

Abstract
Hepatic stellate cells (HSCs) comprise a minor cell population in the liver but serve numerous critical functions in the normal liver and in response to injury. HSCs are primarily known for their activation upon liver injury and producing the collagen-rich extracellular matrix in liver fibrosis. In the absence of liver injury, HSCs reside in a quiescent state, in which their main function appears to be storage of retinoids, or vitamin A containing metabolites. Less appreciated functions of HSCs include amplifying the hepatic inflammatory response and expressing growth factors that are critical for liver development and both the initiation and termination of liver regeneration. Recent single-cell RNA sequencing studies have corroborated earlier studies that HSC activation involves a diverse array of phenotypic alterations and identified unique HSC populations. This review serves to highlight these many functions of HSCs, and briefly describe recent genetic tools that will help to thoroughly investigate the role of HSCs in hepatic physiology and pathology.
Keywords: stellate cell, liver, fibrosis
Graphical Abstract
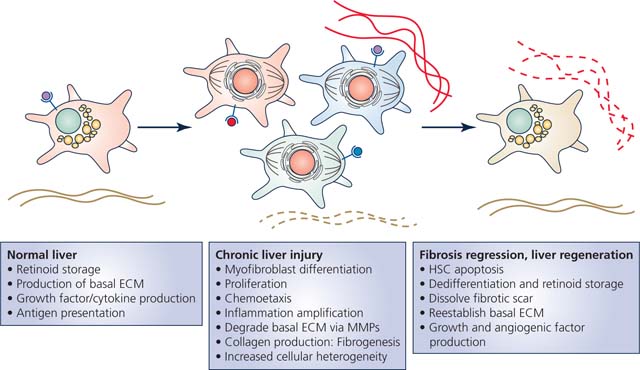
Hepatic stellate cells (HSCs) are best known for their production of fibrosis in response to liver injury, but they display many other phenotypes and vital functions. In the normal liver, HSCs help to orchestrate liver development, produce and maintain the basal extracellular matrix (ECM), produce growth factors and cytokines, present antigens, and serve as the body’s main storage depot for retinoids or vitamin A-containing metabolites. Upon liver injury, HSCs lose the retinoid-containing lipid droplets and differentiate into myofibroblasts and proliferate. Single cell sequencing studies have uncovered that activated HSCs display a high degree of cellular heterogeneity for specific functions. Some are more proliferative, while others are more inflammatory, and others more fibrogenic. If liver injury is resolved, HSC numbers will decrease due to a large degree of apoptosis, and remaining HSCs will at least partially dedifferentiate into more quiescent HSCs. These HSCs help remove the fibrotic scar and reestablish the basal ECM scaffold. If liver regeneration is required, HSCs will coordinate this by building of the basal ECM and production of growth factors, cytokines, and angiogenic factors.
Introduction
Hepatic stellate cells (HSCs) are resident mesenchymal cells located in the perisinusoidal area between endothelial cells and hepatocytes known as the Space of Disse (Figure 1). As non-parenchymal cells, most estimates suggest HSCs comprise between 5–10% of all resident cells within the liver (Giampieri et al., 1981). Recent analyses based on single-cell or single-nuclei isolation and RNA sequencing report as low as 0.3% up to ~15% of isolated liver cells or nuclei express known HSC markers (MacParland et al., 2018; Massalha et al., 2020; Zhu et al., 2020; Andrews et al., 2021; Diamanti et al., 2021; Nault et al., 2021; Payen et al., 2021; Richter et al., 2021). HSCs are predominantly viewed within the scope of producing fibrosis in response to liver injury, as HSCs are the main contributor to hepatic extracellular matrix deposition during all etiologies of liver injury (Mederacke et al., 2013). The central dogma for stellate cells is that they reside in a quiescent state in the normal liver and become “activated” in response to liver injury. This activation mainly arises from oxidative stress and inflammatory signals, causing an array of phenotypic alterations related to cellular activation: enhanced proliferation, differentiation to myofibroblast-like cells, chemotaxis, fibrogenesis, and production of extracellular matrix remodeling enzymes (Friedman, 2008). However, recent evidence suggests heterogeneous populations of HSCs exist with diverse phenotypic responses to activation. Much less is known about the importance of HSCs outside of this fibrotic response to liver injury. Since HSCs comprise such a relatively minor population of cells and until recently it has been challenging to create HSC-specific genetic models, uncovering roles for HSCs in normal hepatic or systemic physiology has been challenging. This review seeks to describe the multitude of HSC functions and shine light on the current lack of understanding regarding the roles of HSCs in normal hepatic physiology.
Figure 1 – Hepatic cellular architecture.
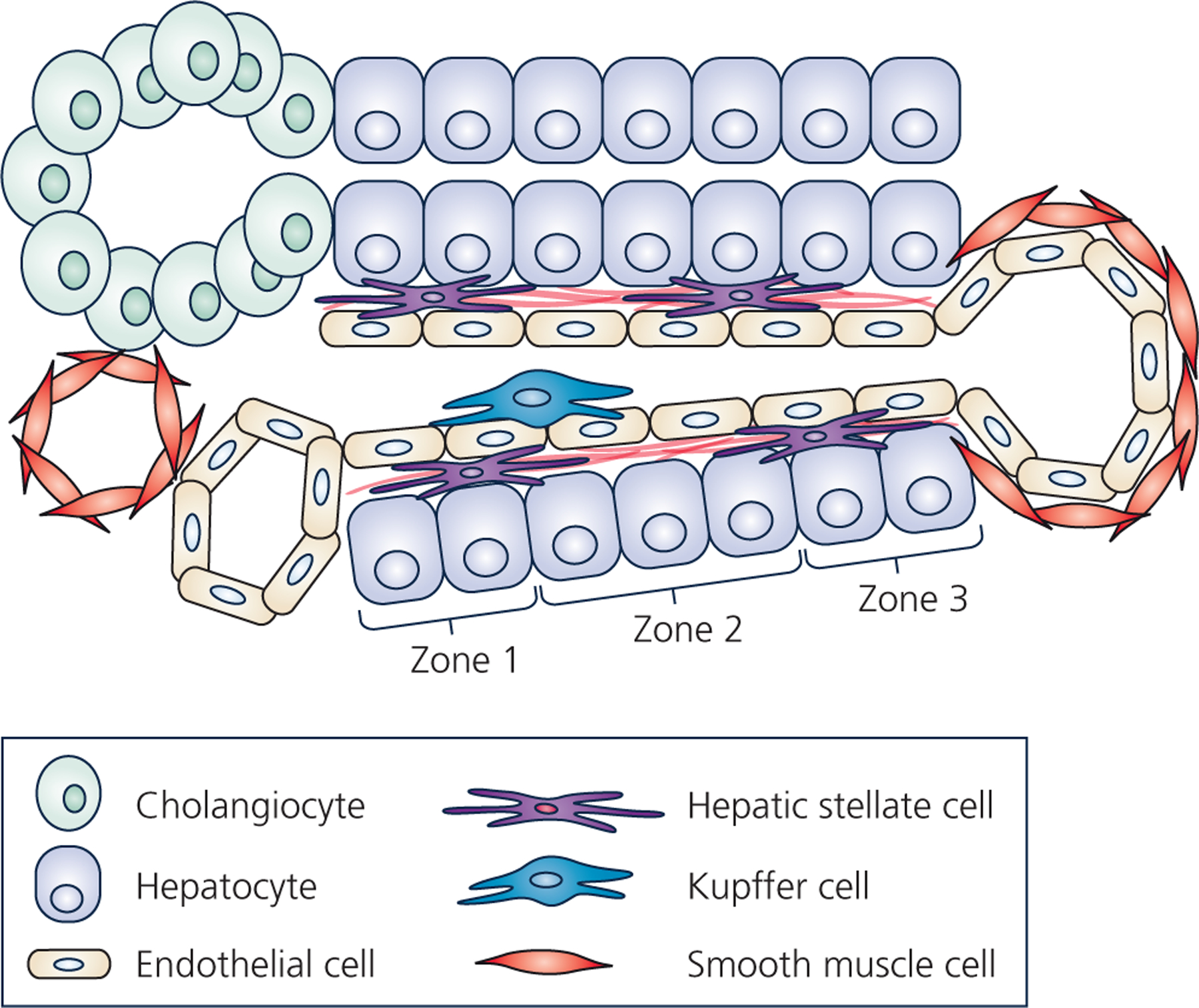
Schematic represents a cross-section of a liver lobule, which contains the complete functional structure and main cell types of the liver. One the left, is the “portal triad” comprised of the portal vein, the hepatic artery, and the bile ductule. The bile ductule is comprised of cholangiocytes (represented as green oval-shaped cells), which collects bile produced by the hepatocytes. Bile ductules ultimately combine into the bile duct which drains bile to be stored in the gallbladder. The hepatic artery (represented as a red smooth-muscle cell-lined vessel) supplies oxygenated blood originating from the celiac artery, whereas the portal vein (represented as an endothelial cell-lined vessel) supplies nutrient- and toxin-rich blood from the stomach, pancreas, gallbladder, and spleen into the liver lobule. The portal vein and hepatic artery drain blood flow into the liver sinusoid, which is lined by liver sinusoidal endothelial cells. Ultimately, blood is collected into the central vein, lined with both endothelial cells and smooth muscle cells. Resident macrophages, known as Kupffer cells (represented as blue cell), reside in the luminal side of the sinusoid. The hepatic stellate cells (HSCs) (represented as peach-colored cells with long projections) reside in the space of Disse between hepatocytes and liver sinusoidal endothelial cells and produce the basement extracellular matrix (black lines near the HSCs in space of Disse). Regions along the length of the sinusoid are commonly referred to as zones, with zone 1 being peri-portal, zone 3 being peri-central vein, and zone 2 residing between. Hepatocytes comprise the hepatic parenchyma (the predominant cell type) and display an incredible array of functions including synthesis of serum proteins, clotting factors, lipoproteins, cholesterol and bile salts, gluconeogenesis and glycogen storage, as well as detoxification. Often these different hepatocyte functions are localized to specific zones along the sinusoid.
The Quiescent Hepatic Stellate Cell
Retinoid Storage and Function
In their inactive or quiescent state, HSCs are characterized by the presence of cytoplasmic lipid droplets. These lipid droplets contain a large amount of vitamin A metabolites, or retinoids, which allowed for the original identification of these cells by Karl Wilhelm von Kupffer using gold chloride staining (Kupffer, 1876). In fact, up to 70–95% of all retinoid storage in the body resides within HSCs (Wake, 1971; Knook et al., 1982; Hendriks et al., 1985). These studies also described that HSC retinoid storage was increased when provided high levels of vitamin A or retinyl esters (Kupffer, 1876; Hendriks et al., 1985). Due to the presence of these lipid droplets, “fat-storing cells” or “lipocytes” were early names for hepatic stellate cells. “Ito cells” was another common name for HSCs after Toshio Ito reinvigorated interest in the cells after Kupffer had concluded they may just be a subset of Kupffer macrophage cells (Reuben, 2002). The retinoids in these droplets are comprised predominantly of retinyl esters of exclusively long-chain acyl moieties (retinyl-palmitate > retinyl-stearate > retinyl-oleate > retinyl-linoleate) (Knook et al., 1982; Hendriks et al., 1985; Yamada et al., 1987). In addition to retinyl esters, HSC lipid droplets contain triglycerides, and small amounts of cholesterol, cholesterol esters, phospholipids, and free fatty acids (Yamada et al., 1987; Moriwaki et al., 1988). While lipid droplet retinoid content varies based on dietary retinoid consumption, altered triglyceride consumption has little effect on HSC triglyceride content (Moriwaki et al., 1988). Strangely, despite the diversity of lipid species stored in these droplets, blocking synthesis of retinyl esters completely prevents the formation of lipid droplets in HSCs (O’Byrne et al., 2005). Retinyl ester synthesis in the liver is dependent on the enzyme lecithin:retinol acyltransferase (LRAT), and LRAT−/− mice completely lack HSC lipid droplets and hepatic retinyl ester storage (Batten et al., 2004; O’Byrne et al., 2005). In LRAT-deficient mice, dietary retinol can still be esterified by intestinal diacylglycerol acyltransferase 1 (DGAT1), and retinyl esters (and retinol) are instead stored primarily in adipose tissue (O’Byrne et al., 2005). The adipose from LRAT−/− mice displayed a 3–4-fold increase in cytosolic retinol-binding protein type III (CRBPIII) (O’Byrne et al., 2005), which is known to be important for retinol uptake by cells (Piantedosi et al., 2005). These adipose tissue retinoid stores were still able to be mobilized when the mice were placed on a retinoid-deficient diet (O’Byrne et al., 2005).
Vitamin A is essential and must be obtained from dietary sources. Vitamin A can be obtained from animal-based foods in the form of retinyl esters, or from plant-based foods in the form of carotenoids which can then be converted to retinol. Retinoids, or more appropriately, the biologically active forms which are retinaldehyde or retinoic acid, have various vital physiologic roles. In 1913, it was discovered that a certain lipid-soluble substance, later identified as vitamin A, affected growth rates independent of dietary carbohydrate, lipid, and protein (McCollum, 1913). Shortly after, it was realized that vitamin A was an important factor in detection of light by the retina (Rosenheim & Drummond, 1925; Wald, 1935), which explains why vitamin A metabolites have the name of retinoids. In addition to supporting growth and vision, retinoids support numerous immune system and reproductive system functions (reviewed in (Haaker et al., 2020)). Thus, retinoid storage in HSCs likely preserves many vital functions for numerous cell types throughout the body.
Due to inexistent hepatic retinoid stores, LRAT-deficient mice were believed to be more prone to developing retinoid deficiency compared to wildtype mice. While no phenotypic symptoms were observed, after only 1 month on a retinoid-deficient diet, LRAT−/− mice had depleted their adipose tissue retinoid stores, serum retinol concentrations and retinol binding protein (RBP) were decreased, and hepatic RBP expression was elevated by 10-fold (O’Byrne et al., 2005). These changes in serum/liver RBP are used as biomarkers of retinoid deficiency when performed in combination with serum vitamin A measurement.
For reasons that remain poorly understood, loss of retinoid storage in HSCs is one of the classic hallmarks of HSC activation. However, it seems this retinoid lipid droplet depletion is a consequence of activation rather than a cause. LRAT deficient mice lacking HSC retinoid stores showed no activation of HSCs compared to wildtype mice (O’Byrne et al., 2005). However, chronic liver diseases resulting in cirrhotic liver failure (involving excessive HSC activation and fibrogenesis) are associated with symptomatic vitamin A deficiency (Mahmood et al., 2008; Venu et al., 2013; Chaves et al., 2015). There is conflicting data regarding whether retinoids can directly alter stellate cell activation. Treatment with retinoic acid has been suggested to decrease HSC proliferation and collagen expression in vitro and in vivo (Davis et al., 1990). However, subsequent studies with 9-cis-retinoic acid and 9,13-di-cis-retinoic acid suggest promotion of fibrosis by activation of plasminogen activator and tumor growth factor β (TGFβ) (Okuno et al., 1997; Okuno et al., 1999). Activated HSCs downregulate LRAT expression, greatly decreasing their ability to form and store retinyl esters (Shmarakov et al., 2019). In summary, retinoid storage is the primary function of quiescent HSCs, and retinoid depletion upon HSC activation can lead to systemic vitamin A deficiency.
Transcriptional Regulation of Adipocyte-like Phenotype
Likely intricately related to the presence of these lipid droplets, quiescent HSCs express a virtually identical transcriptional program to mature adipocytes. Expression of peroxisome proliferator-activated receptor gamma (PPARγ) and sterol regulatory-element-binding protein-1 (SREBP-1c) in HSCs governs fatty acid storage and metabolism. In quiescent cells, PPARγ and SREBP-1c expression is high, but are both dramatically reduced upon HSC activation (She et al., 2005; Tsukamoto, 2005). Enhancement of PPARγ and SREBP-1c expression or activity prevents or reverses HSC activation, suggesting that maintenance of the adipogenic phenotype maintains quiescence (Miyahara et al., 2000; She et al., 2005; Tsukamoto, 2005). Conversely, knockdown of PPARγ in cultured human HSCs significantly increases activation (Tao et al., 2020). Interestingly, despite the complete loss of lipid droplets in LRAT−/− HSCs, these cells display maintained expression of PPARγ and SREBP-1c, perhaps explaining their preserved quiescent phenotype (O’Byrne et al., 2005). HSCs also express the “adipokines” leptin and adiponectin, and adiponectin signaling is antifibrotic (Kamada et al., 2003; Ding et al., 2005). In clinical practice, treating patients with nonalcoholic steatohepatitis (NASH) with the PPARγ agonist pioglitazone significantly reduces hepatic fibrosis (Boettcher et al., 2012; Bril et al., 2018) which could be the result of direct PPARγ activation in the HSCs or via enhanced adiponectin signaling. Cumulatively, these data strongly suggest that maintenance of this adipogenic phenotype prevents HSC activation.
One hypothesis for why the adipogenic phenotype and lipid droplets are lost during activation is that activation requires a great deal of energy, supplied by the metabolism of fatty acids. Activated HSCs display enhanced expression of a transcriptional regulator of fatty acid oxidation enzymes peroxisome proliferator-activated receptor beta (PPARβ) (She et al., 2005). Surprisingly, peroxisome proliferator-activated receptor alpha (PPARα), which also regulates gene expression for fat oxidation enzymes/transporters appears to be reduced in activated HSCs (Miyahara et al., 2000). During activation, fatty acids can be sourced from the hydrolysis of lipid droplet-stored triglycerides, or the de-esterification of the stored retinyl esters (Shmarakov et al., 2019), and these processes are at least partially dependent on autophagy of the lipid droplets (Hernandez-Gea et al., 2012). A number of retinyl ester hydrolases and lipases, including adipose triglyceride lipase (ATGL), patatin-like phospholipase domain-containing protein 3 (PNPLA3), hormone-sensitive lipase (HSL), and carboxyl ester lipase have been shown to be important for retinyl ester release from lipid droplets (Weng et al., 1999; Mello et al., 2008; Pirazzi et al., 2014; Taschler et al., 2015). Inhibiting fatty acid oxidation can block HSC activation (Hernandez-Gea et al., 2012), suggesting that liberating fatty acid fuel from the lipid droplets may be required for HSC activation. However, HSC activation also enhances glucose and amino acid catabolism (reviewed in (Trivedi et al., 2021)). This enhanced glucose utilization does increase mitochondrial oxidation, but more significantly increases glycolytic flux and lactate production, which is believed to directly contribute to activation (Chen et al., 2012). Conversely, reducing mitochondrial pyruvate metabolism, which would theoretically increase intracellular lactate, has been suggested to decrease HSC activation (McCommis et al., 2017).
The Activated Hepatic Stellate Cell
Extensive work in the 1970s-80s described that HSCs were found near the fibrotic collagen fibers in injured livers and were the predominant source of this extracellular matrix in injured livers (McGee & Patrick, 1972; Kent et al., 1976; Okanoue et al., 1983; Martinez-Hernandez, 1984; Friedman et al., 1985; Martinez-Hernandez, 1985). It is now understood that activation of HSCs involves a host of phenotypic changes in addition to extracellular matrix production. Activation of HSCs includes: differentiation from adipogenic to myofibroblast phenotype with loss of lipid droplets and expression of contractile fibers, increased cell proliferation, increased HSC chemotaxis as well as signaling to attract leukocytes, and development of matured rough endoplasmic reticulum to support production of extracellular matrix fibers and matrix remodeling enzymes. Ultimately, all these cellular processes are coordinated to enhance the local accumulation of extracellular matrix capable of forming a scar in areas of liver injury. Unsurprisingly, blocking signaling pathways for protein synthesis via p70S6K knockout or inhibition decreases HSC activation and fibrogenesis (Reiter et al., 2022). As HSC activation and fibrogenesis are not the primary focus of this review, we will refer to several excellent reviews of this topic (Friedman, 2000; Bataller & Brenner, 2005; Zisser et al., 2021). However, we must acknowledge that hepatic fibrosis is a vitally important aspect to the pathology of all chronic liver diseases. The degree of liver fibrosis is the greatest predictor of outcomes in alcoholic liver disease (Lackner et al., 2017), NASH (Hagstrom et al., 2017; Taylor et al., 2020), viral hepatitis (Vergniol et al., 2011), and cholestatic liver injury (Saffioti et al., 2021). Justifiably, there has been tremendous focus on development of anti-fibrotic therapies.
Hepatic Stellate Cell Heterogeneity
Even early studies of HSCs in normal livers described a range of HSC features, primarily based upon liver zonal location. For example, some HSCs lack expression of the diagnostic contractile fiber desmin, while other cells lack extensive vitamin A droplets (Ballardini et al., 1994; Ramm et al., 1995). This heterogeneity led to the hypothesis that some HSCs are more important for retinoid storage, while others are “primed” for quick activation. Single-cell RNA sequencing of non-steatotic human liver samples from all isolated cells, identified 2 HSC populations, that could not be explained by quiescent versus activated signatures (Payen et al., 2021). Instead, one population was characterized by expression of the cell surface proteoglycan GPC3 and the neurotrophic receptor NTRK2, while the other population expressed the dopamine norepinephrine-converting enzyme DBH and the hedgehog signaling modulator HHIP (Payen et al., 2021). Using RNA in situ hybridization, it was determined the GPC3-expressing population resided almost exclusively near the portal and central vein regions, while the DBH-expressing population was more diffuse in the perisinusoidal space other than the portal and central vein (Payen et al., 2021). Gene ontology analyses of these two populations suggested that the GPC3+-cells were characterized by glycosaminoglycan metabolism and elastic fiber constituents, while the DBH+-cells more reminiscent of antigen-presenting cells (Payen et al., 2021). Thus, these quiescent HSCs can be characterized by different spatial zonation and likely important functional differences (Figure 2).
Figure 2 – Functions and heterogeneity of quiescent HSCs.
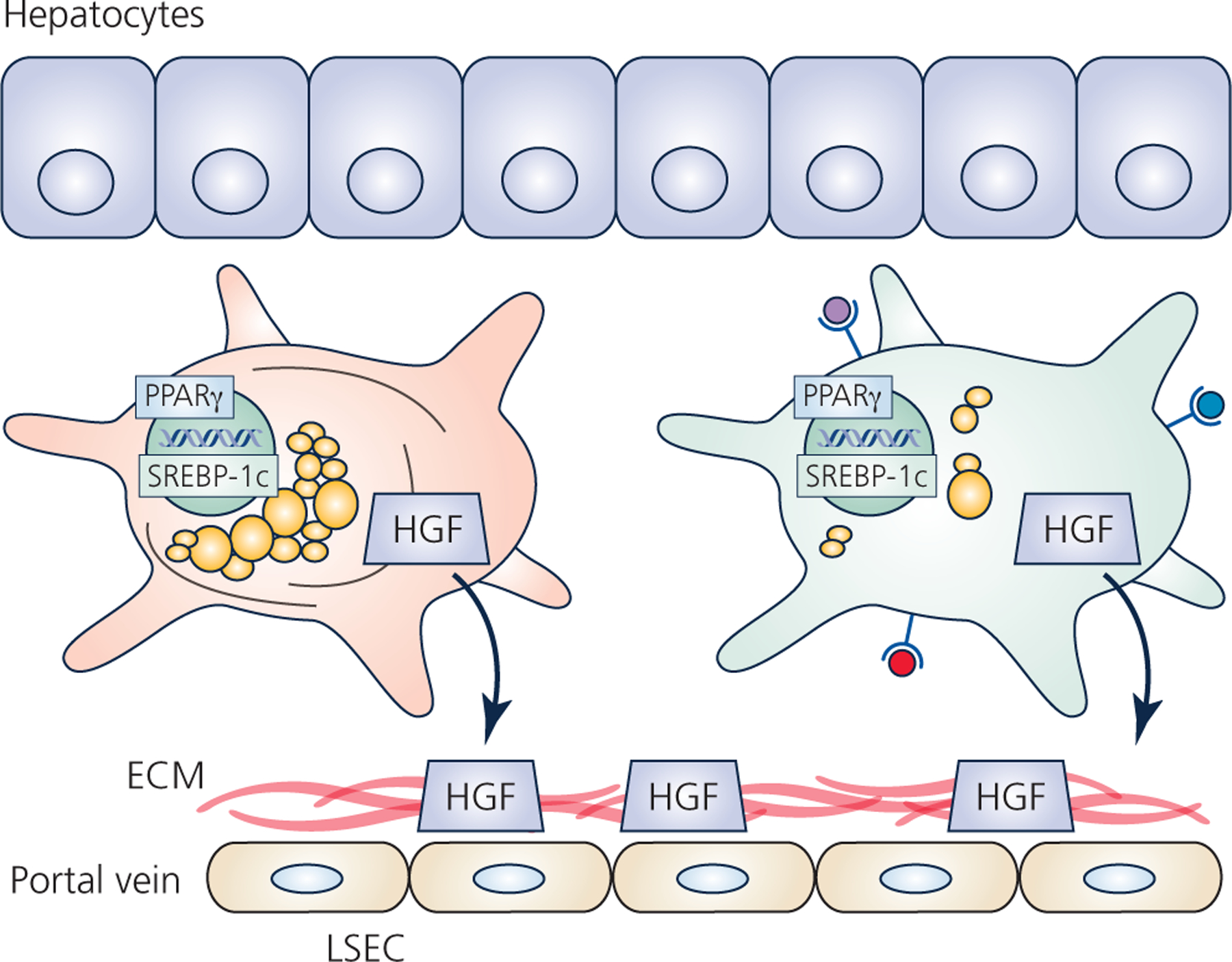
All HSCs reside in the space of Disse between hepatocytes and liver sinusoidal endothelial cells (LSEC). Quiescence is maintained by high expression of the adipogenic transcription factors peroxisome proliferator-activated receptor gamma (PPARγ) and sterol regulatory-element-binding protein-1c (SREBP-1c). Quiescent HSCs also produce and secrete cytokines and growth factors, such as hepatocyte growth factor (HGF) which are sequestered by the basal extracellular matrix (ECM). Physically and functionally different HSCs likely reside in different regions along the portal tract. Early studies described distinct populations of HSCs based on presence/absence of desmin fibers, or abundance of lipid droplets. In addition to these macroscopic differences, single-cell RNA sequencing studies suggest other functional differences in these distinct populations, such as altered glycosaminoglycan synthesis or increased antigen presentation.
HSCs appear to be even more heterogeneous upon activation from liver injury. A classical marker of HSC activation is the expression of the contractile fiber alpha smooth muscle actin (αSMA, gene name ACTA2). However, not all cells that start to express collagen 1α1 (Col1α1) also express αSMA, and vice versa, which was shown in vitro and in vivo using mice expressing a dual reporter with green fluorescent protein expressed from the Col1α1 promoter and red fluorescent protein expressed from the αSMA promoter (Magness et al., 2004). Indeed, while 52% of the cells expressed both activation/fibrogenesis markers, 14% expressed only Col1α1 and 9% expressed only αSMA, and each population displayed distinct patterns of gene expression (Magness et al., 2004). Our appreciation of the diversity of HSC populations has been expanded recently by single-cell RNA sequencing studies. Using a western diet-fed mouse model of NASH, a total of 9 separate HSC populations could be identified by unique gene expression patterns despite all of them expressing many of the classical HSC genes (Rosenthal et al., 2021). In this study, quiescent HSCs from control mice belonged predominantly to 2 separate clusters. HSCs from the NASH livers resided in 6 of the 7 other clusters with one representing the classical activated myofibroblast, but other populations being defined by either a weak activation signature, a proliferation signature, or an inflammatory signature. A third group of mice was placed on the NASH-inducing diet but was then switched back to chow diet to provoke NASH regression. HSCs isolated from this group resided in many of the same clusters as the NASH livers, but also contained a relatively unique population that was more similar to quiescent HSCs, and contained much fewer HSCs from active NASH livers (Rosenthal et al., 2021). In another study of a mouse model of fibrosis caused by the hepatotoxin carbon tetrachloride, single-cell RNA sequencing identified 8 separate HSC populations reflecting different degrees of activation or even some populations displaying enhanced expression of genes important for tissue repair and angiogenesis (Zhang et al., 2021). Altogether, these studies suggest that HSCs, even in the activated state, have an array of functions independent of, or in addition to fibrogenesis.
Hepatic Stellate Cells During Fibrosis Regression
Successful treatment of various chronic liver diseases have revealed the ability to reverse hepatic fibrosis (reviewed in (Kisseleva & Brenner, 2021)). One key question has been if and how HSCs could be inactivated to reduce fibrosis. Indeed, placing culture-activated HSCs onto culture dishes containing a basement membrane-like extracellular matrix can reverse their activation (Gaca et al., 2003). Likewise, treatment with an adipogenic cocktail, or exogenous PPARγ or SREBP-1c expression reverts activated HSCs to quiescent, lipid storing cells (She et al., 2005). More recently, evidence of HSC reversion or inactivation in vivo has also been obtained. Using yellow fluorescent protein (YFP) HSC reporter mice treated with CCl4 or intragastric ethanol but then allowed to recover to significantly resolve the fibrosis showed that many HSCs that were previously activated (YFP+) no longer expressed the activated myofibroblast marker αSMA (Kisseleva et al., 2012). These inactivated cells displayed a much more quiescent gene expression phenotype, with reduced expression of collagens, αSMA, and tissue inhibitor of metalloproteinase 1, and normalized expression of PPARγ (Kisseleva et al., 2012). If a similar experiment is performed in HSC-specific PPARγ knockout mice, regression of liver fibrosis is greatly slowed (Liu et al., 2020), arguing the importance of returning to the adipogenic HSC phenotype to resolve hepatic fibrosis. However, these inactivated HSCs are distinct from quiescent HSCs in uninjured livers, as they appear to be primed for an enhanced fibrotic response upon TGFβ treatment in vitro, or after reinjury with carbon tetrachloride in vivo (Kisseleva et al., 2012). Lastly, single-cell sequencing has also described a relatively unique population of HSCs in livers after removal of a NASH-inducing diet (Rosenthal et al., 2021). Altogether, these studies suggest that activated HSCs can be inactivated during the regression of fibrosis, but these cells likely do not return to a completely quiescent phenotype.
While some activated HSCs become inactivated, it has also been well established that ~50% of activated HSCs undergo apoptosis during fibrosis regression (Iredale et al., 1998; Kisseleva et al., 2012). This apoptotic cell death is driven by activation of extrinsic receptor-mediated pathways such as FAS or TRAIL, enhanced caspase 3 and caspase 8 activity, and upregulation of proapoptotic proteins such as p53 and BAX as well as activation of hepatic natural killer cells (Radaeva et al., 2006; Glassner et al., 2012; Kisseleva et al., 2012). One key characteristic of HSCs that avoid apoptosis and instead inactivate is enhanced expression of the pro-survival genes for heat-shock proteins 1a/b (Kisseleva et al., 2012). Unsurprisingly, artificially inducing HSC apoptosis to deplete HSCs can significantly limit fibrosis progression or resolve hepatic fibrosis (Parsons et al., 2004; Oakley et al., 2005; Puche et al., 2013). Thus, after removal or treatment of the pathologic etiology driving fibrosis, it is clear that hepatic fibrosis can regress by a combination of HSC apoptosis and HSC inactivation.
Roles of Hepatic Stellate Cells Not Directly Related to ECM Deposition
HSCs as Immune Regulators
The common dogma is that stellate cells become activated by sensing inflammatory signals such as TGFβ that arise from liver injury. In actuality, HSCs not only respond to inflammatory signals, but also produce many cytokines, chemokines, and immunomodulatory signals in both normal and injured liver. HSCs produce TGFβ and the TGFβ receptor, and autocrine signaling is critically important for HSC activation (Bissell et al., 1995; Purps et al., 2007). The same is true for many other cytokines and growth factors. Of course, these factors can also act as paracrine signals to other liver cell types and immune cells, altering their activation.
HSCs also express toll-like receptors and can respond to bacterial infection by sensing lipopolysaccharide (Brun et al., 2005). HSCs also act as antigen presenting cells, capable of activating T cells (Winau et al., 2007). Activated stellate cells are also known to express programmed death ligand 1 (PD-L1)(Yu et al., 2004), which can bind the programmed cell death protein 1 (PD-1) receptor on an array of immune cells including macrophages, T cells, and B cells. Overall, these signaling and antigen presenting properties of HSCs help to respond and amplify the immune response from liver injury, which can also propagate HSC activation.
HSCs in Liver Development and Regeneration
Understanding the embryonic origin of HSCs has been difficult, as HSCs express classical gene markers for all three germ layers (endoderm, mesoderm, and ectoderm). Lineage tracing suggests mesoderm origin from the septum transversum, as both Wilms tumor suppressor-expressing and mesoderm posterior 1-expressing cells develop into HSCs (Asahina et al., 2009; Asahina et al., 2011), and HSCs express the mesoderm-specific transcription factor FoxF1 (Kalinichenko et al., 2003). Yet expression of desmin, cytokeratin-7/8, and CD34 suggests endoderm origin, while expression of glial fibrillary acidic protein (GFAP), nestin, neurotrophins, and other markers suggest ectodermal origin from the neural crest (reviewed in (Friedman, 2008)). Desmin and GFAP expression are used as defining markers for identification of HSCs, with αSMA expression considered the defining marker of activated HSCs. It is not entirely known how the pool of HSCs is maintained. It has been suggested that bone marrow-derived cells could contribute to both quiescent and activated HSCs (Baba et al., 2004; Russo et al., 2006), however fate tracing with LratCre and bone-marrow transplantation experiments exclude this premise (Mederacke et al., 2013). Thus, it seems likely that HSCs are liver-resident cells that proliferate to maintain or expand their pool size.
Several lines of evidence suggest that HSCs play significant roles during liver development. HSCs reside in the progenitor cell niche of the liver and are known to produce cytokines and growth factors capable of stimulating various hepatic cell types, including tumor growth factor alpha, epidermal growth factor, hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), stem cell factor, acidic and basic fibroblast growth factors (FGF), macrophage colony-stimulating factor, platelet activating factor, and others (reviewed in (Friedman, 2008)). These factors stimulate the stellate cells themselves in an autocrine manner, but can also stimulate hepatocytes, endothelial cells, cholangiocytes, macrophages, neutrophils, and others. Interestingly, as many as 46% of HSCs transiently express Col1α1 during the postnatal period, yet these HSCs lack αSMA expression and other myofibroblast characteristics and instead maintain quiescent HSC expression phenotypes (Kisseleva et al., 2012). The purpose of this transient collagen expression is not understood.
In comparison to liver development, we have a better understanding of the impact of HSCs during liver regeneration. In most instances of liver injury, liver regeneration is primarily accomplished by re-entry into the cell cycle and proliferation of existing hepatocytes to regain normal liver mass and function (reviewed in (Michalopoulos, 2007)). HSCs produce and secrete HGF on a nearly continuous basis, which is stored in an inactive form within the basal extracellular matrix. Upon liver injury, active HGF is released and can bind its cMet receptor on hepatocytes, biliary cells, and endothelial cells. The crucial role of HSCs in liver regeneration has been demonstrated in several studies in which depletion of HSCs or inhibition of HSC activation has prevented regeneration. In one such study, depletion of stellate cells via gliotoxin injection during a model of acetaminophen-induced liver injury produced a dramatic decrease in proliferating hepatocytes, a decrease in growth factor gene expression, and significantly increased liver necrosis (Shen et al., 2011). Inhibition of HSC activation with L-cysteine was also shown to reduce cell proliferation of virtually all hepatic lineages in partial hepatectomy and 2-acetylaminofluorene model of hepatic injury (Pintilie et al., 2010). Lastly, genetic inhibition of HSC activation also reduces hepatocyte proliferation and worsens liver injury from various etiologies in mice (Kalinichenko et al., 2003; Passino et al., 2007). Altogether, these studies suggest that HSC activation after injury is vital to hepatocyte proliferation during liver regeneration.
Another important aspect to liver regeneration is the formation of new vasculature or angiogenesis. Growth factors are also vital to angiogenesis, and the positioning of HSCs and liver sinusoidal endothelial cells within the Space of Disse allows for direct contact of these cells as well as paracrine signaling. Factors produced by HSCs such as PDGF, TGFβ, FGF, vascular endothelial growth factor, and angiopoietin all enhance angiogenesis (reviewed in (Kitto & Henderson, 2021)).
It appears that HSCs also play an important role in the termination of liver regeneration. During the final phases of regeneration, HSCs rebuild the basal extracellular matrix which can then sequester the high levels of growth factors and prevent them from activating hepatocytes causing them to exit the cell cycle (Block et al., 1996). Additionally, HSCs are a source of TGFβ, which is known to be an inhibitor of cell proliferation. Preventing TGFβ production from activated HSCs has been shown to enhance hepatocyte proliferation in mice after partial hepatectomy (Ebrahimkhani et al., 2011). Thus, HSCs appear to be of vital importance during the early and late stages of hepatic regeneration after injury (Figure 3).
Figure 3 – HSCs during liver injury and regeneration.
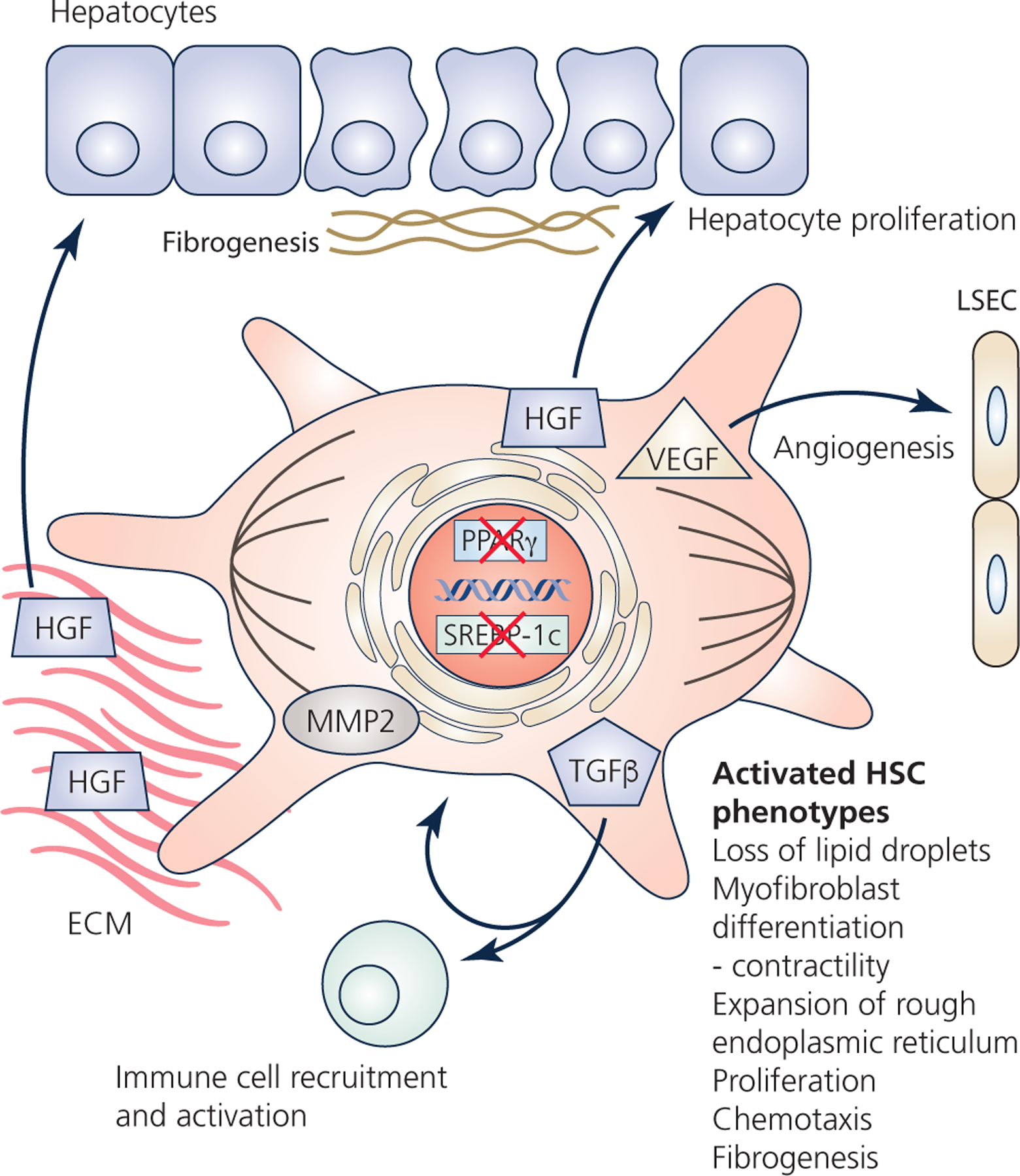
HSCs become activated by liver injury. Activation involves downregulation of the adipogenic transcription factors, and loss of the retinoid-storing lipid droplets. HSCs differentiate into contractile myofibroblasts, proliferate, and can respond to chemotactic signals. A number of secreted factors participate in the response to injury. For example, tumor growth factor beta (TGFβ) activates immune cells, but also enhances HSC activation via autocrine signaling. The HSCs continue to produce hepatocyte growth factor (HGF), but also degrade the basal extracellular matrix (ECM) by matrix metalloproteinase 2 (MMP2), which frees sequestered HGF and stimulates hepatocyte proliferation. HSCs also produce vascular endothelial growth factor (VEGF), as well as fibroblast growth factor, platelet-derived growth factor, and TGFβ which can all enhance angiogenesis. Activated HSCs also synthesize the collagen-rich fibrosis, as well as proteoglycans, glycosaminoglycans, and tissue-inhibitor of matrix metalloproteinases which all participate in the formation of the fibrotic scar. Lastly, HSCs participate in the termination of liver regeneration by re-establishing the basal ECM which sequesters HGF. Likewise, TGFβ from HSCs can inhibit hepatocyte proliferation to help terminate liver regeneration.
HSCs as Pleuripotent Stem Cells?
Another intriguing possibility for HSCs to be involved in response to injury is they may have the capacity to differentiate into other liver cell types, including hepatocytes. It has been suggested that up to 40% of rat HSCs express the stem cell marker CD133, as well as a host of other stem cell markers (Kordes et al., 2007). In culture, these cells were able to be differentiated by cytokines known to differentiate endothelial progenitor cells into cells resembling endothelial cells, and even branched tube-like structures that expressed endothelial nitric oxide synthase and ve-cadherin (Kordes et al., 2007). Additionally, these CD133+ HSCs were shown to be differentiated into hepatocyte-like cells when cultured on collagen with a medium containing specific growth factors/cytokines (Kordes et al., 2007). This in vitro data displays the plasticity of HSC and points to a possible flexibility of HSC to fill in the gaps of liver cells during regeneration. This stemness also appears to exist in vivo, as shown by fate-tracing with GFP-labeled HSCs. Intriguingly, both before and after liver injury, GFP was detected in bile duct cells and sinusoidal cells, demonstrating the differentiation capacity of HSCs (Yang et al., 2008). After these reporter mice were subjected to a methionine and choline deficient diet supplemented with 0.15% ethionine to cause hepatocellular injury and activate stellate cells, GFP+ hepatocytes arose that remained even after return to normal diet and liver injury regression (Yang et al., 2008). However, more recent fate tracing with HSCs genetically labelled by LratCre and ZsGreen suggested that HSCs were not a source of new hepatocytes, cholangiocytes, or endothelial cells after chemical toxin, bile-duct ligation, fatty liver injury, or partial hepatectomy (Mederacke et al., 2013). Thus, while these studies highlight the potential for HSCs to differentiation into other liver cell types, this pleuripotent potential of HSCs is rather controversial.
Future Studies of HSCs in Physiology
While the fibrotic response of HSCs in response to liver injury and their importance to liver regeneration become increasingly understood, the question of how important HSCs are to normal physiology largely remains. As described above, the most widely known significance for HSCs in the normal liver is retinoid storage. Use of HSC depletion models would allow for testing of how livers devoid of HSCs are able to carry out other important processes in hepatic and systemic physiology. These types of depletion models have been previously generated, but unfortunately only studied within the context of injury and regeneration. For example, a specific model of HSC depletion using herpes simplex virus thymidine kinase expressed from the GFAP promoter and treatment with ganciclovir (Puche et al., 2013), is an approach which only works when the cells are proliferating, therefore when they are activated. Additionally, GFAP is also not HSC-specific and also expressed in cholangiocytes in the liver. LratCre-mediated expression of diphtheria toxin receptor has been used to deplete a large majority of HSCs, but this approach was performed mainly to confirm that HSCs comprise the majority of myofibroblasts after liver injury (Mederacke et al., 2013). The HSC-expressed diphtheria toxin receptor model would be ideal for testing HSC depletion in the normal liver. Intriguing questions to ask would be if the liver is still fully capable of performing its numerous detoxifying or biosynthetic functions. HSCs express several cytochrome P450 enzymes (Yamada et al., 1997), but it is unknown how much HSCs contribute to xenobiotic detoxification in relation to hepatocytes. The liver’s biosynthetic functions related to protein production (plasma proteins such as albumin and almost all critical blood clotting factors), maintenance of glycemia (via glycogenolysis and gluconeogenesis), and lipid metabolism (lipoprotein synthesis, fat oxidation, and ketogenesis) are all prominent hepatocyte functions, but could potentially be influenced by nearby HSCs. Hopefully future studies will attempt to shed light on the importance of HSCs to these aspects of normal hepatic physiology, in addition to the response to liver injury.
Conclusion
While stellate cells comprise a minor hepatic cell population by number, evidence is accumulating for their critical importance in response to liver injury. During all etiologies of liver injury, these cells produce the fibrotic scar, respond to and amplify the inflammation signals, and also contribute to all phases of liver regeneration by the production of growth factors and cytokines. Recent single-cell or single-nuclei RNA sequencing studies have established a much greater diversity beyond quiescent versus activated HSCs. Further work is required to ascertain the importance of these numerous HSC populations in response to injury. In the absence of liver injury, the storage of retinoids and production of several growth factors are the best understood functions of HSCs. Future studies are required to truly appreciate if and how HSCs contribute to normal hepatic physiologic functions.
Supplementary Material
Funding
Kyle McCommis is supported by NIH R00 HL136658.
Abbreviations:
- αSMA
alpha smooth muscle actin
- Col1α1
collagen 1 alpha 1
- CRBPIII
cytosolic retinol-binding protein type III
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- HGF
hepatocyte growth factor
- HSC
hepatic stellate cell
- LSEC
liver sinusoidal endothelial cell
- LRAT
lecithin:retinol acyltransferase
- NASH
nonalcoholic steatohepatitis
- PD1
programmed cell death protein 1
- PD-L1
programmed death ligand 1
- PDGF
platelet-derived growth factor
- PPARα
peroxisome proliferator-activated receptor alpha
- PPARβ
peroxisome proliferator-activated receptor beta
- PPARγ
proliferator-activated receptor gamma
- RBP
retinol binding protein
- SREBP-1c
sterol regulatory-element-binding protein-1
- TGFβ
tumor growth factor β
- YFP
yellow fluorescent protein
Biographies

Dakota Kamm was awarded a M.S. in Exercise Physiology from Southern Illinois University-Edwardsville in 2019. Dakota then became a Research Assistant in the laboratory of Dr. Kyle McCommis at Saint Louis University. He is currently a Ph.D. student in the Movement Science program at Washington University in St. Louis.

Kyle McCommis is an Assistant Professor in the Department of Biochemistry & Molecular Biology at the Saint Louis University School of Medicine. His research focuses on mitochondrial and lipid metabolism of cardiometabolic diseases such as heart failure, diabetes, and nonalcoholic fatty liver disease.
Footnotes
Competing interests
The authors have no conflicts of interest to declare.
References
- Andrews TS, Atif J, Liu JC, Perciani CT, Ma XZ, Thoeni C, Slyper M, Eraslan G, Segerstolpe A, Manuel J, Chung S, Winter E, Cirlan I, Khuu N, Fischer S, Rozenblatt-Rosen O, Regev A, McGilvray ID, Bader GD & MacParland SA. (2021). Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity. Hepatol Commun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K, Tsai SY, Li P, Ishii M, Maxson RE Jr., Sucov HM & Tsukamoto H (2009). Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 49, 998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K, Zhou B, Pu WT & Tsukamoto H. (2011). Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53, 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba S, Fujii H, Hirose T, Yasuchika K, Azuma H, Hoppo T, Naito M, Machimoto T & Ikai I. (2004). Commitment of bone marrow cells to hepatic stellate cells in mouse. J Hepatol 40, 255–260. [DOI] [PubMed] [Google Scholar]
- Ballardini G, Groff P, Badiali de Giorgi L, Schuppan D & Bianchi FB. (1994). Ito cell heterogeneity: desmin-negative Ito cells in normal rat liver. Hepatology 19, 440–446. [PubMed] [Google Scholar]
- Bataller R & Brenner DA. (2005). Liver fibrosis. J Clin Invest 115, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W & Palczewski K. (2004). Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem 279, 10422–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell DM, Wang SS, Jarnagin WR & Roll FJ. (1995). Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 96, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block GD, Locker J, Bowen WC, Petersen BE, Katyal S, Strom SC, Riley T, Howard TA & Michalopoulos GK. (1996). Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J Cell Biol 132, 1133–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher E, Csako G, Pucino F, Wesley R & Loomba R. (2012). Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 35, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bril F, Kalavalapalli S, Clark VC, Lomonaco R, Soldevila-Pico C, Liu IC, Orsak B, Tio F & Cusi K. (2018). Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin Gastroenterol Hepatol 16, 558–566 e552. [DOI] [PubMed] [Google Scholar]
- Brun P, Castagliuolo I, Pinzani M, Palu G & Martines D. (2005). Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 289, G571–578. [DOI] [PubMed] [Google Scholar]
- Chaves GV, Peres WA, Goncalves JC & Ramalho A. (2015). Vitamin A and retinol-binding protein deficiency among chronic liver disease patients. Nutrition 31, 664–668. [DOI] [PubMed] [Google Scholar]
- Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont R, Suliman HB, Piantadosi CA & Diehl AM. (2012). Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 143, 1319–1329 e1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BH, Kramer RT & Davidson NO. (1990). Retinoic acid modulates rat Ito cell proliferation, collagen, and transforming growth factor beta production. J Clin Invest 86, 2062–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti K, Inda Diaz JS, Raine A, Pan G, Wadelius C & Cavalli M. (2021). Single nucleus transcriptomics data integration recapitulates the major cell types in human liver. Hepatol Res 51, 233–238. [DOI] [PubMed] [Google Scholar]
- Ding X, Saxena NK, Lin S, Xu A, Srinivasan S & Anania FA. (2005). The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol 166, 1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimkhani MR, Oakley F, Murphy LB, Mann J, Moles A, Perugorria MJ, Ellis E, Lakey AF, Burt AD, Douglass A, Wright MC, White SA, Jaffre F, Maroteaux L & Mann DA. (2011). Stimulating healthy tissue regeneration by targeting the 5-HT(2)B receptor in chronic liver disease. Nat Med 17, 1668–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL. (2000). Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275, 2247–2250. [DOI] [PubMed] [Google Scholar]
- Friedman SL. (2008). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88, 125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL, Roll FJ, Boyles J & Bissell DM. (1985). Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A 82, 8681–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaca MD, Zhou X, Issa R, Kiriella K, Iredale JP & Benyon RC. (2003). Basement membrane-like matrix inhibits proliferation and collagen synthesis by activated rat hepatic stellate cells: evidence for matrix-dependent deactivation of stellate cells. Matrix Biol 22, 229–239. [DOI] [PubMed] [Google Scholar]
- Giampieri MP, Jezequel AM & Orlandi F. (1981). The lipocytes in normal human liver. A quantitative study. Digestion 22, 165–169. [DOI] [PubMed] [Google Scholar]
- Glassner A, Eisenhardt M, Kramer B, Korner C, Coenen M, Sauerbruch T, Spengler U & Nattermann J. (2012). NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest 92, 967–977. [DOI] [PubMed] [Google Scholar]
- Haaker MW, Vaandrager AB & Helms JB. (2020). Retinoids in health and disease: A role for hepatic stellate cells in affecting retinoid levels. Biochim Biophys Acta Mol Cell Biol Lipids 1865, 158674. [DOI] [PubMed] [Google Scholar]
- Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R & Kechagias S. (2017). Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol 67, 1265–1273. [DOI] [PubMed] [Google Scholar]
- Hendriks HF, Verhoofstad WA, Brouwer A, de Leeuw AM & Knook DL. (1985). Perisinusoidal fat-storing cells are the main vitamin A storage sites in rat liver. Exp Cell Res 160, 138–149. [DOI] [PubMed] [Google Scholar]
- Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ & Friedman SL. (2012). Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C & Arthur MJ. (1998). Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 102, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinichenko VV, Bhattacharyya D, Zhou Y, Gusarova GA, Kim W, Shin B & Costa RH. (2003). Foxf1 +/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology 37, 107–117. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, Okamoto Y, Kihara S, Miyagawa J, Shinomura Y, Funahashi T & Matsuzawa Y. (2003). Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 125, 1796–1807. [DOI] [PubMed] [Google Scholar]
- Kent G, Gay S, Inouye T, Bahu R, Minick OT & Popper H. (1976). Vitamin A-containing lipocytes and formation of type III collagen in liver injury. Proc Natl Acad Sci U S A 73, 3719–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T & Brenner D. (2021). Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 18, 151–166. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H, Evans SM, Dillmann W, Glass CK & Brenner DA. (2012). Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A 109, 9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitto LJ & Henderson NC. (2021). Hepatic Stellate Cell Regulation of Liver Regeneration and Repair. Hepatol Commun 5, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knook DL, Seffelaar AM & de Leeuw AM. (1982). Fat-storing cells of the rat liver. Their isolation and purification. Exp Cell Res 139, 468–471. [DOI] [PubMed] [Google Scholar]
- Kordes C, Sawitza I, Muller-Marbach A, Ale-Agha N, Keitel V, Klonowski-Stumpe H & Haussinger D. (2007). CD133+ hepatic stellate cells are progenitor cells. Biochem Biophys Res Commun 352, 410–417. [DOI] [PubMed] [Google Scholar]
- Kupffer KVW. (1876). Ueber Sternzellen der Leber. Briefliche Mitteilung an Prof. Waldyer. Arch mikr Anat 12, 353–358. [Google Scholar]
- Lackner C, Spindelboeck W, Haybaeck J, Douschan P, Rainer F, Terracciano L, Haas J, Berghold A, Bataller R & Stauber RE. (2017). Histological parameters and alcohol abstinence determine long-term prognosis in patients with alcoholic liver disease. J Hepatol 66, 610–618. [DOI] [PubMed] [Google Scholar]
- Liu X, Xu J, Rosenthal S, Zhang LJ, McCubbin R, Meshgin N, Shang L, Koyama Y, Ma HY, Sharma S, Heinz S, Glass CK, Benner C, Brenner DA & Kisseleva T. (2020). Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 158, 1728–1744 e1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, Gupta R, Cheng ML, Liu LY, Camat D, Chung SW, Seliga RK, Shao Z, Lee E, Ogawa S, Ogawa M, Wilson MD, Fish JE, Selzner M, Ghanekar A, Grant D, Greig P, Sapisochin G, Selzner N, Winegarden N, Adeyi O, Keller G, Bader GD & McGilvray ID. (2018). Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 9, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magness ST, Bataller R, Yang L & Brenner DA. (2004). A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology 40, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Mahmood K, Samo AH, Jairamani KL, Ali G, Talib A & Qazmi W. (2008). Serum retinol binding protein as an indicator of vitamin A status in cirrhotic patients with night blindness. Saudi J Gastroenterol 14, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Hernandez A (1984). The hepatic extracellular matrix. I. Electron immunohistochemical studies in normal rat liver. Lab Invest 51, 57–74. [PubMed] [Google Scholar]
- Martinez-Hernandez A (1985). The hepatic extracellular matrix. II. Electron immunohistochemical studies in rats with CCl4-induced cirrhosis. Lab Invest 53, 166–186. [PubMed] [Google Scholar]
- Massalha H, Bahar Halpern K, Abu-Gazala S, Jana T, Massasa EE, Moor AE, Buchauer L, Rozenberg M, Pikarsky E, Amit I, Zamir G & Itzkovitz S. (2020). A single cell atlas of the human liver tumor microenvironment. Mol Syst Biol 16, e9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum EVD M (1913). The necessity of certain lipins in the diet during growth. J Biol Chem 15, 167–175. [Google Scholar]
- McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE, Colca JR & Finck BN. (2017). Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 65, 1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee JO & Patrick RS. (1972). The role of perisinusoidal cells in experimental hepatic fibrogenesis. J Pathol 106, Pvi. [PubMed] [Google Scholar]
- Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP & Schwabe RF. (2013). Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 4, 2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello T, Nakatsuka A, Fears S, Davis W, Tsukamoto H, Bosron WF & Sanghani SP. (2008). Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem Biophys Res Commun 374, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalopoulos GK. (2007). Liver regeneration. J Cell Physiol 213, 286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF Jr., Motomura K, Anania F, Willson TM & Tsukamoto H. (2000). Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem 275, 35715–35722. [DOI] [PubMed] [Google Scholar]
- Moriwaki H, Blaner WS, Piantedosi R & Goodman DS. (1988). Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J Lipid Res 29, 1523–1534. [PubMed] [Google Scholar]
- Nault R, Fader KA, Bhattacharya S & Zacharewski TR. (2021). Single-Nuclei RNA Sequencing Assessment of the Hepatic Effects of 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Cell Mol Gastroenterol Hepatol 11, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, Palczewski K & Blaner WS. (2005). Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J Biol Chem 280, 35647–35657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, May MJ, Millward-Sadler H, Wright MC & Mann DA. (2005). Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology 128, 108–120. [DOI] [PubMed] [Google Scholar]
- Okanoue T, Burbige EJ & French SW. (1983). The role of the Ito cell in perivenular and intralobular fibrosis in alcoholic hepatitis. Arch Pathol Lab Med 107, 459–463. [PubMed] [Google Scholar]
- Okuno M, Moriwaki H, Imai S, Muto Y, Kawada N, Suzuki Y & Kojima S. (1997). Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-beta in liver stellate cells. Hepatology 26, 913–921. [DOI] [PubMed] [Google Scholar]
- Okuno M, Sato T, Kitamoto T, Imai S, Kawada N, Suzuki Y, Yoshimura H, Moriwaki H, Onuki K, Masushige S, Muto Y, Friedman SL, Kato S & Kojima S. (1999). Increased 9,13-di-cis-retinoic acid in rat hepatic fibrosis: implication for a potential link between retinoid loss and TGF-beta mediated fibrogenesis in vivo. J Hepatol 30, 1073–1080. [DOI] [PubMed] [Google Scholar]
- Parsons CJ, Bradford BU, Pan CQ, Cheung E, Schauer M, Knorr A, Krebs B, Kraft S, Zahn S, Brocks B, Feirt N, Mei B, Cho MS, Ramamoorthi R, Roldan G, Ng P, Lum P, Hirth-Dietrich C, Tomkinson A & Brenner DA. (2004). Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology 40, 1106–1115. [DOI] [PubMed] [Google Scholar]
- Passino MA, Adams RA, Sikorski SL & Akassoglou K. (2007). Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science 315, 1853–1856. [DOI] [PubMed] [Google Scholar]
- Payen VL, Lavergne A, Alevra Sarika N, Colonval M, Karim L, Deckers M, Najimi M, Coppieters W, Charloteaux B, Sokal EM & El Taghdouini A. (2021). Single-cell RNA sequencing of human liver reveals hepatic stellate cell heterogeneity. JHEP Rep 3, 100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantedosi R, Ghyselinck N, Blaner WS & Vogel S. (2005). Cellular retinol-binding protein type III is needed for retinoid incorporation into milk. J Biol Chem 280, 24286–24292. [DOI] [PubMed] [Google Scholar]
- Pintilie DG, Shupe TD, Oh SH, Salganik SV, Darwiche H & Petersen BE. (2010). Hepatic stellate cells’ involvement in progenitor-mediated liver regeneration. Lab Invest 90, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, Burza MA, Indiveri C, Ferro Y, Montalcini T, Maglio C, Dongiovanni P, Fargion S, Rametta R, Pujia A, Andersson L, Ghosal S, Levin M, Wiklund O, Iacovino M, Boren J & Romeo S. (2014). PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet 23, 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puche JE, Lee YA, Jiao J, Aloman C, Fiel MI, Munoz U, Kraus T, Lee T, Yee HF Jr. & Friedman SL. (2013). A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 57, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purps O, Lahme B, Gressner AM, Meindl-Beinker NM & Dooley S. (2007). Loss of TGF-beta dependent growth control during HSC transdifferentiation. Biochem Biophys Res Commun 353, 841–847. [DOI] [PubMed] [Google Scholar]
- Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z & Gao B. (2006). Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 130, 435–452. [DOI] [PubMed] [Google Scholar]
- Ramm GA, Britton RS, O’Neill R, Blaner WS & Bacon BR. (1995). Vitamin A-poor lipocytes: a novel desmin-negative lipocyte subpopulation, which can be activated to myofibroblasts. Am J Physiol 269, G532–541. [DOI] [PubMed] [Google Scholar]
- Reiter FP, Ye L, Ofner A, Schiergens TS, Ziesch A, Brandl L, Ben Khaled N, Hohenester S, Wimmer R, Artmann R, He Y, Lee SML, Mayr D, Zhang C, Gerbes AL, Mayerle J, Denk G & De Toni EN. (2022). p70 ribosomal protein S6 kinase is a checkpoint of human hepatic stellate cell activation and liver fibrosis in mice. Cell Mol Gastroenterol Hepatol 13, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A (2002). Ito becomes a star. Hepatology 35, 503–504. [DOI] [PubMed] [Google Scholar]
- Richter ML, Deligiannis IK, Yin K, Danese A, Lleshi E, Coupland P, Vallejos CA, Matchett KP, Henderson NC, Colome-Tatche M & Martinez-Jimenez CP. (2021). Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nat Commun 12, 4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenheim O & Drummond JC. (1925). A Delicate Colour Reaction for the Presence of Vitamin A. Biochem J 19, 753–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal SB, Liu X, Ganguly S, Dhar D, Pasillas MP, Ricciardelli E, Li RZ, Troutman TD, Kisseleva T, Glass CK & Brenner DA. (2021). Heterogeneity of HSCs in a Mouse Model of NASH. Hepatology 74, 667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo FP, Alison MR, Bigger BW, Amofah E, Florou A, Amin F, Bou-Gharios G, Jeffery R, Iredale JP & Forbes SJ. (2006). The bone marrow functionally contributes to liver fibrosis. Gastroenterology 130, 1807–1821. [DOI] [PubMed] [Google Scholar]
- Saffioti F, Hall A, de Krijger M, Verheij J, Hubscher SG, Maurice J, Luong TV, Pinzani M, Ponsioen CY & Thorburn D. (2021). Collagen proportionate area correlates with histological stage and predicts clinical events in primary sclerosing cholangitis. Liver Int 41, 2681–2692. [DOI] [PubMed] [Google Scholar]
- She H, Xiong S, Hazra S & Tsukamoto H. (2005). Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem 280, 4959–4967. [DOI] [PubMed] [Google Scholar]
- Shen K, Chang W, Gao X, Wang H, Niu W, Song L & Qin X. (2011). Depletion of activated hepatic stellate cell correlates with severe liver damage and abnormal liver regeneration in acetaminophen-induced liver injury. Acta Biochim Biophys Sin (Shanghai) 43, 307–315. [DOI] [PubMed] [Google Scholar]
- Shmarakov IO, Jiang H, Liu J, Fernandez EJ & Blaner WS. (2019). Hepatic stellate cell activation: A source for bioactive lipids. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Wu L, Zhang W, Ma WT, Yang GY, Zhang J, Xue DY, Chen B & Liu C. (2020). Peroxisome proliferator-activated receptor gamma inhibits hepatic stellate cell activation regulated by miR-942 in chronic hepatitis B liver fibrosis. Life Sci 253, 117572. [DOI] [PubMed] [Google Scholar]
- Taschler U, Schreiber R, Chitraju C, Grabner GF, Romauch M, Wolinski H, Haemmerle G, Breinbauer R, Zechner R, Lass A & Zimmermann R. (2015). Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim Biophys Acta 1851, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RS, Taylor RJ, Bayliss S, Hagstrom H, Nasr P, Schattenberg JM, Ishigami M, Toyoda H, Wai-Sun Wong V, Peleg N, Shlomai A, Sebastiani G, Seko Y, Bhala N, Younossi ZM, Anstee QM, McPherson S & Newsome PN. (2020). Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 158, 1611–1625 e1612. [DOI] [PubMed] [Google Scholar]
- Trivedi P, Wang S & Friedman SL. (2021). The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab 33, 242–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H (2005). Adipogenic phenotype of hepatic stellate cells. Alcohol Clin Exp Res 29, 132S–133S. [DOI] [PubMed] [Google Scholar]
- Venu M, Martin E, Saeian K & Gawrieh S. (2013). High prevalence of vitamin A deficiency and vitamin D deficiency in patients evaluated for liver transplantation. Liver Transpl 19, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergniol J, Foucher J, Terrebonne E, Bernard PH, le Bail B, Merrouche W, Couzigou P & de Ledinghen V. (2011). Noninvasive tests for fibrosis and liver stiffness predict 5-year outcomes of patients with chronic hepatitis C. Gastroenterology 140, 1970–1979. [DOI] [PubMed] [Google Scholar]
- Wake K (1971). “Sternzellen” in the liver: perisinusoidal cells with special reference to storage of vitamin A. Am J Anat 132, 429–462. [DOI] [PubMed] [Google Scholar]
- Wald G (1935). Carotenoids and the Visual Cycle. J Gen Physiol 19, 351–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng W, Li L, van Bennekum AM, Potter SH, Harrison EH, Blaner WS, Breslow JL & Fisher EA. (1999). Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry 38, 4143–4149. [DOI] [PubMed] [Google Scholar]
- Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, Modlin RL, Liblau RS, Gressner AM & Kaufmann SH. (2007). Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 26, 117–129. [DOI] [PubMed] [Google Scholar]
- Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM & Goodman DS. (1987). Biochemical characteristics of isolated rat liver stellate cells. Hepatology 7, 1224–1229. [DOI] [PubMed] [Google Scholar]
- Yamada T, Imaoka S, Kawada N, Seki S, Kuroki T, Kobayashi K, Monna T & Funae Y. (1997). Expression of cytochrome P450 isoforms in rat hepatic stellate cells. Life Sci 61, 171–179. [DOI] [PubMed] [Google Scholar]
- Yang L, Jung Y, Omenetti A, Witek RP, Choi S, Vandongen HM, Huang J, Alpini GD & Diehl AM. (2008). Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells 26, 2104–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, Lu L & Qian S. (2004). Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 40, 1312–1321. [DOI] [PubMed] [Google Scholar]
- Zhang W, Conway SJ, Liu Y, Snider P, Chen H, Gao H, Liu Y, Isidan K, Lopez KJ, Campana G, Li P, Ekser B, Francis H, Shou W & Kubal C. (2021). Heterogeneity of Hepatic Stellate Cells in Fibrogenesis of the Liver: Insights from Single-Cell Transcriptomic Analysis in Liver Injury. Cells 10, 2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Rostami MR, Zuo WL, Leopold PL & Crystal RG. (2020). Single-Cell Transcriptome Analysis of Mouse Liver Cell-Specific Tropism and Transcriptional Dysregulation Following Intravenous Administration of AAVrh.10 Vectors. Hum Gene Ther 31, 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisser A, Ipsen DH & Tveden-Nyborg P. (2021). Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis-Roles as Putative Treatment Targets? Biomedicines 9, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.