

Abstract
Deciphering immune recognition is critical for understanding a broad range of diseases and for the development of effective vaccines and immunotherapies. Efforts to do so are limited by a lack of technologies capable of simultaneously capturing the complexity of adaptive immune receptor repertoires and the landscape of potential antigens. To address this, we present RAPTR (Receptor-Antigen Pairing by Targeted Retroviruses). RAPTR combines viral pseudotyping and molecular engineering approaches to enable one-pot library on library interaction screens by displaying antigens on the surface of lentiviruses and encoding their identity in the viral genome. Antigen-specific viral infection of cell lines expressing human T or B cell receptors allows readout of both antigen and receptor identities via single-cell sequencing. The resulting system is modular, scalable, and compatible with any cell type. These techniques provide a suite of new tools for targeted viral entry, molecular engineering, and interaction screens with broad potential applications.
Introduction
The adaptive immune system recognizes and responds to the diverse pathogens humans encounter throughout their lives. B cells recognize extracellular antigens via the B cell receptor (BCR), leading to the production of secreted antibodies to inhibit and eliminate pathogens. T cells use T cell receptors (TCRs) to recognize short peptide fragments presented by major histocompatibility complex proteins (pMHCs), enabling antigen-specific coordination of the immune response and elimination of malignant or infected cells. This remarkable ability to sense, respond to, and remember threats is key to any successful adaptive immune response, and accordingly is the cornerstone of successful vaccines and immunotherapies.
The importance of T cell-mediated immunity has led to the development of a series of approaches dedicated to identifying antigens or pMHC-TCR pairs1. Conventional T cell assays, such as ELISPOT or intracellular cytokine staining2, provide direct readouts of T cell function. While these approaches can be highly multiplexed, they do not readily lend themselves to TCR sequencing and are limited in their ability to identify single reactive antigens. More recently, T-Scan and similar approaches have broadened the antigenic scope of functional assays to genome scale3,4, but cannot provide paired receptor information without panning of pre-determined TCRs or iterated steps of target antigen identification followed by sorting of reactive cells for TCR sequencing. Other recent cell-based reporter assays convert pMHCs into the recognition domain of immune signaling complexes5,6, or leverage trogocytosis7 or IL-2 capture8 to identify the pMHC targets of a given TCR. These approaches can identify successful TCR-pMHC interactions for single TCRs from antigen libraries on the scale of 103-104, but require substantial library redundancy, are limited in their ability to multiplex TCRs, and may require multiple rounds of screening. Requiring a discrete experiment per TCR imposes a significant scalability constraint; each individual has a TCR repertoire of as many as ~1012 unique clones1,9,10, and very little overlap is expected between individual repertoires even for people who share common MHC alleles.
Recombinant protein-based screening can broaden either the number of T cell clones or the number of antigens that are feasible to screen. Baculoviral display libraries have enabled screening of ~105 pMHCs by panning of infected Sf9 cells with fluorescently labeled recombinant TCRs11,12. Yeast display of pMHCs enables screening of ~108 unique pMHC antigens13,14, but can only examine a limited number of recombinantly expressed TCRs at a time, and can require significant optimization for each MHC allele. Conversely, recent advances in barcoded pMHC multimers enable screens on the order of 103 antigens in bulk15,16, or hundreds while maintaining receptor-antigen pairing17,18. While such analyses can be performed on polyclonal T cells, they are inherently bottlenecked by several technical limitations: (i) the need to manually assemble individual barcoded multimers; (ii) the ability to correctly identify interactions in large pools of multimers; and (iii) the relatively small set of MHC molecules that have been recombinantly expressed successfully.
Identifying antigenic targets of B cells poses similar limitations. Recent approaches have shown that antigen-BCR pairs can be identified via the oligonucleotide tagging of recombinantly expressed proteins19,20, which is inherently limited in scale. Thus, our understanding of antigen recognition by both arms of the adaptive immune system is currently constrained by significant hurdles to experimental scale.
Deciphering the full complexity of immune recognition requires the ability to screen for interaction pairs while incorporating diversity of both antigen receptors and their targets at the same time. If only the target antigens are known, it is difficult to understand the cellular factors regulating a successful response. On the other hand, if antigen receptors are characterized without knowledge of their specificity, it is difficult to understand which target antigens are important for preventing or eliminating disease. This represents a broader experimental challenge for interaction screens commonly known as “library vs library screening”. The most well-established approach to this problem is yeast two-hybrid21, in which intracellular protein pairs are used to drive expression of a reporter gene. Other previously established approaches include automated individual ELISA screens of recombinantly expressed protein pairs22–24, as well as mass-spectrometry-based identification of interacting pairs25,26. There have been several recent efforts at designing novel systems, including those based on yeast mating or spatial colocalization of DNA-barcoded molecules27,28. However, these approaches can be labor-intensive, are often not suited for complex, extracellular protein complexes such as immune receptors, and may be inefficient at low (i.e. micromolar) affinities. Together, these limitations have thus far rendered such approaches unsuitable for applications such as identifying TCR-pMHC pairs.
To overcome the above limitations, we have developed a technique that combines lentiviral surface display29–31 with a versatile pseudotyping strategy and viral genome engineering to enable one-pot library vs library screening (Figure 1). We demonstrate that our pseudotyping strategy – an engineered fusogen termed VSVGmut co-expressed with a targeting moiety – is general and versatile for both receptor and ligand usage. We leverage these abilities to present Receptor-Antigen Pairing by Targeted Retroviruses (RAPTR), which matches receptors with their cognate antigens based on specific infection of receptor-expressing cells by antigen-displaying viruses. Putative hits can be identified by bulk or single-cell sequencing, enabling screens of single or polyclonal receptors. We demonstrate the feasibility of this approach for both T and B cell receptors, including a library on library screen comprising 96 pMHC antigens and receptors enriched from a library of >450,000 TCRs (thousands of potential interactions).
Figure 1 |. The VSVGmut pseudotyping system is modular and utilizes a broad range of receptors and ligands.
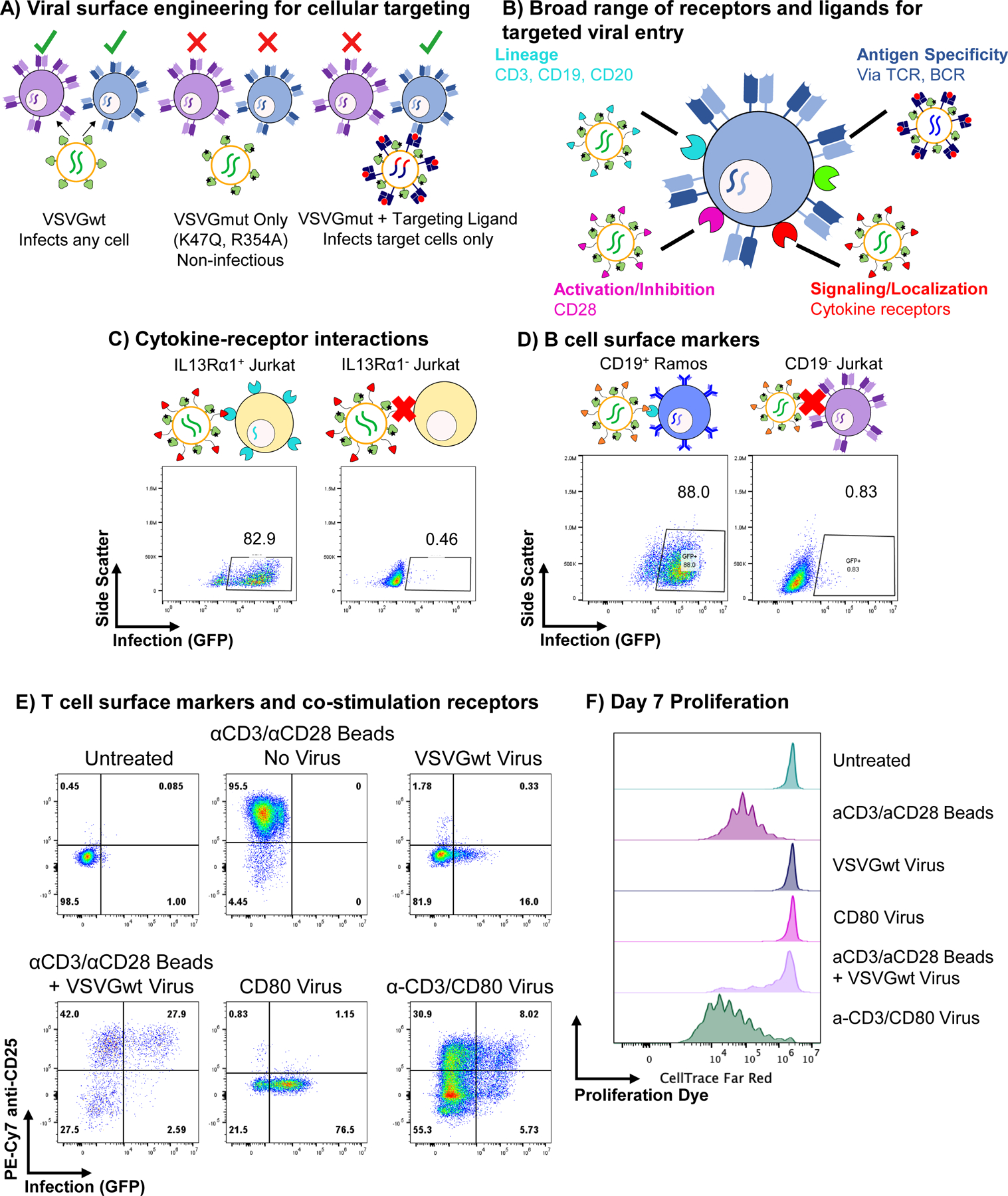
A, Schematic of the VSVGmut (VSVG K47Q, R354A) pseudotyping system enables specific targeting by coexpression of receptor-blinded VSV-G and a modular targeting ligand. B, VSVGmut is compatible with a broad range of receptors and targeting ligands, including immune cell surface markers, signaling and antigen receptors. C, Specific infection of IL-13Rα1-expressing Jurkat cells (left) as compared to parental cell line (IL-13Rα1−, right) via VSVGmut lentiviruses displaying surface tethered IL-13; data representative of 2 biologically independent experiments. D, Specific infection of CD19+ Ramos cells (left), but not CD19− Jurkat cells (right) by VSVGmut pseudotyped viruses displaying an anti-CD19 scFv; data are representative of 2 biologically independent experiments. E, Infection of Primary CD8 T cells (MOI = 1) via lentiviruses displaying anti-CD3 Fab and/or the costimulatory receptor CD80, or VSVGwt viruses with or without anti-CD3/anti-CD28 magnetic beads; data were collected at day 4 post-infection and are representative of three biological replicates. F, Proliferation of Primary CD8 T cells following viral infection described in E, as measured by dilution of cell tracking dye added at Day 0 prior to the addition of virus. Data are for Day 7 post-infection.
Results
VSVGmut pseudotyped lentiviruses enable modular tropism
Developing a scalable pipeline for antigen-receptor screening presents several requirements: (i) a straightforward ability to track both receptor and antigen sequences; (ii) mammalian expression systems to maximize the ability to express complex, multimeric proteins; and (iii) the ability to generate selection reagents without the need to recombinantly express and characterize each protein for every experiment. To address these needs, we turned to lentiviruses, which have decades of precedent for facile molecular manipulations. They can be created at large scale, are already known to infect T cells, and, upon successful infection, leave a permanent record of infection due to their integration into the host genome.
Lentiviruses have been pseudotyped via a number of different strategies to enable their use as biotechnology tools and gene therapy vectors32. Due to its robustness and efficient infection of many cell types, vesicular stomatitis virus G protein (VSVG, referred to as VSVGwt in this manuscript) is the most common pseudotype for laboratory studies and cell manufacturing for clinical applications33. More recently, approaches to enable cell type-specific targeting, via co-expressing receptor-blinded versions of Sindbis virus34–36 or paramyxovirus envelope proteins37–42 with targeting moieties have been described. While these approaches show promise, they have been reported to have strict limitations on targeting ligand and receptor binding topology due to their mechanism of entry.
Since these factors constrain the generality of an interaction screening system, we developed an alternative strategy based upon VSVG. We used the recently described crystal structure of VSVG in complex with its native receptor43, the low-density lipoprotein receptor (LDLR), to engineer VSVGmut, which incorporates the K47Q and R354A mutations reported to ablate affinity for the LDLR family of receptors (Figure 1A). To retarget VSVGmut pseudotyped viruses, we co-expressed a variety of surface-bound molecules during viral production (Figure 1B). To benchmark against established systems38, we used IL-13 as a viral targeting ligand. After testing several surface architectures (Extended Data Figure 1), we found that while many distinct constructs conferred specific infection of IL-13Rα1-expressing cells, the dimerized, surface-tethered IL-13 yielded the most efficient infection while retaining excellent specificity (Figure 1C). We also found that VSVGmut pseudotyped viruses displaying an anti-CD19 single-chain antibody fragment (scFv) efficiently infected CD19+ B cell lines, but not CD19− T cell lines (Figure 1D).
Next, we sought to exploit the unique modularity of the VSVGmut system to develop synergistic targeting strategies. While display of the anti-CD3 Fab UCHT1 yielded only modest infection of Jurkat T cell lines, CD80 mediated robust infection (Extended Data Figure 2A). Although the infection rate for the synergistic strategy was not better than for CD80 alone, we reasoned that the ability to incorporate multiple signals during infection could be useful for engineering primary cells by providing user-defined phenotypic inputs. Therefore, we used these viruses to infect primary CD8 T cells and measured infection and CD25 upregulation as a marker of activation (Figure 1E). We observed that the anti-CD3/CD80 combination viruses both infected and activated cells, but viruses displaying CD80 alone infected but did not activate the cells. We also observed that the anti-CD3/CD80 combination viruses induced robust T cell proliferation, while viruses displaying CD80 alone did not cause proliferation (Figure 1F). To our knowledge, this is the first example of a viral targeting system capable of simultaneously delivering multiple synergistic signals while infecting cells.
Antigen-specific T cell targeting by engineered lentiviruses
Following the observation that anti-CD3 viruses yielded infection of T cells via a component of the TCR complex, we next sought to determine whether we could achieve antigen-specific cell entry via the TCR itself. One potential challenge to repurposing the TCR-pMHC interaction for viral entry is its affinity, which is typically 1–50 μM44. We therefore generated cell lines expressing a range of previously characterized affinity variants for the 1G4 TCR45,46, which recognizes a peptide derived from the NY-ESO-1 cancer testis antigen (SLLMWITQV) presented in the context of HLA-A*02:01, with reported affinities ranging from picomolar to micromolar (Figure 2A). To display these molecules on the virus surface, we expressed them as single-chain trimers47, which are comprised of covalently linked peptide, beta-2-microglobulin, and MHC. We found that pMHC-displaying lentiviruses are able to efficiently infect T cells in an antigen-dependent manner. We observed similar infection efficiency across the tested affinity range, with only a modest reduction for the lowest-affinity variant (Figure 2B). To ensure that TCR-mediated infection was generalizable, we displayed several individual pMHCs as single-chain trimers alongside VSVGmut and used them to infect either Jurkat cells expressing its parental TCR (off-target) or on-target J76 cell lines expressing their cognate TCRs (Figure 2C). We observed specific infection across 3 different TCR-pMHC pairs with minimal background infection. As VSVG enables cell entry via an endocytic route48, and because endocytosis is one of the earliest downstream events following T cell activation49–51, we next sought to determine whether viral infection was accompanied by TCR signaling. We measured CD69 expression following viral infection (Figure 2D), and observed robust CD69 upregulation during TCR-mediated viral entry, but not during off-target combinations or VSVGwt infection (Extended Data Figure 2C). Moreover, TCR-mediated entry was inhibited by dasatinib, a TCR signaling inhibitor52, while TCR-independent infection via VSVGwt was unaffected (Extended Data Figure 2B). Thus, we concluded that pMHC-targeted viruses integrate both binding and signaling as a means of infection. Interestingly, pMHC-based targeting was more efficient than infection by viruses displaying an anti-CD3 Fab. While it is unclear why this is the case, it is possible that the pMHC-based approach is more efficient at inducing TCR signaling when engaged as monomeric proteins, or that the orientation of the pMHC is more favorable for viral entry than the CD3 epitope recognized by the anti-CD3 Fab.
Figure 2 |. TCR-mediated infection is sensitive, specific, and induces signaling.
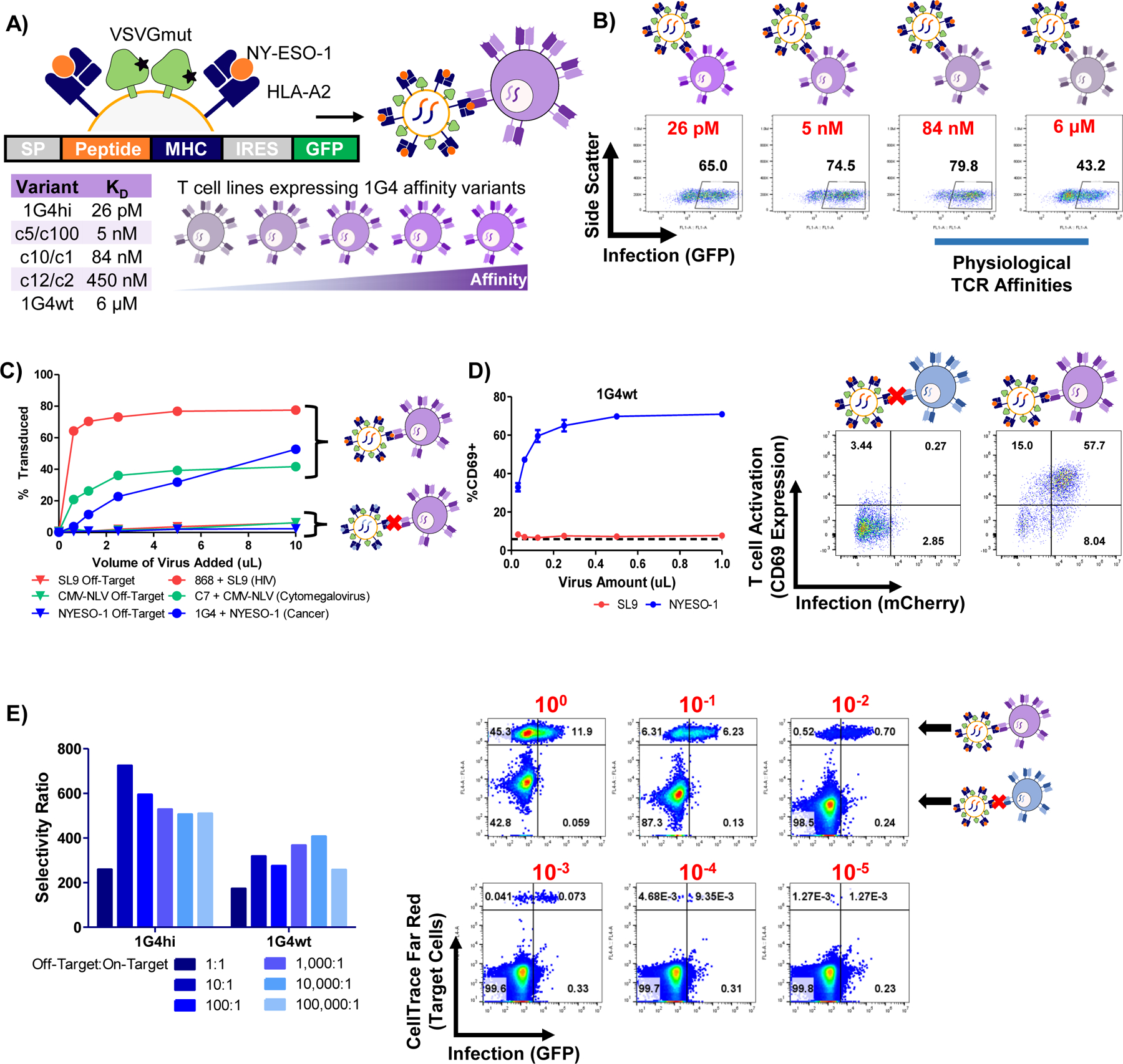
A, Schematic of the 1G4 TCR-HLA-A2/NY-ESO-1 system used for TCR-pMHC characterization. B, Representative infection data for various 1G4 TCR affinity variants by HLA-A2/NY-ESO-1 displaying VSVGmut lentiviruses. C, infection of J76 cells via specific TCR-pMHC interactions via pMHC-displaying VSVGmut lentiviruses for three independent TCR-pMHC pairs (HLA-A2/SL9-displaying viruses infecting 868 TCR-expressing J76 cells; HLA-A2/NLV-displaying viruses infecting C7 TCR-expressing J76 cells; HLA-A2-NY-ESO-1-displaying viruses infecting 1G4 TCR-expressing J76 cells). D, Upregulation of CD69 on J76 cells transduced with the 1G4wt TCR during viral entry; data shown represent mean + SD across three biological replicates, dashed line represents CD69 expression in untreated J76-1G4wt cells. E, Selectivity ratio of on target to off target infection of J76 cells expressing 1G4 TCR variants mixed at indicated ratios with off-target Jurkat cells; representative of three independent experiments, flow plots correspond to 1G4wt TCR-expressing J76 cells with target cell frequencies indicated in red. The selectivity ratio was calculated as the transduction rate of on-target cells divided by the transduction rate of off-target cells.
We next sought to determine the limitations of our system by characterizing its sensitivity and specificity using the 1G4-NY-ESO-1 system. To determine the ability of our approach to discern on-target interactions in complex mixtures, we labeled 1G4-expressing cells with a cell tracking dye, mixed them at varying ratios with unlabeled Jurkat cells, and infected the mixture with NY-ESO-1/HLA-A2 displaying viruses (Figure 2E). We calculated a selectivity ratio of on-target infection relative to off-target infection by dividing the transduction rate of on-target cells by the transduction rate of off-target cells, and observed on-target selectivity greater than 200:1, down to target cell frequencies of 1× 10−5. Thus, we concluded that our system can detect on-target interactions in the micromolar affinity range, even in complex mixtures containing hundreds of thousands of non-target T cells.
A viral packaging system to maintain protein-barcode linkage
To fully enable interaction screening at library scale, we next turned to the challenge of producing the lentiviral libraries. While lentiviral screens have been widely used for functional genomics, standard lentiviral packaging techniques pose inherent limitations that are particularly relevant to interaction screens. The key challenge stems from the fact that at least hundreds of plasmids containing library elements are mixed in each transfected packaging cell (Figure 3A). In functional genomics, this leads to well-described intermolecular recombination of library elements53,54, which is a significant source of noise in combinatorial experiments. To date, solutions to this problem have included plasmid dilution (accompanied by a 100x reduction in viral titer)54, or simply restricting libraries to sizes suitable for arrayed screens55–57. This problem is compounded in our approach, as multiple plasmids entering the same packaging cell would cause multiple different targeting molecules to be expressed on each virus surface, compromising the link between viral genotype and surface phenotype.
Figure 3 |. LeAPS-based lentiviral packaging enables scalable viral library construction.
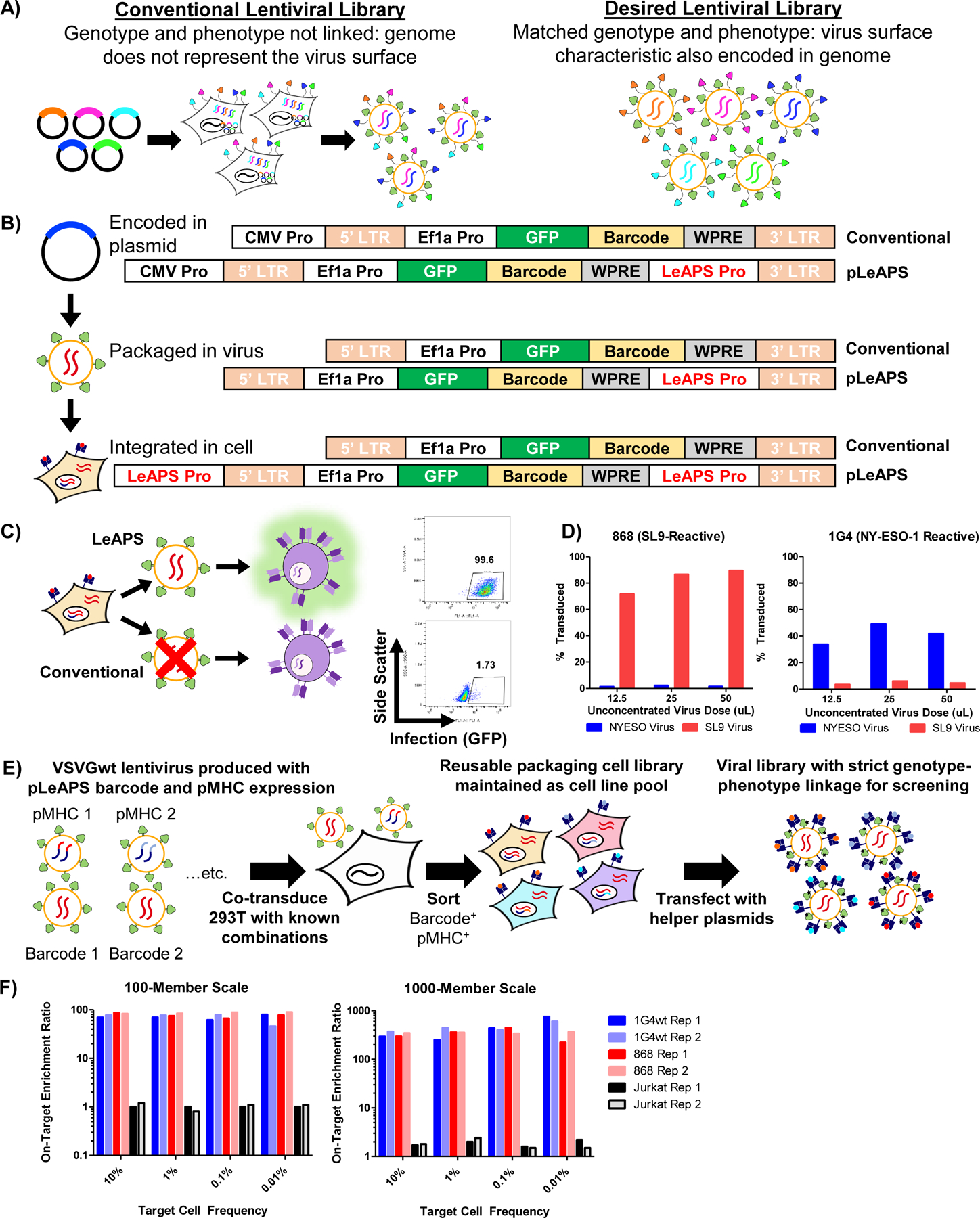
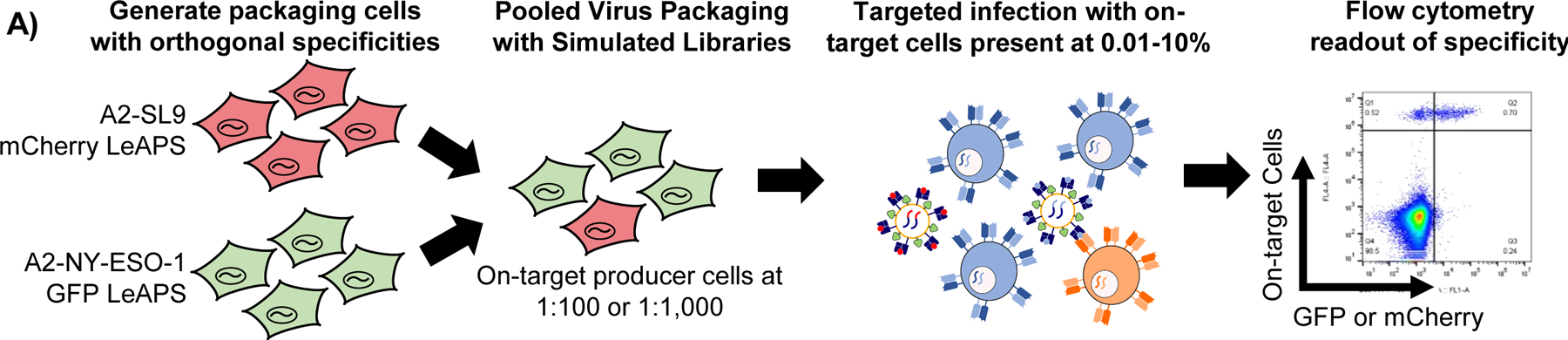
A, Schematic overview of the limitation of conventional lentiviral packaging strategies for surface displayed lentiviral libraries. B, Schematic overview of the mechanism of promoter translocation used in the LeAPS system. C, Packaging cells transduced with viruses containing LeAPS-encoding genomes and then transfected with lentiviral helper plasmids (top), and transduced with viruses containing standard genomes (bottom). D, LeAPS-produced viruses maintain TCR specificity in two independent TCR-pMHC systems,. E, Schematic overview of the implementation of LeAPS for viral library assembly. F, On-target enrichment of libraries using defined mixtures of A2-NY-ESO-1 and A2-SL9 packaging cells to represent 100- or 1,000-variant libraries.
To obviate this issue, we exploited a detail of HIV-1 replication that results in the copying of sequences between the polypurine tract and the 3’ LTR to the 5’ end of the genome during reverse transcription and integration58. There have been several recent reports exploiting this phenomenon to enable genomic screens via copying of CRISPR gRNA cassettes59,60. Here, we insert a promoter to enable re-packaging of the viral genome following infection of a cell61 (Figure 3B). Conventional 3rd generation lentiviral packaging approaches are inherently self-inactivating in part due to an inability to transcribe packageable viral genomes following infection; i.e. the LTRs, which are weak promoters, are truncated, and there is no promoter upstream of the viral genome following successful integration. Our approach, which we term Lentiviruses Activated by Promoter Shuffling (LeAPS), enables generation of lentiviruses from previously transduced cells upon the re-introduction of helper plasmids by placing a strong promoter 5’ of the integrated viral genome, allowing the integrated vector to produce additional viral genomes at high copy number. As a result, we can use lentiviral transduction to ensure a single library member per packaging cell without incorporating additional viral elements into the genome. We first validated that viruses produced with the LeAPS strategy yielded high-titer virus (>106 TU/mL unconcentrated), whereas conventional constructs (without a LeAPS promoter) yielded very little following transduction (Figure 3C). By stably expressing pMHC cassettes in LeAPS-transduced packaging cells, we verified that viruses produced in this manner maintain antigen-specific targeting capability (Figure 3D).
To develop a scalable library assembly strategy, we co-transduced 293T cells with two VSVGwt-pseudotyped viruses: one that drives surface expression of a known pMHC using a conventional lentivirus, and another that uses the LeAPS system to deliver a repackageable genome driving expression of a fluorescent protein and a defined barcode (Figure 3E). Following initial transduction, packaging cells for each library member were pooled and sorted for expression of pMHC and barcode in a single sorting step. This yields a library packaging line that can be used to generate the same lentiviral library indefinitely via single transfections with only helper plasmids. By contrast, conventional approaches require arrayed transfection to produce each library member for each experiment62, which poses a substantial impediment to experimental throughput.
To validate the utility of our approach, we employed a two-component system of the HIV SL9 antigen (SLYNTVATL) paired with a LeAPS-mCherry cassette and the NY-ESO-1 antigen paired with a LeAPS GFP cassette. By mixing the packaging cells for each at different ratios, we assessed the feasibility of screening pMHC libraries at 100-member and 1,000-member scales. To assess the feasibility of a 1,000-member library, we mixed the SL9 and NY-ESO-1 LeAPS packaging cell lines at a ratio of 1:1,000 by cell number, produced virus in pool via transfection with helper plasmids, and used the resulting viral pools for target infections of TCR knock-in T cell lines (Extended Data Figure 3). We then used flow cytometry to quantify signal to noise across different frequencies of on-target cells (Figure 3F). Across both library sizes, and extending from target cell frequencies of 10% down to 0.01%, we observed robust enrichment of on-target interactions, but minimal enrichment of off-target interactions (i.e. no enrichment of either component on Jurkat cells expressing an irrelevant TCR). These results gave us confidence that the screening step would be feasible at scales of at least 1,000 pMHC variants using the LeAPS library generation approach.
Receptor-Antigen Pairing by Targeted Retroviruses (RAPTR)
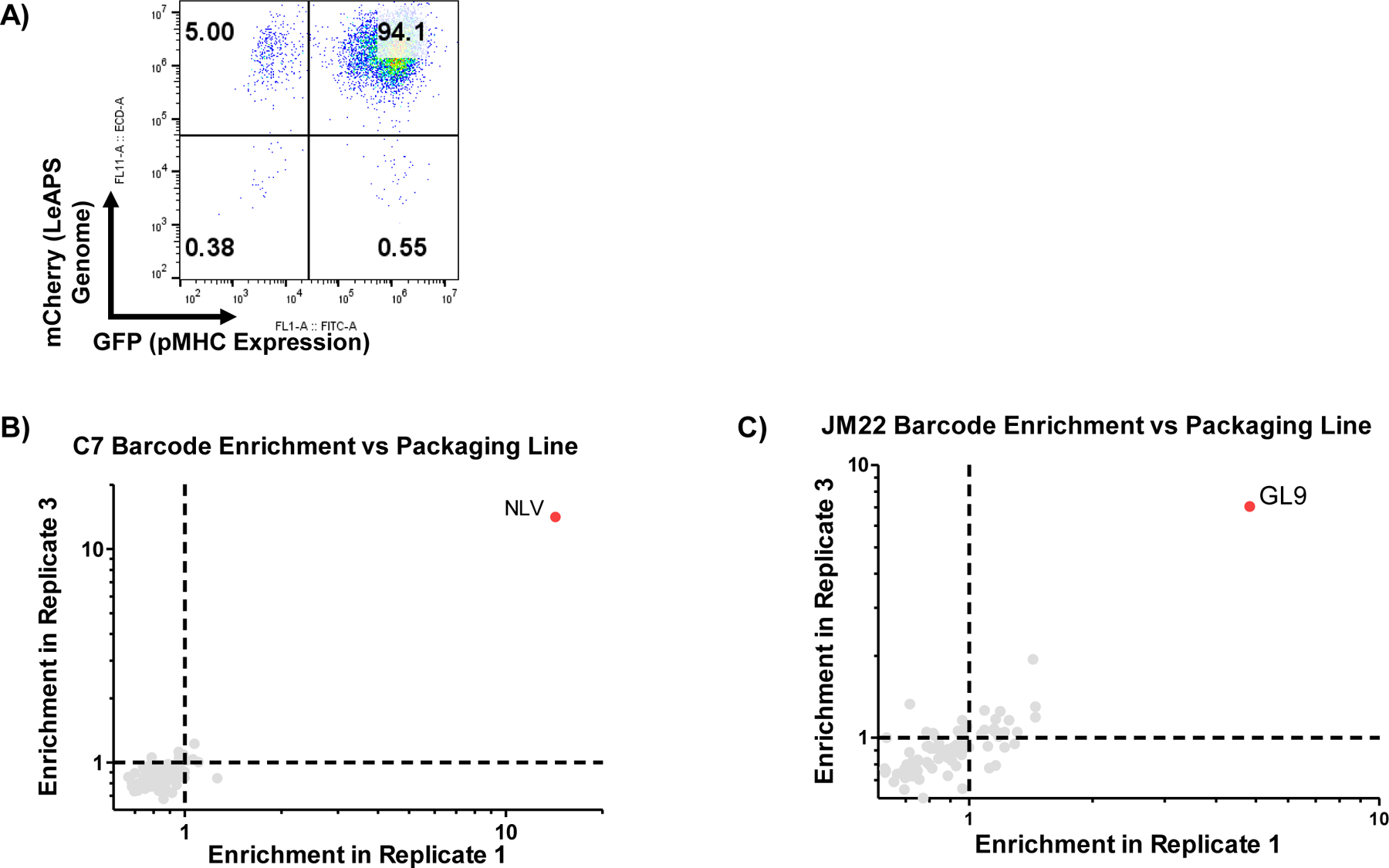
With all the necessary tools for RAPTR validated, we assembled a library of 96 pMHC constructs consisting of known seroprevalent viral or vaccine antigens from the Immune Epitope Database (IEDB)63, and further filtered for binding to HLA-A2 using NetMHC4.164 (Supplementary Tables 1 and 2). We co-transduced each into 293T cells alongside a LeAPS vector encoding a barcode, pooled the resulting cells proportionally, and sorted for cells expressing both barcode (via mCherry) and pMHC (via GFP). We verified that antigen and barcode expression in our packaging cell library was stable after at least 9 passages post-sort (Extended Data Figure 4A). We next used the library to infect a cell line expressing the C7 TCR65, which is known to recognize the CMV peptide NLVPMVATV (known as NLV). Without any cell sorting step required, we sequenced genomic DNA from transduced cells to enumerate barcode frequencies (Figure 4A). We observed a large enrichment of NLV barcode in transduced cells relative to the packaging cell line (Figure 4B), with NLV representing 15% of all reads in transduced cells and no notable enrichment of other sequences. We further validated this approach in C7 replicates (Figure 4C and Extended Data Figure 4B), and in a separate screen of the JM22 TCR 65, which recognizes the GL9 peptide from influenza A (Figure 4D and Extended Data Figure 4C). In all cases, we observed robust enrichment of on-target, but not off-target, barcodes.
Figure 4 |. RAPTR enables facile antigen identification for T cell receptors.
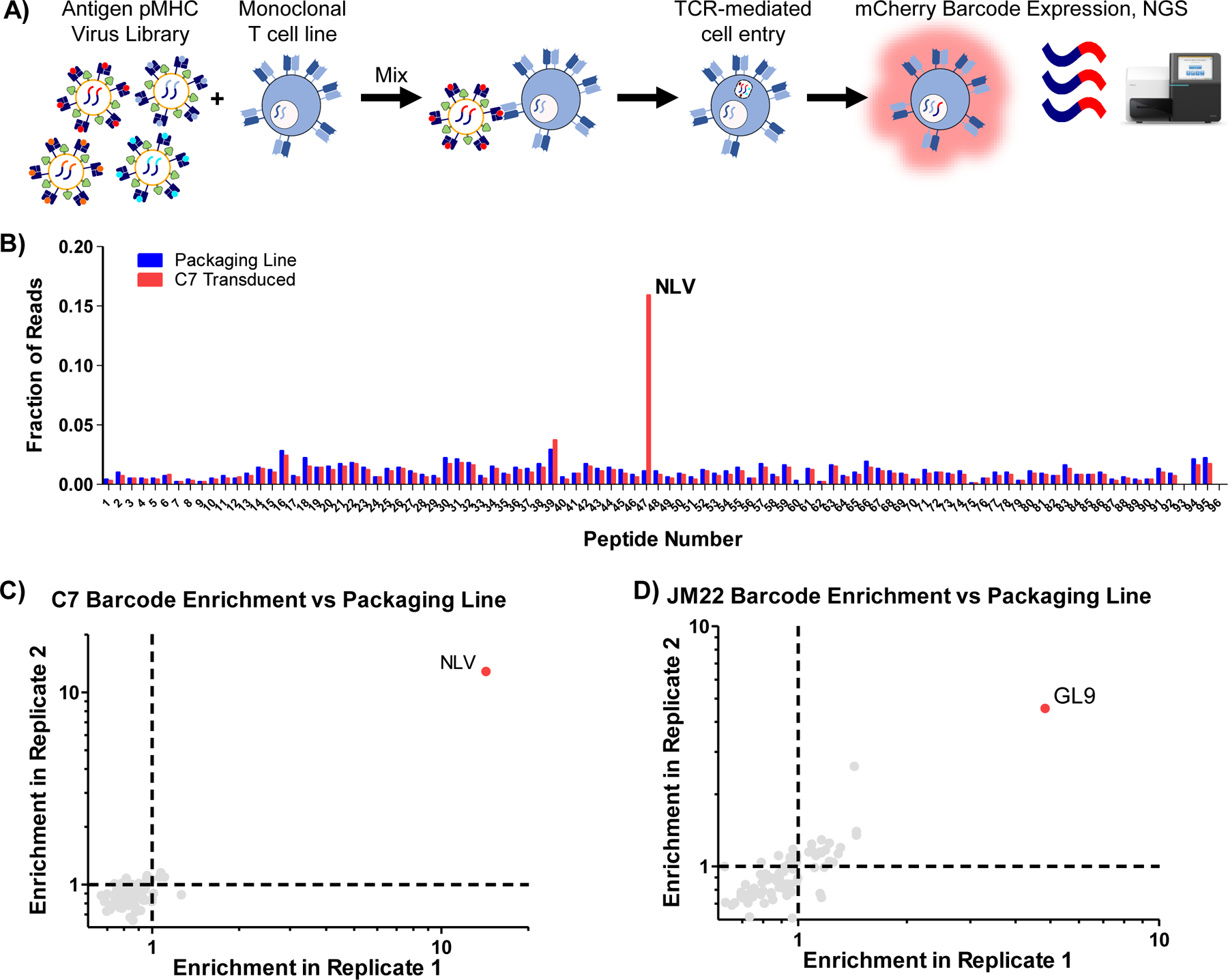
A, Workflow for RAPTR on monoclonal cell lines. The 96-member pMHC-displaying lentiviral library was mixed with J76 cells expressing a known TCR. After 2 days of infection, genomic DNA from unsorted transduced cells was harvested and analyzed by NGS. B, Read fraction of barcodes following infection of C7 TCR-transduced J76 cells. Blue bars represent the read fractions of barcodes from the 293T packaging cell line used to produce the viral library, and provides the unenriched (null) distribution absent TCR selection. The red bars indicate the read fraction in transduced C7-expressing J76 cells, revealing the selectivity of the TCR based on increasing read fraction of the barcode for the cognate ligand (NLV). C, Comparison of antigen enrichment relative to the packaging line upon library infection of C7 TCR-transduced J76 cells across two additional replicates. D, Comparison of antigen enrichment relative to the packaging line upon library infection of JM22 TCR-transduced J76 cells across two replicates.
RAPTR can be adapted to B cell receptors
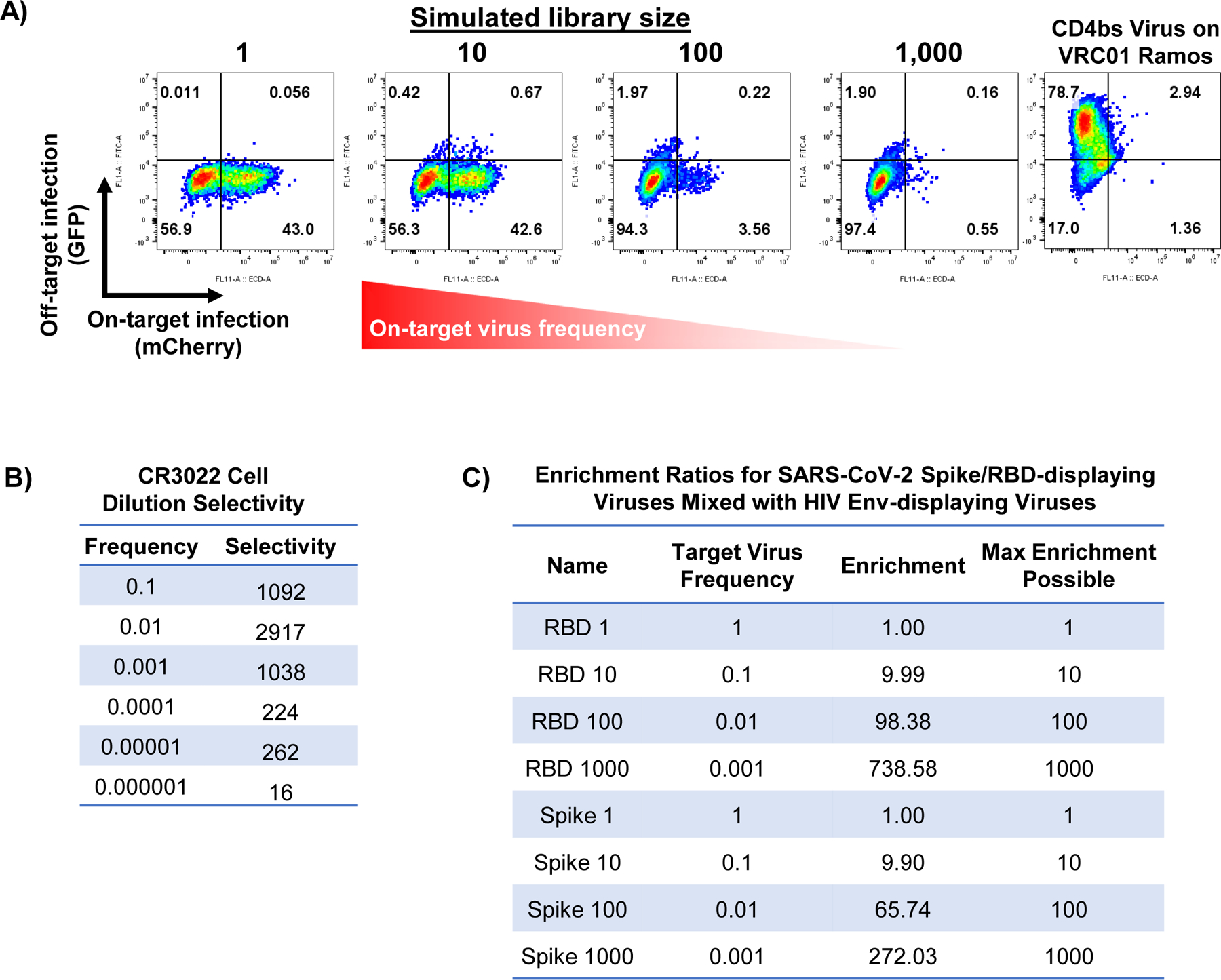
Many of the same constraints that limit TCR-pMHC interaction screens are also shared by efforts to identify B cell antigens at scale. Most approaches rely on barcoded multimers for very few targets, and the largest antigen library reported to date is 9 antigens20. As BCRs are also rapidly endocytosed upon antigen engagement66,67, we reasoned that we could use stabilized viral surface antigens or known immunogens as targeting ligands for viral infection. We thus generated a “hybrid” pseudotype co-expressing either the surface-tethered receptor binding domain (RBD) or the full-length, pre-fusion stabilized spike (S2P) protein from SARS-CoV-2 68 alongside VSVGmut (Figure 5A). We used each of these to achieve efficient, antigen-specific infection of a Ramos cell line expressing CR3022, a B cell receptor clone that is cross-reactive for the RBDs of SARS-CoV-1 and 2 69,70 (Figure 5B). Similarly, hybrid pseudotypes incorporating HIV env CD4 binding site (CD4bs) constructs from diverse clades 71 each infected cell lines expressing the bNab VRC01 72, and a known affinity-reducing mutation (D368R) dramatically reduced, but did not fully abrogate, infection 73.
Figure 5 |. RAPTR enables profiling of B cell reactivity via infection of BCR knock-in cells.
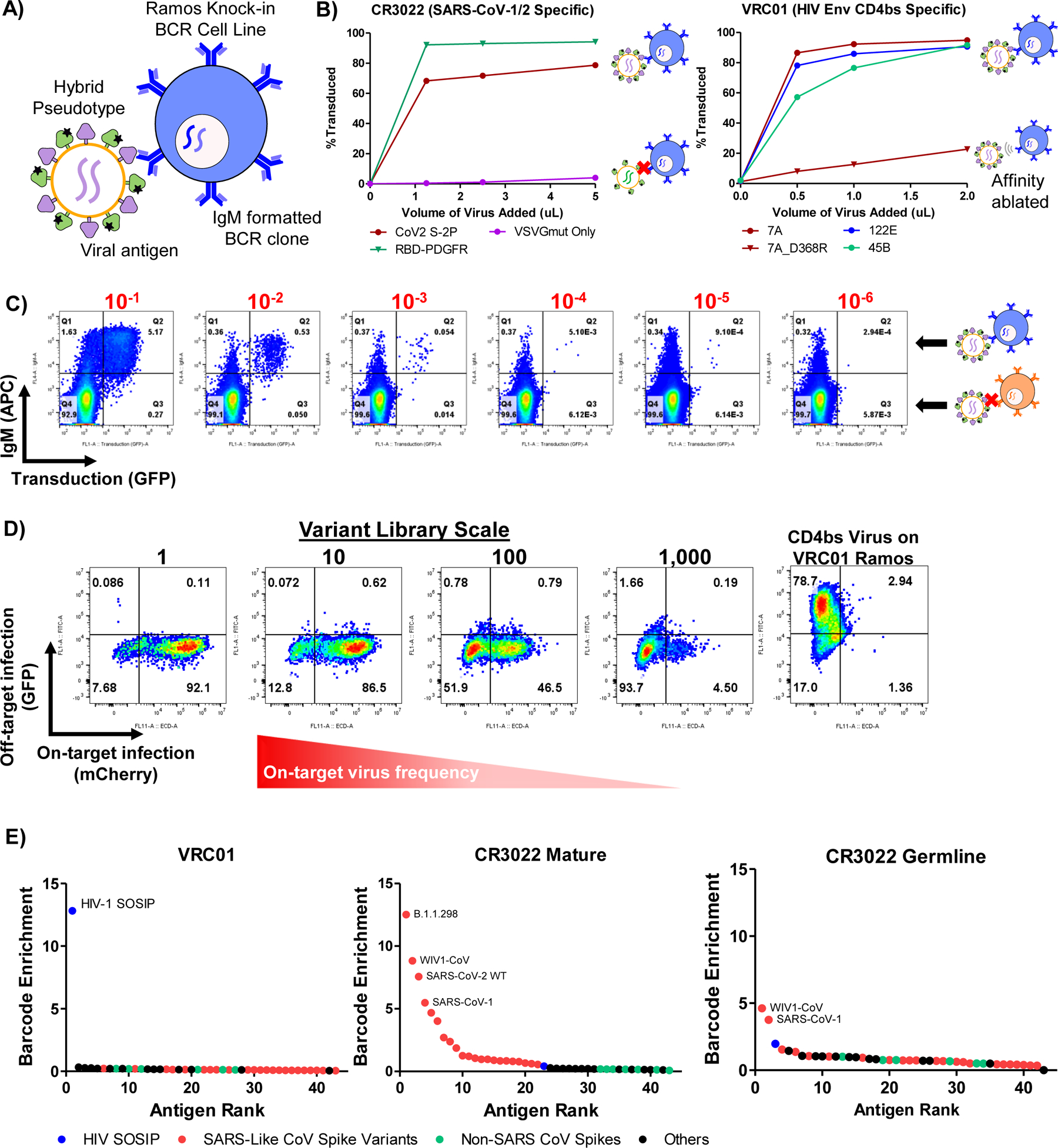
A, Overview of BCR-based targeting via the VSVGmut hybrid pseudotype in which a viral protein is co-expressed with VSVGmut. B, Targeting of Ramos cells expressing the SARS-CoV S protein specific CR3022 BCR via VSVGmut lentiviruses displaying either SARS-CoV-2 spike (2P) or surface-bound SARS-CoV-2 S protein RBD, or VRC01 BCR via env constructs representing multiple clades of HIV; representative data from 2 independent experiments. C, SARS-CoV-2 RBD-displaying viral infection of CR3022 BCR-expressing Ramos cells (on-target, IgM+) mixed into IgM-negative Ramos cells (off-target, IgM−) at target cell frequencies indicated in red, demonstrating selectivity in a mixed population of cells. D, Infection of CR3022 BCR-expressing Ramos cells with mixtures of SARS-CoV-2 RBD (on-target, expressing mCherry) and HIV env (off-target, expressing GFP) viruses; variant library size indicates the ratio of off-target to on-target virus present. Infection of VRC01-expressing Ramos cells with HIV env hybrid pseudotyped viruses (right) demonstrates that the viral particles remain functional. E, Enrichment of antigen barcodes relative the viral packaging cell line following viral glycoprotein library infection of Ramos cells expressing BCRs for VRC01, mature CR3022, or the germline-reverted version CR3022 demonstrating selectivity even for antibody sequences before affinity maturation.
To determine the sensitivity for BCR-mediated cell entry, we mixed Ramos cells expressing either the CR3022 BCR (IgM+) or no BCR (IgM-negative), and tracked on-target cells via surface IgM expression (Figure 5C, Extended Data Figure 5). Using RBD-displaying viruses, we observed specific infection at target cell frequencies at least as rare as 1×10−5, but could not calculate a meaningful selectivity ratio due to a lack of off-target infection. Next, to assess the potential feasible library size, we mixed on-target RBD-displaying viruses encoding GFP with off-target CD4bs-displaying viruses encoding mCherry and used them to infect CR3022 BCR-expressing Ramos cells (Figure 5D, Extended Data Figure 5C). Keeping the total virus amount constant, we were able to robustly detect on-target interactions with minimal off-target infection in mixtures as dilute as 1:1,000 on-target viruses. We observed similar results when displaying SARS-CoV-2 S2P instead of RBD (Extended Data Figure 5). These results gave us confidence in the feasibility of extracting meaningful signal from interaction screens using lentiviral infection via the BCR.
Next, we sought to establish the capabilities of RAPTR for BCR profiling in a true library setting. We assembled a library composed of 21 prefusion-stabilized SARS-CoV-2 spike variants and 22 additional vial surface glycoproteins or immunogens from diverse sources as controls to enable proof of concept experiments (Supplementary Tables 3 and 4). When we used this library to infect cells expressing the VRC01 BCR, we observed clear enrichment of only HIV-1 SOSIP74 (Figure 5E), highlighting the potential for direct receptor de-orphanization. When we infected cells expressing the CR3022 BCR, we observed clear enrichment of SARS-CoV spikes, including multiple SARS-CoV-2 variants. Notably, a version of CR3022 that excluded the mutations acquired via somatic hypermutation yielded enrichment of only SARS-CoV-1 spike and the closely related WIV-1 spike, highlighting the ability of RAPTR to distinguish BCR cross-reactivity even for naïve BCRs that have yet to undergo affinity maturation.
RAPTR enables library-on-library screens
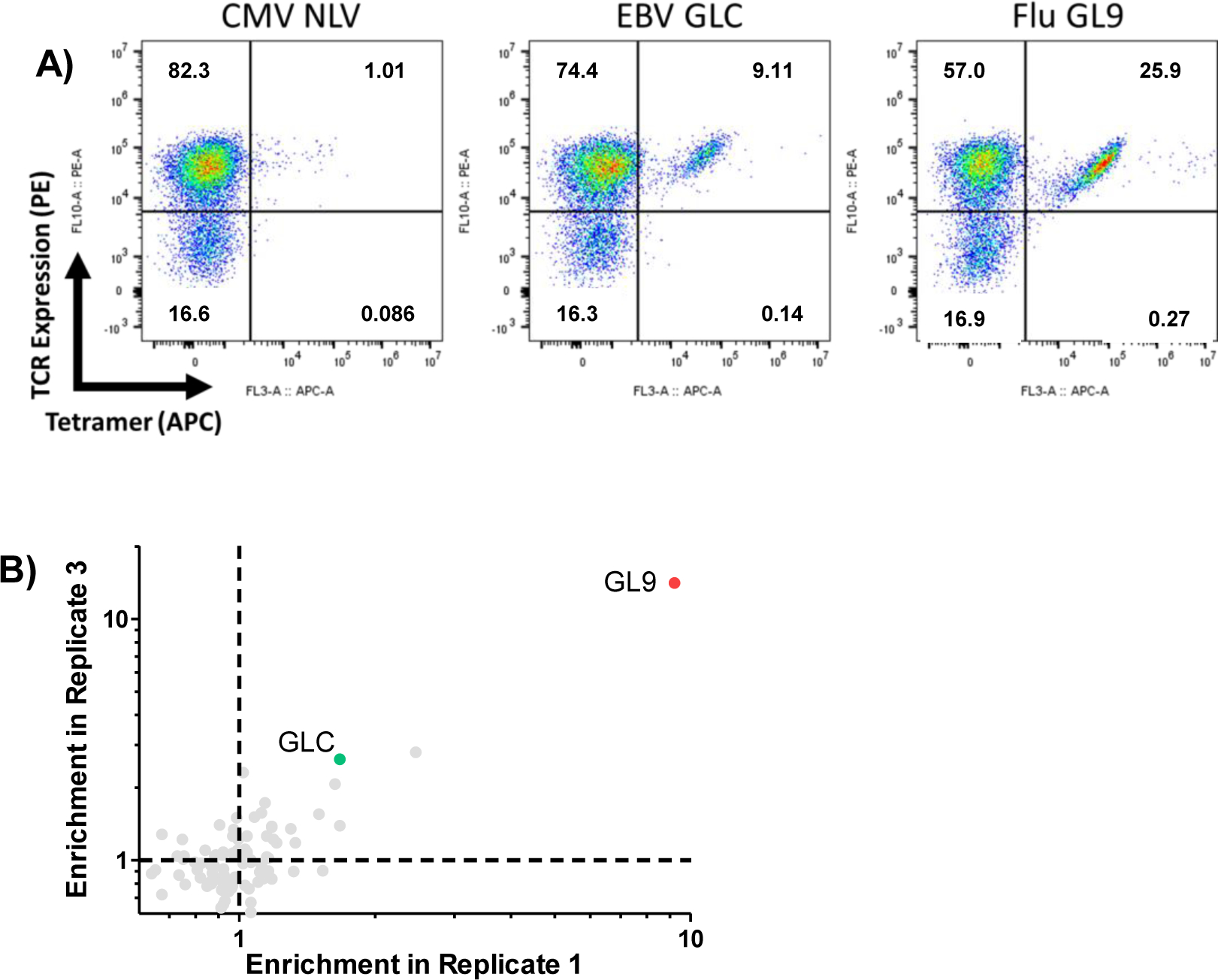
To fully realize the potential of RAPTR for antigen identification, we sought to apply it to library vs library screening by using our 96-member pMHC viral library to pan a previously reported library of >450,000 TCRs75. To simplify the first attempt, we pre-enriched for potentially reactive cells using tetramers for CMV, EBV, and Flu (CEF) antigens presented by HLA-A2, resulting in a polyclonal pool of TCRs with greater prevalence of these specificities, similar to the routine process of pre-expanding cells (Figure 6A). Following transduction and FACS to isolate transduced cells, we used bulk sequencing to identify which antigens were enriched in aggregate. In line with tetramer staining of the untransduced cells, we observed strong enrichment of Flu GL9, and more modest enrichment of EBV GLC (Figure 6B, Extended Data Figure 6).
Figure 6 |. RAPTR scales to polyclonal antigen identification in a single, scalable pipeline.
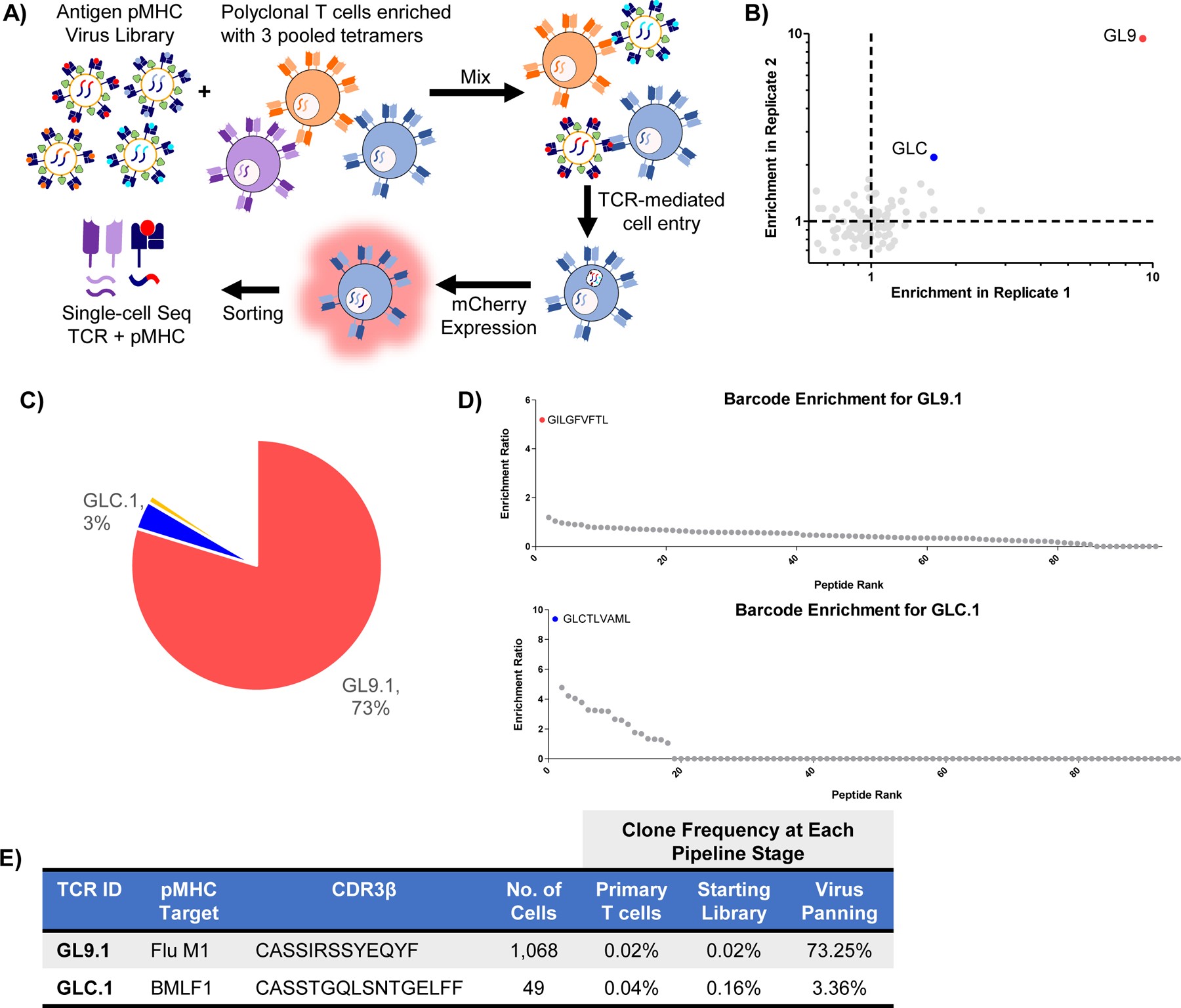
A, Schematic overview of the library on library RAPTR paradigm for TCR antigen identification. The 96-member viral library is mixed with polyclonal TCR-expressing J.RT3 cells; 2 days post-infection, mCherry+ cells were sorted and analyzed by single-cell RNA sequencing to identify expressed TCRs and enumerate integrated pMHC barcodes. B, Bulk sequencing of barcodes across all infected cells following infection of enriched CSS-930 TCR library with viral antigen library shows enrichment of immunodominant EBV and Influenza antigens in two independent infections. C, Clonal frequencies of enriched TCRs in cells analyzed by single cell sequencing; white space represents TCRs found in < 2 cells. D, Enrichment of antigen barcodes in the top two most abundant clones relative to the viral packaging cell line. E, Detailed tracking of TCR clones with matched antigens, including CDR3 sequence, number of cells analyzed, and clonal frequencies at each stage of the pipeline.
With this validation in hand, we performed single-cell RNA sequencing using the 10x Genomics Chromium 5’ chemistry with V(D)J enrichment. We included a targeted primer at the reverse transcription step and a custom amplification protocol to enable recovery of our pMHC barcodes, akin to ECCITEseq and other similar methods76,77. In this experiment, we recovered 1,458 cells, with TCR clonotypes dominated by a few clones (Figures 6C and 6E). We grouped cells of common clonotype together and analyzed pMHC barcode expression to determine enrichment relative to packaging library frequency. The most prevalent TCR clone, GL9.1, demonstrated strong enrichment of GL9 pMHC signal (Figure 6D). Upon literature search, we noted that GL9.1 represents a well-characterized public clonotype known to recognize GL9 peptide presented by HLA-A278.
The next most abundant clone, GLC.1, was identified in a previous screen of this same TCR library by recombinant HLA-A2 GLC dextramer75. Our screen also identified GLC as the target pMHC (Figure 6D). Notably, this was the only clone previously identified by GLC dextramer panning that was validated by monoclonal binding and functional tests. Our approach did not identify any of the previously reported false positives as hits, providing further evidence that RAPTR is an efficient means of identifying T cell antigens that integrates both binding and signaling. Finally, while T cells that recognize NLV are common in HLA-A2+ CMV seropositive individuals79, we did not observe any NLV-reactive cells (Figure 6B). However, this result is consistent with previously reported screening of the same library via orthogonal methods75, and our tetramer staining data (Extended Data Figure 6A).
Discussion
RAPTR is a high-throughput platform for directly linking immune receptors with their cognate antigens. It is based on the combination of several conceptual and technological advances that enable (i) versatile and efficient targeted viral entry via the VSVGmut pseudotyping system; (ii) a method for scalable, reproducible lentiviral library packaging (LeAPS); and (iii) the use of viral entry as a means of screening for interactions. We demonstrated a system for efficient, antigen-specific infection via both the TCR and the BCR that integrates both binding and target cell activation. We then exploited this mode of infection to match receptors with target antigens in complex mixtures and in a library vs library format.
We demonstrated that the VSVGmut pseudotyping system is efficient, specific, and uniquely modular by targeting a variety of cell surface proteins, including cytokine receptors, costimulatory molecules, lineage markers, and both T and B cell receptors. We report here as many different targeting approaches for VSVGmut as have been reported for any other single pseudotyping system32, including the first true antigen-specific infection of lymphocytes and the first targeting approach that can simultaneously deliver multiple signals. In addition, the VSVGmut system produces viruses at higher average titers than other systems while maintaining a high degree of specificity. Whereas most pseudotyping systems have been limited to the use of small, stable, high-affinity targeting ligands (e.g. scFvs or DARPins), VSVGmut is uniquely versatile in ligand usage due to the modular design it permits. We report targeting even at micromolar affinities, whereas the efficiency of infection by paramyxoviral pseudotypes was reported to dramatically decrease in the low nanomolar range80. We observed efficient pMHC-based infection for both anti-viral TCRs (C7, JM22, and the TCRs recovered in the Fig. 6) and 1G4, which recognizes a self-antigen, suggesting that the full range of possible CD8+ antigens should be able to be profiled.
Applications of RAPTR
We have demonstrated the utility of RAPTR for identifying antigens recognized by TCRs and BCRs. For T cell reactivity, our approach could be directly applied to rapidly deorphanize TCRs from existing single-cell studies, which routinely pair TCR sequences with gene expression for cancer, autoimmunity, or infectious disease. Existing techniques, including yeast display, SABRs, and T-Scan, require individual screens to pair each TCR with its cognate antigen, including at least one cell sorting step per TCR. In contrast, RAPTR can be performed on pools of TCRs in a single step, with only one cell sorting step required (and no sorting required for single-TCR experiments). While barcoded pMHC multimers can be built in to single-cell workflows, large antigen pools are inherently difficult to produce and validate, and require an equivalent effort for each new batch. These factors have largely limited their use to only a few specialized labs thus far. RAPTR library production is straightforward and far more scalable in comparison, requiring only a simple transfection per batch following initial library assembly. In principle, these reagents could be made and distributed at large scale, either via sharing the packaging cell line or by leveraging current industrial infrastructure for production of lentiviruses.
For B cells, we envision applying pools of protein variants, such as the one presented here, to large libraries of BCRs. Such an approach would greatly accelerate isolation and optimization of cross-reactive mAbs to a wide range of target classes. Current techniques require the recombinant expression of individual mAbs followed by ELISA for each target of interest, which represents a major bottleneck to experimental scale and scope81. Using RAPTR with BCR knock-in cells, which could themselves be prepared as pooled lentiviral libraries70, could alleviate this bottleneck. For example, even scales smaller than the one we presented would be sufficient to represent every subtype of influenza hemagglutinin and neuraminidase, multiple representatives of each of the major clades of HIV-1, and more. Larger libraries on the scale of 1000s of variants, whose feasibility we demonstrated, could enable detailed antigenic site or epitope mapping for many mAbs at once, a process currently limited to structural studies that are difficult to scale.
Finally, the demonstrated modularity of the system opens the door to many potential applications. Since our approach only requires HEK cell surface expression of the targeting molecule, we hypothesize that RAPTR can be readily adapted to other class I MHC alleles, class II MHC alleles, as well as non-human systems for vaccine and immunotherapy development. This compares favorably to recombinant MHC expression or yeast display, each of which can require allele-specific optimizations and/or mutations to ensure proper folding13,82,83. However, our system is not restricted to immunological applications. In principle, it is applicable to any receptor-ligand system in which one binding partner can be displayed on a lentivirus and the other can be expressed as an endocytic receptor on cells. This may enable large-scale interactome and co-evolution studies that are not readily achievable by existing techniques. Furthermore, the VSVGmut pseudotyping system may be useful as a new platform for the development of efficient, specific gene therapies.
Limitations
As with any technology, RAPTR has several limitations. Our proof of concept studies were performed on ~100-antigen scale, and while our control experiments indicate this screening capability could be readily increased to 1,000s, these scales will still require pre-selection of antigen targets for analysis. Further streamlining library assembly and increasing transduction efficiency will be focuses for enabling larger-scale assays. At present, the LeAPS system involves one-time arrayed transfections and transductions at scales and complexity routinely performed for genomics screens55–57, after which lentiviral library generation can be performed in a straightforward manner by standard transfections of the pooled packaging cell library. As a result, library generation is currently the largest bottleneck to throughput. Due to the one-time nature of the bottleneck, we believe automation will readily enable library sizes of up to 103, but adapting our approach for pooled library generation could aid in further scaling.
In addition, the single-cell sequencing step must be performed in sufficient depth to facilitate acceptable signal to noise determinations. While we chose to use a commercially available platform to ensure broad applicability, recent and future advances in the scale of single-cell analysis84 can improve the utility of RAPTR regardless of platform. Like other genetically encoded techniques, RAPTR also requires prior selection of MHC haplotype(s) to generate libraries. The antigen identification studies we presented were on cell lines with receptors knocked in, rather than primary cells. While the decreasing cost and turnaround time of gene synthesis will make such resources increasingly available, future studies can apply RAPTR directly to primary cells. Moreover, immortalized TCR libraries are becoming increasingly common as library generation methods continue to improve75,85–87, and can serve as renewable resources to conduct deep profiling of low-abundance or irreplaceable samples such as patient-derived tumor infiltrating lymphocytes. Finally, for each of these cases, our data indicate that the ability to enrich for antigen-specific cells at library scale will be beneficial for efficient deorphanization, to ensure that target cells are present at sufficient frequencies for infection and analysis. Existing techniques, including stimulation via peptide pools, should readily achieve this but may require some optimization.
RAPTR is a versatile platform for high-throughput interaction screens that uniquely incorporates diversity of both receptors and ligands in a single assay. The resulting tools will enable detailed studies of antigen recognition for understanding and engineering the adaptive immune response, as well as broader interactome studies.
Online Methods
Ethics statement
This work was reviewed and approved by the MIT Institutional Review Board (Protocol number 1801190804).
Media and Cells
HEK293T cells (ATCC CRL-11268) were cultured in DMEM (ATCC) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologics) and penicillin-streptomycin (Pen/Strep, Gibco).
Jurkat (ATCC TIB-152), J76 cells, and Ramos cells (ATCC CRL-1596) were cultured in RPMI-1640 (ATCC) supplemented with 10% FBS and Pen/Strep. J76 cells88 (PMID 29707134) were a gift from Mirjam Heemskerk and Mark Davis.
CSS-930 TCR library cells were a gift from David Johnson75, and were cultured in RPMI-1640 (ThermoFisher) supplemented with 10% FBS, Pen/Strep, 1x non-essential amino acids (ThermoFisher), 2mM GlutaMAX (ThermoFisher), and 1mM sodium pyruvate (ThermoFisher).
Plasmid Construction
The plasmid pHIV-EGFP was gifted by Bryan Welm & Zena Werb (Addgene plasmid #21373) and pMD2.G and psPAX2 were gifted by Didier Trono (Addgene plasmid #12259 and #12260). pLentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid # 52961). IL-13 (Uniprot ID P35225) residues 35–146 were cloned into the pHIV backbone containing the Igκ leader peptide, the PDGFR stalk and transmembrane domain (Uniprot P09619 residues 449–497) with extracellular linkers listed in Supplementary Figure 1B. The anti-CD19 scFv FMC63 and was cloned into the IgG4 hinge-PDGFR display format in the pMD2 backbone. The anti-CD3 Fab UCHT1 was cloned into the PDGFR stalk only format. Human CD80 (Uniprot P33681, residues 1–273) was cloned into the pMD2 backbone. pMHC single-chain trimers47 were cloned into either the pMD2 backbone for individual infections or the pHIV backbone for library construction. To generate the pLeAPS backbone, the CMV core promoter was cloned into the pLenti backbone between the polypurine tract and the 3’ LTR, analogous to previous work60. For individual infections, SARS-CoV-2 RBD or HIV env CD4bs constructs were cloned into the pMD2 backbone on the PDGFR stalk only display architecture. Prefusion-stabilized SARS-CoV-2 spike (2P) was cloned into the pMD2 backbone for individual infections. For library assembly, viral constructs described in Supplementary Table 4 were cloned into the pLenti backbone, C-terminally fused to GFP via a P2A motif. For pLeAPS barcodes, mCherry with an 8-nt degenerate sequence was cloned into the pLeAPS backbone downstream of the Ef1α core promoter.
Transfection for Lentiviral Production
Lentiviruses were prepared by transient transfection of HEK293T cells with linear 25 kDa polyethylenimine (PEI, Santa Cruz Biotechnology) at a 3:1 mass ratio of PEI to DNA. Briefly, DNA and PEI were diluted in Opti-MEM (ThermoFisher) and mixed to form complexes. Complex formation was allowed to proceed for 15 minutes at room temperature before dropwise addition to cells. Media was changed to complete DMEM + 25 mM HEPES after 3–6 hours.
For individually targeted or VSVGwt viruses, plasmid mass ratios were 5.6:3:3:1 for transfer plasmid to psPAX2.1 to targeting plasmid (when used) to fusogen plasmid (either VSVGmut or VSVGwt). Targeting plasmids contain expression cassettes for virally-displayed ligands in the pMD2 backbone. Total plasmid amounts are indicated in Supplementary Table 5. For LeAPS-based virus production, packaging cells were transfected with a 3:1 mass ratio of psPAX2.1:fusogen.
Viral Purification
Unconcentrated viruses were filtered (0.45μm PES) and used directly. If needed, they were stored at 4°C for up to 2 weeks or at −80°C indefinitely. Concentrated viruses were filtered (0.45μm PES) and concentrated 200x by ultracentrifugation for 90 minutes at 100,000×g at 4°C. The supernatant was discarded and viral pellets were resuspended in Opti-MEM overnight at 4°C.
Single lentiviral infections
Individual infections were carried out with the indicated amounts of virus and cells, in the presence of 8 μg/mL of either polybrene or diethylaminoethyl (DEAE)-dextran (Sigma-Aldrich). After 24h, an additional 1x volume of media was added. Cells were analyzed by flow cytometry 48–72h post-infection for Jurkat or J76 infections, and 24–48h post-infection for Ramos cell infections. If cells were only assessed for viral infection, cells were washed once in FACS buffer (PBS + 0.1% BSA and 1 mM EDTA) before analysis on an Accuri C6 or Cytoflex S flow cytometer. In experiments where an additional cell lineage or activation marker such as CD25 or CD69 were assessed, cells were washed once in FACS buffer, stained for 10 minutes in FACS buffer containing a marker-specific antibody, then washed 2x with FACS buffer before analysis for infection (GFP or mCherry) via flow cytometry.
Human Primary T cell activation and transduction
Peripheral blood mononuclear cells from healthy donors were purified from leukopaks purchased from Stem Cell Technologies using Ficoll-Paque PLUS (GE Healthcare) density gradient centrifugation with SepMate tubes (Stem Cell Technologies) per manufacturer instructions. Primary CD8+ T cells were isolated using EasySep Human CD8+ T Cell Enrichment Kits (Stem Cell Technologies) and cultured in RPMI-1640 (ATCC) supplemented with 10% fetal bovine serum, 100 U/ml penicillin-streptomycin (Corning), and 30 IU/ml recombinant human IL-2 (R&D Systems). Prior to transduction with VSVGwt viruses, T cells were activated using a 1:1 ratio of DynaBeads Human T-Activator CD3/CD28 (Thermo Fisher) for 24 hours, after which 8 μg/mL of polybrene (Santa Cruz Biotechnology) and concentrated lentivirus were added to culture at a multiplicity of infection of 1. For targeted viruses, the same protocol was used, but DynaBeads were omitted. For cell proliferation tracking, cells were stained with CellTrace dye (ThermoFisher) according to the manufacturer’s instructions on day 0, prior to viral infection.
Antigen receptor cell line generation
Lentiviral TCR cassettes were formatted as TCRβ-P2A-TCRα and cloned into the pHIV backbone. TCR KO J76 cells were transduced as described above and sorted based on TCR expression to establish monoclonal cell lines. Ramos BCR cells were established according to the published protocol70.
Lentiviral infections in Mixed Cell Populations
Cells were labeled with CellTrace dyes (ThermoFisher) according to the manufacturer’s instructions, counted, and mixed at the indicated ratios. After labeling, viral infections were carried out as indicated above. After 48 hours, cells were washed in FACS buffer and analyzed via flow cytometry to examine infection in CellTrace+ vs CellTrace− cells. After flow cytometry, the selectivity was calculated as follows:
Generation of LeAPS libraries
To generate LeAPS production cell lines, 293T cells were transduced as outlined above while being seeded at 20% confluency on either 6-well or 96-well plates (ThermoFisher). For 6-well plates, 500 μL each of unconcentrated, VSVGwt-pseudotyped LeAPS barcode and ligand expression viruses were used for infection. 50 μL of each virus was used for 96-well plates.
For libraries, cells were transduced in duplicate. One of the duplicates was assessed by flow cytometry to determine the proportion of cells transduced with both ligand (GFP+) and LeAPS barcode (mCherry+). Cells from the remaining duplicate were then pooled, with each library member normalized to include an equivalent number of ligand-expressing (GFP+) and LeAPS barcode-transduced (mCherry+) cells for each library member. This pool of cells was then sorted based on GFP and mCherry expression to make library packaging cell pools. To produce lentiviruses, library packaging cells were transfected as described above, but using only psPAX2.1 and pMD2-VSVGmut. For library creation, both transfer plasmids and the pMD2 targeting plasmid were omitted, as they were replaced by the LeAPS packaging line. After 48 hours, virus was then collected and concentrated as described above.
Library screening of monoclonal TCR lines
10 μL of concentrated pMHC library virus was used to transduce one million TCR-expressing J76 cells as outlined above. After infection was confirmed by flow cytometry as described above, genomic DNA was isolated using the PureLink Genomic DNA kit (ThermoFisher). Barcode inserts were then amplified via 25 cycles of PCR and submitted for Amplicon-EZ analysis by Genewiz. Enrichment was calculated for each barcode as the fraction of total barcode-containing reads divided by the barcode frequency in the packaging cells.
Library screening of monoclonal Ramos BCR lines
500 μL of unconcentrated viral antigen library was used to transduce one million BCR-expressing Ramos cells as outlined above. After infection was confirmed by flow cytometry, genomic DNA was isolated using the PureLink Genomic DNA kit. Barcode inserts were then amplified via 25 cycles of PCR and submitted for Amplicon-EZ analysis by Genewiz or analyzed via a MiSeq v3 150×150nt PE Nano kit (MIT BioMicroCenter). Enrichment was calculated for each barcode as the fraction of total barcode-containing reads divided by the barcode frequency in the packaging cells.
Tetramer enrichment of TCR libraries
To enrich a polyclonal pool of cells from the CSS-930 TCR library for known specificities, 50 million library cells were co-stained with an anti-TRBC antibody (clone IP26) and a pool of HLA-A2 tetramers (NIH Tetramer Core) presenting the following peptides: NLVPMVATV (NLV), GILGFVFTL (GL9), and GLCTLVAML (GLC), all at 1:800 dilutions. Sorting for double-positive cells was performed on a Sony MA900 FACS.
Library vs library TCR-pMHC screen
10 μL of concentrated pMHC library virus was used to transduce two million tetramer-enriched cells. Cells were sorted for infection based on mCherry expression, and then submitted for analysis using the 10x Genomics Chromium 5’ v2 V(D)J kit with a barcode construct-specific primer spiked in prior to droplet encapsulation (see Supplementary Methods for further details). TCR amplicons were prepared and sequenced according to the manufacturer’s instructions. Following cDNA amplification, mCherry barcodes were enriched via separate PCRs and sequenced on an Illumina MiSeq (150×150nt paired end reads). CellRanger V(D)J was used to assign TCR clone identities for each cell. Cell barcodes were used to match TCRs with associated pMHC barcodes counts.
Antibodies used in flow cytometry
All antibodies were used at a 1:50 dilution from the stock concentration. Gating strategies shown in Extended Data Figure 7.
Software
Graphs were generated using Graphpad Prism (v8). Flow cytometry data were analyzed by FlowJo (10.8.0).
Statistical analysis
Statistical analyses (calculation of standard deviation) were performed using the Prism 8 (GraphPad) software. Sample sizes were not predetermined using statistical methods.
Extended Data
Extended Data Fig. 1. IL-13 Surface Architecture Development.
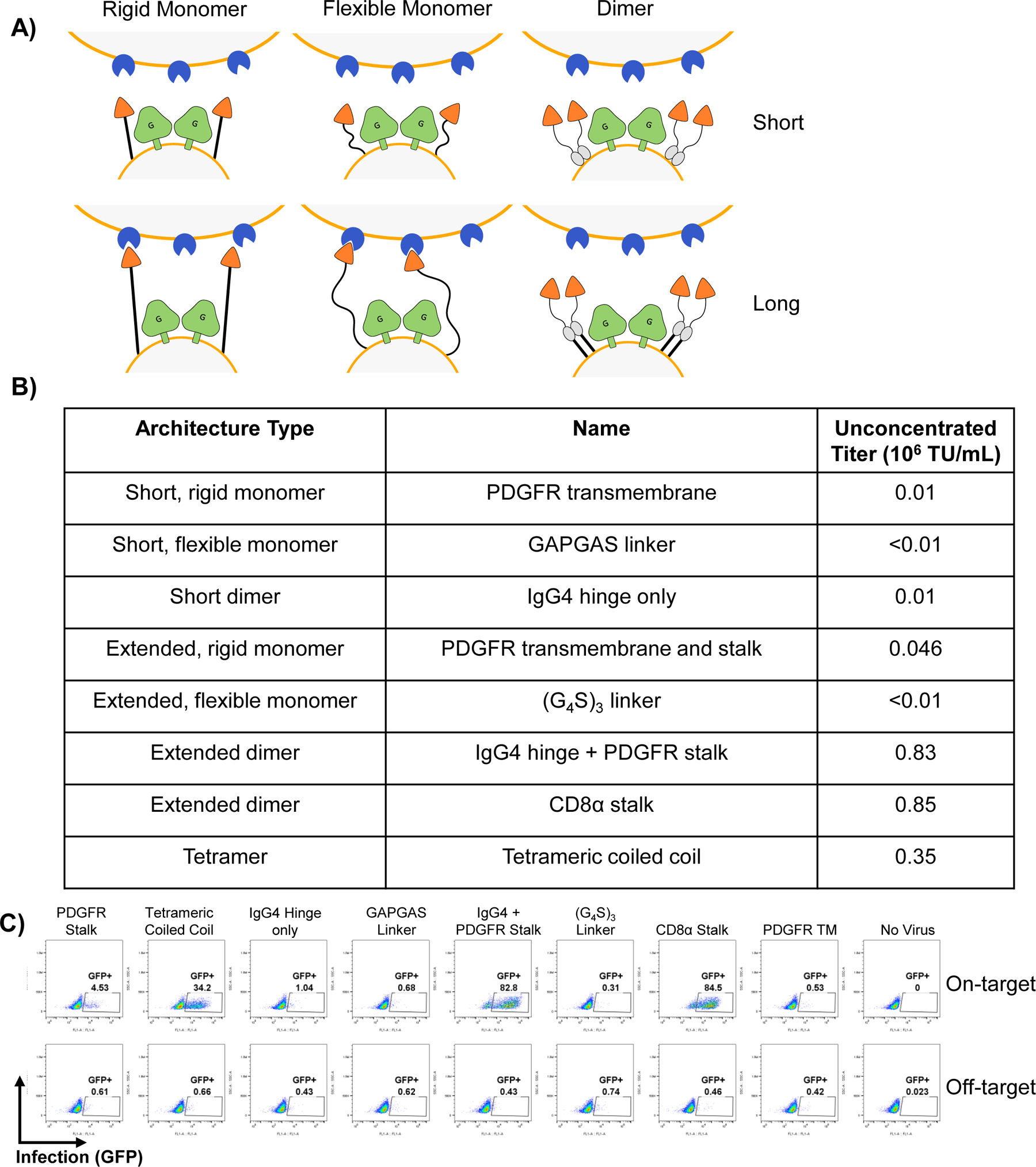
A, Schematic diagram of the different targeting ligand architectures used. B, Summary table with details of each individual construct and titer achieved. C, Flow cytometry plots showing infection of IL13Rα1-expressing Jurkat cells (top, on-target) and parental Jurkat cells which don’t express the receptor (bottom, off-target)
Extended Data Fig. 2. Viral entry via the TCR complex and costimulatory receptors, and inhibition by dasatinib.
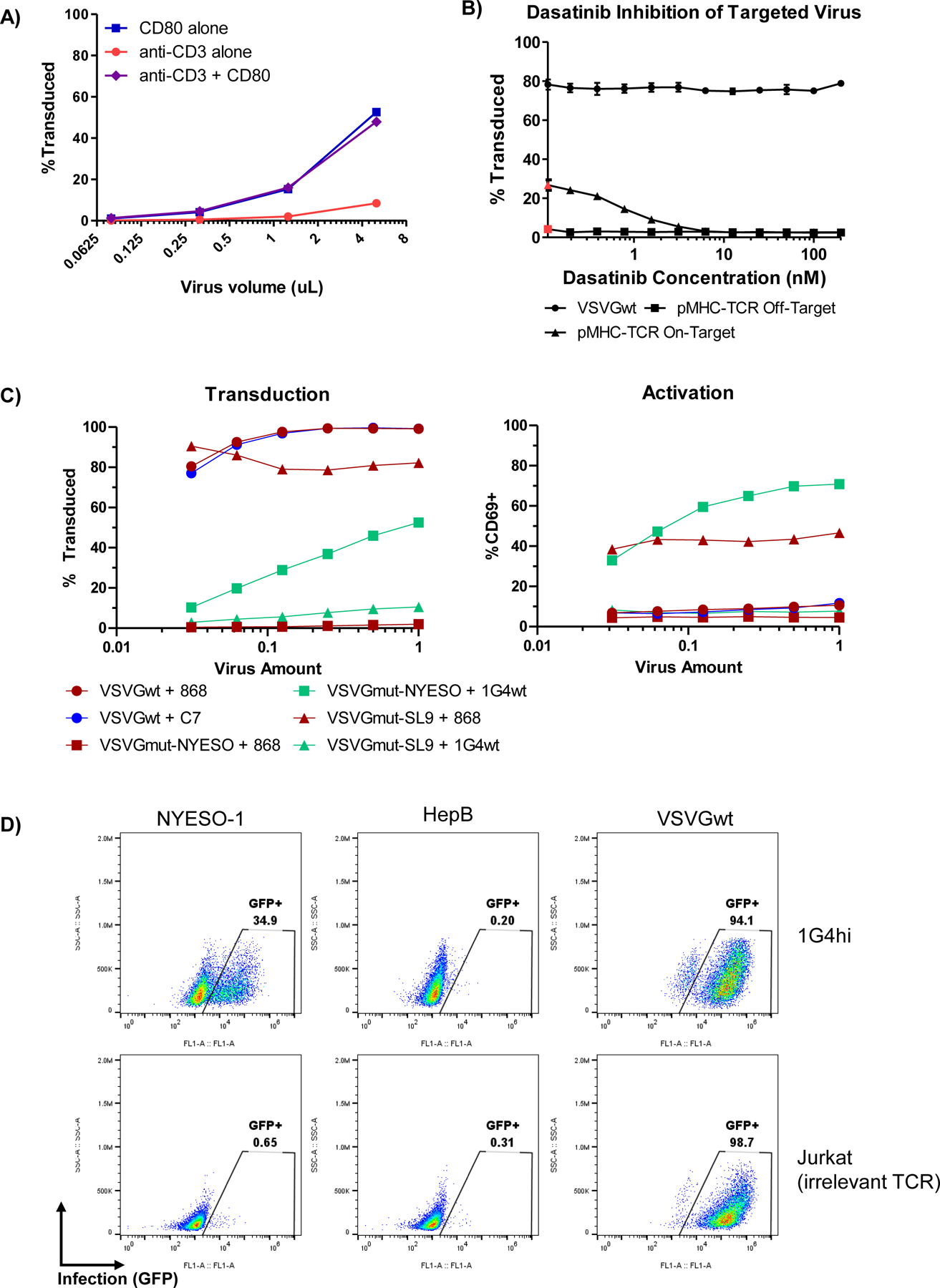
A, 1×105 Jurkat cells were infected with the indicated amount of VSVGmut pseudotyped virus displaying either anti-CD3 Fab, CD80, or a combination of the two; data shown as mean + SD of three biological replicates B, 5×104 J76 cells expressing the 868 TCR were pre-incubated with the indicated concentrations of dasatinib for 2 hours to inhibit TCR signaling prior to virus addition (red dots indicate no dasatinib condition). Next, 5 μL of virus displaying either VSVGmut and HLA-A2-SL9 (on-target), VSVGmut and HLA-A2-NY-ESO-1 (off-target), or VSVGwt were added. Infection was measured 48h post-infection. Results represent the mean and standard deviation of 3 biological replicates. C, The indicated amount of concentrated virus was added to each TCR-expressing J76 cell (100,000 cells/well in a 96-well plate). After 24h, transduction was measured by fluorescent reporter (left) and activation was measured by CD69 upregulation (right) relative to unstimulated cells. D, 500,000 1G4hi-expressing J.RT3 cells or parental Jurkat cells were infected with 500μL of unconcentrated VSVGmut-pseudotyped virus displaying HLA-A2 presenting either NYESO-1 or an irrelevant hepatitis B peptide (SLYAVSPSV), or VSVGwt-pseudotyped virus.
Extended Data Fig. 3. Schematic diagram of LeAPS proof of concept experiments.
Packaging cells transduced with (i) SL9 presented on HLA-A2 and LeAPS-mCherry and (ii) NY-ESO-1 presented on HLA-A2 and LeAPS-GFP were mixed at either 1:100 or 1:1000 for each to represent libraries containing 100 constructs and 1,000 constructs, respectively. These mixtures were then used as packaging lines, transfected with helper plasmids to produce viruses, and used to transduce mixtures of cell tracking dye-labeled J76 cells expressing either 868 TCR or 1G4wt TCR at defined ratios.
Extended Data Fig. 4. Quality control of LeAPS packaging line and reproducibility of infection.
A, LeAPS-barcode expression (mCherry) and pMHC expression (GFP) of the RAPTR 96-member packaging line after 9-passages post-sorting. B, Correlation between infection replicates of the C7 TCR cell line. C, Correlation between infection replicates of the JM22 TCR cell line.
Extended Data Fig. 5. Signal to noise in virus mixtures containing SARS-CoV-2 full spike and HIV env.
A, Infection of CR3022 BCR-expressing Ramos cells with mixtures of SARS-CoV-2 prefusion stabilized spike (on-target, expressing mCherry) and HIV env (off-target, expressing GFP) viruses; variant library size indicates the ratio of off-target to on-target virus present. Infection of VRC01-expressing Ramos cells with HIV env hybrid pseudotyped viruses (right) demonstrates that the viral particles remain functional. B, Quantification of selectivity ratios for the SARS-CoV-2 RBD infections of CR3022 Ramos presented in Figure 5C. Selectivity ratio is calculated as the proportion of on-target cells infected divided by the proportion of off-target cells infected times the frequency of on-target cells (see Methods). C, Quantification of on-target enrichment for mixtures of HIV env and either SARS-CoV-2 RBD (presented in Fig. 5D) or full spike (presented in Supplementary Fig. 5A). Enrichment is calculated by fraction of cells expressing on-target signal divided by fraction of cells expressing off-target signal times on-target virus frequency.
Extended Data Fig. 6. Quality control and reproducibility for the library vs library experiment.
A) Tetramer staining for each of the pooled specificities used for enrichment. B) Correlation between infection replicates in bulk sequencing data for the library vs library experiment
Extended Data Fig. 7. Gating strategies used throughout manuscript.
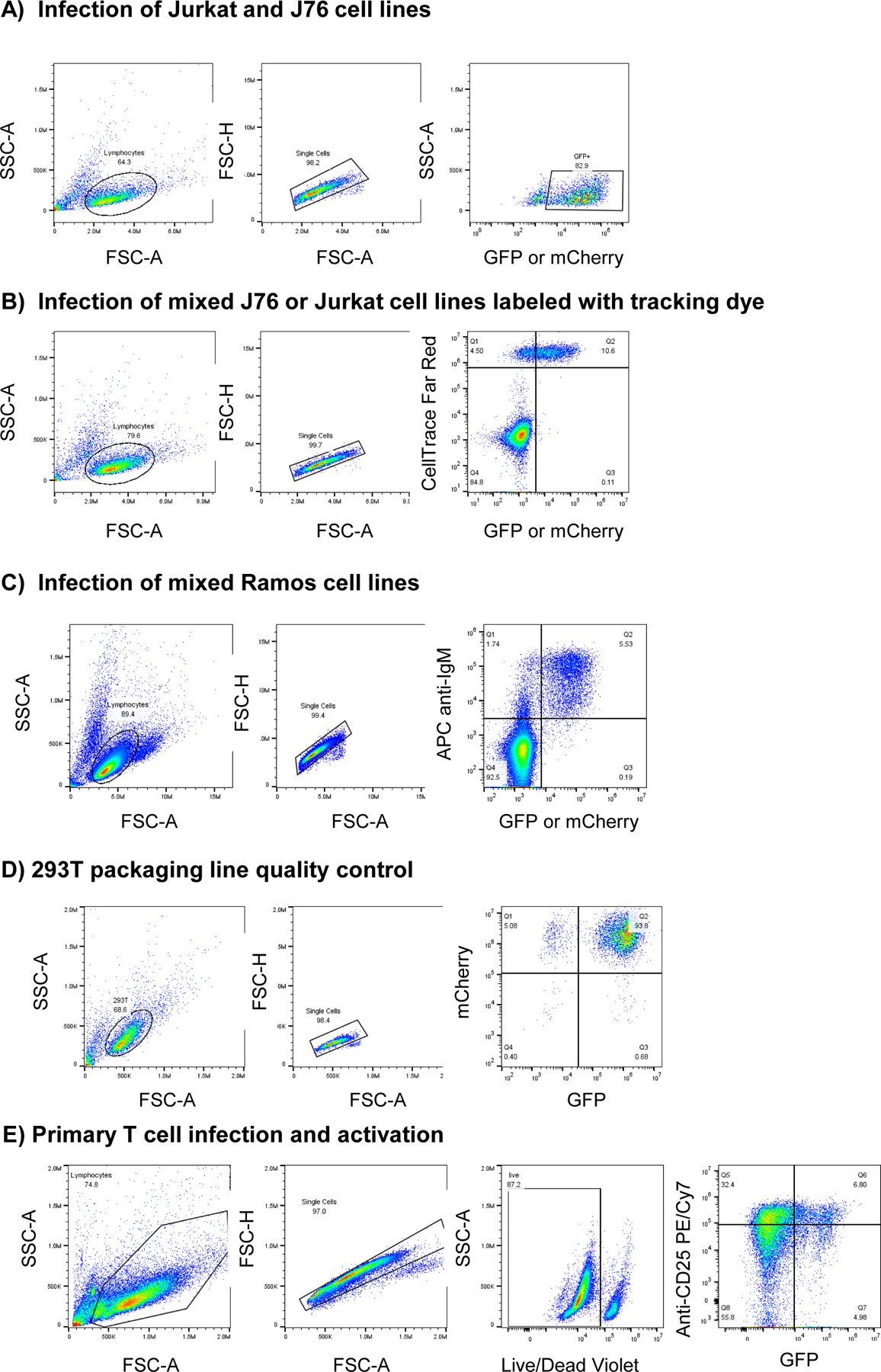
Flow cytometry gating strategies for experiments described.
Supplementary Material
Acknowledgments
We thank the Koch Institute Robert A. Swanson Biotechnology Center for their technical support, especially the Flow Cytometry Facility, MIT BioMicro Center, and High Throughput Sciences Facility. We thank Charlie Whittaker of the Barbara K. Ostrom (1978) Bioinformatics and Computing Facility for helpful discussion and aid in implementing single-cell sequencing data analysis pipelines. We thank Glenn Paradis and Stuart Levine for many helpful discussions and suggestions. We thank Aaron Winkler, Kellie Kravarik, and Marc Wadsworth for helpful discussions and suggestions. We thank the NIH Tetramer Core Facility (contract number 75N93020D00005) for providing pMHC tetramer reagents used in this study.
This work was supported by the Koch Institute Frontier Research Program through the Michael (1957) and Inara Erdei Fund and the Casey and Family Foundation Cancer Research Fund, the Packard Foundation, the Damon Runyon Cancer Research Foundation, the Michelson Medical Research Foundation, Schmidt Futures, Pfizer, Inc., and the Department of Defense (W81XWH757 18-1-0208) to M.E.B.; the National Institutes of Health (DP2AI158126 to M.E.B. and R01AI137057, R01AI153098 to D.L.); a National Science Foundation Graduate Research Fellowship and a Siebel Scholarship to C.S.D.; a Canadian Institutes of Health Research Doctoral Foreign Study Award to S.G.; a graduate research fellowship from the Ludwig Center at MIT’s Koch Institute to E.J.K.; and a Medical Scientist Training Program grant (T32 GM007753) from the National Institute of General Medical Sciences at Harvard Medical School to B.E.S.. M.E.B and S.K.D. were funded by a Technology Impact Award from the Cancer Research Institute and are Pew-Stewart Scholars in Cancer Research. Core facilities in the Koch Institute are partially supported by Cancer Center Support (core) Grant P30-CA14051 from the NCI. This research is additionally supported by the National Research Foundation, Prime Minister’s Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE) program, through Singapore MIT Alliance for Research and Technology (SMART): Critical Analytics for Manufacturing Personalised-Medicine (CAMP) Inter-Disciplinary Research Group.
Footnotes
Competing interests
The lentiviral targeting approach in this manuscript is the subject of US patent applications with M.E.B., S.G., and C.S.D. as co-inventors. M.E.B. is a founder, consultant, and equity holder of Viralogic Therapeutics and Abata Therapeutics, is an equity holder in 3T Biosceinces, has received consulting fees from Repertoire Immune Medicines, and has received research funding from Pfizer, Inc. that partially funded this work. S.K.D. received unrelated research funding from Novartis Pharmaceuticals, Eli Lilly and Company, and Bristol-Myers Squibb, and is a founder, science advisory board member and equity holder in Kojin. M.D. is a science advisory board member for Neoleukin, has research funding from Novartis and Eli Lilly, has received consulting fees from Tillotts Pharma, ORIC Pharmaceuticals, Partner Therapeutics, SQZ Biotech, AzurRx, Mallinckrodt Pharmaceuticals, and Moderna. The remaining authors declare no competing interests.
Code Availability
A sample R workspace for extracting barcode counts based on CITEseq76 is available in the Birnbaum lab code repository (https://github.com/birnbaumlab/Dobson-et-al-2022).
Data Availability
The next-generation sequencing datasets generated during and/or analyzed during the current study are available in the NCBI sequence read archive (Accession Number PRJNA796957). The UniProt database was used to design constructs in this study (https://www.uniprot.org) for determining the peptide sequence of human IL-13 (accession number P35225), human PDGFR (accession number P09619), and human CD80 (accession number P33681). All data generated or analyzed during this study are included in this published article, the sequence read archive, and its supplementary information files.
References
- 1.Joglekar AV & Li G T cell antigen discovery. Nat. Methods 18, 873–880 (2021). [DOI] [PubMed] [Google Scholar]
- 2.McCutcheon M et al. A sensitive ELISPOT assay to detect low-frequency human T lymphocytes. J. Immunol. Methods 210, 149–166 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Kula T et al. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 178, 1016–1028.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma G, Rive CM & Holt RA Rapid selection and identification of functional CD8+ T cell epitopes from large peptide-coding libraries. Nat. Commun 10, 4553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joglekar AV et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods 16, 191–198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisielow J, Obermair F-J & Kopf M Deciphering CD4+ T cell specificity using novel MHC–TCR chimeric receptors. Nat. Immunol 20, 652–662 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Li G et al. T cell antigen discovery via trogocytosis. Nat. Methods 16, 183–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee MN & Meyerson M Antigen identification for HLA class I- and HLA class II-restricted T cell receptors using cytokine-capturing antigen-presenting cells. Sci. Immunol 6, eabf4001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robins HS et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci. Transl. Med 2, 47ra64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arstila TP et al. A direct estimate of the human alphabeta T cell receptor diversity. Science 286, 958–961 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Wang Y et al. Using a baculovirus display library to identify MHC class I mimotopes. Proc. Natl. Acad. Sci. U. S. A 102, 2476–2481 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford F, Huseby E, White J, Marrack P & Kappler JW Mimotopes for alloreactive and conventional T cells in a peptide-MHC display library. PLoS Biol. 2, E90 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birnbaum ME et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gee MH et al. Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell 172, 549–563.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentzen AK et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol 34, 1037–1045 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Bentzen AK et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat. Biotechnol (2018) doi: 10.1038/nbt.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overall SA et al. High throughput pMHC-I tetramer library production using chaperone-mediated peptide exchange. Nat. Commun 11, 1909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S-Q et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol 36, 1156–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiakolas AR et al. Efficient discovery of potently neutralizing SARS-CoV-2 antibodies using LIBRA-seq with ligand blocking. bioRxivorg 2021.06.02.446813 (2021) doi: 10.1101/2021.06.02.446813. [DOI] [Google Scholar]
- 20.Setliff I et al. High-throughput mapping of B cell receptor sequences to antigen specificity. Cell 179, 1636–1646.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fields S & Song O A novel genetic system to detect protein-protein interactions. Nature 340, 245–246 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Wojtowicz WM et al. A human IgSF cell-surface interactome reveals a complex network of protein-protein interactions. Cell 182, 1027–1043.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verschueren E et al. The immunoglobulin superfamily receptome defines cancer-relevant networks associated with clinical outcome. Cell 182, 329–344.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Özkan E et al. An extracellular interactome of immunoglobulin and LRR proteins reveals receptor-ligand networks. Cell 154, 228–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havugimana PC et al. A census of human soluble protein complexes. Cell 150, 1068–1081 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babu M et al. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 489, 585–589 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Younger D, Berger S, Baker D & Klavins E High-throughput characterization of protein-protein interactions by reprogramming yeast mating. Proc. Natl. Acad. Sci. U. S. A 114, 12166–12171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu L et al. Multiplex single-molecule interaction profiling of DNA-barcoded proteins. Nature 515, 554–557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchholz CJ, Duerner LJ, Funke S & Schneider IC Retroviral display and high throughput screening. Comb. Chem. High Throughput Screen 11, 99–110 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Buchholz CJ et al. In vivo selection of protease cleavage sites from retrovirus display libraries. Nat. Biotechnol 16, 951–954 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Schneider RM et al. Directed evolution of retroviruses activatable by tumour-associated matrix metalloproteases. Gene Ther. 10, 1370–1380 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Frank AM & Buchholz CJ Surface-Engineered Lentiviral Vectors for Selective Gene Transfer into Subtypes of Lymphocytes. Molecular therapy. Methods & clinical development 12, 19–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naldini L et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Lei Y, Joo K-I & Wang P Engineering fusogenic molecules to achieve targeted transduction of enveloped lentiviral vectors. J. Biol. Eng 3, 8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Joo K-I, Ziegler L & Wang P Cell type-specific targeting with surface-engineered lentiviral vectors co-displaying OKT3 antibody and fusogenic molecule. Pharm. Res 26, 1432–1445 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang L, Bailey L, Baltimore D & Wang P Targeting lentiviral vectors to specific cell types in vivo. Proc. Natl. Acad. Sci. U. S. A 103, 11479–11484 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frecha C et al. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood 112, 4843–4852 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Ou W et al. Specific targeting of human interleukin (IL)-13 receptor α2-positive cells with lentiviral vectors displaying IL-13. Hum. Gene Ther. Methods 23, 137–147 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Funke S et al. Targeted cell entry of lentiviral vectors. Mol. Ther 16, 1427–1436 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender RR et al. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 12, e1005641 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfeiffer A et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agarwal S et al. In vivo generation of CAR T cells selectively in human CD4+ lymphocytes. Mol. Ther 28, 1783–1794 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikolic J et al. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun 9, 1029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossjohn J et al. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol 33, 169–200 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. The Journal of Immunology 179, 5845–5854 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol 23, 349–354 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Yu YYL, Netuschil N, Lybarger L, Connolly JM & Hansen TH Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. The Journal of Immunology 168, 3145–3149 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Matlin KS, Reggio H, Helenius A & Simons K Pathway of vesicular stomatitis virus entry leading to infection. J. Mol. Biol 156, 609–631 (1982). [DOI] [PubMed] [Google Scholar]
- 49.Alcover A & Alarcón B Internalization and intracellular fate of TCR-CD3 complexes. Crit. Rev. Immunol 20, 325–346 (2000). [PubMed] [Google Scholar]
- 50.Valitutti S, Müller S, Salio M & Lanzavecchia A Degradation of T cell receptor (TCR)-CD3-zeta complexes after antigenic stimulation. J. Exp. Med 185, 1859–1864 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu H, Rhodes M, Wiest DL & Vignali DA On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity 13, 665–675 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Blake S, Hughes TP, Mayrhofer G & Lyons AB The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin. Immunol 127, 330–339 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Xie S, Cooley A, Armendariz D, Zhou P & Hon GC Frequent sgRNA-barcode recombination in single-cell perturbation assays. PLoS One 13, e0198635 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feldman D, Singh A, Garrity AJ & Blainey PC Lentiviral co-packaging mitigates the effects of intermolecular recombination and multiple integrations in pooled genetic screens. bioRxiv (2018) doi: 10.1101/262121. [DOI] [Google Scholar]
- 55.Adamson B et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parnas O et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berger AH et al. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell 30, 214–228 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu W-S & Hughes SH HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med 2, a006882–a006882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.OhAinle M et al. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Broussau S et al. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol. Ther 16, 500–507 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Hill AJ et al. On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vita R et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jurtz V et al. NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang X et al. Structural basis for clonal diversity of the public T cell response to a dominant human Cytomegalovirus Epitope. J. Biol. Chem 290, 29106–29119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malhotra S, Kovats S, Zhang W & Coggeshall KM B cell antigen receptor endocytosis and antigen presentation to T cells require Vav and dynamin. J. Biol. Chem 284, 24088–24097 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dougan SK et al. Antigen-specific B-cell receptor sensitizes B cells to infection by influenza virus. Nature 503, 406–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsieh C-L et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan M et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weaver GC et al. In vitro reconstitution of B cell receptor-antigen interactions to evaluate potential vaccine candidates. Nat. Protoc 11, 193–213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou T et al. Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell 161, 1280–1292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu X et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Y et al. HIV-1 neutralizing antibodies display dual recognition of the primary and coreceptor binding sites and preferential binding to fully cleaved envelope glycoproteins. J. Virol 86, 11231–11241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sliepen K et al. Structure and immunogenicity of a stabilized HIV-1 envelope trimer based on a group-M consensus sequence. Nat. Commun 10, 2355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spindler MJ et al. Massively parallel interrogation and mining of natively paired human TCRαβ repertoires. Nat. Biotechnol 38, 609–619 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mimitou EP et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Replogle JM et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol 38, 954–961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sant S et al. Single-cell approach to influenza-specific CD8+ T cell receptor repertoires across different age groups, tissues, and following influenza virus infection. Front. Immunol 9, 1453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wills MR et al. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol 70, 7569–7579 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hasegawa K et al. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J. Virol 81, 13149–13157 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Utset HA, Guthmiller JJ & Wilson PC Bridging the B cell gap: Novel technologies to study antigen-specific human B cell responses. Vaccines (Basel) 9, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adams JJ et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 35, 681–693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Altman JD & Davis MM MHC-peptide tetramers to visualize antigen-specific T cells. Curr. Protoc. Immunol 115, 17.3.1–17.3.44 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Datlinger P et al. Ultra-high throughput single-cell RNA sequencing by combinatorial fluidic indexing. bioRxiv (2019) doi: 10.1101/2019.12.17.879304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hu Z et al. A cloning and expression system to probe T-cell receptor specificity and assess functional avidity to neoantigens. Blood 132, 1911–1921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oliveira G et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 596, 119–125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fahad AS et al. Immortalization and functional screening of natively paired human T cell receptor repertoires. Protein Eng. Des. Sel 35, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosskopf S et al. A Jurkat 76 based triple parameter reporter system to evaluate TCR functions and adoptive T cell strategies. Oncotarget 9, 17608–17619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The next-generation sequencing datasets generated during and/or analyzed during the current study are available in the NCBI sequence read archive (Accession Number PRJNA796957). The UniProt database was used to design constructs in this study (https://www.uniprot.org) for determining the peptide sequence of human IL-13 (accession number P35225), human PDGFR (accession number P09619), and human CD80 (accession number P33681). All data generated or analyzed during this study are included in this published article, the sequence read archive, and its supplementary information files.