

Abstract
On December 16, 2020, the FDA granted regular approval to margetuximab-cmkb (MARGENZA), in combination with chemotherapy, for the treatment of adult patients with HER2-positive (HER2+) metastatic breast cancer (MBC) who have received two or more prior anti-HER2 regimens, at least one of which was for metastatic disease. Approval was based on data from SOPHIA, a multicenter, randomized, open-label, active controlled study comparing margetuximab to trastuzumab, in combination with chemotherapy. The primary efficacy endpoint was progression-free survival (PFS) by blinded independent central review (BICR). SOPHIA demonstrated a 0.9-month difference in median PFS between the two treatment arms (5.8 months vs 4.9 months, respectively; stratified hazard ratio (HR) 0.76 [95% CI: 0.59, 0.98; p=0.0334]). Overall survival (OS) was immature at data cut off (DCO) date of September 10, 2019. Infusion related reactions (IRRs) are an important safety signal associated with margetuximab plus chemotherapy. In SOPHIA, 13% of patients treated with margetuximab plus chemotherapy reported IRRs of which 1.5% was grade 3. The most commonly reported adverse drug reactions (> 10%) with margetuximab in combination with chemotherapy were fatigue/asthenia, nausea, diarrhea, vomiting, constipation, headache, pyrexia, alopecia, abdominal pain, peripheral neuropathy, arthralgia/myalgia, cough, decreased appetite, dyspnea, IRR, palmar-plantar erythrodysesthesia, and extremity pain. Overall, the favorable risk-benefit profile for margetuximab when added to chemotherapy supported its approval for the intended indication.
Introduction
Breast cancer is the most commonly diagnosed cancer and the leading cause of cancer death in women (1). In the United States (US), more than 270,000 cases are diagnosed annually, with approximately 42,000 deaths (2). Human epidermal growth factor receptor 2 (HER2) is expressed in approximately 15–20% of all breast cancers. HER2-positive (HER2+) disease is associated with a younger patient population and more aggressive tumor biology (3). The 5-year relative survival rate of HER2+ metastatic breast cancer (MBC) ranges from 37 to 44%, depending on clinic-pathologic factors such as hormone-receptor (HR) status (4). In the US, the standard of care for first-line HER2+ MBC is the combination of trastuzumab, pertuzumab, and docetaxel based on trial results from CLEOPATRA (5). Currently, the preferred second-line option is T-DM1 based on EMILIA (6). Until recently, treatment options beyond the second-line were limited and included lapatinib plus capecitabine or trastuzumab combined with single-agent chemotherapy (7, 8). New therapies have become available with the approvals of tucatinib in combination with trastuzumab and capecitabine (9,10), neratinib in combination with capecitabine (11,12), and accelerated approval of fam-trastuzumab deruxtecan (13,14). Although treatment with anti-HER2 therapies improved disease outcomes, HER2+ MBC remains incurable (8). This article summarizes the data and FDA’s review leading to the approval of margetuximab.
Chemistry, Manufacturing, and Control
Margetuximab (MGAH22) is a human/mouse chimeric Fc-engineered IgG1 kappa monoclonal antibody generated in Chinese hamster ovary cells and purified by standard bioprocessing procedures. The variable domains of margetuximab are derived from the same murine precursor as trastuzumab (4D5) to bind to extracellular domain IV of HER2 (15). Margetuximab differs from trastuzumab in that its Fc region has been engineered to increase antibody-dependent cellular cytotoxicity and immune function via five amino acid substitutions designed to mediate increased binding to both allotypes (158F or V) of the activating FcγRIIIa receptor (CD16A) and decreased binding to the inhibitory FcγRIIb receptor (CD32B). Margetuximab was licensed with several manufacturing-related post-marketing commitments (PMCs) pertinent to drug manufacture and control including optimization of the potency assays for IC50 and EC50, drug product (DP) leachables and DP shipping validation studies (15).
Nonclinical Pharmacology and Toxicology
Mechanism of Action
Margetuximab binds to HER2 overexpressed on the tumor cell surface and (a) blocks downstream signaling and shedding of HER2 extracellular domain; (b) inhibits tumor cell proliferation; and (c) mediates tumor cell lysis by engaging immune effector cells (16, 17).
Nonclinical Toxicology
Toxicological assessments with 6- and 13-week repeat-dose studies were conducted in cynomolgus monkeys to evaluate the toxicity of margetuximab. No target organs for margetuximab toxicity were identified nor was an increased release of IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-10, or TNF-α in serum. Compared to vehicle controls, margetuximab treatment resulted in partially reversible decreases in blood CD3-/CD159a+ and CD16+/CD159a+ NK cells. Margetuximab had no effect on male and female reproductive organs in sexually mature cynomolgus monkeys. In an enhanced pre- and postnatal development toxicology study in pregnant cynomolgus monkeys, treatment with margetuximab starting on gestation day 20 until delivery resulted in oligohydramnios, increased incidence of abortion/stillbirth primarily due to hypersensitivity reactions, placental transfer of margetuximab, and delayed kidney development in infants. Oligohydramnios and associated renal toxicity are class effects of HER2-directed antibodies. Based on these data and postmarketing reports of oligohydramnios following use of a HER2-directed antibody during pregnancy, margetuximab can cause fetal harm when administered to a pregnant woman and a Boxed Warning for embryo-fetal toxicity is included in the label.
Clinical Pharmacology
The Clinical Pharmacology review evaluated the acceptability of the proposed dosing regimen, and effect of Fcγ receptor polymorphisms on the response to margetuximab.
The proposed dosing regimen of margetuximab is 15 mg/kg every 3 weeks (Q3W), and is supported by the observed efficacy and safety in SOPHIA, plus supportive pharmacokinetics (PK) and dose-response relationships from two margetuximab studies: CP-MGAH22–01, a Phase 1 dose escalation/expansion in HER2+ carcinoma with no standard therapy and CP-MGAH22–02, a Phase 2 single-arm study in HER2 low expressing relapsed or refractory advanced breast cancer. Exposure-response analyses with margetuximab safety and efficacy are limited to data from patients treated at a single dose level of 15 mg/kg Q3W in SOPHIA. No meaningful exposure-response relationships were identified with key safety endpoints over the exposure range achieved at this dosage, but a statistically significant trend towards longer progression free survival (PFS) with increasing margetuximab exposure was observed. Recommended dosing is based on body weight. No other dose modifications are recommended for intrinsic (e.g., age, sex, race, etc.) or extrinsic factors (e.g., number of prior lines of therapy, concurrent chemotherapy). There is limited or no information regarding the impact of severe renal and moderate or severe hepatic impairment on the safety and PK of margetuximab. However, margetuximab PK is not expected to be markedly altered in these populations. No formal drug‐drug interaction studies have been conducted with margetuximab. No CYP enzyme or transporter-mediated drug drug interactions (DDIs) are expected.
Margetuximab is administered as an IV infusion over 120 minutes for the initial dose, then over a minimum of 30 minutes for all subsequent doses. Safety, tolerability and PK in a non-randomized infusion sub-study in SOPHIA support the proposed 30 minute infusion duration in Cycle 2 and beyond. Modification of the infusion duration in Cycle 2 and beyond did not markedly impact exposure of margetuximab.
Population PK analysis showed no effect of Fc-gamma receptor (FCGR) genotype on margetuximab PK. To further characterize the clinical impact of margetuximab, agreed upon PMC will provide additional information pertinent to CD16A genotype and patient outcome from clinical trials that correlate CD16A genotype with trial efficacy endpoints. Another PMC issued by the FDA is to develop an adequately validated assay for neutralizing antibody and assess the neutralizing antibodies against margetuximab in SOPHIA given that the bioanalytical performance of the current neutralizing antibody assay is inadequate.
Clinical Trial Design:
The efficacy and safety of margetuximab was evaluated in a phase 3, randomized, open-label, study (CP-MGAH22–04, SOPHIA, NCT02492711) (18) which compared margetuximab to trastuzumab, each combined with chemotherapy, in patients with metastatic HER2+ BC previously treated with trastuzumab and pertuzumab, and progressing on or after the most recent therapy (Figure 1). Randomization (1:1) was stratified for chemotherapy (choice of capecitabine, eribulin, gemcitabine, or vinorelbine made prior to randomization), number of of lines of therapy in the metastatic setting, and number of metastatic sites. Treatment was given until disease progression or unacceptable toxicity. The dual primary efficacy endpoints of PFS by blinded independent review (BICR) and overall survival (OS) were analyzed sequentially. A total of three analyses of OS were planned (two interim analyses and one final analysis). Secondary endpoints included investigator-assessed PFS and confirmed objective response rate (ORR) by RECIST version 1.1.
Figure 1: Trial Schema of SOPHIA.
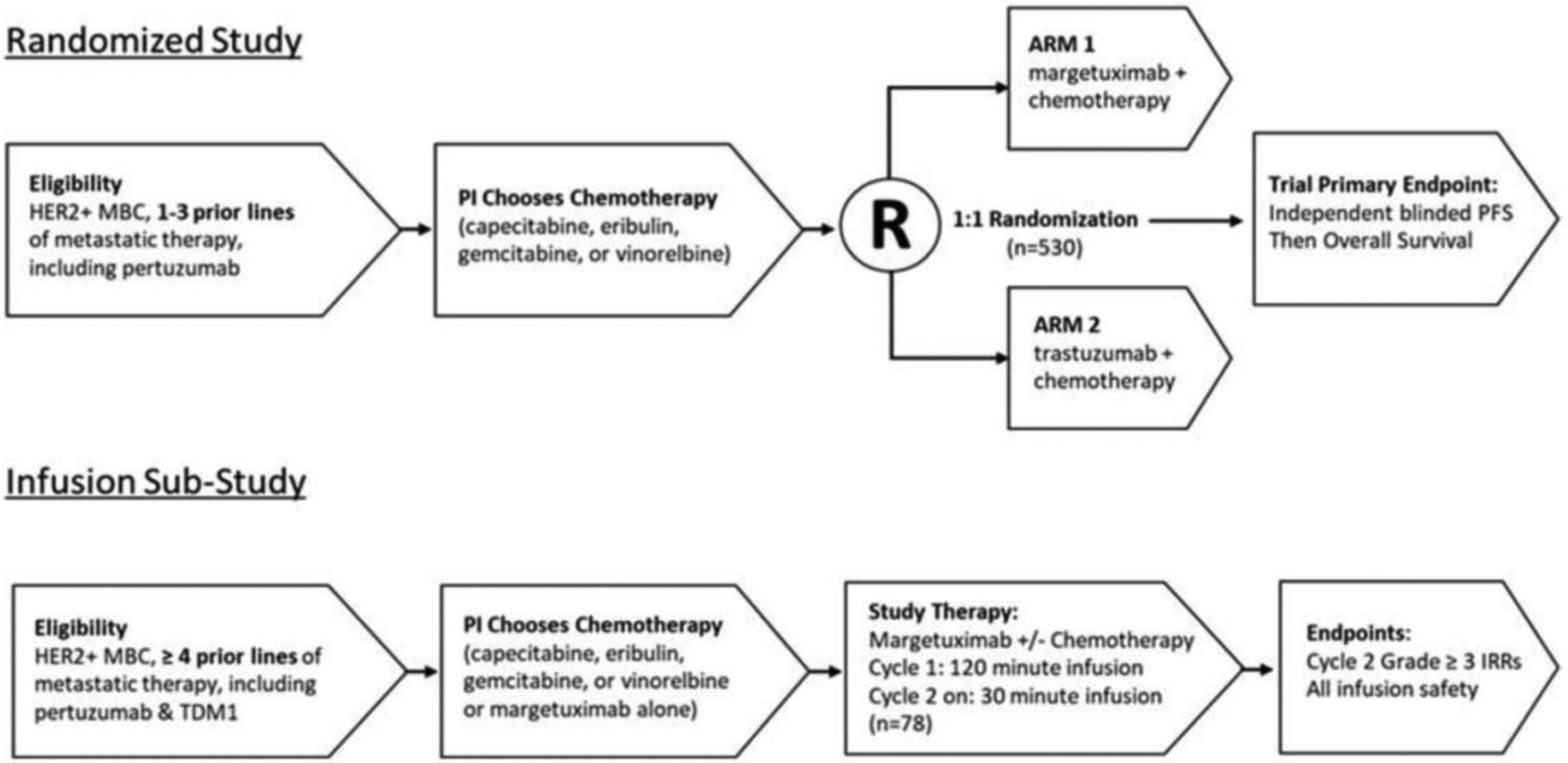
Abbreviations: HER2: human epidermal growth factor receptor 2; IRR: infusion-related reaction; MBC: metastatic breast cancer; PFS: progression-free survival; PI: principal investigator; T-DM1: ado-trastuzumab emtansine.
Source: Margetuximab Assessment Aid submitted to FDA (Study 04 – Schematic of Study Design)24
Results:
A total of 536 patients with pre-treated HER2+ MBC comprised the intent-to-treat (ITT) population. Patient demographics is shown in Table 1. Most variables were well-balanced across the arms with no imbalance likely to have affected the results. Male patients were also eligible although only 3 male patients were enrolled, all received trastuzumab plus chemotherapy. Other than under representation of racial/ethnic minorities (12.1%), the study population of SOPHIA was generally consistent with a U.S. population. Baseline disease characteristics are shown in Table 2. CD16A [CD16A-158F or V], CD32A [CD32A-131H or R] and CD32B [CD32B-232I or T] genotyping was evaluated retrospectively in 506 patients by a validated assay in a central lab (19). Treatment groups were balanced for genotype variants. All patients received prior trastuzumab, all but 1 patient had prior pertuzumab, and 91.2% of patients had prior T-DM1.
Table 1:
Summary of Demographics for SOPHIA (ITT population)
| Margetuximab plus Chemotherapy (N = 266) | Trastuzumab plus Chemotherapy (N = 270) | Total (N = 536) | |
|---|---|---|---|
| Sex, n (%) | |||
| Female | 266 (100.0) | 267 (98.9) | 533 (99.4) |
| Male | 0 | 3 (1.1) | 3 (0.6) |
| Age (years) | |||
| Median | 55 | 56 | 56 |
| Range | 29 – 83 | 27 – 86 | 27 – 86 |
| Race, n (%) | |||
| White | 205 (77.1) | 222 (82.2) | 427 (79.7) |
| Black or African American | 16 (6.0) | 12 (4.4) | 28 (5.2) |
| Asian | 20 (7.5) | 14 (5.2) | 34 (6.3) |
| American Indian or Alaska Native | 0 | 0 | 0 |
| Native Hawaiian or Pacific Islander | 1 (0.4) | 2 (0.7) | 3 (0.6) |
| Other | 24 (9.0) | 20 (7.4) | 44 (8.2) |
| ECOG | |||
| 0 | 149 (56.0) | 161 (59.6) | 310 (57.8) |
| 1 | 117 (44.0) | 109 (40.4) | 226 (42.2) |
Source: Margetuximab Assessment Aid submitted to FDA (Study 04 CSR - Table 14.1.5)24
Table 2:
Baseline Disease Characteristics for SOPHIA (ITT population)
| Margetuximab plus Chemotherapy (N = 266) | Trastuzumab plus Chemotherapy (N = 270) | Total (N = 536) | |
|---|---|---|---|
| Hormone Receptor Status, n (%) | |||
| Positive (ER+ &/or PgR+) | 164 (61.7) | 170 (63.0) | 334 (62.3) |
| Negative (ER- and PgR-) | 102 (38.3) | 98 (36.3) | 200 (37.3) |
| Unknown | 0 | 2 (0.7) | 2 (0.4) |
| HER2 Result * , n (%) | |||
| ISH amplified | 117 (44.0) | 128 (47.4) | 245 (45.7) |
| IHC3+ | 149 (56.0) | 142 (52.6) | 291 (54.3) |
| Prior Pertuzumab, n (%) | |||
| Yes | 266 (100) | 269 (99.6) | 535 (99.8) |
| No | 0 (0) | 1 (0.4) | 1 (0.2) |
| Disease Status at Study Entry, n (%) | |||
| Locally Advanced, Unresectable | 6 (2.3) | 6 (2.2) | 12 (2.2) |
| Metastatic | 260 (97.7) | 264 (97.8) | 524 (97.8) |
| Number of Metastatic Sites, n (%) | |||
| ≤ 2 | 138 (51.9) | 144 (53.3) | 282 (52.6) |
| > 2 | 128 (48.1) | 126 (46.7) | 254 (47.4) |
| Site(s) of Metastases at Study Entry, n (%) | |||
| Bone | 153 (57.5) | 155 (57.4) | 308 (57.5) |
| Lymph Node | 140 (52.6) | 151 (55.9) | 291 (54.3) |
| Lung | 124 (46.6) | 126 (46.7) | 250 (46.6) |
| Liver | 93 (35.0) | 95 (35.2) | 188 (35.1) |
| Breast | 44 (16.5) | 37 (13.7) | 81 (15.1) |
| Skin | 41 (15.4) | 32 (11.9) | 73 (13.6) |
| Brain | 37 (13.9) | 34 (12.6) | 71 (13.2) |
| Pleura | 22 (8.3) | 17 (6.3) | 39 (7.3) |
| Other Visceral | 21 (7.9) | 15 (5.6) | 36 (6.7) |
| Other non-Visceral | 15 (5.6) | 16 (5.9) | 31 (5.8) |
| Peritoneum | 9 (3.4) | 9 (3.3) | 18 (3.4) |
| Adrenal Medulla | 4 (1.5) | 2 (0.7) | 6 (1.1) |
| Ovary | 4 (1.5) | 0 | 4 (0.7) |
Four patients in each group did not have reference laboratory assessments but were confirmed HER2+ on central laboratory testing.
Source: Margetuximab Assessment Aid submitted to FDA (Study 04 CSR - Table 12 and 14.1.16)24
Efficacy:
Efficacy results are shown in Table 3. With data cut off date of October 10, 2018, SOPHIA met its primary endpoint of improved PFS by BICR, with a 0.9 months difference between the treatment arms favoring margetuximab plus chemotherapy (HR 0.76 [95% CI: 0.59, 0.98; p=0.0334]). Subgroup analyses were generally supportive of primary analysis results except for the subgroup of patients homozygous for the CD16A 158V allele (V/V) with a HR of 1.776 (95% CI : 0.871, 3.620).
Table 3:
Efficacy Results in SOPHIA (ITT Population)
| Efficacy Parameter | Margetuximab plus Chemotherapy (N = 266) | Trastuzumab plus Chemotherapy (N = 270) |
|---|---|---|
| Median PFS by BICR a , months (95% CI) | 5.8 (5.52–6.97) | 4.9 (4.17–5.59) |
| HR per Stratified Cox Model (95% CI) | 0.76 (0.59–0.98) | |
| Stratified Log-Rank Test p value (2 sided) | 0.0334 | |
| Median OS b , months (95% CI) | 21.6 (18.86–24.05) | 19.8 (17.54–22.28) |
| HR per Stratified Cox Model (95% CI) | 0.89 (0.69–1.13) | |
| Stratified Log-Rank Test p value (2 sided) | 0.3264 | |
| ORRa,* (95% CI) | 22.1 (17.1, 27.2) | 16.0 (11.6, 20.5) |
| Complete Response | 2.7% | 1.5% |
| Partial Response | 19.5% | 14.5% |
| Median DoR ** , months (95% CI) | 6.1 (4.1, 9.1) | 6.0 (4.0, 6.9) |
| Min-Max | 0.0 – 20.8+ | 0.0 – 15.7+ |
Data cut off 10-Oct-2018;
Data cut off 10-Sep-2019
In response evaluable patients (i.e., with measurable disease at baseline); n=262 in each treatment arm.
Calculated only for responders.
Source: Margetuximab Assessment Aid submitted to FDA (Study 04 CSR – Section 11.4.1.1.3 and Addendum 1 - Section 2.2.2)24
Data were immature for a conclusive result on the co-primary endpoint of OS. At the second interim OS analysis (DCO of 10-Sep-2019), 50% of patient in the ITT population have died, representing 70% of targeted OS events for the final analysis. Investigator-assessed PFS had acceptable concordance with the blinded independent review. ORR was numerically higher for the margetuximab group (22.1% vs. 16.0%). Median duration of response (DOR) was 6.1 months in the margetuximab arm and 6.0 months in the trastuzumab arm.
Safety:
The most frequently reported adverse events (AEs) (≥ 20% patients) on margetuximab were fatigue (57%), nausea (33%), diarrhea (25%), and vomiting (21%). As might be expected for treatment with a regimen that included cytotoxic chemotherapy, the most frequently reported Grade ≥ 3 AEs on margetuximab plus chemotherapy included neutropenia/neutrophil count decreased and fatigue. AEs reported at a higher frequency on margetuximab included infusion related reactions (IRRs), fatigue/asthenia, pyrexia, vomiting, decreased hemoglobin and increased creatinine. Serious adverse reactions (SAEs) occurred in 16% and 18% of patients who received margetuximab plus chemotherapy and trastuzumab plus chemotherapy, respectively.
IRRs are an important adverse reaction in SOPHIA, reported by 13.3% and 3.4% of patients who received margetuximab vs. trastuzumab, respectively. Grade 3 IRRs were reported in 3 margetuximab-treated patients (1.5%). All IRRs resolved within 24 hours irrespective of severity, and were manageable with supportive care. IRRs leading to infusion interruption occurred in 8.7% in patients treated with margetuximab and chemotherapy and 1.1% in patients treated with trastuzumab and chemotherapy. One patient discontinued therapy due to IRRs on margetuximab arm. Of the 35 patients with IRRs on margetuximab, 32 received additional infusions. Shorter margetuximab infusion time did not seem to increase the frequency or severity of IRRs as shown in the infusion substudy where the highest grade IRR was Grade 2 and occurred mostly during cycle 1 (120 minute infusion) rather than in subsequent cycles (30 minute infusion). The prescribing information contains a warning and precaution to inform health care professionals about the risk of IRRs in patients treated with margetuximab.
In SOPHIA, left ventricular (LV) dysfunction was low and reported in similar proportions between the margetuximab and trastuzumab treatment arms, 2.7% and 2.6%, respectively. Treatment with margetuximab has not been studied in patients with a history of clinically significant cardiac disease or LVEF less than 50% prior to initiation of treatment. LVEF is a known class effect of HER2-directed therapies and was included as Warnings and Precautions in FDA labeling (20).
Regulatory Insights
Efficacy and safety were evaluated adequately in SOPHIA, with ancillary safety evidence from Studies −01 and −02, to support approval for the treatment of patients with unresectable or metastatic HER2+ breast cancer who have received two or more prior anti-HER2-based regimens, at least one of which was for metastatic disease.
The review of this BLA utilized the Assessment Aid, a multidisciplinary review template that is a voluntary submission from the applicant to focus and streamline the FDA’s written review on critical thinking and analysis (21).
A major review issue with this application was that the magnitude of improvement in median PFS per BIRC was small. However, for FDA regular approval, there is not a requirement that a therapy be better than available therapies; the efficacy of a drug can be determined based on efficacy above placebo or in a non-inferiority trial. In this case, trastuzumab plus chemotherapy is an active control and margetuximab served as a replacement therapy for trastuzumab. Therefore, there is a reasonable assurance that margetuximab plus chemotherapy confers at least the same amount of benefit as trastuzumab plus chemotherapy, and is likely better than placebo, in this disease setting. Additionally, given the magnitude of benefit was shorter than the imaging interval of 6 weeks, the Applicant conducted a post-hoc interval censored analysis of PFS to assess sensitivity of primary analysis results with respect to imaging interval. These results from the post-hoc sensitivity analysis are consistent with the primary analysis and provide further assurance of robustness of primary analysis results.
An additional review issue was that the subgroup of patients homozygous for the CD16A-158V allele (V/V) appeared to have different outcomes in a direction unfavorable to margetuximab plus chemotherapy compared to the overall study population and other subgroups. This is a mechanistically unexpected finding, because margetuximab is specifically Fc-engineered to provide higher affinity, compared to trastuzumab, to both CD16A alleles (158F, 158V) (16). Further post-hoc analysis accounting for moderate imbalance in prognostic factors did not yield a satisfactory explanation for this observed anomaly. The subgroup analysis result should be interpreted with caution due to a small sample size (n=69), possible imbalances in patient known/unknown characteristics between treatment arms, and no clear mechanistic basis to support the observed heterogeneity. More studies will be required to explain and better understand this apparent incongruity. Additional information pertinent to CD16A F158V genotype and patient outcome from clinical trials that correlate CD16A F158V genotype with trial efficacy endpoints to further characterize the clinical benefit of margetuximab will be provided as part of an agreed upon PMC.
The indication for margetuximab is inclusive of all adults with HER2+ breast cancer to ensure flexibility in the use of this therapy across various patient populations. Historically, men have been excluded from breast cancer trials due to the low incidence of the disease in males and their management in clinical practice is extrapolated from data in women. The FDA has encouraged the inclusion of male patients in breast cancer trials and has released a guidance on this topic, which in part details that even when male enrollment is limited it may be possible to extrapolate findings in cases when no difference between males and females is anticipated based on the mechanism of action of the drug (22). Although all three male participants enrolled in SOPHIA were treated with trastuzumab plus chemotherapy, it is not anticipated that margetuximab plus chemotherapy would have differing efficacy to exclude males from the indication.
Conclusions
Results from SOPHIA demonstrate a favorable benefit-risk profile for margetuximab plus chemotherapy (Table 4) and represents another treatment option for patients with heavily pretreated HER2+ advanced or metastatic breast cancer. This indication is approved based on improvement in PFS over an active control, trastuzumab plus chemotherapy (23). OS is immature at the time of submission, but no detriment was observed for margetuximab plus chemotherapy compared to trastuzumab plus chemotherapy. The final OS analysis will occur after about 385 deaths and will be provided to the FDA to update the prescribing information.
Table 4:
FDA Benefit-Risk Assessment of Margetuximab plus chemotherapy
| Dimension | Evidence and Uncertainties | Conclusion and Reasons |
|---|---|---|
| Analysis of Condition |
|
Advanced or metastatic HER2-positive breast cancer is a serious and life-threatening condition with an ongoing unmet medical need. |
| Current Treatment Options |
|
There is an unmet medical need to improve the outcomes of patients with HER2-positive advanced or metastatic breast cancer whose disease has progressed on available HER2-directed therapies. |
| Benefit |
|
SOPHIA resulted in a statistically significant difference in PFS when margetuximab plus chemotherapy was compared to trastuzumab plus chemotherapy. Although the PFS improvement is only 0.9 months, margetuximab plus chemotherapy represents a replacement therapy for trastuzumab plus chemotherapy and patients may benefit from having an alternative therapy. Overall survival was immature; however no apparent detriment was observed in patients who received margetuximab plus chemotherapy compared to those that received trastuzumab plus chemotherapy. |
| Risk and Risk Management |
|
Margetuximab demonstrated acceptable tolerability for the indicated population with a serious and life-threatening disease. The safe use of margetuximab plus chemotherapy can be managed through appropriate labeling, including boxed warning for left ventricular dysfunction and embryofetal toxicity as well as a warning and precaution for infusion related reactions. No REMS is indicated. |
Source: Margetuximab Assessment Aid, FDA benefit-risk assessment section.24
Footnotes
Note: This is a U.S. Government work. There are no restrictions on its use.
Disclosure of Potential Conflicts of Interest: The authors report no financial interests or relationships with the commercial sponsors of any products discussed in this report.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021. Feb 4. doi: 10.3322/caac.21660. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Female Breast Cancer. Available from: https://seer.cancer.gov/statfacts/html/breast.html [Google Scholar]
- 3.Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LAG, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014; 106(5): dju055 doi: 10.1093/jnci/dju055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Female Breast Cancer Subtypes. Available from: https://seer.cancer.gov/statfacts/html/breast-subtypes.html [Google Scholar]
- 5.Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel in HER2+ metastatic breast cancer. N Engl J Med 2015; 372(8):724–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2+ advanced breast cancer. New Engl J Med. 2012; 367(19):1783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.National Comprehensive Cancer Network (NCCN) Guidelines. Breast Cancer. Available from https://www.nccn.org/professionals/physician_gls/default.aspx.
- 8.Giordano SH, Temin S, Chandarlapaty S, Crews JR, Esteva FJ, Kirshner JJ, et al. Systemic Therapy for Patients With Advanced Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: ASCO Clinical Practice Guideline Update. J Clin Oncol 2018;36(26):2736–40 [DOI] [PubMed] [Google Scholar]
- 9.U.S. Food and Drug Administration: FDA approves tucatinib for patients with HER2+ metastatic breast cancer. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tucatinib-patients-HER2+-metastatic-breast-cancer
- 10.Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N Engl J Med. 2020. Feb 13;382(7):597–609. [DOI] [PubMed] [Google Scholar]
- 11.U.S. Food and Drug Administration: FDA approves neratinib for metastatic HER2+ breast cancer. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neratinib-metastatic-her2-positive-breast-cancer
- 12.Saura C, Oliveira M, Feng YH, Dai MS, Chen SW, Hurvitz SA, et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated With ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J Clin Oncol. 2020. Sep 20;38(27):3138–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.U.S. Food and Drug Administration: FDA approves fam-trastuzumab deruxtecan-nxki for unresectable or metastatic HER2+ breast cancer. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fam-trastuzumab-deruxtecan-nxki-unresectable-or-metastatic-HER2+-breast-cancer
- 14.Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2+ Breast Cancer. The New England journal of medicine 2020;382(7):610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margetuximab approval letter. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/761150Orig1s000ltr.pdf
- 16.Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011;13(6):R123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bang YJ, Giaccone G, Im SA, et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann Oncol. 2017;28(4):855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rugo H, Im SA, Cardoso F, et al. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021;7(4):573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gavin P, Song N, Kim SR, et al. Association of Polymorphisms in FCGR2A and FCGR3A With Degree of Trastuzumab Benefit in the Adjuvant Treatment of ERBB2/HER2–Positive Breast Cancer Analysis of the NSABP B-31 Trial. JAMA Oncol. 2017;3(3):335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margetuximab (MARGENZA™) Prescribing Information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761150s000lbl.pdf
- 21.U.S. Food and Drug Administration: Assessment Aid. Available from: https://www.fda.gov/about-fda/oncology-center-excellence/assessment-aid
- 22.U.S. Food and Drug Administration: Male Breast Cancer: Developing Drugs for Treatment. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/male-breast-cancer-developing-drugs-treatment
- 23.U.S. Food and Drug Administration: FDA approves margetuximab for metastatic HER2-positive breast cancer. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-margetuximab-metastatic-her2-positive-breast-cancer.
- 24.U.S. Food and Drug Administration: BLA 761150 - Margetuximab Assessment Aid.