

Abstract
N-heterocyclic carbenes (NHCs) have emerged as catalysts for the construction of C–C bonds in the synthesis of substituted ketones under single-electron processes. Despite these recent reports, there still remains a need to increase the utility and practicality of these reactions by exploring new radical coupling partners. Herein, we report the synthesis of γ-aryloxyketones via combined NHC/photoredox catalysis. In this reaction, an α-aryloxymethyl radical is generated via oxidation of an aryloxymethyl potassium trifluoroborate salt, which is then added into styrene derivatives to provide a stabilized benzylic radical. Subsequent radical-radical coupling reaction with an azolium radical affords the γ-aryloxy ketone products.
Keywords: N-heterocyclic carbene, acyl azolium, photochemistry, potassium trifluoroborate, radical relay
Graphical Abstract

N-Heterocyclic carbenes (NHCs) have traditionally been used as organocatalysts to achieve umpolung (polarity reversal) reactivity in two-electron processes.[1] The majority of these reactions involve the Lewis basic NHC adding into an aldehyde to generate a Breslow intermediate, which can then act as a nucleophile to react with various sp2-derived electrophiles (e.g., carbonyl groups, imines, conjugate acceptors).[2] More recently, there has been an increase in reports that utilize NHCs as single-electron operators for radical-radical coupling reactions.[3] These NHC-mediated radical reactions are typically based on a single-electron oxidation of the electron-rich Breslow intermediate to yield an azolium radical species, which could undergo radical-radical coupling with numerous radical sources.[4] However, the opposite pathway involving single-electron reduction of an acyl azolium species remains underexplored.[5]
In 2020, we reported a single-electron reduction of an acyl azolium intermediate derived from activated carboxylic acids.[6] The resulting azolium radical species could then undergo radical-radical coupling with an oxidatively generated radical derived from alkyl and benzyl Hantzsch esters to access ketones (Figure 1A). This report highlighted new radical-based NHC reactivity because, a) it allowed for the direct alkylation of the azolium intermediate through an alternative method, and b) included the ability to incorporate aliphatic acid substrates. We have expanded this radical coupling platform[7] to include bis-catecholato silicates,[8] and developed an alkoxycarbonylation reaction utilizing potassium trifluoroborate salts (Figure 1B).[9] Potassium alkyltrifluoroborates offer several attractive features over other oxidative radical precursors including bench stability, chromatography-free synthesis, and high atom economy. [10]
Figure 1.

Previous examples of combined NHC/photoredox processes.
Studer and coworkers have expanded this reductive alkylation NHC catalysis further with their ability to perform a three-component radical relay with acyl fluorides, olefins, and trifluoromethyl radicals (Figure 1C).[11] This work, along with our reports, served as inspiration to develop a complimentary three-component reaction that utilizes oxidatively generated radicals not yet examined in azolium reductive alkylations. We sought to develop this three-component NHC/photoredox catalyzed reaction by surveying aryloxymethyl radicals derived from aryloxymethyltrifluoroborates (Figure 1D).[12] These starting materials are readily accessible and produce a radical that is stabilized by electron donation from the oxygen atom.[13] This 3-component reaction has the ability to generate a 1,4-dioxygenation pattern. While 1,3-[14] and 1,5-dioxygenation[15] patterns are quite common due to their traditional bond disconnection approach, the 1,4-dioxygenation pattern is generally obtained through Umpolung reactivity via a Stetter reaction.[16] Herein we report the synthesis of γ-aryloxy ketones through combined NHC/photoredox catalysis.
To commence our investigation, we surveyed various acyl azolium precursors, including acyl imidazoles and acyl fluorides, and multiple photocatalysts. The well-established NHC precursor Az-1 was chosen as the azolium catalyst while cesium carbonate was selected as the base to be analyzed.[7, 11] While the desired product 4 was observed using both acyl imidazoles and acyl fluorides, we opted to pursue conditions using acyl imidazole 1a due to their ease of preparation and handling.[17] These initial hit conditions provided the desired product in 29% yield using acyl imidazole 1a, aryloxymethyl trifluoroborate salt 2a, styrene (3a) as the olefin, cesium carbonate as the base, NHC precursor Az-1, and the organophotocatalyst 4-CzIPN (PC-1) (Table 1, Entry 1). A brief screen of NHC precursors confirmed that Az-1 was the best azolium catalyst to efficiently afford the γ-aryloxy ketone 4 (Entries 1 and 2, see Supporting Information for more details). It was also found that CsOAc slightly increased the yield compared to other inorganic and organic bases (Entries 2–4). Tuning the electronics of the aryloxymethyl trifluoroborate salt to a more electron rich substrate 2a as well as making styrene the limiting reagent provided a marked increase in yield (Entries 5–8). Finally, reducing the overall concentration of the reaction provided the product in 90% isolated yield (Entry 9). Interestingly, substituting benzoyl fluoride for the acyl donor under the optimized conditions resulted in trace product detected (Entry 10).
Table 1.
Optimization of reaction conditions.

| |||||
|---|---|---|---|---|---|
| entry | Az | Base | [M] | 2 | Yield (%)a,b |
| 1 | 1 | Cs2CO3 | 0.1 | 2a | 29 |
| 2 | 2 | Cs2CO3 | 0.1 | 2a | 20 |
| 3 | 1 | CsOAc | 0.1 | 2a | 32 |
| 4 | 1 | K2CO3 | 0.1 | 2a | 22 |
| 5 | 1 | CsOAc | 0.1 | 2b | 48c |
| 6 | 1 | CsOAc | 0.05 | 2b | 48c |
| 7 | 1 | CsOAc | 0.1 | 2b | 68d |
| 8 | 1 | CsOAc | 0.05 | 2b | 97 (90)d |
| 9 | 1 | CsOAc | 0.05 | 2b | tracee |
| 10 | 1 | CsOAc | 0.05 | 2b | 0f |
| 11 | - | CsOAc | 0.05 | 2b | (30) |
| 12 | 1 | CsOAc | 0.05 | 2b | 0g |
| |||||
Reaction conditions unless otherwise stated: 1a (0.3 mmol), 2 (0.1 mmol), Az (0.015 mmol), base (0.2 mmol), styrene 3a (0.3 mmol), PC (5 μmol), and solvent (0.1 M) for 24 h.
1H NMR yield using 1,3,5-trimethoxybenzene as internal standard (0.33 equiv.) and yield of isolated product is given in parenthesis.
1a (0.3 mmol), 2 (0.2 mmol), 3a (0.1 mmol).
1a (0.3 mmol), 2 (0.5 mmol), 3a (0.1 mmol).
same conditions as [d] except benzoyl fluoride used as acyl starting material (0.3 mmol).
absence of PC-1.
no light.
With our optimized conditions in hand, we first examined the scope of the reaction by altering the substitution on the trifluoroborate (Table 2A). Electronic effects proved to be crucial in this reaction, as the methoxy-derived aryloxymethyl trifluoroborate was uncovered to be the best borate salt (in terms of product yield) while the methyl substituted and neutral phenol derivates only provided product in moderate yields (4b,4c). The bulkier and more electron-rich naphthlene derived borate salt also afforded the desired product 4d in 63% yield. Interestingly, various halogenated aryloxymethyl trifluoroborates were well-tolerated in this reaction in greater than 60% yield (4e–4g). As expected, the electron-withdrawing trifluoromethyl-substituted aryloxymethyl salt produced the product 4h in only 14% isolated yield. Replacing the trifluoromethyl group with an ester group (–CO2Et) failed to afford the respective product, presumably due to the increased oxidation potential of the substrate.
Table 2.

|
See the Supporting Information for rection details.
Yield of isolated product.
PMP = para-methoxy phenyl.
Next, we explored the reaction scope by modifying the substitution on the acyl imidazole (Table 2B). A variety of electron-donating and electron-withdrawing substituents were tolerated on the acyl imidazole. The methoxy-substituted aryl acyl imidazole provided ketone 4i in 66% yield. Methyl groups at the meta- and para-position of the aryl acyl imidazole afforded the desired products 4j-4k in good yield. Changing the substitution to a more electron-withdrawing cyano group also allowed formation of the γ-aryloxyketone 4l. However, the halogenated acyl imidazoles only delivered 4m and 4n in 22% and 38% yield, respectively. While aliphatic substrates have been used in direct coupling reactions via NHC/photoredox cooperative catalysis, they have not yet been employed in a 3-component radical-relay NHC/photoredox catalyzed process.[7, 18] Much to our delight, these reaction conditions were able to tolerate various aliphatic derived acyl imidazoles to generate the ketone products 4o-4q in low-to-good yields.
Lastly, we wanted to examine the scope with respect to the alkene (Table 2C). An electron-rich para-methoxy styrene was able to provide the desired ketone 4s while using 2b as the trifluoroborate in 50% yield. Gratifyingly, we were also able to use the electron-rich methoxy styrene while changing substitution on the trifluoroborate to afford a series of ketones 4t-4w in low-to-moderate yields. Use of meta-methyl styrene generated the ketones with the para-methoxy trifluoroborate salt as well as the napthyl-derived trifluoroborate salt in 67% and 55% yield, respectively. Surprisingly, α-methyl-substituted styrene and more sterically hindered 1,1-diphenylethylene afforded the corresponding ketones 4z-4ab in good yield, showcasing the ability of this methodology to generate quaternary centers. Unfortunately, when the olefins cyclohexene and ethyl vinyl ether were subjected to our reaction conditions, no desired product was observed.
To demonstrate the utility of our products, we hypothesized that the resulting para-methoxy phenyl (PMP) group could be removed to provide γ-hydroxyketones. Thus, exposure of 4a to oxidative conditions using cerium ammonium nitrate (CAN) afforded the 2,3-dihydrofuran 5 in 58% yield (Scheme 1).[19] This heterocyclic motif is embedded in many important natural products as well as medicinally relevant molecules and is considered a useful intermediate for other synthetic transformations.[20] Additionally, employing sodium borohydride as the reducing agent afforded alcohol 6 presumably favoring the Felkin isomer with 11:1 dr and in 96% yield.
Scheme 1.
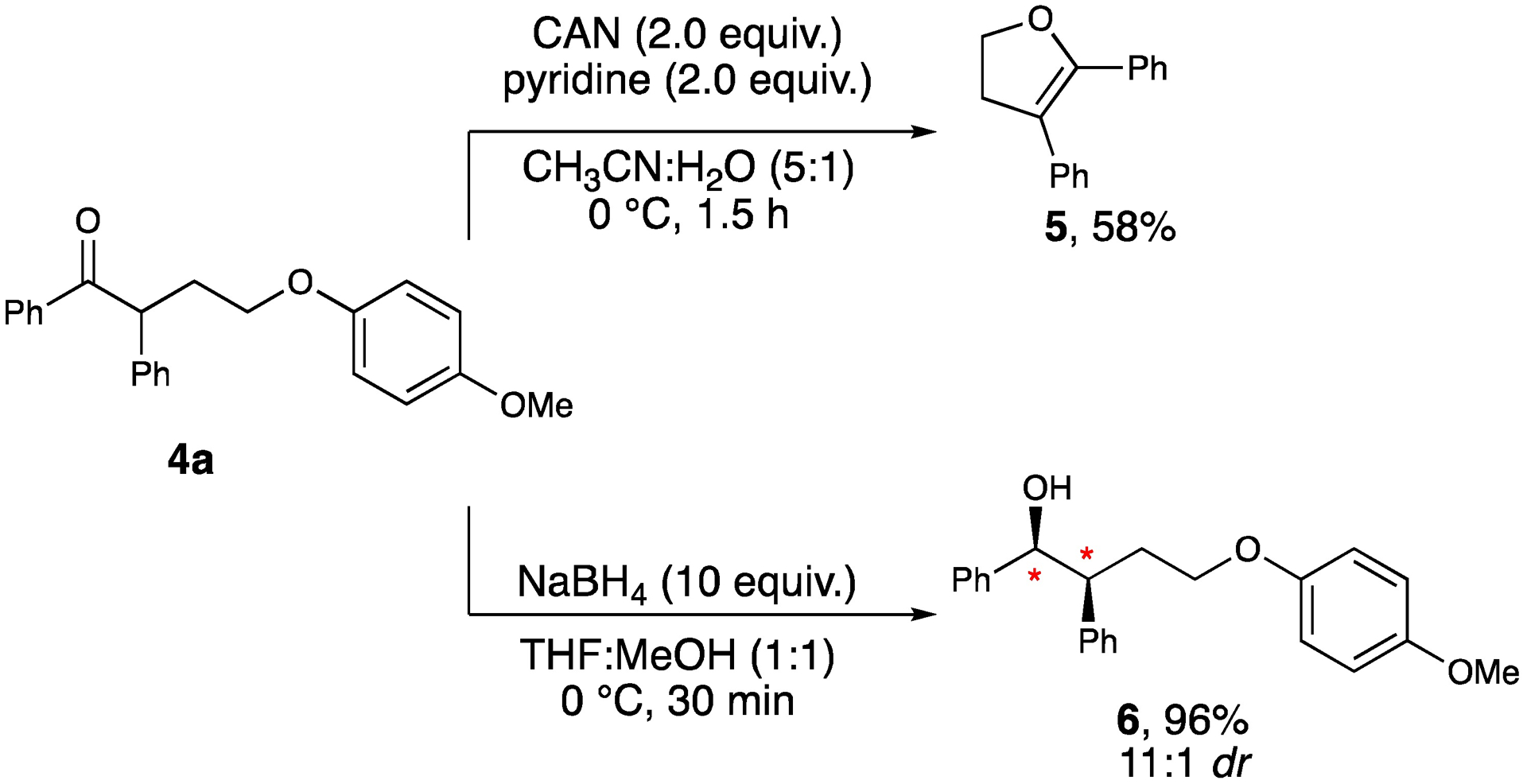
Synthetic transformations with 4a.
To probe the reaction pathway of the overall process, control reactions and radical trapping experiments were performed. No product was observed in the absence of photocatalyst or light suggesting that this reaction is a photocatalyzed process (Table 1, Entries 10 and 12, respectively). Additionally, the reaction did not proceed in the presence of TEMPO as a radical trap. Instead, the aryloxymethyl TEMPO adduct was observed, indicating that a radical process is likely occurring (Scheme 2A). However, it is important to note that while product was detected in the absence of Az-1, it was in much lower quantity, highlighting the importance of the azolium catalyst in this reaction for overall efficiency (Table 1, Entry 11).[5e] The importance of the azolium can also be displayed in Scheme 2B. Isolated azolium species 7 was synthesized and our optimized conditions were employed. Under these conditions, 4a was only isolated in 25% yield. While 7 may be a possible intermediate of the reaction, we attribute the decrease in yield due to the steric and electronic effects of the NHC. Based on these results and other combined NHC/photoredox catalysis reports, we propose the following catalytic cycle (Scheme 2C).[7, 11] The excited state of the photocatalyst (4-CzIPN*) can oxidize the trifluoroborate salt 2 with exposure to LED irradiation. Subsequent fragmentation affords the aryloxymethyl radical I and the reduced photocatalyst.[21] The addition of I into styrene generates a stable benzyl radical intermediate II. Single-electron reduction of the acyl azolium III provides the stabilized azolium radical IV while regenerating the ground-state photocatalyst (4-CzIPN).[22] Based on our previous mechanistic investigations, the steric hindrance and electronic effects from the NHC mesityl group likely guides the final radical-radical coupling between IV and V to afford the desired γ-aryloxy ketone product V.[7]
Scheme 2.

Mechanistic studies and proposed mechanism.
In summary, we have developed a three-component NHC/photoredox catalyzed reaction to synthesize γ-aryloxy ketones. This transformation builds on the emerging area of single-electron carbene catalysis by incorporating oxidatively generated aryloxymethyl radicals. A wide variety of γ-aryloxy ketones can be accessed rapidly with good functional group compatibility as well as use highly substituted styrenes to generate quaternary centers adjacent to the carbonyl group. The key-step in this reaction is the reduction of an acyl azolium to yield a stabilized azolium radical, which can then undergo radical-radical coupling with the aryloxymethyl radical. The utility of these products was showcased in the ability to remove the PMP group to afford 2,3-dihydrofurans. Current studies in our laboratory involve exploring new oxidatively generated radical species with NHC/photoredox chemistry and their applications in synthesis.
Experimental Section
General information
All reactions were carried out under an argon or nitrogen atmosphere in oven-dried glassware with magnetic stirring. All solvents were purified by passing through a bed of activated alumina, dried over 3Å molecular sieves, and then degassed using freeze-pump-thaw method (3–4 cycles). Purification of reaction products was carried out by flash chromatography on Biotage Isolera 4 systems with Ultra-grade silica cartridges. Analytical thin layer chromatography was performed on EM Reagent 0.25 mm silica gel 60-F plates. Visualization was accomplished with UV light. 1H NMR spectra were recorded on AVANCE III 500 MHz w/ direct cryoprobe (500 MHz) spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm). Data are reported as (s = singlet, d = doublet, t = apparent triplet, q = quartet, m = multiplet, b = broad; coupling constant(s) in Hz; integration.) Proton-decoupled 13C NMR spectra were recorded on an AVANCE III 500 MHz w/ direct cryoprobe (126 MHz) spectrometer and are reported in ppm using solvent as an internal standard (CDCl3 at 77.16 ppm). Mass spectra were obtained on a WATERS Acquity-H UPLC-MS with a single quad detector (ESI) or an Agilent 7890 gas chromatograph equipped with a 5975C single quadrupole EI-MS. High-resolution mass spectrometry (HRMS) was obtained using an Agilent 6201 MSLC-TOF (ESI). All photocatalytic reactions were carried out in a SynLED Parallel Photoreactor (465–470 nm) purchased from Sigma-Aldrich. [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (IrdF) was purchased from Strem Chemicals and used without purification. 4-CzIPN was prepared according to the literature procedure. The spectra of 10 known prepared BF3K substrates (2a-2k) were matched with the literatures data.
General synthetic procedure for the synthesis of acyl imidazoles (1)
The appropriate acid (10 mmol, 1.0 equiv) was dissolved in dry dichloromethane (0.3 M), and CDI (carbonyldiimidazole, 15 mmol, 1.5 equiv) was added slowly (caution, exothermic). The resulting mixture was stirred overnight at room temperature. Upon completion, the solution was transferred to a separatory funnel and washed with deionized water (2 × 25 mL), and then the organic layer was dried over Na2SO4. Concentration under reduced pressure afforded the acyl imidazole, which was used in the following reaction without further purification.
General synthetic procedure for BF3K salts (2)
A solution of potassium tert-butoxide (3.0 equiv) in anhydrous THF at 0 °C under N2 was treated with phenol (3.0 equiv). The reaction mixture was stirred at 0 °C for 15 min and then was allowed to warm to room temperature over 30 min. Potassium bromomehtyltrifluoroborate (1.0 equiv) was added in one batch at 0 °C. The resulting reaction mixture was stirred at room temperature for 23 h and 4.5 M KHF2 (2.1 equiv) solution was added. The reaction mixture was stirred at room temperature for 30 min, and the white suspension was added with benzene/toluene (2–3 times). The solvent was removed, and the resulting solid was washed with Et2O followed by hot acetone to yield the pure BF3K salts.
General synthetic procedure for light-driven carbene-catalyzed reactions
All reactions were set up inside a glovebox under N2 atm. To an oven-dried 2-diam vial containing a stir bar was added the respective BF3K salts (2, 5.0 equiv), respective styrene (3, 1.0 equiv), respective acyl imidazole (1, 3.0 equiv), CsOAc (2.0 equiv), Az-1 (15 mol%) and PC-1 (5 mol%). Acetonitrile (0.05 M) was added, and the reaction vial was capped and taken out of the glovebox. Parafilm was wrapped around the cap to prevent air from entering. The vials were placed in a SynLED Parallel Photoreactor (blue LEDs) and the reaction mixture was stirred for 24 h. The reaction progress was monitored by GCMS or LCMS. After the reaction was deemed complete, the reactions were diluted with ethyl acetate and passed through a Celite pad. The yellow filtrate was collected and concentrated. The resulting residue was purified by column chromatography (5%–10% ethyl acetate in hexanes) on silica gel.
Supplementary Material
Acknowledgements
We thank Northwestern University and the National Institute of General Medical Sciences (R35GM136440) for support of this work. The authors thank Ms. Ada Kwong (NU) for assistance with HRMS.
References
- [1].a) Bugaut X, Glorius F, Chem. Soc. Rev 2012, 41, 3511–3522; [DOI] [PubMed] [Google Scholar]; b) Hopkinson MN, Richter C, Schedler M, Glorius F, Nature 2014, 510, 485–496; [DOI] [PubMed] [Google Scholar]; c) Flanigan DM, Romanov-Michailidis F, White NA, Rovis T, Chem. Rev 2015, 115, 9307–9387; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bellotti P, Koy M, Hopkinson MN, Glorius F, Nat. Rev. Chem 2021, 5, 711–725. [DOI] [PubMed] [Google Scholar]
- [2].a) Ukai T, Tanaka R, Dokawa T, J. Pharm. Soc. Jpn 1943, 63, 296–300; [Google Scholar]; b) Breslow R, J. Am. Chem. Soc 1958, 80, 3719–3726; [Google Scholar]; c) Pareek M, Reddi Y, Sunoj RB, Chem. Sci 2021, 12, 7973–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) White NA, Rovis T, J. Am. Chem. Soc 2014, 136, 14674; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Y, Du Y, Huang Z, Xu J, Wu X, Wang Y, Wang M, Yang S, Webster RD, Chi YR, J. Am. Chem. Soc 2015, 137, 2416–2419; [DOI] [PubMed] [Google Scholar]; c) Ishii T, Nagao K, Ohmiya H, Chem. Sci 2020, 11, 5630–5636; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu J, Xing X-N, Huang J-H, Lu L-Q, Xiao W-J, Chem. Sci 2020, 11, 10605–10613; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dai L, Ye S, Chin. Chem. Lett 2020;; f) Mavroskoufis A, Jakob M, Hopkinson MN, ChemPhotoChem 2020, 4, 5147–5153; [Google Scholar]; g) Ohmiya H, ACS Catal 2020, 10, 6862–6869; [Google Scholar]; h) Li Q-Z, Zeng R, Han B, Li J-L, Chemistry – A European Journal 2021, 27, 3238–3250. [DOI] [PubMed] [Google Scholar]
- [4].a) Chen XY, Chen KQ, Sun DQ, Ye S, Chem. Sci 2017, 8, 1936–1941; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishii T, Kakeno Y, Nagao K, Ohmiya H, J. Am. Chem. Soc 2019, 141, 3854–3858; [DOI] [PubMed] [Google Scholar]; c) Ishii T, Ota K, Nagao K, Ohmiya H, J. Am. Chem. Soc 2019, 141, 14073–14077; [DOI] [PubMed] [Google Scholar]; d) Li J-L, Liu Y-Q, Zou W-L, Zeng R, Zhang X, Liu Y, Han B, He Y, Leng H-J, Li Q-Z, Angew. Chem. Int. Ed 2019, 58, 2–10; [Google Scholar]; e) Kim I, Im H, Lee H, Hong S, Chem. Sci 2020, 11, 3192–3197; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liu M-S, Shu W, ACS Catal 2020, 10, 12960–12966; [Google Scholar]; g) Ota K, Nagao K, Ohmiya H, Org. Lett 2020, 22, 3922–3925; [DOI] [PubMed] [Google Scholar]; h) Kusakabe M, Nagao K, Ohmiya H, Org. Lett 2021, 23, 7242–7247; [DOI] [PubMed] [Google Scholar]; i) Matsuki Y, Ohnishi N, Kakeno Y, Takemoto S, Ishii T, Nagao K, Ohmiya H, Nat. Commun 2021, 12, 3848; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Liu M-S, Min L, Chen B-H, Shu W, ACS Catal 2021, 11, 9715–9721; [Google Scholar]; k) Gao Y, Quan Y, Li Z, Gao L, Zhang Z, Zou X, Yan R, Qu Y, Guo K, Org. Lett 2021, 23, 183–189; [DOI] [PubMed] [Google Scholar]; l) Yang H-B, Wan D-H, Org. Lett 2021, 23, 1049–1053; [DOI] [PubMed] [Google Scholar]; m) Chen L, Jin S, Gao J, Liu T, Shao Y, Feng J, Wang K, Lu T, Du D, Org. Lett 2021, 23, 394–399. [DOI] [PubMed] [Google Scholar]
- [5].a) Mavroskoufis A, Rajes K, Golz P, Agrawal A, Ruß V, Götze JP, Hopkinson MN, Angew. Chem. Int. Ed 2020, 59, 3190–3194; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu K, Studer A, J. Am. Chem. Soc 2021, 143, 4903–4909; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng Q-Y, Lezius L, Studer A, Nat. Commun 2021, 12, 2068; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ren S-C, Lv W-X, Yang X, Yan J-L, Xu J, Wang F-X, Hao L, Chai H, Jin Z, Chi YR, ACS Catal 2021, 2925–2934; [Google Scholar]; e) Sato Y, Goto Y, Nakamura K, Miyamoto Y, Sumida Y, Ohmiya H, ACS Catal 2021, 12886–12892; [Google Scholar]; f) Mavroskoufis A, Rieck A, Hopkinson MN, Tetrahedron 2021, 132497; [Google Scholar]; g) Zuo Z, Daniliuc CG, Studer A, Angew. Chem. Int. Ed 2021, 60, 25252–25257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Bay AV, Fitzpatrick KP, Betori RC, Scheidt KA, Angew. Chem. Int. Ed 2020, 59, 9143–9148; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bayly AA, McDonald BR, Mrksich M, Scheidt KA, Proc. Natl. Acad. Sci. U. S. A 2020, 117, 13261–13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bay AV, Fitzpatrick KP, González-Montiel GA, Farah AO, Cheong PH-Y, Scheidt KA, Angew. Chem. Int. Ed 2021, 60, 17925–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Corce V, Chamoreau LM, Derat E, Goddard JP, Ollivier C, Fensterbank L, Angew. Chem. Int. Ed 2015, 54, 11414–11418; [DOI] [PubMed] [Google Scholar]; b) Jouffroy M, Primer DN, Molander GA, J. Am. Chem. Soc 2016, 138, 475–478; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Goddard J-P, Ollivier C, Fensterbank L, Acc. Chem. Res 2016, 49, 1924–1936. [DOI] [PubMed] [Google Scholar]
- [9].Zhu JL, Scheidt KA, Tetrahedron 2021, 92, 132288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Molander GA, Ellis N, Acc. Chem. Res 2007, 40, 275–286; [DOI] [PubMed] [Google Scholar]; b) Yasu Y, Koike T, Akita M, Adv. Synth. Catal 2012, 354, 3414–3420; [Google Scholar]; c) Molander GA, J. Org. Chem 2015, 80, 7837–7848; [DOI] [PubMed] [Google Scholar]; d) Amani J, Sodagar E, Molander GA, Org. Lett 2016, 18, 732–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meng Q-Y, Döben N, Studer A, Angew. Chem. Int. Ed 2020, 59, 19956–19960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]. At the time of preparing this manuscript, Ohmiya and coworkers published a direct-coupling and three-component radical relay utilizing acyl imidazoles and alkylborates. See reference 5f.
- [13].Karakaya I, Primer DN, Molander GA, Org. Lett 2015, 17, 3294–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matsuo J.-i., Murakami M, Angew. Chem. Int. Ed 2013, 52, 9109–9118. [DOI] [PubMed] [Google Scholar]
- [15].a) Michael A, Journal für Praktische Chemie 1887, 35, 349–356; [Google Scholar]; b) Michael A, Journal für Praktische Chemie 1894, 49, 20–25; [Google Scholar]; c) Tokoroyama T, Eur. J. Org. Chem 2010, 2010, 2009–2016. [Google Scholar]
- [16].a) Heravi MM, Zadsirjan V, Kafshdarzadeh K, Amiri Z, Asian J Org. Chem 2020, 9, 1999–2034; [Google Scholar]; b) Kieslich D, Christoffers J, Synthesis 2021, 53, 3485–3496. [Google Scholar]
- [17].Lee A, Scheidt KA, Chem. Commun 2015, 51, 3407–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kakeno Y, Kusakabe M, Nagao K, Ohmiya H, ACS Catal 2020, 10, 8524–8529. [Google Scholar]
- [19].Masaki Y, Yoshizawa K, Itoh A, Tetrahedron Lett 1996, 37, 9321–9324. [Google Scholar]
- [20].a) Friedrichsen W, in Comprehensive Heterocyclic Chemistry II (Eds.: Katritzky AR, Rees CW, Scriven EFV), Pergamon, Oxford, 1996, pp. 351–393; [Google Scholar]; b) Faul MM, Huff BE, Chem. Rev 2000, 100, 2407–2474; [DOI] [PubMed] [Google Scholar]; c) Hou X-L, Yang Z, Yeung K-S, Wong HNC, in Prog. Heterocycl. Chem, Vol. 17 (Eds.: Gribble GW, Joule JA), Elsevier, 2005, pp. 142–171; [Google Scholar]; d) Kilroy TG, O’Sullivan TP, Guiry PJ, Eur. J. Org. Chem 2005, 2005, 4929–4949. [Google Scholar]
- [21].Miyazawa K, Yasu Y, Koike T, Akita M, Chem. Commun 2013, 49, 7249–7251. [DOI] [PubMed] [Google Scholar]
- [22].a) Leifert D, Studer A, Angew. Chem. Int. Ed 2020, 59, 74–108; [DOI] [PubMed] [Google Scholar]; b) Delfau L, Nichilo S, Molton F, Broggi J, Tomás-Mendivil E, Martin D, Angew. Chem. Int. Ed 2021, 60, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.