

SUMMARY
The RhoGEFs Kalirin-7 and Trio are regulators of synaptic plasticity, and their dysregulation is associated with a range of neurodevelopmental and neurodegenerative disorders. Although studies have implicated both Kalirin and Trio in certain diseases, such as tauopathies, they remarkably differ in their association with other disorders. Using unbiased proteomics, we identified interactomes of Kalirin-7 and Trio to ascertain distinct protein association networks associated with their respective function and revealed groups of proteins that preferentially interact with a particular RhoGEF. In comparison, we find Trio interacts with a range of axon guidance and presynaptic complexes, whereas Kalirin-7 associates with several synaptic adhesion molecules. Specifically, we show Kalirin-7 is an interactor of the cell adhesion molecule neuroligin-1 (NLGN1), and NLGN1-dependent synaptic function is mediated through Kalirin-7 in an interaction-dependent manner. Our data reveal not only the interactomes of two important disease-related proteins, but also provide an intracellular effector of NLGN1 function.
In Brief
Paskus et al. use quantitative proteomics to determine the synaptic interactomes of the disease-associated proteins Kalirin-7 and Trio, identifying Kalirin-7 as an interactor of NLGN1. Investigation of this interaction unveils Kalirin-7 as a primary intracellular effector of NLGN1 gain of function.
Graphical Abstract
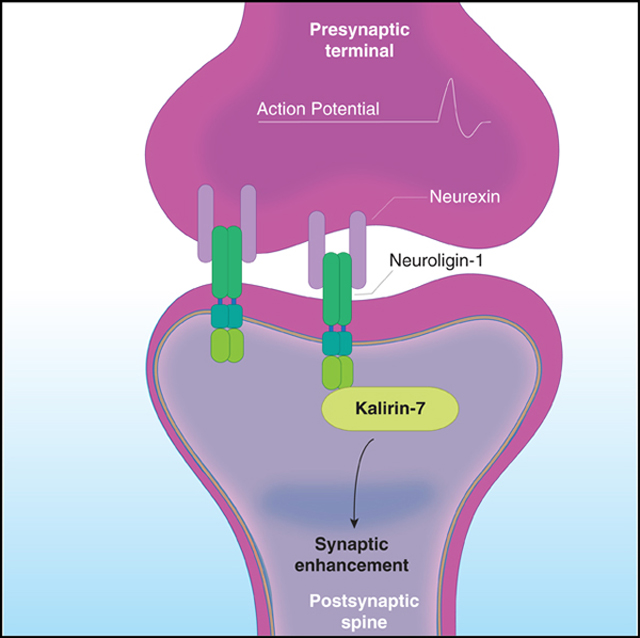
INTRODUCTION
Dendritic spines are composed of a dynamic network of actin filaments, the remodeling of which is an essential feature of the synaptic plasticity that underlies learning and memory (Konietzny et al., 2017; Okamoto et al., 2004). Ras homologous (Rho) family GTPases mediate the assembly of actin filaments and are, as such, central actors in this remodeling. Guanine nucleotide exchange factors (GEFs) are important regulators of Rho protein signaling through catalyzing the exchange of GDP for GTP and are thus critical molecular components in the neuronal processes of synaptic plasticity and in disease (Ba and Nadif Kasri, 2017; Duman et al., 2015; Kiraly et al., 2010). Kalirin and Trio are essential RhoGEFs of the postsynaptic density (PSD), regulating spine dynamics, glutamatergic synaptic transmission, and plasticity (Herring and Nicoll, 2016; Penzes and Jones, 2008). Kalirin and Trio are paralog proteins of the diffuse B cell lymphoma (Dbl) family of GEFs, evolving from a mutual ancestral gene (Kratzer et al., 2019). Kalirin and Trio share ~90% conservation of their respective Rac1 GEF domains, although only ~60% total homology (Schmidt and Debant, 2014). This diversity in non-RhoGEF sequences supports protein interactions that are unique to either Kalirin or Trio and thus establish specialized molecular regulatory mechanisms that govern the subcellular localization, scaffolding function, and/or RhoGEF activity of these proteins (Cook et al., 2014). Elucidating the protein interactions of Kalirin and Trio will facilitate a better understanding of their respective synaptic functions and their discrete and coinciding roles in disease.
Recent work has implicated Kalirin and Trio in a range of neurodegenerative and developmental disorders, such as autism spectrum disorders (ASD), schizophrenia, and Alzheimer’s disease (AD) (Dejanovic et al., 2018; Penzes and Remmers, 2012; Russell et al., 2014; Sadybekov et al., 2017). Although Kalirin and Trio have both been implicated in certain diseases, such as tauopathies, they differ in their association with other complex brain disorders. For example, whole-exome sequencing data have linked Trio, but not Kalirin, to ASD (Sadybekov et al., 2017; Sanders et al., 2015). Additionally, Trio knockout mice are embryonic-lethal, whereas Kalirin knockout mice are viable (Ma et al., 2008; O’Brien et al., 2000). It has been reported previously, however, that Kalirin and Trio may be redundant in function following development, with each protein able to rescue functional deficits produced by loss of the other (Herring and Nicoll, 2016). Taken together, this suggests functional and developmental differences, as well as similarities, in the roles of Kalirin and Trio in synaptic transmission and in disease.
In the present study, we investigated the function of Kalirin-7 (Kal-7) and Trio by performing unbiased analysis of their interactomes using quantitative proteomics. In identifying the Kal-7 and Trio interactomes, we observed several protein families that display differential affinity for a particular synaptic RhoGEF. Strikingly, we find that Kal-7 interacts with neuroligin-1 (NLGN1), a synaptogenic cell adhesion molecule (Jeong et al., 2017). We further demonstrate that NLGN1 not only interacts with Kal-7, but is functionally dependent on it as a downstream regulator in an interaction-dependent manner. We establish the interactomes and comparative analysis between two closely related and essential proteins of the PSD, the results of which reveal a mechanism of action for NLGN1. Furthermore, proteomic data of the interactomes of Kal-7 and Trio will aid in elucidating these proteins’ roles in neurodegenerative and neurodevelopmental disorders.
RESULTS
Quantitative Proteomics Reveals Kalirin-7 and Trio Interactomes
Recent work has shown that Kalirin and Trio serve complementary roles in excitatory synaptic transmission (Herring and Nicoll, 2016). To better understand their respective synaptic functions and roles in disease, we performed unbiased quantitative proteomics to identify their protein interaction complexes. We first generated antibodies to both Kalirin and Trio for the purpose of immunoprecipitating endogenous protein for interactor screening. For Kalirin, we generated a previously described isoform-specific antibody to Kal-7, the major adult and brain-specific species, targeting its divergent C-tail (amino acids 1,625–1,644) that contains a PDZ-interacting motif not present in either Kal-9 or full-length Kal-12 (Figure 1A) (Penzes et al., 2000). For Trio, we generated a specific antibody using an SH3-RhoGEF linking region (amino acids 1,794–1,806) as an epitope that was adequately divergent to the homologous Kalirin region as to eliminate potential cross-reactivity (Figure 1A). We characterized these antibodies on their specificity and ability to selectively enrich. Immunoprecipitating from the crude synaptosomal fraction (P2) of adult rat brain revealed that the Kal-7 antibody selectively enriches this Kalirin isoform, and the Trio antibody abundantly enriches as well (Figures 1B and 1C). Moreover, the Kal-7 antibody selectively enriches this non-denatured isoform from HEK293T cells, but not Kal-9 or Kal-12 (Figure S1D). Likewise, the Trio antibody enriches full-length Trio from HEK293T cells (Figure S1C). Furthermore, the Trio antibody does not recognize Kal-12 (Figure S1A), while the Kal-7 antibody does not recognize other Kalirin isoforms from rat brain lysate (Figure S1B). To screen for interacting partners, we immunoprecipitated using these antibodies to Kal-7 or Trio, with IgG as control, from isolated P2 fractions of adult rat brains and performed quantitative proteomics using label-free liquid chromatography with tandem mass spectrometry (LC-MS/MS) (Figure 1D). We next compared the Kal-7 and Trio interactomes with IgG (Figures S1E and S1F). Both Kal-7 and Trio were abundantly (>1 log2 fold change) and significantly enriched (<p = 0.05), confirming adequate enrichment of target proteins relative to IgG. We noted many hits and protein complexes that were enriched with either Trio or Kal-7 (Figures S1E and S1F; Tables S1 and S2). Nonetheless, to resolve differences in their protein interaction networks, we next compared the Kal-7 interactome against that of Trio (Figure 1E; Table S3). We observed several proteins to be either exclusively or preferentially associated with either Kal-7 or Trio. We find that members of the collapsing response mediator protein family (CRMP1/DPYSL2–5) are highly enriched with Trio, but not with Kal-7 (Figure 1F). This protein family is involved in neurite formation and axon guidance through cytoskeleton modulation and has been shown to be involved in diseases such as Parkinson’s, AD, and schizophrenia (Bader et al., 2012; Cole et al., 2007; Hensley and Kursula, 2016; Khazaei et al., 2014; Quach et al., 2004; Togashi et al., 2019; Wang and Strittmatter, 1996; Zhang et al., 2016). Given we observe both presynaptic and postsynaptic complexes to be associated with Trio, we next explored Trio’s endogenous subcellular localization relative to Kal-7. Using Immunogold electron microscopy in rat hippocampal neurons, we found Trio to be present at both pre- and post-synaptic terminals (Figures 1G and 1H), consistent with our proteomic data. Conversely, we found Kal-7 to be localized to the postsynaptic compartment, with a portion localized close to the membrane (Figures 1G and 1H). Interestingly, the distribution of Kal-7 and Trio in all synaptic compartments appeared to be quite similar (Figure S1G). Kal-7 is known to localize to the postsynaptic density, which is consistent with significant hits from our proteomic analysis. Of prominent Kal-7 specific interactors, the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (HCN1 and HCN2), the cell division cycle 42 (CDC42) downstream effectors CDC42BPA and CDC42BPB, the Rap1 GTPase binding partner cyclase-associated protein 1 (CAP1), and excitingly, all the members of the neuroligin family (NLGN1–3) of postsynaptic cell adhesion molecules were differentially associated and enriched with Kal-7. The association of NLGN family members and the small GTPase regulators CDC42BPA, CDCBPB, and CAP1 with Kal-7 represent a persuasive link between synaptic adhesion and downstream changes in the actin cytoskeleton (Figure 1F).
Figure 1. Quantitative Proteomics Reveal Kalirin-7 and Trio Interactomes.

(A) Schematic alignment of full-length Trio, Kal-12, and Kal-7 depicting unique epitopes targeted for antibody generation. A Trio antibody was generated targeting an unstructured SH3-linking region. A Kal-7 specific antibody was generated targeting the PDZ-ligand.
(B) Immunoprecipitation (IP) of endogenous Kal-7 from the crude synaptic fraction (P2) of adult rat brain demonstrating selective enrichment of Kal-7.
(C) IP of endogenous Trio from rat P2 demonstrating selective enrichment of Trio.
(D) Schematic of the strategy for interactome screening. Adult rat brain homogenates were fractioned to the P2. Endogenous proteins were IPed and label-free LC-MS/MS was performed on 3 independent samples run in tandem per condition (Kal-7, Trio, and IgG).
(E) Volcano plot of protein enrichments between Kal-7 and Trio. Purple indicates proteins differentially enriched with Kal-7 and green indicates proteins enriched with Trio.
(F) Diagram of a subset of significant Kal-7 interactors (purple lines). Dashed-gray lines indicate known or predicted interactions. Exact p value and abundance ratios are found in Table S3.
(G) Distribution of endogenous Kal-7 and Trio at rat hippocampal synapses by ImmunoEM. Kal-7 (purple arrows) is localized to the postsynaptic membrane (KAL7-post), while Trio (green arrows) is localized to both the post- and presynaptic membrane (TRIO-post and TRIO-pre).
(H) Histograms of distance of Trio Immunogold to the postsynaptic membrane (n = 44 particles) and presynaptic membrane (n = 63 particles, 24 synapses) and the distance of Kal-7 to the postsynaptic membrane (n = 46 particles, 14 synapses).
NLGN1 Associates with Kalirin-7
Given the related phenotypes of NLGN1 and Kal-7 in excitatory synaptic transmission, we investigated this interaction further. To confirm if Kal-7 interacts with NLGN1, we first employed a heterologous system for co-immunoprecipitation experiments. We co-expressed HA-NLGN1 with Kal-7 in HEK293T cells and immunoprecipitated with HA antibodies. We found that Kal-7 robustly interacted with NLGN1 in vitro (Figure 2A). Despite NLGN1 not appearing in our Trio-specific interaction screen, we inquired nonetheless as to whether Trio was additionally able to interact with NLGN1, given Trio’s high sequence homology with Kalirin. Surprisingly, coIP experiments in HEK293T cells revealed that Trio could robustly interact with NLGN1 in this heterologous system, which was unexpected given our proteomic data (Figure 2B). Nevertheless, heterologous systems do not represent the CNS environment, so we next tested these interactions in cultured cortical neurons. We subsequently performed co-immunoprecipitation experiments in cultured rat cortical neurons at days in vitro (DIV) 21 from isolated P2 fractions. Immunoprecipitating endogenous Kal-7 co-precipitated NLGN1, demonstrating that Kal-7 interacts with NLGN1 in cultured cortical neurons (Figure S2B). However, immunoprecipitating Trio co-precipitated minimal NLGN1, suggesting these proteins have minimal interaction in cultured neurons (Figure S2C). Furthermore, we tested this interaction in brain lysate by immunoprecipitating endogenous Trio and Kal-7 from adult rat P2 fraction. These experiments demonstrated that Kal-7 robustly interacts with NLGN1 in brain lysate, whereas the interaction with Trio was negligible (Figure 2C and D). We also show that NLGN2 and NLGN3 interact with Kal-7 in brain lysate (Figures S2D and S2E). Furthermore, we validated additional Kal-7 interactors from the literature that have been previously reported to interact, including GluN2A, GluN2B, and GluA1 (Figure 2C) (Kiraly et al., 2011; Xie et al., 2007). It was striking to find that NLGN1 is a more robust interactor as compared to these canonical Kal-7 interacting proteins. Taken together, we demonstrate that Kal-7 interacts with the NLGN family of proteins. Given that our proteomic data indicate that NLGN1 associates with Kal-7, but not Trio, we focused continued studies on the significance of the Kal-7 and NLGN1 relationship. Kalirin has several endogenous isoforms found at synapses that differ in structure and function. This provided us with a physiologically relevant way to perform domain mapping of the interaction with NLGN1 without resorting to arbitrary truncations that disturb normal protein folding and targeting, which may influence protein-protein interactions. We tested the interaction of NLGN1 with full-length Kal-12, Kal-9, Kal-7, and Kal-5 in HEK293T cells expressing both HA-NLGN1 and myc-tagged Kalirin isoforms. Kal-12, Kal-9, and Kal-7 were co-immunoprecipitated with HA-NLGN1 (Figure S2A). However, Kal-5 did not interact with NLGN1 (Figure 2H). This was surprising given that Kal-5 differs from Kal-7 only in that it lacks the N-terminal sec14p domain and the first four spectrin repeats (sec14pSR1:4), but it retains the GEF1 domain and PDZ-interacting motif that targets it to the synapse (Figure 2G). We further validated that this sec14pSR1:4 fragment was sufficient to bind NLGN1 in HEK293T cells (Figure 2I). To test if this interaction was direct, we performed a pull-down using a GST fusion protein of the N-terminal domain (NTD) of Kal-7 incubated with P2 brain lysate that had been denatured by SDS. We consistently observed an interaction with NLGN1 (Figure S2F). However, the reciprocal experiment using GST-NLGN1 C-tail protein was unable to detect binding with Kal-7. It is likely the interaction is direct, but this does not preclude the interaction as being part of a larger complex involving an intermediary protein. Given that we observe a robust interaction between NLGN1 and Kal-7, we next tested if genetic removal of NLGN1 would dysregulate Kal-7 levels in the PSD fraction. We performed biochemical fractionation on NLGN1 knockout brains and compared the level of Kal-7 in the PSD fraction to wild-type (WT). We observed an ~50% reduction in Kal-7 levels in the NLGN1 knockout PSD fraction, but not in the P2, as compared to WT (Figures 2E and 2F), revealing a genetic link between these two proteins.
Figure 2. NLGN1 Associates with Kalirin-7 but Not the Delta-Isoform Kalirin-5.

(A) Immunoblot analysis showing co-immunoprecipitation (coIP) of HA-NLGN1 with Kal-7 in HEK293T cells.
(B) Immunoblot analysis showing coIP of HA-NLGN1 with Trio in HEK293T cells.
(C) Immunoblot analysis showing coIP of endogenous Kal-7 with NLGN1, GluN2A, GluN2B, GluA1, and gephyrin from adult rat P2.
(D) Immunoblot analysis showing coIP of endogenous Trio with NLGN1 and DPYSL2 from adult rat P2.
(E) Immunoblot analysis of P2 and PSD fraction samples from NLGN1 knockout brains versus WT.
(F) Quantification of (E) (Student’s t test).
(G) Schematic alignment depicting Kal-7, Kal-5, and the Sec14p and first four spectrin repeats (Sec14pSR1:4) absent in Kal-5.
(H) Immunoblot analysis showing coIP of HA-NLGN1 with Kal-7 and Kal-5 in HEK293T cells. Kal-5 does not interact with NLGN1.
(I) Immunoblot analysis showing coIP of HA-NLGN1 with Kal-5 and Sec14pSR1:4. The Sec14pSR1:4, which is present in Kal-7 and absent in Kal-5, is sufficient to bind NLGN1.
NLGN1-Mediated Spine Formation Requires RhoGEF Signaling
To determine whether NLGN1-induced spinogenesis depends on Kal-7, we performed spine imaging experiments using rat hippocampal neurons. To restrict our analysis to only NLGN1, and to prevent the influence of NLGN1 heterodimers and Kal-7 interacting with additional NLGN isoforms, we performed imaging experiments on a diminished NLGN background using the previously described exogenous chained microRNA against NLGN1–3 (NLmiRs) (Shipman et al., 2011). Expectedly, reduction of endogenous NLGNs, Kal-7, or Kal-7 and Trio resulted in a decrease in spine number compared to GFP alone, whereas reintroducing a miR-resistant HA-NLGN1 increased spine number dramatically (Bemben et al., 2014) (Figures 3A, 3B, S2G, and S2H). To examine whether Kalirin is needed for NLGN1-induced spinogenesis, we knocked down Kalirin using the previously described miR-Kalirin on a NLGN null background while simultaneously reintroducing NLGN1 (Herring and Nicoll, 2016). Remarkably, we found that knocking down Kalirin inhibited the ability of NLGN1 to augment spine number (Figures 3A and 3B). Given that we observe a moderate amount of Trio interacting with NLGN1, and that we have previously shown Trio and Kalirin to have redundancies, we next knocked down both Kalirin and Trio using the previously described miR-Kalirin and Trioshort hairpin RNA (shRNA) on the NLGN null background while simultaneously reintroducing NLGN1 (Herring and Nicoll, 2016). We observed an additional significant decrease in NLGN1-mediated spine formation, which we attribute to Trio interacting with NLGN1. Taken together, these data indicate that NLGN1-mediated spinogenesis requires Kalirin and, to an extent, Trio.
Figure 3. RhoGEF Signaling Is Required for NLGN1 Overexpression-Mediated Spine Formation.

(A) Confocal images of cultured hippocampal neurons transfected at DIV5 with a combination of GFP, NLmiRs, HA-NLGN1, miR-KAL (Kalirin knockdown (KD), and miR-KAL and TRIO-shRNA (Kalirin/Trio KD). Cells were stained for GFP and HA and imaged at DIV12–DIV14. Scale bar is 20 μm.
(B) Spine number quantification of (A), with a one-way ANOVA and Tukey’s multiple comparison test across independent conditions (n = 3). ±SEM is reflective of total number of observations.
NLGN1’s Influence on Glutamatergic Synapse Function Requires Kalirin-7
We then wanted to determine whether Kal-7/NLGN1 binding specifically is required for the effects of NLGN1 on glutamatergic synapse function. To address this question, we performed electrophysiology experiments using organotypic hippocampal slice cultures (Figure 4A). Increasing postsynaptic NLGN1 expression in CA1 pyramidal neurons results in an increase in glutamatergic synapse number (Boucard et al., 2005). This increase in glutamatergic synapse number results in significant increases in both AMPA receptor- and NMDA receptor-evoked excitatory post-synaptic currents (AMPAR and NMDAR-eEPSCs) (Bemben et al., 2014). Using a biolistic transfection method, we expressed NLGN1 in CA1 pyramidal neurons in DIV1 organotypic hippocampal slice cultures (Figure 4B). Six days after transfection, we employed a dual whole cell voltage clamp technique to record AMPAR and NMDAR-eEPSCs from transfected fluorescent neurons and neighboring untransfected control neurons simultaneously following Schaffer collateral stimulation (Figures 4A and 4B). This approach permits a pairwise, internally controlled comparison of the consequences of the genetic manipulation. As shown previously, we found that overexpression of NLGN1 produces an ~2.5-fold increase in both AMPAR and NMDAR-eEPSC amplitudes (Figures 4C–4E) (Bemben et al., 2014). If the interaction between Kal-7 and NLGN1 is essential for NLGN1 function, then preventing this interaction should prevent NLGN1’s ability to increase AMPAR and NMDAR-eEPSC amplitudes. In the present study, we find that Kal-7, Kal-9, and Kal-12 interact with NLGN1, but Kal-5 does not (Figure S2A). We can therefore eliminate the interaction between NLGN1 and Kalirin in neurons by replacing endogenous Kalirin isoforms with Kal-5. If molecular replacement of endogenous Kalirin isoforms with Kal-5 prevents NLGN1-dependent increases in glutamatergic neurotransmission, this result will demonstrate that NLGN1’s synaptogenic effects depend on an interaction with Kalirin.
Figure 4. Kalirin-7, but Not Kalirin-5, Supports NLGN1 Function.

(A) Electrophysiology recording setup.
(B) Timeline of transfection and recording.
(C and D) Summary of (C) AMPAR and (D) NMDAR-eEPSC amplitudes (±SEM) for each condition (E–K) normalized to their respective neighboring untransfected paired control neurons (black bar). Bars showing the Kalirin and Trio double knockdown phenotype and the single Kalirin knockdown phenotype were previously published (Herring and Nicoll, 2016) and are repeated here for clarity. Significance was determined by Wilcoxon Signed Rank Test in each condition. Wilcoxon rank-sum tests were used to compare across independent conditions (i.e., Kalirin/Trio KD and Kal-7 versus Kalirin/Trio KD and Kal-7 and NLGN1; *p < 0.05; Kalirin/Trio KD and Kal-5 versus Kalirin/Trio KD and Kal-5 and NLGN1; p > 0.05; Kalirin KD and Kal-5 versus Kalirin KD and Kal-5 & NLGN1; p > 0.05). (E–K) Scatterplots showing the individual conditions summarized in (C) and (D). Open circles represent individual paired recordings, and filled circles represent the means ± SEM. The traces show representative currents for each condition, with the transfected cell in color and the control cell in black (vertical scale bars, 20 pA; horizontal scale bars, 20 ms for AMPAR, 50 ms for NMDAR). (E) Relative to paired control neurons, NLGN1 expression on a WT background increased both AMPAR and NMDAR-eEPSCs (AMPAR-eEPSCs, n = 8, *p < 0.05; NMDAR-eEPSCs, n = 9, *p < 0.05). (F) Replacing Kalirin and Trio with Kal-7 produced AMPAR-and NMDAR-eEPSCs that were similar to paired controls (AMPAR-eEPSCs, n = 8, p > 0.05; NMDAR-eEPSCs, n = 8, p > 0.05). (G) AMPAR and NMDAR-eEPSCs were increased by NLGN1 when Kalirin and Trio were replaced by Kal-7 (AMPAR-eEPSCs, n = 8, *p < 0.05; NMDAR-eEPSCs, n = 9, *p < 0.05). (H) Replacement of Kalirin and Trio with Kal-5 produced AMPAR and NMDAR-eEPSCs that were similar to paired controls (AMPAR-eEPSCs, n = 9, p > 0.05; NMDAR-eEPSCs, n = 8, p > 0.05). (I) AMPAR and NMDAR-eEPSCs were not increased by NLGN1 when Kalirin and Trio were replaced by Kal-5 (AMPAR-eEPSCs, n = 8, p > 0.05; NMDAR-eEPSCs, n = 7, p > 0.05). (J) Replacement of Kalirin with Kal-5 produced AMPAR and NMDAR-eEPSCs that were similar to paired controls (AMPAR-eEPSCs, n = 7, p > 0.05; NMDAR-eEPSCs, n = 7, p > 0.05). (K) AMPAR and NMDAR-eEPSCs were not increased by NLGN1 when Kalirin were replaced by Kal-5 (AMPAR-eEPSCs, n = 7, p > 0.05; NMDAR-eEPSCs, n = 6, p > 0.05).
NLGN1 binds to Kal-7 robustly, but a NLGN1 interaction with Trio is also detectable. To specifically analyze the role of Kalirin in NLGN1 function, we conducted our molecular replacement experiments on a Kalirin and Trio double knockdown background. We have shown previously that knocking down endogenous Kalirin and Trio isoforms simultaneously in CA1 pyramidal neurons significantly reduces AMPAR and NMDAR-eEPSCs (Figures 4C and 4D) (Herring and Nicoll, 2016). We have also shown previously, and again in the present study, that replacing endogenous Kalirin and Trio isoforms with a high level of recombinant RNAi-resistant Kal-7 restores normal basal synapse function (Figures 4C, 4D, and 4F) (Herring and Nicoll, 2016). This result demonstrates that recombinant Kal-7, when provided in sufficient quantity, is able to substitute for Trio in supporting basal glutamatergic synapse function. We then asked whether NLGN1 overexpression on this Kal-7 replacement background is able to increase AMPAR- and NMDAR-eEPSC amplitudes. Indeed, we found that coexpressing NLGN1 along with our Kalirin and Trio RNAi’s and Kal-7 for 6 days produced a significant increase in AMPAR-and NMDAR-eEPSC amplitudes compared to paired control cells (Figures 4C, 4D, and 4G). The magnitude of this increase relative to paired control neurons and the Kal-7 replacement condition was nearly identical to that produced by NLGN1 expression on a WT background (Figures 4C and 4D). Together, these data are consistent with Kal-7 supporting NLGN1’s influence on glutamatergic synapse function. We then wanted to know whether NLGN1 can increase glutamatergic neurotransmission when endogenous Kalirin and Trio isoforms are replaced by Kal-5. We found that co-transfection of CA1 pyramidal neurons with our Kalirin and Trio RNAi’s and our RNAi-resistant Kal-5 expression construct results in AMPA- and NMDAR-eEPSCs that are comparable to untransfected neurons (Figures 4C, 4D, and 4H). This result is consistent with normal CA3-CA1 glutamatergic neurotransmission following NLGN1 inhibition in postnatal CA1 pyramidal neurons (Shipman and Nicoll, 2012). We then overexpressed NLGN1 on this Kal-5 replacement background. In marked contrast to Kal-7, replacing Kalirin and Trio with Kal-5 completely blocked the ability of NLGN1 to increase AMPAR- and NMDAR-eEPSC amplitudes compared to either paired control neurons or the Kal-5 replacement condition (Figures 4C, 4D, and 4I). Consistent with our prior imaging data, we also find that replacing Kalirin with Kal-5 in the absence of Trio knockdown prevents NLGN1-mediated increases in glutamatergic synaptic transmission (Figures 4C, 4D, 4J, and 4K). None of our genetic manipulations were found to affect presynaptic glutamate release probability (Figure S3). Taken together, these data demonstrate that prevention of NLGN1’s interaction with Kalirin prevents NLGN1’s influence on glutamatergic synaptic transmission. While our data do not exclude the possibility of Trio-mediated support of NLGN1 function, our data do suggest that Kal-7 is the primary carrier of NLGN1’s influence on glutamatergic synaptogenesis.
DISCUSSION
Cell adhesion molecules are critical components in synapse maturation and function, the best studied being the neuroligin family of adhesion molecules for their ability to induce synapse specialization (Jeong et al., 2017; Chih et al., 2005). How neuroligins regulate and maintain synapses remains largely unknown. Canonical understanding of neuroligin function involves the interaction with presynaptic neurexin (Scheiffele et al., 2000). While interactions with neurexins are important mechanisms of neuroligin function, the role of neuroligins in synaptic plasticity and basal neurotransmission must also involve a network of downstream effector molecules and interactions with the NLGN1 C-tail. It has been suggested that targeting of specific neuroligin isoforms to postsynaptic sites depends on interactions with postsynaptic scaffolding molecules; however, particular protein interactions that have been defined have not shed substantial light on the specific downstream mechanisms of neuroligin function (Giannone et al., 2013). NLGN1 has been shown to promote actin assembly associated with spine enlargement and plasticity, and a direct link between NLGN1 and actin remodeling has been hypothesized to involve the spine-associated Rap GTPase-activating protein (SPAR) (Liu et al., 2016). However, we now show that NLGN1 is able to interact with Kal-7, an activator of Rac1, which drives its ability to strengthen excitatory transmission and modulate spine number. Although numerous interactions with the C-tail of NLGN1 have been described biochemically, the importance of these interactions, as they relate to the function of NLGN1, has remained unclear. Indeed, major scaffolding proteins at the PSD appear not to be required for NLGN1 gain of function. We now show that NLGN1 gain of function is mediated through Kal-7 in an interaction-dependent manner, providing a mechanism of action for NLGN1. Furthermore, it has recently been suggested that the function of NLGN1 in maintaining NMDAR-mediated responses involves only NLGN1’s C-tail (Wu et al., 2019). This finding is consistent with data demonstrating specific reduction in NMDAR currents in the Kal-7KO animal, and knocking down Kal-7 reduces NMDAR EPSCs, whereas knocking down Trio affects only AMPAR EPSCs, suggesting that the NMDAR-mediated responses of NLGN1 may be mediated by Kal-7 (Herring and Nicoll, 2016; Kiraly et al., 2011).
We show that NLGN1 interacts specifically with Kal-7, and not with Kal-5, a delta isoform of Kal-7 that lacks the N-terminal sec14p domain and the first four spectrin-repeats. Analogous to other RhoGEFs, the non-enzymatic domains of Kal-7 regulate its subcellular localization and influence on synaptic structure and function. Fascinatingly, the GEF domains of Kal-7 and Kal-5 are equally active and both retain the PDZ interaction motif that targets both proteins to the synapse, suggesting an additional role for the N-terminal domains of Kal-7 in its ability to control cell dendritic morphology and synapse number, although it is unclear how this occurs and what protein-protein interactions are necessary (Penzes et al., 2001). Given the phenotypic intersection between Kal-7 and NLGN1, it is perhaps likely that not only does NLGN1 function through Kal-7, but Kal-7’s ability to control cell morphology is likewise dependent on NLGN1. It may be that NLGN1 tethers Kal-7 to postsynaptic sites, positioning Kal-7’s GEF domain to respond to changes in celladhesion. Conversely, Kal-7 might act as a scaffold, controlling NLGN1 surface expression or targeting.
Despite distinct functions in development, Kal-7 and Trio do have compensatory functions in the adult (Herring and Nicoll, 2016). Moreover, recent data have implicated Kalirin and Trio together in the pathophysiology of tauopathies. Levels of Kalirin and Trio were reduced in the PSD fractions of the Tau-P301S mouse, suggesting that loss of such GTPase regulatory proteins contribute to spine loss (Dejanovic et al., 2018). This is consistent with prior reports noting decreased levels of Kalirin in AD patients (Mandela and Ma, 2012; Penzes and Remmers, 2012). Interestingly, NLGNs have also been implicated in AD pathophysiology. A rare truncating mutation in NLGN1, resulting in reduced forward trafficking, has been identified in a patient with a familial history of AD (Tristán-Clavijo et al., 2015). NLGN1 loss has also been observed in patients with AD. Given that we show NLGN1 function to be mediated through Kal-7, it may be that loss of function or reduction in NLGN1 dysregulates Kal-7 downstream, resulting in spine destabilization and loss. Interestingly, our proteomic analysis of Kal-7 also reveals a tight association with the 26S proteasome, which has been a therapeutic target in proteotoxic diseases. Further work is needed to elucidate the role of Kal-7, and Trio, in this process.
In conclusion, our proteomic analyses of Kal-7 and Trio has identified unique protein networks between these two genetic paralogs, which undoubtedly coordinate in mediating their particular molecular functions at the synapse and may arbitrate their respective roles in disease and development. By investigating the Kal-7 link to NLGN1 in detail, we have discovered that Kal-7 is critical for the overexpression-mediated synaptogenic function of NLGN1, positioning Kal-7 as a primary intracellular effector of NLGN1 function.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Information and requests for reagents should be directed to the Lead Contact, Katherine W. Roche (rochek@ninds.nih.gov). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
The NINDS Animal Care and Use Committee approved all experimental animal use (protocol #1171). Male and female Sprague Dawley outbred rats (Envigo) and C57BL/6 inbred mice (Charles River) were used at >1 month of age. Rats and mice were housed on a standard 12 hr dark and light cycle.
Neuronal Cultures
Primary hippocampal and cortical neurons were prepared from male and female E18 Sprague Dawley rats (Envigo) following the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. Briefly, the animals were narcotized with CO2 and the embryos removed. Embryonic brains were subsequently dissected out and enzymatically and mechanically dissociated prior to plating on poly-D-lysine coated plates and coverslips. Mixed gender cultures were maintained in Neurobasal Medium (Life Technologies, Cat# 21103–049) supplemented with 2% B-27 (Life Technologies, Cat#17504–044) and 2 mM L-Glutamine (Sigma-Aldrich, Cat# G-7513) at 37 C and 5% CO2. For electrophysiology, 400 μm organotypic hippocampal slice cultures were prepared from P6–9 Sprague Dawley rat pups as described previously (Stoppini et al., 1991). Culture media was exchanged every other day. Sparse biolistic transfections of organotypic slice cultures were carried out on DIV1 as previously described (Schnell et al., 2002). All organotypic slice culture experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the University of Southern California Institutional Animal Care and Use Committee.
METHOD DETAILS
Experimental Constructs and Antibody Generation
For biochemical experiments pCAG-eGFP, human pCAG-Trio-9, mouse pCAG-HA–NLGN1, pEAK10-His-Myc-Kal5, pEAK10-His-Myc-Kal7, pEAK10-His-Myc-Kal9, pEAK10-His-Myc-Kal12, pEAK10-Myc-Sec14pSR1:4 were used. The HA-NLGN1 construct was RNA interference resistant, as previously described (Bemben et al., 2014; Shipman et al., 2011). All Kalirin isoform constructs were purchased from AddGene. The pEAK10-Myc-Sec14pSR1:4 and pGEX-Sec14p constructs were generously provided by Dr. Betty Eipper (Uconn Health). For imaging and electrophysiology experiments we additionally used the previously described pCAG-NLmiRs-GFP, Kal/Trio RNAi (Kal-miR & Trio shRNA), and IRES-HA–NLGN1 (Bemben et al., 2014; Herring and Nicoll, 2016; Shipman et al., 2011). Kal-7 and Kal-5 cDNAs were made RNAi resistant as described previously (Herring and Nicoll, 2016). Rabbit antibodies for Kal-7 and Trio were generated by New England Peptide and were affinity purified using an antigen peptide. For Kal-7 we used amino acids 1625–1644 (CGNLVPRWHLGPGDPFSTYV), and for Trio we used amino acids amino acids 1794–1806 (KKLAHKHKKSREV) (Penzes et al., 2000).
Immunoprecipitation and Immunoblotting
For heterologous co-immunoprecipitation experiments HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (Life Technologies, Cat# 10313–021) supplemented with 5% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and were maintained at 37°C and 5% CO2. Cells were transfected using Lipofectamine 2000 (Invitrogen, Cat# 12566014) and were incubated for 48 h after transfection. Cells were subsequently washed in cold phosphate-buffered saline (PBS) and were lysed in a 1% Triton x-100 lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 1 mM EDTA. Lysates were rocked at 4°C for 1 h and centrifuged in a microfuge at full speed for 20 min. Supernatants were collected and used for immunoprecipitation. For co-immunoprecipitation experiments in primary cortical neurons, and in rat brain, samples were homogenized in cold TEVP buffer containing 320 mM sucrose, 10 mM TrispHCl (pH 7.5), 5 mM EDTA, with protease inhibitors (Roche, Cat#: 11697498001), and phosphatase inhibitor mixture II (Sigma-Aldrich, Cat#: P5726) and III (Sigma-Aldrich, Cat#: P0044). Homogenates were centrifuged at 1000 × g for 10 min at 4°C. The supernatant was centrifuged again at 10000 × g for 20 min at 4°C to obtain the crude synaptosomal fraction (P2). The resulting pellet was solubilized in 200 μL of 1% sodium deoxycholate (DOC) buffer containing 50 mM TrispHCl (pH 8.8) at 37°C for 30 min. The sample was subsequently neutralized with 800 μL of 1% Triton x-100 buffer containing 50 mM TrispHCl (pH 8.0). Lysates were spun down in a refrigerated microfuge at full speed for 20 min and supernatants were collected. For immunoprecipitation from heterologous cells, lysates were incubated with HA-beads overnight at 4°C . For immunoprecipitation of endogenous Kal-7 or Trio, 10 μg of rabbit Kal-7 antibody or Trio antibody was incubated with lysates overnight at 4°C. Protein A-agarose beads (Sigma-Aldrich, Cat#: P3391) were added to samples for 4 h at 4°C. All samples were washed 3 x with 0.1% Triton x-100 lysis buffer, and immunoprecipitated proteins were resuspended in 4 x SDS/PAGE sample buffer and were subsequently subjected to western blotting. Immunoblots were developed by film or digitally, and images were corrected (decreased brightness) to match each other.
Subcellular Fractionation
For mouse brain subcellular fractionation experiments, age matched (> 3 months old) NLGN1 knockout or WT whole brains were homogenized and fractioned to P2 as described above. The resultant supernatant was further centrifuged at 25000 × g for 20 min at 4°C to acquire the synaptic plasma membrane fraction (SPM). 1% Triton x-100 lysis buffer was added to the SPM pellet to resuspended it, and it was incubated with agitation for 30 min at 4°C. The lysate was subsequently centrifuged at 33000 × g for 30 min at 4°C to acquire both a soluble fraction and an insoluble pellet (PSD fraction). The pellet was subsequently solubilized with a 1% sodium dodecyl sulfate (SDS) lysis buffer for 30 min at 37°C. This lysate was further centrifuged at 100,000 × g for 30 min at 4°C to obtain the PSD fraction. Protein assays were performed on all samples and protein levels were normalized between conditions and samples were subjected to western blotting.
GST-Fusion Protein Production
Fusion proteins were made using the previously described protocol (Bemben et al., 2014). Briefly, GST fusion proteins were purified from BL21 cells (Agilent, Cat#: 200132) transformed with pGEX-Sec14p. Cultures were grown at 37°C and 50 μM isopropyl β-D-1-thiogalactopyranoside was added to induce protein expression. Cells were subsequently lysed with a sonicator. Sonicated lysates were incubated with glutathione–Sepharose 4B (GE Healthcare, Cat#: 17075601) for 1 h at 4°C and were subsequently washed with tris-buffered saline (TBS).
Confocal and Widefield Imaging
Hippocampal neurons were cotransfected with GFP, NLmiRs, HA-NLGN1, Kal-miR, and/or Kal-miR & Trio shRNA using Liptofectamine 2000 (Herring and Nicoll, 2016; Shipman et al., 2011). To assess spine density, hippocampal neurons were transfected on DIV5 and on DIV14 neurons were fixed in 4% paraformaldehyde in PBS containing 4% sucrose and permeabilized with 0.25% Triton x-100 in PBS. HA-NLGN-1 was labeled with anti-HA antibodies and GFP was labeled with anti-GFP antibodies. Coverslips were mounted with Invitrogen ProLong Gold Antifade mounting (Cat#: P10144) and imaged on either a Zeiss Axio Imager.M2 research microscope with Apotome.2 or a Zeiss LSM 800 Confocal microscope. Serial optical sections were collected at 0.30 mm intervals and projected. Spines were counted manually, with experimenters blinded to the conditions.
Electron Microscopy
For electron microscopy (EM) immunolabeling, DIV21 dissociated rat hippocampal neurons on coverslips were fixed in 4% paraformaldehyde in 0.1 M PBS for 35 min, washed 4 x with PBS, and were permeabilized and blocked with 0.1% saponin in 5% normal goat serum (NGS) in PBS for 1 hr at room temperature. Neurons were incubated with primary antibodies for 1 hr at room temperature, washed, and incubated with secondary antibodies (Fab conjugated to 1.4 nm Nanogold) in 1% NGS and 0.02% saponin for 1 hr at room temperature. Labeled cells were fixed in 2% glutaraldehyde for 1 hr at room temperature and subsequently stored at 4°C for further EM processing. 1.4 nm Nanogold particles were enlarged in a dark room for ~8min using a silver-enhancement HQ kit (Nanoprobe Inc, Cat#: 2012–45) prior to being rinsed in 0.1 M cacodylate buffer and treated with 0.2% osmium tetroxide for 30 min on ice, followed by 0.25% uranyl acetate overnight. Ascending dehydration occurred in 50, 70, and 90% EtOH, followed by three 7-min rinses with 100% EtOH. Samples were next infiltrated with 1:1 Epon/EtOH for 30 mins, followed by 30 min with 2:1 Epon/EtOH at room temperature, 10 min with 100% Epon at 47°C, and 2 × 1 h with 100% Epon at 47°C. Polymerization occurred in a 50°C oven overnight and samples were subsequently moved into a 60°C oven. The block and coverslip was examined in a light microscope to mark labeled neurons. The glass coverslip was released from the Epon block by a brief immersion in liquid nitrogen and the marked area was cut out with a saw and mounted for thin sectioning. For conventional EM morphological analysis, the section was cut to a thickness of ~70 nm and was grid stained with UA and lead citrate. EM images were acquired on a JOEL-200 CX transmission EM equipped with a bottom mounted CCD camera (2624×2624 pixels, AMT, Woburn, MA). EM images were analyzed using ImageJ (NIH).
Organotypic Hippocampal Slices and Electrophysiology
Sparse biolistic transfections of organotypic slice cultures were carried out on DIV1 as described (Schnell et al., 2002). Construct expression was confirmed by GFP and mcherry co-transfection. Paired whole-cell recordings from transfected neurons and non-transfected control neurons were performed on DIV7 slices. During recording, all slices were maintained in room temperature artificial cerebrospinal fluid (aCSF) saturated with 95% O2/5% CO2. aCSF contained 119 mM NaCl, 2.5 mM KCl, 1mM NaH2PO4, 26.2 mM NaHCO3, 11 mM glucose, 4mM CaCl2, 4 mM MgSO4, supplemented with 5 mM 2-chloroadenosine to dampen epileptiform activity and 0.1mM picrotoxin to block GABA(A) receptors. The internal whole-cell recording solution contained: 135 mM CsMeSO4, 8 mM NaCl, 10 mM HEPES, 0.3mM EGTA, 5 mM QX-314, 4 mM Mg-ATP, and 0.3mM Na-GTP. The internal solution was pH buffered at 7.3–7.4 and Osmolarity was adjusted to 290–295 mOsm. Whole-cell recording were carried out as described (Sadybekov et al., 2017). Synaptic responses were evoked by stimulating with a monopolar glass electrode filled with aCSF in stratum radiatum of CA1. AMPA receptor-mediated current were measured at −70mV. NMDA receptor-mediated current were recorded at +40mV, temporally isolated from AMPAR current by measuring amplitudes 250ms following the stimulus. In most cases AMPAR and NMDAR mediated currents were recorded from the same neuron by changing membrane potential. In the scatterplot, each open circle represents one paired recording, with the y axis for transfected neuron eEPSC amplitude and the x axis for control neuron eEPSC amplitude. When eEPSC amplitudes in control neurons are higher than that of transfected neurons, data points fall below the diagonal line. Paired-pulse ratio was recorded by delivering two stimuli at intervals of 40ms and dividing the peak response of stimulus 2 by the peak response of stimulus 1. No more than one paired recording was performed on a given slice.
Label Free Mass Spectrometry
Endogenous Kal-7 and Trio were immunoprecipitated from the crude P2 fraction as described above, using rabbit IgG as control (3 months old, n = 3 per condition). Samples were analyzed via label free liquid chromatography with tandem mass spectrometry (LC-MS/MS). Samples were reduced with tris(2-carboxyethyl)phosphine (TCEP), alkylated with N-Ethylmaleimide (NEM), and digested with trypsin. Digests were extracted from the gel and desalted with Waters Oasis HLB μElution plate. An UltiMate 3000 RSLC-nano system (Thermo Fisher Scientific) was used for chromatography separation. Peptides were separated on a nano-ES802 column over a 60-minute gradient from 2% to 27% acetonitrile at a flow rate of 300 nl/min. LC MS/MS experiments were performed on an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) in data-dependent acquisition (DDA) mode. The MS resolution was 120K at m/z 400, MS scan range was 300–1500 m/z, the automated gain control (AGC) target was 2 × 10e5. The quadrupole isolation window was 1.4 m/z. Precursors with charge states 2 6 and intensity higher than 1×10e4 within a 3 s cycle between MS1 scans were selected for MS/MS acquisition in the linear ion trap.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and Analysis of Immunocytochemistry and Electrophysiology
For spine number quantification, unique regions from three secondary or tertiary dendrites were randomly selected per individual neuron. Spine number was manually counted using ImageJ (NIH) over a 20 μm distance, and spine number was subsequently normalized to spine per 1 μm. Statistical significance in Figure 3B was derived using a one-way ANOVA (GraphPad Prism 8), with post hoc analysis (Tukey’s multiple comparison test). Statistical significance in Figure S2H was derived using a Mann-Whitney U test. For electrophysiology experiments in Figures 4 and S3, a Wilcoxon Rank Sum Test was used to compare data across independent conditions. Data graphics and detailed statistical analyses were performed with GraphPad Prism 8.
Quantification and Analysis of Immunoblots
Immunoblots were quantified with ImageJ (NIH) using area under the curve. In Figure 2F, Kal-7 intensity was normalized to actin per sample, and values were normalized to WT control. Statistical analysis of PSD and P2 samples was done using a Student’s t test. In Figure S2F, NLGN1–3 intensity as percent protein immunoprecipitated relative to the intensity of Kal-7 immunoprecipitated. Statistical significance was derived from a one-way ANOVA.
Quantification and Analysis of Immunogold EM
For Figures 1G and 1H, distances from centroid of immunogold particles to extracellular side of membrane of pre- and post-synaptic terminals were measured using the built-in tools in ImageJ (NIH). Statistical analysis of the overlaid accumulative distribution of Trio and Kal-7 immunogold particles from EM in Figure S1G was derived using a Kruskal-Wallis test.
Quantification and Analysis of Label Free Mass Spectrometry
Database searches were performed with Proteome Discoverer 2.2 (Thermo Fisher, Cat# 30795) using MASCOT as the search engine. The following parameters were used: trypsin digestion with full specificity; 1 missed cleavages allowed; N-ethylmaleimide on cysteines as fixed modification; oxidation (M) as variable modification, the mass tolerance is 5 ppm for precursor ions and 0.5 Da for fragment ions. The false discovery rate for peptide-spectrum matches (PSMs) was set to 0.05 using Percolator. Only unique peptides were considered for the quantification and intensities were reported with no corrections applied. The search results were filtered by a FDR of 1% at protein level. The label-free quantitation analysis was also performed with Proteome Discoverer 2.2 software. The summed intensity of the unique peptides matched to the protein is used for protein ratio calculation. The maximum fold change allowed is set to 100. The total peptide amount of each sample is used for the normalization. Individual protein data were tested via ANOVA in Figure 1E and in Tables S1, S2, and S3.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit Kalirin [1:1000] | Millipore | Cat#: 07–122 RRID:AB_310380 |
| Rabbit HA [1:2000] | Abcam | Cat#: ab9110 RRID:AB_307019 |
| Rabbit Myc (71D10) [1:5000] | Cell Signaling | Cat#: 2278 |
| Mouse Actin [1:5000] | Abm | Cat#: G043 |
| Rabbit IgG | Millipore | Cat# PP64 RRID:AB_97852 |
| Rabbit Neuroligin-3 [1:1000] | Synaptic Systems | Cat#: 129103 RRID:AB_887748 |
| Mouse Neuroligin-1 [1:1000] | Synaptic Systems | Cat#: 129111 RRID:AB_887747 |
| Mouse Neuroligin-2 [1:2000] | Synaptic Systems | Cat#: 129511 RRID:AB_2619813 |
| Rabbit Trio [1:1000] | Abcam | Cat#: ab194365 RRID:AB_11127474 |
| Mouse Dpysl2 (Crmp2) [1:1000] | Abcam | Cat#: ab62539 RRID:AB_941175 |
| Mouse Gephyrin [1:1000] | Cell Signaling | Cat#: 14304 RRID:AB_2798443 |
| Rabbit Kalirin-7 [1:3000] | This manuscript | Cat#: N/A |
| Rabbit Trio [1:1000] | This manuscript | Cat#: N/A |
| Rabbit Trio [1:1000] | This manuscript | Cat#: N/A |
| Mouse GluN2A [1:1000] | Sigma-Aldrich | Cat#: SAB5200888 RRID:AB_2629501 |
| Mouse GluN2B [1:1000] | Neuromab | Cat#: Q00960 |
| Mouse GluA1 [1:1000] | Antibodies Inc | Cat#: 75327 RRID:AB_2315840 |
| Rabbit GFP [1:1000] | Thermo Fischer | Cat# A11122 RRID:AB_221569 |
| Mouse IgG HRP linked whole antibody [1:5000] | GE Healthcare | Cat#: NA931 RRID:AB_772210 |
| Rabbit IgG HRP linked whole antibody [1:5000] | GE Healthcare | Cat#: NA934 RRID:AB_772206 |
| Anti-mouse IgG (H+L) Alexa Fluor 488 [1:500] | Thermo Fisher | Cat#: A21131 RRID:AB_141618 |
| Anti-mouse IgG (H+L) Alex Fluor 555 [1:500] | Thermo Fisher | Cat#: A21422 RRID:AB_141822 |
| Anti-rabbit IgG (H+L) Alexa Fluor 555 [1:500] | Thermo Fisher | Cat#: A21428 RRID:AB_141784 |
| Anti-rabbit IgG (H+L) Alexa Fluor 488 [1:500] | Thermo Fisher | Cat#:A11034 RRID:AB_2576217 |
| Nanogold®-Fab’ Goat anti-Mouse IgG (H+L) | Nanoprobes Inc. | Cat #2001 |
| Nanogold®-IgG Goat anti-Rabbit IgG (H+L) | Nanoprobes Inc. | Cat #2003 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Lipofectamine 2000 | Thermo Fisher | Cat# 11668019 |
| Protease inhibitor mixture | Roche | Cat#: 11697498001 |
| Phosphatase inhibitor mixture II | Sigma-Aldrich | Cat#: P5726 |
| Phosphatase inhibitor mixture III | Sigma-Aldrich | Cat#: P0044 |
| Glutathione Sepharose 4B | GE Healthcare | Cat#: 17075601 |
| Protein A-Sepharose | Sigma-Aldrich | Cat#: P3391 |
|
| ||
| Cell Lines | ||
|
| ||
| Human embryonic kidney (HEK) 293T | ATTC | Cat#: CRL11268 RRID:CVCL_1926 |
| Escherichia coli: BL21(DE3) | Agilent | Cat#: 200132 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCAG-Trio-9 | Herring and Nicoll, 2016 | N/A |
| pCAG-HA–Nlgn1 | Bemben et al., 2014 | N/A |
| IRES-HA–Nlgn1 | Bemben et al., 2014 | N/A |
| pEAK10-His-Myc-Kalirin-5 | Addgene | Cat#: 25440 RRID:Addgene_25440 |
| pEAK10-His-Myc-Kalirin-7 | Addgene | Cat#: 25454 RRID:Addgene_25454 |
| pEAK10-His-Myc-Kalirin-9 | Addgene | Cat#: 25441 RRID:Addgene_25441 |
| pEAK10-His-Myc-Kalirin-12 | Addgene | Cat#: 25442 RRID:Addgene_25442 |
| pEAK10-Myc-Sec14pSR1:4 | Dr. Betty Eipper (UConn) | N/A |
| pGEX-Sec14p | Dr. Betty Eipper (UConn) | N/A |
| pCAG-NLmiRs-GFP | Bemben et al., 2014 | N/A |
| pCAG-eGFP | Bemben et al., 2014 | N/A |
| Kalirin-miR & Trio shRNA | Herring and Nicoll, 2016 | N/A |
| pCAG-Kalirin7 | Herring and Nicoll, 2016 | N/A |
|
| ||
| Organisms/Strains | ||
|
| ||
| Sprague Dawley® outbred rat | Envigo | Cat#: 002 |
| C57BL/6 inbred mouse | Charles River | Cat#: 027 |
| NLGN1 −/− | Bemben et al., 2014 | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| Adobe Photoshop CC 2018 | Adobe | RRID:SCR_014199 |
| Adobe Illustrator CC 2018 | Adobe | RRID:SCR_010279 |
| ImageJ | NIH | RRID:SCR_003070 |
| GraphPad Prism 8 | Graphpad | RRID:SCR_002798 |
| Proteome Discoverer 2.2 | Thermo Fisher | Cat# 30795 RRID:SCR_014477 |
| Cytoscape | Cytoscape | RRID:SCR_003032 |
Highlights.
Kalirin-7 and Trio have discrete and coinciding protein-protein interaction networks
Kalirin-7 strongly associates with the synaptic adhesion molecule NLGN1
NLGN1 gain of function requires RhoGEF signaling
Binding of Kalirin-7 to NLGN1 is required to support NLGN1 function
ACKNOWLEDGMENTS
We are grateful to Carolyn Smith and the NINDS imaging facility for their assistance. We also thank Christine Winters for providing rat hippocampal neuronal cultures for EM and guidance on Immunogold EM labeling, and we thank Virginia Crocker and Susan Cheng (NINDS) for image processing. We also thank all members of the lab. Research was supported by the NINDS Intramural Research Program (to K.W.R.), the National Institute of Mental Health (MH103398 to B.E.H.), and the Simons and McKnight Foundations (to B.E.H.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.115.
REFERENCES
- Ba W, and Nadif Kasri N (2017). RhoGTPases at the synapse: An embarrassment of choice. Small GTPases 8, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader V, Tomppo L, Trossbach SV, Bradshaw NJ, Prikulis I, Leliveld SR, Lin CY, Ishizuka K, Sawa A, Ramos A, et al. (2012). Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum. Mol. Genet. 21, 4406–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemben MA, Shipman SL, Hirai T, Herring BE, Li Y, Badger JD 2nd, Nicoll RA, Diamond JS, and Roche KW (2014). CaMKII phosphorylation of neuroligin-1 regulates excitatory synapses. Nat. Neurosci. 17, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, and Südhof TC (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, and Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328. [DOI] [PubMed] [Google Scholar]
- Cole AR, Noble W, van Aalten L, Plattner F, Meimaridou R, Hogan D, Taylor M, LaFrancois J, Gunn-Moore F, Verkhratsky A, et al. (2007). Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 103, 1132–1144. [DOI] [PubMed] [Google Scholar]
- Cook DR, Rossman KL, and Der CJ (2014). Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33, 4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, et al. (2018). Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336. [DOI] [PubMed] [Google Scholar]
- Duman JG, Mulherkar S, Tu YK, X Cheng J, and Tolias KF (2015). Mechanisms for spatiotemporal regulation of Rho-GTPase signaling at synapses. Neurosci. Lett. 601, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G, Mondin M, Grillo-Bosch D, Tessier B, Saint-Michel E, Czöndör K, Sainlos M, Choquet D, and Thoumine O (2013). Neurexin-1β binding to neuroligin-1 triggers the preferential recruitment of PSD-95 versus gephyrin through tyrosine phosphorylation of neuroligin-1. Cell Rep. 3, 1996–2007. [DOI] [PubMed] [Google Scholar]
- Hensley K, and Kursula P (2016). Collapsin Response Mediator Protein-2 (CRMP2) is a Plausible Etiological Factor and Potential Therapeutic Target in Alzheimer’s Disease: Comparison and Contrast with Microtubule-Associated Protein Tau. J. Alzheimers Dis. 53, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, and Nicoll RA (2016). Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. USA 113, 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Paskus JD, and Roche KW (2017). Posttranslational modifications of neuroligins regulate neuronal and glial signaling. Curr. Opin. Neurobiol. 45, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazaei MR, Girouard MP, Alchini R, Ong Tone S, Shimada T, Bechstedt S, Cowan M, Guillet D, Wiseman PW, Brouhard G, et al. (2014). Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J. Biol. Chem. 289, 30133–30143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, Eipper-Mains JE, Mains RE, and Eipper BA (2010). Synaptic plasticity, a symphony in GEF. ACS Chem. Neurosci. 1, 348–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, Lemtiri-Chlieh F, Levine ES, Mains RE, and Eipper BA (2011). Kalirin binds the NR2B subunit of the NMDA receptor, altering its synaptic localization and function. J. Neurosci. 31, 12554–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konietzny A, Bär J, and Mikhaylova M (2017). Dendritic Actin Cytoskeleton: Structure, Functions, and Regulations. Front. Cell. Neurosci. 11, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer MC, England L, Apel D, Hassel M, and Borchers A (2019). Evolution of the Rho guanine nucleotide exchange factors Kalirin and Trio and their gene expression in Xenopus development. Gene Expr. Patterns 32, 18–27. [DOI] [PubMed] [Google Scholar]
- Liu A, Zhou Z, Dang R, Zhu Y, Qi J, He G, Leung C, Pak D, Jia Z, and Xie W (2016). Neuroligin 1 regulates spines and synaptic plasticity via LIMK1/cofilin-mediated actin reorganization. J. Cell Biol. 212, 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Kiraly DD, Gaier ED, Wang Y, Kim EJ, Levine ES, Eipper BA, and Mains RE (2008). Kalirin-7 is required for synaptic structure and function. J. Neurosci. 28, 12368–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandela P, and Ma XM (2012). Kalirin, a key player in synapse formation, is implicated in human diseases. Neural Plast. 2012, 728161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien SP, Seipel K, Medley QG, Bronson R, Segal R, and Streuli M (2000). Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 97, 12074–12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, and Hayashi Y (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 7, 1104–1112. [DOI] [PubMed] [Google Scholar]
- Penzes P, and Jones KA (2008). Dendritic spine dynamics–a key role for kalirin-7. Trends Neurosci. 31, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, and Remmers C (2012). Kalirin signaling: implications for synaptic pathology. Mol. Neurobiol. 45, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Alam MR, Kambampati V, Mains RE, and Eipper BA (2000). An isoform of kalirin, a brain-specific GDP/GTP exchange factor, is enriched in the postsynaptic density fraction. J. Biol. Chem. 275, 6395–6403. [DOI] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Sattler R, Zhang X, Huganir RL, Kambampati V, Mains RE, and Eipper BA (2001). The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron 29, 229–242. [DOI] [PubMed] [Google Scholar]
- Quach TT, Duchemin AM, Rogemond V, Aguera M, Honnorat J, Belin MF, and Kolattukudy PE (2004). Involvement of collapsin response mediator proteins in the neurite extension induced by neurotrophins in dorsal root ganglion neurons. Mol. Cell. Neurosci. 25, 433–443. [DOI] [PubMed] [Google Scholar]
- Russell TA, Blizinsky KD, Cobia DJ, Cahill ME, Xie Z, Sweet RA, Duan J, Gejman PV, Wang L, Csernansky JG, and Penzes P (2014). A sequence variant in human KALRN impairs protein function and coincides with reduced cortical thickness. Nat. Commun. 5, 4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadybekov A, Tian C, Arnesano C, Katritch V, and Herring BE (2017). An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 8, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. ; Autism Sequencing Consortium (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, and Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. [DOI] [PubMed] [Google Scholar]
- Schmidt S, and Debant A (2014). Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 5, e29769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt D, and Nicoll R (2002). Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc. Natl. Acad. Sci. U S A 99, 13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL, and Nicoll RA (2012). A subtype-specific function for the extracellular domain of neuroligin 1 in hippocampal LTP. Neuron 76, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL, Schnell E, Hirai T, Chen BS, Roche KW, and Nicoll RA (2011). Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat. Neurosci. 14, 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs P-A, and Muller D (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. [DOI] [PubMed] [Google Scholar]
- Togashi K, Hasegawa M, Nagai J, Tonouchi A, Masukawa D, Hensley K, Goshima Y, and Ohshima T (2019). Genetic suppression of collapsing response mediator protein 2 phosphorylation improves outcome in methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s model mice. Genes Cells 24, 31–40. [DOI] [PubMed] [Google Scholar]
- Tristán-Clavijo E, Camacho-Garcia RJ, Robles-Lanuza E, Ruiz A, van der Zee J, Van Broeckhoven C, Hernandez I, Martinez-Mir A, and Scholl FG (2015). A truncating mutation in Alzheimer’s disease inactivates neuroligin-1 synaptic function. Neurobiol. Aging 36, 3171–3175. [DOI] [PubMed] [Google Scholar]
- Wang LH, and Strittmatter SM (1996). A family of rat CRMP genes is differentially expressed in the nervous system. J. Neurosci. 16, 6197–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Morishita WK, Riley AM, Hale WD, Sudhof TC, and Malenka RC (2019). Neuroligin-1 Signaling Controls LTP and NMDA Receptors by Distinct Molecular Pathways. Neuron 102, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, and Penzes P (2007). Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56, 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JN, Michel U, Lenz C, Friedel CC, Köster S, d’Hedouville Z, Tönges L, Urlaub H, Böhr M, Lingor P, and Koch JC (2016). Calpainmediated cleavage of collapsin response mediator protein-2 drives acute axonal degeneration. Sci. Rep. 6, 37050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.