

Abstract
Antiphospholipid antibodies (aPLs) cause severe autoimmune disease characterized by vascular pathologies and pregnancy complications. Here, we identify endosomal lysobisphosphatidic acid (LBPA) presented by the CD1d-like endothelial protein C receptor (EPCR) as a pathogenic cell surface antigen recognized by aPLs for induction of thrombosis and endosomal inflammatory signaling. The engagement of aPLs with EPCR–LBPA expressed on innate immune cells sustains interferon- and toll-like receptor 7-dependent B1a cell expansion and autoantibody production. Specific pharmacological interruption of EPCR–LBPA signaling attenuates major aPL-elicited pathologies and the development of autoimmunity in a mouse model of systemic lupus erythematosus. Thus, aPLs recognize a single cell surface lipid–protein receptor complex to perpetuate a self-amplifying autoimmune signaling loop dependent on the cooperation with the innate immune complement and coagulation pathways.
One-Sentence Summary:
EPCR mediates pathologies of antiphospholipid antibodies and their interferon-dependent expansion in autoimmunity.
Introduction
Lipid-reactive antibodies transiently appear in infectious diseases, but clonal evolution of persistent autoimmune antiphospholipid antibodies (aPLs) cause the antiphospholipid syndrome (APS), which is characterized by severe thrombo-embolic and microangiopathic complications, pregnancy morbidity, and fetal loss (1). Although APS is the only manifestation of autoimmunity in many patients (primary APS), it also develops in in the context of other autoimmune diseases, in particular systemic lupus erythematosus (SLE) (secondary APS). aPLs display heterogeneous reactivity with a variety of anionic phospholipids and blood proteins, including β2 glycoprotein I (β2GPI) (2). aPL reactivity against β2GPI or against cardiolipin as a prototypic lipid is used to diagnose APS (3), but aPL heterogeneity and cross-reactivities between these antigens appear to develop during clonal evolution (4, 5). This diversity of aPLs has hampered the definition of central mechanisms that cause the spectrum of APS-related pathologies (1, 3, 6). Identification of pathogenic targets for aPLs may yield improved diagnostic approaches as well as insights into the development of autoimmunity in APS.
Monoclonal antibodies with selective reactivity for lipid versus β2GPI induce distinct potentially pathogenic cellular responses (7–11). Lipid-reactive aPLs translocate toll-like receptor (TLR)7/8 from the endoplasmic reticulum to the endosome and thereby sensitize to endosomal proinflammatory signaling of extracellular RNA in monocytes (12). This pathway is independent of LDL-receptor related protein (LRP)8 (8), which is implicated in β2GPI-reactive aPL signaling (13, 14). Lipid-reactive aPLs also cause pathological complications via crosstalk with the innate immune complement and coagulation pathways (15, 16).
The cytokine receptor-like tissue factor (TF) is central to pregnancy morbidity and the prothrombotic monocyte activation induced by aPLs. Together with its ligand protease coagulation factor (F) VIIa, TF initiates coagulation (11, 17). TF is present on quiescent monocytes in complex with its physiological inhibitor, TF pathway inhibitor (TFPI). Only lipid-reactive aPLs induce signaling by disrupting the inhibited TF complex through interaction with an unknown cell surface ligand (fig. S1) (11). TF–FVIIa-generated FXa also engages the endothelial protein C receptor (EPCR), which is encoded by PROCR and is primarily known for its vascular cytoprotective functions. In the context of TF–EPCR signaling, FXa cleaves the protease activated receptor (PAR)2 (18, 19) that is essential for LPS-induced interferon (IFN) responses in dendritic cells (DCs) (20). Because IFN-α is induced by aPLs in DCs (12) and IFN signaling is central to autoimmune diseases (21, 22), we tested the hypothesis that EPCR contributes to APS pathologies and the development of aPL autoimmunity.
Results
EPCR-dependent signaling of aPL
We used benchmark genes for LPS-induced and EPCR-dependent IFN responses previously established in DCs (20) to confirm that inhibitory (clone 1560), but not non-inhibitory (clone1562) α-EPCR antibodies (18) blocked LPS-induced IFN signaling, but not Tnf mRNA induction in monocytes (Fig. 1A and fig. S2A). These α-EPCR antibodies had no apparent agonistic activity, even when combined with the TRL7 agonist R848. Lipid-reactive human monoclonal aPLs HL5B and HL7G represent different stages of aPL hypermutation, the latter having acquired β2GPI cross-reactivity (5). Both of these aPLs rapidly induced Tnf mRNA as well as IFN-regulated genes as effectively as LPS in an EPCR-dependent manner (Fig. 1A). Thus, aPLs engage EPCR on the cell surface to induce pathways related to host defense.
Figure 1: EPCR is an aPL receptor.
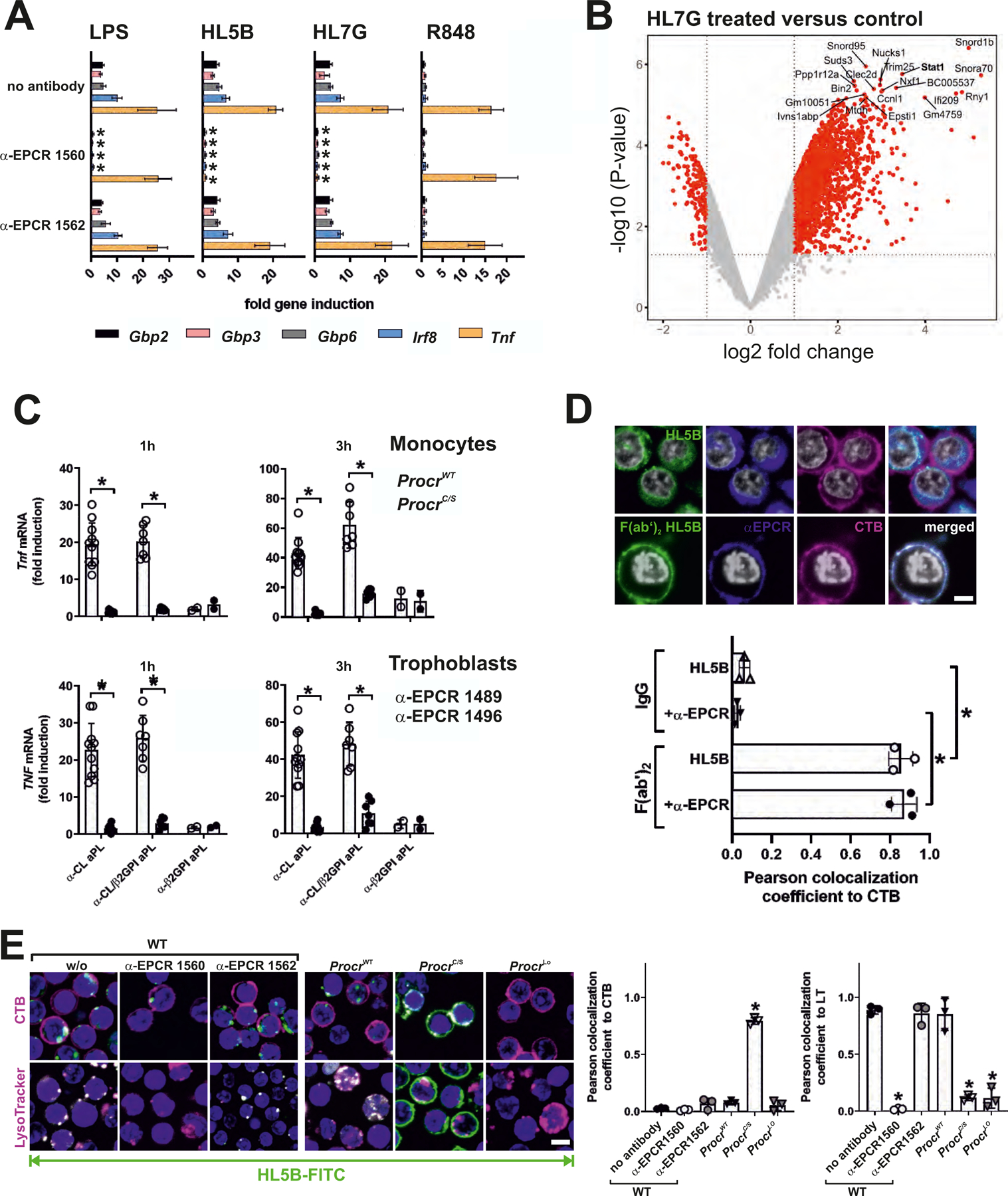
(A) EPCR-dependent induction of IFN-regulated genes and Tnf in mouse monocytes after 1 hour of stimulation with LPS, aPL HL5B, aPL HL7G, or TLR7 agonist R848; relative expression induced by aPLs was normalized to IgG isotype control; n=6, *P<0.05 compared to stimulation without inhibitor; two-way ANOVA, Sidak’s multiple comparisons test. (B) Volcano plot of aPL HL7G-induced transcripts, including Stat1, a known IFN-regulated gene. (C) Induction of Tnf in CD115+ splenic monocytes from ProcrC/S or strain matched ProcrWT mice (upper panel) and human trophoblast cells (lower panel) stimulated for 1 or 3 hours with IgG (100 µg/ml) isolated from APS patients with confirmed reactivity to cardiolipin alone (α-CL), β2GPI alone (α-β2GPI), or dual reactivity (α-CL/β2GP). Human trophoblast cells were pretreated with either non-inhibitory α-EPCR 1489 or inhibitory α-EPCR 1496; *P<0.0001; one-way ANOVA. (D) Live-cell imaging of aPL HL5B IgG or F(ab′)2 colocalization (green) with cholera toxin B (CTB; magenta) or EPCR (blue) using non-inhibitory α-EPCR 1489 in human MM1 cells. Nuclei were stained with Hoechst 33342 (gray). Quantification of colocalization; n=3 ROI (regions of interest) consisting of at least three cells, *P<0.0001; one-way ANOVA. (E) Live-cell imaging of HL5B internalization (green) in monocytes of the indicated mouse strains with CTB or LysoTracker counterstaining (magenta). Nuclei were stained with Hoechst 33342 (blue); bar=5 µm; n=3 ROI, *P<0.0001; one-way ANOVA.
EPCR-dependent aPL signaling was conserved in human monocytes and endothelial cells (fig. S2, B to D). In mouse monocytes, aPL signaling occurred independently of Lrp8 (fig. S2E), a known co-receptor for signaling by EPCR-protein C (23) and by β2GPI-reactive aPLs (10, 13, 14). EPCR has a single intracellular, membrane proximal Cys residue with potential cell signaling functions. Importantly, inactivation of EPCR Cys242 by knock-in mutagenesis to Ser in a mouse line, ProcrC/S mice, was sufficient to prevent aPL signaling (fig. S2E), including the induction of Ifna, Ifnb, Ifng, and Stat1, a prototypic IFN-regulated gene (fig. S2F) selected from a genome-wide profiling of aPL-stimulated mouse monocytes (Fig. 1B). The monocyte transcriptional profile induced by aPL HL7G overlapped with the transcriptional response to the TLR7 agonist R848 and to the TLR3 agonist PolyI:C and the TLR9 agonist CpGB to some extent (fig. S3, A and B). However, aPL HL7G induced an apparently more robust upregulation of these target genes in comparison to R848, in line with prior findings measuring IFN-α secretion (12). Remarkably, unlike EPCR-deficient cells (20), ProcrC/S monocytes showed no defect in LPS signaling (fig. S2F). Thus, EPCR has a distinct signaling function in aPL pathology.
We confirmed these data with IgG fractions isolated from previously characterized APS patients (8, 15). These IgG fractions have three distinct diagnostic reactivities: (1) reactivity with cardiolipin only (similar to aPL HL5B), (2) cross-reactivity with cardiolipin and β2GPI (similar to aPL HL7G), or (3) reactivity with β2GPI alone. Rare APS IgG with β2GPI reactivity alone (αβ2GPI; 2/20 patients) did not induce rapid proinflammatory responses in mouse monocytes or human trophoblast cells (Fig. 1C). By contrast, IgG fractions reactive to lipid, either alone (11/20 patients) or with β2GPI cross-reactivity (7/20 patients), robustly induced TNF in both cell types (Fig. 1C). Lipid-reactive APS IgG signaling was markedly reduced in mouse ProcrC/S monocytes and in human trophoblast cells treated with inhibitory α-EPCR 1496 (Fig. 1C). Thus, EPCR plays an essential species conserved aPL signaling role in innate immune and embryonic cells.
These data suggest that EPCR is a receptor for aPLs in proinflammatory signaling which depends on aPL endosomal translocation. In monocytes, aPL internalization requires aPL-induced dissociation of a cell surface TF–FVIIa–FXa–TFPI complex leading to PAR1–PAR2 heterodimer signaling by thrombin (fig. S1) (11). aPLs also trigger Fc-mediated complement activation, which is coupled to protein disulfide isomerase (PDI)-mediated TF conformational changes leading to TF–FVIIa endosomal trafficking. Incubation with the intact IgG form of aPL HL5B induced internalization of both HL5B and EPCR, which colocalized intracellularly (Fig. 1D). F(ab′)2 fragments of HL5B, which lack the ability to fix complement, bound to the cell surface, but were neither internalized nor induced EPCR internalization (Fig. 1D). Similarly, inhibitors of complement, PDI and coagulation prevent EPCR internalization, as previously shown for TF–FVIIa (fig. S3C). Importantly, whereas aPLs failed to bind EPCR-deficient or EPCR-blocked monocytes, aPLs bound to ProcrC/S monocytes but did not internalize (Fig. 1E). Thus, EPCR serves as receptor for aPL endosomal trafficking.
EPCR cell-surface presentation of endosomal LBPA
Lipid reactive aPL HL5B, but not HL7G inhibited protein C activation on endothelial cells (fig. S4A), although both aPLs efficiently induced TNF. This indicated that aPL signaling is uncoupled from the physiological function of EPCR in anticoagulation. Accordingly, among non-inhibitory antibodies to mouse EPCR screened in this assay, α-EPCR 1682 specifically blocked signaling (Fig. 2A) and internalization, but not surface binding of lipid-reactive aPLs (Fig. 2B). Surprisingly, α-EPCR 1682 did not react with EPCR expressed at normal levels on monocytes from ProcrC/S mice (Fig. 2C). Staining of TfpiΔK1 monocytes, which cannot form a TF–FVIIa–FXa–TFPI complex known to recycle (24), excluded the possibility that α-EPCR 1682 specifically recognized EPCR associated with the inhibited complex. We therefore hypothesized that structural alterations of EPCR caused the differential antibody reactivity.
Figure 2: EPCR presents late endosomal lysobisphosphatidic acid (LBPA) on the cell surface.
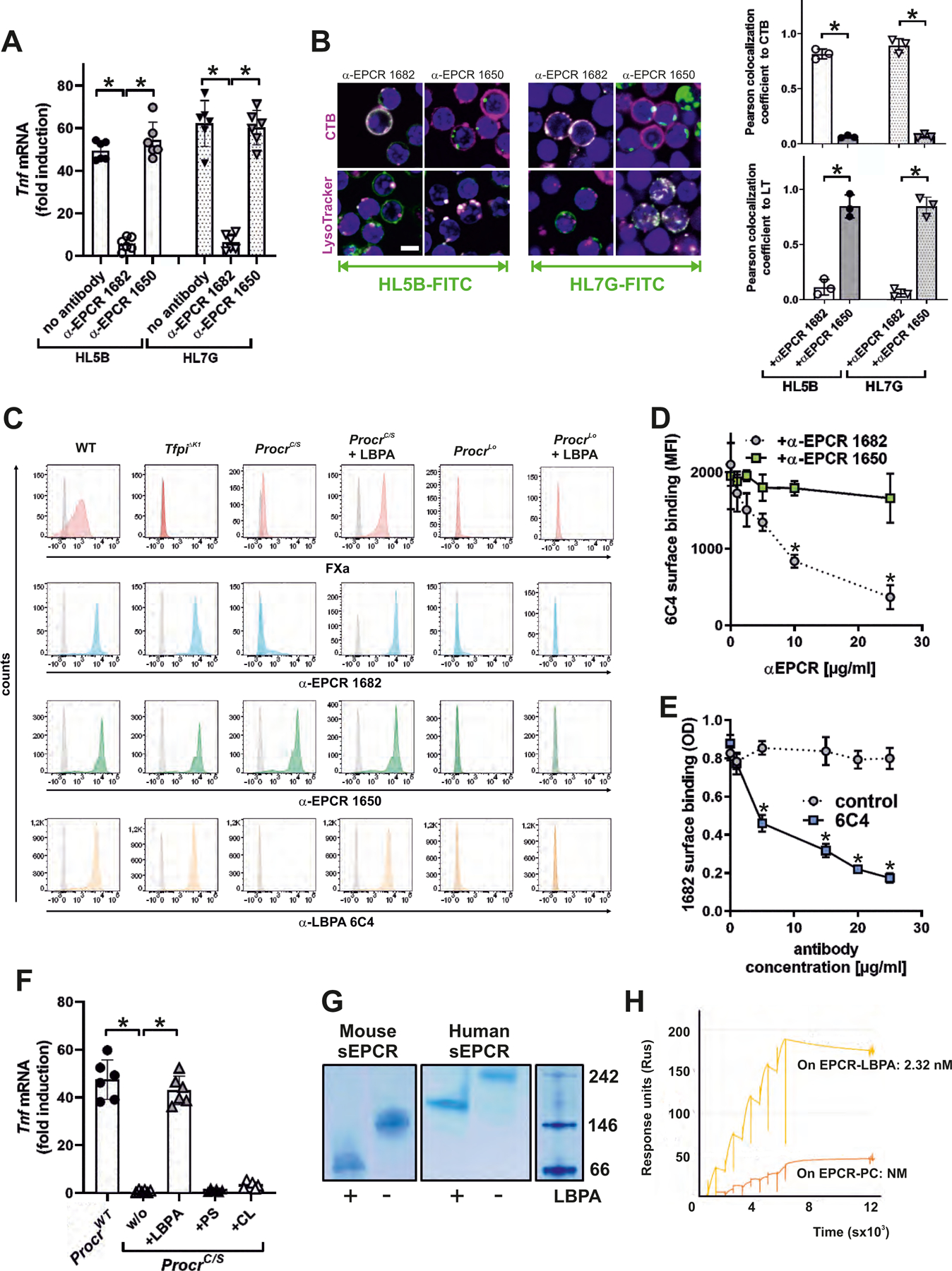
(A) Effect of blocking mouse EPCR with α-EPCR 1682 versus α-EPCR 1650 on Tnf mRNA induction after 3 hours of stimulation of mouse monocytes by aPL HL5B and HL7G; n=6, *P<0.0001; one-way ANOVA. (B) Effect of α-EPCR on aPL HL5B and HL7G (green) internalization in mouse CD115+ spleen monocytes with CTB or LysoTracker counterstaining (magenta). Bar=5 µm. Quantification of colocalization; n=3 ROI consisting of at least three cells, *P<0.0001; one-way ANOVA. (C) Flow cytometric detection of EPCR, LBPA, and FXa as marker for inhibited TF complex formation on CD115+ spleen monocytes isolated from the indicated mouse strains. Effect of pretreatment with 10 µM LBPA for 10 min on surface staining by the indicated antibodies in comparison to isotype control (gray). (D) Competition of α-EPCR 1650 and 1682 for binding of FITC-labeled α-LBPA 6C4 to mouse monocytes; n=3, *P≤0.001; two-way ANOVA, Sidak’s multiple comparisons test. (E) Competition of α-LBPA 6C4 for binding of α-EPCR 1682 to mouse monocytes; n=3, *P≤0.001; two-way ANOVA, Sidak’s multiple comparisons test. (F) Effect of pretreatment with LBPA, cardiolipin (CL), and phosphatidylserine (PS) (10 µM) on aPL HL5B signaling in ProcrC/S monocytes. Induction of Tnf mRNA after 3 hours is shown; n=6, *P<0.0001; one-way ANOVA. (G) LBPA loading of purified mouse or human sEPCR evidenced by faster mobility on native gels. (H) Surface plasmon resonance analysis of aPL HL5B binding to purified human sEPCR or sEPCR–LBPA. The affinity calculation was based on a monovalent binding model because no cooperative binding was evident. Affinity for EPCR with the typical structural lipid phosphatidyl choline (PC) (25) (EPCR-PC) was not measurable (NM).
EPCR function is dependent on structurally bound lipid (25, 26). Because the late endosomal lipid lysobisphosphatidic acid (LBPA) colocalizes with aPLs after internalization (27) and EPCR and aPLs showed similar endosomal trafficking patterns that were prevented by the ProcrC/S mutation (Fig. 1, D and E), we hypothesized that EPCR recycling altered α-EPCR 1682 reactivity by lipid exchange. Supporting the possibility that LBPA replaced the structurally bound lipid of EPCR, non-permeabilized cells expressing EPCR, but not EPCR-deficient or signaling-defective ProcrC/S cells, could be stained with α-LBPA 6C4 (27) (Fig. 2C). Importantly, simply adding LBPA to the culture medium of ProcrC/S, but not EPCR-deficient (Procrlo) cells restored cell surface α-LBPA 6C4 as well as α-EPCR 1682 staining (Fig. 2C). The cell surface presentation of LBPA by EPCR was further substantiated by experiments demonstrating that α-EPCR 1682 specifically prevented binding of α-LBPA 6C4 to mouse monocytes (Fig. 2D). Conversely, α-LBPA 6C4 competed with surface binding of α-EPCR 1682 (Fig. 2E).
The addition of LBPA to ProcrC/S monocytes restored HL5B mediated Tnf mRNA induction. Remarkably, supplementation with the commonly assumed aPL target cardiolipin (CL) or the procoagulant phosphatidylserine (PS) did not restore aPL pro-inflammatory signaling of ProcrC/S cells (Fig. 2F), confirming the functional role of EPCR loading specifically with LBPA. Exposure of purified recombinant human or mouse soluble EPCR (sEPCR) (18) to LBPA yielded a re-purified protein with a marked shift in mobility on native gels (Fig. 2G). Structurally bound phosphatidylcholine in sEPCR was replaced by LBPA after loading (fig. S4B). Thus, LBPA likely occupies the previously identified lipid-binding pocket for phosphatidylcholine in the CD1d-like structural fold of EPCR (25).
Although lipid exchange did not alter competition of sEPCR with protein C activation (fig. S4C), only purified human sEPCR–LBPA tightly bound aPL HL5B in contrast to unmodified sEPCR (Fig. 2H). Lipid-reactive aPL, but not the β2GPI-specific aPL rJGG9 without lipid reactivity (8), recognized heterologous expressed EPCR after loading with LBPA (fig. S4, D and E). Hypermutated aPL HL7G with acquired β2GPI cross-reactivity bound with similar affinity as its precursor HL5B to human and mouse EPCR specifically after loading with LBPA (fig. S4, F and G). Thus, the clonal evolution of aPLs preserves high-affinity binding to the pathogenic target EPCR–LBPA.
Activation of cell-surface acid sphingomyelinase by EPCR–LBPA
It remained unclear how α-EPCR–LBPA 1682 inhibited aPL signaling without directly competing for aPL cell-surface binding (Fig. 2B). Because aPLs rapidly expose extracellular procoagulant PS (11), which is induced by acid sphingomyelinase (ASM) (28), we blocked ASM with desipramine. ASM was required for aPL-mediated PS exposure and the activation of coagulation (Fig. 3A) as well as aPL internalization (Fig. 3B) and signaling (fig. S5A). Various agonists, including thrombin, induce ASM activation and cell surface translocation (29). Within 15 min, aPL maximally stimulated ASM activity in human monocytes in a manner dependent on the activation of coagulation and thrombin-mediated PAR1 cleavage. However, ASM activation was independent of complement, PDI, or integrin trafficking (Fig. 3C), which are required for aPL internalization (fig. S1). This pathway of ASM activation was conserved in the mouse (fig. S5B). Importantly, F(ab′)2 of aPL HL5B also induced ASM activity (Fig. 3C) and promoted the thrombin-dependent appearance of ASM on the cell surface (Fig. 3D). Thus, ASM activation is an early event that precedes complement-dependent aPL internalization.
Figure 3: aPLs induce EPCR–LBPA activation of cell surface ASM.
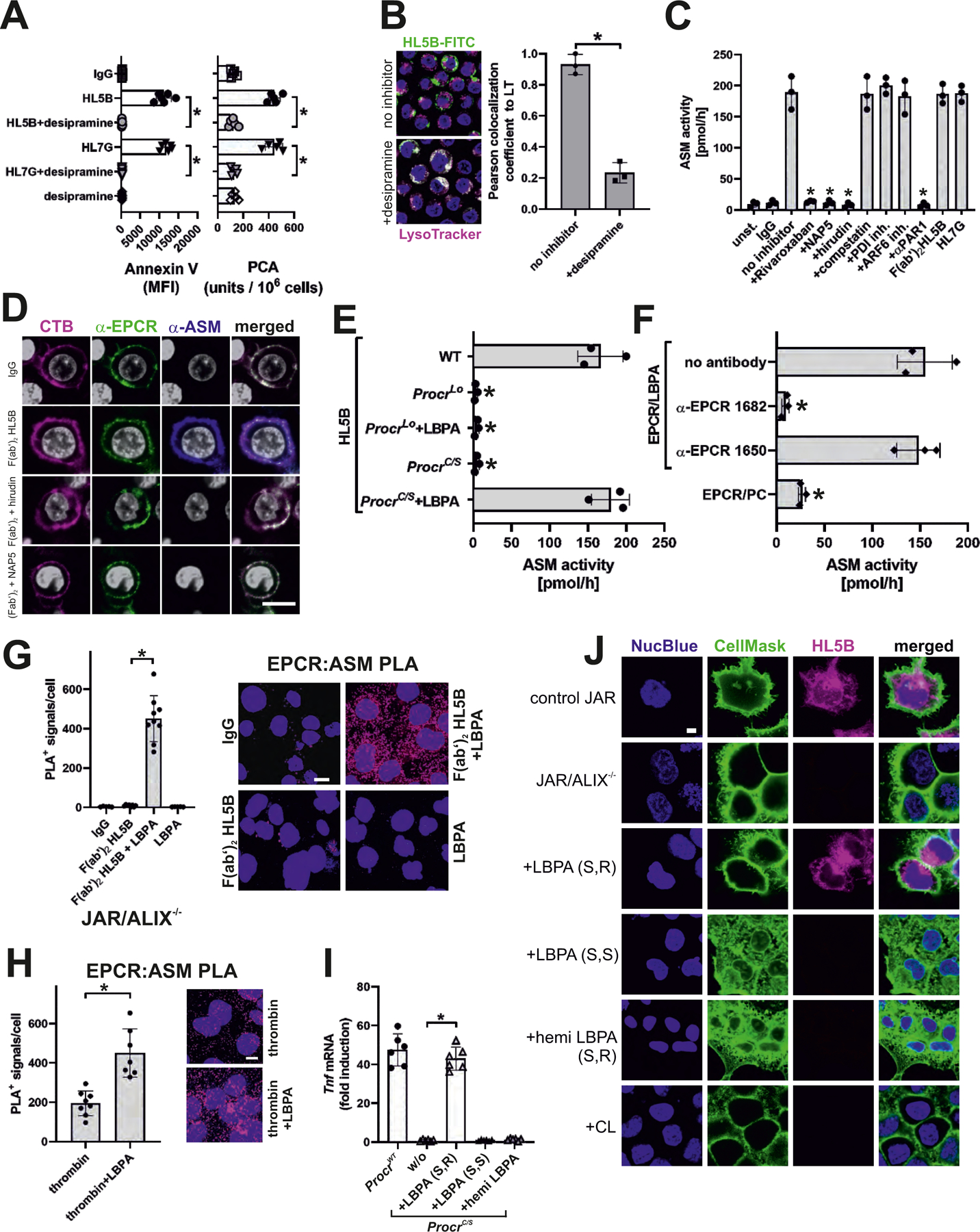
(A) aPL-mediated phosphatidylserine exposure (measured by Annexin V-FITC staining) and aPL-mediated TF activation in MM1 cells (measured as procoagulant activity, PCA) were prevented by desipramine; n=6, *P<0.0001; one-way ANOVA. (B) aPL internalization in MM1 cells is blocked by sphingomyelinase inhibitor desipramine. Bar=5 µm. Quantification of colocalization; n=3 ROI consisting of at least three cells, *P=0.002; t-test. (C) aPL-induced ASM activity in MM1 cells is blocked by inhibitors of FXa (Rivaroxaban, NAP5), thrombin (hirudin), and PAR1 cleavage (αPAR1, ATAP2/WEDE15) but not by inhibitors of complement (compstatin), PDI and ARF6; n=3, *P<0.0001; one-way ANOVA. (D) Surface ASM exposure in MM1 cells after 30 min of stimulation with aPL HL5B F(ab′)2. Scale bar=5 µm. Live-cell imaging of EPCR colocalization (green) with cholera toxin B (CTB; magenta) or ASM (blue). Nuclei were stained with Hoechst 33342 (gray). (E) LBPA (10 µM) loading of mouse ProcrC/S cells enables ASM activation in CD115+ monocytes stimulated with HL5B; n=3, *P<0.0001; one-way ANOVA. (F) ASM activity in unstimulated mouse monocytes lysates after addition of sEPCR–LBPA (2.5 µM) is blocked by α-EPCR–LBPA 1682; n=3, *P<0.0001; one-way ANOVA. (G, H) Proximity ligation assays (PLA) with magenta fluorescence dots for ASM and EPCR on ALIX−/− trophoblast cells after 10 min of stimulation with aPL HL5B F(ab′)2 fragments (G) or thrombin (H) with or without LBPA loading. Nuclei were stained with Hoechst 33342 (blue) Scale bar=25 µm. (I) aPL HL5B signaling in ProcrC/S monocytes was restored by adding 10 µM LBPA (S,R), but not by LBPA (S,S) or hemi LBPA. Stimulation time 3 hours; n=6, *P<0.0001; one-way ANOVA. (J) aPL HL5B internalization in ALIX-deficient JAR cells after pretreatment with the indicated phospholipids at 10 µM. Scale bar=5 µm.
ASM requires LBPA for its activity (30) and ASM activation by aPL was inhibited by α-EPCR–LBPA 1682 (fig. S5B). The extracellular addition of LBPA to ProcrC/S monocytes but not to EPCR-deficient cells restored ASM activation by aPL (Fig. 3E). Thrombin-mediated ASM surface expression activated ASM (fig. S5C). This activation was blocked by extracellular addition of α-EPCR–LBPA 1682. TfpiΔK1 cells express EPCR–LBPA but lack surface FXa required for aPL-induced thrombin generation (Fig. 2C). In line with thrombin-dependent ASM surface translocation, ASM activation in TfpiΔK1 cells was no longer triggered by aPL (fig. S5C). However, direct thrombin stimulation of TfpiΔK1 cells induced ASM activity and this effect was blocked by extracellular addition of α-EPCR–LBPA 1682. Importantly, addition of purified mouse sEPCR–LBPA, but not unmodified sEPCR to cell lysates of unstimulated monocytes efficiently induced ASM activity, which was inhibited by α-EPCR–LBPA 1682 (Fig. 3F). Thus, thrombin signaling mobilizes ASM to the cell surface for stimulation of its activity by EPCR–LBPA.
We next analyzed the proposed pathway in human trophoblast cells by knock-down of ALIX (ALG-2-interacting protein X, PDCD6IP) (fig. S6A), which is required for endosomal organization and trafficking of LBPA (31). ALIX knockdown diminished LBPA cell-surface presentation, but not EPCR expression (fig. S6B) and abolished aPL-induced TNF mRNA expression (fig. S6C). Addition of extracellular LBPA restored aPL signaling in ALIX−/− cells.
Proximity ligation assays (PLA) showed that EPCR and ASM colocalized after stimulation with thrombin or aPL F(ab′)2 HL5B and this interaction was prevented by the thrombin inhibitor hirudin (fig. S6D). In ALIX−/− cells, F(ab′)2 HL5B induced colocalization of ASM and EPCR only after addition of exogenous LBPA (Fig. 3G). In addition, thrombin cell surface recruitment of ASM resulted in increased proximity ligation with EPCR when LBPA was added to ALIX−/− cells (Fig. 3H). Thus, EPCR presents LBPA for direct interaction with cell-surface ASM and thereby promotes ASM-dependent aPL signaling.
Human ALIX−/− trophoblast cells and murine ProcrC/S monocytes provided tools to compare the species conservation of lipid presentation by EPCR. Only the addition of S/R 18:1 LBPA, but not S/S 18:1 LBPA, hemi-S/R LBPA, or cardiolipin restored aPL signaling in ProcrC/S monocytes (Fig. 3I) and aPL HL5B binding to ALIX−/− trophoblast cells (Fig. 3J). Thus, human and mouse EPCR present LBPA with the same selectivity, providing an explanation for the remarkable species cross-reactivity of pathogenic aPL documented in this and other studies.
EPCR contributions to aPL-induced pathologies
aPLs bind to EPCR on the cell surface to induce thrombin–PAR1 signaling which translocates ASM for cell-surface activation by EPCR–LBPA. In turn, the modification of surface lipid by ASM is required for complement-dependent endosomal trafficking and signaling of EPCR-bound aPL (Fig. 4A). The conservation of the signaling mechanism in monocytes and trophoblast cells prompted us to analyze the role of EPCR in a mouse model of aPL-induced fetal loss. Although EPCR plays a pivotal role in maintaining embryonic trophoblast function and survival (32), we found no significant spontaneous embryo loss in ProcrC/S mice or Procrlo mice relative to WT controls (Fig. 4B). However, EPCR signaling-deficient mice were protected from fetal loss induced by lipid-reactive aPL HL5B. These data imply that aPL-EPCR signaling is crucial for pregnancy complications induced by aPL in vivo.
Figure 4: aPL–EPCR signaling promotes fetal loss and thrombosis.
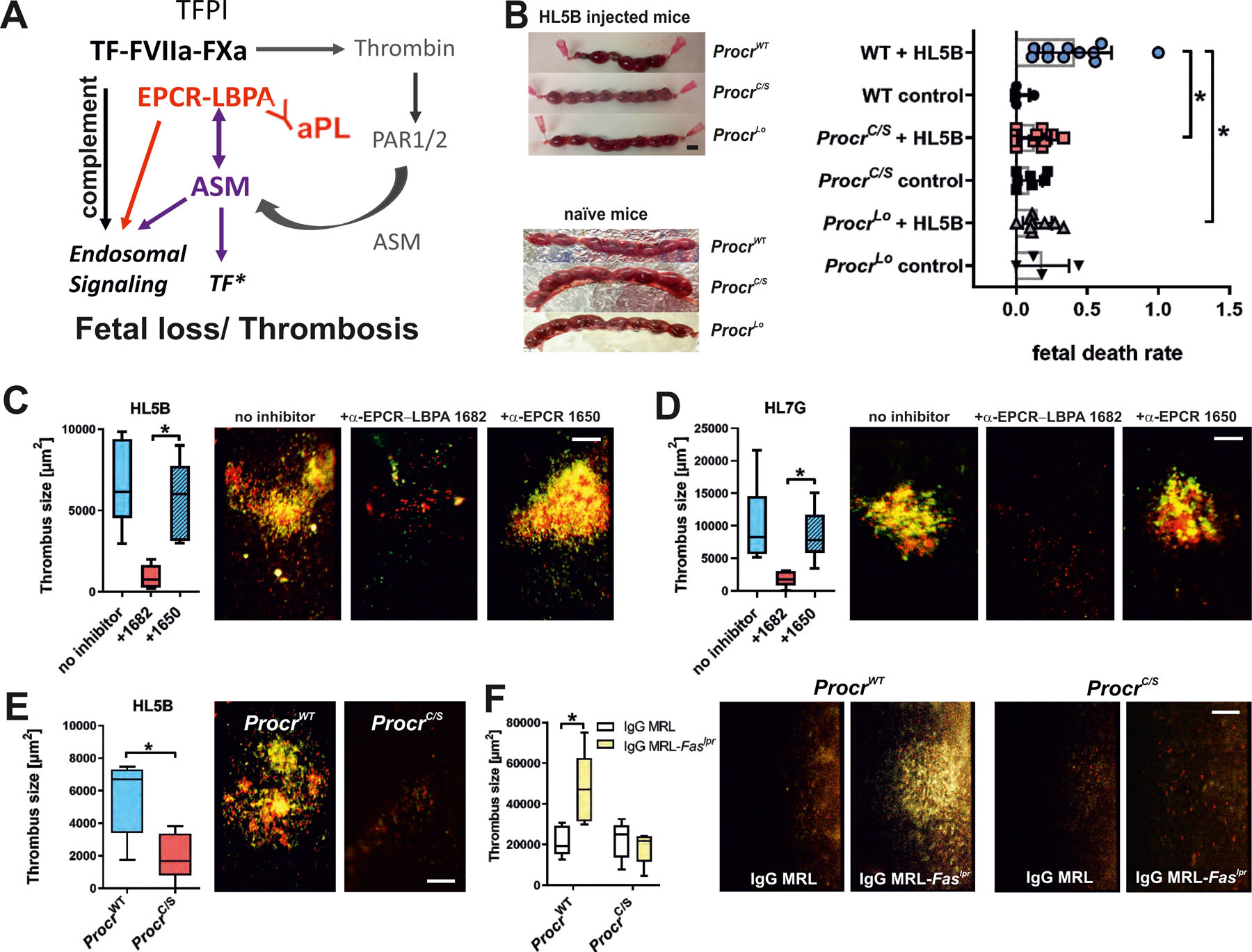
(A) Schematic representation of the role of ASM in aPL signaling and thrombosis induction. See also fig. S1 for description of the initial reactions following aPL-induced dissociation of the inhibited TF complex. (B) Representative uteri of pregnant mice of the indicated strains. Pregnancy loss was scored at day 15.5 p.c. after injection of 100 µg aPL HL5B on days 8 and 12; *P<0.02; one-way ANOVA. Scale bar=5 mm. (C and D) HL5B- or HL7G-induced thrombosis in WT mice treated with the indicated α-EPCR antibodies. Quantification of thrombus size in vena cava inferior 3 hours after aPL injection; median, interquartile range, and range; n=6–11, *P<0.004; one-way ANOVA. Platelets are shown in red and leukocytes are shown in green. (E and F) Thrombosis induction by aPL HL5B (E) or IgG isolated from age-matched 16-week-old MRL/MpJ control mice or MRL-Faslpr mice with lupus-like syndrome (F) in the indicated mouse strains. Quantification of thrombus size 3 hours after aPL injection; median, interquartile range, and range; n=5, *P=0.0025; two-way ANOVA with Sidak’s multiple comparisons test. Platelets are shown in red and leukocytes are shown in green.
We capitalized on the unique inhibitory properties of mouse monoclonal α-EPCR–LBPA 1682 in aPL pathological signaling without abolishing the generation of anti-coagulant activated protein C (Fig. 2A and fig. S4A) and evaluated whether specific targeting of EPCR–LBPA could block monocyte-dependent thrombosis (15, 33). Thrombosis induced by aPL HL5B was markedly attenuated by α-EPCR–LBPA 1682, but not by the isotype matched non-inhibitory α-EPCR 1650 (Fig. 4C). Similarly, the induction of Lrp8-independent thrombosis (fig. S7A) by the dual-reactive aPL HL7G was blocked specifically by α-EPCR–LBPA 1682 (Fig. 4D). ProcrC/S as compared to strain-matched WT controls were also resistant to aPL HL5B-induced thrombosis (Fig. 4E).
In order to assess the broader implications of these findings for autoimmune pathologies, we isolated IgG fractions from MRL-Faslpr mice which develop spontaneously lupus-like autoimmune pathologies and coagulation-related tissue injury (34). At the age of 16 weeks, MRL-Faslpr mice had high IgG titers to cardiolipin, LBPA, prothrombin, protein S, and some reactivity with β2GPI compared to age-matched MRL/MpJ control mice (fig. S7B). MRL-Faslpr IgG induced EPCR–LBPA-dependent IFN responses in monocytes (fig. S7C) and thrombosis (Fig. 4F). The thrombosis induced by these polyreactive IgG was essentially absent in ProcrC/S mice and was comparable to levels observed in WT mice injected with control IgG (Fig. 4F). Thus, EPCR–aPL signaling is required for thrombosis, even when triggered by polyclonal IgG from animals with a complex lupus-like pathology.
Development of autoimmunity by aPL-induced IFN signaling
The upregulation of IFN responses in circulating immune cells is linked to the development of APS (35, 36) and TLR7 contributes to autoimmunity in lupus erythematosus (21, 22, 37). We next asked whether EPCR signaling not only contributes to APS pathologies, but also to the development of autoimmunity. Immunization with lipid-reactive monoclonal or polyclonal antibodies induces the appearance of cardiolipin-reactive antibodies in mice (38, 39). Accordingly, immunization with lipid-reactive aPL HL5B, but not isotype control, produced sustained anti-cardiolipin titers. This response was absent in Tlr7−/− mice, whereas Tlr9−/− mice displayed an enhanced response (Fig. 5A), in line with negative regulation of lupus pathology by TLR9 (37). Thus, aPLs develop in a model of autoimmune disease dependent on endosomal TLR7 signaling.
Figure 5: TLR7 is required for IFN signaling-dependent expansion of B cells producing EPCR–LBPA-reactive aPLs.
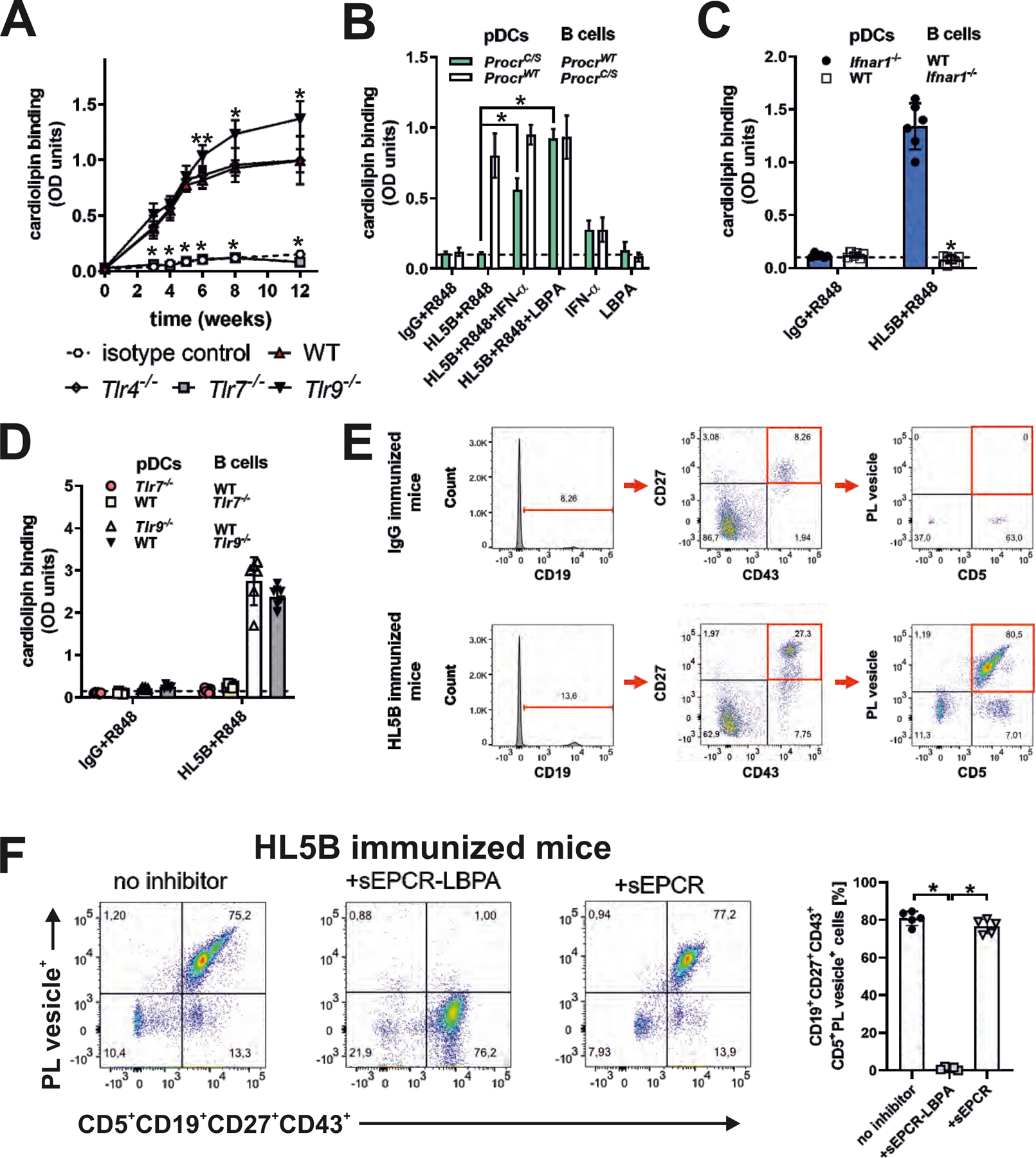
(A) Mice of the indicated genotypes were immunized with aPL HL5B or isotype-matched control IgG and serum anti-cardiolipin titers were determined at the indicated times; n=10, *P<0.005 for Tlr7−/− or Tlr9−/− versus WT; one-way ANOVA. (B to D) Isolated spleen pDCs and B cells from the indicated mouse strains were co-cultured in the presence of TLR7 agonist R848, aPL HL5B, IFN-α, and LBPA as indicated for 8 days, followed by determination of anti-cardiolipin titers; n=6, *P<0.0001; one-way ANOVA. (E) Gating for CD5+CD19+CD27+CD43+ memory type B1a cells and demonstration of their reactivity with negatively charged fluorescent phospholipid vesicles specifically in mice immunized with aPL HL5B, but not isotype-matched IgG. (F) Competition of sEPCR–LBPA, but not sEPCR with phospholipid vesicle binding to B1a cells; n=5, *P<0.0001; one-way ANOVA.
To further analyze this unusual response to immunization with aPLs, we established an in vitro co-culture model. In mouse plasmacytoid DCs (pDCs), aPL HL5B induced much more potently IFN-regulated transcripts dependent on EPCR in comparison to TLR7 agonist R848 (fig. S8A). Stimulation of pDCs by aPLs is also more efficient than TLR7 agonist to induce rapid TLR7-dependent IFN-α secretion (12), which in turn can license B cells for immunoglobulin production without T cell help (40). In pDC–B cell co-cultures, only the combination of aPL HL5B and R848 induced cardiolipin-reactive antibodies of the IgG isotype. The antibodies did not develop when mouse pDCs, but not B cells, were isolated from ProcrC/S mice (Fig. 5B). The addition of LBPA or IFN-α restored the induction of anti-cardiolipin producing B cells in co-cultures with aPL-signaling–deficient ProcrC/S pDCs (Fig. 5B). The appearance of lipid-reactive antibodies required type I IFN receptor expression by B cells, but not pDCs (Fig. 5C). Specifically, the presence of B1a cells was required (fig. S8B), demonstrating that aPLs induce IFN production by pDCs to stimulate B cells.
In this co-culture system, TLR7 was required on both pDCs and B cells, in line with the previously demonstrated requirement of TLR7 for the aPL-mediated induction of IFN-α (12) and a role for TLR7 signaling in B cell expansion. By contrast, the development of aPLs was rather enhanced in co-cultures from mice lacking TLR9 (Fig. 5D), consistent with our in vivo results (Fig. 5A). The synergistic stimulation of anti-cardiolipin production by aPL HL5B and the TLR7 agonist R848 also applied to co-cultures of human pDCs with B cells (fig. S8C). Under these conditions, EPCR was upregulated on pDCs (fig. S8D) and specific antibody inhibition of human EPCR prevented the production of cardiolipin-reactive antibodies (fig. S8C).
We next searched for circulating lipid-reactive B cells in vivo. Immunization with aPLs promoted the appearance of circulating B1a cells (CD5+CD19+CD27+CD43+) reactive with fluorescently labeled negatively charged liposomes (4) (Fig. 5E). Liposome staining of this B1a cell population was prevented by addition of sEPCR–LPBA, but not by unmodified sEPCR (Fig. 5F). Thus, EPCR–LPBA-reactive B1a cells expand in immunized mice that develop anti-cardiolipin titers.
To address the role of EPCR in the development of autoimmunity, we first showed that anti-cardiolipin titers did not develop in immunized ProcrC/S mice in sharp contrast to strain-matched WT controls or β2GPI signaling-deficient Lrp8−/− mice (Fig. 6A). Polyclonal IgG isolated from mice with persistent anti-cardiolipin titers 12 weeks after the beginning of HL5B immunization, but not IgG from immunized ProcrC/S mice, elicited monocyte responses typical for human aPLs or pathogenic IgG from mice developing a lupus-like syndrome (Fig. 6B). Thus, lipid-reactive aPLs not only cause APS pathologies but also expand in vivo in an EPCR-dependent manner.
Figure 6: EPCR–LBPA signaling drives aPL expansion and autoimmune pathology in vivo.
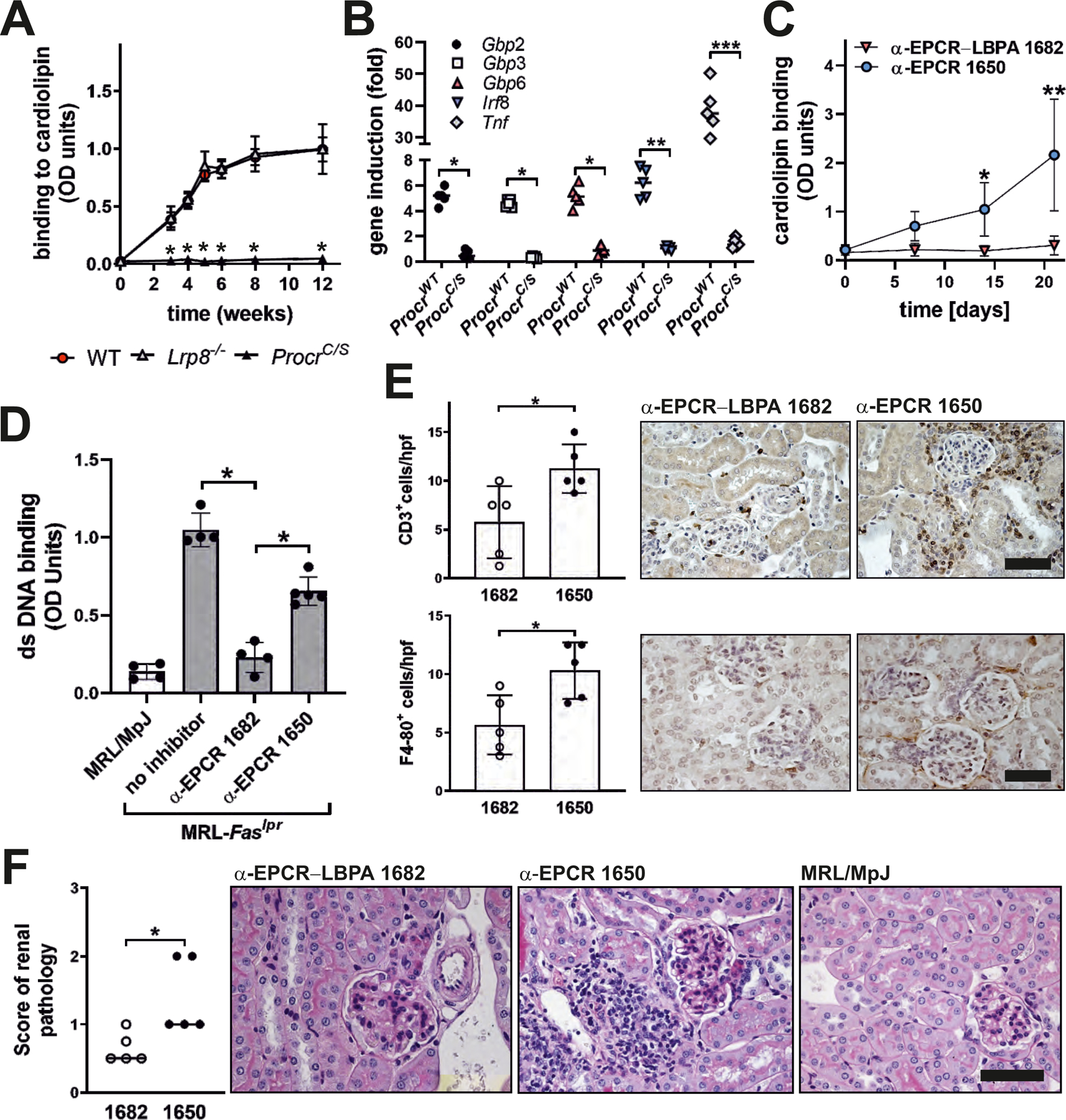
(A) Mice of the indicated genotypes were immunized with aPL HL5B and anti-cardiolipin titer determined at the indicated times; n=10, *P<0.0001 for ProcrC/S vs. WT; one-way ANOVA. (B) IgG isolated 12 weeks after the start of aPL immunization were used to stimulate human MM1 cells for 1 hour for induction of the indicated genes; n=5, *P<0.05, **P=0.011, ***P<0.0001; two-way ANOVA, Sidak’s multiple comparisons test. (C) MRL-Faslpr lupus-prone mice were treated with the indicated α-EPCR antibodies at an age of 4 weeks (day 0) and anti-cardiolipin titers were determined in serum at the indicated time points; n=5, *P=0.03; **P<0.0001; two-way ANOVA, Sidak’s multiple comparisons test. (D) Antibodies to double stranded (ds) DNA were measured in α-EPCR 1650- and α-EPCR–LBPA 1682-treated MRL-Faslpr mice 2 weeks after the last dose or in 6-week-old MRL/MpJ control or MRL-Faslpr mice; n=4–5, *P<0.0001; one-way ANOVA. (E) Immune cell infiltration of α-EPCR-treated MRL-Faslpr mice; n=5, *P<0.025. (F) Renal pathology scores of α-EPCR-treated MRL-Faslpr mice; n=5, *P=0.0317; Mann–Whitney U test. Scale bars=80 µm.
MRL-Faslpr mice develop aPLs spontaneously in early stages of lupus-like immunopathology, which is dependent on TLR7 and amplified in Tlr9−/− mice (37). Based on these similarities with experimental aPL induction, we evaluated the role of EPCR–LBPA signaling for spontaneous aPL development in lupus. MRL-Faslpr mice at an age of 4 weeks were randomized to treatment with α-EPCR–LBPA 1682 or the non-inhibitory, isotype-matched α-EPCR 1650 for 3 weeks. Anti-cardiolipin titers were analyzed weekly. Specific inhibition of EPCR–LBPA completely prevented the development of aPLs (Fig. 6C) as well as double-stranded DNA autoantibodies, which were detectable already in 6-week-old MRL-Faslpr mice but not control MRL/MpJ mice (Fig. 6D). Treatment of MRL-Faslpr mice with α-EPCR–LBPA 1682 not only reduced the development of autoantibodies but also protected from progressive kidney pathology as evidenced by reduced CD3+ and F4/80+ immune cell infiltration in the kidneys (Fig. 6E) and reduced renal pathology scores reflecting glomerular and interstitial damage (Fig. 6F).
In an independent experiment, MRL-Faslpr mice were treated with α-EPCR–LBPA 1682 or α-EPCR 1650 for 6 weeks and analyzed 2 weeks after the end of treatment. Α-EPCR–LBPA 1682 again specifically suppressed serum α-LBPA and α-CL titers to levels seen in aged-matched MRL/MpJ control mice (fig. S9A) and attenuated kidney infiltration of CD45+/F4/80+ immune cells measured by flow cytometry (fig. S9B). These infiltrating myeloid cells expressed IFN-γ (fig. S9C). Albuminuria only developed in mice treated with non-inhibitory α-EPCR 1650, but not with inhibitory α-EPCR–LBPA 1682 or in MRL/MpJ control mice (fig. S9D). Thus, EPCR–LBPA signaling is crucial for both the development of lipid-reactive antibodies as well as, more generally, the development of kidney autoimmune pathology in this endosomal TLR7-dependent animal model.
Discussion
Here we delineate how recognition of a specific cell-surface antigen by aPLs induces complex pathogenic signaling that involves the major humoral innate defense pathways. By binding to EPCR expressed on myeloid cells, aPLs target a crucial toggle switch that links coagulation and innate immune signaling (20). EPCR acts as the co-receptor for TF–FVIIa−FXa–PAR2 signaling and thereby plays a pivotal role in the TLR4-induced IFN response. This function of EPCR is controlled by the anticoagulant protein C pathway, which is altered in carriers of the prevalent FVLeiden prothrombotic mutation (41). The anticoagulant protein S participates in this control of coagulation-dependent IFN signaling and, intriguingly, is one of the proteins for which aPLs develop cross-reactivity during clonal evolution (4, 42). Hypermutated lipid-reactive aPLs may therefore stimulate signaling through recognition of EPCR–LBPA, while simultaneously neutralizing the regulatory function of protein S in innate immune signaling. Genetic polymorphisms in the identified components of the aPL signaling mechanism may help define additional susceptibility risk factors that contribute to clonal evolution of autoantibodies in APS. In addition, diagnostic assays, which specifically measure antibody reactivity to the pathogenic target EPCR–LBPA, can improve precision diagnostic for APS for better risk stratification and therapy in autoimmune disease.
We here demonstrate the surface presentation of an endogenous endosomal lipid, LBPA, by EPCR primarily known for its roles in vascular signaling. The ability of autoantibodies to specifically recognize this receptor–lipid complex constitutes a central mechanism for the pathological consequences as well as development of autoimmune disease in APS. EPCR has a major histocompatibility complex (MHC)-like fold that is shared with the lipid-presenting molecule CD1d. Lipid exchange and loading through endosomal recycling is a prominent feature of CD1 molecules and plays important roles in host defense, malignancy, and the development of autoimmune disease (43–45). Since the coagulation signaling function of EPCR also activates major innate antiviral and antimicrobial responses, it appears that an evolutionarily conserved mechanism of host defense perpetuates autoimmunity and ultimately produces the major clinical manifestations of APS and lupus.
Materials and Methods
Mice
Mice with EPCR carboxyl terminal residue 242 Cys mutated to Ser (ProcrC/S mice) were generated by knock-in mutagenesis of exon 4 isolated from the 129 strain, using a modified construct employed for Procrtm2Cte targeting (46). This strain (ProcrWT) was used as control for ProcrC/S mice after backcrossing to C57BL/6J. We further used TfpiK1flfl(47) crossed with LysMcre mice, Lrp8−/− mice (48) backcrossed onto C57BL/6J, B6.129S1-Tlr7tm1Flv/J (JAX stock #008380) (49), C57BL/6J-Tlr9M7Btlr/Mmjax (JAX stock # 34329), Ifnar1−/− mice (50), Procrlo mice (51), MRL/MpJ control (MRL/MpJ, JAX stock # 000485), and MRL-Faslpr mice with spontaneous lupus-like pathology (MRL/MpJ-Faslpr, JAX stock # 006825). Experiments in age- and sex-matched mice received approval of the TSRI IACUC (#08–0009) and the Landesuntersuchungsamt Rheinland-Pfalz, Koblenz, Germany (23177–07/G14–1043/G19–0189).
Materials
Table S1 lists antibodies used. aPLs: Human monoclonal aPLs HL5B, HL7G, JGG9 and unreactive isotype control were described previously (5, 8, 15, 52, 53). The IgM aPL JGG9 was recombinantly expressed as IgG1 (rJGG9). α-EPCR: In addition to previously described α-EPCR (18, 54), EPCR-deficient mice were immunized with recombinant mouse EPCR for generating mouse anti-mouse EPCR antibodies by hybridoma technology. Antibodies with reactivity to immobilized EPCR were purified endotoxin-free from culture supernatants on a GammaBind Plus Sepharose PG column by 0.1 M acetic acid pH 3 elution. After neutralization, antibody was sterile-filtered in HBS pH 7.4 and stored frozen. Other reagents used are listed in tables S2 and S3.
Cell signaling assays
aPL signaling was evaluated in established monocyte, MonoMac-1 (MM1) (DSMZ, ACC252), endothelial, and trophoblast (JAR) cell (DSMZ, ACC462) models (11, 12, 15) cultured in RPMI, 10% FCS. Mouse monocytes were isolated from spleens on α-CD115 beads. Human CD14+ monocytes were isolated from Buffy coats of healthy donors. Inhibitors were added 15 min before stimulation with agonists, as listed in table S4. Coagulation inhibitors and EPCR antibodies were previously characterized in cell-signaling assays (11, 18, 55). EPCR-dependent signaling readouts were based on transcript changes in LPS-stimulated DCs (20) and genome wide expression analysis of aPL-stimulated monocytes. Total RNA was reverse transcribed for real-time PCR on the iCycler iQ thermal cycler (Bio-Rad) and normalization to GAPDH levels using primer sequences shown in table S5.
Microarray analysis of aPL response
RNA from ProcrWT monocytes stimulated for 3 hours with IgG control, HL7G, R848, CpG B, or Poly I:C was processed with GeneChip™ WT PLUS Reagent Kit and hybridized on a GeneChip™ Mouse Gene 2.0 ST Array (Affymetrix). Data normalization by Robust Multi-array Average (RMA) and analysis of differentially expressed probe sets by Tukey’s Bi-weight average algorithm and Analysis of Variance (ANOVA) used the Affymetrix Expression and Transcriptome Analysis Console Software. Statistical analysis for differential expression by limma linear modeling (56) and empirical Bayes statistics used a false discovery rate <0.05. Hierarchical clustering of relative expression with complete linkage also used R version 3.6.3.
Cell-binding assays
CD115+ splenocytes, trophoblast cells, or EPCR-transfected CHO cells (in Ham’s F12 medium, 10% FCS) loaded or not with 10 µM LPBA for 30 minutes (18, 57) were incubated for 15 min with FITC or POD labeled aPLs, FITC-labeled α-EPCR or α-LBPA (6C4). Fluorescence or optical densities were detected after one PBS washing step in a microplate reader. ALIX was deleted in JAR cells with gRNA vectors in the Origen kit with Viromer Red as transfection reagent. After puromycin selection, ALIX knockdown was confirmed by protein immunoblotting.
Functional assays
Endosomal ROS was detected with the fluorescent probe H2DCFDA by flow cytometry on a FACS Canto in CD11b+CD115+PI− cells, as described (11). Clotting activity and phosphatidylserine (PS) exposure by annexin 5 staining on monocytes were determined as described (15). Protein C activation was determined on Bend3 mouse endothelial cells (ATCC, CRL-2299) with 1 nM thrombin and 250 nM protein C. Inhibition of EPCR-dependent protein C activation was defined by reactions with inhibitory α-EPCR 1560 (18). ASM activity was determined with the ASM Assay (Echelon Biosciences) in cell lysates prepared by three freeze–thaw cycles.
Characterization of recombinant EPCR
Human and mouse sEPCR were expressed in Drosophila melanogaster S2 cells and purified by nickel-nitrilotriacetic acid affinity chromatography and MonoQ ion-exchange chromatography (18). Purified protein was mixed with LBPA, repurified on MonoQ, and lipid exchange confirmed on NativePAGE 4 to 16% Bis-Tris gels. Lipid loading was quantified by extracting sEPCR solutions with methanol (1:3 v/v) followed by injection onto an Acquity UPLC system with a Waters Acquity BEH column. LBPA and phosphatidylcholine were quantified after specific fragmentation on a Waters XEVO TQ-XS tandem mass spectrometer. aPL HL5B binding was measured by surface plasmon resonance after capture of human sEPCR on a lipid bilayer made of PC-SMA-PG-cholesterol-DOGS (50–20-5–20-5) immobilized on L1 chip surface. Presented experimental data are representative of three independent repeats and were fitted with the BIAevaluation3.2 package using global fitting and comparing a Langmuir 1:1 model with a bivalent analyte model.
Co-culture experiments
pDCs and B cell cocultures used human B cells from buffy coats or from murine spleen cells isolated with CD19 Micro Bead kit or mouse B1a cell isolation kit (table S3). B1a-depleted B cells were further purified by capturing α-CD19-APC-stained cells with α-APC MicroBeads. pDCs were isolated with negative selection kits for mouse or human pDCs. pDCs (5×104 cells/ml) and B cells (5×105 cells/ml) were cocultured in RPMI, 10% FCS for 8 days. α-CL titers were determined in supernatants.
aPL ELISAs
ELISA plates were coated with 2 µg/ml phospholipid in ethanol at 4°C. After blocking with 1% Tween 20, 40 µg/ml of IgG fractions, mouse sera, or culture supernatant was added for 1 hour. For protein reactivity, Immulon 2HB plates were coated with 2 µg/ml purified human coagulation factors, blocked with non-fat dry milk in TBS, and reacted with IgG diluted in TBS, 0.5% BSA. Binding was quantified with HRP-coupled anti-IgG secondary antibodies and TMB substrate.
Confocal microscopy
Imaging was on a Zeiss LSM 710 NLO confocal laser scanning microscope with a 1.4 oil DIC M27 63X plan apochromat objective (Zeiss) and built-in detector. All images were collected using the multi-track function of the Zeiss Zen software to avoid bleed-through. Internalization of aPL was visualized after incubation of cells for 30 min with 500 ng/ml labeled HL5B or IgG in the presence of 50 nM Lysotracker and α-EPCR and nuclear counterstaining with Hoechst 33342. Plasma membrane was stained on ice with 1 µg/ml cholera toxin B (CTB) after stimulation. For ASM exposure, cells were stimulated for 15 min with F(ab′)2 aPL HL5B and then stained on ice for 30 min with CTB, FITC-labeled α-EPCR 1489, rabbit α-ASM followed by α-rabbit 647, and Hoechst 33342 for live-cell imaging.
Proximity ligation assay (PLA) was performed with Duolink in situ PLA kit (Sigma-Aldrich) as described (58). JAR cells cultured on glass coverslips were stimulated for 10 min, fixed in methanol, and blocked with 2.5% normal horse serum. PLA was determined in cells stained with mouse α-EPCR 1489 and rabbit α-ASM and species-specific secondary antibodies with unique short DNA strands for enzymatic ligation and rolling circle amplification. We quantified numbers of discrete fluorescent spots per cell with Image J software in 30 fields of images acquired with the same exposure settings and gain of laser on a Leica SP8 confocal microscope with HC PL APO CS2 63X/1.40 oil objective and Leica DFC9000 GTC sCMOS camera.
Flow cytometry
Cell surface staining used directly labeled antibodies or indirect detection with species specific probes (11, 59). Internalization of proteins surface-stained with FITC-labeled antibodies was determined after quenching with 0.4% trypan blue on a FACS Canto I. aPL-producing B cells were detected in fresh EDTA blood of immunized mice by phospholipid vesicles (PL) staining (4). Data analysis used FlowJo 7.2 software (TreeStar Inc).
Inferior vena cava (IVC) thrombosis model
aPL-amplified thrombosis (60) was induced with aPL HL5B (1 μg) or 40 μg IgG purified from 16-week-old MRL/MpJ control mice (MRL/MpJ) or MRL-Faslpr (MRL-MpJ-Faslpr) mice with severe lupus-like pathology. IgG was injected via jugular catheter into 8–12 week-old male mice 1 hour before flow reduction by IVC ligation over a transiently positioned spacer (0.26 mm). Rhodamine B-labeled platelets and acridine orange to label leukocytes were infused for imaging thrombus formation on a high‐speed wide‐field Olympus BX51WI fluorescence microscope with long‐distance condenser, 10X (NA 0.3) water immersion objective with a monochromator (MT 20E; Olympus Deutschland GmbH, Hamburg, Germany), and a charge‐coupled device camera (ORCA‐R2; Hamamatsu Photonics, Hamamatsu City, Japan). Thrombi typically developed at sites where the vena cava inferior was manipulated with forceps and image analysis was performed with Realtime Imaging System eXcellence RT (Olympus Deutschland GmbH) (11, 15, 60).
aPL induced pregnancy loss
Mice were mated overnight and challenged by i.p. injection of 100 μg aPL HL5B on day 8.5 and 12.5 p.c.. At day 15.5 p.c., pregnant ProcrWT, ProcrC/S, or Procrlo mice were euthanized for scoring fetal loss macroscopically. Spontaneous fetal loss was scored in unchallenged mice. Fetal death rate was determined as the percentage of partially resorbed or missing embryos in each pregnancy.
Autoimmune disease models
Experimental APS was induced by immunization with 1 µg of aPL or control IgG in complete Freund’s adjuvant into one footpad (38, 39). After 2 weeks a booster with incomplete Freund’s adjuvant was injected into the contralateral footpad. Serum α-CL titers were measured by ELISA. The role of EPCR in lupus-like autoimmune disease was evaluated in the MRL-Faslpr model (61). MRL-Faslpr mice were randomized at an age of 4 weeks to receive treatment with 25 µg per gram of body weight i.p. α-EPCR 1650 or α-EPCR 1682 every 72 hours for 3 weeks and euthanized 5 weeks after initiation of treatment. Serum titers of α-CL and α-dsDNA were determined. Serum anti-dsDNA antibodies were measured on pre-coated plates with sera diluted 1:10 in PBS and detected using an HRP-coupled anti-IgG secondary antibody. In a repeat experiment, mice were randomized at an age of 4 weeks, treated for 6 weeks, and analyzed 2 weeks later for antibody titers and kidney pathology.
Paraffin sections (4 µm) stained with periodic acid–Schiff reagent were scored for glomerulus pathology in 20 glomeruli per kidney: 0:”normal” (15 to 25 cells per glomerular cross section (GCS); 1:”mild” (hypercellularity: 26 to 35 cells per GCS); 2:”moderate” (hypercellularity: 36 to 45 cells per GCS, segmental and/or diffuse proliferative changes, hyalinosis), 3:”severe” (hypercellularity: 46 to 60 cells per GCS, segmental or global sclerosis, necrosis, crescent formation). Interstitial/tubular pathology was assessed semi-quantitatively on a scale of 0 to 3 in 10 randomly selected high-power fields. Glomerular and interstitial/tubular scores were averaged to obtain the renal pathology score. CD68 and CD3 cell infiltration was quantified in stained sections and randomly selected high-power fields (61). Single-cell suspensions from kidneys obtained by mechanical disruption and 70-μm filtration were analyzed for immune cell composition on a FACSCalibur (Becton Dickinson, San Jose, CA). Albumin in spot-urine samples was detected with mouse albumin ELISA (Bethyl Laboratories) and normalized to creatinine content with the Creatine Assay Kit (Creative BioMart).
Statistics
Data shown are mean and standard deviation (SD), unless otherwise indicated. GraphPad Prism 7 was used for group comparisons with Student’s t test following Shapiro–Wilk test for normal distribution, Mann–Whitney U test for non-normally distributed group comparisons, or one-way ANOVA and multi-comparison correction (Dunnett’s), unless otherwise indicated.
Supplementary Material
Acknowledgments:
We thank P. Wilgenbus, S. Reyda, and G. Carlino for assistance in managing the mouse colonies and Federico Marini for advice on statistics.
Funding:
This study was funded by the National Institutes of Health (NHLBI, HL060742 and UM1-HL120877), the Humboldt Foundation of Germany (Humboldt Professorship Ruf), the Federal Ministry of Education and Research Germany (BMBF 01EO1003 and 01EO1503), the German Center for Cardiovascular Research (DZHK), and the German Research Foundation (MU4233/1–1, SFB1292).
Footnotes
Competing interests: A provisional patent application for the discoveries described in the paper has been filed (US 62/955,060).
Data and materials availability: Microarray data are available in the Gene Expression Omnibus (GEO) under the accession number GSE161575. All other data associated with this study are in the main text or supplementary materials. Materials are available from the corresponding authors upon request and may require simple academic material transfer agreements in accordance with institutional policies.
References and notes:
- 1.Giannakopoulos B, Krilis SA, The pathogenesis of the antiphospholipid syndrome. N. Engl. J Med 368, 1033–1044 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Ruff WE et al. , Pathogenic Autoreactive T and B Cells Cross-React with Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe 26, 100–113 e108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia D, Erkan D, Diagnosis and Management of the Antiphospholipid Syndrome. N Engl J Med 378, 2010–2021 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Lieby P et al. , The clonal analysis of anticardiolipin antibodies in a single patient with primary antiphospholipid syndrome reveals an extreme antibody heterogeneity. Blood 97, 3820–3828 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Prinz N, Hauser F, Lorenz M, Lackner KJ, von Landenberg P, Structural and functional characterization of a human IgG monoclonal antiphospholipid antibody. Immunobiology 216, 145–151 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Schreiber K et al. , Antiphospholipid syndrome. Nat Rev Dis Primers 4, 17103 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Wu M, Barnard J, Kundu S, McCrae KR, A novel pathway of cellular activation mediated by antiphospholipid antibody-induced extracellular vesicles. J. Thromb. Haemost 13, 1928–1940 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller-Calleja N et al. , Antiphospholipid antibody-induced cellular responses depend on epitope specificity : implications for treatment of antiphospholipid syndrome. J. Thromb. Haemost 15, 2367–2376 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Hollerbach A, Muller-Calleja N, Canisius A, Orning C, Lackner KJ, Induction of tissue factor expression by anti-beta2-glycoprotein I is mediated by tumor necrosis factor alpha. J Thromb Thrombolysis 49, 228–234 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Urbanus RT, Pennings MT, Derksen RH, de Groot PG, Platelet activation by dimeric beta2-glycoprotein I requires signaling via both glycoprotein Ibalpha and apolipoprotein E receptor 2’. J Thromb Haemost 6, 1405–1412 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Muller-Calleja N et al. , Tissue factor pathway inhibitor primes monocytes for antiphospholipid antibody-induced thrombosis. Blood 134, 1119–1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prinz N et al. , Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood 118, 2322–2332 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Romay-Penabad Z et al. , Apolipoprotein E receptor 2 is involved in the thrombotic complications in a murine model of the antiphospholipid syndrome. Blood 117, 1408–1414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulrich V et al. , ApoE Receptor 2 Mediation of Trophoblast Dysfunction and Pregnancy Complications Induced by Antiphospholipid Antibodies in Mice. Arthritis Rheumatol 68, 730–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller-Calleja N et al. , Complement C5 but not C3 is expendable for tissue factor activation by cofactor-independent antiphospholipid antibodies. Blood Adv 2, 979–986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redecha P et al. , Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood 110, 2423–2431 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G, Activation of neutrophils by the Tissue Factor-Factor VIIa-PAR2 axis mediates fetal death in antiphospholipid syndrome. J Clin Invest 118, 3453–3461 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Disse J et al. , The Endothelial Protein C Receptor Supports Tissue Factor Ternary Coagulation Initiation Complex Signaling through Protease-activated Receptors. J Biol. Chem 286, 5756–5767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graf C et al. , Myeloid cell-synthesized coagulation factor X dampens antitumor immunity. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang HP et al. , EPCR-dependent PAR2 activation by the blood coagulation initiation complex regulates LPS-triggered interferon responses in mice. Blood 125, 2845–2854 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sang A et al. , Innate and adaptive signals enhance differentiation and expansion of dual-antibody autoreactive B cells in lupus. Nat Commun 9, 3973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giannakopoulos B et al. , Deletion of the antiphospholipid syndrome autoantigen beta2-glycoprotein I potentiates the lupus autoimmune phenotype in a Toll-like receptor 7-mediated murine model. Arthritis Rheumatol 66, 2270–2280 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Sinha RK et al. , Apolipoprotein E Receptor 2 Mediates Activated Protein C-Induced Endothelial Akt Activation and Endothelial Barrier Stabilization. Arterioscler. Thromb. Vasc. Biol 36, 518–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ott I et al. , Reversible regulation of tissue factor-induced coagulation by glycosyl phosphatidylinositol-anchored tissue factor pathway inhibitor. Arterioscler. Thromb. Vasc. Biol 20, 874–882 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Oganesyan V et al. , The crystal structure of the endothelial protein C receptor and a bound phospholipid. J. Biol. Chem 277, 24851–24854 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Sagaseta J et al. , sPLA2-V inhibits EPCR anticoagulant and antiapoptotic properties by accommodating lysophosphatidylcholine or PAF in the hydrophobic groove. Blood 119, 2914–2921 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi T et al. , A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392, 193–197 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Pendurthi UR, Rao LVM, Sphingomyelin encrypts tissue factor: ATP-induced activation of A-SMase leads to tissue factor decryption and microvesicle shedding. Blood Adv 1, 849–862 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenkins RW, Canals D, Hannun YA, Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal 21, 836–846 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oninla VO, Breiden B, Babalola JO, Sandhoff K, Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J. Lipid Res 55, 2606–2619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuo H et al. , Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303, 531–534 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Isermann B et al. , The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat. Med 9, 331–337 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Subramaniam S et al. , Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 129, 2291–2302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moratz C et al. , Regulation of systemic tissue injury by coagulation inhibitors in B6.MRL/lpr autoimmune mice. Clin Immunol 197, 169–178 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Perez-Sanchez C et al. , Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann. Rheum. Dis 74, 1441–1449 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Palli E, Kravvariti E, Tektonidou MG, Type I Interferon Signature in Primary Antiphospholipid Syndrome: Clinical and Laboratory Associations. Front Immunol 10, 487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christensen SR et al. , Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Pierangeli SS, Harris EN, Induction of phospholipid-binding antibodies in mice and rabbits by immunization with human beta 2 glycoprotein 1 or anticardiolipin antibodies alone. Clin Exp Immunol 93, 269–272 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bakimer R et al. , Induction of primary antiphospholipid syndrome in mice by immunization with a human monoclonal anticardiolipin antibody (H-3). J Clin Invest 89, 1558–1563 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poeck H et al. , Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood 103, 3058–3064 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Liang HP et al. , Coagulation factor V mediates inhibition of tissue factor signaling by activated protein C in mice. Blood 126, 2415–2423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oosting JD et al. , Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: an explanation for their pathogenic mechanism? Blood 81, 2618–2625 (1993). [PubMed] [Google Scholar]
- 43.Moody DB et al. , T cell activation by lipopeptide antigens. Science 303, 527–531 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Zhou D et al. , Lysosomal glycosphingolipid recognition by NKT cells. Science 306, 1786–1789 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Mattner J et al. , Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 434, 525–529 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Gu JM et al. , Disruption of the endothelial cell protein C receptor gene in mice causes placental thrombosis and early embryonic lethality. J. Biol. Chem 277, 43335–43343 (2002). [DOI] [PubMed] [Google Scholar]
- 47.White TA et al. , Endothelial-derived tissue factor pathway inhibitor regulates arterial thrombosis but is not required for development or hemostasis. Blood 116, 1787–1794 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trommsdorff M et al. , Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Lund JM et al. , Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 101, 5598–5603 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schaupp L et al. , Microbiota-Induced Type I Interferons Instruct a Poised Basal State of Dendritic Cells. Cell 181, 1080–1096 e1019 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Castellino FJ et al. , Mice with a severe deficiency of the endothelial protein C receptor gene develop, survive, and reproduce normally, and do not present with enhanced arterial thrombosis after challenge. Thromb. Haemost 88, 462–472 (2002). [PubMed] [Google Scholar]
- 52.Buschmann C et al. , Generation and characterization of three monoclonal IgM antiphospholipid antibodies recognizing different phospholipid antigens. Ann N Y Acad Sci 1051, 240–254 (2005). [DOI] [PubMed] [Google Scholar]
- 53.von Landenberg C, Lackner KJ, von Landenberg P, Lang B, Schmitz G, Isolation and characterization of two human monoclonal anti-phospholipid IgG from patients with autoimmune disease. J. Autoimmun 13, 215–223 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Liaw PCY, Mather T, Oganesyan N, Ferrell G, Esmon CT, Identification of the protein C/activated protein C binding sites on the endothelial cell protein C receptor. J. Biol. Chem 276, 8364–8370 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Schaffner F et al. , Endothelial Protein C Receptor Function in Murine and Human Breast Cancer Development. PLoS. ONE 8, e61071-. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritchie ME et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Disse J, Ruf W, Endothelial protein C receptor is required for tissue factor ternary complex signaling in the mouse. J Thromb Haemost 9, 2516–2518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madhusudhan T et al. , Signal integration at the PI3K-p85-XBP1 hub endows coagulation protease activated protein C with insulin-like function. Blood 130, 1445–1455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamikubo Y et al. , Selective factor VIII activation by the tissue factor-factor VIIa-factor Xa complex. Blood 130, 1661–1670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manukyan D et al. , Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J. Thromb. Haemost 14, 1011–1020 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Menke J et al. , Programmed death 1 ligand (PD-L) 1 and PD-L2 limit autoimmune kidney disease: distinct roles. J Immunol 179, 7466–7477 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Norledge B, Petrovan RJ, Ruf W, Olson A, The tissue factor/factor VIIa/factor Xa complex: a model built by docking and site-directed mutagenesis. Proteins 53, 640–648 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Baugh RJ, Broze GJ Jr., Krishnaswamy S, Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J. Biol. Chem 273, 4378–4386 (1998). [DOI] [PubMed] [Google Scholar]
- 64.Ahamed J, Belting M, Ruf W, Regulation of tissue factor-induced signaling by endogenous and recombinant tissue factor pathway inhibitor 1. Blood 105, 2384–2391 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Langer F et al. , Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood 121, 2324–2335 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rothmeier AS et al. , Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood 131, 674–685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rothmeier AS et al. , Tissue Factor Prothrombotic Activity Is Regulated by Integrin-arf6 Trafficking. Arterioscler Thromb Vasc Biol 37, 1323–1331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.