

Abstract
The NLRP3 inflammasome is a critical component of the innate immune system that is activated by microbial infections and cellular stress signals. The molecular mechanism of NLRP3 inflammasome activation remains not fully understood. As an NLRP3-interacting partner, NEK7 has emerged as a critical mediator for NLRP3 inflammasome activation. In contrast to NEK7, NEK6, the closely related member of the NEK family, does not support NLRP3 inflammasome activation. Here, we show that the mouse NEK7 catalytic domain, which shares high sequence identity with the counterpart of NEK6, mediates its interaction with NLRP3 and inflammasome activation in mouse macrophages. Within their catalytic domains, a single amino acid residue at a corresponding position (R121NEK7, Q132NEK6) differentiates their function in NLRP3 inflammasome activation. Surprisingly, the substitution of the glutamine residue to arginine residue at position 132 confers NEK6 the ability of NLRP3 binding and inflammasome activation in mouse macrophages. Furthermore, our results suggest a structural pocket surrounding the residue R121 of NEK7 that is essential for NLRP3 binding and inflammasome activation.
Introduction
Inflammasomes are supramolecular complexes that form intracellularly in response to microbial infections and cellular stress signals (1, 2). After sensing pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), a subset of pattern recognition receptors (PRRs), such as members from the nucleotide-binding and oligomerization domain (NOD)-like receptor family, absent-in-melanoma 2 (AIM2) or pyrin, initiate inflammasome assembly through the interaction with the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) adaptor, which in turn leads to caspase-1 activation (3-5). Activated caspase-1 processes the proinflammatory cytokines interleukin (IL)-1β and IL-18 into their mature forms and cleaves GSDMD to generate active N-terminal fragments, which subsequently form membrane pores for cytokine release and pyroptotic cell death (6-8). Among the six well-documented inflammasomes, the NLRP3 inflammasome is the most investigated one in the past decades (9, 10). Activation of the NLPR3 inflammasome is important for host defense against microbial infections (11). However, its aberrant activation contributes to the pathogenesis of several inflammatory diseases including cryopyrin-associated periodic syndromes, Alzheimer’s disease, type 2 diabetes, gout, and atherosclerosis (12). In most experimental settings, NLRP3 inflammasome activation requires two signals: priming and activation (13, 14). The priming signal is provided by toll-like receptor (TLR) ligands or endogenous cytokines, which upregulate the expression of NLRP3 and pro-IL-1β. The activation signal is induced by a range of seemingly unrelated stimuli such as ATP, pore-forming toxins (e.g., nigericin), and particulate matter (e.g., silica and uric acid crystals). Although the exact nature of the proximal signal for NLRP3 activation remains unknown, most NLRP3 stimuli induce K+ efflux which is critical for NLRP3 inflammasome activation (15).
The NLRP3 inflammasome is regulated by a growing list of NLRP3-interacting proteins including SGT1, HSP90, TXNIP, MAVS, MARK4, MIF, NEK7, DDX3X, and RACK1 (16-24). Among these interacting partners, NEK7, a mitotic Ser/Thr protein kinase from the NEK family, has emerged as a critical mediator for NLRP3 inflammasome activation in macrophages (23-25). Furthermore, the kinase activity of NEK7 is not required for this function, suggesting a novel mechanism for NEK7-mediated NLRP3 activation (23, 24). A recent structural study of the NLRP3-NEK7 complex reveals that the NEK7 catalytic domain interacts with both the LRR and NACHT domains of NLRP3 (26). However, the detailed mechanism by which NEK7 mediates NLRP3 activation remains unclear as the NLRP3-NEK7 complex was inactive (26).
NEK7 is closely related to NEK6 in the NEK family (27, 28). NEK7 and NEK6 share an overall 87% sequence identity within their catalytic domains but are highly divergent in their N-terminal extension preceding the catalytic domain (27, 29). In contrast to NEK7, NEK6 is not involved in NLRP3 inflammasome activation (23, 24). Although several residue differences within the second half of their catalytic domains have been suggested to account for this functional difference, the exact structural determinant of NEK7 that differentiates it from NEK6 in NLRP3 inflammasome activation remains unclear (26). Furthermore, NEK9, an upstream kinase for both NEK6 and NEK7, has been suggested to prevent NLRP3 activation by sequestering NEK7 from NLRP3 during the mitosis phase (24, 26). In this study, we found that the NEK7 catalytic domain is required for NLRP3 binding and inflammasome activation. A single corresponding amino acid residue within their catalytic domains (R121NEK7, Q132NEK6) differentiated NEK7 from NEK6 in NLRP3 inflammasome activation. A NEK6 mutant with an arginine substitution at position 132 restored NLRP3 inflammasome activation in NEK7-deficient macrophages. Although this NEK7 R121Q mutation enhanced its binding to NEK9, NEK9 depletion did not rescue defective NLRP3 inflammasome activation in NEK7R121Q knock-in macrophages. Furthermore, our results from alanine-screening mutagenesis suggest the existence of a NEK7 structural pocket containing this residue R121 that is critical for NLRP3 binding and inflammasome activation.
Materials and Methods
Cell culture
Immortalized bone marrow-derived macrophages (iBMDMs) were generated as previously described (30). iBMDMs were cultured in IMDM (Thermo Fisher, 12440053) supplemented with 10% FBS (Thermo Fisher, 16000044), 2 mM L-glutamine (Thermo Fisher, 25030081), and 1x Antibiotic-Antimycotic (Thermo Fisher, 15240062). NEK7-deficient (NEK7 KO) iBMDMs were generated by the CRISPR-Cas9 gene-editing system as previously described (23). HEK 293T cells were cultured in DMEM (Thermo Fisher, 11960044) supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate (Thermo Fisher, 11360070), and 1x Antibiotic-Antimycotic. HEK 293T cells and iBMDMs were tested to be free of mycoplasma contamination.
Reagents, plasmids, and transfection
Anti-Flag (A00187-200), Anti-HA (A01244-100), Anti-Actin (A00730-100), protein G resin (L00209) were purchased from GenScript. Anti-NLRP3 (AG-20B-0014-C100) was purchased from Adipogen. Anti-GSDMD (ab209845) and Anti-NEK9 (ab138488) were purchased from Abcam. Anti-ASC (67824) was purchased from Cell Signaling Technology (CST). Anti-mouse Caspase-1 was a kind gift from Dr. Gabriel Núñez (University of Michigan Medical School). Ultra-pure LPS (tlrl-pb5lps), Nano-SiO2 (tlrl-sio), poly(dA:dT)/lyovec (tlrl-patc) were purchased from InvivoGen. Nigericin (481990) and EDTA-free protease inhibitor cocktail (11873580001) were purchased from Sigma. Salmonella strain SL1344 was a kind gift originally from Dr. Denise Monack (Stanford University). The constructs for NLRP3-Flag, NLRP3-SFP, and NEK7-HA have been previously described (23). Mouse NEK6 from a cDNA clone (Harvard plasmid, MmCD00315333) was cloned into the pHIV-EGFP vector (Addgene, 21373). Genes for NEK7-NEK6 chimeras (NEK7-(N)-NEK6-(C) and NEK6-(N)-NEK7-(C)) were synthesized by GenScript. NEK7 or NEK6 mutations were generated by the QuikChange® mutagenesis kit from Agilent (200523). pLKO.1 constructs for NEK9 shRNA1 (TRCN0000027597) and NEK9 shRNA2 (TRCN0000027595) were purchased from Sigma. HEK 293T cells were plated into 6-well tissue culture plates (6.25 x 105 cells per well) overnight. Plasmids expressing Flag-tagged NLRP3, SFP-tagged NLRP3, HA-tagged NEK7, NEK6, NEK7/NEK6 chimeras, NEK7 or NEK6 mutants were single- or co-transfected into HEK 293T cells by Lipofectamine LTX (Thermo Fisher, 15338100) for 16 h.
Immunoprecipitation and Pull-down assay
Following transient transfection, HEK 293T cells were lysed in ice-cold lysis buffer (50 mM Tris, pH 7.4, 2 mM EDTA, 150 mM NaCl, 0.5% Triton X-100, 1x EDTA-free protease inhibitor cocktail), and clarified by centrifugation (12,000g) at 4°C for 10 min. Pre-cleared cell lysates were incubated with an anti-Flag (1:200) antibody at 4°C overnight. The proteins bound by the antibody were pulled down with protein G resin and subjected to immunoblotting analysis. For the pull-down of NLRP3-SFP by streptavidin beads, pre-cleared cell lysates were incubated with streptavidin agarose resin (ThermoFisher, 20359) on a rotator at 4°C for 4 hr. Beads were washed 3 times with lysis buffer before being analyzed by immunoblot.
Reconstitution of NEK7, NEK6, or NEK7/NEK6 chimeras in NEK7 KO iBMDMs
Lentiviruses expressing HA-tagged NEK7, NEK6, or NEK7-NEK6 chimera were packaged in HEK 293T cells. NEK7 KO iBMDMs were then transduced with lentiviruses. After 3-4 days, transduced cells were sorted by flow cytometry using EGFP as a marker. The expression of reconstituted proteins was determined by immunoblotting with an anti-HA antibody.
Inflammasome activation
iBMDMs were plated at 5 x 105 cells per well of 12-well plates with IMDM containing reduced serum concentration (1% FBS). One day later, the culture medium was replaced with 0.5 mL serum-free IMDM per well. Cells were primed with 200 ng ml−1 ultrapure LPS for 4 h followed by stimulation with 5 μM nigericin (1 h), Nano-SiO2 (200 μg ml−1, 4 h), poly(dA:dT) (2 μg ml−1, 4 h), Salmonella (m.o.i. = 10, 2h). After stimulation, supernatants were collected, and cells were directly lysed with 150 μl of 2X sample buffer (Biorad, 1610737). Equal amounts of cell lysates and supernatants were combined together for immunoblotting.
Measurement of IL-1β by ELISA
Cytokine IL-1β in the supernatants collected from triplicate wells was measured with ELISA kits (R&D Systems, DY401-05) according to the manufacturer’s instructions.
ASC speck staining
iBMDMs were plated on an 8-well permanox chamber slide (Thermo Scientific, 12-565-22) overnight. Cells were primed with 200 ng ml−1 LPS for 4 h, then stimulated with 5 μM nigericin (1 h) or transfected with 2 μg ml−1 poly(dA:dT) (4 h). After stimulation, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with PBS buffer containing 5% BSA. Cells were stained with anti-ASC antibody (CST, 67824; 1:200 dilution) and Alexa-Fluor-488 conjugated secondary antibody (Thermo Fisher, A-11008; 1:1,000 dilution). Slides were then prepared with antifade mounting medium containing DAPI (Vectorlabs, H-1200-10). Cell images were taken using a Nikon E-800 fluorescence microscope system and analyzed by ImageJ.
NEK7R121Q knock-in and NEK9 knockdown in macrophages
Alt-R® CRISPR-Cas9 crRNA for mouse NEK7 (targeting sequence: TTACCTTTATCATTCTGGAGAGG), tracrRNA (1072533), HDR donor oligos (Sense: GTCAGAAAAACAAATGCATGAACAAACTTACTTTTATCATTTGGGAGAGGTCACCAGCATCTGCTAACTCCAAAACTATGTTCA; Antisense:TGAACATAGTTTTGGAGTTAGCAGATGCTGGTGACCTCTCCCAAATGATAAAAGTAAGTTTGTTCATGCATTTGTTTTTCTGAC), Cas9 nuclease (1081059) and HDR Enhancer (1081072) were purchased from IDT and the RNP complexes were prepared according to the manufacturer’s protocol. The RNP complexes were delivered into iBMDMs by electroporation with nucleofector kit V (Lonza, VCA-1003). Cell clones harboring NekR121Q homologous alleles were identified by sequencing. Lentivirus expressing NEK9 shRNA or control shRNA (Addgene 1864) were packaged in HEK 293T cells and used to transduce macrophages. Puromycin-resistant macrophages were used for experiments.
Statistical analysis
No statistical method was used to predetermine sample sizes or include or exclude samples. Data are expressed as mean ± SEM. Statistical analysis was performed using unpaired two-tailed Student’s t-test or one-way ANOVA with GraphPad. A p value less than 0.05 was considered statistically significant.
Results
The catalytic domain of NEK7, but not NEK6, mediates NLRP3 binding and inflammasome activation
NEK7 and NEK6 are closely related members in the NEK family and share 87% sequence identity within their catalytic domains (27). However, NEK6 is not involved in NLRP3 inflammasome activation in macrophages (23, 24). A recent structural study of the NLRP3-NEK7 complex suggests that the second part (residues 260-302) of the NEK7 catalytic domain, which slightly differs from NEK6, might contribute to their differential role in NLRP3 inflammasome activation (26). Besides those residue differences within the catalytic domains, NEK7 and NEK6 are highly divergent in a short N-terminal extension (NTE) preceding their catalytic domains: residues 1-33 for NEK7; residues 1-43 for NEK6, which has been suggested to differentially regulate these kinases (Supplemental Fig. 1) (31). This NTE was not visible in the recently reported structure of the NLRP3-NEK7 complex (26). Whether this NTE contributes to NLRP3 inflammasome activation and therefore differentiates NEK7 from NEK6 in this function remains unknown. We constructed hemagglutinin (HA) –tagged NEK7, NEK6, and NEK7-NEK6 chimeras with different combinations of the NTE and catalytic domain [Nek6(N)-Nek7(C), Nek7(N)-Nek6(C)] (Fig. 1A). Each construct was co-transfected with Flag-tagged NLRP3 into human embryonic kidney (HEK) 293T cells. In agreement with previous findings, NEK7, not NEK6, was co-immunoprecipitated with NLRP3 (Fig. 1B). Importantly, the chimera containing the NEK7 catalytic domain, albeit at a lower level of expression, also co-immunoprecipitated with NLRP3, while the chimera containing the NEK6 catalytic domain failed to do so. This suggests that the NEK7 catalytic domain (residues 34-302) is responsible for its interaction with NLRP3 (Fig. 1B).
FIGURE 1.
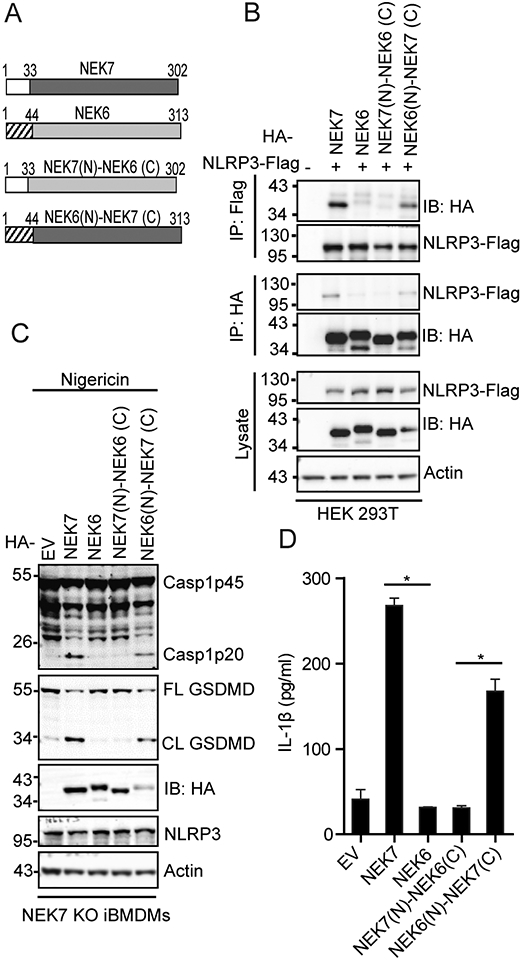
The catalytic domain of NEK7 mediates NLRP3 binding and inflammasome activation. (A) Schematic presentation for domains of NEK7, NEK6, and NEK7-NEK6 chimeras. The N-terminal domain of NEK7 contains the first 33 amino acid residues. The N-terminal domain of NEK6 contains the first 44 amino acid residues. The left amino acid residues comprise the catalytic domains for NEK7 and NEK6, respectively. (B) HA-tagged mouse NEK7, NEK6, or NEK7-NEK6 chimera was co-expressed with Flag-tagged NLRP3 in HEK 293T cells. Cell lysates were immunoprecipitated with anti-Flag or anti-HA antibody and immunoblotted with indicated antibodies. (C) NEK7 KO iBMDMs were reconstituted with HA-tagged NEK7, NEK6, or NEK7-NEK6 chimera by lentiviral transduction. Macrophages were stimulated with 5 μM nigericin for 1 h after LPS priming. Mixtures of cell lysates and supernatants were immunoblotted with indicated antibodies. EV, empty vector. Actin was used as an internal control. (D) Measurement of IL-β in the supernatants from nigericin-stimulated macrophages by ELISA. Data are representative of three experiments. Error bars in D show the SEM of triplicate wells. *p < 0.05.
Next, we sought to examine whether the NEK7 catalytic domain can support NLRP3 inflammasome activation. NEK7, NEK6, or a NEK7-NEK6 chimera was reconstituted into NEK7 KO iBMDMs. Both NEK7 and the NEK7-NEK6 chimera containing the catalytic domain of NEK7 restored NLRP3 inflammasome activation, as indicated by the appearance of caspase-1 p20, GSDMD cleavage, and IL-1β secretion in macrophages after LPS priming and nigericin treatment (Fig. 1C, 1D). In contrast, both NEK6 and the chimera containing the catalytic domain of NEK6 failed to activate the NLRP3 inflammasome (Fig. 1C, 1D). Collectively, these results indicate that the NEK7 catalytic domain is the structural determinant for NLRP3 binding and inflammasome activation.
The arginine residue at position R121 of NEK7 is critical for NLRP3 binding and inflammasome activation
We previously showed that residues 1-212 of NEK7 support its binding to NLRP3 (23). Therefore, we postulated that residues 34 - 212 within the NEK7 catalytic domain might differentiate between NEK7 and NEK6 in NLRP3 binding. Therefore, a series of NEK7 mutants (R35Q; 57GVP/RKT: G57R_V58K_P59T; A77Q; D84G; 98YA/LD: Y98L_A99D; R121Q; H125Y; K198E) were made to replace single or several differential residues of NEK7 with the corresponding residues of NEK6 (Supplemental Fig. 1). Most NEK7 mutants (R35Q, 57GVP/RKT, A77Q, D84G, 98YA/LD, K198E) have comparable interaction with NLRP3 as compared with wild-type NEK7 (Fig. 2A). Notably, the replacement of residue R121NEK7 with the Q132NEK6 markedly reduced the interaction between NEK7 and NLRP3 (Fig 2A). Additionally, the replacement of residue H125NEK7 to tyrosine also reduced the NLRP3-NEK7 interaction to a less extent (Fig. 2A). To test whether those replacements affect NLRP3 inflammasome activation, we reconstituted NEK7 KO iBMDMs with wild-type and mutant NEK7. After LPS priming and nigericin treatment, macrophages reconstituted with each of NEK7 mutants, including R35Q, 57GVP/RKT, A77Q, D84G, 98YA/LD, and K198E mutants, showed comparable levels of caspase-1 activation as compared with wild-type NEK7-reconstituted macrophages (Supplemental Fig. 2A). However, NLRP3 inflammasome activation was markedly reduced in macrophages with NEK7 R121Q mutant after LPS priming and treatment with nigericin or Nano-SiO2, as indicated by caspase-1 activation, GSDMD cleavage, and IL-1β secretion (Fig. 2B, 2C). Macrophages reconstituted with NEK7 H125Y also showed a slightly reduced level of caspase-1 activation (Supplemental Fig. 2B). In contrast, those reconstituted macrophages showed no discernible defect in Salmonella-induced NLRC4 inflammasome activation and poly(dA:dT)-induced AIM2 inflammasome activation (Fig. 2B, 2C). Accordingly, as compared to macrophages with wild-type NEK7, macrophages reconstituted with R121Q had fewer ASC specks in response to nigericin, although they both had comparable ASC speck formation in response to poly(dA:dT) (Fig. 2D, 2E). To further confirm the role of R121 of NEK7 in inflammasome activation, we introduced this mutation (R121Q) into the endogenous NEK7 protein through the CRISPR-Cas9 gene-editing system. Sequencing results confirmed that both Nek7 alleles in macrophages had this intended mutation (Supplemental Fig 3). In line with our reconstitution experiments, NEK7R121Q knock-in (KI) macrophages failed to induce caspase-1 activation and IL-1β secretion in response to LPS priming and nigericin treatment, although they have comparable levels of caspase-1 activation and IL-1β secretion in response to Salmonella infection as compared with wild-type parent macrophages (Fig. 2F, 2G).
FIGURE 2.

The residue R121 of NEK7 is critical for NLRP3 binding and inflammasome activation. (A) SFP-tagged NLRP3 (NLRP3-SFP) was co-expressed with HA-tagged NEK7 mutants containing swapped residue(s) from NEK6 at the corresponding position(s) in HEK 293T cells. Cell lysates were pulled downed (PD) with streptavidin beads and immunoblotted with indicated antibodies. EV, empty vector. (B) NEK7 KO iBMDMs were reconstituted with wild-type NEK7 or NEK7 mutant (R121Q) by lentiviral transduction. Macrophages were primed with LPS and stimulated with PBS (mock), 5 μM nigericin (1 h), 200 μg/ml Nano-SiO2 (4 h), 4 μg/ml poly(dA:dT) (4 h) or Salmonella (m.o.i= 10, 2h). Mixtures of cell lysates and supernatants were immunoblotted with indicated antibodies. (C) Measurement of IL-β in the supernatants from stimulated macrophages by ELISA. (D) ASC immunostaining in wild-type or mutant NEK7-reconstituted macrophages primed with LPS and stimulated with PBS, nigericin, or poly(dA:dT). Scale bars, 10 μm. (E) Quantification of ASC specks from D. The percentage of ASC speck-containing cells was calculated from three different fields with at least 100 cells each. (F) Parent (WT) or NEK7R121Q knock-in mutant (KI) macrophages were stimulated with PBS, nigericin (5 μM, 1h), or Salmonella (m.o.i=10, 2h) after LPS priming. Mixtures of cell lysates and supernatants were immunoblotted with indicated antibodies. (G) Measurement of IL-β in the supernatants from the stimulated parent (WT) or NEK7R121Q knock-in mutant (KI) macrophages by ELISA. Actin was used as an internal control. Data are representative of three experiments. Error bars in C, E, and G show the SEM of triplicates. *p < 0.05.
Substitution of the glutamine residue to arginine residue at position 132 confers NEK6 the ability of NLRP3 binding and inflammasome activation
Given our data showing that the arginine residue at position 121 of NEK7 is critical for NLRP3 binding and inflammasome activation, we next asked whether the replacement of the corresponding glutamine residue of NEK6 with the arginine residue can confer NLRP3 binding by NEK6. In addition, we also included a NEK6 mutant in which the nearby tyrosine residue (Y136) was replaced with the corresponding histidine residue of NEK7. In contrast to the wild-type NEK6 and Y136H mutant, NEK6 Q132R mutant (NEK6Q132R) bound to NLRP3 when co-expressed with flag-tagged NLRP3 in HEK 293T cells, suggesting that this single amino acid residue differentiates between NEK7 and NEK6 in NLRP3 binding (Fig. 3A). Since other differential residues, especially in the second half of the catalytic domain, exist between NEK7 and NEK6, we asked whether this single substitution mutation is sufficient to restore NLRP3 inflammasome activation in NEK7 KO iBMDMs. Wild-type NEK6 did not restore caspase-1 activation and IL-1β secretion in macrophages after LPS priming and nigericin treatment (Fig. 3B, 3C). However, macrophages with NEK6 Q132R mutant showed caspase-1 activation and IL-1β secretion (Fig. 3B, 3C). Furthermore, we detected ASC speck formation in NEK7 KO iBMDMs reconstituted with NEK6 Q132R mutant, but not with wild-type NEK6 (Fig. 3D). Taken together, these results indicate that the arginine residue at position 121 of NEK7 is the key structural determinant for NLRP3 inflammasome activation and differentiates between NEK7 and NEK6 in this pathway.
FIGURE 3.

The Q132R mutation confers NEK6 the ability of NLRP3 binding and inflammasome activation. (A) HA-tagged mouse wild-type (WT), mutant NEK6 Q132R, or Y136H was expressed alone or with Flag-tagged mouse NLRP3 in HEK 293T cells. Cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with indicated antibodies. (B) NEK7 KO iBMDMs were reconstituted with HA-tagged mouse wild-type (WT) or Q132R mutant NEK6 by lentiviral transduction. Macrophages were stimulated with 5 μM nigericin for 1 h after LPS priming. Mixtures of cell lysates and supernatants were immunoblotted with indicated antibodies. EV, empty vector. Actin was used as an internal control. (C) Measurement of IL-β in the supernatants from nigericin-stimulated macrophages by ELISA. (D) ASC immunostaining in wild-type (WT) or Q132R mutant NEK6-reconstituted macrophages after nigericin stimulation. Scale bars, 10 μm. Data are representative of three experiments. Error bars in C show the SEM of triplicate wells. *p < 0.05.
NEK9 deletion fails to rescue NLRP3 inflammasome activation in NEK7R121Q knock-in macrophages
The NEK7 region surrounding the R121 residue has been shown to interact with NEK9, an upstream kinase that regulates NEK6 and NEK7 during mitosis (32). Furthermore, the sequestering of NEK7 by NEK9 is suggested to prevent NLRP3 inflammasome activation during mitosis (24, 26). We postulated that the residue at this position (R121NEK7, Q132NEK6) might affect their binding to NEK9 and thus differentiate their role in NLRP3 inflammasome activation. To test this, we examined the interaction between NEK9 and NEK7, NEK6, NEK7 R121Q, or NEK6 Q132R. When expressed in HEK 293T cells, NEK7 had a weaker interaction with NEK9 as compared to NEK6. NEK7 R121Q mutation increased the interaction between NEK7 and NEK9 (Fig. 4A). In contrast, NEK6 Q132R mutation reduced the interaction between NEK6 and NEK9 (Fig. 4A). To examine how NEK9 affects NLRP3 inflammasome activation in macrophages, we knocked down NEK9 expression in both wild-type macrophages and NEK7R121Q Knock-in macrophages via short hairpin RNA interference (Fig. 4B). Reduction of NEK9 did not trigger NLRP3 inflammasome activation in macrophages after LPS priming (Fig. 4B, 4C). After nigericin treatment, wild-type macrophages with NEK9 knockdown showed comparable caspase-1 activation and IL-1β secretion (Fig. 4B, 4C). Importantly, NEK9 knockdown in NEK7R121Q knock-in macrophages did not rescue caspase-1 activation or IL-1β secretion after nigericin treatment (Fig. 4B, 4C).
FIGURE 4.
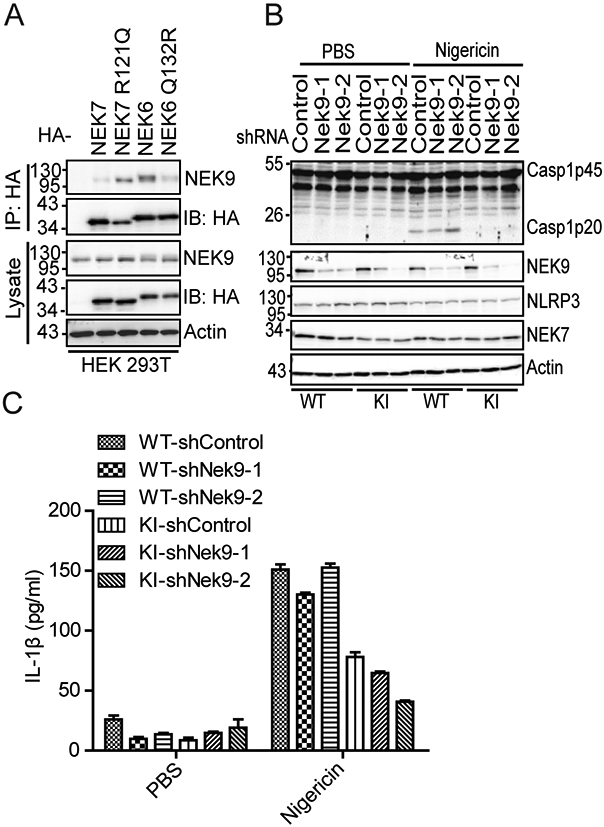
NEK9 knockdown fails to restore defective NLRP3 inflammasome activation caused by NEK7 R121Q mutation. (A) HA-tagged NEK7, NEK7 R121Q, NEK6, or NEK6Q132R was co-expressed with NEK9 in HEK 293T cells. Cell lysates were immunoprecipitated with an anti-HA antibody and immunoblotted with indicated antibodies. (B) Parent (WT) or NEK7R121Q knock-in mutant (KI) macrophages were treated with control shRNA or two individual NEK9-targeting shRNAs. Macrophages were stimulated with PBS or 5 μM nigericin after LPS priming. Cell lysates were immunoblotted with indicated antibodies. (C) Measurement of IL-β in the supernatants from PBS or nigericin-stimulated macrophages after the treatment of indicated shRNAs by ELISA. Data are representative of three experiments. Error bars in C show the SEM of triplicate wells.
The basic property of the amino acid residue at position 121 of NEK7 is critical for NLRP3 binding and inflammasome activation
The residue R121 of NEK7 has a positively charged side chain while the residue Q132 of NEK6 has a polar uncharged side chain. The interaction between NLRP3 and NEK7 is suggested to be mediated by electrostatic complementarity (26). Therefore, we sought to examine whether the basic property of residue R121NEK7 is critical for NLRP3 binding and inflammasome activation. We found that replacement of the arginine residue at this position with alanine residue (R121A) abolished its interaction with NLRP3, similar to the R121Q mutation (Fig. 5A). In contrast, the NEK7 mutant with the lysine residue as the replacement (R121K) had a comparable level of interaction with NLRP3 as wild-type NEK7 (Fig. 5A). Furthermore, NEK7 KO iBMDM reconstituted with NEK7R121A failed to induce caspase-1 activation, GSDMD cleavage, and IL-1β secretion after LPS priming and nigericin treatment, similar to macrophages with NEK7R121Q (Fig. 5B, 5C). In contrast, reconstitution with NEK7R121K restored caspase-1 activation, GSDMD cleavage, and IL-1β secretion after LPS priming and nigericin treatment (Fig. 5B, 5C). These data indicate that the basic property of the residue at position 121 of NEK7 is critical for NLRP3 binding and inflammasome activation.
FIGURE 5.
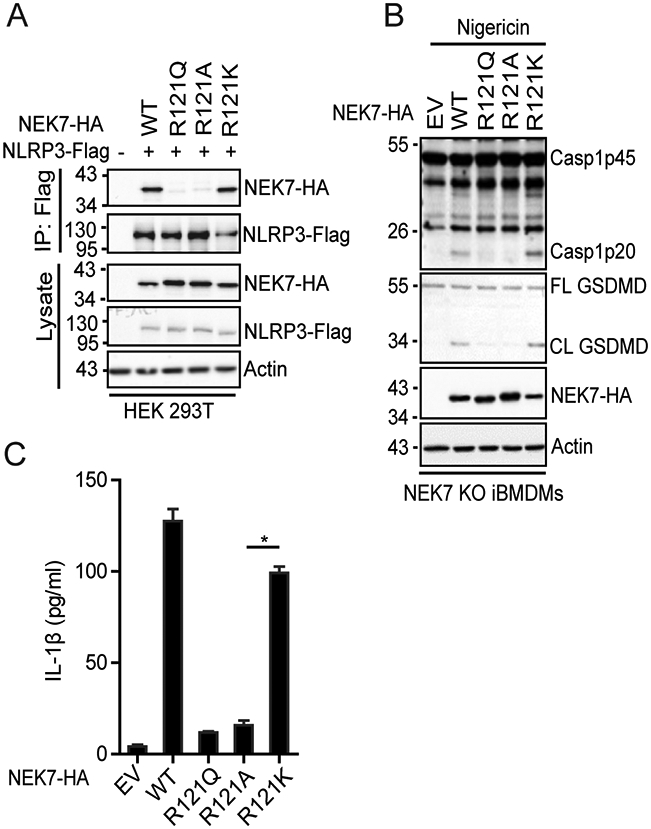
The basic property of R121NEK7 is critical for NLRP3 binding and inflammasome activation. (A) Flag-tagged NLRP3 was co-expressed with HA-tagged wild-type or mutant NEK7 in HEK 293T cells. Cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with indicated antibodies. (B) NEK7 KO iBMDMs were reconstituted with HA-tagged mouse wild-type or mutant NEK7 by lentiviral transduction. Macrophages were stimulated with 5 μM nigericin for 1 h after LPS priming. Mixtures of cell lysates and supernatants were immunoblotted with indicated antibodies. (C) Measurement of IL-β in the supernatants from nigericin-stimulated macrophages by ELISA. EV, empty vector. Actin was used as an internal control. Data are representative of three experiments. Error bars in C show the SEM of triplicate wells. *p < 0.05.
A potential structural pocket of NEK7 for NLRP3 binding and inflammasome activation
Since R121NEK7 is critical for NLRP3 binding and inflammasome activation, we examined the surrounding region of this residue in the available crystal structure of NEK7. R121 is located at a shallow surface pocket found at the catalytic domain of NEK7 (29) (Fig. 6A). The base of this pocket is made up of residues M122, Y141, and I169, while R121 forms one side of the pocket with residues D118 and H125 (29) (Fig. 6A). We performed alanine-scanning mutagenesis on residues within or surrounding this NEK7 pocket. Mutation of the pocket-forming residues, including R121A, M122A, Y141A, and I169A, significantly reduced NEK7-NLRP3 interaction, caspase-1 activation, or IL-1β production (Fig. 6B-D). Except G117A, mutation of the peripheral residues, including D118A, L119A, and H125A had no or modest effects on its interaction with NLRP3, but significantly inhibited caspase-1 activation and IL-1β secretion (Fig. 6B-D). Of note, NEK7 T170A mutation did not affect NEK7-NLRP3 interaction but caused a significant reduction in caspase-1 activation and IL-1β secretion, in agreement with its side chain facing outside and not directly involved in forming the pocket (Fig. 6B-D). These data indicate that this NEK7 pocket containing residue R121 is essential for NLRP3 binding and inflammasome activation.
FIGURE 6.

Identification of a potential NEK7 structural pocket for NLRP3 binding and inflammasome activation. (A) A close view of the region containing the residue R121 of NEK7. (B) Flag-tagged NLRP3 was co-expressed with HA-tagged wild-type or mutant NEK7 in HEK 293T cells. Cell lysates were immunoprecipitated with anti-Flag antibody and immunoblotted with indicated antibodies. (C) NEK7 KO iBMDMs were reconstituted with HA-tagged mouse wild-type or mutant NEK7 by lentiviral transduction. Macrophages were stimulated with 5 μM nigericin for 1 h after LPS priming. Mixtures of cell lysates and supernatants were immunoblotted with indicated antibodies. (D) Measurement of IL-β in the supernatants from nigericin-stimulated macrophages by ELISA. EV, empty vector. Actin was used as an internal control. Data are representative of three experiments. Error bars in D show the SEM of triplicate wells. *p < 0.05.
Discussion
The NLRP3 inflammasome is a critical component of innate immunity and when dysregulated contributes to the pathogenesis of several inflammatory diseases (10). As an NLRP3-interacting protein, NEK7 has emerged as a key mediator for its activation (23-25). NEK6, although closely related to NEK7, does not support NLRP3 inflammasome activation. In this study, our results show that the catalytic domains of NEK7 and NEK6 determine their requirement in NLRP3 binding and inflammasome activation. We further show that a single residue at position 121 of the NEK7 catalytic domain is critical for NLRP3 inflammasome activation and the basic property of this arginine residue differentiates NEK7 from NEK6 in this function. In addition, our results suggest the existence of a NEK7 structural pocket that is essential for NEK7-NLRP3 interaction and inflammasome activation.
What is the structural determinant of NEK7 that differentiates it from NEK6 in NLRP3 binding and inflammasome activation? Previous studies have found that the catalytic domain of NEK7 interacts with NLRP3 and residues 1-212 are sufficient for this interaction (23, 24, 26). It is unknown whether the divergent N-terminal extension of NEK7 is involved in NLRP3 inflammasome activation and therefore differentiates NEK7 from NEK6 in this function. In consistence with a previous finding that the deletion of the NEK7 N-terminus resulted in significantly reduced expression of NEK7 in HEK 293T cells, our attempts to express the NEK7 catalytic domain alone in macrophages have not been successful (data not shown) (24). Nevertheless, using NEK7-NEK6 chimeras containing different combinations of the catalytic domain and N-terminal extension, we found that the catalytic domain of NEK7 mediated NLRP3 binding and inflammasome activation as only the chimera containing the NEK7 catalytic domain bound to NLRP3 and restored NLRP3 inflammasome activation in macrophages. NEK7 has been shown as a centrosomal protein and mediates NLRP3 inflammasome activation at the centrosome (18, 33, 34). Beyond the scope of this study, it might be possible that the N-terminal extension, both from NEK7 and NEK6, plays a role in their centrosomal localization and, in the case of NEK7, contributes to NLRP3 inflammasome activation.
Structural analysis of the NEK7-NLRP3 complex has revealed that residues within the C-lobe of NEK7 catalytic domain, including Q129, R131, and R136, have largest buried surfaces in the interface (26). Notably, those involved residues are also conserved at the corresponding positions of NEK6. Since there is no crystal structure available for NEK6, it is unknown whether those NEK6 residues assume a different conformation in comparison with NEK7. The R121 of NEK7 has been suggested to mediate the NEK7-NLRP3 interaction through the hydrogen bonding or charged interaction, as the R121E mutation slightly reduces NLRP3 binding by NEK7 in vitro binding experiments with recombinant proteins (26). How this R121E mutation affect NLRP3 inflammasome activation in macrophages has not been further explored. We have extended this finding by showing that this residue R121 of NEK7 is critical for NLRP3 binding and inflammasome activation. Change of this arginine residue to the glutamine residue of NEK6 drastically reduced the NLRP3-NEK7 interaction and therefore failed to rescue NLRP3 inflammasome activation in NEK7 KO macrophages; conversely, substitution of the glutamine residue to the arginine residue in NEK6 conferred NEK6 the ability of NLRP3 binding and inflammasome activation. Thus, we have demonstrated that this single amino acid residue within NEK7 catalytic domain differentiated NEK7 from NEK6 in NLRP3 inflammasome activation. The basic property of this residue is the key feature to support this NEK7 function as the lysine residue substitution at this position support NLRP3 binding and inflammasome activation. Our findings on this critical residue are consistent with a recent study that an anti-inflammatory compound BBR inhibits NLRP3 inflammasome activation in macrophages and in vivo by targeting this R121 residue via the hydrogen bonding (35). Furthermore, our alanine-screening experiments on residues surrounding this R121 residue suggest the existence of a NEK7 structural pocket for NLRP3 binding and inflammasome activation. Although they are not indicated in the interface of the NLRP3-NEK7 complex, residues including M122, Y141, and I169 appear to be essential for the NLRP3-NEK7 interaction in cells as alanine substitutions of those residues completely abolished this interaction. A recent report shows that recombinant NEK7 dissociates the native oligomeric form of NLRP3 (NLRP3 cages) (36). In the future, it will be interesting to examine whether those residues within this NEK7 pocket are involved in the interaction between NEK7 and NLRP3 cages, which might assume an interface different from the NLRP3-NEK7 complex. Alternatively, alanine substitutions of those residues might indirectly alter the NEK7-NLRP3 interaction through those reported residues, such as R121, Q129, R131, and R136 of NEK7. Notably, alanine substitution of residue D180, L119, H125, or T170, has no or modest effect on the NLRP3-NEK7 interaction, but caused significant defect in NLRP3 inflammasome activation, suggesting mutation of those residues might affect NEK7 conformation required for NLRP3 activation following the NLRP3-NEK7 interaction. Similar to our observation, Sharif et al. have reported that a NEK7 mutation (E280R) did not affect the interaction of NEK7 with NLRP3, but impaired NLRP3 activation (26). Further defining this R121-containig structural pocket of NEK7 in the presence of the prototype compound BBR might help the development of novel therapeutic compounds that target NEK7-NLRP3 interaction to treat NLRP3-driven diseases.
NEK9 interacts with both NEK6 and NEK7 and activates them during mitosis (37, 38). Recent studies suggest that NEK9 might compete with NLRP3 for NEK7 and therefore inhibit NLRP3 inflammasome activation during mitosis (24, 26). Furthermore, a crystal structure reveals that NEK9 also binds to the C-lope of NEK7 catalytic domain containing the residue R121 (32). Structural modeling by Sharif et al. suggests that the interaction of NEK7 with NEK9 interferes with its interaction with NLRP3 (26). Our findings indicated that NEK7 R121Q mutation enhanced its interaction with NEK9 but failed to support NLRP3 binding and inflammasome activation. In contrast, NEK6 Q132R mutation reduced its interaction with NEK9, but conferred the ability of NLRP3 binding and inflammasome activation. However, NEK9 knockdown did not rescue the activity of NEK7 R121Q mutant in NLRP3 inflammasome activation, suggesting different structural requirements for NEK7 binding to NLRP3 and NEK9 in this overlapping binding region.
In summary, we have demonstrated that the catalytic domain of NEK7 is required for NLRP3 inflammasome activation and a single residue R121 of NEK7 within this catalytic domain differentiates NEK7 from NEK6 in NLRP3 inflammasome activation. Furthermore, our findings suggest the existence of a structural pocket of NEK7 surrounding the residue R121 for NLRP3 binding and inflammasome activation. This NEK7 structural pocket might serve as a target for future development of therapeutic compounds for NLRP3-driven diseases.
Supplementary Material
Key Points.
NEK7 catalytic domain is required for NLRP3 activation.
A single residue differentiates between NEK7 and NEK6 in NLRP3 activation.
A NEK7 structural pocket is essential for NLRP3 activation.
Acknowledgments
We would like to thank Dr. Gabriel Núñez (the University of Michigan) for providing an anti-caspase-1 antibody, NEK7 KO iBMDMs, and other important reagents. We would like to thank Dr. Adrian Stecula (Atomwise) for suggestions on NEK7 residues involved in NLRP3-NEK7 interaction. We would also like to thank Dr. Jessica Back and Eric Van Buren at the Microscopy, Imaging, and Cytometry Resources Core of Wayne State University for cell sorting.
This work was supported by funding from the National Institutes of Health AI148544 and by Wayne State Startup funds (to Y.H.). The Microscopy, Imaging, and Cytometry Resources Core is supported in part by NIH Center grant P30 CA22453 to the Karmanos Cancer Institute and R50 CA251068-01 to Kamiar Moin, Wayne State University.
Abbreviations used in this article:
- iBMDMs
immortalized mouse bone marrow-derived macrophages
- NLRP3
NOD-like receptor family pyrin domain-containing 3
- ASC
apoptosis-associated speck-like protein containing a caspase-recruitment domain
- GSDMD
gasdermin D
- WT
wild-type
- shRNA
short hairpin RNA
- m.o.i.
multiplicity of infection
References
- 1.Schroder K, and Tschopp J. 2010. The Inflammasomes. Cell 140: 821–832. [DOI] [PubMed] [Google Scholar]
- 2.Martinon F, Burns K, and Tschopp J. 2002. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell 10: 417–426. [DOI] [PubMed] [Google Scholar]
- 3.Rathinam VA, Vanaja SK, and Fitzgerald KA. 2012. Regulation of inflammasome signaling. Nat Immunol 13: 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamkanfi M, and Dixit VM. 2014. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022. [DOI] [PubMed] [Google Scholar]
- 5.Broz P, and Dixit VM. 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nature reviews. Immunology 16: 407–420. [DOI] [PubMed] [Google Scholar]
- 6.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 7.Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, and Dixit VM. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526: 666–671. [DOI] [PubMed] [Google Scholar]
- 8.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J. 2015. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25: 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelley N, Jeltema D, Duan Y, and He Y. 2019. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanson KV, Deng M, and Ting JP. 2019. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature reviews. Immunology 19: 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anand PK, Malireddi RK, and Kanneganti TD. 2011. Role of the nlrp3 inflammasome in microbial infection. Front Microbiol 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo H, Callaway JB, and Ting JP. 2015. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutterwala FS, Haasken S, and Cassel SL. 2014. Mechanism of NLRP3 inflammasome activation. Annals of the New York Academy of Sciences 1319: 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Hara H, and Nunez G. 2016. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, and Nunez G. 2013. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38: 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayor A, Martinon F, De Smedt T, Petrilli V, and Tschopp J. 2007. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nature Immunology 8: 497–503. [DOI] [PubMed] [Google Scholar]
- 17.Zhou RB, Tardivel A, Thorens B, Choi I, and Tschopp J. 2010. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunology 11: 136–U151. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, Masters L, James JR, Shi Y, Meng G, and Mallat Z. 2017. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nature communications 8: 15986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, Fan H, Creed SJ, Deen NS, Traore DAK, Mueller I, Stanisic D, Baiwog FS, Skene C, Wilce MCJ, Mansell A, Morand EF, and Harris J. 2018. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nature communications 9: 2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, and Germain RN. 2013. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153: 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan Y, Zhang L, Angosto-Bazarra D, Pelegrin P, Nunez G, and He Y. 2020. RACK1 Mediates NLRP3 Inflammasome Activation by Promoting NLRP3 Active Conformation and Inflammasome Assembly. Cell Rep 33: 108405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, Sharma BR, Nourse A, King SV, Pitre A, Burton AR, Pelletier S, Gilbertson RJ, and Kanneganti TD. 2019. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573: 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y, Zeng MY, Yang D, Motro B, and Nunez G. 2016. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, Lyon S, Scott L, Quan J, Sun Q, Russell J, Arnett S, Jurek P, Chen D, Kravchenko VV, Mathison JC, Moresco EM, Monson NL, Ulevitch RJ, and Beutler B. 2015. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, and Hornung V. 2016. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem 291: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Nunez G, Mao Y, and Wu H. 2019. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570: 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kandli M, Feige E, Chen A, Kilfin G, and Motro B. 2000. Isolation and characterization of two evolutionarily conserved murine kinases (Nek6 and nek7) related to the fungal mitotic regulator, NIMA. Genomics 68: 187–196. [DOI] [PubMed] [Google Scholar]
- 28.Fry AM, O'Regan L, Sabir SR, and Bayliss R. 2012. Cell cycle regulation by the NEK family of protein kinases. Journal of cell science 125: 4423–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards MW, O'Regan L, Mas-Droux C, Blot JM, Cheung J, Hoelder S, Fry AM, and Bayliss R. 2009. An autoinhibitory tyrosine motif in the cell-cycle-regulated Nek7 kinase is released through binding of Nek9. Mol Cell 36: 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blasi E, Mathieson BJ, Varesio L, Cleveland JL, Borchert PA, and Rapp UR. 1985. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature 318: 667–670. [DOI] [PubMed] [Google Scholar]
- 31.Minoguchi S, Minoguchi M, and Yoshimura A. 2003. Differential control of the NIMA-related kinases, Nek6 and Nek7, by serum stimulation. Biochem Biophys Res Commun 301: 899–906. [DOI] [PubMed] [Google Scholar]
- 32.Haq T, Richards MW, Burgess SG, Gallego P, Yeoh S, O'Regan L, Reverter D, Roig J, Fry AM, and Bayliss R. 2015. Mechanistic basis of Nek7 activation through Nek9 binding and induced dimerization. Nature communications 6: 8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S, Lee K, and Rhee K. 2007. NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem Biophys Res Commun 360: 56–62. [DOI] [PubMed] [Google Scholar]
- 34.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, Sharif H, Hu JJ, Evavold CL, Kagan JC, Schmidt FI, Fitzgerald KA, Kirchhausen T, Li Y, and Wu H. 2020. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng Q, Deng H, Li Y, Fan T, Liu Y, Tang S, Wei W, Liu X, Guo X, Jiang J, Wang Y, and Song D. 2021. Berberine Directly Targets the NEK7 Protein to Block the NEK7-NLRP3 Interaction and Exert Anti-inflammatory Activity. J Med Chem 64: 768–781. [DOI] [PubMed] [Google Scholar]
- 36.Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, and Wu H. 2021. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belham C, Roig J, Caldwell JA, Aoyama Y, Kemp BE, Comb M, and Avruch J. 2003. A mitotic cascade of NIMA family kinases. Nercc1/Nek9 activates the Nek6 and Nek7 kinases. J Biol Chem 278: 34897–34909. [DOI] [PubMed] [Google Scholar]
- 38.de Souza EE, Meirelles GV, Godoy BB, Perez AM, Smetana JH, Doxsey SJ, McComb ME, Costello CE, Whelan SA, and Kobarg J. 2014. Characterization of the human NEK7 interactome suggests catalytic and regulatory properties distinct from those of NEK6. J Proteome Res 13: 4074–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.