

Abstract
Bile acids play crucial roles in host physiology by acting as both detergents that aid in digestion and as signaling molecules that bind to host receptors. Gut bacterial bile salt hydrolase (BSH) enzymes perform the gateway reaction leading to the conversion of host-produced primary bile acids into bacterially modified secondary bile acids. Small molecule probes that target BSHs will help elucidate the causal roles of these metabolites in host physiology. We previously reported the development of a covalent BSH inhibitor with low gut permeability. Here, we build on our previous findings and describe the development of a second-generation gut-restricted BSH inhibitor with enhanced potency, reduced off-target effects, and durable in vivo efficacy. Structure-activity relationship (SAR) studies focused on the bile acid core identified a compound, AAA-10, containing a C3-sulfonated lithocholic acid scaffold and an alpha-fluoromethyl ketone warhead as a potent pan-BSH inhibitor. This compound inhibits BSH activity in mouse and human fecal slurry, bacterial cultures, and purified BSH proteins and displays reduced toxicity against mammalian cells compared to first generation compounds. Oral administration of AAA-10 to wild-type mice for 5 days resulted in a decrease in the abundance of the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA) in the mouse GI tract with low systemic exposure of AAA-10, demonstrating that AAA-10 is an effective tool for inhibiting BSH activity and modulating bile acid pool composition in vivo.
Metabolites derived from the human gut microbiota have been implicated as causal agents in the maintenance of host health and the progression of disease.1 Advances in metabolomics, sequencing technologies, and the development of genetic tools have facilitated the identification of bacterial metabolites and the biosynthetic pathways responsible for their production. However, the lack of specific tools to control the levels of these metabolites in complex microbial communities has hindered our ability to interrogate the roles of these metabolites in host physiology. Encouragingly, the recent development of small molecule modulators of bacterial metabolites has revealed the potential of microbiota-targeted therapies to treat disease, including colon cancer, cardiovascular disease, and Parkinson’s disease.2–4
Bile acids are one large class of molecules that undergo substantial metabolism by gut bacteria.5 While bile acids have long been studied for their detergent properties,6,7 recent findings have illustrated the key role that these metabolites play as signaling molecules. Specific bile acids act as ligands for host nuclear hormone receptors (NhRs) and G-protein-coupled receptors (GPCRs), thereby affecting host metabolic and immunomodulatory processes.8–11 Disruption of bile acid homeostasis has been suggested to contribute to the initiation and progression of disease, including cancer, obesity, and hypercholesterolemia,8,12–15 underscoring the need for tools that control the levels of these metabolites in vivo.
Host-produced primary bile acids are conjugated to taurine or glycine in the mammalian liver, stored in the gallbladder, and secreted into the small intestine post-prandially where they act as detergents that facilitate digestion. In the lower GI tract, resident bacteria chemically modify these metabolites, producing a large class of molecules called secondary bile acids. Before these modifications can occur, the C24 amide of conjugated bile acids must be hydrolyzed, a gateway reaction that is carried out exclusively by gut bacterial bile salt hydrolases (BSHs) (EC 3.5.1.24) (Figure 1a).16 BSHs are widespread in human gut bacteria and have been identified in members of 12 different phyla, including Bacteroidetes and Firmicutes, the two dominant phyla in the human gut.17 Recent studies have found that BSH abundance or activity are correlated with human diseases, including inflammatory bowel diseases, type 2 diabetes, and cardiovascular disease.17–19 The causal role of BSH activity in host physiology, however, remains unclear. For example, studies involving antibiotic-treated and germ-free mice colonized with BSH-containing or BSH-deficient bacteria20,21 or conventional mice treated with non-selective small molecules22,23 have reported conflicting results about the effects of BSH activity on host metabolism. An inhibitor that targets a wide array of BSHs but exhibits limited off-target effects against bacterial and host cells would allow for the selective in vivo modulation of bile acid composition, shifting the bile acid pool toward conjugated bile acids and decreasing the abundance of deconjugated and secondary bile acids. Such a tool could be utilized in fully colonized animals and would provide valuable information about how bile acids affect host physiology.
Figure 1. Targeting gut bacterial BSHs.
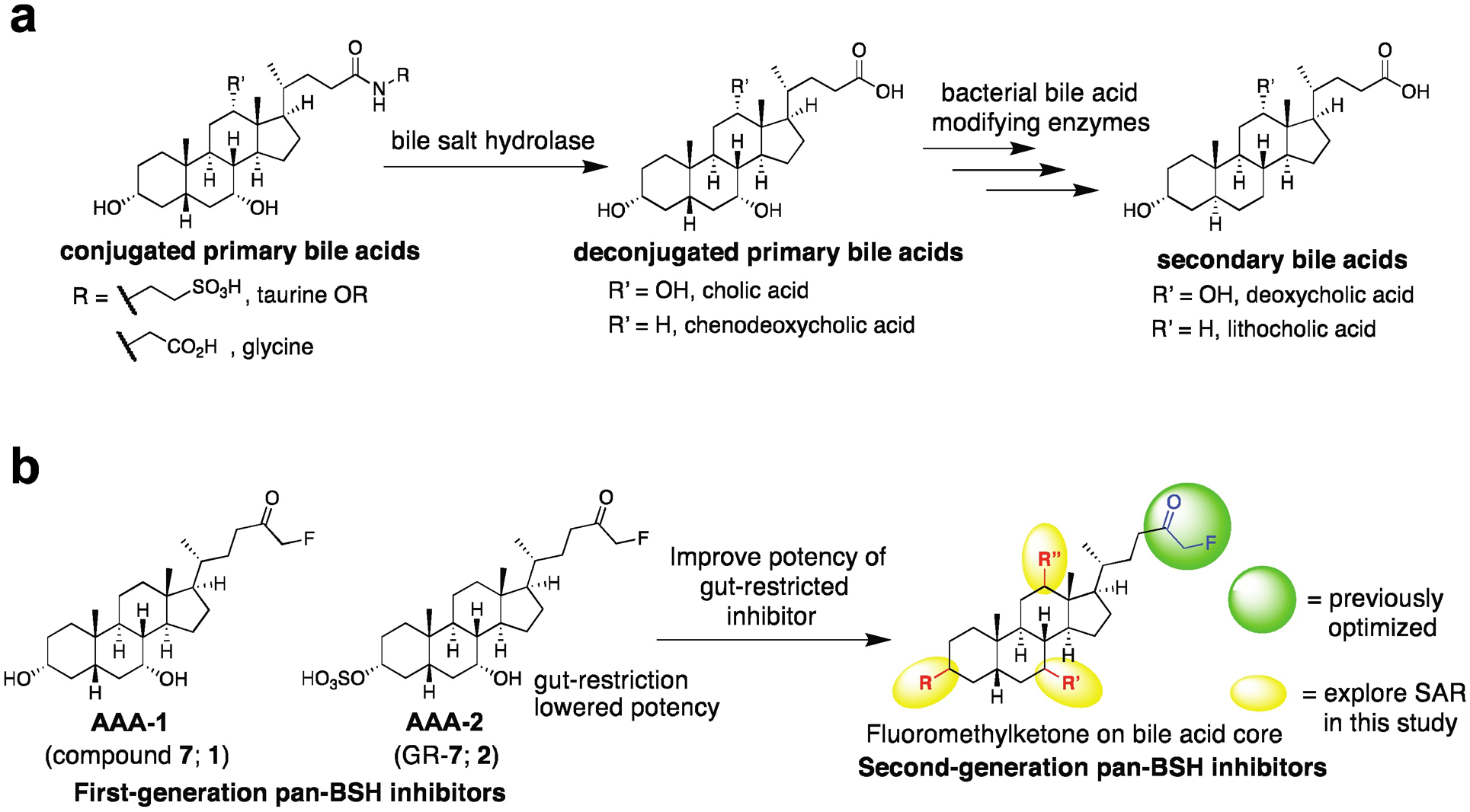
a, Bacterial bile salt hydrolases (BSHs) perform the gateway reaction leading from host-produced conjugated primary bile acids to bacterially modified secondary bile acids. b, Development of second-generation BSH inhibitors starting from previously reported covalent pan-BSH inhibitors. Sulfonation of AAA-1 at the C3-OH position previously resulted in an inhibitor with low systemic exposure but decreased potency (AAA-2). Here, SAR studies focusing on the bile acid core were performed with the goal of yielding a second-generation pan-BSH inhibitor with improved potency.
In prior work, we took advantage of the nucleophilicity of the highly conserved active site N-terminal cysteine residue (Cys2) in BSHs to develop first-in-class covalent pan-BSH inhibitors (Figure 1b).24 In this study, we screened electrophilic warheads appended to the core of chenodeoxycholic acid (CDCA), an abundant human bile acid that is recognized by a broad spectrum of BSHs.20,25,26 This work established an alpha-fluoromethylketone (FMK)-containing molecule, compound 7 (referred to here as AAA-1, 1), as a potent and selective pan-BSH inhibitor. Treatment of conventional mice with a single dose of AAA-1 allowed us to inhibit BSH activity and shift the in vivo bile acid pool toward host-produced bile acids for one day. We also showed that appending a sulfonate27 to the C3 hydroxyl group resulted in gut-restriction of the inhibitor, a change that limited the systemic exposure of this compound (GR-7, referred to here as AAA-2, 2). These studies demonstrated the potential of alpha-fluoromethyl ketone-containing inhibitors to target BSHs in vivo.
To increase the utility of BSH inhibitors for use in vivo as well as to overcome several limitations in our prior study, we sought to develop second-generation inhibitors. The gut-restricted inhibitor AAA-2 exhibited lower potency than AAA-1, motivating the synthesis of new lead compounds. Moreover, while we previously demonstrated proof-of-principle that a gut-restricted inhibitor could affect BSH activity in vivo, we did not demonstrate a shift in the in vivo bile acid pool in our prior work. Finally, to demonstrate the potential utility of these compounds in animal models, we sought to show that a gut-restricted inhibitor could shift the bile acid pool over a multi-day period. Here, we have built on our previous findings and report the development of a second-generation gut-restricted BSH inhibitor with enhanced potency, reduced off-target effects, and multi-day in vivo efficacy. Our structure-activity relationship (SAR) studies focused on the bile acid core, and we identified a lithocholic acid core-based inhibitor, AAA-10 (8) (Figure 2), as a potent pan-BSH inhibitor through screening against conventional mouse fecal slurries, bacterial cultures, and recombinant proteins. This compound is not antibacterial, displays reduced toxicity against mammalian cells compared to AAA-1 and AAA-2, and does not affect signaling through the farnesoid X receptor (FXR) or Takeda G-protein receptor 5 (TGR5), key bile acid-mediated receptors. Finally, we demonstrate that AAA-10 (8) can modulate the in vivo bile acid pool for 5 days, resulting in the decreased abundance of the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA).
Figure 2. Library of sulfonated inhibitors.
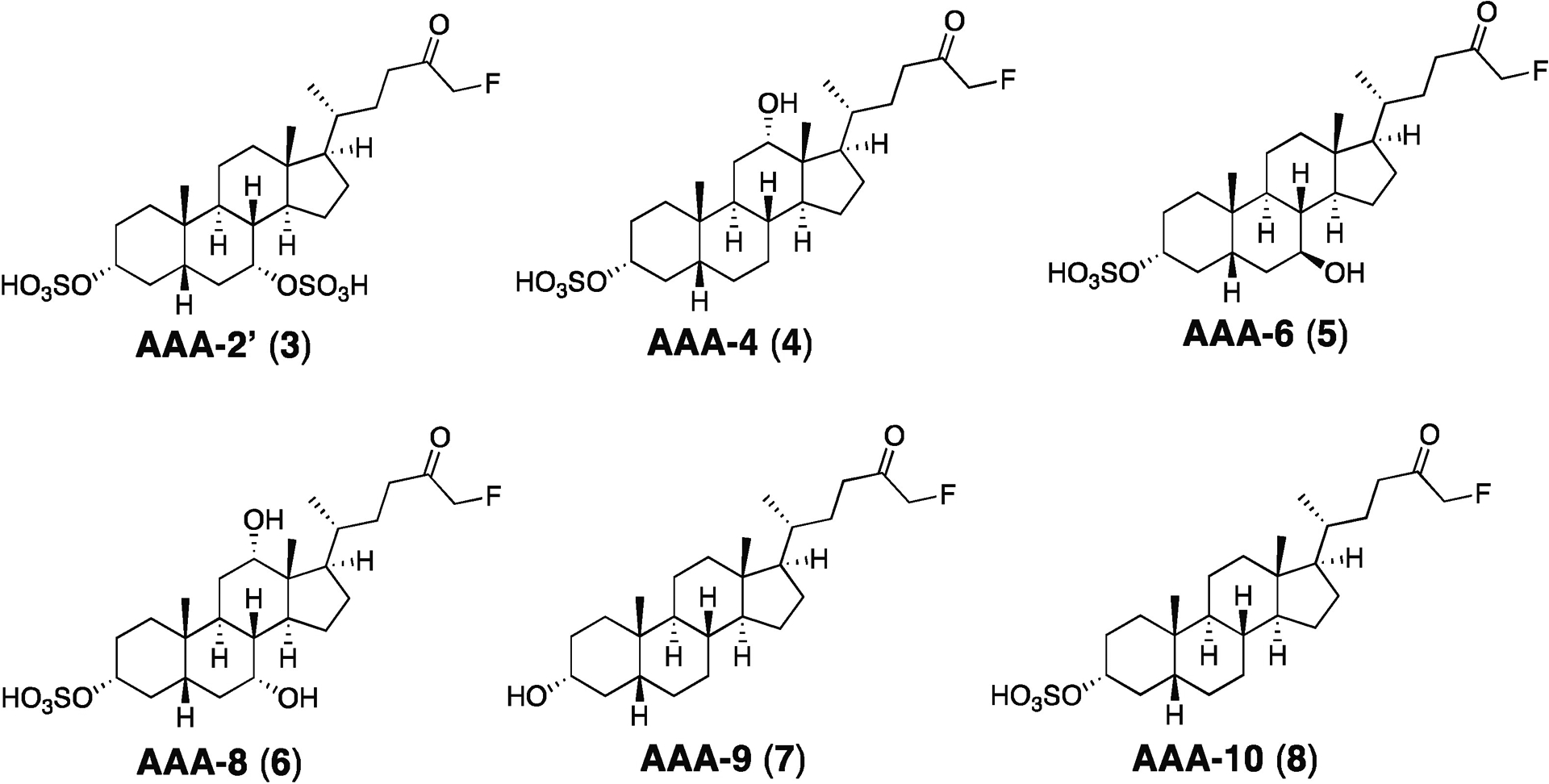
A small library of inhibitors was generated with SAR focused on incorporating the cores of naturally occurring bile acids found in both mouse and humans while maintaining an α-fluoromethyl ketone electrophile.
RESULTS AND DISCUSSION
Synthesis of BSH inhibitor candidates
In prior work, sulfonation of AAA-1 (1) at C3, a position that is exposed to solvent in the co-crystal structure of this compound with the BSH from the gut bacterium Bacteroides thetaiotaomicron (B. theta), increased the solubility of this compound and limited its systemic exposure.24,28 The resultant compound AAA-2, (2), however, was less potent than AAA-1.24 With the goal of improving potency while still maintaining gut restriction, we decided to append the optimized FMK warhead on naturally occurring bile acid cores found in both the murine and human gut (CDCA, DCA, ursodeoxycholic acid (UDCA), cholic acid (CA), and LCA). These compounds could then be sulfonated at the C3 position to produce second-generation inhibitor candidates (Figure 1B).
In order to expedite the synthesis of the library, an optimized protecting group-free synthesis was developed (Scheme S1).29 Following activation of the unprotected bile acid with carbonyldiimidazole (CDI), addition of magnesium benzyl fluoromalonate provided the fluoro beta-ketoester. Removal of the benzyl group followed by decarboxylation under hydrogenation conditions provided the FMK compounds. Finally, the sulfonate group was installed using SO3.pyridine. Because the C3-α-OH group on the bile acid core is more sterically accessible than the C7 and C12 α-alcohols, the sulfonation reactions proceeded selectively to provide the candidate C3-sulfonated inhibitors (Figure 2, 3–6, 8).
Library screen in conventional mouse feces
We previously reported the use of a wild-type conventional mouse fecal slurry assay23 to identify pan-inhibitors of BSHs.24 Because fecal slurry should contain BSHs from nearly all bacteria in the distal region of the murine GI tract, demonstrating inhibition of BSH activity in this assay represents an important benchmark that all inhibitor candidates should meet. We therefore utilized this assay as the first screen in the process of developing second-generation BSH inhibitors. Inhibitor candidates were added to fresh feces obtained from conventional wild-type mice (C57Bl/6J) and suspended in buffer under reducing conditions (Figure 3a). To facilitate identification of an inhibitor with enhanced potency compared to AAA-2, the first-generation gut-restricted inhibitor, compounds were intentionally tested at 10 μM, a concentration at which neither AAA-1 nor AAA-2 completely inhibits enzyme activity.24 After 30 min, glycine-conjugated deuterated chenodeoxycholic acid (GCDCA-d4) was added and its conversion to the deconjugated product CDCA-d4 was quantified using ultra-high performance liquid chromatography-mass spectrometry (UPLC-MS).
Figure 3. Identification of AAA-10 as a second-generation pan-BSH inhibitor.
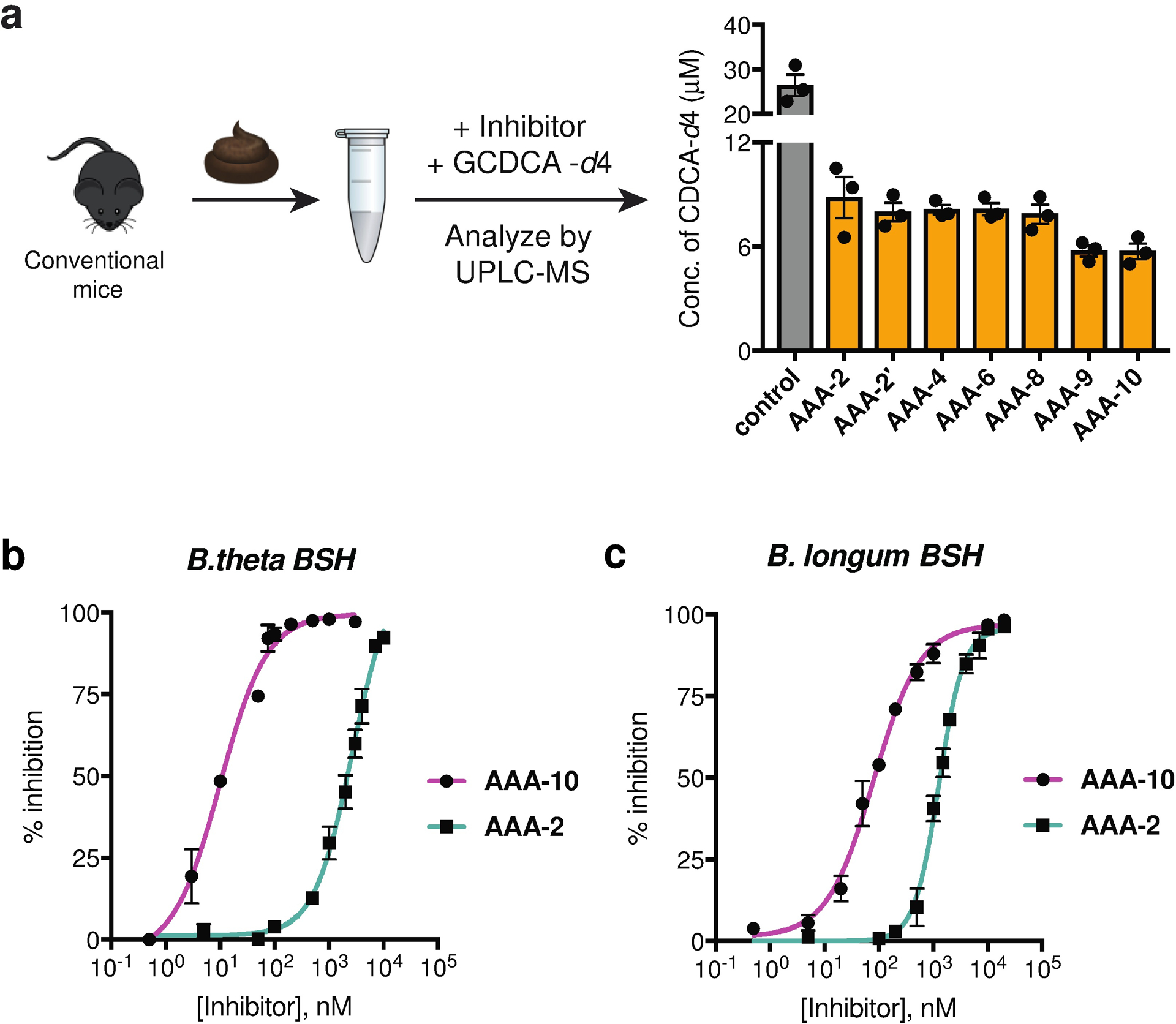
a, Assay design for screening the inhibitor library. Screening in fresh mouse feces identified AAA-10 as a potent second-generation pan-inhibitor of BSHs. Inhibitors were tested at a concentration of 10 μM. Sulfonation did not reduce the potency of AAA-10 compared to AAA-9. Assay was performed three times independently with similar results. Figure shows representative results from one assay. b and c, AAA-10 is more potent than AAA-2 against recombinant BSHs. Comparison of AAA-10 and AAA-2 dose-response curves against Bacteroides thetaiotaomicron (B. theta, accession number AA077193.1) and Bifidobacterium longum (B. longum, accession number AAF67801.1) BSHs using tauro-ursodeoxycholic acid (TUDCA) and tauro-deoxycholic acid (TDCA) as the respective substrates. See Table S1 for comparison of IC50 values of AAA-1, AAA-2 and AAA-10. For b and c, Graphpad was used to fit IC50 curves. All assays were performed in biological triplicate, and data are presented as mean ± s.e.m.
Mono-sulfonated inhibitor candidates containing DCA, UDCA, and CA cores (AAA-4 (4), AAA-6 (5), and AAA-8 (6), respectively) inhibited BSH activity but were not more potent than AAA-2 in this assay (Figure 3A). The C3, C7-disulfonated derivative AAA-2’ (3) was also equipotent to AAA-2. Notably, the LCA core-based analog AAA-10 was more potent than both AAA-2 and the inhibitor candidates AAA-2’, AAA-4, AAA-6, and AAA-8. AAA-10 was equipotent to its unsulfonated analog AAA-9 (7), indicating that C3-sulfonation did not hinder BSH inhibitory activity (Figure 3a). AAA-10 also inhibited BSH activity in mouse feces when TCA-d4 was used as a substrate (Figure S1a–b).
Mice fed a high-fat diet (HFD) possess higher levels of bile acids, including conjugated bile acids, than mice fed a chow diet.30 Because increased substrate concentration may increase in vivo BSH activity, we also evaluated the ability of AAA-10 to inhibit the enzyme activity in feces obtained from HFD-fed mice. We found that AAA-10 inhibited BSH activity in this assay as effectively as AAA-1, our most potent first-generation BSH inhibitor (Figure S1c). Together, our data suggest that AAA-10 is a potent inhibitor of BSHs found in the murine gut.
AAA-10 inhibits recombinant BSHs and is more potent than AAA-2
To further characterize the potency of AAA-10 compared to AAA-1 and AAA-2, we determined the half maximal inhibitory concentration (IC50) values of both AAA-10 and AAA-2 against purified recombinant B. theta (Gram-negative) and B. longum (Gram-positive) BSHs and compared these values to the IC50 values for AAA-1 which had been determined in our previous work24 (Figure 3b–c). The IC50 values were evaluated using a conjugated bile acid substrate for which the enzymes demonstrated the best hydrolytic efficiency (TUDCA and TDCA, respectively).24 AAA-10 exhibited an IC50 value of 10 nM against B. theta rBSH and 80 nM against B. longum rBSH, demonstrating that AAA-10 was ~250 fold more potent than AAA-2 against B. theta rBSH and ~15 fold more potent against B. longum rBSH (Table S1). Compared to AAA-1, AAA-10 was ~40 fold more potent against B. theta rBSH and equally potent against B. longum rBSH (Table S1). The increased potency of AAA-10 against B. theta rBSH, a selective BSH, compared to both first-generation inhibitors highlights the potential of this compound to target BSHs which might otherwise be difficult to inhibit. These data demonstrate that we have developed a second-generation sulfonated inhibitor with increased potency compared to first-generation compounds.
AAA-10 inhibits BSH activity in bacterial cultures
Next, we evaluated the ability of AAA-10 to inhibit enzyme activity in growing cultures of BSH-containing bacteria using three Gram-negative and three Gram-positive strains found in the human gut (Figure 4a). Each bacterial culture was diluted to pre-log phase and co-incubated with 100 μM of AAA-10 and 100 μM of an equimolar mixture of taurine-conjugated bile acids that are abundant in the murine gallbladder and small intestine (tauro-betamuricholic acid (TβMCA), taurocholic acid (TCA), TUDCA, and TDCA).31 Bacteria were then allowed to grow into stationary phase over 24 h. Because bacteria vary in their ability to metabolize different conjugated bile acids, this approach provides an unbiased way of testing the inhibitory activity of AAA-10. After 24 h, percent deconjugation was determined by quantifying bile acid concentrations in bacterial cultures by UPLC-MS. AAA-10 exhibited near-complete inhibition of enzyme activity in all six bacterial cultures (<8% deconjugation) (Figure 4a, Figure S2). AAA-10 displayed equivalent inhibitory activity to AAA-1, except in the case of C. perfringens, where AAA-10 inhibited deconjugation to a greater extent than AAA-1 (6% vs 22% deconjugation, respectively).24
Figure 4. AAA-10 inhibits BSH activity in bacterial cultures without exhibiting antibacterial effects.
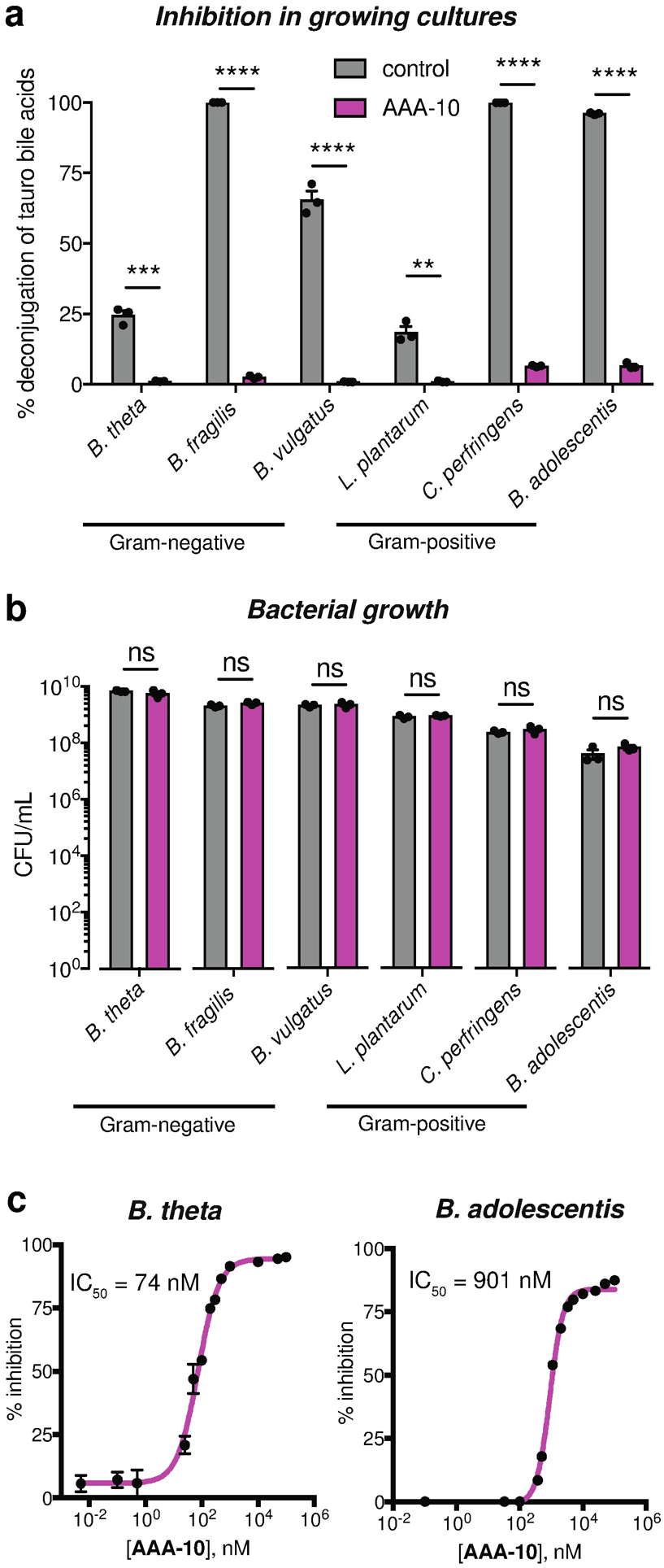
a, AAA-10 inhibits bacterial BSH activity. The BSH inhibitory activity of AAA-10 (100 μM) against three Gram-negative (B. theta VPI-5482, Bacteroides fragilis ATCC 25285, and Bacteroides vulgatus ATCC 8482) and three Gram-positive (Lactobacillus plantarum WCFS1, Clostridium perfringens ATCC 13124, and Bifidobacterium adolescentis L2–32) human gut bacteria using 100 μM taurine-conjugated bile acids (TβMCA, TCA, TUDCA and TDCA, 25 μM each) as substrates was evaluated. BSH activity was quantified as percent deconjugation of tauro-conjugated bile acids at 24 h as determined by UPLC–MS (for absolute concentrations of substrates and products recovered, see Figure S2). b, AAA-10 did not affect bacterial cell viability. At the end of the assay in (a), the bacteria were plated to determine cell viability. c, AAA-10 is a nanomolar inhibitor of bacterial BSHs. Dose-response curves of AAA-10 against B. theta and B. adolescentis cultures were generated using tauro-ursodeoxycholic acid (TUDCA) and tauro-deoxycholic acid (TDCA) as substrates, respectively. For a, and b, two-tailed Student’s t-test were performed. For c, Graphpad was used to fit IC50 curves. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant. All assays were performed in biological triplicate, and data are presented as mean ± s.e.m.
To demonstrate that the BSH inhibitory activity of AAA-10 in bacterial cultures was not due to growth inhibition, we evaluated the colony forming units in aforementioned bacterial cultures treated with AAA-10. We found that this compound did not significantly affect the growth of any of the tested bacterial strains at a concentration of 100 μM (Figure 4b).
We also determined the IC50 values of AAA-10 against B. theta (Gram-negative) and B. adolescentis (Gram-positive) whole cell cultures. For this purpose, we used a single conjugated bile acid against which the enzyme demonstrated the highest deconjugation efficiency (TUDCA and TDCA, respectively). While the IC50 value for AAA-10 against B. adolescentis was higher than the previously reported value for AAA-1 (901 nM versus 108 nM, respectively), AAA-10 displayed a lower IC50 value against B. theta than AAA-1 (74 nM versus 427 nM, respectively)24 (Figure 4c). These data are consistent with our results using purified protein and show that AAA-10 is the most potent inhibitor of the B. theta BSH yet developed. Together, these results demonstrate that AAA-10 is a nanomolar inhibitor of gut bacterial BSHs that does not display anti-bacterial properties.
AAA-10 displays limited off-target effects on mammalian cells
At high in vivo concentrations, bile acids have been shown to disrupt cell membranes and can induce apoptosis in mammalian cells15,32,33. Because AAA-10 is based on a bile acid scaffold, we evaluated the toxicity of AAA-10 on intestinal cells as well as its off-target effects on host bile acid receptors. Human intestinal Caco-2 cells were differentiated in transwell inserts to form a polarized monolayer with tight junctions34 (Figure S3a). Incubation of these cells with AAA-1, AAA-2 or AAA-10 showed that while AAA-1 and AAA-2 (100 μM) negatively affected the cell viability, AAA-10 did not have an effect on the cell viability at this concentration (Figure 5a). AAA-10 also had no effect on the viability of human liver cells (Hep-G2) at 100 μM or 500 μM concentrations (Figure S3b). We next determined whether BSH inhibitors affected intercellular tight junctions by measuring the passive diffusion of FITC-dextran (4 kDa) from the apical to the basolateral chamber of the transwells containing differentiated Caco-2 cells treated apically with our compounds. AAA-10 did not appear to damage epithelial integrity at 100 μM or 500 μM concentrations, while AAA-1, AAA-2, and AAA-9 increased FITC-d permeability by over 85% (~1.5–3 fold) at a concentration of 100 μM (Figure 5b). In order to test the gut-restricted properties of AAA-10, we also quantified the amount of inhibitor in the apical and basolateral chambers in these transwell assays. We have previously shown that bile acids, including LCA, pass through Caco-2 monolayers.35 In contrast, while we were able to detect AAA-10 in the apical chamber, no inhibitor was detected in the basolateral chamber 16 h after apical application, indicating that AAA-10 does not pass through an epithelial monolayer (Figure 5c–d).
Figure 5. AAA-10 is not toxic to mammalian cells and not a ligand for FXR or TGR5.
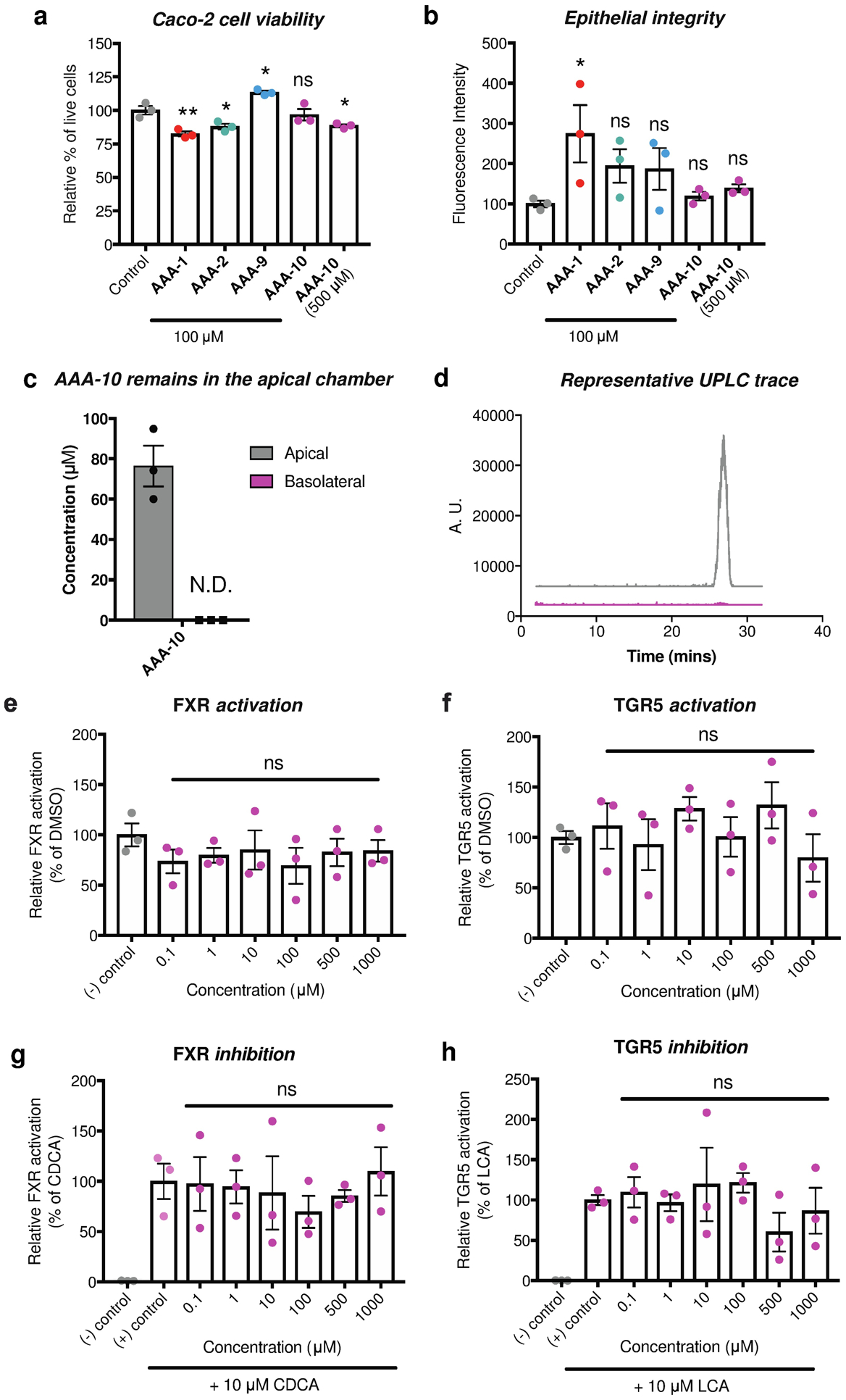
a, Incubation of differentiated Caco-2 cells with AAA-10 (100 μM) did not result in toxicity, while incubation with an equivalent concentration of AAA-1 and AAA-2 resulted in decreased cell viability. b, AAA-10 did not damage epithelial tight junctions at 100 μM or 500 μM, while treatment with AAA-1 resulted in loss of epithelial integrity. Epithelial junction integrity was determined by measuring the transport of 4 kDa FITC-dextran from the apical to the basolateral chamber. c, AAA-10 did not pass through an epithelial monolayer in an in vitro transwell assay (for assay setup see Figure S3a). Passage of the molecule from apical chamber to basolateral chamber was quantified by UPLC-MS. d, Representative UPLC-MS extracted ion chromatogram (EIC) traces of apical and basolateral chamber showing that no AAA-10 was detected in the basolateral chamber. e and f, FXR and TGR5 agonist activity was measured by incubating Caco-2 cells with varying concentrations of AAA-10 overnight. g and h, FXR and TGR5 antagonist activity was measured by incubating Caco-2 cells with varying concentrations of AAA-10 overnight in the presence of 10 μM of the FXR agonist chenodeoxycholic acid (CDCA) or 10 μM of the TGR5 agonist lithocholic acid (LCA), respectively. For a-b and e-h, one-way ANOVA followed by Dunnett’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant. All assays were performed in biological triplicate, and data are presented as mean ± s.e.m.
Bile acids can signal through the host receptors FXR and TGR5, thereby affecting host metabolism and immune function.36 Incubation of Caco-2 cells with increasing concentrations of AAA-10 revealed that this compound did not act as an agonist of FXR or TGR5 (Figures 5e–f). Incubation of Caco-2 cells with either CDCA (FXR agonist) or LCA (TGR5 agonist) followed by treatment with increasing concentrations of AAA-10 revealed that this compound did not antagonize FXR or TGR5 (Figures 5g–h). Collectively, these data suggest that AAA-10 is a potent pan-BSH inhibitor with low epithelial permeability that exhibits reduced off-target effects on host cells compared to the first-generation inhibitors AAA-1 and AAA-2.
AAA-10 reduces secondary bile acid abundance in vivo
We next evaluated the ability of AAA-10 to inhibit BSH activity and modulate bile acid levels in vivo. Wild-type C57Bl/6J mice fed ad libitum were gavaged once daily with AAA-10 at a dose of 30 mg/kg for 5 days (Figure 6a). The inhibitor was administered at 6 pm to coincide with the start of the dark photoperiod when mice exhibit increased food consumption.37 Fecal BSH activity was significantly decreased on days 2 and 6 in AAA-10-treated mice compared to vehicle-treated mice (Figure 6b and Figure S4a), indicating that we were able to achieve durable BSH inhibition in vivo using AAA-10. We next performed cecal and fecal bile acid pool analysis. To account for the fact that variations in bile acid concentrations are known to occur both between both across subjects and within a subject over time, we represented bile acid levels as abundances.38–40 Cecal abundances of DCA and LCA were significantly lowered in the AAA-10-treated group (Figures 6c and Figure S5). DCA and LCA are secondary bile acids that are produced exclusively by gut bacteria.5 Cecal AAA-10 concentration was also negatively correlated with cecal concentrations of both DCA and LCA (Figures S4b–c). In addition, the abundances of DCA and LCA were decreased in feces each day starting on day 2 and overall in feces throughout the course of the study in AAA-10-treated mice compared to vehicle-treated mice (Figures 6d–e, Figure S4d and Figure S6). Together, these findings indicate that AAA-10 treatment resulted in a sustained reduction in secondary bile acids in vivo over the period of study.
Figure 6. AAA-10 reduces secondary bile acid abundance in vivo and inhibits BSH activity in human feces.
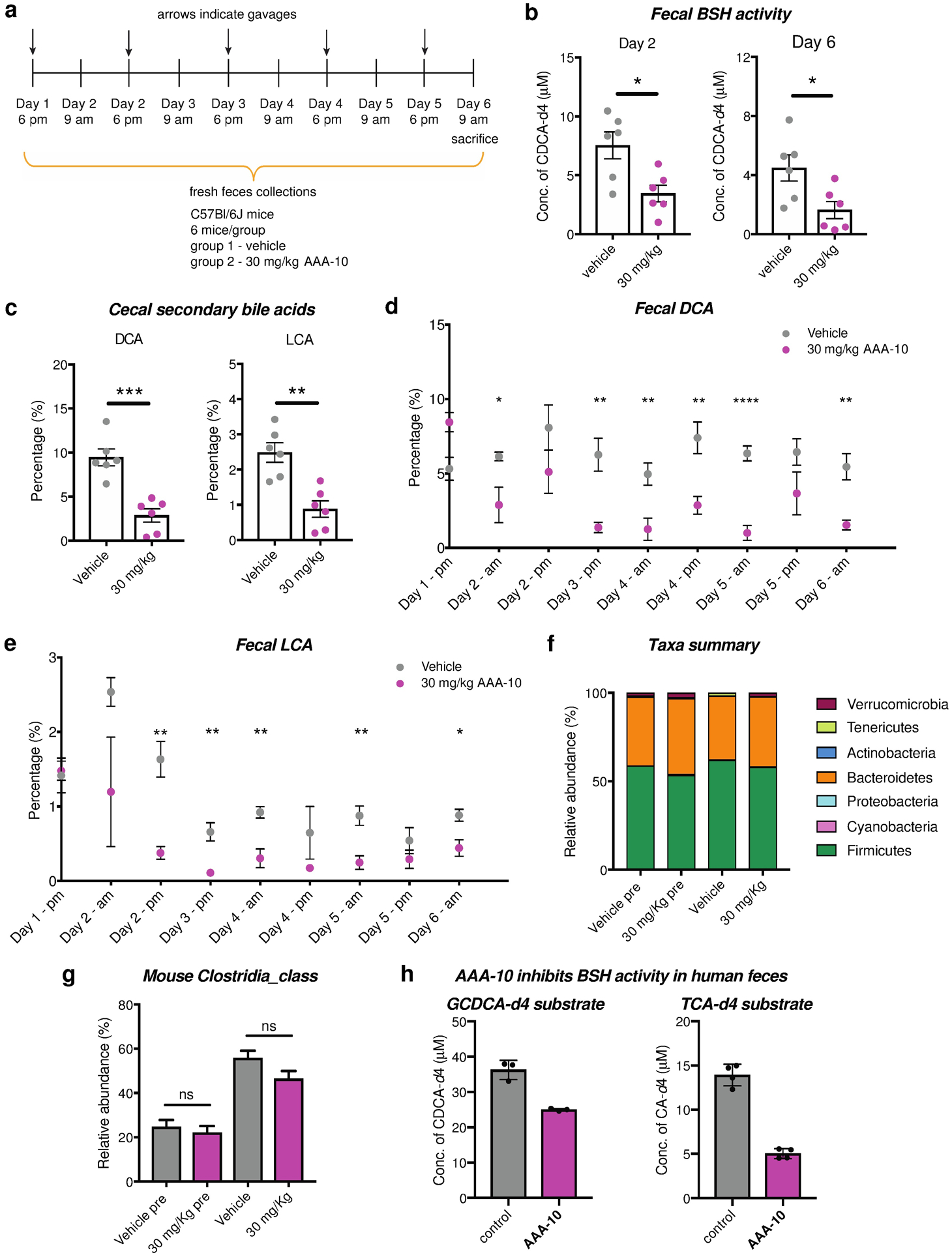
a, In vivo study design. C57Bl/6J mice fed ad libitum were orally gavaged with AAA-10 (30 mg/kg) once daily for 5 days. Feces were collected daily and utilized to evaluate bile acid changes and BSH activity. Mice were sacrificed 15h after the final gavage. b, AAA-10-treated mice exhibited decreased BSH activity compared to vehicle-treated mice in fresh feces collected on days 2 and 6. c, Percentages of the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA) were reduced in cecal contents of mice treated with AAA-10. d and e, Analysis of fecal bile acid contents over the period of the study showed that abundances of the two secondary bile acids DCA and LCA were consistently decreased throughout the experiment. f, Stacked bar plot showing mean relative abundances of phylum-level taxa in mouse samples. g, Relative abundance of class Clostridia in samples. n=6 per group, Vehicle pre and AAA-10 pre are pretreatment mouse fecal samples; Vehicle and AAA-10 are post-treatment mouse cecal samples. h, AAA-10 inhibited BSH activity in human fecal slurry over a period of 2 h (n ≥ 3). For b-e, n=6 mice/group, two-tailed Welch’s t test was performed. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant. For g, ns=not significant, one-way ANOVA followed by Tukey’s multiple comparisons test). All data are presented as mean ± s.e.m.
Cecal and total fecal bile acid concentrations were significantly increased in AAA-10-treated compared to vehicle-treated mice. This increase was driven by increased levels of conjugated and primary bile acids (Figure S5 and Figure S6). Concentrations of total bile acids and conjugated and primary bile acids were also increased in plasma, although the changes were not statistically significant (Figure S7). We observed an increase in the conjugated bile acid TβMCA, a potent FXR antagonist,31 both in feces 15h after the first gavage (Figure S6b) and in cecal contents (Figure S5b). FXR inhibition increases expression of the cytochrome P450 7A1 gene (Cyp7A1), which encodes the rate-limiting enzyme in the production of bile acids from cholesterol, thereby increasing bile acid synthesis.31,41 Future work investigating transcriptional- and protein-level changes may provide evidence to support the hypothesis that a TβMCA-mediated loop is driving the increase in bile acids levels observed following inhibitor treatment.
AAA-10 treatment did not significantly affect microbial community composition at the phylum level as indicated by 16S recombinant DNA sequencing (Figure 6f). Moreover, AAA-10 treatment did not significantly affect the relative abundance of the phylum Firmicutes, the class Clostridia, the order Clostridiales, the family Clostridiaceae, or the genus Clostridium (Figure 6g and Figure S8a). We also showed that AAA-10 did not affect the in vitro viability of Clostridum scindens ATCC 35704 and Clostridium scindens VPI 12708, two known 7α-dehydroxylating bacteria (Figure S8b).5 Together, these data suggest that AAA-10 is not decreasing LCA or DCA abundance in vivo by reducing the levels of Clostridia that produce these molecules.
Finally, to evaluate the gut permeability of AAA-10, we quantified the levels of this compound in cecal contents and plasma at sacrifice and in feces over the course of the experiment (Figures S4e–g). We observed a mean value of 276 picomol/mg wet mass (~276 μM) of AAA-10 in cecal contents and a range of 66–2087 picomol/mg (~66–2087 μM) in feces. In contrast, five of the six mice exhibited undetectable levels of AAA-10 in plasma (Figure S4g). In a separate experiment in which mice were sacrificed 4 hours after the final gavage, we detected mean AAA-10 concentrations of 128 picomol/mg wet mass (~128 μM) in cecal contents and 12 nM in plasma (Figures S4h–i). Together, these data indicate that AAA-10 displays high colonic exposure and low gut permeability.
AAA-10 inhibits BSH activity in human feces
To test whether AAA-10 is a pan-inhibitor of BSHs found in the human gut, we incubated feces from a human donor with 20 μM AAA-10 for 30 min and then added either GCDCA-d4 or TCA-d4 as substrate. After incubating for 2 h, we quantified conversion to deconjugated product (CDCA-d4 or CA-d4, respectively), using UPLC-MS. We observed that AAA-10 significantly inhibited deconjugation of both substrates, indicating that AAA-10 is capable of inhibiting BSHs found in the distal human gut (Figure 6h).
AAA-10 itself could also be metabolized by gut bacteria, reducing its efficacy. To investigate this possibility, we determined the stability of AAA-10 in mouse and human fecal suspensions (Figure S9). We found that 97% and 81% (mean values) of AAA-10 was recovered after incubation with mouse and human feces, respectively, suggesting that the inhibitor does not undergo substantial off-target degradation by gut bacteria.
CONCLUSION
The studies reported herein were initiated with the goal of improving the potency of the first-generation gut-restricted inhibitor AAA-2. Structure-activity relationship studies that focused on incorporating the carbon scaffolds of different abundant bile acids into our inhibitor design led to the identification of a second-generation inhibitor, AAA-10. This compound is more potent than AAA-2 in vitro and exhibited an improved off-target effects profile compared to AAA-1 and AAA-2. The structure of AAA-10 is based on the core of LCA, a bile acid that contains a single hydroxyl group at C3. In previous work, we characterized the BSH activity of 20 abundant human gut Bacteroidetes species against glyco- and tauro-conjugated bile acids.20 Glyco-lithocholic acid (GLCA) and tauro-lithocholic acid (TLCA) were effectively deconjugated (>90% conversion) by all BSH-containing Bacteroidetes species tested. In contrast, all other bile acids were incompletely deconjugated (<70%) by two or more of the species tested. Taken together, these results suggest that the LCA core may be effective as a scaffold for BSH inhibitors because it is recognized as a substrate by a range of gut bacteria. Future studies testing the deconjugating ability of a variety of Gram-positive and Gram-negative strains against a panel of conjugated bile acid substrates may further elucidate the substrate scope of gut bacterial BSHs and thus aid in next-generation inhibitor design.
We also demonstrated that AAA-10 inhibits BSH activity in vivo and decreased the abundance of the secondary bile acids DCA and LCA in feces and cecal contents. DCA and LCA are known to play crucial roles in host physiology. On the one hand, DCA and LCA are strongly associated with colon cancer development in patients, and evidence indicates that these compounds promote carcinogenesis in the colon and liver.13,14,42–44 On the other hand, DCA has been shown to limit growth of the pathogen Clostridium difficile,45 and recent work has shown that LCA induces the production of the anti-diabetic metabolite cholic acid-7-sulfate.35 The ability to modulate the abundance of these compounds in vivo in fully colonized animals will facilitate investigations of the roles of these molecules in host physiology.
The origin of the increase in total bile acid levels following AAA-10 treatment warrants further investigation. Notably, humans possess little to no muricholic acids,38 and therefore, we would not expect increased bile acids following BSH inhibition in humans. Future work administering BSH inhibitors to cytochrome P450 2c (Cyp2c) knock-out mice, which lack the cytochrome responsible for muricholic acid synthesis by murine liver and therefore possess a “humanized” bile acid pool,46 may help to better predict how BSH inhibition will affect the bile acid pool and host responses in human subjects. Our data indicate that AAA-10 can inhibit BSH activity in feces from a healthy human donor. Further studies using feces from an array of human subjects will help reveal whether AAA-10 is an effective BSH inhibitor across different human microbiota communities.
Looking ahead, it will be valuable to consider whether long-term use of BSH inhibitors in vivo affects microbial community composition. In addition, once-daily dosing via oral gavage may not be optimal in the case of ad libitum feeding, and further optimization of a strategy for inhibitor administration may be required. Nonetheless, our data indicate that we have developed a potent, non-toxic BSH inhibitor that modulates the in vivo bile acid pool, shifting the bile acid pool away from DCA and LCA. Because bile acids are absorbed and recirculated to the liver via the portal vein,5,47 BSH inhibitors will facilitate investigations of how bile acids are causally involved in the initiation and progression of both liver and GI tract disorders, including inflammatory bowel diseases, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), liver cirrhosis, and liver and colon cancer.17,19 Demonstration of prevention or amelioration of disease phenotypes in animals would suggest that BSHs could be targeted in a therapeutic context to treat human disease.
METHODS
Additional details for materials and methods are provided in the Supporting Information. The 16S rDNA datasets analyzed in the manuscript are available through the NCBI under BioProject ID PRJNA729977.
Screen of inhibitors in conventional mouse feces.
BSH activity in fecal pellets was quantified using a modified version of a published method.23,24 Fresh feces were collected from wildtype adult male C57Bl6/J or C57Bl6/N mice. Fecal pellets (approximately 10–20 mg) were broken into fine particles in buffer (PBS with 0.25 mM TCEP) to obtain a concentration of 100 mg ml−1. This solution was further diluted to a concentration of 1 mg ml−1. Inhibitors were then added at the indicated concentrations and incubated at 37 °C for 30 mins. 100 μM glycochenodeoxycholic acid-d4 (GCDCA-d4) or or taurocholic acid-d4 (TCA-d4) was added to the mixture and incubated at 37 °C for 18 h. The tubes were then frozen in dry ice for 5 mins and upon thawing were diluted with an equal volume of HPLC grade methanol. The slurry was centrifuged at 12,500 g for 10 mins. The supernatant was removed into a clean Eppendorf tube and centrifuged again. The supernatant was transferred to MS vials and samples were analyzed as per the method described in “UPLC-MS Analysis” (see Supporting Information). The concentration of product detected from these assays was reported directly.
BSH inhibition in bacterial cultures.
Bacterial cultures were diluted to OD600 of 0.1 in 4 ml BHI+, containing 100 μM taurine conjugated bile acid pool (TβMCA, TCA, TUDCA and TDCA, 25 μM each) and 100 μM AAA-10. These cultures were then grown anaerobically at 37 °C. After 21 h, serial dilutions were plated on BHI+ agar to determine cell viability (CFU ml−1). 1 ml of the entire bacterial culture was acidified to pH = 1 using 6M HCl followed by addition of 2 ml ethyl acetate and vortexed. The cultures were spun down in a centrifuge at 2,500 g for 5 mins to obtain better separation. The organic layer was then removed and the aqueous layer was extracted again using 2 ml of ethyl acetate. The dried organic extracts were resuspended in 1:1 methanol:water and transferred to mass spec vials and analyzed as per the method described in “UPLC-MS Analysis”. The obtained concentrations of bile acids were used to determine percent deconjugation.
Luciferase reporter assay.
Luminescence was measured using the Dual-Luciferase Reporter Assay System (Promega Corporation) according to manufacturer’s instructions. Cells were washed gently with PBS and lysed in PLB from the kit. Luminescence was measured using a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA) at the ICCB-Longwood Screening Facility at HMS. Luminescence was normalized to Renilla luciferase activity and percentage relative luminescence was calculated compared to DMSO control.
Epithelial permeability assay.
Undifferentiated Caco-2 cells were seeded in 24-well plate transwells (0.4 μM pore size, Costar) at 200,000 cells per transwell. Media was changed on days 4, 8, 12, 16, and 18 to differentiate Caco-2 cells in vitro.35 On day 21, fully differentiated and polarized cells were used for FITC-dextran permeability assay. Briefly, inhibitors were added in PBS at indicated concentrations to the apical chamber of the transwells containing differentiated Caco-2 cells and incubated overnight. The apical chamber of the transwells contained a volume of 100 μl PBS with inhibitor or DMSO control, while the basolateral chamber contained 500 μl of PBS. Caco-2 epithelial integrity was assayed by measuring passive diffusion of 4 kDa FITC-Dextran (Sigma Aldrich) added at a concentration of 5 μM to the apical chamber. Diffusion from the apical to basolateral side was measured by fluorescence reading in PBS on the basolateral side of the transwell system using a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA) at the ICCB-Longwood Screening Facility at HMS. Fluorescence reading was normalized to the DMSO control. Transport of inhibitors from the apical to basolateral compartment was measured by drying basolateral media under vacuum and resuspending contents in methanol prior to injecting in the UPLC-MS.
BSH inhibition and bile acid pool modulation in vivo.
Male C57BL/6J mice obtained from Jackson laboratories were maintained under a strict 12 h/12 h light/dark cycle and under constant temperature (21 ± 1 °C) and humidity (55–65%). All experiments were conducted on 13–14 week-old mice. Mice were maintained on a standard chow diet (Purina LabDiet, catalog no. 5008) for the duration of the experiment and fed ad libitum. Mice were split into two groups of six mice each and were gavaged once daily at the beginning of the dark phase with either 200 μl of 95% PBS/ 5% DMSO containing 10% captisol (w/v) (vehicle group) or with 200 μl of 95% PBS/ 5% DMSO containing 10% captisol (w/v) and AAA-10 at a concentration of 3.75 mg ml-1 (treatment group) for five consecutive days. For the fecal pellet collection, each mouse was transferred to a temporary cardboard cage for a several minutes until a fecal pellet was produced.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to S. Blacklow, N. Gray, D. Scott, J. Hatcher, J. Wang, J. Clardy, and members of the Devlin and Clardy groups for helpful discussions. We would like to acknowledge the Blacklow and Kruse labs for help with equipment and reagents and L. Yao for help with 16s rRNA sequencing. We thank K. Schoonjans (Ecole polytechnique fédérale de Lausanne-EPFL) for the FXR reporter plasmid and the ICCB-Longwood Screening Facility for use of their fluorescent plate reader. We thank the scientists at Bienta, Enamine for help with in vivo pharmacokinetics experimental design and execution and the Massachusetts Host-Microbiome Center for 16S rRNA sequencing service. We thank D. Paik and J. Huh (HMS) for providing fresh mouse feces and E. Alm and T. Nguyen (MIT and the Center for Microbiome Informatics and Therapeutics) for providing human feces. Images from Biorender were used in the graphical abstract.
Funding
This research was supported National Institutes of Health (NIH) grant R35 GM128618 (A.S.D), an Innovation Award from the Center for Microbiome Informatics and Therapeutics at MIT (A.S.D), a grant from Harvard Digestive Diseases Center (supported by NIH grant 5P30DK034854-32) (A.S.D), a John and Virginia Kaneb Fellowship (A.S.D), a Quadrangle Fund for the Advancement and Seeding of Translational Research at Harvard Medical School (Q-FASTR) grant (A.S.D), and an HMS Dean’s Innovation Grant in the Basic and Social Sciences (A.S.D). S.N.C. acknowledges an American Heart Association Postdoctoral Fellowship. M.D.M. acknowledges an NSF Graduate Research Fellowship (DGE1745303).
Footnotes
Supporting Information
Materials and methods (including detailed synthetic protocols and characterization data), Figures S1–9, Table S1, Scheme S1.
The authors declare the following competing financial interest(s): A. Sloan Devlin is an ad hoc consultant for Takeda Pharmaceuticals and Axial Therapeutics. The other authors declare that no competing interests exist.
REFERENCES
- (1).Donia MS, and Fischbach MA (2015) Small molecules from the human microbiota. Science 349, 1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh L-A, Mani S, and Redinbo MR (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, Barrington WT, Russell MW, Reed JM, Duzan A, Lang JM, Fu X, Li L, Myers AJ, Rachakonda S, DiDonato JA, Brown JM, Gogonea V, Lusis AJ, Garcia-Garcia JC, and Hazen SL (2018) Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med 24, 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ, and Balskus EP (2019) Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ridlon JM, Kang D-J, and Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J. Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- (6).Hofmann AF (1963) The function of bile salts in fat absorption. The solvent properties of dilute micellar solutions of conjugated bile acids. Biochem. J 89, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Roda A, Hofmann AF, and Mysels KJ (1983) The influence of bile salt structure on self-association in aqueous solutions. J. Biol. Chem 258, 6362–6370. [PubMed] [Google Scholar]
- (8).Fiorucci S, and Distrutti E (2015) Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol Med 21, 702–714. [DOI] [PubMed] [Google Scholar]
- (9).Schaap FG, Trauner M, and Jansen PLM (2014) Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11, 55–67. [DOI] [PubMed] [Google Scholar]
- (10).Pols TWH, Puchner T, Korkmaz HI, Vos M, Soeters MR, and de Vries CJM (2017) Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLOS ONE (Rottenberg ME, Ed.) 12, e0176715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hang S, Paik D, Yao L, Kim E, Jamma T, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, Zheng Y, Longman RS, Rastinejad F, Devlin AS, Krout MR, Fischbach MA, Littman DR, and Huh JR (2019) Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Begley M, Hill C, and Gahan CGM (2006) Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol 72, 1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ridlon JM, Wolf PG, and Gaskins HR (2016) Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 7, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, Korangy F, Wang XW, Trinchieri G, and Greten TF (2018) Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ajouz H, Mukherji D, and Shamseddine A (2014) Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol 12, 164–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ridlon JM, Harris SC, Bhowmik S, Kang D-J, and Hylemon PB (2016) Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7, 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Song Z, Cai Y, Lao X, Wang X, Lin X, Cui Y, Kalavagunta PK, Liao J, Jin L, Shang J, and Li J (2019) Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Parasar B, Zhou H, Xiao X, Shi Q, Brito IL, and Chang PV (2019) Chemoproteomic Profiling of Gut Microbiota-Associated Bile Salt Hydrolase Activity. ACS Cent Sci 5, 867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jia B, Park D, Hahn Y, and Jeon CO (2020) Metagenomic analysis of the human microbiome reveals the association between the abundance of gut bile salt hydrolases and host health. Gut Microbes 11, 1300–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, DiBenedetto N, Mina AI, Banks AS, Bry L, and Devlin AS (2018) A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Joyce SA, MacSharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, Hill C, and Gahan CGM (2014) Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. U.S.A 111, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, and Gonzalez FJ (2013) Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun 4, 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Xie C, Jiang C, Shi J, Gao X, Sun D, Sun L, Wang T, Takahashi S, Anitha M, Krausz KW, Patterson AD, and Gonzalez FJ (2017) An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 66, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Adhikari AA, Seegar TCM, Ficarro SB, McCurry MD, Ramachandran D, Yao L, Chaudhari SN, Ndousse-Fetter S, Banks AS, Marto JA, Blacklow SC, and Devlin AS (2020) Development of a covalent inhibitor of gut bacterial bile salt hydrolases. Nature Chemical Biology 16, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tanaka H, Hashiba H, Kok J, and Mierau I (2000) Bile salt hydrolase of Bifidobacterium longum-biochemical and genetic characterization. Appl. Environ. Microbiol 66, 2502–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang Z, Zeng X, Mo Y, Smith K, Guo Y, and Lin J (2012) Identification and Characterization of a Bile Salt Hydrolase from Lactobacillus salivarius for Development of Novel Alternatives to Antibiotic Growth Promoters. Appl. Environ. Microbiol 78, 8795–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Strott CA (2002) Sulfonation and molecular action. Endocr Rev 23, 703–732. [DOI] [PubMed] [Google Scholar]
- (28).Alnouti Y (2009) Bile Acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol. Sci 108, 225–246. [DOI] [PubMed] [Google Scholar]
- (29).Palmer JT, and Inc P (1994) Process for forming a fluoromethyl ketone.
- (30).Zheng X, Huang F, Zhao A, Lei S, Zhang Y, Xie G, Chen T, Qu C, Rajani C, Dong B, Li D, and Jia W (2017) Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice. BMC Biol 15, 120–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, and Bäckhed F (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17, 225–235. [DOI] [PubMed] [Google Scholar]
- (32).Perez M-J, and Briz O (2009) Bile-acid-induced cell injury and protection. World J. Gastroenterol 15, 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Glinghammar B, Inoue H, and Rafter JJ (2002) Deoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and the transcription factors NF-kB and AP-1. Carcinogenesis 23, 839–845. [DOI] [PubMed] [Google Scholar]
- (34).Ferruzza S, Rossi C, Scarino ML, and Sambuy Y (2012) A protocol for differentiation of human intestinal Caco-2 cells in asymmetric serum-containing medium. Toxicol In Vitro 26, 1252–1255. [DOI] [PubMed] [Google Scholar]
- (35).Chaudhari SN, Luo JN, Harris DA, Aliakbarian H, Yao L, Paik D, Subramaniam R, Adhikari AA, Vernon AH, Kiliç A, Weiss ST, Huh JR, Sheu EG, and Devlin AS (2020) A microbial metabolite remodels the gut-liver axis following bariatric surgery. Cell Host & Microbe 317, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wahlström A, Sayin SI, Marschall H-U, and Bäckhed F (2016) Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 24, 41–50. [DOI] [PubMed] [Google Scholar]
- (37).Ellacott KLJ, Morton GJ, Woods SC, Tso P, and Schwartz MW (2010) Assessment of feeding behavior in laboratory mice. Cell Metab. 12, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Setchell KD, Lawson AM, Tanida N, and Sjövall J (1983) General methods for the analysis of metabolic profiles of bile acids and related compounds in feces. J. Lipid Res 24, 1085–1100. [PubMed] [Google Scholar]
- (39).Setchell KD, Ives JA, Cashmore GC, and Lawson AM (1987) On the homogeneity of stools with respect to bile acid composition and normal day-to-day variations: a detailed qualitative and quantitative study using capillary column gas chromatography-mass spectrometry. Clin Chim Acta 162, 257–275. [DOI] [PubMed] [Google Scholar]
- (40).Duparc T, Plovier H, Marrachelli VG, Van Hul M, Essaghir A, Ståhlman M, Matamoros S, Geurts L, Pardo-Tendero MM, Druart C, Delzenne NM, Demoulin J-B, van der Merwe SW, van Pelt J, Bäckhed F, Monleon D, Everard A, and Cani PD (2017) Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 66, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, Liu Y, Gavrilova O, Patterson AD, and Gonzalez FJ (2015) Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 6, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Reddy BS, Narasawa T, Weisburger JH, and Wynder EL (1976) Promoting effect of sodium deoxycholate on colon adenocarcinomas in germfree rats. J. Natl. Cancer Inst 56, 441–442. [DOI] [PubMed] [Google Scholar]
- (43).Narisawa T, Magadia NE, Weisburger JH, and Wynder EL (1974) Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-N-nitrosoguanidine in rats. J. Natl. Cancer Inst 53, 1093–1097. [DOI] [PubMed] [Google Scholar]
- (44).Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Hattori M, Honda K, Ishikawa Y, Hara E, and Ohtani N (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- (45).Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MRM, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, and Pamer EG (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, Wolf CR, Henderson CJ, and Gonzalez FJ (2016) Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res 57, 2130–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).van de Peppel IP, Verkade HJ, and Jonker JW (2020) Metabolic consequences of ileal interruption of the enterohepatic circulation of bile acids. Am. J. Physiol. Gastrointest. Liver Physiol 319, G619–G625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.