics table. These data, reflecting a subset of both ENGAGE and EMERGE cohorts (n = 1,087), included 19 Black or African American participants (6 assigned to placebo, 6 assigned to aducanumab 3 mg/kg, 1 assigned to aducanumab 6 mg/kg, and 6 assigned to aducanumab 10 mg/kg).28 It is not known whether available efficacy and safety data can be directly translated to patients who were not well represented within clinical trial populations.
Infrastructure Demands
The safe prescription of aducanumab will require substantial investment in human and physical infrastructure. The approval of therapies for patients with early symptomatic AD are expected to markedly increase demand for clinicians with appropriate training in the assessment and staging of patients with AD and interpretation of AD biomarker results. Clinics may need to increase capacity for lumbar punctures to assess CSF-based measures of Aβ, particularly in rural communities where PET imaging is less available. Clinical capacity will also need to grow to accommodate follow-up visits to assess treatment response and evaluate potential AEs. This increase in demand will further strain the neurology workforce, potentially compromising access to care, patient outcomes, and career satisfaction.42 Demands will likewise increase for outpatient infusion center space, specialized nursing services to ensure safe administration of aducanumab and patient monitoring, and radiologists and neurologists trained to diagnose ARIA.30
Coverage Considerations
The Institute for Clinical and Economic Review assigned a rating of “insufficient” for whether aducanumab provides a net health benefit for patients. Results of the cost-effectiveness analysis suggested small overall health gains, justifying drug prices of $2,950–$8,360 per year if traditional cost-effectiveness thresholds were applied.43 This figure contrasts with the manufacturer-established price of aducanumab. Although aducanumab was originally marketed at $4,312 per infusion ($56,000 per year for a person who weighs 74 kg),44 the price was reduced by 50% 6 months after launch ($28,000 per year).45 Even with this price adjustment, the average annual cost of treatment is likely to exceed $75,000 when costs associated with clinical assessments, investigations, and medication administration are incorporated.
It is unclear what proportion of costs will be covered by payers. After the announcement that the FDA will investigate the accelerated approval process,46 several large academic centers as well as the Department of Veterans Affairs publicly stated they will not add aducanumab to their formularies.47 The Centers for Medicare & Medicaid Services initiated a national coverage determination analysis to determine whether Medicare will establish a national Medicare coverage policy for monoclonal antibodies targeting amyloid for the treatment of AD. Draft recommendations were published in early 2022, indicating the intent not to cover aducanumab outside of a randomized controlled trial approved by the Centers for Medicare & Medicaid Services; final recommendations are expected by April 2022.48
Patient Preferences
Patients living with AD and their families have varying views on aducanumab. Patients who pursue treatment may do so while balancing the controversy surrounding efficacy, safety, and cost of aducanumab against the reality of a living with an incurable, relentlessly progressive neurodegenerative disease.40 Those who choose not to pursue treatment may weigh uncertain benefit against the possibility of AEs and certainty of high direct and indirect costs measured in dollars, time, and inconvenience associated with frequent infusions, clinic visits, and ancillary tests. Many patients fall somewhere in the middle, seeking additional information and provider input to guide decision making. The principles of shared decision making emphasize the importance of soliciting patient and caregiver perspectives surrounding treatment and the incorporation of these values and goals in treatment decisions.49 A focused understanding of available data is key to this process, including recognition of limitations in study design or trial conduct that influence extrapolation of study findings to specific patients or patient groups (Table 4).
Table 4.
Selected Topics to Include When Facilitating Shared Decision-making Discussions Concerning Aducanumab

Other Considerations
Safety-Related Follow-up
The FDA recommends “enhanced clinical vigilance for ARIA…during the first 8 doses of treatment.”13 If symptoms or signs suggestive of ARIA are detected, clinical evaluation and brain MRI are indicated. Prescribing indications recommend that brain MRI be performed at baseline (within 1 year of initiating aducanumab) and prior to the 7th and 12th monthly infusions of aducanumab.13 Optimal MRI parameters include serial completion in the same scanner with the same imaging protocols, inclusion of sequences to detect microhemorrhages, and review by clinicians or radiologists trained in ARIA detection. Susceptibility-weighted imaging sequences (or equivalent) are preferred over gradient-echo T2*-weighted imaging (or similar) sequences, given increased sensitivity for microhemorrhages.50 The detection of cerebral microbleeds also increases with MRI field strength (Figure 2).51
Figure 2. Detection of Microhemorrhages Varies by Magnet Strength and Imaging Technique.
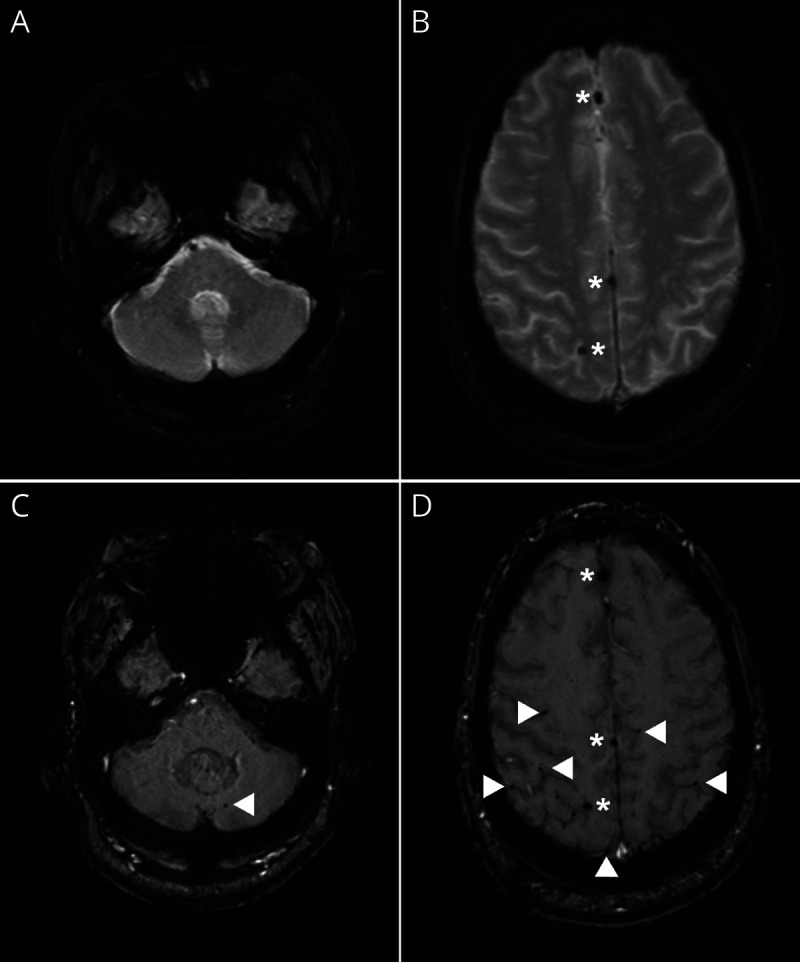
(A, B) Gradient-echo MRI obtained on a 1.5T scanner discloses 3 microhemorrhages. (C, D) Susceptibility-weighted imaging on a 3T scanner discloses multiple additional areas of increased susceptibility consistent with microhemorrhages in the anterior and posterior fossae (arrowheads). Scans completed on the same day in the same patient. Images displayed in radiologic convention. Images courtesy of Dr. T.L.S. Benzinger (Washington University in St. Louis, Missouri).
Multiple microhemorrhages or localized superficial siderosis on baseline MRI suggest underlying cerebral amyloid angiopathy,52 which may intimate an increased ARIA risk. Patients with localized superficial siderosis, >4 microhemorrhages, or brain hemorrhage >1 cm on pretreatment brain MRI were excluded from clinical trials of aducanumab.27 Aducanumab safety is not established in these patients.
In aducanumab-treated patients, the emergence of micro- or macrohemorrhages or superficial siderosis may indicate ARIA. In lieu of specific studies on the topic, experience and protocols successfully applied in phase 3 studies of aducanumab may be extrapolated to inform management of symptomatic or asymptomatic ARIA (Table 5). Although no established pharmacologic therapy exists for symptomatic ARIA, response to pulse steroids has been documented in severe cases (Class IV, case studies)53; corticosteroids were the most commonly administered medications for ARIA in ENGAGE/EMERGE.28
Table 5.
Strategies to Manage Symptomatic and Asymptomatic ARIA Extrapolated From Phase 3 Clinical Trials26

APOE Genotype
APOE ε4 carriers treated with aducanumab 10 mg/kg in ENGAGE/EMERGE were more likely to experience clinically significant ARIA in phase 3 studies.12,13,27 When known (from clinical or direct-to-consumer testing), this information may influence risk stratification, although prescribing instructions do not mandate APOE genotyping.13
Treatment Cessation
The optimal duration of treatment with aducanumab is unknown. Primary outcomes in ENGAGE/EMERGE were assessed after 78 weeks of treatment, but the FDA did not include treatment duration in the prescribing instructions.13 Pending new data, decisions on treatment duration will likely be guided by coverage considerations, access (e.g., related to cost, ability to attend monthly infusions over long periods), side effects, perceptions of efficacy, and shared decision making.40 In the absence of AEs, clinical- or biomarker-based criteria may also inform decisions concerning treatment cessation.
Approach to Dementia Care
Patients in community-based cohorts commonly receive a diagnosis of “unspecified dementia,” with a minority accessing dementia specialists within 5 years of diagnosis of dementia.54 The approval of a first-in-class antiamyloid therapy for patients with early symptomatic AD (and prospect of future approvals55) is expected to change this, establishing the importance of early recognition of symptomatic AD. This shift aligns with patient and caregiver expressed preference for dementia disclosure,56 but will come at the cost of increased demand for clinical evaluations and AD-specific biomarkers.
A team-based approach will be required to meet the demands for clinical evaluations, testing, and treatment. Community-based practitioners will play an important role in screening patients with mild memory complaints and identifying those who may be considered for treatment with approved antiamyloid agents. Formalized partnerships between community-based providers and subspecialty centers may allow interested patients to receive an expedited evaluation that includes review of eligibility criteria for treatment, individualized risk stratification, and counseling concerning the potential risks, benefits, and practical implications associated with specific treatment plans. Community-based practitioners will also be essential to support the longitudinal follow-up of treated patients, including urgent/emergent assessment for potential drug-related AEs, and to ensure that patients can access dementia care in their local neighborhoods. This point is especially important when discussing therapies that must be administered every 4 weeks to patients who may depend on caregivers for wayfinding and transportation. Although coverage decisions and special access programs have been touted as strategies to ensure access to diagnostics and treatments for qualified patients, including underserved populations,57 even the best designed program is likely to exacerbate the financial burden borne by patients and families and to divert scarce resources that could have been applied to other programs aiming to improve the quality of life for people living with dementia.
Suggestions for Future Research
The FDA stated that continued approval of aducanumab may be contingent upon verification of clinical benefit in confirmatory trials.13 Biogen plans to conduct a phase 4 (postmarket) trial to demonstrate the clinical benefit of aducanumab in patients with early symptomatic AD, including patients from diverse ethnicities (ICARE AD-US [International Collaboration for Real-World Evidence in Alzheimer's Disease: A Prospective Real-World Observational Study of Aducanumab-avwa in Patients With Alzheimer's Disease in the US]).58 Expanded clinical experience with aducanumab may also prompt other studies leveraging pragmatic trial designs to inform the safety and efficacy of aducanumab in broader populations. Until these data are available, clinicians, patients, and payers are likely to grapple with questions including whether aducanumab should be provided, to whom, and for how long. These questions and others (Table 4) should be incorporated within shared decision-making discussions.
Future research is needed to determine whether aducanumab-related reductions in cerebral amyloid burden translate to clinically meaningful outcomes. Additional studies are required to inform the safety and efficacy of aducanumab in patients excluded from EMERGE/ENGAGE who are likely to also be underrepresented in phase 4 studies. These include individuals with autosomal dominant AD-causing mutations (e.g., variants in APP, PSEN1, PSEN2), trisomy 21 (Down syndrome), atypical clinical presentations (e.g., posterior cortical atrophy, primary progressive aphasia), or concurrent pathology (e.g., Lewy body disease, TDP-43, vascular disease). The translatability of trial-ready efficacy measures (e.g., CDR-SB, ADAS-Cog 13) represents another priority area, acknowledging the need for efficacy data that inform outcomes prioritized by patients and caregivers, including maintenance of independence (including driving), quality of life, and survival. Absent high-quality data to inform these questions, uncertainty surrounding the optimal use of aducanumab in clinical practice will remain.
Disclaimer
Clinical practice guidelines, practice advisories, systematic reviews, and other guidance published by the American Academy of Neurology and its affiliates are assessments of current scientific and clinical information provided as an educational service. The information (1) should not be considered inclusive of all proper treatments or methods of care, or as a statement of the standard of care; (2) is not continually updated and may not reflect the most recent evidence (new evidence may emerge between the time information is developed and when it is published or read); (3) addresses only the question(s) specifically identified; (4) does not mandate any particular course of medical care; and (5) is not intended to substitute for the independent professional judgment of the treating provider, as the information does not account for individual variation among patients. In all cases, the selected course of action should be considered by the treating provider in the context of treating the individual patient. Use of the information is voluntary. The AAN provides this information on an “as is” basis, and makes no warranty, expressed or implied, regarding the information. The AAN specifically disclaims any warranties of merchantability or fitness for a particular use or purpose. The AAN assumes no responsibility for any injury or damage to persons or property arising out of or related to any use of this information or for any errors or omissions.
Conflict of Interest
All AAN guideline authors must meet the stipulations outlined in the AAN's Conflict of Interest Policy59 in order to participate on a guideline development panel. This policy is described further in the 2017 AAN Clinical Practice Guideline Development Manual.20
Acknowledgment
The authors thank Jennifer Blough, who was seated to the author panel but withdrew prior to project start, for her contributions to the framing of objectives for this Evidence in Focus product from a care partner perspective.
Glossary
- AAN
American Academy of Neurology
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAS-Cog 13
Alzheimer's Disease Assessment Scale–Cognitive Subscale
- ADCS-ADL-MCI
Alzheimer's Disease Cooperative Study–Activities of Daily Living scale adapted for Mild Cognitive Impairment
- AE
adverse event
- ARIA
amyloid-related imaging abnormalities
- ARIA-E
amyloid-related imaging abnormalities with edema/effusion
- ARIA-H
amyloid-related imaging abnormalities with edema/effusion with microhemorrhage/hemosiderosis
- CDR-SB
Clinical Dementia Rating Sum of Boxes
- FDA
Food and Drug Administration
- IgG1
immunoglobulin G1
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- SUVR
standardized uptake value ratio
Appendix. Authors
Study Funding
The AAN used a virtual process for development of this product; there was no reimbursement for travel costs as a result. No targeted funding was provided for this study.
Disclosure
G.S. Day is supported by a career development grant from the NIH (K23AG064029), owns stock (>$10,000) in ANI Pharmaceuticals (a generic pharmaceutical company), and serves as a topic editor for DynaMed (EBSCO), as a consultant for Parabon Nanolabs Inc, and as the Clinical Director of the Anti-NMDA Receptor Encephalitis Foundation (Canada, uncompensated). N. Scarmeas is supported by grants from the National Institute on Aging (R01AG061008), by Novo Nordisc (industry-sponsored phase III clinical trial: local recruitment center, funds paid to institution), and by Albert Einstein College of Medicine (Chair of Data Safety Monitoring Board of an NIH-sponsored clinical trial, personal fees). R Dubinsky is involved in ENROLL-HD, a natural history study funded by CHDI, and in PROOF-HD, an industry-sponsored study to slow progression of Huntington disease. K. Coerver is a consultant for GE Health Care Systems. A. Mostacero and B. West report no disclosures relevant to the manuscript. S.R. Wessels is an employee of the American Academy of Neurology (AAN). M.J. Armstrong is supported by grants from the National Institute on Aging (P30AG047266, R01AG068128) and the Florida Department of Health (grant 20A08) and was previously supported by a grant from the Agency for Healthcare Research and Quality (K08HS24159), serves as a consultant for the Parkinson Foundation on a grant from PCORI, serves as an investigator for a Lewy Body Dementia Association Research Center of Excellence, serves on the level of evidence editorial board for Neurology® and related publications (uncompensated), previously received compensation from the AAN for work as an evidence-based medicine methodology consultant, and has received honoraria for teaching at conferences for the AAN and the International Parkinson and Movement Disorder Society, participating in creating educational materials for the Parkinson Foundation, and serving on data safety monitoring boards for the Alzheimer's Clinical Trials Consortium/Alzheimer's Therapeutic Research Institute and Alzheimer's Disease Cooperative Study. Go to Neurology.org/N for full disclosures.
References
- 1.Alzheimer’s Association. 2021 Alzheimer's disease facts and figures. Alzheimers Dement 2021;17:327-406. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer’s Association. 2020 Alzheimer's disease facts and figures. Alzheimers Dement Epub 2020 10 Mar.
- 3.Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 2015;18:794-799. [DOI] [PubMed] [Google Scholar]
- 4.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992;256:184-185. [DOI] [PubMed] [Google Scholar]
- 5.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and 'wingmen.' Nat Neurosci 2015;18:800-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sperling R, Henley D, Aisen PS, et al. Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: a truncated randomized phase 2b/3 clinical trial. JAMA Neurol 2021;78:293-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer's disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimer Res Ther 2021;13:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med 2021;384:1691-1704. [DOI] [PubMed] [Google Scholar]
- 9.Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med 2018;378:321-330. [DOI] [PubMed] [Google Scholar]
- 10.Ackley SF, Zimmerman SC, Brenowitz WD, et al. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ 2021;372:n156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature 2016;537:50-56. [DOI] [PubMed] [Google Scholar]
- 12.US Food and Drug Administration. ADUHELM (aducanumab-avwa) injection [package insert]. 2021. accessdata.fda.gov/drugsatfda_docs/label/2021/761178s000lbl.pdf
- 13.US Food and Drug Administration. ADUHELM (aducanumab-avwa) injection, updated [package insert]. 2021. biogencdn.com/us/aduhelm-pi.pdf
- 14.Rabinovici GD. Controversy and progress in Alzheimer's disease: FDA approval of aducanumab. N Engl J Med 2021;385:771-774. [DOI] [PubMed] [Google Scholar]
- 15.Alexander GC, Knopman DS, Emerson SS, et al. Revisiting FDA approval of aducanumab. N Engl J Med 2021;385:769-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planche V, Villain N. US Food and Drug Administration approval of aducanumab: is amyloid load a valid surrogate end point for Alzheimer disease clinical trials? JAMA Neurol 2021;78(11):1307-1308. [DOI] [PubMed] [Google Scholar]
- 17.Knopman DS, Perlmutter JS. Prescribing aducanumab in the face of meager efficacy and real risks. Neurology 2021;97:545-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neurology AAN. Aducanumab resources [online]. 2021. aan.com/tools-and-resources/aducanumab-resources/
- 19.Chiong W, Tolchin BD, Bonnie RJ, et al. Decisions with patients and families regarding aducanumab in Alzheimer disease, with recommendations for consent: AAN position statement. Neurology Epub 2021 Nov 17. [DOI] [PubMed] [Google Scholar]
- 20.Gronseth GS, Cox J, Gloss D, et al. Clinical Practice Guideline Process Manual, 2017 ed. American Academy of Neurology; 2017. [Google Scholar]
- 21.National Library of Medicine. PubMed clinical queries [online]. 2021. pubmed.ncbi.nlm.nih.gov/clinical/
- 22.Wilczynski NL, McKibbon KA, Walter SD, Garg AX, Haynes RB. MEDLINE clinical queries are robust when searching in recent publishing years. J Am Med Inform Assoc 2013;20:363-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrero J, Williams L, Stella H, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer's disease. Alzheimers Dement 2016;2:169-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevigny J, Chiao P, Bussiere T, et al. Addendum: the antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature 2017;546:564. [DOI] [PubMed] [Google Scholar]
- 25.Chiao P, Bedell BJ, Avants B, et al. Impact of reference and target region selection on amyloid PET SUV ratios in the phase 1b PRIME study of aducanumab. J Nucl Med 2019;60:100-106. [DOI] [PubMed] [Google Scholar]
- 26.Andrews JS, Desai U, Kirson NY, Zichlin ML, Ball DE, Matthews BR. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer's disease clinical trials. Alzheimers Dement 2019;5:354-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biogen. Combined FDA and Applicant PCNS Drugs Advisory Committee Briefing Document. Published November 6, 2020. Accessed September 2, 2021. fda.gov/media/143502/download
- 28.Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 2022;79:13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sperling RA, Jack CR Jr., Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement 2011;7:367-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barakos J, Sperling R, Salloway S, et al. MR imaging features of amyloid-related imaging abnormalities. AJNR Am J Neuroradiol 2013;34:1958-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belluck P. Concerns grow over safety of Aduhelm after death of patient who got the drug. New York Times 2021 Nov 22. nytimes.com/2021/11/22/health/aduhelm-death-safety.html [Google Scholar]
- 32.European Medicines Agency. Aduhelm [online]. 2021. ema.europa.eu/en/medicines/human/summaries-opinion/aduhelm
- 33.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer disease centers, 2005-2010. J Neuropathol Exp Neurol 2012;71:266-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sevigny J, Suhy J, Chiao P, et al. Amyloid PET screening for enrichment of early-stage Alzheimer disease clinical trials: experience in a phase 1b clinical trial. Alzheimer Dis Assoc Disord 2016;30:1-7. [DOI] [PubMed] [Google Scholar]
- 35.Rabinovici GD, Gatsonis C, Apgar C, et al. Association of amyloid positron emission tomography with subsequent change in clinical management among Medicare beneficiaries with mild cognitive impairment or dementia. JAMA 2019;321:1286-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmqvist S, Zetterberg H, Mattsson N, et al. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015;85:1240-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaw LM, Arias J, Blennow K, et al. Appropriate use criteria for lumbar puncture and cerebrospinal fluid testing in the diagnosis of Alzheimer's disease. Alzheimers Dement 2018;14:1505-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the amyloid imaging task force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer's Association. Alzheimers Dement 2013;9:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson TS, Ayanian JZ, Souza J, Landon BE. Representativeness of participants eligible to be enrolled in clinical trials of aducanumab for Alzheimer disease compared with Medicare beneficiaries with Alzheimer disease and mild cognitive impairment. JAMA 2021;326:1627-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis 2021;8:398-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manly JJ, Glymour MM. What the aducanumab approval reveals about Alzheimer disease research. JAMA Neurol 2021;78:1305-1306. [DOI] [PubMed] [Google Scholar]
- 42.Majersik JJ, Ahmed A, Chen IA, et al. A shortage of neurologists: we must act now: a report from the AAN 2019 transforming leaders program. Neurology Epub 2021 Apr 30. [DOI] [PubMed] [Google Scholar]
- 43.Lin GA, Whittington MD, Synnott PG, et al. Aducanumab for Alzheimer's disease: effectiveness and value: draft evidence report: 2021. icer.org/assessment/alzheimers-disease-2021/ [DOI] [PMC free article] [PubMed]
- 44.Biogen and Eisai. Biogen and Eisai launch multiple initiatives to help patients with Alzheimer’s disease access ADUHELM™ . 2021. globenewswire.com/news-release/2021/06/07/2243021/0/en/Biogen-and-Eisai-launch-multiple-initiatives-to-help-patients-with-Alzheimer-s-disease-access-ADUHELM.html [Google Scholar]
- 45.Endpoints News. Biogen slashes Aduhelm price in half, foreshadows significant layoffs as sales continue to stagnate. Endpoints Company 2021. endpts.com/biogen-slashes-aduhelm-price-in-half-foreshadows-significant-layoffs-as-sales-continue-to-stagnate/ [Google Scholar]
- 46.US Department of Health and Human Services. Review of the FDA's Accelerated Approval Pathway: report OIG-12-21-01. US Department of Health and Human Services; 2021. [Google Scholar]
- 47.Reuters. Veterans Health Administration turns down Biogen Alzheimer's drug. Reuters 2021. www.reuters.com/business/healthcare-pharmaceuticals/us-veterans-health-administration-turns-down-biogen-alzheimers-drug-2021-08-11/ [Google Scholar]
- 48.Centers for Medicare & Medicaid Services. Monoclonal antibodies directed against amyloid for the treatment of Alzheimer's disease. 2022. cms.gov/medicare-coverage-database/view/ncacal-decision-memo.aspx?proposed=Y&NCAId=305
- 49.Armstrong MJ, Shulman LM, Vandigo J, Mullins CD. Patient engagement and shared decision-making: what do they look like in neurology practice? Neurol Clin Pract 2016;6:190-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng AL, Batool S, McCreary CR, et al. Susceptibility-weighted imaging is more reliable than T2*-weighted gradient-recalled echo MRI for detecting microbleeds. Stroke 2013;44:2782-2786. [DOI] [PubMed] [Google Scholar]
- 51.Haller S, Vernooij MW, Kuijer JPA, Larsson EM, Jager HR, Barkhof F. Cerebral microbleeds: imaging and clinical significance. Radiology 2018;287:11-28. [DOI] [PubMed] [Google Scholar]
- 52.Graff-Radford J, Lesnick TG, Mielke MM, et al. Cerebral amyloid angiopathy burden and cerebral microbleeds: pathological evidence for distinct phenotypes. J Alzheimers Dis 2021;81:113-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.VandeVrede L, Gibbs DM, Koestler M, et al. Symptomatic amyloid-related imaging abnormalities in an APOE epsilon4/epsilon4 patient treated with aducanumab. Alzheimers Dement 2020;12:e12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drabo EF, Barthold D, Joyce G, Ferido P, Chang Chui H, Zissimopoulos J. Longitudinal analysis of dementia diagnosis and specialty care among racially diverse Medicare beneficiaries. Alzheimers Dement 2019;15:1402-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loftus P. Eli Lilly asks FDA to approve Alzheimer's drug. Wall Street J 2021;2021. [Google Scholar]
- 56.Armstrong MJ, Gronseth GS, Day GS, et al. Patient stakeholder versus physician preferences regarding amyloid PET testing. Alzheimer Dis Assoc Disord 2019;33:246-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mehta P, Syrop I, Singh JR, Kirschner J. Systematic review of the efficacy of particulate versus nonparticulate corticosteroids in epidural injections. PM&R 2017;9:502-512. [DOI] [PubMed] [Google Scholar]
- 58.Biogen, Eisai. AD-US Study, the First Real-World Observational Phase 4 Study in Alzheimer's Disease at AAIC 2021. 2021. investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-announce-design-aduhelm-icare-ad-us-study-first [Google Scholar]
- 59.American Academy of Neurology. AAN's conflict of interest policy. 2021. aan.com/about-the-aan/organizational-policies