

Abstract
BACKGROUND & AIMS:
In upper airway cells, T helper 2 cytokines that signal through interleukin-4 (IL-4) receptor-α have been shown to stimulate eotaxin-3 secretion via a nongastric proton pump (ngH+,K+ATPase). To seek novel targets for eosinophilic esophagitis (EoE) treatments, we evaluated ngH+,K+ATPase expression in EoE squamous cells, and explored molecular pathways involved in eotaxin-3 secretion by IL-4 receptor-α signaling.
METHODS:
ngH+,K+ATPase expression in EoE cells was evaluated by quantitative real-time polymerase chain reaction and Western blotting. IL-4–stimulated eotaxin-3 secretion was measured by enzyme-linked immunosorbent assay after treatment with omeprazole, SCH 28080 (potassium-competitive acid blocker), ethylene glycol-bis(β-aminoethyl)-N,N,N′,N′-tetraacetoxymethyl ester (calcium chelator), 2-aminoethoxydiphenyl borate (inhibitor of endoplasmic reticulum calcium release), verapamil, and diltiazem (L-type calcium channel inhibitors). Intracellular calcium transients were measured by Fluo-4 fluorescence. Key experiments were confirmed in EoE primary cells and in RNA sequencing datasets from mucosal biopsies of patients with EoE and controls.
RESULTS:
EoE cells expressed ngH+,K+ATPase messenger RNA and protein. Omeprazole and SCH 28080 decreased IL-4–stimulated eotaxin-3 secretion. IL-4 increased intracellular calcium transients, and IL-4–stimulated eotaxin-3 secretion was blocked by ethylene glycol-bis(β-aminoethyl)-N,N,N′,N′-tetraacetoxymethyl ester, 2-aminoethoxydiphenyl borate, verapamil, and diltiazem. The combination of omeprazole and verapamil suppressed IL-4–stimulated eotaxin-3 secretion more than either agent alone. EoE biopsies expressed higher ngH+,K+ATPase and exhibited more calcium signaling than controls.
CONCLUSIONS:
EoE cells express a nongastric proton pump that mediates T helper 2 cytokine–stimulated eotaxin-3 secretion. IL-4 induces calcium release from the endoplasmic reticulum and calcium entry via L-type calcium channels, increasing intracellular calcium that contributes to eotaxin-3 secretion by EoE cells. L-type calcium channel inhibitors block T helper 2 cytokine–stimulated eotaxin-3 secretion, suggesting a potential role for these agents in EoE treatment.
Keywords: Proton Pump Inhibitors, Verapamil, Diltiazem, Potassium-Competitive Acid Blockers
Graphical Abstract
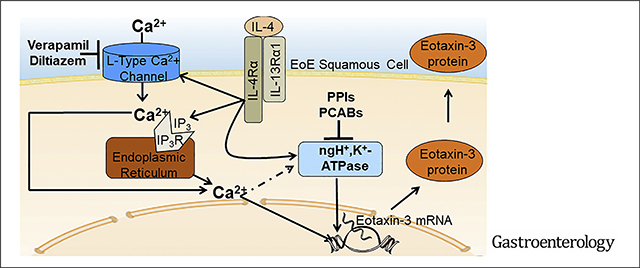
Eosinophilic esophagitis (EoE) is a modern malady that has rapidly emerged as a leading cause of dysphagia and food impaction in young adults.1 In EoE, food and aeroallergens trigger a type 2 immune response with increased esophageal expression of T helper 2 (Th2) cytokines, such as interleukin (IL)-5, IL-4, and IL-13.2 IL-5, a critical regulator of eosinophilia, is involved in eosinophil production, activation, and recruitment.3 The overexpressed IL-4 and IL-13 in EoE stimulate esophageal squamous cells to produce eotaxin-3, a potent eosinophil chemoattractant that draws activated eosinophils to the esophagus, where they can degranulate and release toxic products that contribute to esophageal damage and remodeling.4
Presently, treatment options for EoE are limited in number, and no single treatment is effective in all patients. In their recent guideline on management of EoE, the American Gastroenterological Association and Joint Task Force on Allergy-Immunology sanction only 3 EoE treatments outside of clinical trials: proton pump inhibitors (PPIs), swallowed topical glucocorticoids, and diet.5 Only 40%–50% of patients with EoE treated with PPIs and two-thirds of those treated with swallowed topical glucocorticoids achieve clinical and histologic remission.6–8 EoE remission rates exceeding 90% have been reported for elemental diets,9 but such diets are expensive, unpalatable, and unacceptable to many patients. Empiric elimination diets, which are successful in only 50%–70% of patients, are restrictive and cumbersome, requiring numerous follow-up endoscopies to document efficacy and identify food triggers.9,10 Clearly, there is a need for more safe and effective EoE therapies.
Clinical trials of biologic agents that target IL-5 and IL-13 have demonstrated significant histologic improvement but only limited symptomatic benefit for patients with EoE,11–13 and agents targeting tumor necrosis factor–α and IgE do not appear to be effective EoE treatments.13 Among the most promising biologic agents for EoE is dupilumab, a fully humanized monoclonal antibody directed against the IL-4 receptor-α (IL4Rα) through which both IL-4 and IL-13 signal to increase eotaxin-3 production by esophageal squamous cells.14,15 In a multicenter, randomized, placebo-controlled phase 2 study, dupilumab demonstrated significant efficacy in improving both clinical and histopathologic features of EoE.14 Elucidation of downstream nodes in the IL4Rα signal transduction pathway might reveal novel molecular targets for EoE therapies.
IL-4 and IL-13 signaling through IL4Rα results in phosphorylation and dimerization of STAT6, which enters the nucleus to bind and activate the eotaxin-3 gene promoter. In earlier studies, we elucidated a potential molecular mechanism for PPI responsiveness in EoE by showing that omeprazole blocks Th2 cytokine–stimulated secretion of eotaxin-3 in esophageal squamous cells via induction of chromatin modifications in the eotaxin-3 gene promoter that block its binding to STAT6.4,16 A recent study has shown that Th2 cytokines stimulate eotaxin-3 secretion in upper airway cells by activating a nongastric proton pump (ngH+,K+ATPase) that is susceptible to inhibition by PPIs and potassium-competitive acid blockers (P-CABs).17 Th2 cytokine signaling through IL4Rα has been shown to increase intracellular calcium in human tonsillar B lymphocytes,18 and increases in intracellular calcium are known to stimulate gastric H+,K+ATPase activity in parietal cells.19,20 We have explored whether esophageal squamous cells from patients with EoE express ngH+,K+ATPase that can be targeted to prevent Th2 cytokine–stimulated eotaxin-3 secretion. We have elucidated how Th2 cytokine signaling through IL4Rα increases intracellular calcium and have explored whether targeting molecules involved in intracellular calcium regulation can block Th2 cytokine–stimulated eotaxin-3 secretion in esophageal squamous cells from patients with EoE.
Materials and Methods
Squamous Epithelial Cell Lines and Primary Cultures
We used previously characterized, non-neoplastic, telomerase-immortalized, esophageal squamous cell lines established from mucosal biopsies of patients with EoE (EoE1-T and EoE2-T) and of a patient with gastroesophageal reflux disease (NES-G4T).4,21 We also established 9 primary cultures of esophageal squamous cells from mucosal biopsies of patients with EoE (8 male) using techniques described previously.4 The male predominance of study patients is due to male predominance of EoE, and recruitment primarily from the Department of Veterans Affairs. These studies were approved by Institutional Review Boards of Dallas Veterans Affairs Medical Center and Baylor Scott & White Research Institute. Primary cultures and cell lines were co-cultured with a fibroblast feeder layer and maintained at 37°C in a 5% CO2 incubator. For individual experiments, cells were seeded equally into collagen IV–coated wells (BD Biosciences, San Jose, CA) (without fibroblast feeder layers) and maintained in neutral pH (7.4) in a supplemented 3:1 mixture of Dulbecco’s modified Eagle medium/Ham’s F12 medium as described previously.22 Short tandem repeat analysis confirmed the identity of cell lines used in this study.
Bioinformatics Analysis of RNA Sequencing Datasets
Raw fastq files from 2 independent datasets (GSE41687 and GSE11334123,24) were downloaded from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) using sratoolkit (http://ncbi.github.io/sra-tools/). Aligned BAM files from a third dataset (GSE5864025) were acquired from the author, and reads were counted against genome version GRCh37 to match the genome used in the original publication. A list of analyzed samples (23 patients with EoE and 21 controls) are provided in Supplementary Table 1.
Air–Liquid Interface Cell Culture System
Primary EoE cells were seeded (1 × 105 cells per well) on a 0.4-μm pore-size permeable membrane support (Corning, Corning, NY), and grown to confluence over 3 days in the presence of Dulbecco’s modified Eagle medium/F12 (1.8 mM calcium). On day 4, medium was removed from the inner chamber of the support system to expose the cells to air, leading to epithelial stratification.26 On day 9, medium containing IL-4 (5 ng/mL) with and without omeprazole (50 μM), verapamil (50 μM), or the combination of both was added to the lower (basal) chamber for 48 hours.
Cytokine Stimulation of Esophageal Squamous Cells and Inhibition of the Nongastric H+,K+ATPase (ATP12A)
IL-4 and IL-13 stimulate eotaxin-3 secretion in esophageal squamous cells but, because our earlier studies documented more robust exotaxin-3 secretion with IL-4 than with IL-13, we used IL-4 as our primary study Th2 cytokine.4,16 Key experiments were confirmed using IL-13. Cells were stimulated with 5 ng/mL of IL-4 or 50 ng/mL of IL-13 (R&D Systems, Minneapolis, MN) for 24 or 48 hours.
In initial experiments, we attempted to knock down ngH+,K+ATPase (ATP12A) in EoE cell lines and primary cells by small interfering RNA (SMARTpool: ON-TARGETplus ATP12A siRNA; Dharmacon, Cambridge, MA) using single and multiple transfection protocols, with messenger RNA (mRNA) and protein assays performed 48 and 96 hours after transfection. Although we consistently found reductions in ATP12A mRNA by quantitative real-time polymerase chain reaction at both post-transfection time points (with greater reductions at 48 than at 96 hours), Western blots showed no decrease in ATP12A protein levels. We also attempted to knockout ATP12A by CRISPR using 2 different guide RNAs (CRISPR guide RNA3 and guide RNA4; GenScript, Piscataway, NJ). Once again, we found substantial reductions in ATP12A mRNA, but no decrease in ATP12A protein in our cell lines or primary cells. Because we were unable to knock down or knockout ATP12A protein, we instead used a PPI and a P-CAB to inhibit ngH+,K+ATPase pharmacologically.
In addition to its well-known effect of inhibiting gastric H+,K+ATPase (ATP4A), the PPI omeprazole also inhibits ngH+,K+ATPase.17 The P-CAB SCH 28080 is a highly selective, competitive inhibitor of the high-affinity K+ site of both gastric and nongastric H+,K+-ATPases.27,28 To inhibit the ngH+,K+ATPase in EoE cells, we used omeprazole (10 and 50 μM)4,16 or SCH 28080 (1–50 μM)17,28 (both from Sigma, St Louis, MO). Omeprazole was acid-activated in medium with pH 5.5 for 30 minutes, followed by a 1:1 dilution in neutral pH growth medium; non–omeprazole-treated cells in the same experiment had a similar 1:1 dilution of pH 5.5 and neutral pH growth media to control for this condition. Ranitidine, an inverse agonist of the histamine H2 receptor (HRH2), blocks gastric H+,K+-ATPase activity indirectly (ie, without binding to the enzyme).29 We studied the effects of ranitidine (1–50 μM30,31) and famotidine (0.1–50 μM32; both HRH2 blockers from Sigma) in EoE cells. In preliminary studies, EoE cells exhibited no signs of stress when exposed to these inhibitors at our selected doses and durations. EoE cell lines and primary EoE cells were treated with omeprazole, SCH 28080, ranitidine, or famotidine for 2 hours before addition of cytokines, and inhibitors remained in the media throughout the period of cytokine stimulation.
Inhibitors of Calcium Signaling
To inhibit calcium signaling, cells were treated with ethylene glycol-bis(β-aminoethyl)-N,N,N′,N′-tetraacetoxymethyl ester (EGTA-AM), plasma membrane-permeable calcium chelator (1–50 μM),33 2-aminoethoxydiphenyl borate (2-APB), inhibitor of inositol 1,4,5,-trisphosphate (IP3)–mediated calcium release from the endoplasmic reticulum (at 50 and 100 μM),34 or verapamil and diltiazem, L-type calcium channel inhibitors in concentrations ranging from 1 to 200 μM35–37 (all inhibitors from Sigma). Cells were treated with inhibitors for 2 hours before addition of cytokines, and inhibitors remained in the media throughout the period of cytokine stimulation. In preliminary studies, EoE cells exposed to 2-APB 100 μM exhibited morphologic signs of cell stress; therefore, we used 2-APB only at 50 μM concentration. Cells exhibited no signs of stress when exposed to the other inhibitors at our selected doses and durations.
Intracellular Calcium Measurements
Cells were seeded (104 cells per well) into a black 96-well plate with a clear bottom (BD Falcon) and incubated at 37°C in 5% CO2 overnight.38 The next day, cells were stimulated with 5 ng/mL of IL-4 (BioVendor) for 2 hours in culture medium and then loaded with 5 μM Fluo-4 AM for 25 minutes in the dark. After washing, cells were treated with 50 μM verapamil (AdipoGen, San Diego, CA), 1 μM nifedipine, or 10 μM diltiazem (both from Enzo, San Diego, CA), for 10 minutes in the dark.35,36 Inhibitors remained in bath solutions during intracellular calcium measurements. Fluo-4 fluorescence was recorded as an indicator of intracellular calcium transients using a Hamamatsu digital camera C11440 complemented with a Leica DMi8 inverted microscope (Leica, Germany) with 20× objective (NA 0.75). Time-lapse images of live cells were captured every 5 seconds for 5 minutes and analyzed by Image J (version 1.52u; National Institutes of Health). All analyses were performed in 2 independent experiments with no fewer than 5 views containing no fewer than 5 cells for each time period.
Additional details are provided in the Supplementary Materials and Methods and Supplementary Tables 1 and 2.
Results
Esophageal Squamous Epithelial Cells From Patients With Eosinophilic Esophagitis Express Messenger RNA and Protein for ATP12A (Nongastric H+,K+ATPase ) and the Histamine H2 Receptor
Th2 cytokines have been shown to stimulate secretion of eotaxin-3 in upper airway cells by activating ATP12A, the ngH+,K+ATPase that is susceptible to inhibition by PPIs and PCABs.17 It is well established that HRH2 blockers interfere with pathways that activate ATP4A (the gastric H+,K+ATPase), but it is not clear whether these agents interfere with Th2 cytokine–stimulated pathways that activate ATP12A.29 Using quantitative real-time polymerase chain reaction and Western blotting, we found that both EoE1-T and EoE2-T express mRNA and protein for ATP12A and for HRH2 (Figure 1A and C); mRNA expression of ATP4A (the gastric H+,K+ATPase) was minimal (approximately 100- to 1000-fold lower than HRH2 and ATP12A, respectively) in both EoE cell lines (Figure 1A). RNA-sequencing datasets available from 23 patients with EoE and 21 control subjects23–25 demonstrated significantly higher ATP12A expression in patients with EoE compared with control subjects (Figure 1B). After 48 hours of IL-4 stimulation, there were no substantial differences in ATP12A protein levels in EoE cell lines or in primary EoE cells compared with those levels in unstimulated cells (Figure 1D).
Figure 1.
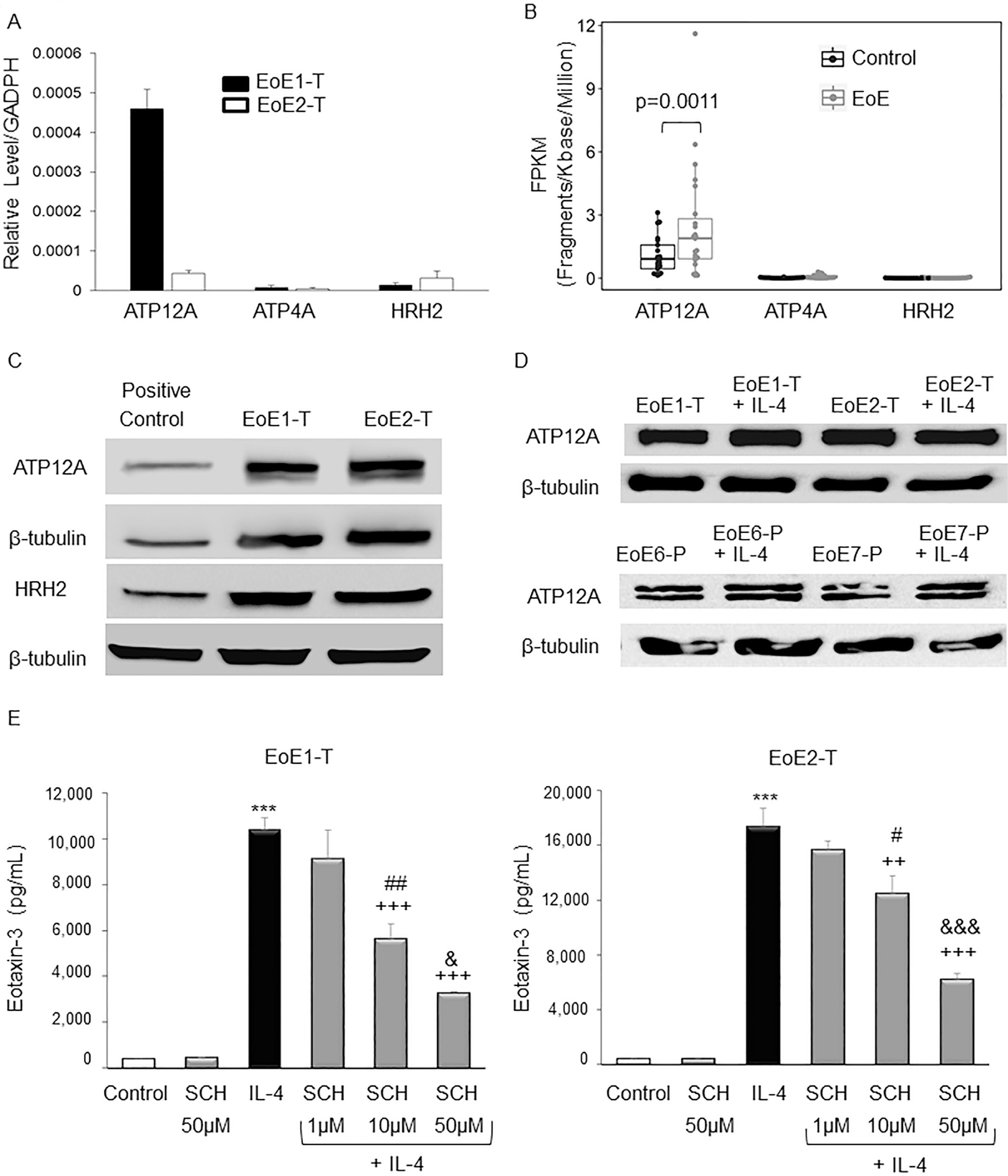
EoE cells express ATP12A (nongastric proton pump) and HRH2. (A) Representative quantitative real-time polymerase chain reactions demonstrating relative levels of ATP12A, ATP4A (gastric proton pump), and HRH2 mRNAs. Bar graphs depict the mean ± SEM from 1 experiment performed in triplicate. (B) RNA-sequencing gene expression represented by FPKM values of ATP12A, ATP4A, and HRH2 mRNAs in 23 patients with EoE and 21 controls from GSE58640, GSE41687, and GSE113341 datasets. Compared with controls, ATP12A expression was significantly increased in patients with EoE; P values from multiple datasets were calculated using DESeq2 and combined using weighted Stouffer’s method. (C) Representative Western blots demonstrating levels of ATP12A and HRH2 proteins EoE cells; 293T cells served as a positive control. (D) Representative Western blots demonstrating levels of ATP12A proteins in EoE cell lines and in primary EoE cells (EoE6-P and EoE7-P) with and without IL-4 stimulation. (E) SCH 28080 decreases IL-4–stimulated eotaxin-3 secretion by EoE cells in a dose-dependent manner. Representative eotaxin-3 enzyme-linked immunosorbent assays 48 hours after IL-4 treatment in cells with and without SCH 28080 (SCH). Bar graphs depict the mean ± SEM from 3 separate samples. ***P ≤ .001 compared with control; ++P ≤ .01, +++P ≤ .001 compared with IL-4; #P ≤ .05, ##P ≤ .01 compared with IL-4 + SCH 1 μM; &P ≤ .05, &&&P ≤ .001 compared with IL-4 + SCH 10 μM; 1-way analysis of variance.
A Potassium-Competitive Acid Blockers (SCH 28080) and a Histamine H2 Receptor Blocker (Ranitidine) Decrease Interleukin-4–Stimulated Eotaxin-3 Protein Secretion in Eosinophilic Esophagitis Cells
In earlier studies, we showed that omeprazole significantly decreased the secretion of eotaxin-3 stimulated by Th2 cytokines in EoE1-T and EoE2-T cells.4 As in those earlier studies, we found that IL-4 significantly increased secretion of eotaxin-3 by EoE1-T and EoE2-T cells, and that omeprazole blocked eotaxin-3 secretion induced by IL-4 (Figures 1E and 2). SCH 28080 (50 μM) and ranitidine alone (50 μM) had no effect on the low baseline levels of eotaxin-3 secretion (Figure 1E and 2). Treatment with 10 and 50 μM doses of SCH 28080 or a 50-μM dose of ranitidine significantly decreased IL-4–stimulated eotaxin-3 protein secretion in both cell lines (Figure 1E and 2). Omeprazole and SCH 28080 caused greater suppression of IL-4–stimulated eotaxin-3 secretion than equivalent doses of ranitidine (Figure 2). To determine whether the inhibitory effect of ranitidine was unique to this drug, we studied effects of famotidine on IL-4–stimulated eotaxin-3 secretion (Supplementary Figure 1). Famotidine alone (50 μM) had no effect on the low baseline levels of eotaxin-3 secretion in EoE2-T cells. Treatment with the 0.1 μM dose of famotidine caused a minimal, but statistically significant, reduction in IL-4–stimulated eotaxin-3 secretion; eotaxin-3 secretion was not decreased by famotidine at dose ≥1 μM. These findings demonstrate that inhibition of ngH+,K+ATPase with omeprazole or SCH 28080 decreases IL-4–stimulated eotaxin-3 secretion in esophageal squamous cells from patients with EoE. Ranitidine also decreases IL-4–stimulated eotaxin-3 secretion, but to a lesser degree than omeprazole or SCH 28080; this reduction appears to be unique to ranitidine, and not a class effect of HRH2 blockers.
Figure 2.
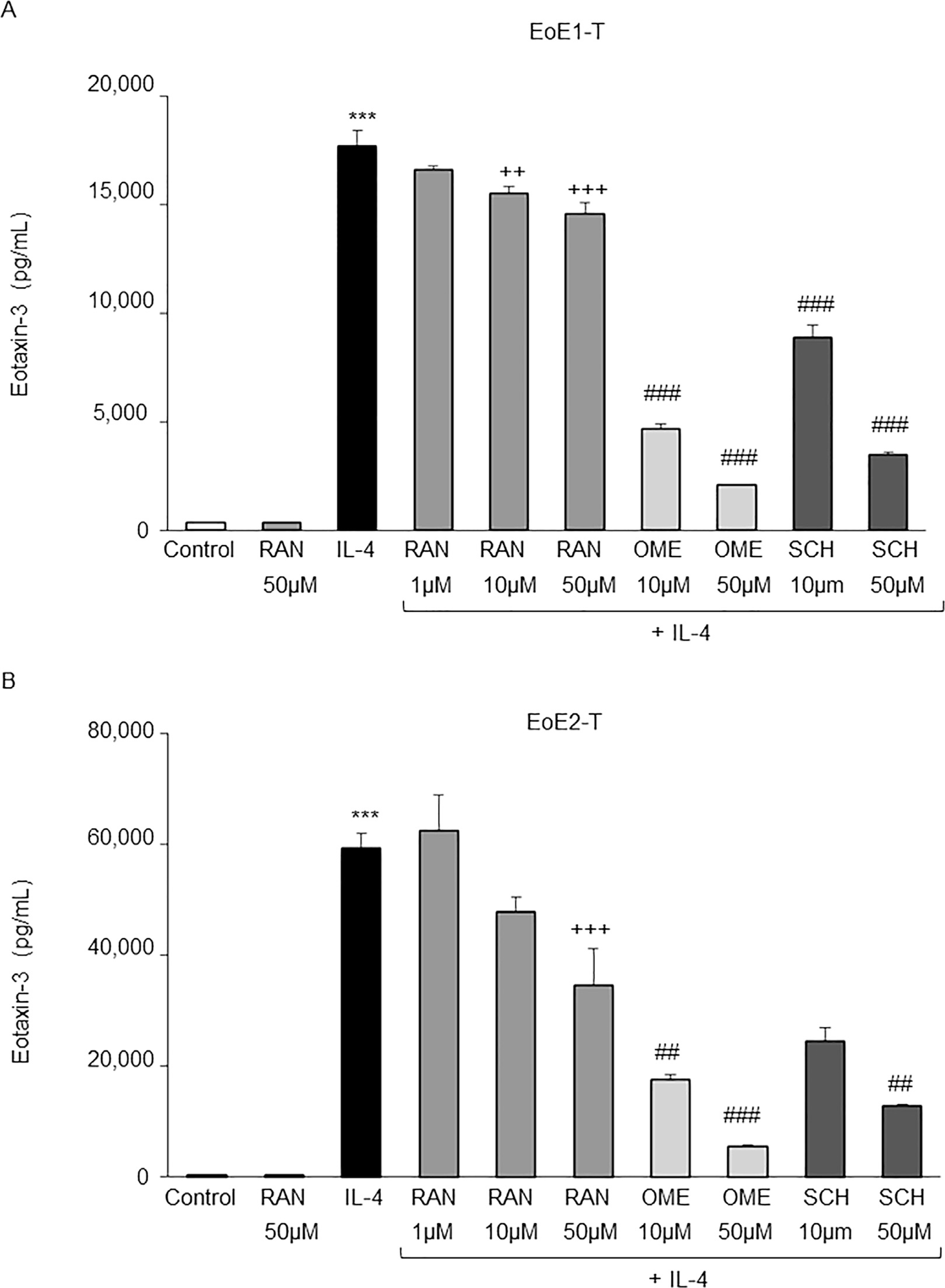
Omeprazole (OME), SCH 28080 (SCH), and ranitidine (RAN) decrease eotaxin-3 secretion stimulated by IL-4 in EoE cells. Bar graphs show results of representative enzyme-linked immunosorbent assays for eotaxin-3 at 48 hours after IL-4 treatment with and without RAN, OME, or SCH in (A) EoE1-T and (B) EoE2-T cells. Bar graphs depict the mean ± SEM from 3 separate samples. ***P ≤ .001 compared with control; ++P ≤ .01; +++P ≤ .001 compared with IL-4; ##P ≤ .01, ###P ≤ .001 compared with IL-4 + RAN 50 μM; 1-way analysis of variance.
Baseline and Interleukin-4–Stimulated Eotaxin-3 Protein Secretion Are Dependent on Both Nongastric H+,K+ATPase Activity and Intracellular Calcium
After demonstrating that ngH+,K+ATPase is involved in the secretion of eotaxin-3 stimulated by IL-4 in EoE cells, we next sought to determine the underlying molecular pathways. We explored a role for calcium because increases in intracellular calcium are known to stimulate gH+,K+ATPase activity in parietal cells,19,20 and IL-4 has been shown to increase intracellular calcium in human tonsillar B lymphocytes.18 EoE2-T and NES-G4T cells were stimulated with IL-4 for 2 hours, loaded with Fluo-4 for 25 minutes, and fluorescence signal intensity was recorded every 5 seconds for 5 minutes (Figure 3A). Few calcium oscillations were observed in untreated EoE2-T or NES-G4T cells, but IL-4 treatment caused a marked increase in those oscillations in EoE2-T cells and an increase, albeit to a lesser extent, in those oscillations in NES-G4T cells (Figure 3B and Supplementary Videos 1, 2, 4, and 5). The percentage of EoE2-T and NES-G4T cells with calcium oscillations increased significantly with IL-4 treatment (Figure 3C); these findings were confirmed in EoE1-T cells (Supplementary Figure 2 and Supplementary Videos 6 and 7).
Figure 3.
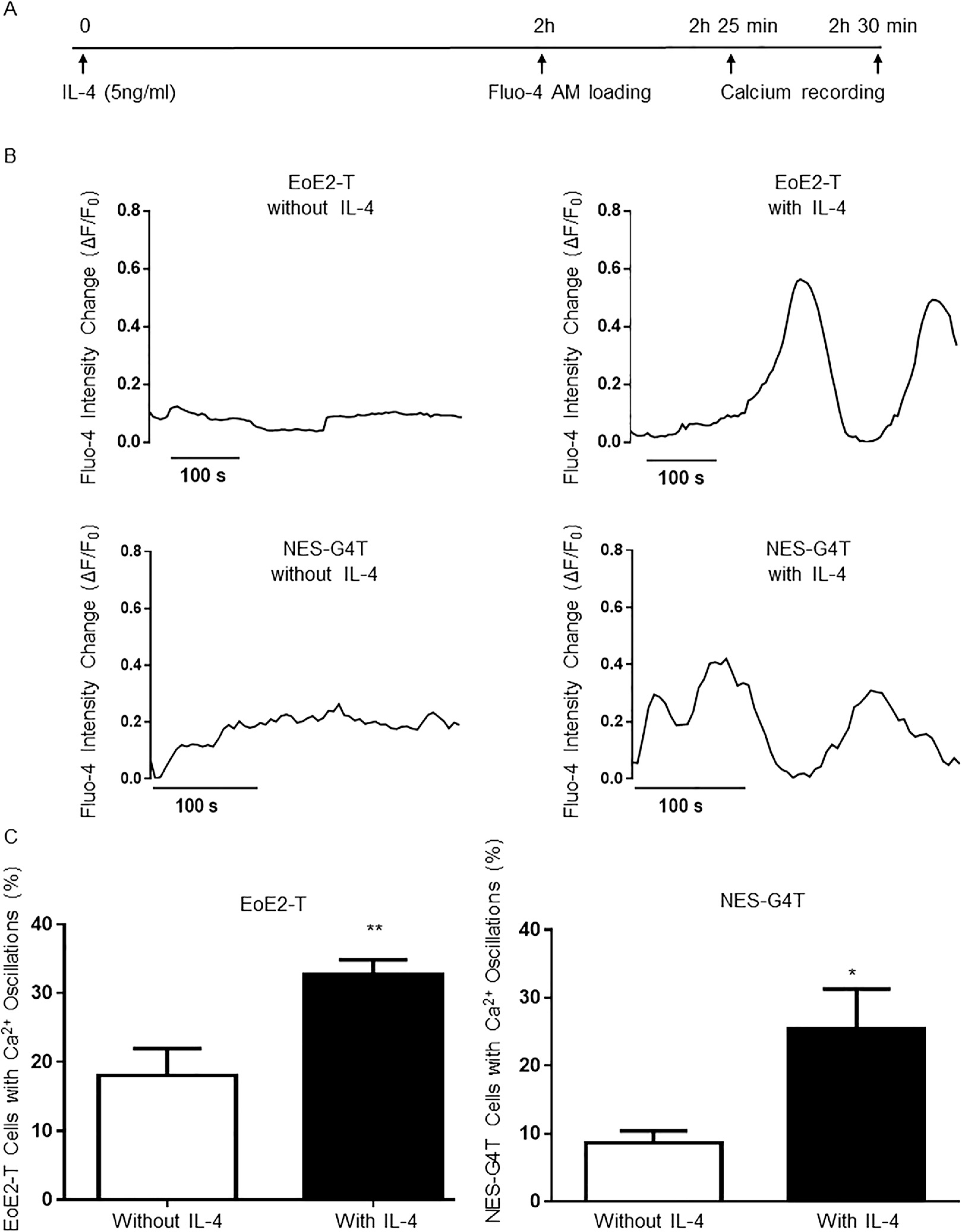
IL-4 induces intracellular calcium oscillations in EoE2-T and NES-G4T cells. (A) Diagram of the experimental protocol. (B) Representative curves of intracellular calcium oscillations (measured as changes in Fluo-4 fluorescence [ΔF/F0]) in single EoE2-T and NES-G4T cells without and with IL-4 treatment. Time scale bar: 100 seconds. (C) Percentage of EoE2-T and NES-G4T cells with calcium (Ca2+) oscillations without and with IL-4 treatment. Bar graphs depict the mean ± SEM from 2 independent experiments, with at least 5 views. **P < .01; *P < .05 compared with non–IL-4 treated cells; unpaired Student t test.
EoE cells were next treated with EGTA-AM, a plasma membrane-permeable calcium chelator, to assess whether calcium chelation affected IL-4–stimulated eotaxin-3 secretion. EGTA-AM alone at 50 μM had no effect on baseline eotaxin-3 secretion (Figure 4A). Treatment with 10 and 50 μM doses of EGTA-AM significantly decreased IL-4–stimulated eotaxin-3 protein secretion in a dose-dependent fashion (Figure 4A). EoE2-T cells were next treated with the combination of 50 μM SCH 28080 and 50 μM EGTA-AM (Figure 4B). As in our previous experiments, both SCH 28080 and EGTA-AM alone significantly decreased IL-4–stimulated eotaxin-3 secretion. However, the combination of SCH 28080 and EGTA-AM resulted in significantly greater suppression of IL-4–stimulated eotaxin-3 secretion than either agent alone (Figure 4B). The combination of SCH 28080 and EGTA-AM also significantly decreased baseline eotaxin-3 secretion, although neither agent alone affected baseline eotaxin-3 secretion. Thus, baseline eotaxin-3 secretion and increases in eotaxin-3 protein secretion stimulated by IL-4 in EoE esophageal squamous cells are dependent on both ngH+,K+ATPase activity and intracellular calcium.
Figure 4.
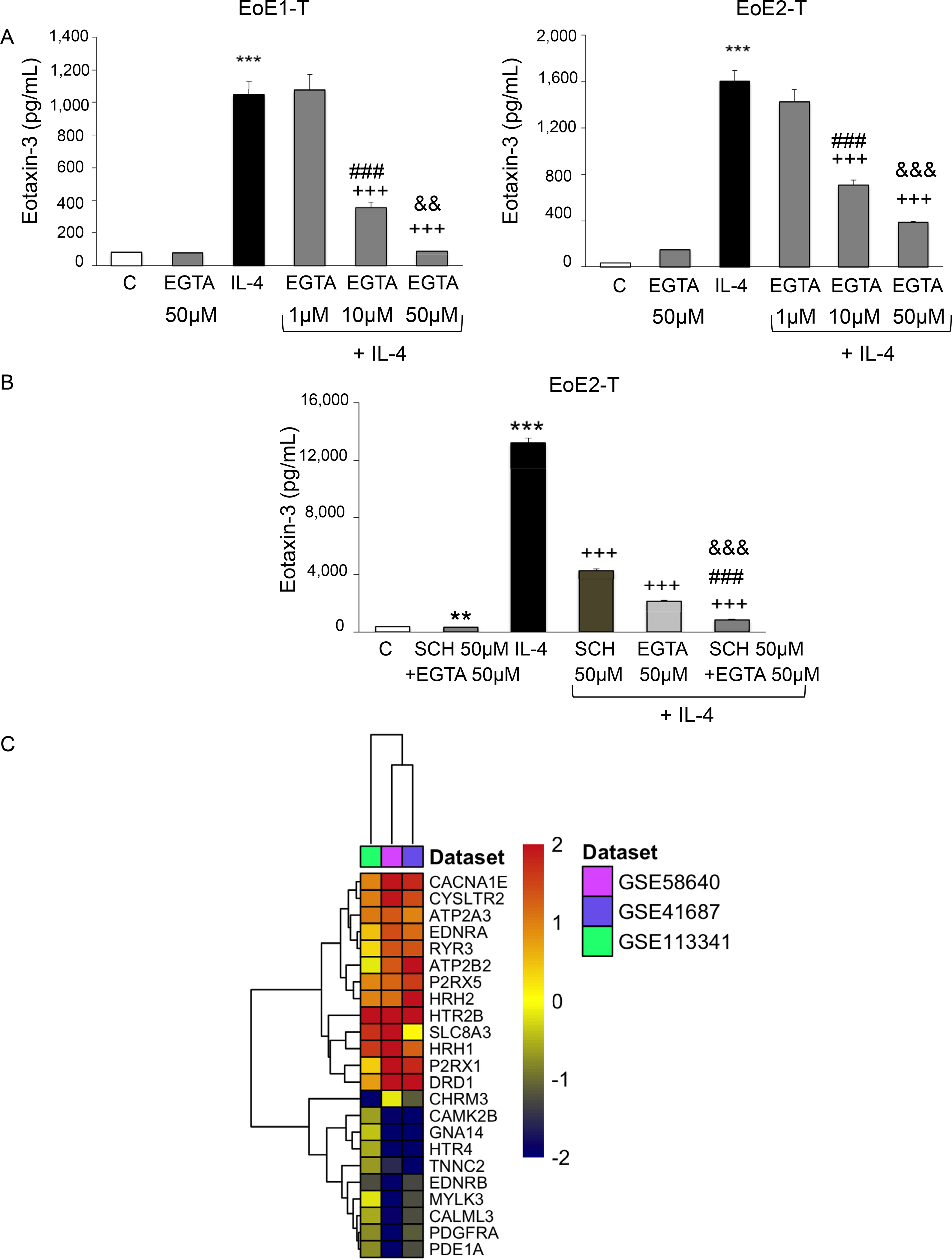
Intracellular calcium chelation by EGTA decreases IL-4–stimulated eotaxin-3 secretion in a dose-dependent fashion in EoE cells. (A) Representative eotaxin-3 enzyme-linked immunosorbent assays (ELISA) 48 or 24 hours after IL-4 treatment in EoE-1T or EoE2-T cells, respectively, with and without EGTA. Bar graphs depict the mean ± SEM from 3 separate samples. ***P ≤ .001 compared with control; +++P ≤ .001 compared with IL-4; ###P ≤ .001 compared with IL-4 + EGTA 1 μM; &&P ≤ .01, &&&P ≤ .001 compared with IL-4 + EGTA 10 μM; 1-way analysis of variance. (B) The combination of SCH 28080 (SCH) with EGTA results in significantly greater suppression of IL-4–stimulated eotaxin-3 secretion in EoE2-T cells than either agent alone. Representative eotaxin-3 ELISA assays 24 hours after IL-4 treatment in cells with and without SCH 28080 and/or EGTA. Bar graphs depict the mean ± SEM from 3 separate samples. **P ≤ .01, ***P ≤ .001 compared with control; +++P ≤ .001 compared with IL-4; ###P ≤ .001 compared with IL-4 + SCH 50 μM; &&&P ≤ .001 compared with IL-4 + EGTA 50 μM; 1-way analysis of variance. (C) Heatmap analysis of 23 differentially expressed genes in the Kyoto Encyclopedia of Genes and Genomes calcium signaling pathway; data are from 3 RNA sequencing databases (GSE58640, GSE41687, and GSE113341) comparing mucosal biopsies of 23 patients with EoE with those of 21 controls. Red or blue color corresponds to up- or down-regulation of gene expression in patients with EoE compared with controls, respectively.
Esophageal Mucosa of Patients With Eosinophilic Esophagitis Exhibits Enrichment for Calcium Signaling Pathway Genes
Esophageal mucosa of patients with EoE expresses ngH+,K+ATPase (Figure 1B). To determine whether calcium signaling might be enhanced in the esophagus of patients with EoE, we used 3 RNA-sequencing datasets of esophageal mucosal biopsies obtained from 23 patients with EoE and 21 healthy control subjects23–25 and performed a meta-analysis on gene expression comparing patients with EoE with control subjects. Among those genes that were differentially expressed, there was significant enrichment for those in the Kyoto Encyclopedia of Genes and Genomes39,40 calcium signaling pathway in the esophageal mucosa of patients with EoE compared with control subjects (P = .0042; Figure 4C).
Interleukin-4 Increases Intracellular Calcium Signaling by Both Releasing Calcium From the Endoplasmic Reticulum and Increasing Extracellular Calcium Entry Through L-Type Calcium Channels in Eosinophilic Esophagitis Cells
Intracellular calcium levels can increase either when calcium sequestered in the endoplasmic reticulum is released into the cytoplasm or when extracellular calcium enters cells through various calcium channels. To explore the former mechanism, cells were treated with IL-4 with and without 2-APB (an inhibitor of IP3-mediated calcium release from the endoplasmic reticulum) at a concentration of 50 μM (a dose known to inhibit IP3-mediated calcium release from endoplasmic reticulum in other cell types).34 2-APB significantly decreased IL-4–stimulated eotaxin-3 secretion in both EoE1-T and EoE2-T cells (Figure 5A). This finding shows that calcium released from the endoplasmic reticulum of EoE cells contributes to IL-4–induced eotaxin-3 secretion.
Figure 5.
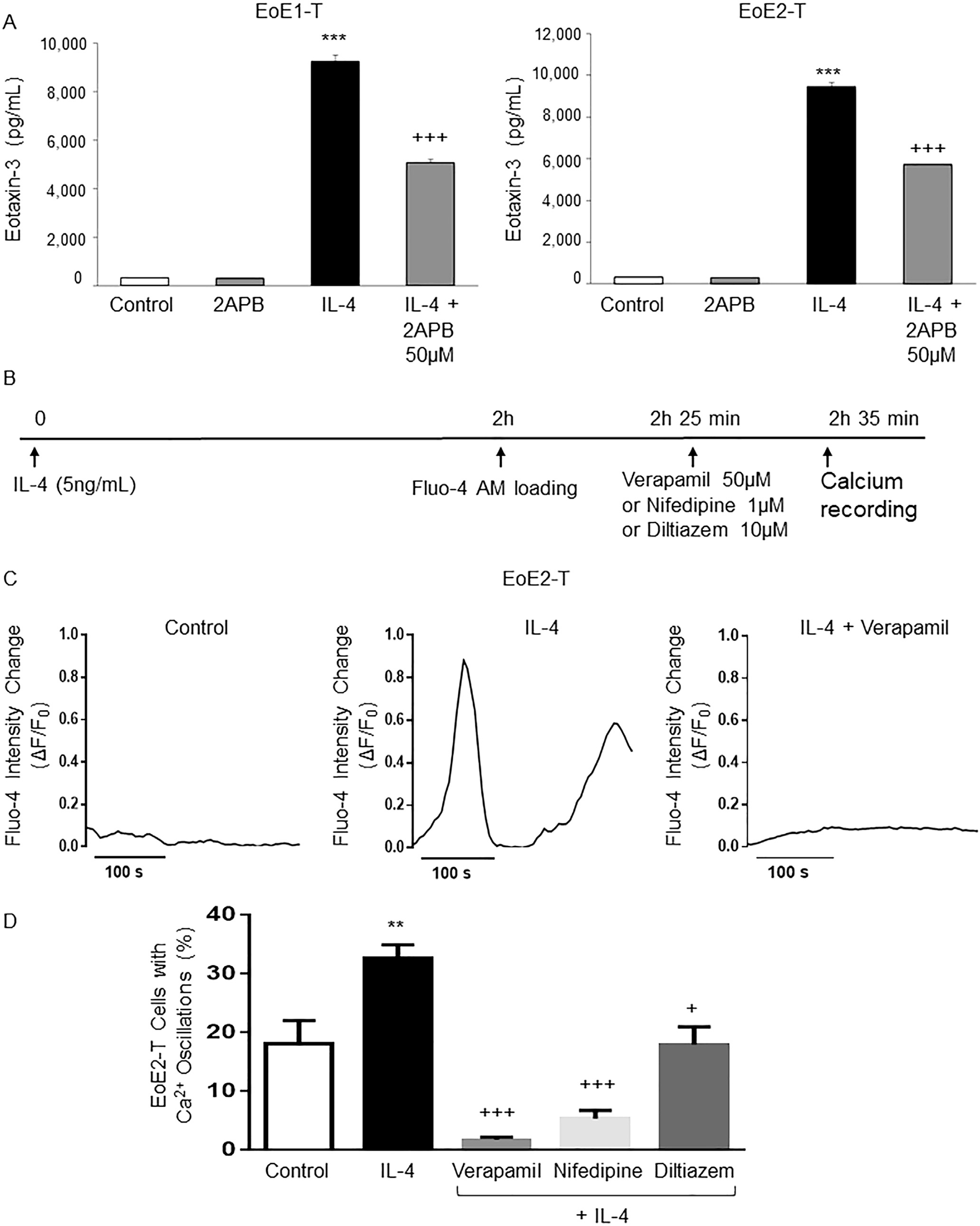
IL-4 stimulates intracellular calcium release from the endoplasmic reticulum, and stimulates extracellular calcium entry through L-type calcium channels in EoE2-T cells. (A) Representative eotaxin-3 enzyme-linked immunosorbent assay 24 hours after IL-4 treatment in cells with or without 2APB. Bar graphs depict the mean ± SEM from 3 separate samples. ***P ≤ .001 compared with control; +++P ≤ .001 compared with IL-4; 1-way analysis of variance. (B) Diagram of the experimental protocol. (C) Representative curves of intracellular calcium oscillations (measured as changes in Fluo-4 fluorescence [ΔF/F0]) in single EoE2-T cells after IL-4 treatment without and with verapamil. (D) Percentage of EoE2-T cells with calcium (Ca2+) oscillations after IL-4 treatment without and with verapamil, nifedipine, or diltiazem. Bar graphs depict the mean ± SEM from at least 5 separate views. **P ≤ .01 compared with control; +P ≤ .05, +++P ≤ .001 compared with IL-4; 1-way analysis of variance.
In gastric parietal cells, calcium entry through L-type calcium channels has been shown to activate gH+,K+ATPase.41 To demonstrate effects on calcium entry via L-type channels, EoE cells were stimulated with IL-4 for 2 hours, loaded with Fluo-4 for 25 minutes, followed by addition of the L-type calcium channel antagonists verapamil (50 μM), nifedipine (1 μM), or diltiazem (10 μM), and measurements of fluorescence intensity signals were performed 10 minutes later (Figure 5B).35,36 Verapamil suppressed IL-4–stimulated intracellular calcium oscillations in EoE2-T (Figure 5C and Supplementary Videos 1–3). All 3 L-type calcium channel antagonists significantly reduced intracellular calcium oscillations in IL-4–stimulated EoE2-T (Figure 5D). These finding suggest that IL-4 stimulates intracellular calcium release from the endoplasmic reticulum and extracellular calcium entry through L-type calcium channels in EoE cells.
Verapamil and Diltiazem Block T Helper 2 Cytokine–Induced Increases in Eotaxin-3 Protein Secretion in Eosinophilic Esophagitis Cell Lines and in Primary Eosinophilic Esophagitis Cells
Next, we determined whether inhibition of extracellular calcium entry by verapamil and diltiazem would suppress IL-4–stimulated eotaxin-3 secretion. Treatment with verapamil at concentrations ≥10 μM or diltiazem at concentrations ≥100 μM significantly decreased IL-4–stimulated eotaxin-3 protein secretion in both EoE cell lines in a dose-dependent fashion (Figure 6A and B); verapamil alone at 50 μM or diltiazem alone at 100 or 200 μM had no effect on baseline eotaxin-3 secretion (Figure 6A and B). Using EoE2-T cells, we confirmed that eotaxin-3 secretion stimulated by IL-13 also was blocked by verapamil 50 μM and diltiazem at concentrations ≥100 μM (Supplementary Figure 3). Using primary esophageal squamous cells from 8 patients with EoE, we confirmed the effects of verapamil on IL-4–stimulated eotaxin-3 secretion. As in the EoE cell lines, treatment with 50 μM verapamil significantly decreased IL-4–stimulated eotaxin-3 protein secretion in primary EoE cells (Figure 6C).
Figure 6.
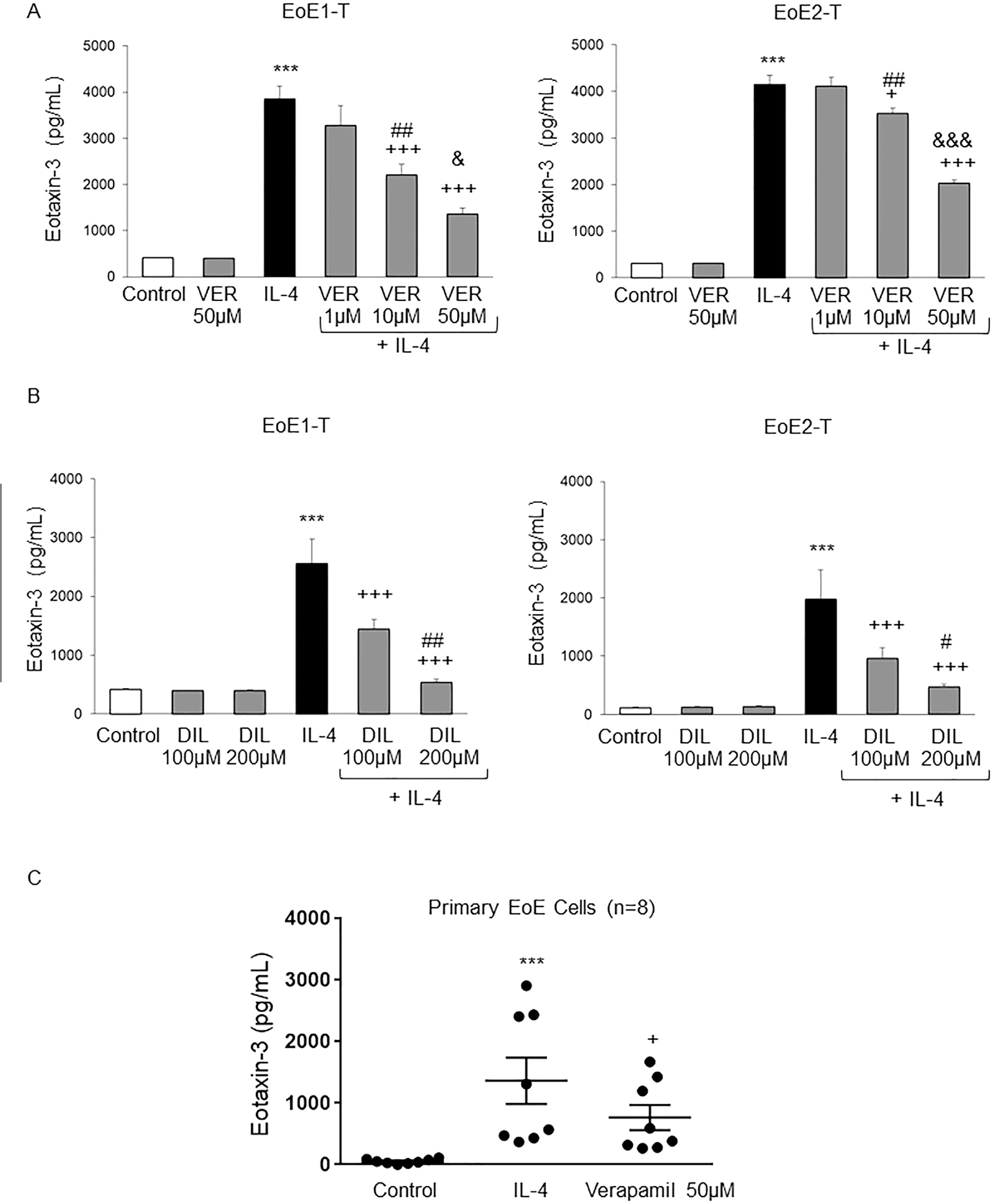
Verapamil (VER) and diltiazem (DIL) block Th2 cytokine–stimulated increases in eotaxin-3 secretion in EoE cells. (A) Representative eotaxin-3 enzyme-linked immunosorbent assays (ELISAs) at 48 or 24 hours in EoE1-T and EoE2-T, respectively after IL-4 treatment with and without VER. Bar graphs depict the mean ± SEM from 3 separate samples. ***P ≤ .001 compared with control; +P ≤ .05, +++P ≤ .001 compared with IL-4; ##P ≤ .01 compared with IL-4 + VER 1 μM; &P ≤ .05, &&&P ≤ .001 compared with IL-4 + VER 10 μM; 1-way analysis of variance (ANOVA). (B) Representative eotaxin-3 ELISAs at 48 or 24 hours in EoE1-T and EoE2-T, respectively after IL-4 treatment with and without DIL. Bar graphs depict the mean ± SEM from 3 separate samples. ***P ≤ .001 compared with control; +++P ≤ .001 compared with IL-4; #P ≤ .05, ##P ≤ .01 compared with IL4 + DIL 100 μM; 1-way ANOVA. (C) VER blocks IL-4–stimulated increases in eotaxin-3 secretion in primary esophageal squamous cell cultures from patients with EoE. Eotaxin-3 ELISA assays at 24 hours in primary cell cultures after IL-4 treatment with and without VER 50 μM. Solid lines depict the mean ± SEM from all 8 male patients with EoE. ***P ≤ .001 compared with control; +P ≤ .05 compared with IL-4; repeated-measures ANOVA.
Omeprazole and Verapamil in Combination Cause Greater Suppression of Interleukin-4–Induced Eotaxin-3 Protein Secretion Than Either Agent Alone in Eosinophilic Esophagitis Cells
After noting that the combination of SCH 28080 and EGTA-AM caused greater suppression of IL-4–induced eotaxin-3 secretion than either agent alone (Figure 4B), we explored the effects of combining omeprazole with verapamil on Th2 cytokine–stimulated eotaxin-3 secretion in EoE2-T, primary EoE2-air–liquid interface (ALI), and primary EoE10-ALI cells. Omeprazole 50 μM and verapamil 50 μM, alone or in combination, had no effect on baseline eotaxin-3 secretion in any of our EoE cells (Figure 7A–C and Supplementary Figure 4). As in previous experiments, both omeprazole and verapamil alone significantly decreased IL-4–stimulated eotaxin-3 secretion, but the combination of the 2 agents caused significantly greater suppression than either agent alone in EoE2-T, primary EoE2-ALI, and primary EoE10-ALI (Figure 7A–C and Supplementary Figure 4). A schematic model summarizing mechanisms elucidated by our study is provided in Figure 7D.
Figure 7.
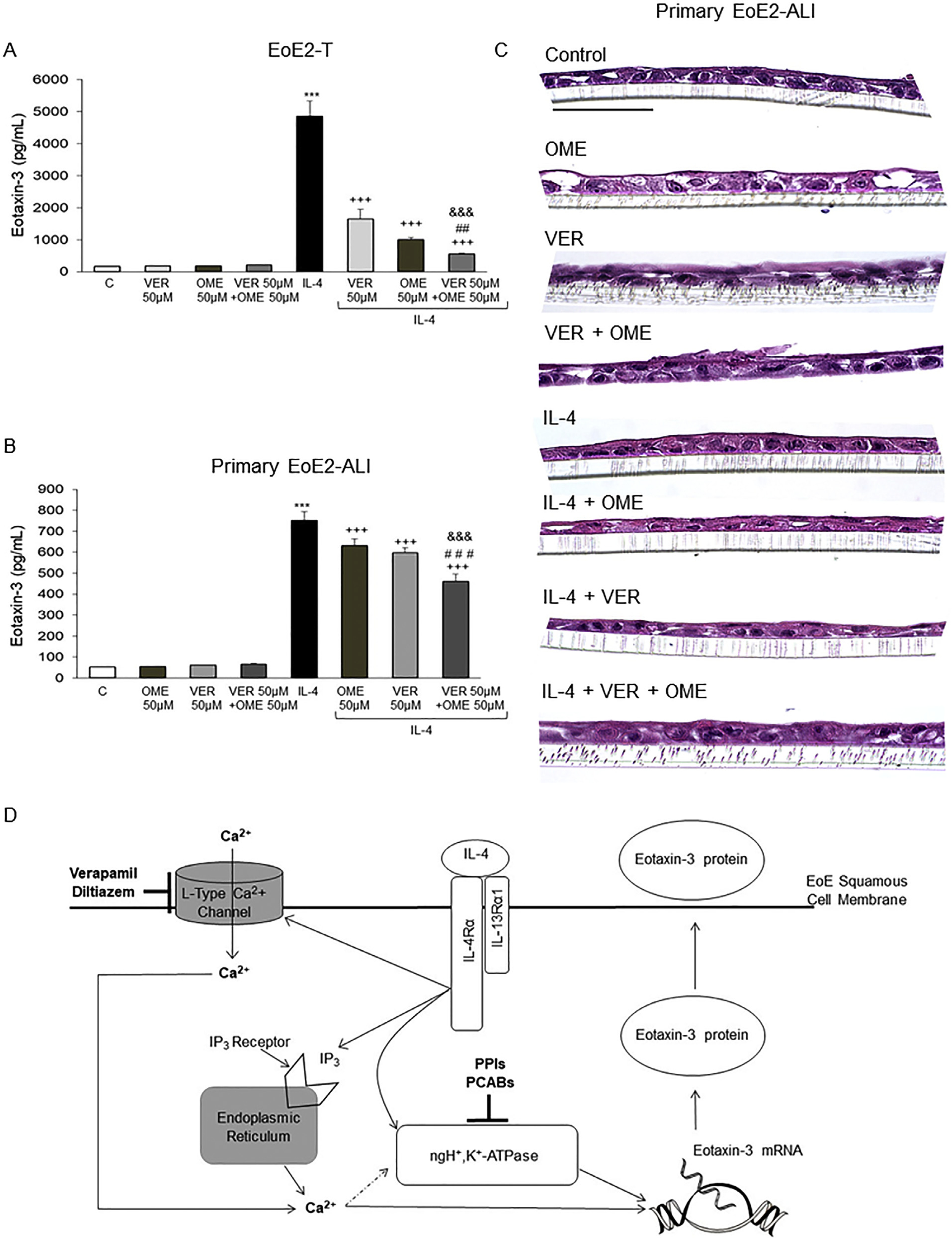
Omeprazole (OME) in combination with verapamil (VER) has a significantly greater suppressive effect on IL-4–stimulated eotaxin-3 secretion than either agent alone in EoE2-T and primary EoE2-ALI cells. Representative eotaxin-3 enzyme-linked immunosorbent assays 24 hours after IL-4 treatment in (A) EoE-2T and (B) 48 hours after IL-4 treatment in primary EoE2-ALI cells with and without OME, VER, or the combination of both. Bar graphs depict the mean ± SEM from 3 separate samples for EoE2-T and from ALI cultures performed at least in triplicate from 1 male EoE patient. ***P ≤ .001 compared with control; +++P ≤ .001 compared with IL-4; ##P ≤ .01, ###P ≤ .001 compared with IL-4 + VER 50 μM; &&&P ≤ .001 compared with IL-4 + OME 50 μM; 1-way analysis of variance. (C) Representative H&E staining of primary EoE2-ALI cultures 48 hours after IL-4 treatment with and without OME, VER, or the combination of both. Scale bar: 75 μm. (D) Diagram demonstrating mechanisms whereby VER and diltiazem (L-type calcium channel blockers) in combination with PPIs and PCABs (inhibitors of the nongastric proton pump) block Th2 cytokine–stimulated eotaxin-3 secretion by esophageal squamous cells. IL-4 and IL-13 bind to the type II IL-4 receptor containing the IL4Rα subunit. This binding activates IL4Rα-mediated calcium signaling by causing IP3-mediated calcium release from the endoplasmic reticulum and by increasing entry of extracellular calcium through L-type calcium channels on the plasma membrane. The increase in intracellular calcium leads to an increase in eotaxin-3 production and secretion, at least in part via effects on activity of the nongastric proton pump. Verapamil and diltiazem block extracellular calcium entry through L-type calcium channels, thereby reducing intracellular calcium signaling, whereas the PPIs and the P-CABs inhibit nongastric proton pump activity, thus reducing Th2 cytokine–stimulated eotaxin-3 secretion. Agents that inhibit intracellular calcium signaling downstream of IL4Rα activation alone or in combination with inhibitors of the nongastric proton pump might have a therapeutic role in EoE (dashed arrow; potential mechanism based on literature review41).
Discussion
We have shown that esophageal squamous cells from patients with EoE express mRNA and protein for the ngH+,K+ATPase and for the HRH2. We have demonstrated that blocking ngH+,K+ATPase in those cells with the PPI omeprazole or with the P-CAB SCH 28080 significantly decreased eotaxin-3 secretion stimulated by Th2 cytokines, as did ranitidine to a lesser degree. Our experiments indicate that baseline and IL-4–stimulated eotaxin-3 protein secretion in EoE squamous cells are dependent on both ngH+,K+ATPase activity and increases in intracellular calcium. RNA-sequencing datasets confirmed significantly higher expression of ngH+,K+ATPase and enrichment of genes involved in calcium signaling in the esophageal mucosa of patients with EoE compared with healthy controls. IL-4 increases intracellular calcium both by stimulating IP3-mediated calcium release from the endoplasmic reticulum and by increasing extracellular calcium entry through L-type calcium channels in EoE cells. We have found that L-type calcium channel inhibitors significantly blocked Th2 cytokine–mediated increases in eotaxin-3 secretion in EoE squamous cell lines and in primary esophageal squamous cells from patients with EoE. Finally, we have shown that omeprazole and verapamil in combination caused greater suppression of IL-4–stimulated eotaxin-3 secretion in EoE cells than either agent alone.
PPIs are well known to block gastric acid secretion by binding covalently to sulfhydryl groups of the gastric H+,K+ATPase in parietal cells, an enzyme encoded by the ATP4A gene.42 However, a number of cells including kidney, prostate, lung, and nasal epithelial cells express ngH+,K+ATPase encoded by the ATP12A gene, and ngH+,K+ATPase also is susceptible to inhibition by PPIs.17,43–45 Min et al17 showed that IL-13 stimulated eotaxin-3 secretion in human bronchial and sinonasal epithelial cells by activating their ngH+,K+ATPase, and that such eotaxin-3 secretion could be blocked by PPIs and PCABs. Our studies now demonstrate that esophageal squamous cells from patients with EoE express mRNA and protein for ATP12A, but exhibit only minimal expression of mRNA for ATP4A.
Despite our attempts to genetically silence ATP12A gene expression, we could not reduce ATP12A protein expression in our EoE cell lines or primary cells. Without a genetic approach to inhibit gene expression, the major issue that arises is loss of selectively in targeting the protein of interest. To address this issue, we used the P-CAB SCH 28080, which is a highly selective, competitive inhibitor of the high-affinity K+ site of both gastric and nongastric H+,K+-ATPases; SCH 28080 has been used extensively to investigate functions of H+,K+ATPase in vitro.27,28,46 Like PPIs, P-CABs also inhibit gastric and nongastric H+,K+ATPases, but through a different biochemical mechanism than PPIs.27,28 P-CABs disable H+,K+ATPase by binding ionically, rather than covalently, to the enzyme’s high-affinity K+ site, thereby blocking the K+ binding required to pump hydrogen ions (protons) out of the cell in exchange for K+ ions.46 Although commercial development of SCH 28080 was halted due to its hepatotoxicity, the P-CABs revaprazan and vonoprazan are used clinically in Asian countries,47 and vonoprazan has been shown to improve symptoms and esophageal eosinophilia in Japanese patients with EoE who were refractory to PPIs.48 We have found that SCH 28080 significantly suppresses IL-4–stimulated eotaxin-3 secretion in EoE esophageal squamous cells, supporting the notion that inhibition of the esophageal ngH+,K+ATPase underlies the beneficial effects of P-CABs and PPIs for patients with EoE.
PPIs are metabolized primarily by CYP2C19, a cytochrome P450 enzyme with multiple polymorphisms that affect its metabolic activity. A recent study on the long-term efficacy of PPIs for patients with EoE found that a CYP2C19 rapid metabolizer genotype was an independent predictor for loss of PPI responsiveness.49 The fact that P-CABs are not metabolized primarily by CYP2C19 is a potential advantage for their long-term use in EoE, but P-CABs are not yet available for use in Western countries.
In addition to the eosinophil-predominant inflammation that characterizes the EoE esophagus, esophageal biopsies from patients with EoE often exhibit increased numbers of intact and degranulated mast cells, as well as up-regulation of mast cell-associated genes.50,51 Mast cell granules are especially rich in histamine, which is known to activate gastric H+,K+ATPase in the stomach indirectly when histamine binds the parietal cell H2 receptor to activate adenylate cyclase and increase cAMP production.52,53 We found that EoE esophageal squamous cells express HRH2, in agreement with an earlier study by Merves et al.30 We also found that the HRH2 blocker ranitidine significantly decreased IL-4–stimulated eotaxin-3 secretion in EoE cells, but to a much lesser extent than omeprazole or SCH 28080, while famotidine had little or no effect on eotaxin-3 secretion. Although there is a popular notion that HRH2 blockers are not effective treatments for EoE,30 there are virtually no published data to substantiate that notion. Our findings provide little support for the use of HRH2 blockers in EoE.
Because the Th2 cytokines that stimulated ngH+,K+ATPase-mediated eotaxin-3 expression in our EoE cells are known to signal predominantly through IL-4Rα,15 we explored molecules downstream of IL-4Rα that might link these effects. We investigated a role for calcium because increases in intracellular calcium are known to stimulate gH+,K+ATPase activity in parietal cells,19,20 and IL-4 has been shown to increase intracellular calcium in human tonsillar B lymphocytes.18 Using Fluo-4–based calcium imaging and an intracellular calcium chelator, we demonstrated that IL-4 increased calcium signaling, which resulted in increased eotaxin-3 secretion by esophageal squamous cells from patients with EoE. In support of these data in EoE cells in vitro, we found that mucosal biopsies from patients with EoE were enriched for genes involved in calcium signaling compared with healthy control subjects. Our experiments using 2-APB and L-type calcium channel antagonists demonstrated that IL-4 treatment increases intracellular calcium oscillations in EoE cells by stimulating IP3-mediated release of calcium sequestered in the endoplasmic reticulum, and by increasing extracellular calcium entry through L-type calcium channels.
The L-type calcium channel blockers verapamil and diltiazem are well-tolerated, time-tested medications that have been used widely to treat hypertension and angina pectoris.54 Drug repurposing, the establishment of new indications for drugs already in clinical use, is a practice that has gained new momentum in the last decade.55 Because those drugs are already available and their safety has already been established, the time and expense required for their application in clinical trials for new conditions is considerably less than that for new drugs. A limitation of our study is that verapamil and diltiazem were used in concentrations higher than those achieved in plasma with conventional dosing for the treatment of hypertension or cardiac disorders. Nevertheless, our findings that verapamil and diltiazem block Th2 cytokine–stimulated eotaxin-3 production in EoE esophageal squamous cells, and that the combination of omeprazole and verapamil was more effective in this regard than either agent alone, suggest that perhaps L-type calcium channel antagonists might be repurposed to treat EoE.
In conclusion, we have shown that esophageal squamous cells from patients with EoE express a nongastric proton pump and the HRH2, and that their secretion of eotaxin-3 stimulated by a Th2 cytokine can be blocked by a PPI, a P-CAB and, to a lesser degree, by ranitidine but not famotidine. IL-4 causes an increase in intracellular calcium oscillations by stimulating calcium release from the endoplasmic reticulum and its entry via L-type calcium channels. In support of our in vitro findings, mucosal biopsies from patients with EoE demonstrate significantly higher gene expression of ngH+,K+ATPase and enrichment of calcium signaling genes compared with those from healthy controls. Moreover, this Th2 cytokine–induced increase in intracellular calcium contributes to eotaxin-3 secretion, which can be blocked by L-type calcium channel inhibitors. Finally, we have demonstrated that the combination of a PPI and an L-type calcium channel inhibitor provides greater suppression of Th2 cytokine–stimulated eotaxin-3 secretion than either agent alone. These findings have revealed novel potential EoE therapeutic targets downstream of IL4Rα, and suggest a potential novel role for L-type calcium channel blockers that might be repurposed for the treatment of EoE.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
In esophageal squamous cells from patients with eosinophilic esophagitis (EoE), Th2 cytokines that signal through IL-4 receptor alpha (IL4Rα) stimulate secretion of the eosinophil chemoattractant eotaxin-3. In upper airway cells, a recent study found that Th2 cytokines stimulate eotaxin-3 secretion by activating a non-gastric proton pump (ngH+,K+ATPase) susceptible to inhibition by PPIs and potassium-competitive acid blockers (P-CABs).
NEW FINDINGS
Esophageal squamous cells from EoE patients express ngH+,K+ATPase, and their eotaxin-3 secretion stimulated by IL-4 can be blocked by a PPI, a P-CAB and, to a lesser extemt, by ranitidine. IL-4-stimulated eotaxin-3 secretion in EoE cells is mediated in part by increased calcium entry via L-type calcium channels, and this eotaxin-3 secretion can be blocked by L-type calcium channel inhibitors verapamil and diltiazem.
LIMITATIONS
This study was performed using human EoE cells in culture, and using verapamil and diltiazem in relatively high concentrations. Further clinical studies in EoE patients are warranted.
IMPACT
Our findings suggest that inhibition of esophageal ngH+,K+ATPase might underlie the beneficial effects of PPIs and P-CABs for patients with EoE, and suggest a potential role for L-type calcium channel inhibitors in the treatment of EoE.
Abbreviations used in this paper:
- ALI
air–liquid interface
- 2APB
2-aminoethoxydiphenyl borate
- EGTA-AM
ethylene glycol-bis(β-aminoethyl)-N,N,N′,N′-tetraacetoxymethyl ester
- EoE
eosinophilic esophagitis
- HRH2
histamine H2 receptor
- IL
interleukin
- IL4Rα
IL-4 receptor-α
- IP3
inositol 1,4,5,-trisphosphate
- mRNA
messenger RNA
- ngH+
K+ATPase, nongastric proton pump
- P-CAB
potassium competitive acid blocker
- PPI
proton pump inhibitor
- Th2
T helper 2
Footnotes
Conflicts of interest
The authors disclose no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2021.02.016.
References
- 1.Dellon ES, Hirano I. Epidemiology and natural history of eosinophilic esophagitis. Gastroenterology 2018; 154:319–332.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn JLM, Shoda T, Caldwell JM, et al. Esophageal type 2 cytokine expression heterogeneity in eosinophilic esophagitis in a multisite cohort. J Allergy Clin Immunol 2020;145:1629–1640.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulkerson PC, Schollaert KL, Bouffi C, et al. IL-5 triggers a cooperative cytokine network that promotes eosinophil precursor maturation. J Immunol 2014;193:4043–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng E, Zhang X, Huo X, et al. Omeprazole blocks eotaxin-3 expression by oesophageal squamous cells from patients with eosinophilic oesophagitis and GORD. Gut 2013;62:824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirano I, Sharaf R, Stollman N, et al. Spotlight: treatment of eosinophilic esophagitis (EoE). Gastroenterology 2020;158:1788. [DOI] [PubMed] [Google Scholar]
- 6.Lucendo AJ, Arias A, Molina-Infante J. Efficacy of proton pump inhibitor drugs for inducing clinical and histologic remission in patients with symptomatic esophageal eosinophilia: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2016;14:13–22.e1. [DOI] [PubMed] [Google Scholar]
- 7.Hirano I, Furuta GT. Approaches and challenges to management of pediatric and adult patients with eosinophilic esophagitis. Gastroenterology 2020;158:840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laserna-Mendieta EJ, Casabona S, Savarino E, et al. Efficacy of therapy for eosinophilic esophagitis in real-world practice. Clin Gastroenterol Hepatol 2020; 18:2903–2911.e4. [DOI] [PubMed] [Google Scholar]
- 9.Arias A, Gonzalez-Cervera J, Tenias JM, et al. Efficacy of dietary interventions for inducing histologic remission in patients with eosinophilic esophagitis: a systematic review and meta-analysis. Gastroenterology 2014; 146:1639–1648. [DOI] [PubMed] [Google Scholar]
- 10.Molina-Infante J, Arias A, Alcedo J, et al. Step-up empiric elimination diet for pediatric and adult eosinophilic esophagitis: the 2–4-6 study. J Allergy Clin Immunol 2018;141:1365–1372. [DOI] [PubMed] [Google Scholar]
- 11.Straumann A, Conus S, Grzonka P, et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut 2010;59:21–30. [DOI] [PubMed] [Google Scholar]
- 12.Hirano I, Collins MH, Assouline-Dayan Y, et al. RPC4046, a monoclonal antibody against IL13, reduces histologic and endoscopic activity in patients with eosinophilic esophagitis. Gastroenterology 2019;156:592–603.e10. [DOI] [PubMed] [Google Scholar]
- 13.Greuter T, Hirano I, Dellon ES. Emerging therapies for eosinophilic esophagitis. J Allergy Clin Immunol 2020; 145:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirano I, Dellon ES, Hamilton JD, et al. Efficacy of dupilumab in a phase 2 randomized trial of adults with active eosinophilic esophagitis. Gastroenterology 2020; 158:111–122.e10. [DOI] [PubMed] [Google Scholar]
- 15.Wills-Karp M, Finkelman FD. Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci Signal 2008;1:pe55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Cheng E, Huo X, et al. Omeprazole blocks STAT6 binding to the eotaxin-3 promoter in eosinophilic esophagitis cells. PLoS One 2012;7:e50037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Min JY, Ocampo CJ, Stevens WW, et al. Proton pump inhibitors decrease eotaxin-3/CCL26 expression in patients with chronic rhinosinusitis with nasal polyps: possible role of the nongastric H,K-ATPase. J Allergy Clin Immunol 2017;139:130–141.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finney M, Guy GR, Michell RH, et al. Interleukin 4 activates human B lymphocytes via transient inositol lipid hydrolysis and delayed cyclic adenosine monophosphate generation. Eur J Immunol 1990;20:151–156. [DOI] [PubMed] [Google Scholar]
- 19.Nandi J, King RL, Kaplan DS, et al. Mechanisms of gastric proton pump inhibition by calcium channel antagonists. J Pharmacol Exp Ther 1990;252:1102–1107. [PubMed] [Google Scholar]
- 20.Berglindh T, Sachs G, Takeguchi N. Ca2+-dependent secretagogue stimulation in isolated rabbit gastric glands. Am J Physiol 1980;239:G90–G94. [DOI] [PubMed] [Google Scholar]
- 21.Zhang HY, Zhang X, Chen X, et al. Differences in activity and phosphorylation of MAPK enzymes in esophageal squamous cells of GERD patients with and without Barrett’s esophagus. Am J Physiol Gastrointest Liver Physiol 2008;295:G470–G478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morales CP, Gandia KG, Ramirez RD, et al. Characterisation of telomerase immortalised normal human oesophageal squamous cells. Gut 2003;52:327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler JC, Vanoni S, Zeng C, et al. 17β-Estradiol protects the esophageal epithelium from IL-13-induced barrier dysfunction and remodeling. J Allergy Clin Immunol 2019;143:2131–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kottyan LC, Davis BP, Sherrill JD, et al. Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat Genet 2014;46:895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherrill JD, Kiran KC, Blanchard C, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014;15:361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiran KC, Rothenberg ME, Sherrill JD. In vitro model for studying esophageal epithelial differentiation and allergic inflammatory responses identifies keratin involvement in eosinophilic esophagitis. PLoS One 2015;10:e0127755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaisser F, Beggah AT. The nongastric H+-K+-ATPases: molecular and functional properties. Am J Physiol 1999; 276:F812–F824. [DOI] [PubMed] [Google Scholar]
- 28.Beil W, Hackbarth I, Sewing KF. Mechanism of gastric antisecretory effect of SCH 28080. Br J Pharmacol 1986; 88:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monczor F Mechanisms of inverse agonism at histamine H(2) receptors—potential benefits and concerns. Inflammopharmacology 2006;14:89–96. [DOI] [PubMed] [Google Scholar]
- 30.Merves J, Chandramouleeswaran PM, Benitez AJ, et al. Altered esophageal histamine receptor expression in eosinophilic esophagitis (EoE): implications on disease pathogenesis. PLoS One 2015;10:e0114831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okajima K, Harada N, Uchiba M. Ranitidine reduces ischemia/reperfusion-induced liver injury in rats by inhibiting neutrophil activation. J Pharmacol Exp Ther 2002;301:1157–1165. [DOI] [PubMed] [Google Scholar]
- 32.Schepp W, Miederer SE, Ruoff HJ. Effects of hormones (calcitonin, GIP) and pharmacological antagonists (ranitidine and famotidine) on isolated rat parietal cells. Regul Pept 1985;12:297–308. [DOI] [PubMed] [Google Scholar]
- 33.Abramov AY, Duchen MR. Measurements of threshold of mitochondrial permeability transition pore opening in intact and permeabilized cells by flash photolysis of caged calcium. Methods Mol Biol 2011;793:299–309. [DOI] [PubMed] [Google Scholar]
- 34.Maruyama T, Kanaji T, Nakade S, et al. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem 1997;122:498–505. [DOI] [PubMed] [Google Scholar]
- 35.Dierkes PW, Wende V, Hochstrate P, et al. L-type Ca2+ channel antagonists block voltage-dependent Ca2+ channels in identified leech neurons. Brain Res 2004; 1013:159–167. [DOI] [PubMed] [Google Scholar]
- 36.Wyse-Jackson AC, Roche SL, Ruiz-Lopez AM, et al. Progesterone analogue protects stressed photoreceptors via bFGF-mediated calcium influx. Eur J Neurosci 2016;44:3067–3079. [DOI] [PubMed] [Google Scholar]
- 37.Nomura Y, Asano M, Ito K, et al. Superficial sarcoplasmic reticulum calcium buffering of resting, voltage-dependent Ca++ influx in rat femoral arterial smooth muscle. J Pharmacol Exp Ther 1996;279:830–837. [PubMed] [Google Scholar]
- 38.Cui C, Chang Y, Zhang X, et al. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett 2018;432:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of Genes And Genomes. Nucleic Acids Res 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanehisa M Toward understanding the origin and evolution of cellular organisms. Protein Sci 2019;28:1947–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Remy C, Kirchhoff P, Hafner P, et al. Stimulatory pathways of the calcium-sensing receptor on acid secretion in freshly isolated human gastric glands. Cell Physiol Biochem 2007;19:33–42. [DOI] [PubMed] [Google Scholar]
- 42.Shin JM, Munson K, Vagin O, et al. The gastric HK-ATPase: structure, function, and inhibition. Pflugers Arch 2009;457:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coakley RD, Grubb BR, Paradiso AM, et al. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci U S A 2003; 100:16083–16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altman KW, Kinoshita Y, Tan M, et al. Western blot confirmation of the H+/K+-ATPase proton pump in the human larynx and submandibular gland. Otolaryngol Head Neck Surg 2011;145:783–788. [DOI] [PubMed] [Google Scholar]
- 45.Pestov NB, Korneenko TV, Shakhparonov MI, et al. Loss of acidification of anterior prostate fluids in Atp12a-null mutant mice indicates that nongastric H-K-ATPase functions as proton pump in vivo. Am J Physiol Cell Physiol 2006;291:C366–C374. [DOI] [PubMed] [Google Scholar]
- 46.Andersson K, Carlsson E. Potassium-competitive acid blockade: a new therapeutic strategy in acid-related diseases. Pharmacol Ther 2005;108:294–307. [DOI] [PubMed] [Google Scholar]
- 47.Oshima T, Miwa H. Potent potassium-competitive acid blockers: a new era for the treatment of acid-related diseases. J Neurogastroenterol Motil 2018; 24:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishimura N, Kinoshita Y. Eosinophilic esophagitis in Japan: focus on response to acid suppressive therapy. J Gastroenterol Hepatol 2018;33:1016–1022. [DOI] [PubMed] [Google Scholar]
- 49.Molina-Infante J, Rodriguez-Sanchez J, Martinek J, et al. Long-term loss of response in proton pump inhibitorresponsive esophageal eosinophilia is uncommon and influenced by CYP2C19 genotype and rhinoconjunctivitis. Am J Gastroenterol 2015;110:1567–1575. [DOI] [PubMed] [Google Scholar]
- 50.Dellon ES, Chen X, Miller CR, et al. Tryptase staining of mast cells may differentiate eosinophilic esophagitis from gastroesophageal reflux disease. Am J Gastroenterol 2011;106:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abonia JP, Blanchard C, Butz BB, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol 2010;126:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siracusa MC, Kim BS, Spergel JM, et al. Basophils and allergic inflammation. J Allergy Clin Immunol 2013; 132:789–801; quiz 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amin K The role of mast cells in allergic inflammation. Respir Med 2012;106:9–14. [DOI] [PubMed] [Google Scholar]
- 54.Calcium Channel Blockers. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases, 2012. [Google Scholar]
- 55.Talevi A, Bellera CL. Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opin Drug Discov 2020; 15:397–401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.