

Abstract
We have used structure-based design techniques to introduce the drug O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphe-nylamino) phenyl] 1-N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO), which is efficiently metabolized to potentially cytolytic nitric oxide by the π isoform of glutathione S-transferase, an enzyme expressed at high levels in many tumors. We have used mouse embryo fibroblasts (MEFs) null for GSTπ (GSTπ−/−) to show that the absence of GSTπ results in a decreased sensitivity to PABA/NO. Cytotoxicity of PABA/NO was also examined in a mouse skin fibroblast (NIH3T3) cell line that was stably transfected with GSTπ and/or various combinations of γ-glutamyl cysteine synthetase and the ATP-binding cassette transporter MRP1. Overexpression of MRP1 conferred the most significant degree of resistance, and in vitro transport studies confirmed that a GSTπ-activated metabolite of PABA/NO was effluxed by MRP1 in a GSH-dependent manner. Additional studies showed that in the absence of MRP1, PABA/NO activated the extracellular-regulated and stress-activated protein kinases ERK, c-Jun NH2-terminal kinase (JNK), and p38. Selective inhibition studies showed that the activation of JNK and p38 were critical to the cytotoxic effects of PABA/NO. Finally, PABA/NO produced antitumor effects in a human ovarian cancer model grown in SCID mice.
Glutathione S-transferases (GSTs) are a family of enzymes that promote the conjugation of the sulfur atom of glutathione (GSH) to an electrophilic center of endogenous and exogenous compounds, thereby increasing their aqueous solubility and subsequent excretion (Armstrong, 1997). The efflux of GS-conjugates from the cell is achieved by multidrug resistance-associated protein (MRP1), a member of the ATP-binding cassette transporter superfamily (Cole et al., 1992). GSTs constitute one component of the maintenance of cellular GSH homeostasis. In turn, intracellular GSH contributes toward redox balance, and the variety of pathways that synthesize or use GSH influence this homeostasis. The isozyme GSTπ is a widely studied member of this family because its expression is frequently elevated in many human tumors and is involved in the development of resistance to several anticancer drugs (O’Brien and Tew, 1996; Townsend and Tew, 2003). Such factors have contributed to the recent efforts to target GSTs as a primary objective in the drug discovery process (Rosario et al., 2000; Townsend et al., 2002).
We seek to turn GSTπ overexpression to the tumor’s disadvantage by designing substrates that release the established cytolytic agent nitric oxide (NO) on metabolism by GSTπ. The compounds we are exploring with this goal in mind are the O2-aryl diazenium diolates, electrophilic species shown (Saavedra et al., 2001) to transfer their aryl groups to attacking nucleophiles with cogeneration of ions that spontaneously release NO at physiological pH (Fig. 1). If these agents do in fact produce abundant NO selectively within the tumor, they could contribute to chemotherapy by: arylating and thus irreversibly consuming intracellular GSH; inhibiting DNA synthesis; showering the overexpressing, highly metabolizing cells and their neighbors with toxic reactive nitrogen/oxygen intermediates; and inhibiting enzymes capable of preventing or repairing cellular damage. An example of such an agent is JS-K, recently shown to suppress the growth of human leukemia and prostate cancer xenografts in mice. However, JS-K was found to react with GSH to produce NO an order of magnitude more rapidly under catalysis by the GSTα than by GSTπ (Shami et al., 2003). We have been working to reverse this order of reactivity, with the goal of improving our ability to target potentially cytotoxic NO to the tumor while avoiding collateral injury of normal tissue in which the prevalent α isoform is required for maintenance of cellular homeostasis.
Fig. 1.
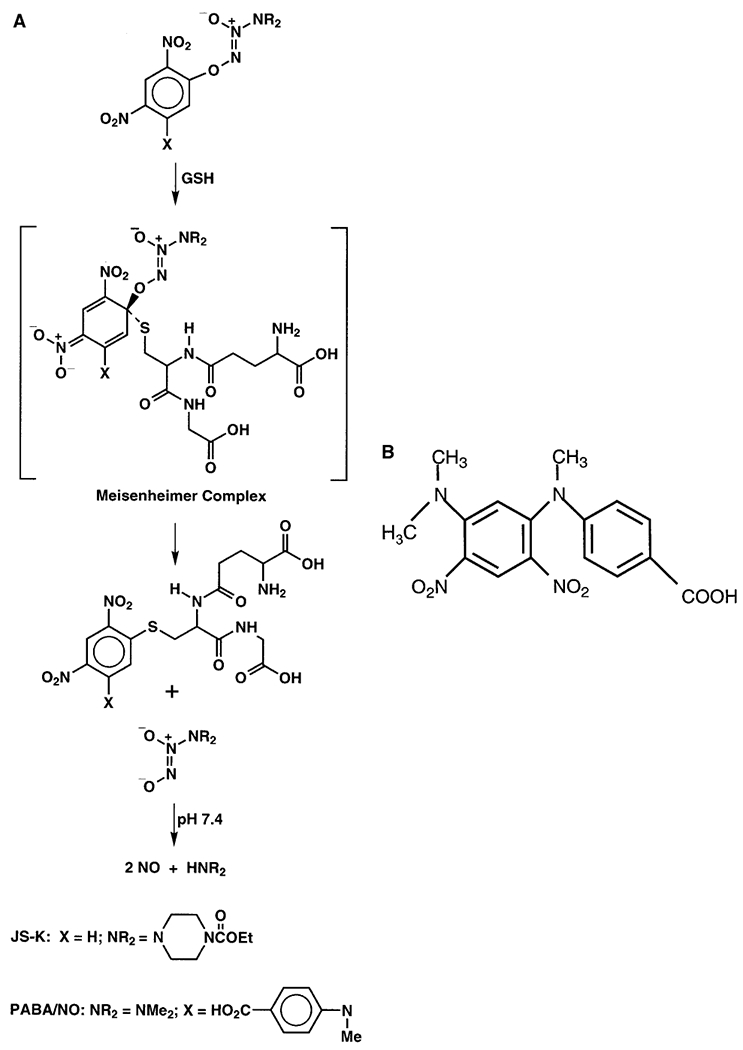
Proposed mechanism by which JS-K and PABA/NO generate NO on activation by GSH. B, structure of non-NO-releasing by-product of PABA/NO, JS-39-94.
Herein, we describe the structure-based design of PABA/NO, an NO-releasing prodrug that is metabolized more efficiently by GSTπ than by GSTα. We show further that PABA/NO and/or its metabolites cause the activation of the extracellular-regulated and stress-activated protein kinases. The parent drug and/or its metabolites can be actively effluxed by MRP1, a transporter that provides resistance to the NO-mediated effects of PABA/NO. Overall, these data imply that cells with high levels of GSTπ, but low MRP1 content, may be ideal targets for induction of cytotoxicity.
Materials and Methods
Molecular Modeling for the Design of PABA/NO.
The Meisenheimer complexes formed by GSH and PABA/NO for GSTπ and GSTα were built based on those for JS-K (Shami et al., 2003). The models were subject to geometry optimization and docked into the active sites of GSTπ (Prade et al., 1997) and GSTα (Sinning et al., 1993), respectively. Model building was carried out with computer graphics program O (Jones and Kjeldgaard, 1997) and energy minimization with Crystallography & NMR System software (Brünger et al., 1998). Illustrations were generated with software packages Grasp (Nicholls et al., 1991), Molscript (Kraulis, 1991), and Raster3D (Merritt and Bacon, 1997).
Preparation of 4-[N-(2,4-Dinitro-5-fluorophenyl)-N-methyl-]aminobenzoic acid.
A solution of 2.5 g (0.0123 mol) of 1,5-difluoro-2,4-dinitrobenzene in 25 ml of tetrahydrofuran (THF) was placed in a 100-ml round-bottomed flask. To this was added 2.6 g (0.025 mol) of anhydrous sodium carbonate. To the stirred mixture was added a solution of 1.86 g (0.0123 mol) of 4-(N-methylamino)benzoic acid in 12 ml of THF. The reaction mixture was stirred for 48 h at room temperature. The THF was removed on a rotary evaporator, and the residue was taken up in dichloromethane and washed with water. The organic solution was dried over sodium sulfate, filtered through magnesium sulfate, and evaporated under vacuum to give 3.6 g of the desired product as an orange solid, which was recrystallized from ethanol: m.p. 133 to 134°C; 1H NMR δ 2.95 (s, 3 H), 6.62 (d, 2 H), 7.49 (d, 1 H), 7.99 (d, 2 H), 8.93 (d, 1 H); UV (ethanol) λmax (ϵ) 232 nm (38 mM−1 cm−1) and 314 nm (52 mM−1 cm−1). Anal. (C14H10N3O6F): C, 50.16; H, 3.01; N, 12.53. Found: C, 50.16; H, 3.02; N, 12.46.
Preparation of PABA/NO.
To a partial solution of 2.6 g (7.7 mmol) of 4-[N-(2,4-dinitro-5-fluorophenyl)-N-methyl]aminobenzoic acid in 10 ml of THF and 10 ml of tert-butyl alcohol was added 10 ml of 5% aqueous sodium bicarbonate. A solution of 1.4 g (0.016 mol) of sodium 1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate in 10 ml of 5% sodium carbonate was added dropwise, and the resulting mixture was stirred at room temperature for 72 h, concentrated under vacuum, and extracted with aqueous sodium bicarbonate. The aqueous portion was washed with dichloromethane, acidified with 10% aqueous hydrochloric acid, and extracted with dichloromethane. The organic solution was dried as described above and evaporated under vacuum to give 1.4 g of PABA/NO as an orange solid. The solid was purified by recrystallization from ethanol to give orange plates: m.p. 146 to 148°C; 1H NMR δ 2.94 (s, 3 H), 3.28 (s, 6 H), 6.61 (d, 2 H), 7.55 (s, 1 H), 8.01 (d, 2 H), 8.92 (s, 1 H); UV (ethanol) λmax (ϵ) 220 nm (15.5 mM−1 cm−1), and 316 nm (22.2 mM−1 cm−1). Anal. (C16H16N6O8): C, 45.72; H, 3.84; N, 19.99. Found: C, 45.61; H, 3.88; N, 19.90.
It should be noted that PABA/NO showed a tendency to remain supersaturated in aqueous solutions, only to separate unexpectedly from the aqueous phase as a fine precipitate. At 37°C in pH 7.4 phosphate buffer, the apparent maximum solubility was 1.7 μM.
Determination of Metabolism and NO Release from PABA/NO by Human GSTs.
Purified preparations of recombinant human GSTα and GSTπ were obtained from PanVera (Madison, WI). All other chemicals were from Sigma (St. Louis, MO). The activity of GSTs toward 1-chloro-2,4-dinitrobenzene was determined as described by Habig et al. (1974), before activity measurements with PABA/NO to ensure that the enzyme preparations were catalytically fully active. For activity measurement toward PABA/NO, the 1-ml reaction mixture at 37°C contained 100 mM potassium phosphate buffer, pH 7.4, 0.2 to 1 mM GSH, 1 μM PABA/NO, and an appropriate amount of GST isoenzyme protein. The reaction was started by the addition of PABA/NO, and the rate of reaction was monitored by measuring the decrease in absorbance of PABA/NO at 315 nm accompanying its reaction with GSH as well as the accumulation of GS-adduct at 322 nm.
Chemiluminescence detection and quantification of NO evolving from the reactions of PABA/NO, and JS-K for comparison, were conducted using an NO-specific chemiluminescence detector (Thermal Energy Analyzer model 502A; Thermedics, Analytical Instrument Division, Waltham, MA) essentially as described previously (Keefer et al., 1996). Briefly, 1.8 ml of 100 mM potassium phosphate buffer, pH 7.4, was sprayed with inert gas until a steady detector response was established. GSH and GSTs in 100 μl of buffer were added to a final concentration of 200 μM GSH and 0.5 μg of GST in 2 ml, as indicated. The NO release profile was followed at 37°C for 10 to 20 min after injecting PABA/NO at a final concentration of 0.5 μM to start the reaction. The resulting curve was integrated to quantify the amount of NO released per mole of drug. Identical conditions were used for JS-K except that 2.5 μg of GST per 2 ml was used. Of a theoretical yield of 2 mol per mole, GSTπ released 0.56 mol of NO per mole of PABA/NO, whereas the same amount of GSTα released only 0.11 mol/mole. JS-K under the same conditions had the reverse enzyme activity; GSTπ released 0.11 mol of NO per mole of JS-K, whereas GSTα released 0.61 mol NO/mol.
Materials and Cell Lines.
Generation of GSTπ−/− mice, extraction of the MEF cell lines from GSTπ+/+ and GSTπ−/− mice, and details of the transfected NIH3T3 cell lines were described previously (Henderson et al., 1998; Rosario et al., 2000; Ruscoe et al., 2001). Cell lines were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum, 100 μg/ml streptomycin, 100 U/ml penicillin, and 2 mM l-glutamine at 5% CO2 and 37°C. [3H]Leukotriene C4 (LTC4) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Unlabeled LTC4, ATP, creatine phosphate, and creatine phosphokinase were obtained from Sigma Chemical Co. (St. Louis, MO). The mitogen-activated protein kinase inhibitors SP600125, SB202190, and U0126 were obtained from Calbiochem (La Jolla, CA). GSH (reduced) was obtained from MP Biomedicals (Aurora, OH).
Preparation of Plasma Membrane Vesicles and Transport Studies.
Membrane vesicles were prepared by the nitrogen cavitation method as described previously (Cornwell et al., 1986), frozen in liquid nitrogen, and stored at −80°C until use. Transport experiments were performed using the rapid filtration method as described previously (Leier et al., 1996). Experiments were carried out in transport buffer containing 30 μg of membrane vesicles, 0.25 M sucrose, 10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 4 mM ATP, 10 mM phosphocreatine, 100 μg/ml creatine phosphokinase, and 50 nM [3H]LTC4, with or without unlabeled substrate, in a total volume of 50μl. Reactions were carried out at room temperature and stopped by the addition of 1 ml of ice-cold stop solution (0.25M sucrose, 100 mM NaCl, and 10 mM Tris-HCl, pH 7.4). Samples were passed, under vacuum, through 0.22-μm Durapore membrane filters (Millipore, Bedford, MA) pre-equilibrated in transport buffer. The filters were washed twice with 3 ml of ice-cold stop solution. Radioactivity was measured by the use of a liquid scintillation counter.
Protein Purification of GSTπ.
The cDNA encoding the human GSTP1-1 was obtained by American Type Culture Collection (Manassas, VA) and amplified as described previously (Chang et al., 1999). The polymerase chain reaction product (650 bp) was ligated into the pCRT7-TOPO vector and transformed into TOP10F’ Escherichia coli cells. Plasmid DNA from a positive transformant was isolated and sequenced. The construct was transformed into Escherichia coli (strain BL-21) and expressed in Luria broth medium containing 50 μg/ml ampicillin and 1 M isopropyl β-d-thiogalactoside. Recombinant GSTπ protein was purified using a standard Ni2+ column for His-tagged proteins and dialyzed against 2 liters of 50 mM Tris-HCl buffer, pH 7. The purified protein was confirmed by a single polypeptide band of 23 kDa on a Coomassie-stained SDS-polyacrylamide gel as well as by Western blot analysis using an anti-human GSTπ antibody from BD Transduction Laboratories (Lexington, KY) (data not shown).
Immunoblot Analysis.
After treatment, cells were collected and washed twice in ice-cold phosphate-buffered saline. Pellets were resuspended in lysis buffer (20 mM Tris-HCl, pH 7.5, 15 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerophosphate) and incubated for 30 min on ice. Phosphatase inhibitors (5 mM NaF and 1 mM Na3VO4) and 1× protease inhibitor cocktail (Sigma) were added fresh to the lysis buffer. Lysates were sonicated for 10 sec and centrifuged for 30 min at 10,000g at 4°C. Protein concentrations were assayed with the Bradford reagent (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein were separated on 10% SDS-polyacrylamide gels and transferred overnight onto nitrocellulose membranes (Bio-Rad). Protein expression was determined using specific primary and secondary antibodies. Briefly, after transfer, membranes were incubated in 10% blocking buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% bovine serum albumin, 0.1% Tween 20, 1× protease inhibitors (Sigma), 5 mM NaF, and 1 mM Na3VO4) for 30 min at room temperature then further incubated with various antibodies diluted in 5% blocking buffer for 1 h. Antibodies were purchased from the following sources: anti-active-c-Jun N-terminal kinase (JNK) and −p38 (Promega, Madison, WI); phospho-specific extracellular signal-regulated kinase (ERK) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-JNK2 (BD Biosciences PharMingen, San Diego, CA); anti-p38 and anti-ERK2 (Santa Cruz Biotechnology). The blots were washed and incubated with the appropriate secondary antibodies, conjugated with horseradish peroxidase, in 5% blocking buffer for 1 h, washed, and developed with ECL detection reagents (Amersham Biosciences).
Protein Nitration.
After treatment, cells were lysed in hot 2× SDS sample buffer (100 mM Tris-HCl, pH 6.8, 4% SDS, and 20% glycerol) and boiled for 10 min. After centrifugation at room temperature for 10 min at 10,000g, 1.5-μl (5–10 μg) samples were spotted onto a nitrocellulose membrane, left to dry, and probed as described above in immunoblot analysis using the anti-nitrotyrosine antibody from Upstate Biotechnology (Lake Placid, NY).
Cytotoxicity Assays.
Cells were plated onto 24-well plates at a density of 100,000 cells per well in 1 ml of medium. After attachment, increasing concentrations of drug were added and the cells were maintained in drug for 48 h. After this period of drug exposure, cell survival was determined using a Guava personal cytometer (Guava Technologies) according to the manufacturer’s instructions.
Annexin V Assay.
Cells were plated onto six-well plates at a density of 200,000 cells/well in 2 ml of medium. The next day, increasing concentrations of drug were added to the cells for 2 h. Cells were collected by trypsinization and pelleted by centrifugation for 10 min at 500g at 4°C. After washing with 1 ml of ice-cold 1× Nexin buffer (Guava Technologies), cells were resuspended in 100 μl of Nexin buffer. After labeling with Annexin V-phycoerythrin and 7-amino-actinomycin, the proportion of live, apoptotic, and necrotic cells was determined on a Guava Personal Cytometer (Guava Technologies, Hayward, CA) according to the manufacturer’s instructions.
In Vivo Tumor Experiments.
Six-week-old female SCID mice were obtained from and housed in the Animal Resource Facility of the Fox Chase Cancer Center. Animal care was provided in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals (NIH Publication 86-23, 1985). Food and water were provided ad libitum. For the injections, 0.5 × 106 cells (A2780) were resuspended in phosphate-buffered saline, mixed 1:1 in Matrigel (BD Biosciences) and injected subcutaneously into the flanks. Each mouse received one injection in each flank. Twenty-four mice were injected with A2780 human ovarian cancer cells. After the cells were injected both sets of mice were divided into three groups of eight mice each. Group 1 received weekly s.c. injections of 2.5 mg/kg cisplatin, group 2 received biweekly injections of 3.36 mg/kg PABA/NO, and group 3 received biweekly injections of 0.8% DMSO. The mice were weighed weekly. Tumor size was measured biweekly with calipers. The animals were sacrificed 6 to 7 weeks after the tumor cells were injected.
Data Analysis.
Tumor volume was calculated using the formula: width × length × [(width × length)/2] × 0.5236. Mean values and S.E. were computed for each group. These data were analyzed for statistically significant differences between groups with Student’s t test. Differences were considered statistically significant if the p was <0.05.
Results
Unlike JS-K, PABA/NO Is Metabolized to NO Efficiently by GSTπ.
JS-K is a much better substrate for GSTα than GSTπ (Shami et al., 2003). We attempted to reverse the isoenzyme preference of JS-K and develop a GSTπ-selective JS-K derivative. Molecular modeling of the Meisenheimer complex formed in GST-catalyzed addition of GSH to JS-K (Shami et al., 2003) suggested that replacing the piperazine ring of JS-K with a smaller amino group might improve its accommodation in the active center of GSTπ (Fig. 2A), whereas adding a sterically demanding group at the 5-position of the 2,4-dinitrophenyl ring should diminish its suitability as a GSTα substrate (Fig. 2B) and, at the same time, further improve its accommodation in the active center of GSTπ (Fig. 2A). PABA/NO was thus designed on the basis of JS-K with its piperazine system substituted by a dimethylamine and the addition of an N-methyl-N-(p-carboxyphenyl) group at the 5-position of the 2,4-dinitrophenyl ring (Fig. 1A). As illustrated in Fig. 2 (C and D), GSTπ accommodates the Meisenheimer complex of PABA/NO very well, but GSTα seems to have serious steric conflicts with the N-methyl-N-(p-carboxyphenyl) group of the transition state complex. Based on molecular modeling, we predicted that GSTπ should be more effective than GSTα for catalyzing the GSH conjugation of PABA/NO.
Fig. 2.
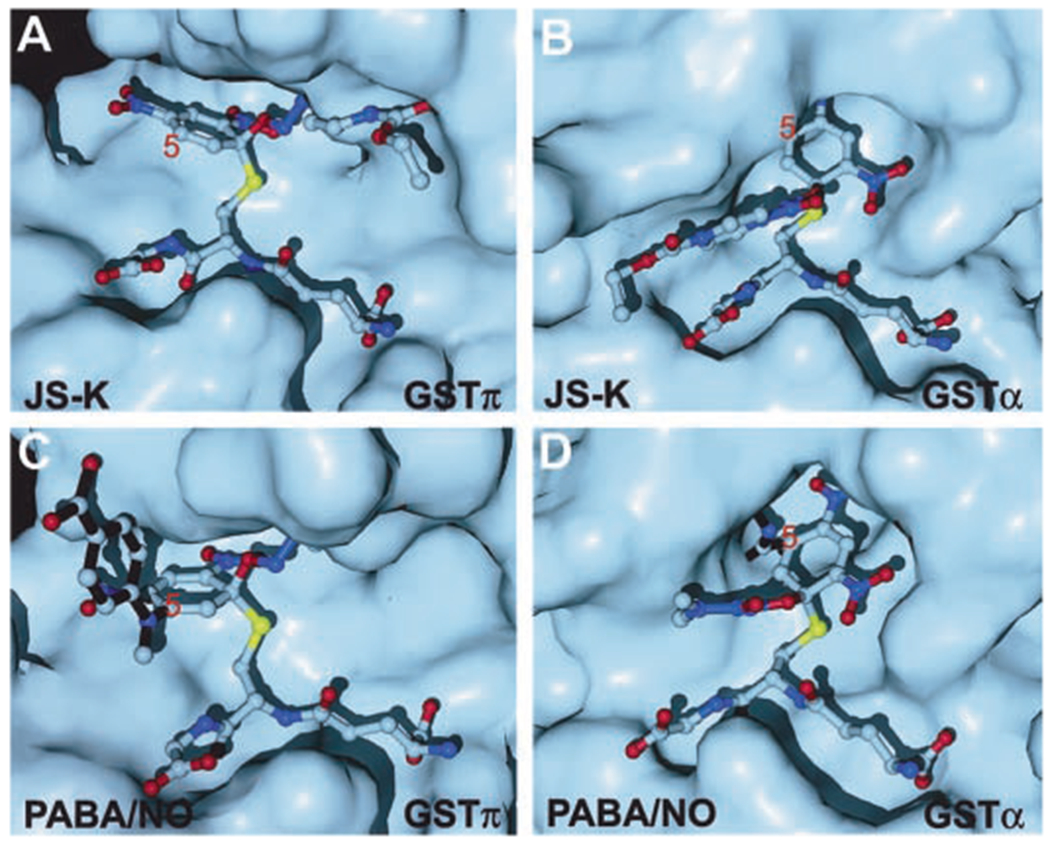
Molecular surface representation of GSTπ and GSTα with ball- and-stick models of the Meisenheimer complexes formed by GSH and NO-releasing prodrugs. A, GSTπ in complex with the Meisenheimer complex of JS-K (Shami et al., 2003). B, GSTα in complex with the Meisenheimer complex of JS-K (Shami et al., 2003). C, GSTπ in complex with the Meisenheimer complex of PABA/NO (this study). D, GSTα in complex with the Meisenheimer complex of PABA/NO (this study). In C and D, the N-methyl-N-(p-carboxyphenyl)amino group is highlighted with black sticks (bonds).
This prediction was confirmed by synthesizing PABA/NO and studying its reactivity with GSH in the presence of the two GSTs. Whereas the yields of NO in the metabolism of JS-K by GSTα and π were 30% and 5%, respectively, those seen with PABA/NO under the same conditions (except for the GST concentration—see Materials and Methods) were 5% and 30%, respectively. The results are consistent with the conclusion of the modeling studies that PABA/NO is metabolized much better by GSTπ than by GSTα thus, the isoenzyme preference of JS-K is reversed by molecular modifications based on the structural information of the transition state.
GSTπ Is Involved in the Cytotoxicity of PABA/NO.
To assess the importance of GSTπ in determining PABA/NO’s activity, a mouse embryo fibroblast (MEF) cell line null for GSTπ (GSTπ−/−) was tested for sensitivity to PABA/NO and compared with the parental wild-type strain (GSTπ+/+). The IC50 value of the GSTπ+/+ cells was 26 ± 13 μM and was significantly lower than that of the GSTπ−/− cells (39 ± 15 μM), resulting in an approximate 2-fold increase in resistance to PABA/NO in the GSTπ−/− cells (Fig. 3). This result is consistent with our previous data with another GSTπ-activated prodrug, TLK286 (Rosario et al., 2000).
Fig. 3.
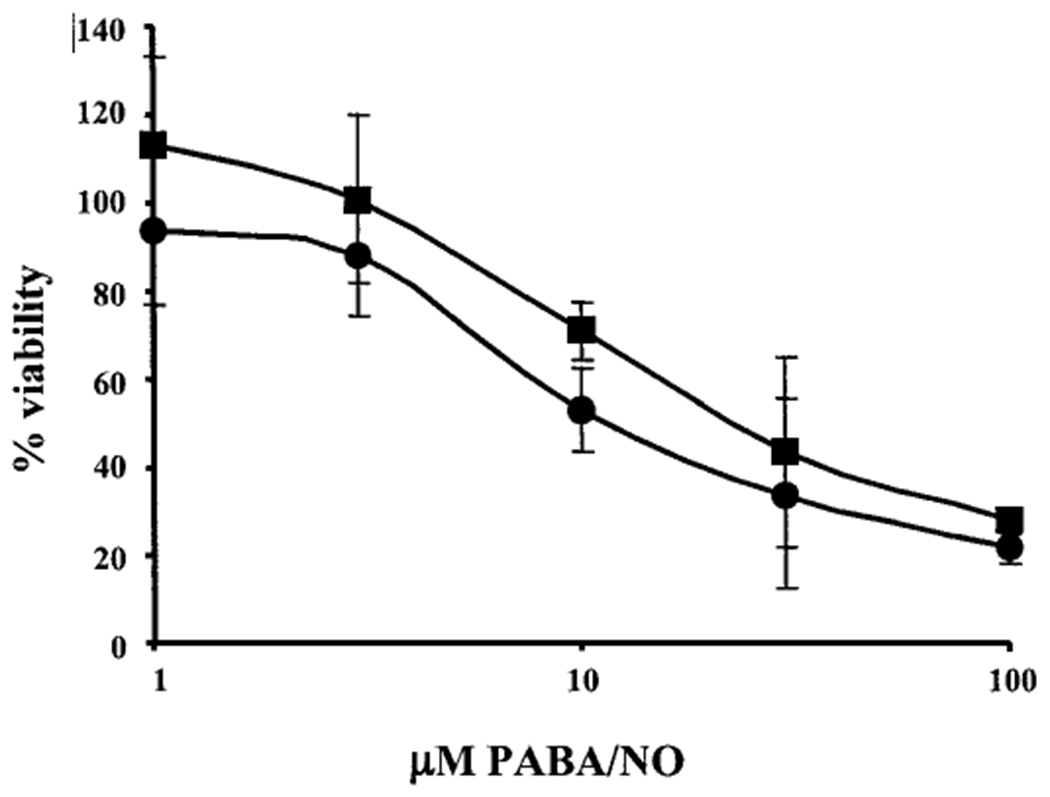
GSTπ−/− MEF cells are less sensitive to PABA/NO than the parental GSTπ+/+. Cell survival of GSTπ−/− (■) and GSTπ+/+ (●) was assessed 48 h after drug exposure, as detailed under Materials and Methods. IC50 values of 26 ± 13 μM for GSTπ+/+ and 39 ± 15 μM for GSTπ−/− were calculated. Data are represented as means ± S.D. of three independent experiments.
MRP1 Protects Cells from PABA/NO Cytotoxicity.
To characterize further the cytotoxic effects of PABA/NO, we studied the effects of the drug on cells stably transfected with various components of the GSH detoxification pathway (O’Brien et al., 2000). Stable transfected lines contained GSTπ and/or γ-GCS (regulatory plus catalytic subunits) and/or MRP1. Table 1 shows a summary of the IC50 values obtained for PABA/NO in the various transfectant cell lines. Whereas the greatest enhancement in resistance to PABA/NO was conferred by MRP1 expression alone (4.6-fold), both GSTπ and γGCS expression influenced the response. For example, GSTπ caused a slight increase in sensitivity, whereas ΰGCS gave a 1.5-fold increase in resistance, although the differences were not found to be significant statistically (p > 0.05). Indeed, a significant increase in resistance was found only when both γGCS and GSTπ were overexpressed (11 ± 1.5 versus 19 ± 2.1 μM), which could be caused by an increase in the cell’s defenses against NO. Increased intracellular GSH levels result from γGCS overexpression (O’Brien et al., 2000), and this influences the transport properties of MRP1 (Karwatsky et al., 2003). In combination with MRP1, GSTπ produced an abrogation of resistance, although again this was not found to be statistically significant compared with the overexpression of MRP1 alone (p > 0.05). However, the overexpression of γGCS in combination with MRP1 led to an increase in sensitivity to PABA/NO compared with MRP1 alone. The levels of MRP1 may be the rate-limiting factor in this case; therefore, the increase in endogenous levels of GSH may increase the rate of NO release from PABA/NO, which overcomes the rate of efflux by MRP1.
TABLE 1.
Summary of the IC50 values for the effect of PABA/NO on NIH3T3 cells transfected with GSTπ, γGCS, MRP1, or appropriate combinations compared with sham-transfected parental cells (3T3/pc)
| IC50 | Ratio | |
|---|---|---|
| 3T3/pc | 11 ± 1.5 | 1.0 |
| 3T3/GCS | 16 ± 3.6 | 1.5 |
| 3T3/GST | 10 ± 1.6 | 0.9 |
| 3T3/GCS/GST | 19 ± 2.1 | 1.7 |
| 3T3/MRP | 51 ± 6.5 | 4.6 |
| 3T3/MRP/GCS | 22 ± 1.3 | 2.0 |
| 3T3/MRP/GST | 39 ± 3.0 | 3.5 |
| 3T3/MRP/GCS/GST | 46 ± 6.3 | 4.2 |
Data are represented as means ± S.D. of three independent experiments.
The effects of PABA/NO were also determined by the Annexin V assay. The data show that PABA/NO causes apoptosis in cells after only a short exposure to the drug (Fig. 4). The proposed mechanism of action for PABA/NO is the production of NO formed from the release of the NONOate by GSTπ (Fig. 1A). To investigate the possible cytotoxic effects of the parent ‘carrier’ molecule, cytotoxicity assays were performed on the proposed ‘nontoxic’ by-product (JS-39-94) of PABA/NO (Fig. 1B). The IC50 value of NIH3T3 wild-type (WT) cells after treatment with JS-39-94 was 248 ± 10.1 μM and was significantly higher than that of the same cells treated with PABA/NO (11 ± 1.5 μM). This gives a >20-fold less toxic effect for JS-39-94 compared with the full drug, suggesting that the release of NO is responsible for cell death.
Fig. 4.
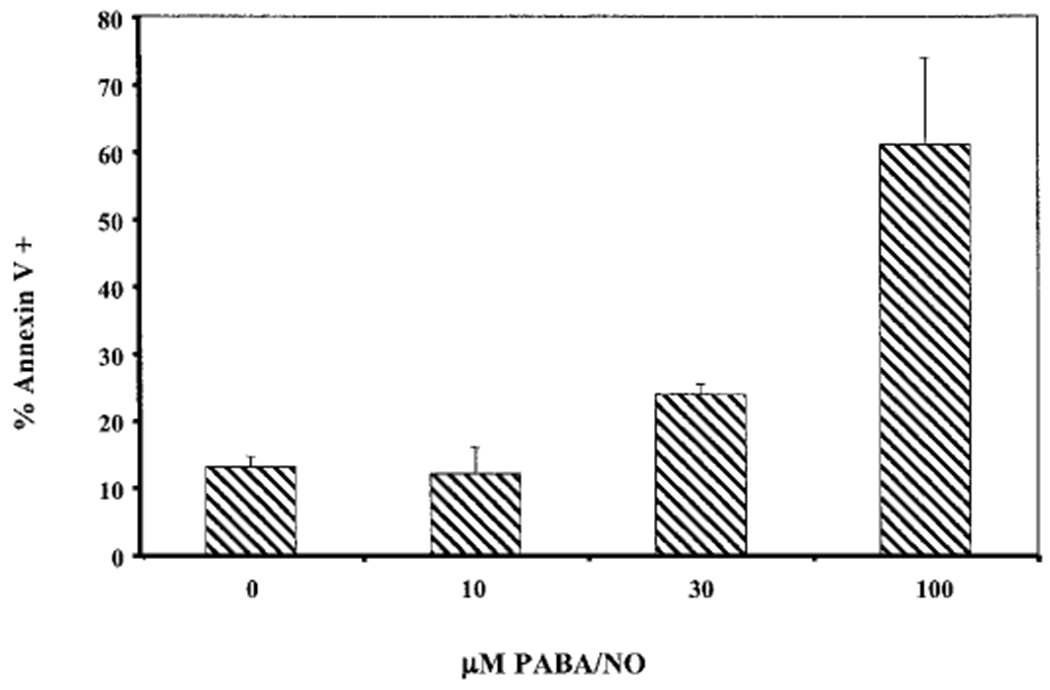
PABA/NO causes cells to undergo apoptosis. Annexin V staining of WT cells was assessed 2 h after drug exposure as detailed under Materials and Methods.
PABA/NO Treatment Leads to Protein Nitration.
Reactive nitrogen/oxygen species can covalently modify a variety of biomolecules. A primary product of protein nitration is nitrotyrosine, the consequence of stable modification of tyrosine residues by NO in a suitably oxidizing environment (Tien et al., 1999). Because a proposed mechanism for cytotoxicity of PABA/NO is the release of NO, we sought to determine whether cells exposed to PABA/NO resulted in an increase in protein nitration. To investigate this, cells were exposed to PABA/NO in both a time- and dose-dependent manner. Entire cell lysates were extracted, and protein nitration was confirmed by dot blot analysis using an anti-nitrotyrosine antibody. Our data show an increase in protein nitration in both a dose- and time-dependent manner in NIH3T3 (WT) cells after drug treatment; in MRP1 transfected cells (3T3/MRP1), however, lower amounts of nitration were observed (Fig. 5 and data not shown). These data and subsequent transport assays (see below) suggest that the decrease in intracellular protein nitration in MRP1-overexpressing cells can be attributed to enhanced efflux of PABA/NO by the transporter.
Fig. 5.
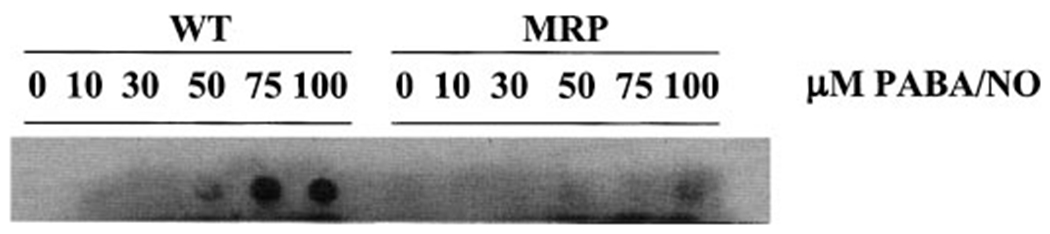
PABA/NO causes protein nitration in WT but not MRP1 overexpressing cells in a dose-dependent manner. Cells were treated for 30 min with the indicated concentrations of PABA/NO.
Activation of JNK, p38, and ERK by PABA/NO.
Three groups of mammalian mitogen-activated protein kinases include the ERKs, the JNKs, and p38 (Boulton et al., 1991; Kyriakis et al., 1994; Lee et al., 1994). Each maybe activated (i.e., phosphorylated) in response to diverse stimuli, but all three respond to treatment with NO (Lander et al., 1996; Camps et al., 1998). As such, the phosphorylation and therefore the activation status of JNK, p38, and ERK were investigated in NIH3T3 WT and MRP1-overexpressing (MRP1) cells after drug treatment. Figure 6 shows that PABA/NO treatment resulted in both a time-(A) and dose-dependent (B) catalytic activation of JNK, p38, and ERK in these cells, whereas the total protein for each kinase was not altered. Although the response of each kinase was different, collective analysis of the data shows a >8-fold increase in activation of each of these pathways, at some point within the dose and time response to PABA/NO. These data imply a link between kinase activation and pharmacological activity. Although the higher drug concentrations were cytotoxic, the short-term treatment schedules ameliorated any concern of a primary cytotoxic event.
Fig. 6.
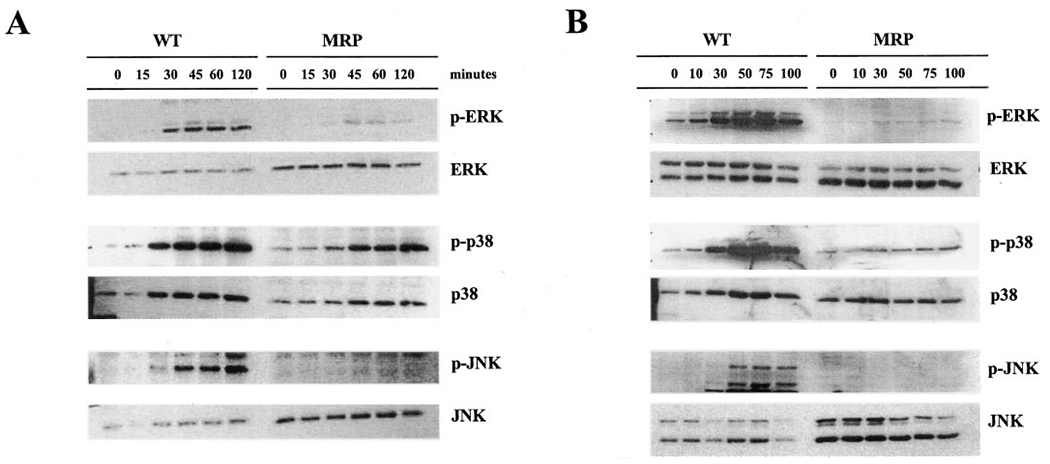
Activation of the ERK, p38, and JNK mitogen-activated protein kinases by treatment with PABA/NO. A, cells were treated with 30 μM PABA/NO for the indicated times. B, cells were treated with the indicated concentrations of PABA/NO for 60 min. 20 μg of protein was loaded in each lane. The protein expression levels of ERK, p38, and JNK were unchanged with the addition of PABA/NO (bottom). Even loading of protein was confirmed by probing for actin (data not shown).
The activation of ERK, p38, or JNK, as measured by phosphorylation via Western blot analysis, was decreased or not detected in cells that over-expressed MRP1 (Fig. 6). No more than a 2-fold increase in activation of the pathways was detected after treatment with PABA/NO either with time or dose in cells that overexpressed MRP1, with the exception of the p38 pathway, which showed a maximal 5-fold increase in activation in response to time. However, this activation was still significantly lower than that observed in the parental WT cells. These data provide further evidence that PABA/NO and/or its metabolites are effectively effluxed by MRP1 before kinase activation.
JNK and p38 Inhibitors Confer Resistance to PABA/NO.
The data generated suggest a plausible role for ERK, JNK, or p38 activation in the cytotoxic effects of PABA/NO. To examine this link, specific kinase inhibitors, SP600125 (JNK) (Bennett et al., 2001), SB202190 (p38) (Lee and Young, 1996), and U0126 (mitogen-activated protein kinase kinase/ERK) (Favata et al., 1998) were tested in combination with PABA/NO. Table 2 shows that inhibition of JNK (SP600125) and p38 (SB202190) resulted in a significant decrease in the cytotoxic effects of PABA/NO (IC50, 17.3 ± 2.8 and 16.1 ± 1.5 μM, respectively) compared with PABA/NO alone (IC50, 9.8 ± 1.9 μM). 50 value. These data suggest that both JNK and p38, but not ERK, influence the cytotoxic effects of PABA/NO, and the diminished activation of these two proteins is consistent with the resistant phenotype observed in the MRP1-overexpressing cells.
TABLE 2.
Summary of the IC50 values for the effect of PABA/NO on NIH3T3 cells in the presence of the JNK (SP600125), p38 (SB202190), or MEK/ERK (U0126) inhibitors
Statistically significant differences from DMSO-treated controls were detected. Data are presented as means ± S.D. of three independent experiments
P < 0.005
P < 0.05.
PABA/NO Inhibits LTC4 Transport.
The expression of MRP1 enhances the resistance to PABA/NO (Table 1). A plausible explanation for this result is that MRP1 can efflux PABA/NO or a metabolite thereof responsible for its cytotoxicity. Because radiolabeled PABA/NO is unavailable, this hypothesis was investigated by competition studies with LTC4, whose GS-conjugate is a substrate with high affinity for MRP1 (Leier et al., 1994). Inside-out membrane vesicles were prepared from NIH3T3/MRP1 cells, and ATP-dependent uptake of [3H]LTC4 was measured as a function of time (Fig. 6). Transport of [3H]LTC4 was not altered in the presence of either PABA/NO or GSH alone. However, complete inhibition of [3H]LTC4 transport was observed when PABA/NO and GSH were added together (0.22 versus 0.03 pmol/mg; Fig. 6A). These data suggest that activation of PABA/NO is required for LTC4 transport inhibition. In support of this hypothesis, Fig. 6B shows that the addition of GSTπ with PABA/NO and GSH potentiates the inhibitory effect of PABA/NO and GSH (0.56 versus 0.35 pmol/mg). These data are consistent with the activation of the drug by GSTπ as well as the efflux of a GS-conjugate by MRP1.
PABA/NO Treatment Delays Tumor Growth in SCID Mice.
We compared the efficacy of PABA/NO and cisplatin in SCID mice using human ovarian cancer cell line A2780. This cell line, when transplanted into mice, maintains a level of expression of GSTπequivalent to that seen in human ovarian tumors, in which GSTπ is the major isoform present in most cases (Schisselbauer et al., 1992). Figure 8 shows the tumor growth rate over 44 days after DMSO, cisplatin, or PABA/NO treatment. Analysis of the growth rate showed that cisplatin and PABA/NO treatment resulted in a significant growth delay compared with DMSO-treated animals (p < 0.05). Tumors in the DMSO-treated animals reached an average volume of 2.57 ± 0.84 cm3 in 44 days, whereas tumors in the PABA/NO treatment group reached 0.37 ± 0.11 cm3 in 44 days. The significant growth delay p < 0.05) observed in the PABA/NO treatment versus DMSO is comparable with cisplatin treatment, where average volume was 0.28 ± 0.06 cm3 at the same time point (Fig. 8).
Fig. 8.
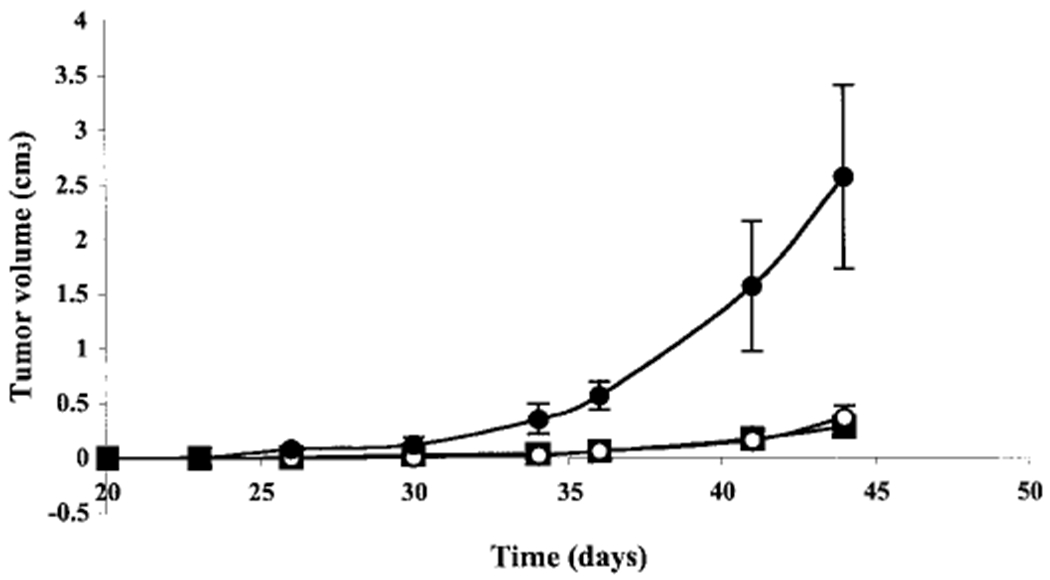
Treatment with PABA/NO delays growth of A2780 human ovarian tumors in SCID mice. The average tumor volumes ± S.E. in control (●), PABA/NO (엯), and cisplatin (■) treated mice are reported. Mice were treated with 0.8% DMSO (control), 3.36 mg/kg PABA/NO and 2.5 mg/kg cisplatin, respectively.
We analyzed the potential toxicity of PABA/NO with respect to weight loss and renal function. No statistical difference was observed in the average percentage change in body weight from day 1 to day 45 in PABA/NO (106% ± 0.07 increase) versus DMSO (115% ± 0.08 increase). Renal damage was assessed by measuring serum creatinine levels and found to be equivalent in animals treated with PABA/NO and DMSO (data not shown). These data suggest that PABA/NO treatment has an effective anticancer activity in a human ovarian cancer xenograft model with no corresponding weight loss or renal toxicity.
Discussion
The frequent occurrence of high GSTπ expression levels in a variety of human cancers and its emergence in drug-resistant disease has led to a targeting of this enzyme for drug development. This fact, together with the knowledge that NO has therapeutic potential, provided a rationale for the design of the NO-releasing GSTπ-activated prodrug, PABA/NO. The GSTπ-catalyzed conjugation of GSH to the parent molecule of PABA/NO is proposed to release a diazeniumdiolate ion, leading to the subsequent release of NO (Fig. 1). The principle of GSTπ activation of prodrugs has been successfully applied elsewhere. For TLK286, the sulfhydryl of the GSH conjugate has been oxidized to a sulfone. The active site of GSTπ has a critical tyrosine residue that promotes a β-elimination reaction cleaving the drug and releasing an alkylating phosphorodiamidate (Lyttle et al., 1994). TLK286 is the first member of GSTπ-activated prodrugs and has achieved successful preclinical and clinical development (Townsend et al., 2002; Rosen et al., 2003). TLK286 differs from PABA/NO in the activation of the parent compound. Other NO donors of the diazeniumdiolate class are known to release NO in an enzyme-catalyzed manner, with activation by cytochrome P450 (Saavedra et al., 1997) or esterases (Saavedra et al., 2000) as opposed to the here proposed use of GSTs. The proposed mechanism of activation of PABA/NO is through the GST-catalyzed formation of a GS-conjugate (Fig. 1).
To establish PABA/NO as a GSTπ-activated prodrug, we used a cell model system established from GSTπ−/− mice (Henderson et al., 1998). In the absence of GSTπ expression we showed a decreased sensitivity of GSTπ−/− MEFs to PABA/NO. These data are consistent with previous results confirming the specific activation of TLK286 (Rosario et al., 2000). Although the quantitative effect is not large, cytotoxicity will be influenced by the fact that GSTπ−/− cells express other isoforms of GSTs that may activate PABA/NO at less efficient rates. In addition, slow spontaneous activation of PABA/NO may occur through noncatalytic GSH conjugation. Other components involved in the GST detoxification pathways were analyzed in a second model system with stably transfected NIH3T3 cell lines overexpressing GSTπ, γGCS, and MRP1. This system confirmed the role of GST and GSH in the activation of PABA/NO. Forced expression of γGCS and MRP1 gave insight into a potential mechanism of resistance toward PABA/NO. Specifically, the greatest increase in resistance to PABA/NO was conferred by MRP1 transfection. Such data support the hypothesis that MRP1, an efflux pump with affinity for GS-conjugates, is involved in the removal of PABA/NO and/or an active metabolite(s). Inside-out MRP1 vesicles provide a means to study this concept further. Radiolabeled PABA/NO (or its metabolites) is not available, and this promoted an effort to study inhibition of transport of a GS-conjugate and known substrate of MRP1, LTC4. Inhibition of LTC4 transport was observed in the presence of both PABA/NO and GSH but not with either substrate alone. Additional studies confirmed that GSTπ further stimulated PABA/NO-induced inhibition of LTC4 transport and, together with the previous data, suggest that the active breakdown products of PABA/NO are substrates for, and are effluxed by, MRP1. When viewed with the data showing that overexpression of MRP1 prevents activation of the extracellular-regulated and stress-activated protein kinase cascades, these results affirm that MRP1 is an important resistance factor for PABA/NO. Whether in situ cellular resistance to PABA/NO can be mediated by increased expression of MRP1 will be answered by selection of a resistant cell line, a project that is currently under development.
The efficacy of PABA/NO was confirmed in vivo using a human ovarian cancer model in SCID mice. We have shown that a nontoxic dose of PABA/NO leads to a significant growth delay. The difference in tumor volume after 44 days was nearly 7-fold. These results are comparable with those seen with one of the most commonly used anti-cancer agents, cisplatin. Selective activation of PABA/NO via GSTπ may provide a therapeutic advantage to cisplatin, because GSTπ-overexpression is associated with a decrease in platinum efficacy.
Our data show that PABA/NO induces p38, JNK, and ERK, but pathway inhibition studies indicate that p38 and JNK are the two most critical pathways through which PABA/NO (or its metabolites) exert an apoptotic effect in these cells. This observation carries greater significance, because GSTπ has a critical function by serving as an endogenous negative regulatory switch for these same regulatory kinase pathways by binding to and inhibiting JNK activity. Mechanistically, JNK activity has been shown to be stimulated upon dissociation of the GSTπ:JNK complex (Adler et al., 1999). GSH peptidomimetics can interfere with protein-protein interactions and lead to JNK activation (Adler et al., 1999; Yin et al., 2000; Wang et al., 2001). Because of the diverse role of NO in various physiological processes, replacement or augmentation of endogenous NO by exogenous NO-releasing drugs has provided the foundation for a broad range of therapeutic applications (Napoli and Ignarro, 2003). NO has been well documented in the modulation of the apoptotic process in a number of different cell types. The choice of inhibition or induction of cell death seems to be dependent on the amount, duration, and the site of NO production and on the cell type and state (Umansky and Schirrmacher, 2001). In support of both cellular antioxidant and pro-oxidant actions of NO in vivo, it has been reported that low doses of NO protect cells against peroxide-induced death, whereas higher doses result in increased killing (Joshi et al., 1999). Our data demonstrate a dose- and time-dependent effect for drug-induced activation of the kinases. MRP1 expression abrogates and/or delays the effect, primarily as a consequence of reducing the effective intracellular concentration of PABA/NO and/or its metabolites. The threshold for direct NO-mediated effects can be altered by MRP1 expression. Whether kinase activation occurs as a result of direct NO interaction (e.g., nitrosylation/nitration of residues in JNK) or is caused by the effects of PABA/NO (or metabolites including GS-NO) on the GSTπ-JNK complex remains to be shown.
In summary, the reduction in nitrotyrosine formation and the lack of PABA/NO activation of the stress response pathways in an MRP1 over-expressing cell line identifies this transporter as one of the critical factors leading to drug resistance. Together with the inhibition of vesicle LTC4 transport in the presence of PABA/NO, GSTπ, and GSH, these results suggest that the active metabolites of PABA/NO effluxed by MRP1 still contain the NO moiety. Although PABA/NO is not yet a finished drug candidate, our in vitro and in vivo anti-tumor data suggest that it is a good lead compound for further structure activity and drug discovery efforts.
Fig. 7.
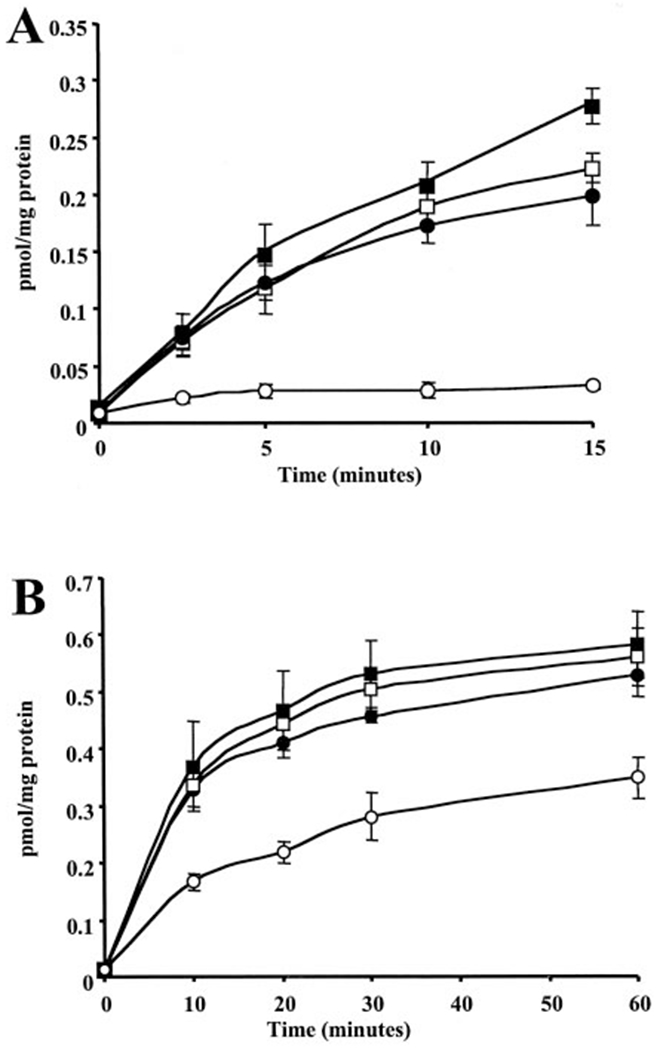
Transport of [3H]LTC4 in the presence of ATP. Uptake of [3H]LTC4 into MRP1 ‘inside-out’ vesicles was measured in the presence of [3H]LTC4 alone (□), 25 μM GSH (■), 25 μM PABA/NO (●), or 25 μM GSH and 25 μM PABA/NO (엯) (A) or 500 nM GSH and 1 μg GSTπ (■), 50 μM PABA/NO and 1 μg GSTπ (●), 50 μM PABA/NO and 500 nM GSH (□), or 500 nM GSH, 50 μM PABA/NO, and 1 μg GSTπ (엯) (B), as detailed under Materials and Methods. Data are represented as means ± S.D. of three independent experiments.
Acknowledgments
This project was funded in part through National Cancer Institute contract N01-CO12400 and by National Institutes of Health grant CA53893 (to K.D.T.).
ABBREVIATIONS:
- GST
glutathione S-transferase
- GSH
glutathione
- MRP
multidrug resistance-associated protein
- PABA/NO
O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl] 1-N,N-dimethylamino)diazen-1-ium-1,2-diolate
- THF
tetrahydrofuran
- LTC4
leukotriene C4
- JNK
c-Jun N-terminal kinase
- ERK
extracellular signal-regulated kinase
- MEF
mouse embryo fibroblasts
- JS-K
O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate
- γGCS
γ-glutamyl cysteine synthetase
- WT
wild-type
- SP600125
anthra[1,9-cd]pyrazol-6[2H]-one 1,9-pyrazoloanthrone
- SB202190
4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole
- U0126
1,4-diamino-2,3-dicyano-1,4-bis(2-aminophynyltio)butadiene
- DMSO
dimethyl sulfoxide
Footnotes
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
References
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ,Wolf CR, et al. (1999) Regulation of JNK signaling by GSTp. EMBO (Eur Mol Biol Organ) J 18:1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RN (1997) Structure, catalytic mechanism and evolution of the glutathione transferases. Chem Res Toxicol 10:2–18. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98:13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, De-Pinho RA, Panayotatos N, Cobb MH, and Yancopoulos GD (1991) ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65:663–675. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr 54:905–921. [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, and Arkinstall S (1998) Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science (Wash DC) 280:1262–1265. [DOI] [PubMed] [Google Scholar]
- Chang M, Bolton JL, and Blond SY (1999) Expression and purification of hexahistidine-tagged human glutathione S-transferase P1–1 in Escherichia coli. Protein Expr Purif 17:443–448. [DOI] [PubMed] [Google Scholar]
- Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AMV, and Deeley RG (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science (Wash DC) 258:1650–1654. [DOI] [PubMed] [Google Scholar]
- Cornwell MM, Gottesman MM, and Pastan IH (1986) Increased vinblastine binding to membrane vesicles from multidrug-resistant KB cells. J Biol Chem 261:7921–7928. [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, et al. (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273:18623–18632. [DOI] [PubMed] [Google Scholar]
- Habig WH, Pabst MJ, and Jakoby WB (1974) Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139. [PubMed] [Google Scholar]
- Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, and Wolf CR (1998) Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc Natl Acad Sci USA 95:5275–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA and Kjeldgaard M (1997) Electron-density map interpretation. Methods Enzymol 277:173–208. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Ponthier JL, and Lancaster JR (1999) Cellular antioxidant and prooxidant actions of nitric oxide. Free Radic Biol Med 27:1357–1366. [DOI] [PubMed] [Google Scholar]
- Karwatsky J, Daoud R, Cai J, Gros P, and Georges E (2003) Binding of a photoaffinity analogue of glutathione to MRP1 (ABCC1) within two cytoplasmic regions (L0 and L1) as well as transmembrane domains 10–11 and 16–17. Biochemistry 42:3286–3294. [DOI] [PubMed] [Google Scholar]
- Keefer LK, Nims RW, Davies KM, and Wink DA (1996) “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol 268:281–293. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 24:946–950. [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, and Woodgett JR (1994) The stress-activated protein kinase subfamily of c-Jun kinases. Nature (Lond) 369:156–160. [DOI] [PubMed] [Google Scholar]
- Lander HM, Jacovina AT, Davis RJ, and Tauras JM (1996) Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem 271:19705–19709. [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature (Lond) 372:739–746. [DOI] [PubMed] [Google Scholar]
- Lee JC and Young PR (1996) Pole of CSBP/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J Leukoc Biol 59:152–157. [DOI] [PubMed] [Google Scholar]
- Leier I, Jedlitschky G, Buchholz U, Cole SP, Deeley RG, and Keppler D (1994) The MRP gene encodes an ATP dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem 269:27807–27810. [PubMed] [Google Scholar]
- Lyttle MH, Satyam A, Hocker MD, Bauer KE, Caldwell CG, Hui HC, Morgan AS, Mergia A, and Kauvar LM (1994) Glutathione-S-transferase activates novel alkylating agents. J Med Chem 37:1501–1507. [DOI] [PubMed] [Google Scholar]
- Merritt EA and Bacon DJ (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol 277:505–524. [DOI] [PubMed] [Google Scholar]
- Napoli C and Ignarro LJ (2003) Nitric oxide-releasing drugs. Annu Rev Pharmacol Toxicol 43:97–123. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, and Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11:281–296. [DOI] [PubMed] [Google Scholar]
- O’Brien ML, Kruh GD, and Tew KD (2000) The influence of coordinate overexpression of glutathione phase II detoxification gene products on drug resistance. J Pharmacol Exp Ther 294:480–487. [PubMed] [Google Scholar]
- O’Brien ML and Tew KD (1996) Glutathione and related enzymes in multidrug resistance. Eur J Cancer 32:967–978. [DOI] [PubMed] [Google Scholar]
- Prade L, Huber R, Manoharan TH, Fahl WE, and Reuter W (1997) Structures of Class pi glutathione S-transferase from human placenta in complex with substrate, transition-state analogue and inhibitor. Structure 5:1287–1295. [DOI] [PubMed] [Google Scholar]
- Rosario LA, O’Brien ML, Henderson CJ, Wolf R, and Tew KD (2000) Cellular response to a glutathione S-transferase P1–1 activated prodrug. Mol Pharmacol 58:167–174. [DOI] [PubMed] [Google Scholar]
- Rosen LS, Brown J, Laxa B, Boulos L, Reiswig L, Henner WD, Lum RT, Schow SR, Maack CA, Keck JG, et al. (2003) Phase I study of TLK286 (glutathione S-transferase P1–1 activated glutathione analogue) in advanced refractory solid malignancies. Clin Cancer Res 9:1628–1638. [PubMed] [Google Scholar]
- Ruscoe JE, Rosario LA, Wang T, Gate L, Arifoglu P, Wolf CR, Henderson CJ, Ronai Z, and Tew KD (2001) Pharmacologic or genetic manipulation of glutathione S-transferase P1–1 (GSTπ) influences cell proliferation pathways. J Pharmacol Exp Ther 298:339–345. [PubMed] [Google Scholar]
- Saavedra JE, Billiar TR, Williams DL, Kim Y-M, Watkins SC, and Keefer LK (1997) Targeting Nitric Oxide (NO) Delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-α-induced apoptosis and toxicity in the liver. J Med Chem 40:1947–1954. [DOI] [PubMed] [Google Scholar]
- Saavedra JE, Shami PJ, Wang LY, Davies KM, Booth MN, Citro ML, and Keefer LK (2000) Esterase-sensitive nitric oxide donors of the diazeniumdiolate family: in vitro antileukemic activity. J Med Chem 43:261–269. [DOI] [PubMed] [Google Scholar]
- Saavedra JE, Srinivasan A, Bonifant CL, Chu J, Shanklin AP, Flippen-Anderson JL, Rice WG, Turpin JA, Davies KM, and Keefer LK (2001) The secondary amine/nitric oxide complex ion R(2)N[N(O)NO](−) as nucleophile and leaving group in S9N)Ar reactions. J Org Chem 66:3090–3098. [DOI] [PubMed] [Google Scholar]
- Schisselbauer JC, Hogan WM, Buetow KH, and Tew KD (1992) Heterogeneity of glutathione S-transferase enzyme and gene expression in ovarian carcinoma. Pharmacogenetics 2:63–72. [DOI] [PubMed] [Google Scholar]
- Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, et al. (2003) JS-K, a glutathione/glutathione s-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther 2:409–417. [PubMed] [Google Scholar]
- Sinning I, Kleywegt GJ, Cowan SW, Reinemer P, Dirr HW, Huber R, Gilliland GL, Armstrong RN, Ji X, Board PG, et al. (1993) Structure determination and refinement of human alpha class glutathione transferase A1–1 and a comparison with the mu and pi class enzymes. J Mol Biol 232:192–212. [DOI] [PubMed] [Google Scholar]
- Tien M, Berlett BS, Levine RL, Chock PB, and Stadtman ER (1999) Peroxynitrite-mediated modification of proteins at physiological carbon dioxide concentration:pH dependence of carbonyl formation, tyrosine nitration and methionine oxidation. Proc Natl Acad Sci USA 96:7809–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Shen H, Staros AL, Gate L, and Tew KD (2002) Efficacy of a glutathione S-transferase pi-activated prodrug in platinum-resistant ovarian cancer cells. Mol Cancer Ther 1:1089–1095. [PMC free article] [PubMed] [Google Scholar]
- Townsend DM and Tew KD (2003) Cancer drugs, genetic variation and the glutathione S-transferase gene family. Am J Pharmacogenomics 3:157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umansky V and Schirrmacher V (2001) Nitric oxide-induced apoptosis in tumor cells. Adv Cancer Res 82:107–131. [DOI] [PubMed] [Google Scholar]
- Wang T, Arifoglu P, Ronai Z, and Tew KD (2001) Glutathione S-transferase P1–1 (GST P1–1) inhibits c-Jun NH2 terminal kinase (JNK1) signaling through interaction with the carboxyl terminus. J Biol Chem 276:20999–21003. [DOI] [PubMed] [Google Scholar]
- Yin Z, Ivanov V, Habelhah H, Tew KD, and Ronai Z (2000) Glutathione S-transferase pi elicits protection against hydrogen peroxide-induced cell death via coordinated regulation of stress kinases. Cancer Res 60:4053–4057. [PubMed] [Google Scholar]