

Abstract
Background
Cancer stem cells (CSC) define a population of rare malignant cells endowed with ‘stemness’ properties, such as self-renewing, multipotency and tumorigenicity. They are responsible for tumor initiation and progression, and could be associated with resistance to immunotherapies by negatively regulating antitumor immune response and acquiring molecular features enabling escape from CD8 T-cell immunity. However, the immunological hallmarks of human lung CSC and their potential interactions with resident memory T (TRM) cells within the tumor microenvironment have not been investigated.
Methods
We generated a non-small cell lung cancer model, including CSC line and clones, and autologous CD8+CD103+ TRM and CD8+CD103− non-TRM clones, to dissect out immune properties of CSC and their susceptibility to specific T-cell-mediated cytotoxic activity.
Results
Unlike their parental tumor cells, lung CSC are characterized by the initiation of an epithelial-to-mesenchymal transition program defined by upregulation of the SNAIL1 transcription factor and downregulation of phosphorylated-GSK-3β and cell surface E-cadherin. Acquisition of a CSC profile results in partial resistance to TRM-cell-mediated cytotoxicity, which correlates with decreased surface expression of the CD103 ligand E-cadherin and human leukocyte antigen-A2-neoepitope complexes. On the other hand, CSC gained expression of intercellular adhesion molecule (ICAM)-1 and thereby sensitivity to leukocyte function-associated antigen (LFA)-1-dependent non-TRM-cell-mediated killing. Cytotoxicity is inhibited by anti-ICAM-1 and anti-major histocompatibility complex class I neutralizing antibodies further emphasizing the role of LFA-1/ICAM-1 interaction in T-cell receptor-dependent lytic function.
Conclusion
Our data support the rational design of immunotherapeutic strategies targeting CSC to optimize their responsiveness to local CD8+CD103+ TRM cells for more efficient anticancer treatments.
Keywords: CD8-positive T-lymphocytes; immune evation; immunity; lymphocytes, tumor-infiltrating; tumor microenvironment
Key messages.
Cancer Stem cells (CSC) often develop resistance to immunotherapies by negatively regulating antitumor T-cell response.
They are a putative target of CD8+CD103+ resident memory T (TRM) cells within the tumor microenvironment; however, the interaction of CD8+ TRM with CSC, and the consequences of CSC feature acquisition on their elimination by TRM cells have not been investigated.
Our data demonstrate that the initiation of an epithelial-to-mesenchymal transition program, mainly via a decreased surface expression of the CD103 integrin ligand E-cadherin, permits to CSC to resist to specific CD8+CD103+ TRM cell-mediated cytotoxicity and potentially also to cancer immunotherapy.
Introduction
Cancer stem cells (CSC) or cancer initiating cells (CIC) are rare neoplastic cells capable of generating phenotypically and functionally diverse cancer cell subtypes. Accumulating evidence indicates that CSC undergo self-renewal and thereby drive growth and progression of tumors.1 These cancer cells are involved in multiple malignancy processes, including neovascularization, resistance to treatments, escape from the antitumor immunity and dissemination.2 CSC were initially described in hematological malignancies as displaying a CD34+CD48− profile.3 They were then identified in several human solid tumors and were defined by expression of cell surface biomarkers, such as CD166 for lung and ovarian cancers,4 CD44 for gastric, prostate and colon cancers,5 and CD133 for brain, colon and pancreatic cancers.6 The C-X-C chemokine receptor type 4 (CXCR4) chemokine receptor was also characterized as a CSC marker in multiple cancer types, including non-small cell lung cancer (NSCLC), and its interaction with the ligand CXCL12 has been described to play a role in CSC maintenance and tumor development.7 In breast cancer, CSC were found to display a CD44+/CD24−/low profile,8 and in ovarian cancer, a OCT4+MYC+NANOG+ profile.9 Increased expression of epithelial-to-mesenchymal transition (EMT) transcription factors, such as OCT4 and SOX2, contributes to the stemness properties of CSC; and OCT4-reactive T cells targeting CSC have been isolated from patients with cancer.9 Several other CSC markers, such as epithelial cell adhesion molecule (EpCAM) and aldehyde dehydrogenase (ALDH) have been reported, and high ALDH activity has been used to target CSC in various solid tumors.10
One of the major properties of CSC is resistance to conventional chemotherapy and radiotherapy, and promotion of tumor relapse and metastasis. As a result, several approaches have been developed to eliminate CSC, such as by targeting CSC niches. Accordingly, strategies inhibiting angiogenesis via neutralization of vascular endothelial growth factor (VEGF) or its receptor (VEGFR) have been elaborated.11 As EMT is a key characteristic of CSC, inhibitors of EMT transcription factors, such as TWIST, SNAIL and SLUG, or of GSK-3β have been proposed to block their stemness properties.12 Immunotherapeutic approaches have also been designed to specifically destroy CSC by developing dendritic cell (DC)-based cancer vaccines targeting ALDHhigh cells.13 In this context, CSC-DC vaccines induced protective immune responses toward CSC resulting in inhibition of tumor growth and metastasis. Moreover, a DNA vaccine targeting a specific tumor antigen has been reported to attenuate CSC behavior and metastasis, and to increase breast cancer sensitivity to chemotherapy.14 Additional therapeutic strategies targeting CSC have been designed, such as with the use of natural killer (NK) cells.15 However, escape from NK cell-mediated cytotoxicity leading to lung metastasis was observed. T-cell-based immunotherapies have also been initiated in immunodeficient mouse models bearing human tumor xenografts. In this setting, adoptive transfer of CD8+ T cells primed in vitro with CSC eliminated CD133+ brain tumor stem-like initiating cells in an antigen-specific manner.16 In another study, adoptive transfer of ALDH1A1-specific cytotoxic T lymphocytes (CTL) resulted in destruction of ALDHbright CSC and inhibition of cancer progression and metastases.10 Unfortunately, such strategies also faced tumor evasion challenges through alterations in antigen presentation to specific CD8+ T lymphocytes.17
CSC are a putative target of CD8+CD103+ resident memory T (TRM) cells within the tumor microenvironment (TME) where they may coexist. TRM frequently infiltrate human solid tumors and play an important role in antitumor CD8 T-cell immunity18 and response to immunotherapies.19 However, the interaction of CD8+ TRM cells with CSC and the consequences of CSC feature acquisition on their recognition and elimination by specific intratumoral CTL have not been investigated. In this report, we show that lung CSC display partial resistance to mutant α-actinin-4-specific CD8+CD103+ TRM-cell-mediated cytotoxicity associated with decreased surface expression of the CD103 ligand E-cadherin and neoantigen-presenting molecules, human leukocyte antigen (HLA)-A2. In contrast, induction of intercellular adhesion molecule (ICAM)-1, the ligand of leukocyte function-associated antigen (LFA)-1, results in CSC responsiveness to CD8+CD103− non-TRM-cell killing. Thus, targeting EMT to sensitize CSC to CD8+CD103+ TRM within the TME may improve current cancer immunotherapies.
Materials and methods
Tumor cell lines, CSC and autologous T-cell clones
The IGR-Heu cell line was derived from a tumor sample of patient Heu suffering from a NSCLC.20 Tumor cells were cultured in adherent conditions in conventional Dulbecco’s Modified Eagle Medium (DMEM)/F-12 medium (Life Technologies, cat# 31331093) supplemented with 10% fetal calf serum (FCS; Life Technologies, cat# A3840402). To obtain sphere cultures, IGR-Heu cells were cultured in ultra-low attachment Corning cell culture flasks (cat#3814 or cat#3815) or 6-well plates (cat# 3471) at 105 cells/mL in serum-free DMEM/F12 medium, supplemented with a commercial hormone mix including N2 supplement (100X) (at 1%, Gibco Life Technologies, cat# 17502001), human epithelial growth factor (EGF) at 20 ng/mL (Miltenyi, hEGF, premium grade, cat# 130-093-825) and human fibroblast growth factor-2 (FGF-2) at 20 ng/mL (Miltenyi, hFGF-2, premium grade, cat# 130-093-564,14000000000), which was termed CSC medium. Floating sphere cultures were expanded in the same medium, and the obtained cell line, named Heu-CSC, was tested for ALDH1 and Hoechst expression. ALDH-positive/Hoechst-negative cells were sorted using a FACS Vantage (BD Biosciences) and seeded at 1 cell/well in ultra-low attachment round-bottom microplates in CSC medium. Two clones were selected for ALDH expression and Hoechst exclusion, and maintained in culture as above. Twice a week, cells were gently dissociated with Trypsin (Gibco) and 0.5 mM EDTA at 37°C for 5-to-15 min and plated in fresh CSC medium in ultra-low attachment flasks.
The Heu171 T-cell clone (TRM), specific to the HLA-A2-restricted mutant α-actinin-4 epitope, was isolated from tumor-infiltrating lymphocytes (TIL) as previously reported.20 The H32-22 T-cell clone (non-TRM) was isolated from autologous peripheral blood lymphocytes (PBL) after stimulation with IGR-Heu cells and sorting with mutant α-actinin-4 peptide-HLA-A2 tetramers.21
Hoechst/ALDH test and soft agar experiments
Tumor cells and CSC (1×106 cells) were resuspended in 1 mL of RPMI 1640 medium (Invitrogen) supplemented with 4% FCS and 10 mM HEPES at 37°C. Next, 5 μg of Hoechst 33342 (Invitrogen, H3570) was added to each sample, and cells were incubated at 37°C for 1 hour. After centrifugation, cells were incubated with Aldefluor substrate for 25 min at 37°C and 5% CO2. Cells were then suspended in 200 μL of phosphate-buffered saline (PBS) (1X) plus 2% FCS and 2 μL of propidium iodide before flow cytometry analysis (LSRII, Becton Dickinson).
For soft agar experiments, single-cell suspensions were prepared by treating monolayer-cultured IGR-Heu cells and spheroid Heu-CSC, CSC-1 and CSC-2 with trypsin (Gibco, cat #25300054) for 5 min with gentle shaking, and rinsed with PBS. Cells were then suspended in DMEM/F-12 medium supplemented with 10% FCS, 2 mM L-glutamine, 1 mM sodium pyruvate, antibiotics (50 U/mL penicillin and 50 µg/mL streptomycin) and 1% ultroser G (Pall) at a density of 2×105/ mL. Single cells were plated on agar-coated 6-well plates. A layer (2 mL/well) of soft agar at 0.5% was added to plates. After solidification, a second layer (3 mL/well) of soft agar at 0.25% containing 1000 cells was added to each well. Plates were then incubated at 37°C, 5% CO2 for 15–22 days for sphere formation, and fresh medium was renewed every 3–4 days, before labeling with crystal violet at 0.05% for 3 hours. Cell colonies were counted at 10× microscope.
Mice and in vivo experiments
Nude mice were bred and maintained at the animal facility of Gustave Roussy and treated in accordance with institutional animal guidelines.
For each experiment, groups of 5–10 female mice, 8–12 weeks of age, received 2×105 IGR-Heu cells or CSC subcutaneously in the right flank. Tumor volume was measured twice a week using a caliper and estimated using the following formula: length 1/2×width×thickness (mm3). For survival analyses, mice with a tumor volume greater than the limit of 200 mm3 were euthanized and counted as dead.
Antibodies and flow cytometry analysis
For tumor cell and CSC phenotyping, anti-CD24 (SN3, Invitrogen MHCD2404, 1/100), anti-CD34 (AC136, Miltenyi 130-113-182, 1/100), anti-CD44 (DB105, Miltenyi 130-113-896, 1/100), anti-CD166 (3A6, BD 562131, 1/100), anti-CXCR4 (12G5, Biolegend 306510, 1/100), anti-VEGF (23410, R&D IC2931A, 1/100), anti-epidermal growth factor receptor (EGFR) (AY13, Biolegend 352913, 1/100), anti-major histocompatibility complex class I (MHC-I) (W6/32, Biolegend 311429, 1/100), anti-HLA-A2 (BB7.2, BD 558570, 1/100), anti-programmed death-ligand 1 (PD-L1) (29E.2A3, Biolegend 329721, 1/100), anti-programmed death-ligand 2 (PD-L2) (MIH18, Biolegend 345515, 1/100), anti-B7-1 (2D10, Biolegend 305207, 1/100), anti-B7-2 (BU63, Biolegend 374215, 1/100), anti-E-cadherin (67A4, Biolegend 324114, 1/100) and anti-ICAM-1 (HA58, Biolegend 353113, 1/100) monoclonal antibodies (mAb) were used.
For T-cell phenotyping, anti-CD8a (RPA-T8, Biolegend 301014, 1/200), anti-CD11a (Miltenyi 130-105-480), anti-CD49a (TS2/7, Invitrogen 46-9490-41, 1/100), anti-CD69 (FN50, Biolegend 310914, 1/100), anti-CD103 (Ber-ACT8, Biolegend 350206, 1/100), anti-PD-1 (EH12-2H7, Biolegend 329923, 1/100), anti-CTLA-4 (14D3, Invitrogen 12-1529-42, 1/100), anti-VEGFR2 (7D4-6, Biolegend 359903, 1/50) and anti-TGFBR2 (W17055E, Biolegend 399703, 1/50) mAb were used.
Phenotypic studies were performed by direct or indirect immunofluorescence, and analyzed using a BD Fortessa flow cytometer. Data were processed using FlowJo software (Treestar).
Western blot analysis and quantification of transforming growth factor-β
Total protein extracts were obtained by lysis of 5×105 cells in ice-cold lysis buffer supplemented with a cocktail of anti-proteases and anti-phosphatases as described.22 Equivalent amounts of protein extracts were denatured, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Blots were incubated for 30 min in Tris-buffered saline containing 0.1% Tween 20% and 5% non-fat dry milk, and then overnight at 4°C with rabbit anti-NANOG (Santa Cruz, clone H155, cat# sc33759), anti-SNAIL1 (Cell Signaling, clone L70G2, cat# 3895S), anti-SOX2 (Cell Signaling, clone D6D9, cat# 3579), anti-GSK-3β (Cell Signaling, clone D75D3, cat# 5676) and antiphosphorylated (phospho)-GSK-3β ser9 (Cell Signaling, cat# 9336S) mAb, followed by appropriate secondary horseradish peroxidase-conjugated antibodies.
For quantification of total and active transforming growth factor (TGF)-β, tumor cells and CSC were plated at 1×107 cells in T75 culture flasks for 24 hours, then starved in 15 mL of serum-free medium for additional 48 hours. The conditioned medium was then harvested and concentrated 70× using Vivaspin Turbo 15 RC 10K (Startorius). Total and active TGF-β were measured using LEGENDplex multi-analyte assay kit according to the manufacturer’s instructions (Biolegend, cat# 740450).
RNA sequencing and analyses
For RNA sequencing (RNA-seq), total RNA was extracted from tumor cells and CSC; 150,000 cells per sample were processed using a single cell RNA purification kit (Norgen) according to the manufacturer’s instructions. RNA (RNA Integrity Score ≥
≥ 7.0) was checked using an Agilent 2100 Bioanalyzer and quantified using Qubit (Invitrogen), as described.19 Libraries were barcoded, purified, pooled in equal concentrations and subjected to paired-end sequencing on a HiSeq-2000 sequencer (Illumina).
7.0) was checked using an Agilent 2100 Bioanalyzer and quantified using Qubit (Invitrogen), as described.19 Libraries were barcoded, purified, pooled in equal concentrations and subjected to paired-end sequencing on a HiSeq-2000 sequencer (Illumina).
Quality control was performed on raw FastQ files with FastQC (V.0.11.8) and quality reports were gathered with MultiQC (V.1.10.1).23 Abundance estimation was performed with Salmon (V.0.9.0) using GENCODE (GRCh38, V.34) annotation.24 This pipeline was powered by both Snakemake and SnakemakeWrappers.25 Quantification results were aggregated with tximport (V.1.14.0) and differential gene analysis was performed using voom function of limma (V.3.42.0) as described.26 Gene set enrichment analysis (GSEA) on differentially expressed genes was performed with clusterProfiler (V.3.14.0), and immunological signature gene sets from MSigDB (C5, HALLMARK, V.7.4). Hallmark signatures enriched in CSC were defined using results from GSEA under a false discovery rate q value of 0.1.
Cytotoxicity experiments
Cytotoxic activity of the T-cell clones was measured by a conventional 4 hour 51Cr-release assay using triplicate cultures. The autologous IGR-Heu cell line and CSC cell line and clones were used as targets in cytotoxicity experiments at indicated effector to target (E:T) ratios.
Cytotoxicity inhibition was assessed by pre-incubating target cells with anti-ICAM-1, anti-MHC-I mAb or isotype control for 1 hour at room temperature before addition of effector cells at a 5:1 E:T ratio. Supernatants were then transferred to LumaPlateTM-96 wells (PerkinElmer, Boston, Massachusetts, USA), dried down and counted on Packard’s TopCount NXT. Per cent-specific cytotoxicity was calculated as described.21
Statistical analyses
Statistical significance was determined with the two-way analysis of variance test with Bonferroni correction or with the two-tailed Student’s t-test using GraphPad Prism (GraphPad software).
Data availability
The RNA-based next-generation sequencing (RNA-seq) dataset comparing the transcriptome of CSC with parental tumor cells are available from the corresponding author on request.
Results
Generation of CSC cell line and clones
To study the molecular and immune properties of human lung CSC, we used an in vitro method developed to enrich for CSC subsets through the generation of cancer spheroids.27 To this aim, we selected the NSCLC cell line IGR-Heu (E-cadherin+ICAM-1−), to which we have previously isolated specific T-cell clones from autologous TIL (Heu171, TRM) and PBL (H32-22, non-TRM).20 21 The Heu-CSC cell line obtained from IGR-Heu cells was sorted for ALDH+/Hoechst− cells and cloned. Among the obtained 30 clones, we retained two representative clones, CSC-1 and CSC-2. Compared with the parental cell line, Heu-CSC, CSC-1 and CSC-2 display dramatic morphology changes with loss of adhesive capacity and formation of spheroids (figure 1A).
Figure 1.
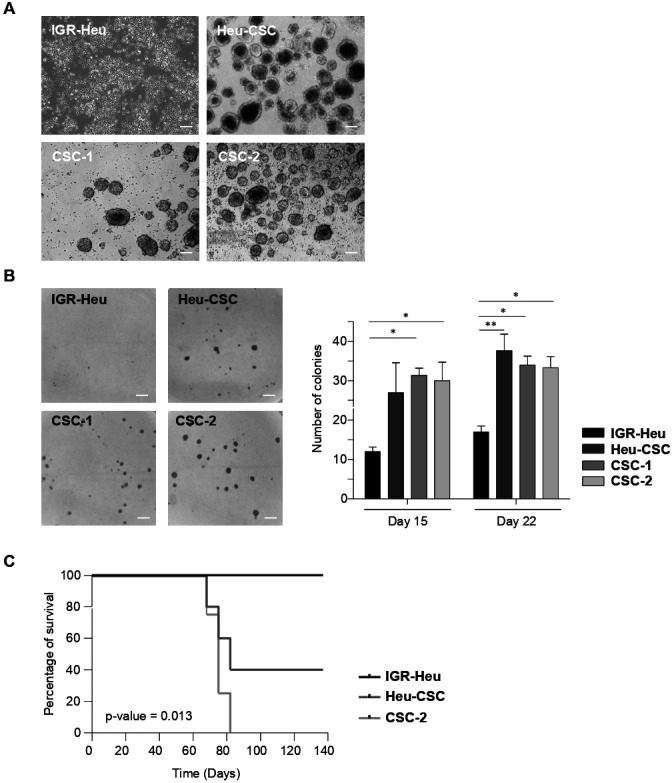
The IGR-Heu cell line and the derived cancer stem cell (CSC) display distinct phenotypic and cell transformation features. (A) Morphology changes of IGR-Heu, Heu-CSC, CSC-1 and CSC-2. Morphologies of cultured cancer cells were observed by phase-contrast light microscope. Objective: 10×; scale bar: 50 µm. Data are from one representative experiment out of four. (B) Clonogenic potential of IGR-Heu and CSC. IGR-Heu, Heu-CSC, CSC-1 and CSC-2 were grown in soft agar, and colonies were monitored by phase-contrast light microscope. Scale bar: 0.25 cm. Right, number of colonies counted at days 15 and 22. Bars represent means±SEM (n=3). P value was determined by two-way analysis of variance test. *P<0.05, **p<0.01. Data are from one representative experiment out of three. (C) Kaplan-Meier curves showing survival of nude mice engrafted with IGR-Heu or CSC (n=5 mice per group). Data are representative of two independent experiments. P value was determined by log-rank test.
Next, we evaluated the cellular transformation potential of parental tumor cells and derived CSC using a soft agar in vitro method. CSC grew more efficiently in soft agar than IGR-Heu cells (figure 1B), with the number of colonies observed at days 15 and 22 of culture being 2.1–2.6 higher for Heu-CSC, CSC-1 and CSC-2 than for parental cells. We then investigated cancer cell tumorigenicity in vivo by grafting IGR-Heu, Heu-CSC and CSC-2 into nude mice and monitoring animal survival. CSC grew more rapidly in immunodeficient mice than IGR-Heu cells, resulting in animals being sacrificed more rapidly (figure 1C). These results support the conclusion that CSC have stem-like properties.
Phenotypic and molecular characterization of lung CSC
To further characterize the generated lung CSC, we assessed the expression of biomarkers known to be expressed by human CIC. Immunofluorescence analyses indicated that Heu-CSC, CSC-1 and CSC-2 expressed low levels of the hematopoietic stem cell marker CD34, and the adhesion molecules CD44 and Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1 or CD31), but not the IGR-Heu cell line (figure 2A). In contrast, all cancer cells were found to express CD24, CD166, CXCR4 and VEGF at similar levels. Expression of additional cell surface markers, such as EGFR, was weaker in CSC than in IGR-Heu cells (figure 2A). Notably, all cancer cells failed to express CD133, CD117, CD40, CD40L, LFA3 (CD58), EpCAM and FasL (CD95L), and expression of CD95 (Fas) and CD105 was similar (online supplemental figure S1A).
Figure 2.

Phenotypic and molecular characterization of lung cancer stem cells (CSC). (A) Flow cytometry profiles of CD34, CD44, PECAM-1 (CD31), CD24, CD166, C-X-C chemokine receptor type 4 (CXCR4), vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) in IGR-Heu, Heu-CSC, CSC-1 and CSC-2. Percentages of positive cells and mean immunofluorescence intensity (MFI) (in parentheses) are shown. (B) Western blot analysis of NANOG, SNAIL1 and SOX2 proteins in IGR-Heu and CSC. Right, normalization of proteins relative to F-actin. (C) Western blot analysis of total GSK-3β and phospho-GSK-3β proteins in IGR-Heu, Heu-CSC, CSC-1 and CSC-2. Right, ratio of phospho-GSK-3β to total GSK-3β. Data are from one representative experiment out of three. (D) Flow cytometry profiles of major histocompatibility complex class I (MHC-I) and HLA-A2 on IGR-Heu, Heu-CSC, CSC-1 and CSC-2. Percentage of positive cells and MFI (in parentheses) are included. (E) Flow cytometry analyses of E-cadherin and intercellular adhesion molecule (ICAM)-1 expression on IGR-Heu, Heu-CSC, CSC-1 and CSC-2. Percentage of positive cells and MFI are shown.
jitc-2022-004527supp001.pdf (6MB, pdf)
We then performed western blot analyses to assess expression of transcription factors involved in EMT, a key characteristic of CSC. Expression of SNAIL1 transcription factor was increased in Heu-CSC and CSC-2 compared with IGR-Heu cells. In contrast, NANOG and SOX2 were expressed at lower levels in CSC than in parental tumor cells (figure 2B). Moreover, the ratio of the inactivated form of GSK-3β, phospho-GSK-3β, to total GSK-3β was weaker in CSC that in IGR-Heu (figure 2C). These results indicate that human lung CSC can be distinguished from parental tumor cells by increased expression of SNAIL1 and decreased phosphorylation of GSK-3β.
CSC and parental tumor cells display different E-cadherin and ICAM-1 surface expression
Next, we evaluated expression of surface molecules involved in antigen presentation and in T-cell adhesion/co-stimulation. Immunofluorescence analyses indicated that IGR-Heu, Heu-CSC and CSC-1 expressed similar levels of MHC-I molecules, except for CSC-2 which expressed lower levels. Surface expression of antigen-presenting molecules HLA-A2 is decreased in CSC as compared with parental cells (figure 2D). Both the percentage of HLA-A2+ cells and mean immunofluorescence intensity (MFI) were lower for CSC than IGR-Heu cells. Expression of HLA-B/C molecules is weak in all cancer cells (online supplemental figure 1B). All cancer cells failed to express the PD-1 ligands PD-L1 and PD-L2 and CD28/CTLA-4 ligands B7-1 (CD80) and B7-2 (CD86) (online supplemental figure S1C).
We then examined expression of surface molecules required for a cohesive interaction between TRM cells and epithelial target cells, the ligands of CD103 and LFA-1 integrins, E-cadherin and ICAM-1, respectively. Immunofluorescence analyses showed that E-cadherin adhesion molecule is more strongly expressed on IGR-Heu cells than on CSC suggesting the initiation of an EMT program by tumor stem-like cells (figure 2E). In contrast, ICAM-1 surface expression is much higher on CSC than on IGR-Heu cells, with MFI approximately threefold to ninefold higher on CSC than on parental cells (figure 2E). These results show that IGR-Heu tumor cells and derived CSC display different adhesion molecule surface expression levels that may impact their sensitivity to autologous CTL-mediated killing.
Lung CSC display an EMT signature
To gain further insight into the transcriptional profile of lung CSC, we performed RNA-seq of Heu-CSC, CSC-1 and IGR-Heu cells. Gene expression analyses carried out with an adjusted p value ≤10−4 revealed a large number of genes (n=1575) differentially expressed in CSC and parental cancer cells (figure 3A, online supplemental figure S2). Among the 826 upregulated and 749 downregulated genes, we identified gene signatures characteristic of cell signaling, development, differentiation, transport, cell motility and adhesion and clusters of genes related to EMT (online supplemental table S1).
Figure 3.
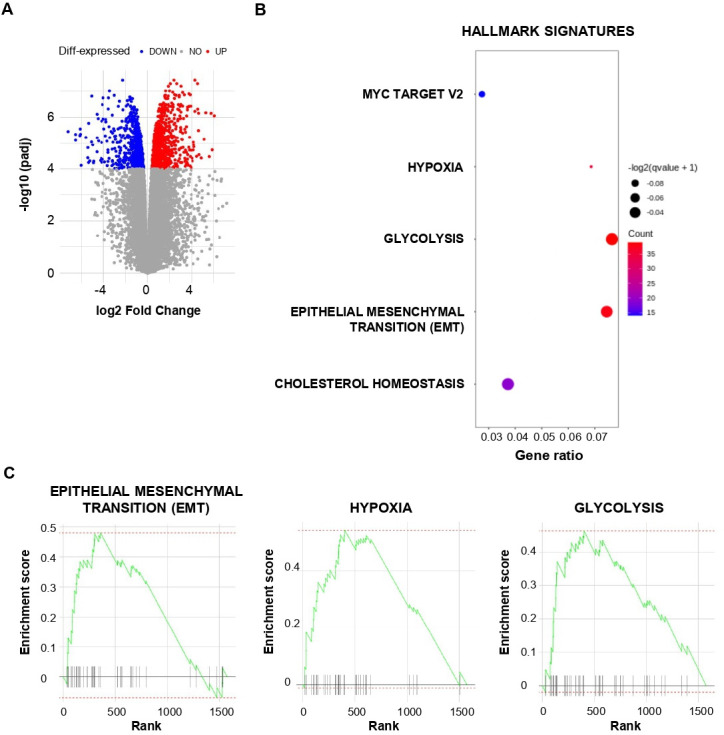
Transcriptional analysis of lung cancer stem cell (CSC). (A) Volcano plot of differential gene expression in CSC versus IGR-Heu analyzed by RNA-based next-generation sequencing (RNA-seq). Points are colored according to their average expression in all data sets. Red points are for genes upregulated in CSC, and blue points for downregulated genes. The expression difference is considered significant for an adjusted p value <10−4. (B) Dot plot showing the results of gene set enrichment analysis (GSEA) with a false discovery rate q value <0.1 using HALLMARK terms (MSigDB, HALLMARK, V.7.4). Each dot plot demonstrates enriched terms in transcriptome of CSC. The size of the dot represents the adjusted p value, and color represents the count of enriched genes. (C) GSEA of the gene set from Hallmark signatures (MSigDB, HALLMARK, V.7.4) in the transcriptome of CSC relative to IGR-Heu (n=3). Enrichment score for the gene set as the analysis ‘walks down’ the ranked list of genes; the position of gene set members (black vertical lines) in the ranked list of genes and the value of ranking metric are shown. A set of EMT genes enriched in the top-ranking genes is shown by a green line.
jitc-2022-004527supp002.pdf (166.3KB, pdf)
We then defined hallmark signatures enriched in CSC using results from GSEA. Hallmark signatures corresponding to Myc target V2, hypoxia, glycolysis, EMT and cholesterol homeostasis were enriched in CSC compared with parental cells (figure 3B, C, online supplemental table S2). Hallmark signatures corresponding to hypoxia, glycolysis and EMT were also enriched in CSC-2 compared with IGR-Heu cells (online supplemental figure S3A). Among EMT signature genes, we observed upregulation of FN1, PLOD1 and PLOD3, TGFB1, TGFBR3, THBS1 and VEGFA, which encode fibronectin, procollagen-lysine,2-oxoglutarate 5-dioxygenase 1 and procollagen-lysine,2-oxoglutarate 5-dioxygenase 3, TGF-β, thrombospondin 1 and VEGFα, respectively (online supplemental table S2). The CDH3 gene, which encodes the P-cadherin adhesion molecule, a marker of epithelial-to-mesenchymal hybrid state or partial EMT, is upregulated in CSC (fold change (FC)=6) as compared with IGR-Heu. In contrast, expression of the VIM gene, encoding vimentin, a marker of more advanced mesenchymal differentiation, is only slightly upregulated in CSC (FC=1.5), and expression of CDH2, encoding N-cadherin, another marker of mesenchymal differentiation, is slightly downregulated. Moreover, expression of the EMT-transcription factor gene TWIST1 is upregulated in CSC (FC=1.9) as compared with parental cells. Expression levels of the CDH1 gene, encoding E-cadherin, is similar in IGR-Heu and CSC clones, and slightly upregulated in Heu-CSC (online supplemental figure S3B). This result supports the hypothesis that E-cadherin expression on CSC is regulated by a post-transcriptional mechanism. Expression of ICAM1 is also increased in Heu-CSC, and at a lower extent in CSC-1, compared with parental cells (online supplemental figure S3B). HLA-A*02, HLA-B and HLA-C-encoding genes are expressed in all cancer cells (online supplemental figure S3C), suggesting a post-transcriptional regulation of HLA molecules in CSC. With regard to hypoxia, we detected upregulation of EFNA3 (encoding ephrin A3); HSPA1A and HSPB1 (encoding HSP family A (HSP70) member 1A and HSP family B (small) member 1, respectively), PTGS1 (encoding prostaglandin-endoperoxide synthase 1), LOXL2 (encoding lysyl oxidase like 2) and VEGFA transcripts (online supplemental table S2). Similar studies conducted with the molecular signature database MSigDB showed enrichment in genes related to mesenchymal cell differentiation and mesenchyme development, regulation of apoptotic signaling, cell morphogenesis and growth, cell adhesion and junction and stem cell differentiation (online supplemental figure S4AB). These data suggest that by initiating an EMT/hypoxia program, NSCLC CSC evade TRM-cell recognition and destruction.
jitc-2022-004527supp003.xls (38.5KB, xls)
CSC evade TRM-cell-mediated cytotoxicity by downregulating cell surface E-cadherin
To investigate the functional consequence of adhesion and recognition molecule differential expression, we tested susceptibility of CSC to autologous T-cell-mediated killing using the CD8+CD103+ (Heu171, TRM) and the CD8+CD103− (H32-22, non-TRM) CTL clones specific to the HLA-A2-restricted mutant α-actinin-4 peptide. Initial phenotypic studies confirmed that the TIL clone (Heu171) expressed the TRM surface marker CD103 (αEβ7 integrin), but not the PBL clone (H32-22). Both clones expressed the activation marker CD69, but at a higher level in TRM than in non-TRM. The TRM and non-TRM clones also expressed LFA-1 (αLβ2) integrin, the T-cell inhibitory receptor PD-1, the TGF-β receptor TGFBR2 and the VEGF receptor VEGFR2, but not CD49a integrin and CTLA-4 (figure 4A).
Figure 4.

Resident memory T (TRM) and non-TRM phenotypic profiles and susceptibility of cancer stem cell (CSC) to cytotoxic T lymphocyte (CTL)-mediated killing. (A) Flow cytometry analyses of CD8, LFA-1, CD103, CD49a, CD69, PD-1, CTLA-4, TGFBR2 and VEGFR2 on CD8+CD103+ TRM (Heu171) and CD8+CD103- non-TRM (H32-22) clones. Mean immunofluorescence intensity (MFI) are in parentheses.(B) Cytotoxic activity of the TRM clone (Heu171) towards autologous IGR-Heu, Heu-CSC, CSC-1 and CSC-2 target cells. Percent of specific lysis are shown at indicated effector to target (E:T) ratios. (C) Quantification of transforming growth factor (TGF)-β in conditioned media from IGR-Heu, Heu-CSC, CSC-1 and CSC-2 by multi-analyte flow assay. Ratios of active/total TGF-β normalized to IGR-Heu are included. Results are presented as mean±SEM of duplicates. (D) Cytotoxicity of the non-TRM clone (H32-22) towards autologous IGR-Heu, Heu-CSC, CSC-1 and CSC-2 target cells. (E) Inhibition of TRM-cell-mediated killing. CSC-1 cells are preincubated in the presence of isotype control, anti-major histocompatibility complex class I (MHC-I) or anti-intercellular adhesion molecule 1 (ICAM-1) neutralizing monoclonal antibody (mAb) and then CTL were added at 5:1 E:T ratio. Symbols represent replicates and horizontal bars represent means±SEM (n=3). P value was determined by two-way analysis of variance test. *p<0.05, **p<0.01,***p<0.001,****p<0.0001. Data are from one representative experiment out of three.
Cytotoxicity experiments indicated that Heu-CSC, CSC-1 and CSC-2 were less efficiently killed by the TRM clone than parental tumor cells (figure 4B). Partial resistance of CSC to TRM cell-mediated cytolytic activity was associated with decreased surface expression of E-cadherin (figure 2E) and correlated with an increased production of active TGF-β (figure 4C). Indeed, Heu-CSC, CSC-1 and CSC-2 displayed an active/total TGF-β ratio around 1.5-fold to 3-fold higher than parental cells. As expected, chromium release assays showed that the E-cadherin+ICAM-1− IGR-Heu cell line was resistant to CD8+CD103- non-TRM lymphocytes (figure 4D). In contrast, Heu-CSC, CSC-2 and at a greater extent CSC-1 target cells were sensitive to the non-TRM-cell clone (figure 4D). Killing of CSC by the TRM-cell clone is associated with expression of ICAM-1 (figure 2E) and is inhibited by anti-ICAM-1 and anti-MHC-I blocking mAb (figure 4E). In contrast, anti-ICAM-1 neutralizing mAb had no effect on TRM-cell-mediated cytotoxicity toward IGR-Heu cells (online supplemental figure S4C). These results explain the partial susceptibility of CSC to TRM-cell-mediated cytotoxicity linked to induction of ICAM-1 and its interaction with LFA-1 on effector T cells. They also further emphasize the involvement of EMT, in particular decreased surface expression of E-cadherin, and downregulation of HLA-A2-antigenic peptide complexes in resistance of CSC to T-cell receptor (TCR)-dependent TRM-cell-mediated cytolysis.
Discussion
In this study, we show that NSCLC CSC display a transcriptomic profile characteristic of EMT. Among genes characteristic of EMT, THBS1 and VEGFA, encoding thrombospondin 1 and VEGFα, respectively, were upregulated in CSC as compared with parental tumor cells. Thrombospondin 1 is an activator of TGF-β through maturation of LAP-TGF-β, suggesting a role in the maintenance of CSC.28 VEGFα promotes EMT and hypoxia in various cancer types, and is a target of the hypoxia-inducible factors (HIF).29 Compared with parental tumor cells, human lung CSC display an increased expression of the EMT marker SNAIL1 transcription factor and decreased phosphorylation of GSK-3β and surface expression of the epithelial cell marker E-cadherin. Expression and stability of SNAIL proteins is regulated by GSK-3β, and a correlation between SNAIL expression level and phosphorylation of GSK-3β was observed in SNAIL-overexpressing NSCLC cells.30 GSK-3β also plays a central role in Wnt/β-catenin pathway and is implicated in the maintenance stem cells and CSC. Accumulating evidence indicates that SNAIL is activated in EMT to regulate gene transcription and expression of the transcription factor NANOG, which is implicated in stemness, emergence of CSC and inhibition of antitumor immune response.31 32 Moreover, a high level of SNAIL is associated with metastasis in NSCLC, and SNAIL-expressing cells display characteristics of CSC and of cells that have undergone EMT, with increased migration, chemoresistance and sphere forming properties.30
RNA-seq analyses of the present study revealed an increase in TGFB1, TGFBR1 and TGFBR3 gene expression in CSC as compared with parental tumor cells. Enhanced production of active TGF-β by CSC was also observed at the protein level. This cytokine is the main mediator of EMT, and upregulated TGF-β expression is often associated with a tumorous EMT program. TGF-β initiates intracellular signaling pathway by binding to its receptors, TGFBR1 and TGFBR2, and activating SMAD2 and SMAD3 transcription factors, which together activate transcription of genes, such as VEGFA and the EMT transcription factors, SNAIL, SLUG and TWIST.33 EMT transcription factors form complexes with SMAD4 to regulate target genes, including repression of the CDH1 gene, which encodes E-cadherin. TGF-β has been shown to increase stem-like properties in breast cancer, and inhibition of TGF-β signaling prevents the development of chemotherapy-resistant CSC.34 This cytokine is also an immunosuppressive mediator frequently used by malignant cells to escape from the immune system.35 36 TGF-β also participates in CD8 T-cell exclusion from the TME and in T-cell dysfunction.37 Paradoxically, TGF-β is involved in expression of the ITGAE gene, which encodes CD103 integrin subunit, in activated CD8+ T lymphocytes21 38 and in differentiation and persistence of CD103+CD8+ TRM in epithelial tissues.39 Moreover, we previously reported that TGF-β signaling intersects with CD103 bidirectional signals to promote CD8 T-cell accumulation in epithelial tumor regions and antitumor TRM-cell functions.40
Our present data show that lung CSC surface express lower levels of E-cadherin (CD324) than parental tumor cells. This adhesion molecule is expressed on epithelial cells forming homotypic bonds between adjacent cells, thereby preventing invasiveness of carcinoma cells.41 Progression of epithelial tumors is characterized by the capacity of cancer cells to overcome cell-cell adhesion in particular by downregulating E-cadherin.42 Reduced expression of E-cadherin during cancer development and metastatic invasion is frequently observed in epithelial tumors.43 Decreased expression of E-cadherin on CSC plasma membrane is most likely linked to a post-transcriptional, membrane trafficking regulation mechanism. In this regard, it has been reported that E-cadherin undergoes endocytosis, sorting and recycling, controlling its cell membrane expressing level.44 45 These results suggest that downregulation of E-cadherin expression on CSC surface result in escape from specific antitumor T-cell immunity provided locally by CD8+ TRM cells. In this regard, the heterophilic adhesive interaction between E-cadherin and CD103 plays a crucial role in retention of CD103+ T cells in epithelial tissues,46 and thus in intratumoral immune surveillance. We have previously shown that the expression level of CD103 on lung tumor-specific T-cell clones correlates with their capacity to kill autologous E-cadherin+ICAM-1- tumor cells, and that killing is abrogated by small interfering RNA targeting E-cadherin.21 47 Along the same lines, our present data indicate that downregulation of E-cadherin on CSC results in dramatic inhibition of their susceptibility to specific CD8+CD103+ TRM-cell-mediated cytotoxicity. The E-cadherin-CD103 interaction plays a major role in maturation of the immune synapse formed between CD103+ TRM cells and E-cadherin+ICAM-1- tumor cells and in the antitumor CTL response.47 In this respect, we have previously demonstrated that interaction of E-cadherin on cancer cells with CD103 on specific TRM cells triggers polarization of cytotoxic granules at the contact area between T cells and tumor cells and subsequent exocytosis, leading to target cell death.21 Moreover, we and others have demonstrated that CD8+CD103+ TRM cells play a key role in antitumor CD8 T-cell immunity, and that this TIL subset can be used as a prognostic factor for survival in NSCLC.18 These tumor-resident T lymphocytes are also involved in response to therapeutic cancer vaccines48 and immune checkpoint blockade immunotherapy.19
While lung CSC downregulate surface expression of E-cadherin, we show here that they upregulate a panel of genes encoding diverse adhesion molecules, including L1 CELL ADHESION MOLECULE (L1CAM), ICAM5 and ICAM1. L1CAM (also known as CD171) is expressed in CSC where it appears to play a role in stemness and biological processes associated with CSC, such as EMT, poor prognosis and resistance to treatments.49 Remarkably, we previously observed an increased expression of ICAM-1 on lung cancer cells upon inhibition of AXL, a member of the TAM (Tyro3, Axl and Mer) receptor tyrosine kinase family associated with cancer cell plasticity and mesenchymal cell drug resistance; this correlated with attenuation of immune resistance and improved survival of patients with NSCLC.50 Furthermore, ICAM-1 plays a major role in T-cell-mediated cytotoxicity by engaging productive ligation with LFA-1 and thereby promoting tight adhesion of CTL to target cells.51 Interaction of ICAM-1 on antigen-presenting cells with LFA-1 on effector cells is a prerequisite for strengthening the interaction between CTL and target cells, and directing released cytolytic granules to the surface of tumor cells inducing their destruction. In the absence of both ICAM-1/LFA-1 and E-cadherin/CD103 adhesion, polarization of cytotoxic granules and their delivery into the target are compromised. Therefore, blocking the LFA-1/ICAM-1 interaction with anti-ICAM-1 neutralizing antibodies led to inhibition of CSC killing by specific CTL. In contrast, transduction of IGR-Heu with ICAM-1 enhanced both TRM and non-TRM clone-mediated killing.47 It should be noted that TRM and non-TRM clones express TGRBR2 and VEGFR2, excluding their implication in the observed differential activities. Whether the data obtained in this study could be generalized to other cancer types need to be further investigated.
The GSEA of NSCLC CSC also revealed a gene signature characteristic of hypoxia. Hypoxia, frequently associated with TME, contributes to CSC development. In this regard, HIF have been shown to promote CSC survival in a hypoxic TME, such as by upregulating OCT4.31 HIF, in particular HIF-1α, increases stemness and multidrug resistance in colorectal cancer, and downregulation of HIF-2α is able to inhibit CSC stemness and to induce CSC apoptosis.52 These data highlight the strong relationship between EMT and hypoxia in regulating CSC phenotype and functions. Importantly, hypoxia negatively regulates expression of MHC-I molecules in a HIF-dependent manner, thereby inhibiting tumor cell recognition and destruction by CTL.53 Downregulation of MHC-I has been described in CSC and was associated with resistance to T-cell-mediated cytotoxicity and evasion from CD8 T-cell immunity.54 Similarly, our results revealed that lung CSC display decreased surface expression of HLA-A2 neoepitope-presenting molecules, which likely contributes to partial resistance to specific CD8+ T cells. Downregulation of HLA molecules on CSC is likely governed by a post-transcriptional mechanism, such as via alterations in antigen processing machinery. Indeed, defects in transporter associated with antigen processing (TAP) play a major role by inducing a sharp decrease in surface expression of MHC-I/β2m-peptide complexes, enabling malignant cells to escape from CTL-mediated killing.55 In this regard, we previously reported that IGR-Heu cells display low levels of TAP, and that restoration of their expression increased recognition by α-actinin-4-specific CTL.56 Kinases, such as EGFR and MEK1, have also been described as negative regulators of MHC-I expression and antigen presentation machinery in multiple cancers.57 Quiescent stem cells were reported to evade T-cell killing via downregulation of the antigen presentation machinery, including MHC-I and TAP.17 Unlike previous reports,58 our data did not reveal increased expression of the T-cell inhibitory ligands PD-L1/PD-L2 on CSC, excluding their involvement in resistance to specific CTL. These immune checkpoint ligands were more strongly expressed on CSC than on conventional cancer cells, and the frequency of PD-L1+ CSC correlated negatively with that of CD4+ T cells in patients with NSCLC. Moreover, a higher percentage of PD-L1+ CSC was observed in patients with progressive disease suggesting resistance of CSC to conventional therapies.59
Overall, our data demonstrate that human lung CSC initiate an EMT signature characterized by upregulation of specific EMT transcription factors and downregulation of E-cadherin adhesion protein at the plasma membrane. They also show decreased surface expression of HLA-A2 antigen-presenting molecules, which together with alterations in CD103 ligand expression, is associated with resistance to tumor neoepitope-specific CD8+CD103+ TRM cells. In contrast, lung CSC acquired expression of ICAM-1 rendering them sensitive to LFA-1-dependent TCR-mediated killing. These data support the conclusion that targeting CSC EMT in combination with immune checkpoint blockade holds promise for future NSCLC immunotherapy approaches.
Acknowledgments
We thank Nathalie Droin for her help with RNA sequencing and Sarah Rio for her help with CSC culture. We also thank Yann Lecluse from the cytometry facility Plateforme d’Imagerie-Cytométrie of Gustave Roussy for his help with flow cytometry analyses.
Footnotes
Twitter: @Stephane.TERRY1
Contributors: Conception and design: SCo, ID, GG and FM-C. Development of methodology: SCo, ID, GG and FM-C. Acquisition of data (providing animals, acquiring and managing patients, providing facilities, etc): SCo, ID, GG, AC and FM-C. Analysis and interpretation of data (eg, statistical analysis, biostatistics, computational analysis): SCo, ID, AC, MD and FM-C. Writing, reviewing and/or revision of the manuscript: SCo, ID, CC, ST, SCh, MD and FM-C. Administrative, technical and material support (ie, reporting and organizing data, constructing databases): SCo, ID, CC, MD and FM-C. Study supervision: FM-C. FM-C acts as the guarantor for the overall content.
Funding: This work was supported by grants from the INSERM, Association pour la Recherche sur le Cancer (ARC; grant number: SIGN’IT20181007792), the French Institut National du Cancer (INCa; PLBIO016-080 grant number 10557) and Bristol-Myers Squibb (BMS Foundation for Research in Immuno-Oncology, France). ID was a recipient of a MENRT fellowship from the French Ministry of Research and the ARC; SC is a recipient of a fellowship from ARC.
Competing interests: No, there are no competing interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request. The RNA-seq dataset comparing the transcriptome of CSC with parental tumor cells are available from the corresponding author on request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Experiments in mice were performed in accordance with guidelines established by the institutional animal committee (CEEA no. 2017-81-12717).
References
- 1.O'Brien CA, Kreso A, Jamieson CHM. Cancer stem cells and self-renewal. Clin Cancer Res 2010;16:3113–20. 10.1158/1078-0432.CCR-09-2824 [DOI] [PubMed] [Google Scholar]
- 2.Noh KH, Kim BW, Song K-H, et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest 2012;122:4077–93. 10.1172/JCI64057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645–8. 10.1038/367645a0 [DOI] [PubMed] [Google Scholar]
- 4.Kim DK, Ham MH, Lee SY, et al. CD166 promotes the cancer stem-like properties of primary epithelial ovarian cancer cells. BMB Rep 2020;53:622–7. 10.5483/BMBRep.2020.53.12.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu P, Clanton DJ, Snipas TS, et al. Characterization of a subpopulation of colon cancer cells with stem cell-like properties. Int J Cancer 2009;124:1312–21. 10.1002/ijc.24061 [DOI] [PubMed] [Google Scholar]
- 6.Maccalli C, Rasul KI, Elawad M, et al. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol 2018;53:189–200. 10.1016/j.semcancer.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.López-Gil JC, Martin-Hijano L, Hermann PC, et al. The CXCL12 crossroads in cancer stem cells and their niche. Cancers 2021;13:469. 10.3390/cancers13030469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.García Bueno JM, Ocaña A, Castro-García P, et al. An update on the biology of cancer stem cells in breast cancer. Clin Transl Oncol 2008;10:786–93. 10.1007/s12094-008-0291-9 [DOI] [PubMed] [Google Scholar]
- 9.Di J, Massuger LFAG, Duiveman-de Boer T, et al. Functional OCT4-specific CD4+ and CD8+ T cells in healthy controls and ovarian cancer patients. Oncoimmunology 2013;2:e24271. 10.4161/onci.24271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visus C, Wang Y, Lozano-Leon A, et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8⁺ T cells. Clin Cancer Res 2011;17:6174–84. 10.1158/1078-0432.CCR-11-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beck B, Driessens G, Goossens S, et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 2011;478:399–403. 10.1038/nature10525 [DOI] [PubMed] [Google Scholar]
- 12.Vijay GV, Zhao N, Den Hollander P, et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res 2019;21:37. 10.1186/s13058-019-1125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ning N, Pan Q, Zheng F, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res 2012;72:1853–64. 10.1158/0008-5472.CAN-11-1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanzardo S, Conti L, Rooke R, et al. Immunotargeting of antigen xCT attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res 2016;76:62–72. 10.1158/0008-5472.CAN-15-1208 [DOI] [PubMed] [Google Scholar]
- 15.Wang B, Wang Q, Wang Z, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res 2014;74:5746–57. 10.1158/0008-5472.CAN-13-2563 [DOI] [PubMed] [Google Scholar]
- 16.Brown CE, Starr R, Martinez C, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res 2009;69:8886–93. 10.1158/0008-5472.CAN-09-2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agudo J, Park ES, Rose SA, et al. Quiescent tissue stem cells evade immune surveillance. Immunity 2018;48:271–85. 10.1016/j.immuni.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djenidi F, Adam J, Goubar A, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 2015;194:3475–86. 10.4049/jimmunol.1402711 [DOI] [PubMed] [Google Scholar]
- 19.Corgnac S, Malenica I, Mezquita L, et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched with Tc17. Cell Rep Med 2020;1:100127. 10.1016/j.xcrm.2020.100127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Echchakir H, Mami-Chouaib F, Vergnon I, et al. A point mutation in the alpha-actinin-4 gene generates an antigenic peptide recognized by autologous cytolytic T lymphocytes on a human lung carcinoma. Cancer Res 2001;61:4078–83. [PubMed] [Google Scholar]
- 21.Le Floc'h A, Jalil A, Vergnon I, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 2007;204:559–70. 10.1084/jem.20061524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Floc'h A, Jalil A, Franciszkiewicz K, et al. Minimal engagement of CD103 on cytotoxic T lymphocytes with an E-cadherin-Fc molecule triggers lytic granule polarization via a phospholipase Cgamma-dependent pathway. Cancer Res 2011;71:328–38. 10.1158/0008-5472.CAN-10-2457 [DOI] [PubMed] [Google Scholar]
- 23.Ewels P, Magnusson M, Lundin S, et al. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8. 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frankish A, Diekhans M, Ferreira A-M, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47:D766–73. 10.1093/nar/gky955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 2015;4:1521. 10.12688/f1000research.7563.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006;9:391–403. 10.1016/j.ccr.2006.03.030 [DOI] [PubMed] [Google Scholar]
- 28.Atanasova VS, Russell RJ, Webster TG, et al. Thrombospondin-1 is a major activator of TGF-β signaling in recessive dystrophic epidermolysis bullosa fibroblasts. J Invest Dermatol 2019;139:1497–505. 10.1016/j.jid.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 29.Luo M, Hou L, Li J, et al. VEGF/NRP-1axis promotes progression of breast cancer via enhancement of epithelial-mesenchymal transition and activation of NF-κB and β-catenin. Cancer Lett 2016;373:1–11. 10.1016/j.canlet.2016.01.010 [DOI] [PubMed] [Google Scholar]
- 30.Liu C-W, Li C-H, Peng Y-J, et al. Snail regulates Nanog status during the epithelial-mesenchymal transition via the Smad1/Akt/GSK3β signaling pathway in non-small-cell lung cancer. Oncotarget 2014;5:3880–94. 10.18632/oncotarget.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasmim M, Noman MZ, Lauriol J, et al. Hypoxia-dependent inhibition of tumor cell susceptibility to CTL-mediated lysis involves NANOG induction in target cells. J Immunol 2011;187:4031–9. 10.4049/jimmunol.1101011 [DOI] [PubMed] [Google Scholar]
- 32.Mao C-P, Wu T, Song K-H, et al. Immune-mediated tumor evolution: Nanog links the emergence of a stem like cancer cell state and immune evasion. Oncoimmunology 2014;3:e947871. 10.4161/21624011.2014.947871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003;113:685–700. 10.1016/S0092-8674(03)00432-X [DOI] [PubMed] [Google Scholar]
- 34.Bhola NE, Balko JM, Dugger TC, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest 2013;123:1348–58. 10.1172/JCI65416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005;8:369–80. 10.1016/j.ccr.2005.10.012 [DOI] [PubMed] [Google Scholar]
- 36.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554:538–43. 10.1038/nature25492 [DOI] [PubMed] [Google Scholar]
- 37.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. 10.1038/nature25501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Asady R, Yuan R, Liu K, et al. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med 2005;201:1647–57. 10.1084/jem.20041044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackay LK, Wynne-Jones E, Freestone D, et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 2015;43:1101–11. 10.1016/j.immuni.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 40.Boutet M, Gauthier L, Leclerc M, et al. TGFβ signaling intersects with CD103 integrin signaling to promote T-lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res 2016;76:1757–69. 10.1158/0008-5472.CAN-15-1545 [DOI] [PubMed] [Google Scholar]
- 41.Frixen UH, Behrens J, Sachs M, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 1991;113:173–85. 10.1083/jcb.113.1.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perl AK, Wilgenbus P, Dahl U, et al. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998;392:190–3. 10.1038/32433 [DOI] [PubMed] [Google Scholar]
- 43.Vleminckx K, Vakaet L, Mareel M, et al. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991;66:107–19. 10.1016/0092-8674(91)90143-M [DOI] [PubMed] [Google Scholar]
- 44.Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell 2004;15:176–88. 10.1091/mbc.e03-05-0319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le TL, Yap AS, Stow JL. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol 1999;146:219–32. [PMC free article] [PubMed] [Google Scholar]
- 46.Cepek KL, Shaw SK, Parker CM, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature 1994;372:190–3. 10.1038/372190a0 [DOI] [PubMed] [Google Scholar]
- 47.Franciszkiewicz K, Le Floc'h A, Boutet M, et al. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res 2013;73:617–28. 10.1158/0008-5472.CAN-12-2569 [DOI] [PubMed] [Google Scholar]
- 48.Nizard M, Roussel H, Diniz MO, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun 2017;8:15221. 10.1038/ncomms15221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giordano M, Cavallaro U. Different Shades of L1CAM in the pathophysiology of cancer stem cells. J Clin Med 2020;9:1502. 10.3390/jcm9051502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terry S, Abdou A, Engelsen AST, et al. AXL targeting overcomes human lung cancer cell resistance to NK- and CTL-mediated cytotoxicity. Cancer Immunol Res 2019;7:1789–802. 10.1158/2326-6066.CIR-18-0903 [DOI] [PubMed] [Google Scholar]
- 51.Anikeeva N, Somersalo K, Sims TN, et al. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc Natl Acad Sci U S A 2005;102:6437–42. 10.1073/pnas.0502467102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Y-A, Chen Y-F, Bao Y, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A 2018;115:E5990–9. 10.1073/pnas.1801348115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sethumadhavan S, Silva M, Philbrook P, et al. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS One 2017;12:e0187314. 10.1371/journal.pone.0187314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018;18:469. 10.1186/s12885-018-4389-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leibowitz MS, Andrade Filho PA, Ferrone S, et al. Deficiency of activated STAT1 in head and neck cancer cells mediates TAP1-dependent escape from cytotoxic T lymphocytes. Cancer Immunol Immunother 2011;60:525–35. 10.1007/s00262-010-0961-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durgeau A, El Hage F, Vergnon I, et al. Different expression levels of the TAP peptide transporter lead to recognition of different antigenic peptides by tumor-specific CTL. J Immunol 2011;187:5532–9. 10.4049/jimmunol.1102060 [DOI] [PubMed] [Google Scholar]
- 57.Brea EJ, Oh CY, Manchado E, et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol Res 2016;4:936–47. 10.1158/2326-6066.CIR-16-0177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsu J-M, Xia W, Hsu Y-H, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun 2018;9:1908. 10.1038/s41467-018-04313-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raniszewska A, Vroman H, Dumoulin D, et al. PD-L1+ lung cancer stem cells modify the metastatic lymph-node immunomicroenvironment in nsclc patients. Cancer Immunol Immunother 2021;70:453–61. 10.1007/s00262-020-02648-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-004527supp001.pdf (6MB, pdf)
jitc-2022-004527supp002.pdf (166.3KB, pdf)
jitc-2022-004527supp003.xls (38.5KB, xls)
Data Availability Statement
The RNA-based next-generation sequencing (RNA-seq) dataset comparing the transcriptome of CSC with parental tumor cells are available from the corresponding author on request.
Data are available on reasonable request. The RNA-seq dataset comparing the transcriptome of CSC with parental tumor cells are available from the corresponding author on request.