

Abstract
Mutations in the RYR1 gene, encoding ryanodine receptor 1 (RyR1), are a well-known cause of Central Core Disease (CCD) and Multi-minicore Disease (MmD). We screened a cohort of 153 patients carrying an histopathological diagnosis of core myopathy (cores and minicores) for RYR1 mutation. At least one RYR1 mutation was identified in 69 of them and these patients were further studied. Clinical and histopathological features were collected. Clinical phenotype was highly heterogeneous ranging from asymptomatic or paucisymptomatic hyperCKemia to severe muscle weakness and skeletal deformity with loss of ambulation. Sixty-eight RYR1 mutations, generally missense, were identified, of which 16 were novel. The combined analysis of the clinical presentation, disease progression and the structural bioinformatic analyses of RYR1 allowed to associate some phenotypes to mutations in specific domains. In addition, this study highlighted the structural bioinformatics potential in the prediction of the pathogenicity of RYR1 mutations. Further improvement in the comprehension of genotype–phenotype relationship of core myopathies can be expected in the next future: the actual lack of the human RyR1 crystal structure paired with the presence of large intrinsically disordered regions in RyR1, and the frequent presence of more than one RYR1 mutation in core myopathy patients, require designing novel investigation strategies to completely address RyR1 mutation effect.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40478-022-01357-0.
Keywords: RYR1-related myopathies, Central core disease, Multi-minicore disease, Genotype–phenotype correlations, Neuromuscular disorder, Protein modelling
Introduction
Core myopathy are clinically and genetically heterogenous congenital myopathies defined by the presence on skeletal muscle biopsy of “cores” with focally reduced oxidative activity, lack of mitochondria and variable degrees of myofibrillar disruption [1]. Based on the histological findings on muscle biopsy, core myopathies were traditionally classified in Central Core Disease (CCD), where the cores, in transverse sections of the myofibers, are single in the centre of the fibre and span through all the length of type 1 muscle fibre, and Multi-minicore Disease (MmD) with multiple and less defined cores without specificity for fibre type [2, 3]. This binary classification of core myopathies is becoming reductive since in the same muscle biopsy both CC and Mm may exist, a transition of the main histopathology features [4] or the presence with cores of other structural abnormalities in muscle fibres (i.e. rods [5, 6]) is common. To add to this complexity, new entities are entering in the picture. The Dusty Core Disease (DuCD) is quite recent, and it is defined by irregular areas of myofibrillar disorganization with reddish-purple granular material depositions that show uneven oxidative staining and are devoid of ATPase activity [7]. Dusty cores, in contrast with central cores, have no clear borders and are not round/ovoidal in shape [7].
The core myopathies are genetically heterogeneous and histopathology may suggest the underlying gene defect only in CCD, mostly due to the ryanodine receptor 1 gene (RYR1), and DuCD due to biallelic RYR1 mutations. In cores-rods myopathy and MmD mutations in a mixture of different genes have been reported including RYR1 [5], SELENON, MYH2, MYH7, TTN, CCDC78, UNC45B, ACTN2, and MEGF10, NEB, ACTA1, KBTBD13, CFL2, TRIP4, and TNNT1[8].
CCD is clinically characterized by hypotonia, motor developmental delay and generalized muscle weakness of variable degree. Distribution of weakness is typically proximal, with prominent involvement of the hip girdle and axial muscles. Bone and joint disorders, such as congenital hip dislocation, kyphoscoliosis, pes cavus, thoracic deformities, joint contractures are often present. The age of onset is extremely variable, ranging from foetal akinesia—which has been primarily reported associated with recessive inheritance [9], or birth onset—with hypotonia and floppy infant syndrome, childhood onset with motor developmental delay, but also adult onset with variable clinical presentation, often within the same family [3]. The penetrance of the disease is variable and genotype–phenotype correlations have yet to be fully clarified [10]. The association with malignant hyperthermia (MH), a life-threatening pharmaco-genetic disorder triggered by exposure of susceptible individuals to inhalational anesthetics and succinylcholine, is strong, though variable, and all patients suffering from core myopathies should be considered susceptible [11].
Both CCD and MH are primarily caused by mutations in RYR1, which encodes ryanodine receptor 1 (RyR1). RyR1, located on the sarcoplasmic reticulum membrane, is a Ca2+ channel comprising of four identical subunits [12], each one of 5,038 amino acids, with a molecular weight of 563.5 kDa [13]. The monomers delimit a central ion-conducting pore. RyR1, interacts with the dihydropyridine receptor (DHPR), and it is involved in the excitation contraction coupling, that ensures skeletal muscle contraction upon stimulation [14, 15].
The RyR1 protein is made of a cytoplasmic shell (also known as “foot”) and domains constituting the RyR1 channel and the activation core. The foot comprises: the N-terminal domains (NTD-A, NTD-B, NTD-C), the bridge solenoid (BSol), the junctional solenoid (JSol)—which connects NTD-C to BSol, the SP1a/ryanodine receptor domains (SPRY1-SPRY3) and the RyR repeats pairs (RY1&2, RY3&4). The RyR1 channel portion and the activation core comprise several structural domains: the shell-core linker peptide, CaM, and JSol binding sites (SCLP), the core solenoid (CSol), the thumb and forefingers domain (TaF), the auxiliary transmembrane helices (TMx), the pseudo voltage sensor domain (pVSD), the channel pore domain, made of six transmembrane helices, and the C-terminal domain (CTD) [16]. This region is directly responsible for the Ca2+-mediated channel activation [12, 17]. Moreover, given its crucial role in muscle physiology, RyR1 activity is finely regulated through a wide variety of post-translational modifications of the channel, including oxidation, phosphorylation[18], and the binding of ligands. In particular, calmodulin (CaM), calstabin1 (previously known as FK506-binding protein 12, FKBP12) and CLIC2[19] interact with the CTD, and triadin, junctin, and calsequestrin with the cytoplasmic shell [12, 19].
Autosomal dominant RYR1 mutations are identified in approximately 90% of CCD and are mostly localized in three hotspots: the cytoplasmic N-terminus (hotspot 1; amino acid 35–614), the central domain (hotspot 2; amino acid 2163–2458) and the C-terminus (hotspot 3; amino acid 4550- 4940) [3, 20]. On the basis of functional studies, distinct molecular mechanisms were proposed to explain how specific RYR1 mutations could result in core myopathies and/or in MH [21]. The clinical spectrum of MmD is also variable and depends on the genetic background [22]. MmD patients may manifest hypotonia and proximal muscle weakness, ophthalmoparesis, atrophy of the small muscles of the hands. Severe neonatal-onset form have been identified [22–25].
MmD may be associated with recessive mutations in the selenoprotein N1 gene (SEPN1, also known as SELENON) [26],—as well as other genes such as MYH2 [27, 28], UNC45B [29], MYH7 [30], TTN [31], MEGF10 [32], SECISBP2, ACTA1, ACTN2, CCD78 [33], and FXR1 [34]—but mutations in RYR1 had been increasingly described [22, 23, 25, 35]. Here we report a large series of Italian patients affected by RYR1-related core myopathy characterized at the clinical, histopathological and molecular level. Structural in silico modelling of RyR1 to predict pathogenicity of the identified RYR1 mutations has been utilized for in-depth genotype–phenotype correlations.
Materials and methods
Patients
An Italian network of tertiary referral Centers for congenital myopathies had been established in 2008 to collect detailed clinical, morphological, and genetic data in a large group of Italian patients. One hundred fifty-three patients were selected according to muscle histopathology consistent with cores or minicores myopathy and chosen for RYR1 mutations analyses. Ethics committees of all participating centers approved the study and written informed consent was obtained from all patients or their legal guardians, in accordance with the ethical standards of the 1975 Declaration of Helsinki, revised in 2013. Clinical evaluation was performed at each referring Center according to a standardized protocol, including the collection of family history, disease onset and progression data, functional abilities, muscle strength and severity of contractures when present. Electrocardiogram, echocardiogram, pulmonary function test results were collected for each patient. Based on disease severity, patients were classified as: “asymptomatic” if they came to medical attention for CK elevation, without symptoms of neuromuscular disease; “paucisymptomatic” if they complained of myalgias, and/or muscle cramps but not overt muscle weakness. In this subset of patients, the presence of minor osteoarticular alterations was allowed; “myopathic” if muscle weakness was present. This last subset was further categorized according to the MRC score of proximal and distal muscles in the upper and lower limbs in mild (MRC score ≥ 4 in all muscle tested), moderate (MRC ≤ 3 in one proximal muscle) and severe (MRC ≤ 3 in two or more proximal muscles). The age of onset in symptomatic patients was defined as “congenital” if clinical symptoms appeared before 12 months of age; “early” if symptoms were present before age of 20; “adult” if symptoms appeared after 20 years of age.
Muscle biopsy analysis
Muscle biopsies, done at the time of diagnosis, were reviewed, when available, at each referring centre by an expert myopathologist. Transverse cryosections of muscle biopsies stained with NADH-TR (nicotinamide adenine dinucleotide dehydrogenase-tetrazolium reductase), COX (cytochrome C oxidase) or SDH (succinate dehydrogenase), haematoxylin–eosin or Gomori’s trichrome were examined on upright microscope (Olympus BX60, Tokyo, Japan). All fibres with central nuclei, cores in type 1 or type 2 muscle fibres, rods, and the number of type 1 fibres were counted in 3 independent microscopic fields (at a magnification of 10). The results had been expressed as mean percentage.
RYR1 mutations analysis
Genomic DNA was extracted from peripheral blood according to standard procedure. The entire coding sequence of RYR1 (NG_008866.1) was amplified from patient genomic DNA and analysed by Sanger sequencing. Frequency of mutation in the hotspots was assessed with proportion test performed with R v. 3.5.3.
Homology modelling of the human RyR1 3D structure
The human RyR1 sequence (accession code: P21817) was obtained from Uniprot [36] and used to perform a Blastp search against the Protein Data Bank [37]. The 3D structure of the RyR1 orthologous protein from Oryctolagus cuniculus (PDB ID 5T15; electron microscopy structure, resolved at 3.6 Å) was selected as a template to build the model based on its high sequence identity and coverage with the human RyR1 (identities = 96.91%, coverage = 72%). As this structure presents multiple not resolved segments, the human RyR1 sequence was aligned against the template with Jalview [38] using T-Coffee [39] algorithm (default parameters) to identify the protein domains. Six comparative models of the target sequence were built by MODELLER [40] using the model single module. Models were then evaluated for value of discrete optimized protein energy using the DOPE method integrated in MODELLER. Model number 2 was selected as the best final model. Overall structure quality was further assessed with QMEANDisCo [41] and MolProbity [42]. The MobiDB-lite [43] was used to exactly identify and map intrinsically disordered regions boundaries mostly overlapping segments not resolved in the template structure. The model presents a good geometry with 90.09% of residues in the most Ramachandran favoured regions while the 2.05% are marked as outliers. The Rama distribution Z-score calculated for the entire model is -0.98 ± 0.11 (Goal: < 2) whereas the number of bad bonds is estimated to 0.01% of total. To avoid introduction of unwanted artifacts there was no attempt made to manually adjust the backbone torsion angles. The final 3D model corresponds to the human RyR1 segments spanning residues 24–1272, 1432–1924, 2057–2562, 2734–2940, 3645–4250, 4546–5034.
Bioinformatics analyses of RYR1 mutations
An integrative in silico pipeline was used to evaluate pathogenic impact of RYR1 mutations described in Additional file 1: Fig. S1. In details, a total of 45 RyR1 homologous sequences were retrieved by PSI-BLAST [44] search (default parameters) against the UniProt sequence database and aligned with T-Coffe (default parameters) [39] to derive a conservation score used each residue. This conservation score was then used as a preliminary filter to evaluate impact of substitutions found in patients. The ELM [45] database was used to search and map functional linear motifs, while FELLS [46] was used to derive structure features. Structural inspection was performed with Chimera [47], while effect of mutations on the human RyR1 3D structure was evaluated with BLUUES [48] and RING2.0 [49]. Stability and pathogenicity assessment were carried out using a consensus approach including predictions from Polyphen2.0 [50], SIFT [51], Mupro [52], Mutationassessor [53] (Additional file 1: Fig. S1). The new Intronic variant has been analysed with two independent algorithms for splice signal detection: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) and Berkeley Drosophila Genome Project (BDGP, https://www.fruitfly.org/seq_tools/splice.html).
Statistical analyses
In order to evaluate associations between the presence of mutations in specific domains and several phenotypic traits (foetal hypokinesia, hypotonia at birth, arthrogryposis, respiratory insufficiency, dysphagia, delayed independent ambulation, congenital hip dislocation, facial weakness, muscle wasting, ocular involvement, bulbar involvement, contractures, foot deformities, scoliosis/rigid spine, cardiac abnormalities, cognitive issues), for each pair of protein domain and phenotypical trait, a 2 × 2 table was devised, with rows indicating presence vs. absence of mutations in that domain, and columns indicating presence vs. absence of the phenotypic trait. P-values < 0.05 are reported (without correction for multiple testing) and should be interpreted as descriptive.
Results
At least one RYR1 mutation was identified in 69 core myopathy patients and these have been further studied (Table 1).
Table 1.
Genetic details of RYR1-affected individuals
| ID/Family | Nucleotide change | Amino acid change | Family history | Exon/Intro | Affected domain | Variant classificationa | References |
|---|---|---|---|---|---|---|---|
| 1/1 | c.212C > A | p.Ser71Tyr | No | E 3 | NTD-A | Pathogenic | [54, 55] |
| c.6847A > C | p.Asn2283His | E 42 | BSol | Pathogenic | [54] | ||
| 2/2 | c.467G > A | p.Arg156Lys | No | E 6 | NTD-A | Pathogenic | [55, 56] |
| 3/3 | c.487C > T | p.Arg163Cys | AD | E 6 | NTD-A | Pathogenic | [57, 58] |
| 4/3 | c.487C > T | p.Arg163Cys | AD | E 6 | NTD-A | Pathogenic | [57, 58] |
| 5/3 | c.487C > T | p.Arg163Cys | AD | E 6 | NTD-A | Pathogenic | [57, 58] |
| 6/4 | c.487C > T | p.Arg163Cys | AD | E 6 | NTD-A | Pathogenic | [57, 58] |
| 7/5 | c.1021G > A | p.Gly341Arg | AR | E 11 | NTD-B | Pathogenic | [59, 60] |
| c.1021G > A | p.Gly341Arg | E 11 | NTD-B | Pathogenic | [59, 60] | ||
| 8/6 | c.1209C > G | p.Ile403Met | No | E 12 | NTD-B | Pathogenic | [61] |
| 9/7 | c.1250 T > C | p.Leu417Pro | No | E 13 | NTD-C | Likely pathogenic | [62] |
| 10/8 | c.1840C > T | p.Arg614Cys | AD | E 17 | NTD-C | Pathogenic | [63, 64] |
| 11/9 | c.3301G > A | p.Val1101Met | No | E 25 | SPRY2/SPRY3 | Likely pathogenic | [65] |
| c.14473C > T | p.Arg4825Cys | E 100 | Pore | Pathogenic | [66] | ||
| 12/10 | c.4711A > G | p.Ile1571Val | No | E 33 | SPRY2/SPRY3 | Likely benign | [62, 67] |
| c.7373G > A | p.Arg2458His | E 46 | BSol | Pathogenic | [68] | ||
| c.10097G > A | p.Arg3366His | E 67 | BSol | Likely pathogenic | [67] | ||
| c.10259 + 7G > A | p.( =) | I 67 | BSol | Benign | dbSNP: rs143752962 | ||
| c.11798A > G | p.Tyr3933Cys | E 86 | CSol | Pathogenic | [67, 69] | ||
| 13/11 | c.4711A > G | p.Ile1571Val | No | E 33 | SPRY2/SPRY3 | Likely benign | [62, 67] |
| c.10097G > A | p.Arg3366His | E 67 | BSol | Likely pathogenic | [67] | ||
| c.11708G > A | p.Arg3903Gln | E 85 | CSol | Pathogenic | [55] | ||
| c.11798A > G | p.Tyr3933Cys | E 85 | CSol | Pathogenic | [67, 69] | ||
| 14/12 | c.4816C > T | p.Arg1606Cys | No | E 33 | SPRY2/SPRY3 | Pathogenic | [70] |
| 15/13 | c.5510A > C | p.Gln1837Pro | AD | E 34 | JSol | Pathogenic | This paper |
| 16/14 | c.6488G > A | p.Arg2163His | No | E 39 | BSol | Pathogenic | [57] |
| 17/15 | c.6502G > A | p.Val2168Met | AR | E 39 | BSol | Pathogenic | [60] |
| c.7372C > T | p.Arg2458Cys | E 46 | BSol | Pathogenic | [68] | ||
| 18/16 | c.7025A > G | p.Asn2342Ser | No | E 43 | BSol | Pathogenic | [71] |
| c.14659C > T | p.His4887Tyr | E 102 | Pore | Pathogenic | [72] | ||
| 19/17 | c.7048G > A | p.Ala2350Thr | AD | E 44 | BSol | Pathogenic | [73] |
| 20/17 | c.7048G > A | p.Ala2350Thr | AD | E 44 | BSol | Pathogenic | [73] |
| 21/18 | c.7085A > G | p.Glu2362Gly | AD | E 44 | BSol | Pathogenic | [55] |
| c.13513G > C | p.Asp4505His | E 92 | pVSD | Pathogenic | [74, 75] | ||
| 22/19 | c.7304G > A | p.Arg2435His | AD | E 45 | BSol | Pathogenic | [60] |
| 23/20 | c.7523G > A | p.Arg2508His | No | E 47 | BSol | Pathogenic | [20, 55] |
| 24/21 | c.9293G > T | p.Ser3098Ile | No | E 63 | BSol | Pathogenic | This paper |
| c.14645C > T | p.Thr4882Met | E 101 | Pore | Pathogenic | [76] | ||
| 25/22 | c.10097G > A | p.Arg3366His | AD | E 67 | BSol | Likely pathogenic | [67] |
| c.11798A > G | p.Tyr3933Cys | E 86 | CSol | Pathogenic | [67, 69] | ||
| 26/23 | c.11708G > A | p.Arg3903Gln | No | E 85 | CSol | Pathogenic | [55] |
| 27/24 | c.11708G > A | p.Arg3903Gln | AR | E 85 | CSol | Pathogenic | [55] |
| c.11708G > A | p.Arg3903Gln | E 85 | CSol | Pathogenic | [55] | ||
| 28/24 | c.11708G > A | p.Arg3903Gln | AR | E 85 | CSol | Pathogenic | [55] |
| c.11708G > A | p.Arg3903Gln | E 85 | CSol | Pathogenic | [55] | ||
| 29/25 | c.13724A > C | p.Asn4575Thr | No | E 94 | pVSD | Pathogenic | [77] |
| 30/26 | c.13910C > T | p.Thr4637Ile | No | E 95 | pVSD | Pathogenic | [78] |
| 31/27 | c.14209C > T | p.Arg4737Trp | AD | E 98 | pVSD | Pathogenic | [79, 80] |
| 32/27 | c.14209C > T | p.Arg4737Trp | AD | E 98 | pVSD | Pathogenic | [79, 80] |
| 33/28 | c.14582G > A | p.Arg4861His | No | E 101 | Pore | Pathogenic | [66, 78] |
| 34/29 | c.14680G > C | p.Ala4894Pro | AD | E 102 | Pore | Pathogenic | [81] |
| 35/29 | c.14680G > C | p.Ala4894Pro | AD | E 102 | Pore | Pathogenic | [81] |
| 36/29 | c.14680G > C | p.Ala4894Pro | AD | E 102 | Pore | Pathogenic | [81] |
| 37/29 | c.14680G > C | p.Ala4894Pro | AD | E 102 | Pore | Pathogenic | [81] |
| 38/30 | c.14690G > A | p.Gly4897Asp | No | E 102 | Pore | Pathogenic | [82, 83] |
| 39/31 | c.14693 T > C | p.Ile4898Thr | AD | E 102 | Pore | Pathogenic | [83–85] |
| 40/31 | c.14693 T > C | p.Ile4898Thr | AD | E 102 | Pore | Pathogenic | [83–85] |
| 41/32 | c.14693 T > C | p.Ile4898Thr | AD | E 102 | Pore | Pathogenic | [83–85] |
| 42/33 | c.14693 T > C | p.Ile4898Thr | No | E 102 | Pore | Pathogenic | [83–85] |
| 43/34 | c.14695G > A | p.Gly4899Arg | AD | E 102 | Pore | Pathogenic | [85, 86] |
| 44/35 | c.14818G > A | p.Ala4940Thr | AD | E 103 | Pore | Pathogenic | [78] |
| 45/36 | c.14928C > G | p.Phe4976Leu | AR | E 104 | CTD | Pathogenic | [87] |
| c.14928C > G | p.Phe4976Leu | E 104 | CTD | Pathogenic | [87] | ||
| 46/36 | c.14928C > G | p.Phe4976Leu | AR | E 104 | CTD | Pathogenic | [87] |
| c.14928C > G | p.Phe4976Leu | E 104 | CTD | Pathogenic | [87] | ||
| 47/37 | c.472_474delGAA | p.Glu158del | No | E 6 | NTD-A | Pathogenic | This paper |
| 48/38 | c.1690 T > C | p.Tyr564His | No | E 16 | NTD-C | Benign | [85] |
| c.2930C > T | p.Thr977Met | E 24 | RY1&2 | Pathogenic | dbSNP: rs375865052 | ||
| 49/39 | c.3866G > A | p.Arg1289Gln | No | E 28 | SPRY2/SPRY3 | Pathogenic | This paper |
| 50/40 | c.3901C > T | p.Arg1301Cys | No | E 28 | SPRY2/SPRY3 | Pathogenic | dbSNP: rs745920741 |
| c.5360C > T | p.Pro1787Leu | E 34 | JSol | Benign | [56] | ||
| 51/41 | c.3935C > T | p.Pro1312Leu | No | E 28 | SPRY2/SPRY3 | Benign | This paper |
| 52/42 | c.4949 T > C | p.Leu1650Pro | No | E 34 | SPRY2/SPRY3 | Pathogenic | This paper |
| c.6352C > T | p.Arg2118Trp | E 39 | JSol | Pathogenic | [88] | ||
| c.14918C > T | p.Pro4973Leu | E 104 | CTD | Likely pathogenic | [79] | ||
| 53/43 | c.6178G > T | p.Gly2060Cys | No | E 38 | JSol | Benign | [54, 56] |
| c.13691G > A | p.Arg4564Gln | E 94 | pVSD | Pathogenic | [7] | ||
| 54/44 | c.6352C > T | p.Arg2118Trp | AR | E 39 | JSol | Pathogenic | [88] |
|
c.14918C > T c.10347 + 1G > A |
p.Pro4973Leu IVS68 + 1G > A |
E 104 I 68 |
CTD – |
Likely pathogenic Pathogenic |
[79] [88] |
||
| 55/45 | c.6377G > A | p.Arg2126Gln | No | E 39 | JSol | Pathogenic | [89] |
| c.11813G > A | p.Gly3938Asp | E 86 | CSol | Pathogenic | [89] | ||
| c.15022G > C | p.Glu5008Gln | E 106 | CTD | Pathogenic | This paper | ||
| 56/46 | c.6617C > T | p.Thr2206Met | No | E 40 | BSol | Pathogenic | [60] |
| c.10537A > G | p.Thr3513Ala | E 71 | BSol | Pathogenic | This paper | ||
| 57/47 | c.9145C > T | p.Leu3049Phe | No | E 61 | BSol | Pathogenic | This paper |
| 58/48 | c.10516C > A | p.Gln3506Lys | No | E 71 | BSol | Pathogenic | This paper |
| 59/49 | c.11609-2A > G | IVS83-2A > G | AD | I 83 | – | Uncertain significance | This paper |
| 60/49 | c.11609-2A > G | IVS83-2A > G | AD | I 83 | – | Uncertain significance | This paper |
| 61/50 | c.13952A > G | p.His4651Arg | AD | E 95 | pVSD | Pathogenic | dbSNP: rs118192139 |
| 62/50 | c.13952A > G | p.His4651Arg | AD | E 95 | pVSD | Pathogenic | dbSNP: rs118192139 |
| 63/51 | c.14815G > T | p.Asp4939Tyr | AD | E 103 | Pore | Pathogenic | This paper |
| 64/51 | c.14815G > T | p.Asp4939Tyr | AD | E 103 | Pore | Pathogenic | This paper |
| 65/52 |
c.12063insAC c.2709_2711delCCC |
p.Asp4021GlufsX4 p.His903del |
AR |
E 88 E 22 |
CSol RY1&2 |
Pathogenic Pathogenic |
This paper This paper |
| 66/53 | c.7080insG | p.Pro2361AlafsX2 | No | E 44 | BSol | Likely pathogenic | This paper |
| 67/54 | c.14510delA | p.Gln4837ArgfsX3 | AD | E 100 | Pore | Pathogenic | [56] |
| 68/54 | c.14510delA | p.Gln4837ArgfsX3 | AD | E 100 | Pore | Pathogenic | [56] |
| 69/55 | c.4711A > G | p.Ile1571Val | No | E 33 | SPRY2/SPRY3 | Likely benign | [62, 67] |
| c.9407delT* | p.Leu3136Argfs | E 63 | BSol | Pathogenic | This paper |
aVariant classification based on literature or bioinformatic analyses. ID: patient’s number; AR: autosomal recessive inheritance; AD: autosomal dominant inheritance; E: exon; I: Intron; NTD-A: N-terminal domain A; NTD-B: N-terminal domain B; NTD-C: N-terminal domain C; BSol: bridge solenoid; JSol: junctional solenoid; SPRY1-SPRY3: SP1a/ryanodine receptor domain; RY1&2: RyR repeats pairs; CSol: core solenoid; pVSD: pseudo voltage sensor domain; CTD: C-terminal domain
RYR1 mutations
Sixty-eight different nucleotide variations in RYR1 sequence were identified in 69 patients from 55 unrelated families. As expected, mutations were localized with higher frequency in the 3 hotspot regions, than in the rest of the sequence (p < 0.0001). Variations included 59 missense mutations, 3 splice site variants, 4 small frameshift insertion or deletion, and two in-frame deletion of a single amino acid (Table 1). Among the 68 mutations, 16 were novel (Table 1). Twenty-three patients carried more than one RYR1 variant (Table 1); of these patients, 13 (57%) were isolated case, 2 (9%) patients had an autosomal dominant (AD) and 8 (35%) an autosomal recessive (AR) inheritance pattern. Unfortunately, parents of patients were not available for analyses and the phase of the mutations was not assessed, with the exception of patients #17 and #69 who carry mutations known to be transmitted in cis [77, 90].
Muscle histopathology
Muscle biopsy, done at time of diagnosis, was reviewed in 52/69 patients. Of the remaining 17 patients, 10 were familial cases where only a relative had the muscle biopsy done and in 7 patients muscle biopsy, consistent with core myopathy according to the referring physician, was not available for review. Quadriceps was biopsied in 45 patients, deltoid in 3, biceps and triceps in one each, while there was no information about the site of biopsy for 2 subjects. For all the samples, only transverse sections were available and analysed. All biopsies but one (#31, Additional file 3: T2) showed cores in a variable number of type 1 muscle fibres (ranging from 8 to 100%). Patient #31 had cores in 39% of type 2 muscle fibres only. In type 1 fibres, cores were centrally located (central cores, CC) in 67% of muscle biopsies, multiple minicores (Mm) and CC were present in 15% and Mm alone in 17% of muscle biopsies. CC and Mm were mostly observed in type 1 muscle fibres, whereas a minority of patients showed CC and Mm in type 2 fibres. No dusty cores were observed in this cohort of patients (Fig. 1).
Fig. 1.
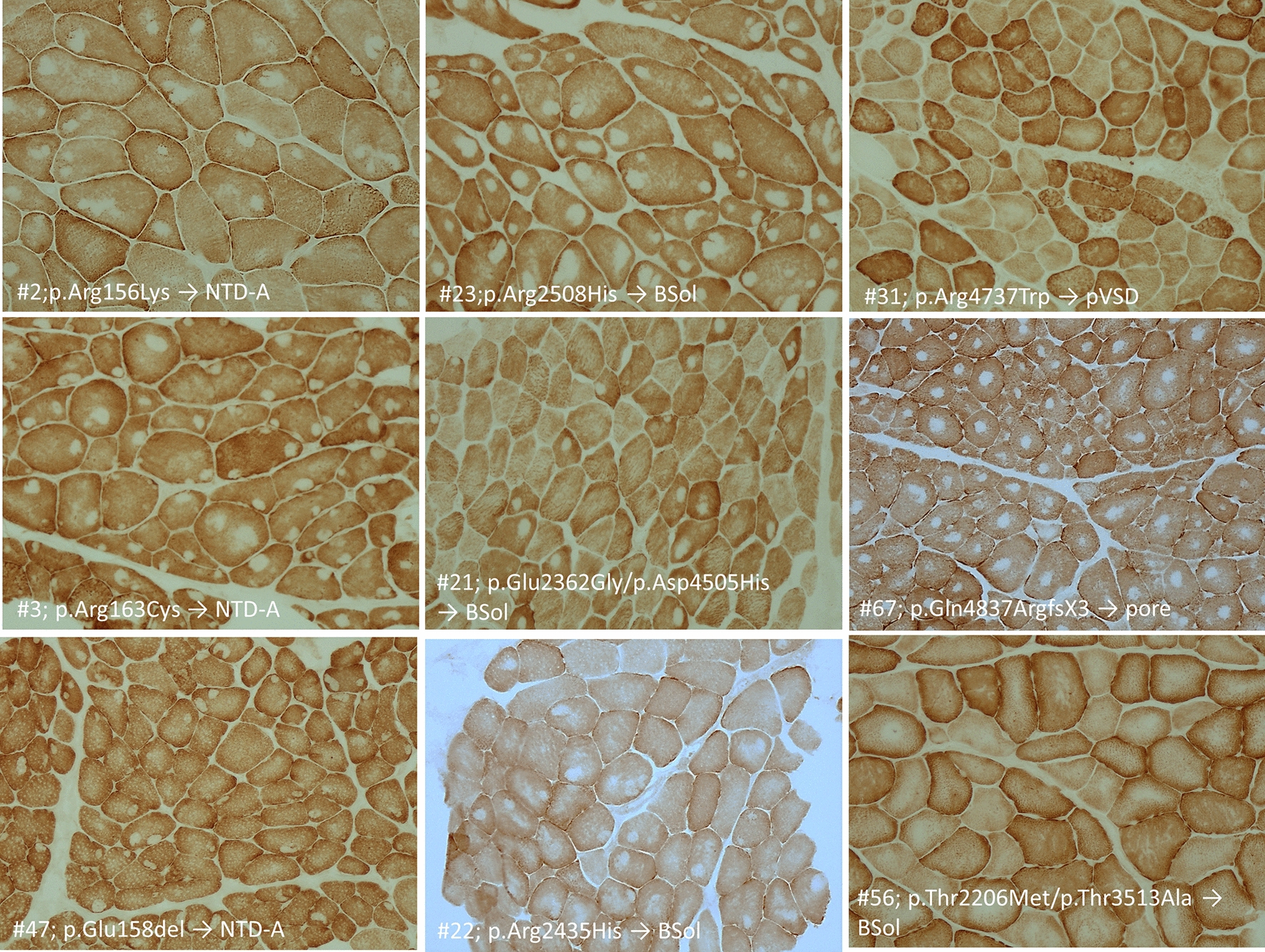
Muscle biopsy stained with COX from patients with core myopathy. In each panel is reported the patient #, the RYR1 mutation and RyR1 protein domain where the mutation is located. Central core (CC) (single or multiple, centrally or peripherally located) are shown in patient #2, 3, 47, 23, 21, 22, 67 and 56. In patient #56 a minority of muscle fibers showed CC where minicores (Mm) (multiple, small randomly distributed areas with focal loss of mitochondrial activity) were more abundant. Patient #22 showed both CC and Mm where patient #31 muscle biopsy showed only Mm. Neither the RYR1 mutation nor the RyR1 protein domain predict the ratio between CC and Mm
Type 1 fibre predominance (defined as more than 45% of type 1 fibres) [91], ranging between 50–100%, was observed in 44 muscle biopsies (44/50; 88%) and increase in central nuclei (> 3% of fibres) was detected in 33 (33/50; 66%). An increase in perimysial and endomysial connective tissue was mild in 24 (24/51; 47%), severe in 6 (6/51; 12%) and absent in 20 (20/51; 39%) muscle biopsies. Only one biopsy (#44, Additional file 3: T2) showed rods in hypotrophic fibres associated with CC. Two muscle biopsies were obtained from patient #14. The first, performed at 5 years of age, showed no cores while the following biopsy, done 7 years later, showed CC in 100% of type 1 fibres (data not shown).
Clinical description
The studied cohort comprised 27 (39%) males and 42 females, aged from 2 to 72 years at the time of the last neurological evaluation (Additional file 4: T3). Thirty-eight/69 (55%) patients had a positive family history: 8 (8/38; 21%) AR and 30 (30/38; 79%) AD; family history was not available for 31 individuals (Table 1). In order to verify if mutations located in specific RyR1 domains were associated with more severe phenotype, we grouped patients based on the domain affected by the RYR1 mutation and utilized clinical data at last evaluation for comparison. Subjects carrying more than one pathogenic mutation were described in each mutated domain group (Fig. 2 and Additional file 2: T1). Variations predicted or known to be benign were excluded from the analysis. Frame-shift mutations have been studied as separate group, independently from the affected domain. Results are summarized in Fig. 2 and Additional file 2: T1, and detailed clinical data are reported in Additional file 3: T2.
Fig. 2.

Histogram showing the proportion of patients manifesting specific phenotypes. Patients are grouped based on the mutated domain; each domain is colored following the legend on the top of the figure. A. Manifestations during pregnancy or at the birth. B. Muscular manifestations. C. Osteoarticular manifestations. D. Respiratory involvement
NTD-A domain mutations. In our cohort 7 patients had mutations in NTD-A domain, only one of them (patient #47) had a severe clinical course. She presented at birth as a floppy infant, independent ambulation was acquired at 16 months and at 12 years of age the patient had a severe proximal and distal muscle weakness. Patient #1 had a congenital onset with hip dislocation, but an overall mild clinical course and at age 16 years the patient has a mild myopathy and retains the ability to walk and run. Muscle weakness was reported by 57% of patients but disease progression was stationary in all but one. The association between mutations in NTD-A with facial weakness and delayed independent ambulation was nominally significant (Fisher exact test p = 0.039, p = 0.038, respectively).
NTD-B domain mutations. Two patients had mutations in NTD-B domain. Both patients presented at birth with arthrogryposis. Patient #7 had a severe AR myopathy with proximal and distal muscle weakness but was last seen at 3 years of age, while patient #8, who had delayed independent ambulation at 24 months of age had a subsequent slowly progressive disease and at age 57 years she is still ambulant with a moderate myopathy. The association between the presence of NTD-B mutation and arthrogryposis was nominally significant (Fisher exact test p = 0.013).
NTD-C domain mutations. Two patients had mutations in this domain. These patients had an early or congenital onset disease, with steady progression and severity ranging from mild to moderate. Muscle wasting, facial weakness, ophthalmoparesis, scoliosis and respiratory involvement were present in 1/2 patients.
Ry1&2 domain mutations. Two patients (#48, #65) carried a pathogenic RYR1 mutation in the Ry1&2 domain. Both patients presented as floppy infant with dysphagia at birth. The patients had a mild to severe disease with bulbar involvement and respiratory insufficiency. Mutations in this domain were associated with foetal hypokinesia (p = 0.031), respiratory insufficiency (p = 0.007) and bulbar involvement (p = 0.010).
SPRY2/SPRY3 domain mutations. Five sporadic patients belong to this group. Early onset and slowly progressive or steady disease characterize this subset of patients. One patient was diagnosed because of CK elevation and at 48 years-of-age remains paucisymptomatic (patient #50).
JSol domain mutations. Four patients carried mutations in JSol domain. Two of them presented with foetal hypokinesia and delayed independent ambulation, 2/6 presented as floppy infant with dysphagia. All showed muscle weakness, 3/4 facial weakness, and 3/4 muscle wasting.
BSol domain mutations. Sixteen patients had mutations in BSol domain. The onset was congenital in 5, early in 6, adult in 3 and in 2/16 asymptomatic patients hyperCKemia prompted diagnosis. Two patients had a severe weakness and one (patient #57) required not-mechanical ventilation. Ocular involvement was shown by 3/16 patients. Presence of mutations in this domain was associated with delayed independent ambulation (p = 0.008) and foetal hypokinesia (p = 0.030).
CSol domain mutations. Seven patients had mutations in the CSol domain. The onset was congenital in 5 and early in 2patients. The severity of the disease was variable ranging from mild to severe. Six/7 patients had proximal muscle weakness and 5 also facial muscles involvement. Two patients had a severe, slowly progressive, myopathy. Contractures, foot deformities and scoliosis were frequent.
pVSD domain mutations. Eight patients belonged to this group. Two patients had an adult onset and 6/8 had a congenital or early onset disease. A positive association with hypotonia at birth (“floppy infant” presentation, p = 0.043) was observed. Five patients had a moderate myopathy, 2 a mild myopathy, and only one patient was paucisymptomatic. Only one patient (#53) had a neonatal onset and at age 2 is still not ambulant.
Pore domain mutations. Seventeen patients carried mutations in the pore domain. Seven presented as floppy infant, and 6 with congenital hip dislocation. Thirteen patients had a congenital (6) or early onset (7), one patient was identified through family screening and was asymptomatic at 1 < 6 years of age and for 2 patients no information were available regarding disease onset. Eight had a mild, 5 moderate and 3 a severe myopathy. Four patients showed facial muscle weakness, and 3 muscle wasting. Nine patients had scoliosis. There was a statistical association with foetal hypokinesia (p = 0.027), presence of contractures (p = 0.023), and foot deformities (0.010).
CTD domain mutations. Five patients carried CTD domain mutations. The onset was congenital in 4 and early in one patient. The myopathy was moderate in 4 patients and severe in 1. Four patients showed facial weakness and 2 muscle wasting. Mutations in this domain were associated with foetal hypokinesia (p = 0.038) and dysphagia (p = 0.006).
Frame-shift RYR1 mutations. We identified 5 patients, belonging to 4 families, carrying frame-shift mutations. The onset of symptoms was heterogeneous comprising congenital (1), early (2), and adult (2). Muscle weakness was moderate in 2, mild in 4 and one patient was paucisymptomatic. Two patients had a moderate myopathy, and one of them had also rhinolalia and dysphagia.
RYR1 mutations bioinformatic analyses
Four RyR1 chains assemble to form a single calcium release channel and multiple 3D structures derived from cryomicroscopy studies describing their organization are available in the literature. To investigate the effect of mutations in our dataset, we generated a homology model of the human RyR1 monomer using the orthologous 3D structure from rabbit (identities = 96.91%, coverage = 72%). Resulting model presents an overall good geometry with 90.09% of residues occupying the Ramachandran favoured regions. The 2.05% of residues are estimated to occupy unfavourite positions. Visual inspection shown that most of them localize at the borders of regions predicted as intrinsically disordered. Pairing these data with a number of bad bonds corresponding to 0.01%, we suggest that our model is good enough to assess the effect of mutations found in patients. In details, mutations were classified as “pathogenetic from literature” if already reported so, “pathogenetic from function” based on in silico studies, “likely benign” based on literature data and/or bioinformatics analyses, and “truncating, frameshift and intronic” based on their effect on the protein sequence.
RYR1 mutations “pathogenetic from literature”
Thirty-eight mutations were identified in this subset.
NTD-A, NTD-B, and NTD-C domains. Mutations falling into RyR1 N-terminal region (amino acid 35–614) are causative of both MH and CCD [92]. We found 7 known mutations in this region (p.Ser71Tyr [54, 55], p.Arg156Lys [56], p.Arg163Cys [57, 58], p.Gly341Arg [59, 60], p.Ile403Met [61], p.Leu417Pro [62], p.Arg614Cys [63, 64]). Structural investigations of these variants suggested that they may promote destabilization of MIR folding domain, thus predisposing to pathological phenotypes (Figs. 3, 4). According to literature, the region between amino acids 1272–1455 is responsible for interaction with the II-III loop of DHPR, which is an intrinsically unstructured protein acting as calcium channel gating activator [93]. Previous experimental validation demonstrates that this region is essential for skeletal muscle contraction in vivo [94]. We hypothesize that the mutations in this region promote secondary structure rearrangement impairing the DHPR function.
JSol, BSol and CSol domains. The impairment of DHPR was was predicted also for p.Arg2118Trp [88] and p.Arg2126Trp [89], as these mutations occurs in a RyR1 region known to interact with DHPR as well [93]. The pathogenicity of p.Arg2118Trp may also be due to the mutation’s proximity to the calmodulin binding site on RyR1 [88]. Mutations p.Arg2163His [57], p.Val2168Met [60], p.Thr2206Met [60] and p.Asn2283His [54] are located in a conserved region of RyR1 central domain. Functional studies demonstrated that these mutations result in increased caffeine sensitivity and altered calcium handling in cells [54, 57, 60]. RyR1 has other regulative modules such as the 2350–2458 segment, known to promote the channel inactivation [95, 96]. The effect of mutations in this region is related to the loss of interaction with calstabin1, a cis–trans peptidyl-prolyl-isomerase, required for the physiological gating of the channel [95]. Due to its specific function, mutations occurring in this region, such as p.Asn2342Ser [71], p.Ala2350Thr [73], p.Glu2362Gly [55], p.Arg2435His [60], p.Arg2458Cys [68], p.Arg2458His [68] and p.Arg2508His [20, 55] are predicted to be likely pathogenetic, due to their possible ability to inactivate the channel gating. Moreover, stability predictors evaluated all the variants as destabilizing. Several predictors found p.Arg3366His [67] and p.Tyr3933Cys [67] to be pathogenic, both mutations involving conserved RyR1 regions. In particular, p.Tyr3933Cys resides in the RyR/IPR3 homology associated domain which is involved in Ca2 + regulation [67]. A similar effect may be hypothesized also for p.Arg3903Gln [55].
pVSD, Pore and CTD domains. The C-terminal region of RyR1 is known to bind CaM and to act as selectivity filter, regulating the number and chemical property of passing ions [97, 98]. The region forms a transmembrane domain and amino acids mutations in this area resulted commonly as pathogenic prone, disrupting the channel activity [98]. As expected [16, 98], six transmembrane helices were correctly predicted between residues 4285–4302, 4329–4346, 4351–4368, 4563–4580, 4794–4811 and 4838–4855. The region between residues 4300–4850 is known to be an important regulative element for RyR1 activation/inactivation switch [98–100], with several known pathological mutations falling within. The analysis of six mutations (i.e. p.Asp4505His [74, 75], p.Asn4575Thr [77], p.Thr4637Ile [78], p.His4651Arg [78], p.Arg4737Trp [79, 80] and p.Arg4825Cys [66]) in the interval 4505–4825 (Figs. 2, 3), suggested a potentially pathological phenotype, affecting the apoCAM binding domain functionality and the ions flux balance regulation. A detailed study of the effect of p.Asp4505His is reported by Groom et al. [75]. This mutation affects the RYR1-divergent region 1 (D1; amino acids 4254–4631) [101], deletion of the majority of this region (Δ4274–4535) potentiates voltage-gated Ca2 + release and enhances channel’s sensitivity to activation by DHPR [102]. Thus, the p.Asp4505His mutation may enhance RyR1 release-channel sensitivity to activation by disrupting the integrity of the D1-negative regulatory module [75]. We found several mutations in RyR1 transmembrane region that had been previously associated to core myopathies, i.e. p.Arg4861His [66, 78], p.Thr4882Met [76], p.His4887Tyr [72], p.Ala4894Pro [81], p.Gly4897Asp [82, 83], p.Ile4898Thr [83–85], p.Gly4899Arg [85, 86], p.Ala4940Thr [78], p.Pro4973Leu [79] and p.Phe4976Leu [87].
Fig. 3.

Schematic representation of RyR1 monomer functional domains. RyR1 sequence is presented as yellow bar with functional regions represented as colored boxes. Domain structural organization is presented on top. Dotted lines highlight regulative regions where red implies inhibition and green activation of the channel. Arrows represent the position for each mutation (frameshift mutations not shown). New mutations modelled in silico are wrote in red and marked with * when predicted as pathogenic, in green marked with # when benign
Fig. 4.

Cartoon representation of the human RyR1 structure. Front and top views of RyR1 tetramer assembly. Different colors represent each monomer. On right side, isolated RyR1 monomer colored by functional domains with mutations noted and grouped accordingly (frameshift mutations not shown). New mutations modelled in silico are wrote in red and marked with * when predicted as pathogenic, in green marked with # when benign
3.2.2 “Pathogenetic from function” RYR1 mutations
We modelled fifteen variants, which were either novel, or not previously studied (11 variants) or with unclear effect on the RyR1 channel from literature (4 variants) (i.e. p.Val1101Met [65], p.Gly3938Asp [89], p.Arg4564Gln [7], p.Arg1606Cys [70]). The human RyR1 (Uniprot code: P21817) was analysed with a collection of different bioinformatics tools to evaluate the impact of amino acid mutations on the RyR1 protein function.
NTD-A domain. The mutation p.Glu158del localizes in a flexible loop connecting two β-strands at the beginning of the MIR2 domain. Pathogenicity predictors suggest that this mutation reduces the domain stability. Further, multiple mutations in this area are already known to promote both MH and CCD [92].
RY1&2 domain. The mutation p.Thr977Met was predicted as damaging. Thr977 is exposed to the solvent and structural investigation suggests its variation in methionine to destabilize the correct RY1&2 (amino acids 850–1054) domain folding. Previous observations proposed that this repeated region, together with the SPRY domains, acts as calstabin1 interacting region [103]. A pathological phenotype 1n patients harbouring the p.Thr977Met may be associated with impairment of this interaction.
SPRY2/SPRY3 domain. The p.Val1101Met is predicted as probably damaging as Val1101 engages hydrophobic interactions with Phe1089 and Phe1091 stabilizing the SPRY2 domain spanning residues 1055–1241. According to literature, the region within residues 1272–1455 is responsible for interaction with the II-III loop of DHPR [94]. Experimental validation demonstrated that the region is relevant for skeletal muscle contraction in vivo [94], suggesting that variants p.Arg1289Gln and p.Arg1301Cys may interfere with the DHPR-mediated regulation of RyR1. A damaging effect on the protein structure is predicted for p.Arg1606Cys. Structural inspection shows Arg1606 to form electrostatic interactions with both Asp1111 and Glu1113. At the structural level, our analysis suggests that this mutation alter correct localization of the SPRY2 domain inducing local protein unfolding. Damaging effect is also predicted for p.Leu1650Pro. This position is well conserved among eukaryotes indicating it may play a relevant structural and/or functional role. Our structural investigation showed Leu1650 to form multiple van der Waals interactions with residues from the adjacent chain, suggesting that its mutation can negatively affect RyR1 tetramerization, yielding a functionally reduced channel. Mutation p.Gln1837Pro localizes in a short alpha helix segment connecting two longer helices spanning residues 1803–1852. The entire segment is part of a larger junctional solenoid domain. As proline is known to destabilize secondary structure, we believe the mutation induces a local unfolding of this domain.
BSol and CSol domains. Mutations p.Leu3049Phe, p.Lys3098Ile, p.Gln3506Lys and p.Thr3513Ala localize in a long disordered region important for RyR1 association with CaM (Fig. 2). All of them are predicted as damaging, possibly interfering with CaM mediated RyR1 regulation. A different pathogenic effect is predicted for p.Gly3938Asp. The Gly3938 localizes at the internal binding interface between RyR1 monomers directly facing the residue Glu79 from an adjacent monomer chain. The repulsive interaction introduced by p.Gly3938Asp is rather predicted to lower RyR1 complex stability than having a negative effect on monomer structure.
pVSD, Pore and CTD domains. The region between residues 4300–4850 is known to be an important regulative element for the RyR1 activation/inactivation switch [98–100], with several known pathological mutations falling within. In particular, p.Arg4564Gln impairs an electrostatic interaction between Arg4564 and Tyr4792. The analysis of mutations localizing into the interval 4505–5008, i.e. p.Asp4939Tyr and p.Glu5008Gln, predict them as damaging and suggest a potentially pathological phenotype, affecting the apoCAM binding domain functionality and the ions flux balance regulation. The evidence is also in agreement with [104].
3.2.4 “Likely benign” RYR1 mutations
NTD-A domain. In silico pathogenicity prediction suggests p.Tyr564His to be tolerated. A modest structural impact is also predicted by investigating the residue interacting network around Tyr564 (Fig. 3). Indeed, investigation shows p.Tyr564His to localize in the N-terminal solenoid spanning residues 393–627. Based on our model, this residue is partially exposed and modestly involved in the tandem repeat domain folding. We thus suggest this mutation to be likely neutral.
SPRY2/SPRY3 domain. As for p.Tyr564His, sequence conservation analysis shows that Pro1312 is only limitedly conserved among different species, reinforcing the idea that mutation p.Pro1312Leu may be tolerated. Another possible explanation of this result is that stability predictors fail to address p.Pro1312Leu pathogenicity as Pro1312 localizes in an intrinsically disordered region. The p.Ile1571Val [62, 67] variant is reported to be benign.
JSol domain and intronic region. p.Pro1787Leu [56], p.Gly2060Cys [54, 56, 105], and c.10259 + 7G > A are reported to be benign variants.
Truncating and frameshift RYR1 mutations
We found four frameshift mutations i.e. p.Pro2361AlafsX2, p.Leu3136Argfs, p.Gln4837ArgfsX3 and p.Asp4021GlufsX4. In our series, a predicted altered quantitative level of RyR1 is consistent with stable or slowly progressive mild/moderate myopathy, suggesting that the level of RyR1 protein may be critical for the normal excitation–contraction coupling [56, 60]. The c.10347 + 1G > A mutation had already been described as probably damaging. The mutation likely abolishes the donor splice site, producing a shorter or unstable mRNA and it was predictably damaging by in silico analyses [88]. Finally, we found also a new intronic variant i.e. c.11609-2A > G, localized in a highly conserved region. Different bioinformatic tools predicted that the mutation causes the loss of a splicing-acceptor site in intron 83. The mutation likely leads to the production of an aberrant transcript with the partial out-of-frame retention of intron 84.
Discussion
In this paper we collected clinical, histological and molecular data from a large cohort of core myopathies patients carrying RYR1 mutations with the intent to use in silico modelling to evaluate the possible pathogenetic impact of the identified RYR1 variants, and to verify if variants targeting to specific RyR1 domain are associated to a more severe phenotype.
We used a candidate gene approach using RYR1 Sanger sequencing to screen a large cohort of core myopathy patients. One hundred fifty-three patients were enrolled and RYR1 mutations were identified in 69 (45%) of them. The lack of detailed histopathological information in most of the original cohort of patients, did not allow to assess the overall prevalence of RYR1 mutations in core and multi-minicore diseases. Nevertheless, since CCD is reported to be mainly due to RYR1 mutation [20], while MmD are associated with mutations in variety of genes (RYR1, but also SEPN1, and less frequently in MYH2, UNC45B, MYH7, TTN, MEGF10, SECISBP2, ACTA1, ACTN2, CCD78, and FXR1) [8] we can hypothesize in our original cohort a majority of MmD patients carrying mutations in genes other than RYR1. The mutational approach in our study has some limitations. First, we can not rule out deep intronic or non-canonical splice site variants, since the primers were designed to cover only exons and canonical splice sites. With our methods the non-canonical splicing events, that are emerging to be numerous and often tissue specific, have been missed and therefore also their potential role in core myopathies may be under recognized. Second, we cannot exclude to have missed some RYR1 variants because of some intrinsically pitfalls in Sanger sequencing (i.e. the quality of sequence in the first 15 to 40 bases where the primers bind, or quality of sequencing in long reads, etc.), and finally, RYR1 Sanger sequencing does not allow to look for mutations in other known genes possibly involved in core myopathies. The knowledge of all deleterious alleles in a given patients may contribute to the understanding of the role of the mutational load in disease phenotype expression.
The cohort of patients studied confirmed clinical heterogeneity in RYR1-related core myopathy, even if the selection criteria we used to enrol patients in this study were chosen to obtain a homogeneous group of patients adding to the presence of RYR1 mutations a histopathological diagnosis of core myopathy.
RYR1-related AD CCD presents with a variable clinical spectrum including congenital (47%), early (38%) and adult (14%) onset manifestations. A delay in reaching motor milestones was frequently described (30 patients), and only one patient is still not walking at the last evaluation at 2 years of age. In patients with adult onset, symptoms and signs at clinical presentation included cramps and myalgias, fatigue and muscle weakness prevalent in the lower limbs.
Distribution of weakness was symmetrical, both proximal and distal, in the pelvic and shoulders girdles in 19% of patients and in a minority included also axial muscle weakness. Facial weakness, configuring the typical myopathic face in congenital myopathy, was present in 35% of patients. Spine and joint contractions were frequent in our cohort.
As previously reported [56, 62, 106], patients with recessive RYR1 mutations (8) were overall more severe than AD cases. Onset was congenital in all, arthrogryposis (37.5% vs 0%), floppy infant presentation (62.5% vs 17%), respiratory insufficiency (37.5% vs 0%) and hip dislocation (29% vs 15%) at onset were more frequent in AR than AD cases (Additional file 3: T2). However, even in the AR cases disease progression was variable (stable in 4 patients, slowly progressive in 3 and one patient showed improvement over time). At last evaluation (range 3–40 years) all patients were ambulant but one, who was able to walk only with support at 16 years of age. Overall disease phenotype was severe/moderate with frequently associated foot deformities (50%) and scoliosis (75% of patients).
Thirty-one patients in our cohort were isolated cases. Unfortunately, parents were not available to unequivocally establish the inheritance pattern. Based on the inheritance pattern of previously reported mutations 3 cases were likely AR CCD (i.e. patient #1 carrying two mutations previously reported in trans [54]; patient #9 [62] and 38 [82] carrying a RYR1 mutation previously reported in AR cases in compound heterozygosity [62, 82]), and 6 likely AD CCD (i.e. patient #8 [61], #14 carrying a mutation previously reported in severe AD centronuclear myopathy [70], #29 [77], #30 and #33 harbouring mutations previously reported in core/rod myopathy [75] and in congenital myopathy with uniformity of type 1 muscle fibres [80] and patient #42 [83]). Three patients carried RYR1 mutations previously associated to AD MH (i.e. #2 [55], #16 [57] and #26 [55]) (Table 1; Additional file 3: T2). In the remaining cases either novel mutations or previously unreported association of various RYR1 mutation do not allow to infer the phase of the variants. All 3 putative AR cases had a congenital onset and mild to severe myopathy associated with contractures or scoliosis according to the rule of more severe phenotype in AR vs AD cases.
It is interesting to note that at least 15 patients in our cohort carried mutations associated to MH. MH is usually linked to minimal or absent muscle weakness and muscle biopsy may, or may not, show cores. Given our selection criteria, all patients presented cores at muscle biopsy and 3/15 showed moderate and 6/15 mild muscle weakness. Two patients (#16 and #26) presented a congenital onset (with arthrogryposis, floppiness, hip dislocations in patient #16 and foetal hypokinesia, dysphagia in patient #26), delayed independent ambulation (30 and 24 months respectively) and an overall mild myopathy with stationary progression over time but associated with joint contractures, scoliosis and pes cavus. In these two patients, given the severe clinical picture, it is likely that a second RYR1 mutation was not detected. Only two patients were asymptomatic (in one diagnosis was reached because of a positive family history and in the second in the work up of a hyperCKemia) and two were paucisymptomatic (myalgia, cramps). Among patients carrying mutations previously associated to MH, at least three had a MH reaction during anaesthesia: patient #3 during pes cavus surgery, patient #21 during surgery for congenital muscular torticollis and patient # 23 during spinal arthrodesis for scoliosis. Patient #3 and #21 had an early onset moderate myopathy with typical features of congenital myopathy, where patient #23 was paucisymptomatic and was studied for hyperCKemia. Given the selection bias in our cohort of patients we were not able to assess either the percentage of MH patients presenting with muscle weakness nor the percentage showing core at histopathology. It is possible to speculate that the presence of core at muscle biopsy predisposes to the development of muscle weakness, but a large number of patients is needed to reach a definite conclusion. On the other hand, patients in our cohort may simply express the clinical variability associated to RYR1 mutations. For example, family 3, carrying the well-known p.Arg163Cys mutation associated with MH, displayed a wide phenotypic spectrum. Among the members of this family, one individual (patient #3) presented with an early onset moderate myopathy with muscle wasting, contractures, foot deformities and scoliosis, patient#4 had an adult onset mild myopathy, while patient #5 came to medical attention for asymptomatic hyperCKemia.
The reasons of this intrafamilial variability are not known but genetic modifiers in genes other than RYR1 may be responsible for the clinical variability.
Five patients (belonging to four families) in our cohort carried heterozygous small frame-shift deletion or insertion mutations causing a premature stop codon predicted to result in a reduced level of RyR1. Recently, it has been shown in a knocked-in mouse for a RYR1 single allele frame-shift mutation, that the level of RYR1 transcript and RyR1 protein are decreased without affecting the other SR proteins. The mouse also showed a mild reduction of muscle performance and decreased muscle strength, suggesting that hypomorphic RYR1 mutation should result in a mild clinical phenotype because of a functional reserve of RyR1 protein [107]. Indeed, all six patients followed the postulated rule of a mild reduction in muscle strength.
The RYR1 gene carries a large number of polymorphisms as well as causative mutations: there are 1,783 public variants vs 843 unique public variants reported in LOVD database (https://databases.lovd.nl/shared/genes/RYR1; accessed June 2021) making the assessment of pathogenicity of the identified variant a crucial issue. We identified 68 RYR1 variants and 16 were novel.
We used an integrative in silico approach to evaluate the pathogenic impact of some of the RYR1 mutations identified. Our approach included an in-depth literature search along with a systematic bioinformatics analysis of some of the RYR1 variants identified that allowed us to determine the molecular details supporting the pathogenicity of several novel RYR1 mutations. Despite our promising data, several hurdles have to be considered: the lack of the human RyR1 crystal structure paired with the presence of large intrinsically disordered regions, the large size of the protein and its composition of multiple functional and regulative elements, all pointing to a difficult assessment of a single variant.
In heterozygous patients the protein may be constitute of variable proportion of mutated monomers, the ratio wild-type/mutated monomers could be directly linked to the disease severity but assessing stoichiometry in single patient it is not feasible at the moment. On the other hand, different mutations lead to different pathogenic mechanisms, such as haploinsufficiency, inactivation of regulative sites, impairment of complex assembly and alterations in the channel’s gating properties. To add to the complexity, the genetic background may modify the pathological effect of the RYR1 mutations. For example, variations in other genes associated with core myopathies or known to be RyR1 interactors (i.e. FKBP1B, TRDN, ASPH, FKBP1A, STAC3, CACNA1S, CACNA1C, CACNA1A, NOS1, CALM1) or even other unknow genes may present single nucleotide polymorphism that cause subtle modification on the protein product but sufficient to modify the phenotype. This hypothesize phenomenon is well represented by the phenotypical variability among member of the same family. Finally, as mentioned before, epigenetic modifications of RYR1 may also play a role [108] or the methylation status of genes implicated in cytosolic Ca2+ buffering or trafficking of the Na+/Ca2+ exchanger [109].
The difficult interpretation of the pathogenicity of RYR1 variant is reported also by Johnston and colleagues, who focused their attention on RYR1 mutations causing malignant hyperthermia. They revised the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMP/AMP) criteria for variant interpretation highlighting the difficulty to define general rules to estimate the effect of a mutations [110].
From a myopathological point of view the majority of patients (67%) showed at muscle biopsies core lesions in type 1 muscle fibres, 17% of patients had multiple minicores in both type 1 and type 2 muscle fibres and 15% of muscle biopsies showed both central core and multiple minicores. In a subset of patients, the presence of cores and minicores was confirmed with ultrastructural electron microscopy (EM) studies but EM was not formally investigated in all patients. No correlations were detected among the number of fibres with core and clinical severity. It is interesting to note that the only patient who had 2 muscle biopsy done at age 5 and 12 years showed a dramatic modification of muscle morphology. The first muscle biopsy did not show cores while the biopsy taken at 12 years of age disclosed central core in 100% of type 1 fibres. These observations are in line with the hypothesis that core lesions may not represent a primary developmental abnormality but could be secondary to a maintained abnormal contraction caused by any excitation–contraction coupling defect as previously reported [23].
Conclusions
Considering all the limitations detailed above we were not able to correlate specific genotype to phenotype, but focusing on protein domains we suggest that, in our cohort, mutations in some domains are more frequently associated with specific phenotypes. For example, the pore domain is associated with foetal hypokinesia, contractures and foot deformities. Moreover, it is of interest that all patients (#48 and #65) carrying mutations in Ry1&2 presented with particularly severe phenotype, as this domain is associated with foetal hypokinesia, bulbar involvement and respiratory insufficiency. However, the low number of patients do not allow definitive conclusions at this point.
Supplementary Information
Additional file 1: Bioinformatics pipeline used for mutations effect prediction.
Additional file 2: Affected RyR1 domains and clinical description.
Additional file 3: Histological description of core myopathies patients.
Additional file 4: Patients RYR1 mutations and clinical description.
Acknowledgements
We acknowledge support from Telethon Network of Genetic BioBank (GTB12001D to E.P) and the Eurobiobank network. EP, LB, CF, CB authors of this publication are members of the European Reference Network for Neuromuscular Diseases – Project ID N° 870177.
Abbreviations
- RyR1
Ryanodine receptor 1 protein
- RYR1
Ryanodine receptor 1 gene
- CCD
Central Core Disease
- MmD
Multi-minicore Disease
- MH
Malignant hyperthermia
- DHPR
Dihydropyridine receptor
- NTD-A
N-terminal domain A
- NTD-B
N-terminal domain B
- NTD-C
N-terminal domain C
- BSol
Bridge solenoid
- JSol
Junctional solenoid
- CSol
Core solenoid
- SPRY
SP1a/ryanodine receptor domain
- RY1&2
RyR repeats pairs 1 and 2
- RY3&4
RyR repeats pairs 3 and 4
- TaF
Thumb and forefingers domain
- TMx
Transmembrane helices
- pVSD
Pseudo voltage sensor domain
- CTD
C-terminal domain
- SEPN1
Selenoprotein N1 gene
- SELENON
Selenoprotein N1 gene
- NADH-TR
Nicotinamide adenine dinucleotide dehydrogenase-tetrazolium reductase
- COX
Cytochrome C oxidase
- SDH
Succinate dehydrogenase
- AD
Autosomal dominant
- AR
Autosomal recessive
- CC
Central cores
- Mm
Multiple minicores
Author contributions
Conceptualization: E.P., C. B.; Data Curation: A. F., D. C.; Supervision: E. P., C. B.; Writing—Original Draft Preparation: A. F, E.P. C. B. G. M.; Formal Analysis: D. C., G.M., D. S., L.B., S. T., F. M. S.; Funding Acquisition, E. P., C. B. Writing—review & editing, A. F., D. C., C. F., V. C., G. A., A. D'A., L. M., F. M., M. P., G. T., D. S., L. B., R. B., P. B., F. F., E. S. B., G. C., S. M., T. M., I. M., C. P., A. B., A. D., V. N., A. P., M. G., C. D., E. R., E. M., G. M., S. T., F. S., C. B. and E. P.
Funding
This work was supported by grants from Italian Telethon UILDM grant (GUP08005) and from a Ministry of Health research grant (RF‐2013‐02359065).
Availability of data and materials
All data generated or analysed during this study are included in this published article [Table 1, Additional file 2: T1, Additional file 3: T2, Additional file 4: T3].
Declarations
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki, and ap-proved by the Institutional Ethic Committee at each participating center. We report here the Ethics Committee approvals of the two senior authors: the study was approved by the Ethical Committee of the Istituto Giannina Gaslini Genova on March 10, 2009 (number 567 DSc/fg) and by the Ethical Committee of the University of Padova—Hospital on May 11, 2009 (number 1879P/0025745).
Consent for publication
Written informed consent has been obtained from the patients to publish this paper.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Fusto and Denise have Cassandrini contribute equally. Elena Pegoraro and Claudio Bruno have contributed equally.
Contributor Information
Aurora Fusto, Email: aurora.fusto@unipd.it.
Denise Cassandrini, Email: dcassandrini@fsm.unipi.it.
Chiara Fiorillo, Email: chiara.fiorillo@edu.unige.it.
Valentina Codemo, Email: valentina.codemo@gmail.com.
Guja Astrea, Email: astreaguja@gmail.com.
Adele D’Amico, Email: adele2.damico@opbg.net.
Lorenzo Maggi, Email: lorenzo.maggi@istituto-besta.it.
Francesca Magri, Email: francescam.magri@gmail.com.
Marika Pane, Email: marika.pane@policlinicogemelli.it.
Giorgio Tasca, Email: giorgiotasca81@gmail.com.
Daniele Sabbatini, Email: daniele.sabbatini@phd.unipd.it.
Luca Bello, Email: luca.bello@unipd.it.
Roberta Battini, Email: roberta.battini@fsm.unipi.it.
Fabiana Fattori, Email: fabiana.fattori@opbg.net.
Enrico Silvio Bertini, Email: enricosilvio.bertini@opbg.net.
Giacomo Comi, Email: giacomo.comi@unimi.it.
Sonia Messina, Email: sonia.messina@unime.it.
Tiziana Mongini, Email: tizianaenrica.mongini@unito.it.
Isabella Moroni, Email: isabella.moroni@istituto-besta.it.
Chiara Panicucci, Email: ch.panicucci@gmail.com.
Angela Berardinelli, Email: angela.berardinelli@mondino.it.
Alice Donati, Email: m.donati@meyer.it.
Vincenzo Nigro, Email: vinnigro@gmail.com.
Antonella Pini, Email: antonella.pini@isnb.it.
Melania Giannotta, Email: melania.giannotta@isnb.it.
Claudia Dosi, Email: dosiclaudia@gmail.com.
Enzo Ricci, Email: enzo.ricci@policlinicogemelli.it.
Eugenio Mercuri, Email: eumercuri@gmail.com.
Giovanni Minervini, Email: giovanni.minervini@unipd.it.
Silvio Tosatto, Email: silvio.tosatto@unipd.it.
Filippo Santorelli, Email: filippo3364@gmail.com.
Claudio Bruno, Email: claudio2246@gmail.com.
Elena Pegoraro, Email: elena.pegoraro@unipd.it.
References
- 1.Shy GM, Magee KR. A new congenital non-progressice myopathy. Brain Narnia. 1956;79:610–621. doi: 10.1093/brain/79.4.610. [DOI] [PubMed] [Google Scholar]
- 2.De Cauwer H, Heytens L, Martin J-J (2002) Workshop report of the 89th ENMC International Workshop: Central Core Disease, 19th-20th January 2001, Hilversum, The Netherlands. Neuromuscul Disord [Internet] 12:588–95. 10.1016/S0960-8966(02)00002-0 [DOI] [PubMed]
- 3.Jungbluth H, Sewry CA, Muntoni F. Core myopathies. Semin Pediatr Neurol [Internet]. 2011;18:239–249. doi: 10.1016/j.spen.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Sewry CA, Müller C, Davis M, Dwyer JSMM, Dove J, Evans G, et al. The spectrum of pathology in central core disease. Neuromuscul Disord. 2002;12:930–938. doi: 10.1016/S0960-8966(02)00135-9. [DOI] [PubMed] [Google Scholar]
- 5.Scacheri PC, Hoffman EP, Fratkin JD, Semino-Mora C, Senchak A, Davis MR, et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 2000;55:1689–1696. doi: 10.1212/WNL.55.11.1689. [DOI] [PubMed] [Google Scholar]
- 6.Monnier N, Romero NB, Lerale J, Nivoche Y, Qi D, MacLennan DH, et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9:2599–2608. doi: 10.1093/hmg/9.18.2599. [DOI] [PubMed] [Google Scholar]
- 7.Garibaldi M, Rendu J, Brocard J, Lacene E, Fauré J, Brochier G, et al. “Dusty core disease” (DuCD): expanding morphological spectrum of RYR1 recessive myopathies. Acta Neuropathol Commun [Internet] 2019;7:3. doi: 10.1186/s40478-018-0655-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogasawara M, Nishino I. A review of core myopathy: central core disease, multiminicore disease, dusty core disease, and core-rod myopathy. Neuromuscul Disord [Internet]. 2021;31:968–977. doi: 10.1016/j.nmd.2021.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Romero NB, Monnier N, Viollet L, Cortey A, Chevallay M, Leroy JP, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain [Internet]. 2003;126:2341–2349. doi: 10.1093/brain/awg244. [DOI] [PubMed] [Google Scholar]
- 10.Avila G, Dirksen RT. Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J Gen Physiol [Internet] 2001;118:277–290. doi: 10.1085/jgp.118.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Litman RS, Griggs SM, Dowling JJ, Riazi S. Malignant hyperthermia susceptibility and related diseases. Anesthesiology. 2018;128:159–167. doi: 10.1097/ALN.0000000000001877. [DOI] [PubMed] [Google Scholar]
- 12.Yan Z, Bai X, Yan C, Wu J, Li Z, Xie T, et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature [Internet]. 2015;517:50–55. doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve [Internet] 2006;33:715–731. doi: 10.1002/mus.20512. [DOI] [PubMed] [Google Scholar]
- 14.Van Petegem F, Van. Ryanodine Receptors: Structure and Function. 2012;287:31624–31632. doi: 10.1074/jbc.R112.349068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fusto A, Moyle LALA, Gilbert PMPM, Pegoraro E (2019) Cored in the act: the use of models to understand core myopathies. Dis Model Mech [Internet] 12:dmm041368 [DOI] [PMC free article] [PubMed]
- 16.des Georges A, Clarke OB, Zalk R, Yuan Q, Condon KJ, Grassucci RA, et al. Structural basis for gating and activation of RyR1. Cell. 2016;167:145–157. doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santulli G, Lewis D, des Georges A, Marks AR, Frank J, Ryanodine receptor structure and function in health and disease. Subcell Biochem. 2018;87:329–352. doi: 10.1007/978-981-10-7757-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Posterino GS, Cellini MA, Lamb GD (2003) Effects of oxidation and cytosolic redox conditions on excitation—contraction coupling in rat skeletal muscle. pp 807–823 [DOI] [PMC free article] [PubMed]
- 19.Dulhunty AF, Beard NA, Casarotto MG. Recent advances in understanding the ryanodine receptor calcium release channels and their role in calcium signalling. F1000Research [Internet] 2018;7:1000. doi: 10.12688/f1000research.16434.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu S, Moreno Ibarra CA, Malicdan MCV, Murayama K, Ichihara Y, Kikuchi H, et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain [Internet]. 2006;129:1470–1480. doi: 10.1093/brain/awl077. [DOI] [PubMed] [Google Scholar]
- 21.Kraeva N, Zvaritch E, Rossi AE, Goonasekera SA, Zaid H, Frodis W, et al. Novel excitation-contraction uncoupled RYR1 mutations in patients with central core disease. Neuromuscul Disord [Internet]. 2013;23:120–132. doi: 10.1016/j.nmd.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jungbluth H. Multi-minicore disease. Orphanet J Rare Dis. 2007;2:1–11. doi: 10.1186/1750-1172-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreiro A, Monnier N, Romero NNB, Leroy J-PJ, Bönnemann C, Haenggeli C-AC, et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51:750–759. doi: 10.1002/ana.10231. [DOI] [PubMed] [Google Scholar]
- 24.Jungbluth H, Zhou H, Hartley L, Halliger-Keller B, Messina S, Longman C, et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology. 2005;65:1930–1935. doi: 10.1212/01.wnl.0000188870.37076.f2. [DOI] [PubMed] [Google Scholar]
- 25.Jungbluth H, Müller CR, Halliger-Keller B, Brockington M, Brown SC, Feng L, et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology [Internet]. 2002;59:284–287. doi: 10.1212/WNL.59.2.284. [DOI] [PubMed] [Google Scholar]
- 26.Ferreiro A, Quijano-Roy S, Pichereau C, Moghadaszadeh B, Goemans N, Bönnemann C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71:739–749. doi: 10.1086/342719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tajsharghi H, Hammans S, Lindberg C, Lossos A, Clarke NF, Mazanti I, et al. Recessive myosin myopathy with external ophthalmoplegia associated with MYH2 mutations. Eur J Hum Genet. 2013;22:801–808. doi: 10.1038/ejhg.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lossos A, Baala L, Soffer D, Averbuch-Heller L, Dotan S, Munnich A, et al. A novel autosomal recessive myopathy with external ophthalmoplegia linked to chromosome. Brain. 2005;128:42–51. doi: 10.1093/brain/awh338. [DOI] [PubMed] [Google Scholar]
- 29.Donkervoort S, Kutzner CE, Hu Y, Lornage X, Rendu J, Stojkovic T, et al. Pathogenic variants in the myosin chaperone UNC-45B cause progressive myopathy with eccentric cores. Am J Hum Genet Cell Press. 2020;107:1078–1095. doi: 10.1016/j.ajhg.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cullup T, Lamont PJ, Cirak S, Damian MS, Wallefeld W, Gooding R, et al. Mutations in MYH7 cause multi-minicore disease (MmD) with variable cardiac involvement. Neuromuscul Disord [Internet]. 2012;22:1096–1104. doi: 10.1016/j.nmd.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Chauveau C, Bonnemann CG, Julien C, Kho AL, Marks H, Talim B, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23:980–991. doi: 10.1093/hmg/ddt494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyden SE, Mahoney LJ, Kawahara G, Myers JA, Mitsuhashi S, Estrella EA, et al. Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics. 2012;13:115–124. doi: 10.1007/s10048-012-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kazamel M, Milone M. Congenital myopathy with a novel SELN missense mutation and the challenge to differentiate it from congenital muscular dystrophy. J Clin Neurosci [Internet] 2019;62:238–239. doi: 10.1016/j.jocn.2018.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Estañ MC, Fernández-Núñez E, Zaki MS, Esteban MI, Donkervoort S, Hawkins C, et al. Recessive mutations in muscle-specific isoforms of FXR1 cause congenital multi-minicore myopathy. Nat Commun [Internet]. 2019;10:1–19. doi: 10.1038/s41467-019-08548-9]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jungbluth H, Dowling JJ, Ferreiro A, Muntoni F. 182nd ENMC International Workshop: RYR1-related myopathies, 15–17th April 2011, Naarden, The Netherlands. Neuromuscul Disord [Internet]. 2012;22:453–462. doi: 10.1016/j.nmd.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Bateman A. UniProt: A worldwide hub of protein knowledge. Nucl Acids Res [Internet] 2019;47:506–515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res [Internet] 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics [Internet] 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol [Internet] 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 40.Webb B, Sali A. Protein structure modeling with moDELLER. Methods Mol Biol [Internet]. 2017;1654:39–54. doi: 10.1007/978-1-4939-7231-9_4. [DOI] [PubMed] [Google Scholar]
- 41.Studer G, Rempfer C, Waterhouse AM, Gumienny R, Haas J, Schwede T. QMEANDisCo—distance constraints applied on model quality estimation. Bioinf [Internet]. 2020;36:1765–1771. doi: 10.1093/bioinformatics/btz828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res [Internet]. 2007;35:1. doi: 10.1093/nar/gkm484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Necci M, Piovesan D, Clementel D, Dosztányi Z, Tosatto SCE. MobiDB-lite 30: fast consensus annotation of intrinsic disorder flavours in proteins. Bioinformatics [Internet] 2020;36:5533–5534. doi: 10.1093/bioinformatics/btaa1045. [DOI] [PubMed] [Google Scholar]
- 44.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res [Internet]. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puntervoll P, Linding R, Gemünd C, Chabanis-Davidson S, Mattingsdal M, Cameron S, et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res [Internet] 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piovesan D, Walsh I, Minervini G, Tosatto SCE. FELLS: fast estimator of latent local structure. Bioinformatics [Internet] 2017;33:1889–1891. doi: 10.1093/bioinformatics/btx085. [DOI] [PubMed] [Google Scholar]
- 47.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera? A visualization system for exploratory research and analysis. J Comput Chem [Internet] 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 48.Walsh I, Minervini G, Corazza A, Esposito G, Tosatto SCE, Fogolari F. Bluues server: electrostatic properties of wild-type and mutated protein structures. Bioinformatics [Internet]. 2012;28:2189–2190. doi: 10.1093/bioinformatics/bts343. [DOI] [PubMed] [Google Scholar]
- 49.Piovesan D, Minervini G, Tosatto SCE. The RING 2.0 web server for high quality residue interaction networks. Nucl Acids Res [Internet]. 2016;44:367–374. doi: 10.1093/nar/gkw315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet [Internet] 7:7.20 [DOI] [PMC free article] [PubMed]
- 51.Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucl Acids Res [Internet] 2012;40:452–457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Prot Struct Funct Bioinforma [Internet] 2006;62:1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- 53.Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:1. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou H, Yamaguchi N, Xu L, Wang Y, Sewry C, Jungbluth H, et al. Characterization of recessive RYR1 mutations in core myopathies. Hum Mol Genet. 2006;15:2791–2803. doi: 10.1093/hmg/ddl221. [DOI] [PubMed] [Google Scholar]
- 55.Galli L, Orrico A, Lorenzini S, Censini S, Falciani M, Covacci A, et al. Frequency and localization of mutations in the 106 Exons of the RYR1 gene in 50 individuals with malignant hyperthermia. Hum Mutat. 2006;913:1–9. doi: 10.1002/humu.9442. [DOI] [PubMed] [Google Scholar]
- 56.Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat [Internet] 2006;27:977–989. doi: 10.1002/humu.20356. [DOI] [PubMed] [Google Scholar]
- 57.Robinson RL, Brooks C, Brown SL, Ellis FR, Halsall PJ, Quinnell RJ, et al (2002) RYR1 Mutations Causing Central Core Disease Are Associated With More Severe Malignant Hyperthermia In Vitro Contracture Test Phenotypes. 97:88–97 [DOI] [PubMed]
- 58.Yang T, Riehl J, Esteve E, Matthaei KI, Goth S, Allen PD, et al. Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock-in mouse. Anesthesiology. 2006;105:1164–1175. doi: 10.1097/00000542-200612000-00016. [DOI] [PubMed] [Google Scholar]
- 59.Yang T, Ta TA, Pessah IN, Allen PD. Functional defects in six ryanodine receptor isoform-1 (RyR1) mutations associated with malignant hyperthermia and their impact on skeletal excitation-contraction coupling. J Biol Chem. 2003;278:25722–25730. doi: 10.1074/jbc.M302165200. [DOI] [PubMed] [Google Scholar]
- 60.Monnier N, Kozak-Ribbens GG, Krivosic-Horber RR, Nivoche Y, Qi D, Kraev N, et al. Correlations Between Genotype and Pharmacological, Histological, Functional, and Clinical Phenotypes in Malignant Hyperthermia Susceptibility. Hum Mutat. 2005;26:413–425. doi: 10.1002/humu.20231. [DOI] [PubMed] [Google Scholar]
- 61.Quane KA, Healy JMSS, Keating KE, Manning BM, Couch FJ, Palmucci LM, et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet Nature Publishing Group. 1993;5:51–55. doi: 10.1038/ng0993-51. [DOI] [PubMed] [Google Scholar]
- 62.Klein A, Lillis S, Munteanu I, Scoto M, Zhou H, Quinlivan R, et al. Clinical and Genetic Findings in a Large Cohort of Patients with Ryanodine Receptor 1 Gene-Associated Myopathies. Hum Mutat [Internet] 2012;33:981–988. doi: 10.1002/humu.22056. [DOI] [PubMed] [Google Scholar]
- 63.Gillard EF, Otsu K, Fujii J, Khanna VK, Leon SDE, Derdemezi J, et al. A Substitution of Cysteine for Arginine 614 in the Ryanodine Receptor Is Potentially Causative of Human Malignant Hyperthermia. Genomics. 1991;755:751–755. doi: 10.1016/0888-7543(91)90084-R. [DOI] [PubMed] [Google Scholar]
- 64.Hogan K, Couch F, Powers PA, Gregg RG. A cysteine-for-arginine substitution (R614C) in the human skeletal muscle calcium release channel cosegregates with malignant hyperthermia. Anesth Analg [Internet]. 1992;75:441–448. doi: 10.1213/00000539-199209000-00022. [DOI] [PubMed] [Google Scholar]
- 65.Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al (2017) Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med [Internet] 9:eaal5209 [DOI] [PMC free article] [PubMed]
- 66.Monnier N, Romero NB, Lerale J, Landrieu P, Nivoche Y, Fardeau M, et al. Familial and sporadic forms of central core disease are associated with mutations in the C-terminal domain of the skeletal muscle ryanodine receptor. Hum Mol Genet. 2001;10:2581–2592. doi: 10.1093/hmg/10.22.2581. [DOI] [PubMed] [Google Scholar]
- 67.Kraeva N, Heytens L, Jungbluth H, Treves S, Voermans N, Kamsteeg E, et al. Compound RYR1 heterozygosity resulting in a complex phenotype of malignant hyperthermia susceptibility and a core myopathy. Neuromuscul Disord [Internet] 2015;25:567–576. doi: 10.1016/j.nmd.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Tong J, Oyamada H, Demaurex N, Grinstein S, Mccarthy TV, Maclennan DH. Caffeine and Halothane Sensitivity of Intracellular Ca2+ Release Is Altered by 15 Calcium Release Channel ( Ryanodine Receptor ) Mutations Associated with Malignant Hyperthermia and / or Central Core Disease. J Biol Chem. 1997;272:26332–26339. doi: 10.1074/jbc.272.42.26332. [DOI] [PubMed] [Google Scholar]
- 69.Gillies RL, Bjorksten AR, Davis M, Sart DDU, Dqg F, Wr DJU, et al. Identification of genetic mutations in Australian malignant hyperthermia families using sequencing of RYR1 hotspots. Anaesth Intensive Care United States. 2008;36:391–403. doi: 10.1177/0310057X0803600311. [DOI] [PubMed] [Google Scholar]
- 70.Wilmshurst JM, Lillis S, Zhou H, Pillay K, Henderson H, Kress W, et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol [Internet] 2010;68:717–726. doi: 10.1002/ana.22119. [DOI] [PubMed] [Google Scholar]
- 71.Zullo A, Klingler W, De Sarno C, Ferrara M, Fortunato G, Perrotta G, et al. Functional characterization of ryanodine receptor (RYR1) sequence variants using a metabolic assay in immortalized B-lymphocytes. Hum Mutat. 2009;30:575–589. doi: 10.1002/humu.20991. [DOI] [PubMed] [Google Scholar]
- 72.Herasse M, Parain K, Marty I, Monnier N, Kaindl AM, Leroy JP, et al. Abnormal distribution of calcium-handling proteins: a novel distinctive marker in core myopathies. J Neuropathol Exp Neurol [Internet]. 2007;66:57–65. doi: 10.1097/NEN.0b013e31802d47ce. [DOI] [PubMed] [Google Scholar]
- 73.Sambuughin N, Nelson TE, Jankovic J, Xin C, Meissner G, Mullakandov M, et al. Identification and functional characterization of a novel ryanodine receptor mutation causing malignant hyperthermia in North American and South American families. Neuromuscul Disord [Internet] 2001;11:530–537. doi: 10.1016/S0960-8966(01)00202-4. [DOI] [PubMed] [Google Scholar]
- 74.Malandrini A, Orrico A, Gaudiano C, Gambelli S, Galli L, Berti G, et al. Muscle biopsy and in vitro contracture test in subjects with idiopathic HyperCKemia. Anesthesiology. 2008;109:625–628. doi: 10.1097/ALN.0b013e3181862a0d. [DOI] [PubMed] [Google Scholar]
- 75.Groom L, Muldoon SM, Tang ZZ, Brandom BW, Bayarsaikhan M, Bina S, et al (2011) Identical de novo mutation in the type 1 ryanodine receptor gene associated with fatal, stress-induced malignant hyperthermia in two unrelated families [Internet]. Anesthesiology Lippincott Williams and Wilkins, p 938–45. http://links.lww.com/ALN/A771 [DOI] [PMC free article] [PubMed]
- 76.von der Hagen M, Kress W, Hahn G, Brocke KS, Mitzscherling P, Huebner A, et al. Novel RYR1 missense mutation causes core rod myopathy. Eur J Neurol [Internet] 2008;15:31–32. doi: 10.1111/j.1468-1331.2008.02094.x. [DOI] [PubMed] [Google Scholar]
- 77.Suman M, Sharpe JA, Bentham RB, Kotiadis VN, Menegollo M, Pignataro V, et al. Inositol trisphosphate receptor-mediated Ca2+ signalling stimulates mitochondrial function and gene expression in core myopathy patients. Hum Mol Genet [Internet] 2018;27:2367–2382. doi: 10.1093/hmg/ddy149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davis MRR, Haan E, Jungbluth H, Sewry C, North K, Muntoni F, et al. Principal mutation hotspot for central core disease and related myopathies in the C-terminal transmembrane region of the RYR1 gene. Neuromuscul Disord Elsevier Ltd. 2003;13:133–142. doi: 10.1016/S0960-8966(02)00216-X. [DOI] [PubMed] [Google Scholar]
- 79.Galli L, Orrico A, Cozzolino S, Pietrini V, Tegazzin V, Sorrentino V. Mutations in the RYR1 gene in Italian patients at risk for Malignant Hyperthermia: evidence for a cluster of novel mutations in the C-terminal region. Cell Calcium [Internet]. 2002;32:143–151. doi: 10.1016/S0143-4160(02)00138-0. [DOI] [PubMed] [Google Scholar]
- 80.Johannsen S, Treves S, Müller CR, Mögele S, Schneiderbanger D, Roewer N, et al. Functional characterization of the RYR1 mutation p.Arg4737Trp associated with susceptibility to malignant hyperthermia. Neuromuscul Disord [Internet]. 2016;26:21–25. doi: 10.1016/j.nmd.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 81.Sato I, Wu S, Ibarra Moreno CA, Hayashi YK, Fujita H, Tojo M, et al. Congenital neuromuscular disease with uniform type 1 fiber and RYR1 mutation. Neurology. 2008;70:114–122. doi: 10.1212/01.wnl.0000269792.63927.86. [DOI] [PubMed] [Google Scholar]
- 82.Amburgey K, Bailey A, Hwang JH, Tarnopolsky MA, Bonnemann CG, Medne L, et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis. 2013;8:117. doi: 10.1186/1750-1172-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gu M, Zhang S, Hu J, Yuan Y, Wang Z, Da Y, et al. Novel RYR1 missense mutations in six Chinese patients with central core disease. Neurosci Lett [Internet]. 2014;566:32–35. doi: 10.1016/j.neulet.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 84.Lynch PJ, Tong J, Lehane M, Mallet A, Giblin L, Heffron JJA, et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci USA. 1999;96:4164–4169. doi: 10.1073/pnas.96.7.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Treves S, Anderson AA, Ducreux S, Divet A, Bleunven C, Grasso C, et al. Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul Disord [Internet]. 2005;15:577–587. doi: 10.1016/j.nmd.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 86.Tilgen N, Zorzato F, Halliger-Keller B, Muntoni F, Sewry C, Palmucci LM, et al. Identification of four novel mutations in the C-terminal membrane spanning domain of the ryanodine receptor 1: association with central core disease and alteration of calcium homeostasis. Hum Mol Genet [Internet]. 2001;10:2879–2887. doi: 10.1093/hmg/10.25.2879. [DOI] [PubMed] [Google Scholar]
- 87.Martin F, Kana V, Mori AC, Fischer D, Parkin N, Boltshauser E, et al. Neurofibromatosis type 1 (NF1) with an unusually severe phenotype due to digeny for NF1 and ryanodine receptor 1 associated myopathy. Eur J Pediatr. 2014;173:1691–1694. doi: 10.1007/s00431-014-2314-6. [DOI] [PubMed] [Google Scholar]
- 88.Fattori F, Maggi L, Bruno C, Cassandrini D, Codemo V, Catteruccia M, et al. Centronuclear myopathies: genotype–phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol [Internet] 2015;262:1728–1740. doi: 10.1007/s00415-015-7757-9. [DOI] [PubMed] [Google Scholar]
- 89.Kraeva N, Riazi S, Loke J, Frodis W, Lou CM, Nolan K, et al. Ryanodine receptor type 1 gene mutations found in the Canadian malignant hyperthermia population. Can J Anesth Can d’anesthésie [Internet]. 2011;58:504–513. doi: 10.1007/s12630-011-9494-6. [DOI] [PubMed] [Google Scholar]
- 90.Klingler W, Heiderich S, Girard T, Gravino E, Heffron JJA, Johannsen S, et al. (2014) Functional and genetic characterization of clinical malignant hyperthermia crises : a multi-centre study [DOI] [PMC free article] [PubMed]
- 91.Staron RS, Hagerman FC, Hikida RS, Murray TF, Hostler DP, Crill MT, et al. Fiber type composition of the vastus lateralis muscle of young men and women. J Histochem Cytochem [Internet]. 2000;48:623–629. doi: 10.1177/002215540004800506. [DOI] [PubMed] [Google Scholar]
- 92.Treves S, Jungbluth H, Muntoni F, Zorzato F. Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm. Curr Opin Pharmacol [Internet] 2008;8:319–326. doi: 10.1016/j.coph.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 93.Proenza C, Brien JO, Nakai J, Mukherjee S, Allen PD, Beam KG. Identification of a region of RyR1 that participates in allosteric coupling with the alfa1s (CaV11) II–III loop. J Biol Chem. 2002;277:6530–6535. doi: 10.1074/jbc.M106471200. [DOI] [PubMed] [Google Scholar]
- 94.Cui Y, Tae H-S, Norris NC, Karunasekara Y, Pouliquin P, Board PG, et al. A dihydropyridine receptor α1s loop region critical for skeletal muscle contraction is intrinsically unstructured and binds to a SPRY domain of the type 1 ryanodine receptor. Int J Biochem Cell Biol [Internet]. 2009;41:677–686. doi: 10.1016/j.biocel.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 95.Gaburjakova M, Gaburjakova J, Reiken S, Huang F, Marx SO, Rosemblit N, et al. FKBP12 binding modulates ryanodine receptor channel gating. J Biol Chem. 2001;276:16931–16935. doi: 10.1074/jbc.M100856200. [DOI] [PubMed] [Google Scholar]
- 96.Avila G, Hui E, Perez CF, Allen PD, Dirksen RT. FKBP12 binding to RyR1 modulates excitation-contraction coupling in mouse skeletal myotubes. J Biol Chem. 2003;278:22600–22608. doi: 10.1074/jbc.M205866200. [DOI] [PubMed] [Google Scholar]
- 97.Gillespie D, Xu L, Wang Y, Meissner G. (De)constructing the ryanodine receptor: modeling ion permeation and selectivity of the calcium release channel. J Phys Chem B [Internet] 2005;109:15598–15610. doi: 10.1021/jp052471j. [DOI] [PubMed] [Google Scholar]
- 98.Hamada T, Bannister ML, Ikemoto N. Peptide probe study of the role of interaction between the cytoplasmic and transmembrane domains of the ryanodine receptor in the channel regulation mechanism. Biochemistry [Internet] 2007;46:4272–4279. doi: 10.1021/bi061557f. [DOI] [PubMed] [Google Scholar]
- 99.Chen SRW, Zhang L, Maclennang DH. Antibodies as probes for Ca2+ activation sites in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1993;268:13414–13421. doi: 10.1016/S0021-9258(19)38666-1. [DOI] [PubMed] [Google Scholar]
- 100.Treves S, Scutari E, Robert M, Groh S, Ottolia M, Prestipino G, et al. Interaction of S100A1 with the Ca2+ release channel (ryanodine receptor) of skeletal muscle. Biochemistry [Internet]. 1997;36:11496–11503. doi: 10.1021/bi970160w. [DOI] [PubMed] [Google Scholar]
- 101.Du GG, Khanna VK, MacLennan DH. Mutation of Divergent Region 1 Alters Caffeine and Ca 2+ Sensitivity of the Skeletal Muscle Ca 2+ Release Channel (Ryanodine Receptor) J Biol Chem [Internet]. 2020;75:11778–11783. doi: 10.1074/jbc.275.16.11778. [DOI] [PubMed] [Google Scholar]
- 102.Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH. Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1) Proc Natl Acad Sci [Internet] 2002;99:16725–16730. doi: 10.1073/pnas.012688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yuchi Z, Yuen SMWK, Lau K, Underhill AQ, Cornea RL, Fessenden JD, et al. Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nat Commun [Internet] 2015;6:7947. doi: 10.1038/ncomms8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yamaguchi N, Prosser BL, Ghassemi F, Xu L, Pasek DA, Eu JP, et al. Modulation of sarcoplasmic reticulum Ca2+ release in skeletal muscle expressing ryanodine receptor impaired in regulation by calmodulin and S100A1. Am J Physiol Cell Physiol [Internet] 2011;300:C998–1012. doi: 10.1152/ajpcell.00370.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zorzato F, Jungbluth H, Zhou H, Muntoni F, Treves S. Critical Review Functional Effects of Mutations Identified in Patients with Multiminicore Disease. IUBMB Life. 2007;59:14–20. doi: 10.1080/15216540601187803. [DOI] [PubMed] [Google Scholar]
- 106.Rocha J, Taipa R, Melo Pires M, Oliveira J, Santos R, Santos M. Ryanodine myopathies without central cores—clinical, histopathologic, and genetic description of three cases. Pediatr Neurol. 2014;51:275–278. doi: 10.1016/j.pediatrneurol.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 107.Elbaz M, Ruiz A, Eckhardt J, Pelczar P, Muntoni F, Boncompagni S, et al. Quantitative reduction of RyR1 protein caused by a single-Allele frameshift mutation in RYR1 ex36 impairs the strength of adult skeletal muscle fibres. Hum Mol Genet [Internet] 2019;28:1872–1884. doi: 10.1093/hmg/ddz025. [DOI] [PubMed] [Google Scholar]
- 108.Zhou H, Brockington M, Jungbluth H, Monk D, Stanier P, Sewry CA, et al. Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. Am J Hum Genet [Internet]. 2006;79:859–868. doi: 10.1086/508500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ponsuksili S, Trakooljul N, Basavaraj S, Hadlich F, Murani E, Wimmers K. Epigenome-wide skeletal muscle DNA methylation profiles at the background of distinct metabolic types and ryanodine receptor variation in pigs. BMC Genomics [Internet] 2019;20:492. doi: 10.1186/s12864-019-5880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Johnston JJ, Dirksen RT, Girard T, Gonsalves SG, Hopkins PM, Riazi S, et al. Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genet Med [Internet]. 2021;23:1288–1295. doi: 10.1038/s41436-021-01125-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Bioinformatics pipeline used for mutations effect prediction.
Additional file 2: Affected RyR1 domains and clinical description.
Additional file 3: Histological description of core myopathies patients.
Additional file 4: Patients RYR1 mutations and clinical description.
Data Availability Statement
All data generated or analysed during this study are included in this published article [Table 1, Additional file 2: T1, Additional file 3: T2, Additional file 4: T3].