

Graphical abstract
Keywords: TNBC, Differential gene expression, Distant-metastasis free survival, Prognostic gene signatures, POU2AF1, S100B
Abbreviations: BrCa, Breast cancer; TNBC, Triple negative breast cancer; ER, Oestrogen Receptor; PR, Progesterone receptor; HER2, Human epidermal growth factor receptor 2; EMT, Epithelial to mesenchymal transition; OS, Overall survival; ML, Machine learning; GEO, Gene expression omnibous; DMFS, Distasnt metastasis free survival; PCA, Principal component analysis; RFE, Recursive feature elimination; SVM, Support vector machine; kNN, k Nearest neighbors; COSMIC, The catalogue of somatic mutations in cancer; DE, Differential Expression; RF, Random forest; ROC, Receiver operating characteristics curve; AUC, Area under the ROC curve; FDR, False discovery rate; NSCLC, Non small cell lung carcinoma; CX-25, Complete XgBoost top 25; DX-20, Driver XgBoost top 20; KM, Kaplan Meier
Abstract
Tumor heterogeneity and the unclear metastasis mechanisms are the leading cause for the unavailability of effective targeted therapy for Triple-negative breast cancer (TNBC), a breast cancer (BrCa) subtype characterized by high mortality and high frequency of distant metastasis cases. The identification of prognostic biomarker can improve prognosis and personalized treatment regimes. Herein, we collected gene expression datasets representing TNBC and Non-TNBC BrCa. From the complete dataset, a subset reflecting solely known cancer driver genes was also constructed. Recursive Feature Elimination (RFE) was employed to identify top 20, 25, 30, 35, 40, 45, and 50 gene signatures that differentiate TNBC from the other BrCa subtypes. Five machine learning algorithms were employed on these selected features and on the basis of model performance evaluation, it was found that for the complete and driver dataset, XGBoost performs the best for a subset of 25 and 20 genes, respectively. Out of these 45 genes from the two datasets, 34 genes were found to be differentially regulated. The Kaplan-Meier (KM) analysis for Distant Metastasis Free Survival (DMFS) of these 34 differentially regulated genes revealed four genes, out of which two are novel that could be potential prognostic genes (POU2AF1 and S100B). Finally, interactome and pathway enrichment analyses were carried out to investigate the functional role of the identified potential prognostic genes in TNBC. These genes are associated with MAPK, PI3-AkT, Wnt, TGF-β, and other signal transduction pathways, pivotal in metastasis cascade. These gene signatures can provide novel molecular-level insights into metastasis.
1. Introduction
Breast cancer (BrCa) has outstripped lung cancer to be the world's most frequently diagnosed cancer, accounting for 11.7 percent of all cases reported worldwide. In 2020, nearly 2.3 million women were diagnosed with breast cancer, while 6,84,996 deaths were observed globally [1]. In India, the world's second-most populous country, BrCa accounts for 13.5 percent of all cancer types, according to the 2020 WHO Global Cancer Observatory report (https://gco.iarc.fr/today/home). BrCa is a complex disease, characterised by a wide range of cell populations and genetic changes causing bottlenecks in clinical treatments, due to its complex etiology and primitive insights into biology of its origin and development [2], [3]. The prognosis and response to the cancer treatment are influenced by a number of characteristics such as histological grade, tumour type and size, lymph node metastasis, and cell receptors [4]. Molecular subtyping of breast cancer, based on classical immunohistochemistry markers is focused on the basis of cell receptors such as ER, PR, and HER2, which play a prominent role in clinical decision making [5]. The St. Gallen (2013) International Breast Cancer Conference established a new definition for the breast cancer classification based on the molecular markers: luminal A (ER+, PR+, HER2-, low Ki67+), luminal B (ER+, HER2-, high Ki67+/PR- OR ER+, HER2+, any Ki67/any PR), Erb-B2 overexpression (HER2 overexpressed or amplified, ER and PR absent), basal-like TNBC (ER-, PR-, HER2-) (Fig. 1) [6].
Fig. 1.

Schematic showing molecular subtypes of breast cancer. These subtypes can be classified based upon the expression of hormone receptors (ER and PR) receptor, tyrosine kinase, HER2 and Ki-67 biomarker.
Among all the breast cancer subtypes, TNBC is the most aggressive subtype and accounts for 10–20% of all breast cancer cases [7]. TNBC has a high recurrence rate, a high potential risk for metastasis, and poor clinical prediction. It is also defined as an obstinate breast cancer owing to its resistance to treatments [8]. Furthermore, it has the highest rate of distant metastasis, poor prognosis and correlates with the shortest overall survival (OS) rate [9]. There is no specific targeted molecular therapy for TNBC, as no pathway-specific targets and biomarkers have been identified for TNBC. Therefore, chemotherapy and surgical resection are the only available effective treatments for TNBC [10], [11]. Moreover, even with these treatments, TNBC patients with tumors confined to the breast and lymph nodes have been reported to encounter distant metastasis within five years [12].
TNBC metastasis accompanies a complex cascade of biological events, including genetic and epigenetic changes, extracellular matrix invasion, angiogenesis, intravasation into blood vessels which allows the survival of tumor cells, their extravasation into distant tissues, and eventually the development of tumor at a distant site [13]. This progression also involves epithelial to mesenchymal transition (EMT) driven by various signal transduction pathways such as Wnt, Notch, and TGF-β [14]. The PI3K and NF-kB signal transduction pathways are involved in tumor proliferation and survival of tumor cells thereby assisting in tumor proliferation at a distant site in the metastasis cascade [15]. The metastasized cancer is incurable due to the resistance of transformed tumor cells to the currently available treatments, which also shortens the survival time of the patients [16], [17], [18]. A majority of TNBC patients die due to the metastatic behavior of the tumor rather than primary tumor growth. However, a better understanding can help us gain further insights into the identification of novel prognostic factors. Hence, identifying new prognostic factors for distant metastasis-free survival of TNBC patients may help identify novel therapeutic targets and improve their distant metastasis-free survival time.
With the recent developments in the field of AI, its evaluation and use in healthcare sector has increased, even in medical oncology. The expanding medical data and the developing AI technologies have exhibited enormous potential for improving cancer diagnosis and prognosis by identifying potential biomarkers [19]. Recently, Villemin and coworkers employed machine learning and reported an EMT-related splicing signature capable of subclassifying the basal-like triple negative tumours [20]. Similarly, Kothari and others implemented machine learning approach and identified two potential prognostic gene signatures, TBC1D9 (TBC1 domain family member 9) and MFGE8 (Milk Fat Globule- EGF factor 8 protein) with a potential to be developed as therapeutic targets too [21].
This study integrates machine learning and systems biology-based approach to identify putative TNBC associated genes, which can potentially serve as prognostic markers. Primarily, we performed a meta-analysis of the Gene Expression Omnibus (GEO) datasets (8 datasets – 5 TNBC and 3 non-TNBC) to identify the genes that can differentiate the TNBC from other BrCa subtypes (non-TNBC) using machine learning (ML). Furthermore, the gene signatures obtained in the feature sets were compared and analysed with the differentially regulated genes. The differentially regulated genes were selected for distant metastasis-free survival analysis (DMFS). And finally, their role as potential prognostic biomarkers were explored using DMFS analysis along with their pathway enrichment analysis.
2. Materials and methods
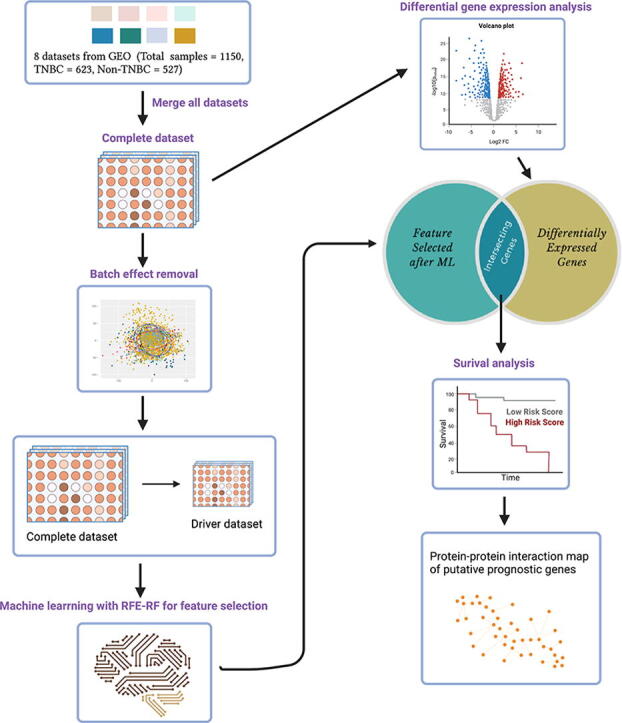
A summary of the workflow applied in this study, from the dataset acquisition from NCBI-GEO to the pathway enrichment analysis of potential prognostic genes are depicted in Fig. 2 and described in the following section.
Fig. 2.

Schematic representation of the bioinformatics workflow. We leverage the existence of multiple microarray gene expression datasets in a platform (GPL570) with differences in gene expression values unique to TNBC in the context of meta-analysis and systems biology, merging and batch-effect removal, encompassing TNBC and non-TNBC gene expression values (N = 1150, TNBC = 623 and non-TNBC = 527 microarray expression profiling datasets) (1 and 2). Genes differentiating TNBC from non-TNBC, leveraging RFE-RF feature selection (3) and differential expression (3) integrated into a framework to nominate the genes for distant metastasis-free survival analysis using KM plotter (4). Furthermore, we used the STRING database to decipher the role of genes nominated by survival analysis and their interacting partners in the cellular machinery (5).
2.1. Microarray dataset download and pre-processing
The raw CEL files of the gene expression profiles and corresponding clinical information of eight independent GEO datasets, comprising 623 TNBC and 527 non-TNBC samples, were downloaded from NCBI GEO (https://www.ncbi.nlm.nih.gov/gds) (Supplementary Table 1). Since publicly available TNBC datasets are limited, we selected all the datasets comprising more than or equal to fifty samples for training and external validation dataset. Datasets comprising less than fifty samples were excluded from the study as they would have increased the batch-effect without providing a significant number of samples. All the downloaded datasets were from the same platform (Affymetrix Human Genome U133 Plus 2.0 Array) to mitigate the effects caused by different platforms and analyze the same set of genes. All the datasets were first read via the ReadAffy function in the affy package for downstream processing. Then, the fRMA method was applied to the read dataset using the fRMA package for background correction and probe to gene mapping [22], [23]. We preferred fRMA over the other available methods as it allows individual analysis of microarrays or in small batches before combining the data for meta-analysis.
Table1.
Selected gene features after ML with their log Fold change and Kaplan-Meier Log Rank P-value.
| CX-25 | DX-20 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | p.Value | logFC | adj.P.Val | Gene Expression | Kaplan-Meier Log rank P (TNBC) |
Gene | p-Value | logFC | adj.P.Val | Gene Expression | Kaplan-Meier Log rank P (TNBC) |
| TTYH1 | 2.10E-53 | 1.51 | 1.96E-52 | Upregulated | 0.38 | AR | 1.87E-86 | −2.34 | 5.48E-85 | Downregulated | 0.57 |
| PTGFR | 2.73E-25 | 0.64 | 9.56E-25 | FALSE | 0.18 | DACH1 | 3.00E-85 | −2.61 | 8.54E-84 | Downregulated | 0.66 |
| PPP4R4 | 5.01E-17 | −0.63 | 1.27E-16 | FALSE | 0.56 | ERBB4 | 1.07E-116 | −3.14 | 1.00E-114 | Downregulated | 0.31 |
| PTPRZ1 | 1.34E-17 | 0.90 | 3.48E-17 | FALSE | 0.83 | ESR1 | 3.49E-106 | −3.90 | 2.08E-104 | Downregulated | 0.38 |
| PTX3 | 2.87E-64 | 2.25 | 3.94E-63 | Upregulated | 0.14 | FOXA1 | 5.49E-201 | −4.32 | 1.14E-196 | Downregulated | 0.41 |
| PCDH8 | 2.67E-14 | 0.60 | 6.04E-14 | FALSE | 0.42 | PGR | 5.25E-78 | −2.63 | 1.18E-76 | Downregulated | 0.23 |
| BCHE | 0.001 | 0.25 | 0.0013 | FALSE | 0.4 | AFF3 | 1.99E-77 | −2.59 | 4.37E-76 | Downregulated | 0.91 |
| S100B | 9.48E-93 | 2.02 | 3.48E-91 | Upregulated | 0.0068 | BCL11A | 9.48E-93 | 1.54 | 3.48E-91 | Upregulated | 0.0038 |
| CXCL5 | 3.69E-24 | 1.006 | 1.24E-23 | Upregulated | 0.92 | POU2AF1 | 5.17E-102 | 2.54 | 2.63E-100 | Upregulated | 0.0033 |
| SFRP1 | 1.09E-136 | 3.633 | 2.36E-134 | Upregulated | 0.2 | ZNF521 | 1.11E-119 | 2.48 | 1.15E-117 | Upregulated | 0.59 |
| PCDH20 | 2.58E-30 | −0.78 | 1.08E-29 | FALSE | 0.025 | MUC16 | 2.05E-37 | 1.41 | 3.93E-36 | Upregulated | 0.65 |
| TFF1 | 4.02E-107 | −3.75 | 2.49E-105 | Downregulated | 0.86 | CDKN2A | 2.91E-55 | 1.39 | 2.89E-54 | Upregulated | 0.98 |
| LRRC31 | 4.21E-43 | −1.276 | 2.76E-42 | Downregulated | 0.4 | TGFBR2 | 5.89E-62 | 1.29 | 7.44E-61 | Upregulated | 0.63 |
| SLC44A4 | 2.85E-66 | −1.695 | 4.17E-65 | Downregulated | 0.19 | WIF1 | 6.42E-26 | 1.13 | 2.30E-25 | Upregulated | 0.45 |
| ZIC1 | 1.18E-51 | 2.05 | 1.04E-50 | Upregulated | 0.53 | NRK | 8.33E-31 | −0.89 | 3.55E-30 | FALSE | 0.75 |
| CAPN6 | 8.07E-65 | 1.90 | 1.13E-63 | Upregulated | 0.99 | COL2A1 | 2.38E-09 | 0.79 | 4.33E-09 | FALSE | 0.38 |
| HORMAD1 | 3.91E-48 | 2.155 | 3.03E-47 | Upregulated | 0.98 | LRP1B | 3.79E-18 | −0.58 | 1.01E-17 | FALSE | 0.78 |
| BBOX1 | 3.89E-62 | 1.899 | 4.96E-61 | Upregulated | 0.31 | S100A7 | 5.48E-11 | 1.13 | 1.08E-10 | Upregulated | 0.42 |
| CT83 | 5.90E-35 | 1.626 | 2.93E-34 | Upregulated | 0.052 | WNK4 | 2.95E-49 | −1.06 | 2.39E-48 | Downregulated | 0.32 |
| PGR | 5.25E-78 | −2.628 | 1.18E-76 | Downregulated | 0.23 | TNFRSF17 | 1.18E-49 | 1.36 | 1.47E-48 | Upregulated | 0.028 |
| ABCC11 | 1.27E-36 | −1.44 | 6.64E-36 | Downregulated | 0.44 | ||||||
| LY6D | 9.36E-47 | 1.31 | 6.96E-46 | Upregulated | 0.96 | ||||||
| SERPINA6 | 3.16E-20 | −0.77 | 9.16E-20 | FALSE | 0.16 | ||||||
| CCN6 | 1.20E-05 | 0.32 | 1.80E-05 | FALSE | 0.7 | ||||||
| ATP7B | 1.71E-126 | −1.703 | 2.49E-124 | Downregulated | 0.16 | ||||||
Further, using the R cbind function, we merged all the eight datasets and annotated the probesets to their corresponding Entrez gene ID using the biomaRT package in R/Bioconductor [24]. However, many probesets do not have their corresponding Entrez gene IDs; hence these were removed. The multiple probesets with the identical Entrez gene IDs were collapsed into one representative row using the function collapseRows with the default MaxMean method in the WGCNA package [25]. The dataset was normalized using the quantile normalization function, which eliminates any technical variability. In addition, it re-distributes the expression intensities of all the samples so that they have a similar distribution [26].
2.2. Batch-effect removal
Merging multiple datasets into one dataset introduces batch-effects or non-biological variations. To overcome batch-effects, adjustments were made using the Empirical Bayes algorithm implemented in the ComBat function in the SVA package [27], [28]. Principal Component Analysis (PCA) was performed (using the R prcomp function) to validate the batch-effect correction in ComBat-transformed data to collapse high-dimensional data into the first two components, which were visualized using the R ggbiplot package. After batch-effect removal the Entrez gene IDs were further mapped to their corresponding external gene names.
The merged dataset obtained after pre-processing and batch-effect removal give rise to an initial dataset comprising of 1150 samples and 20,756 features, henceforth referred as the complete dataset.
2.3. Machine learning
2.3.1. Pre-processing for machine learning
After eliminating the batch-effect, the dataset was used for training the supervised machine learning algorithms. The z-score normalization algorithm was used to standardize the dataset. In machine learning, standardization is extensively used for feature scaling to get the various features to a similar scale. Standardization centres all the feature columns with mean 0, the standard deviation to 1, and conserves the valuable information about the outliers.
2.3.2. Feature selection
In a dataset spreadsheet, input variables are the columns, and samples are the rows for the model training to predict the target variable. We presume columns to be dimensions on an n-dimensional space and rows to be the points within that space. Many columns indicate a high dimensional space, comprising of small or non-representative points in that space. A higher-dimensional space could significantly decrease a prediction model's performance with too many input variables due to the curse of dimensionality.
The process of obtaining a subset from original input variables based on the feature selection algorithm to select the most appropriate features from the dataset for predictive modelling is called feature selection [29]. It also lowers the computational cost and, in some cases, improves the accuracy of the predictive model.
Primarily, we employed Pearson’s correlation coefficient algorithm which is a filter-based approach to reduce redundant features from the complete dataset, comprising of 1150 samples and 20,756 features. Further, we implemented the Recursive Feature Elimination (RFE) algorithm to select the most relevant features for building the efficient predictive model for our target variable. It is a wrapper-type selection algorithm for regression and classification problems and internally, it uses filter-based feature selection. It initiates searching all the features in the training dataset by fitting the given machine learning algorithm in the core, ranking all features by importance, removing the least relevant features, and re-fitting the model. RFE with Random Forest classifier (RFE-RF) as an estimator was implemented using python.
We obtained the top 20, 25, 30, 35, 40, 45, 50 features with 1150 samples, using RFE to train and evaluate the accuracy of five machine learning algorithms, namely Support Vector Machines (SVM), k-Nearest Neighbors (kNN), Logistic Regression, Decision Tree, Random Forest, and XGBoost for binary classification of TNBC versus non-TNBC [30], [31], [32], [33], [34], [35].
2.3.3. Training classification models
We trained different machine learning algorithms with the selected features to generate efficient classifiers after pre-processing, feature selection, and class imbalance treatment. We employed five supervised machine learning algorithms- SVM, kNN, Decision Tree, Random Forest, and XGBoost.
SVM investigates the data and recognizes patterns and decision boundaries by constructing hyperplanes in multi-dimensional space to discretize various classes [36]. kNN performs k-means clustering and then uses the nearest neighbor samples for classification [37]. The Decision Tree can be described as a tree-structure classifier, in which internal nodes represent features, while the leaf nodes represent the targets [38]. It uses multiple algorithms to determine classification. Random Forest is an ensemble algorithm that employs multiple decision trees for classification [31]. XGBoost is a high-speed, high-performance implementation of gradient boosted decision trees [30].
2.3.4. Validation using internal testing dataset
SVM, kNN, Logistic Regression, Decision Tree, Random Forest, and XGBoost were trained on top features obtained using RFE-RF and further validated using stratified 5-fold cross-validation. The accuracy, precision-recall, and F1 score of the training models were compared.
2.3.5. Validation using external testing dataset
We obtained three independent GPL570 platform GEO datasets from the NCBI GEO to further validate model performance (Supplementary Table 2). The external datasets were pre-processed in the same manner as mentioned in the “Microarray Dataset download and pre-processing” and the “batch-effect removal” section, as mentioned above. External independent validation of all the ML algorithms was performed using this external dataset.
2.3.6. Cancer driver gene expression-based model
Machine learning models were also trained by filtering the training datasets to include cancer driver genes exclusively. The list of cancer drivers was compiled using three separate databases: COSMIC, IntOGen, and Bailey. COSMIC, the Catalogue of Somatic Mutations in Cancer, is a widely used database of expert-curated cancer driver genes [39]. IntOGen collects and explores somatic mutations in thousands of tumors genomes to search for cancer driver genes [40].
Further, Bailey and the group used 26 computational tools to identify 299 driver genes and their implications for cancer cell types and anatomical sites [41]. Primarily, we merged the three driver gene lists to create a single list containing only genes unique to all three. Then we data mined our complete dataset to identify the driver genes, obtained above, and created a driver dataset comprising of 1150 samples and 919 features and performed all the steps mentioned above under the machine learning section to build a model based on the expression of these driver genes.
2.4. Differential gene expression analysis
After quantile normalization and removal of low variant genes across all the samples using gene filter, the complete dataset comprising of 1150 samples and 20,756 gene features were used to employ differential gene expression analysis [42]. Further, using the limma package, we identified DE genes in the dataset, using the statistical criteria: logFC > 1; and p < 0.05 [43].
2.5. Survival analysis using KM plotter
The Kaplan-Meier survival analysis was performed for patients with StGallen (2013) basal intrinsic breast cancer subtype of the differentially expressed genes found in ML-identified genes, as discovered in our study, using KM plotter, an open-source web-based tool [44]. TNBC samples were primarily divided into high and low mRNA expression groups for each gene to perform DMFS analysis. In addition, the Cox regression analysis was used to validate further the association between predictive genes and the patient's survival outcome. For survival analysis and Cox regression analysis, a p-value of <0.05 was considered statistically significant.
2.6. Network analysis of prognostic genes
The putative prognostic genes identified in our study were mapped on proteome-wide Homo sapiens interactome downloaded from the STRING (v.11.0b) [45]. The first interacting nodes of each targeted gene were selected and regulatory networks were consecutively constructed using Cytoscape. After that, using the MCODE plugin in Cytoscape, clustering for each regulatory network was examined and top modules with our genes of interest, based on two centrality parameters such as degree and betweenness were selected for further analysis [46]. Finally, the interacting proteins for each gene from top clusters were subjected to pathway enrichment analysis using the KEGG database [47], [48], [49].
2.7. Statistical analysis
All the analyses were conducted three times and the data were pooled together. A moderated paired t-test was employed to evaluate the significance of differences between TNBC and non-TNBC. Statistical significance of p < 0.05 was used as the cut-off for each statistical analysis.
2.8. Software and package information
R and packages: R: 4.0.3, affy: 1.68.0, Biobase: 2.50.0, frma: 1.42.0, hgu133plus2frmavecs: 1.5.0, ggbiplot: 0.55, genefilter: 1.72.1, ggplot: 3.3.4, preprocessCore: 1.52.0, sva: 3.38.0, impute: 1.64.0, WGCNA: 1.70–3, fastcluster: 1.2.3, dynamicTreeCut: 1.63–1, limma: 3.44.3, biomart: 2.44.4, dplyr: 1.0.6, plotly: 4.9.4, tidyverse: 1.3.1, gridExtra: 2.3.
Python and modules: Python: 3.8.5, numpy: 1.19.2, pandas: 1.1.3, seaborn: 0.11.0, sklearn: 0.24.1, matplotlib: 3.3.2, conda 4.10.3.
3. Results
3.1. Uncovering gene panel demarcating TNBC from other BrCa subtypes and preliminary selection of genes correlated with TNBC
Machine learning algorithms facilitate the biomarker panel selection as the algorithms are extraordinarily capable of learning intricated interrelations within high-dimensional data. After background noise correction, all the datasets (8 datasets − 5 TNBC and 3 non-TNBC) were pooled, followed by normalization. The microarray gene expression profile encompassing 20,790 gene products across 1150 BrCa tumor samples (including 623 TNBC samples) was corrected for batch-effects, using ComBat stemming from the datasets pooling.
The batch-effect removal was further validated using PCA. The first two principal components of ComBat analysis demonstrate the data that apprehended the most variance (Fig. 3) [50]. Duplicate gene symbols were eliminated after mapping each Entrez gene ID to its corresponding gene symbol, leading to 20,756 gene products. The 20,756 genes were feature variables for training the machine learning classifiers to predict the targets (TNBC and non-TNBC). For ML classifiers, these 20,756 genes were employed in two different strategies. In the first one, all the genes were used for machine learning (complete dataset). In the second strategy, only a subset of 919 driver genes (listed in the cancer driver databases), driver dataset, was used for ML training. Both the datasets were standardized employing the z-score standardization after splitting the datasets into training and testing (in the ratio 80:20).
Fig. 3.

PCA plots illustrating batch-effect removal. Validation of batch-effect removal was demonstrated using PCA plot. a. Before employing ComBat. b. After employing ComBat. Different colors represent different datasets. Before batch-effect removal all the datasets form separate clusters, while after the batch-effect removal no such separate clusters were observed.
Our datasets have many feature variables (20,756 and 919) that were likely to overfit the training datasets and eventually perform poorly on any external data. Therefore, we employed two feature selection methods, namely Pearson’s correlation coefficient scores and Recursive Feature Elimination with Random Forest (RFE-RF) classifier at its core to eliminate the dataset's extraneous and redundant feature variables. Feature variables or genes with a correlation coefficient higher than or equal to 0.85 were eliminated, leaving 19,493 and 913 genes, respectively. Further, RFE-RF retrieved the top 20, 25, 30, 40, 45, and 50 feature variables from the datasets mentioned above.
3.2. Validation using the internal testing dataset
The filtered genes in both the datasets were employed for model building using different machine learning algorithms (SVM with rbf kernel, kNN, Decision Tree, Random Forest, and XGBoost). The top 20, 25, 30, 35, 40, 45, and 50 gene features for the complete dataset achieved an average accuracy, ranging from 0.91 to 0.97 for all the machine learning classifiers. kNN achieved the lowest accuracy ranging from 0.88 to 0.93, while the XGBoost performed comparatively better than all the other classifiers with accuracy ranging between 0.89 and 0.99 (Supplementary Table 3).
For the driver dataset trained classifiers, the top 20, 25, 30, 40, 45, and 50 gene features achieved an average accuracy ranging from 0.91 to 0.94 for all the machine learning classifiers. The decision tree classifier achieved the lowest accuracy, ranging from 0.86 to 0.90, while the XGBoost again performed better than all the other classifiers with accuracy ranging from 0.94 to 0.97 (Supplementary Table 4).
3.3. Validation using external independent dataset
We further evaluated the performance of all the classifiers on the external independent dataset. We achieved the highest accuracy of 0.94 for the XGBoost classifier enriched with the top 25 gene features for the complete gene expression dataset with an AUC of 0.99 for ROC and for the PR curve (Fig. 4a, Fig. 4c; Supplementary Table 3). Thus, we adopted the XGBoost classifier of the top 25 gene features for the complete gene expression dataset (CX-25: Complete XGBoost top 25) with an accuracy of 0.99 in the internal testing dataset and 0.94 in the external testing dataset (Fig. 4b, Fig. 4d; Supplementary Table 3).
Fig. 4.

ROC and precision-recall curve of the complete dataset after employing ML. ROC curve of all machine learning algorithms on complete external (a) and internal validation dataset (b). The precision-recall curve of all machine learning algorithms on complete external (c) and internal validation dataset (d). XGBoost outperforms all the other ML classifiers with ROC-AUC of 0.99 in internal validation, 0.99 in external validation, and PR-AUC of 0.98 in internal and in the external validation dataset.
In the external driver dataset, we achieved the highest accuracy of 0.92, enriched with the top 20 gene features for the XGBoost classifier with an AUC of 0.98 for ROC and PR curve (Fig. 5a, Fig. 5c; Supplementary Table 4). While for the internal driver dataset, the XGBoost classifier achieved an accuracy of 0.97 with an AUC of 0.99 for ROC for the PR (Fig. 5b, Fig. 5d; Supplementary Table 4). With an accuracy of 0.97 on the internal testing dataset and 0.92 on the external testing dataset, we chose the XGBoost classifier of the top 20 gene features for the driver dataset (DX-20: Driver XGBoost top 20).
Fig. 5.

ROC and precision-recall curve of driver dataset after employing ML. ROC curve of all machine learning algorithms on driver external (a) and complete internal validation (b) dataset. The precision-recall curve of all machine learning algorithms on driver external (c) and internal validation (d) dataset. XGBoost outperforms all the other ML classifiers with ROC-AUC of 0.99 in internal validation, 0.979 in external validation, and PR-AUC of 0.98 in internal and in the external validation dataset.
3.4. Survival analysis of the differentially expressed genes from CX-25 and DX-20
A moderated t-test followed by Benjamini-Hochberg FDR estimation was carried out to analyze differential gene expression among TNBC tumor subtype patients (N = 623) and non-TNBC tumor subtype patients (N = 527). The statistical significance of FDR < 0.05 was kept as the cut-off. Volcano plots in Fig. 6 reports the differentially expressed genes between TNBC and non-TNBC subtypes (Table 1). The genes retrieved after implementing machine learning (CX-25 and DX-20) are highlighted in the volcano plot (Fig. 6).
Fig. 6.

Volcano plots of differentially expressed genes in TNBC. TNBC patients were compared with non-TNBC patients’ group. Log fold change is plotted, and gene transcripts with log fold change > 1 were coloured blue and with log fold change < 1 are coloured green. Differentially expressed genes from the top 25 and top 20 genes obtained after applying ML on CX-25 (a) and DX-20 (b) are depicted. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In CX-25, TFF1, PGR, LRRC31, SLC44A4, ATP7B, and ABCC11 are downregulated, while TTYH1, CT83, HORMAD1, ZIC1, CAPN6, BBOX1, PTX3, S100B, CXCL5, SFRP1, and LY6D are upregulated; in DX-20, AR, FOXA1, ESR1, PGR, AFF3, DACH1, WNK4, and ERBB4 are downregulated, while BCL11A, ZNF521, POU2AF1, MUC16, S100A7, WIF1, CDKN2A, TNFRSF17, and TGFBR2 are upregulated (Table 1). PGR is common in CX-25 and DX-20.
After the differential gene expression analysis, all the above-selected genes were analyzed to test their association with distant metastasis-free survival (DMFS) time (Fig. 7). Kaplan-Meier DMFS analysis identified high- and low-expressed genes that significantly differentiate the patients bifurcated by distant metastasis-free survival ability in the TNBC tumor subtype [51] (Table 1).
Fig. 7.

Survival analysis reveals the significant association between CT83, BCL11A, S100B and POU2AF1 and distant metastasis-free survival in TNBC. In each of the Kaplan-Meier plot, the red line demonstrates the survival of patients in the higher expression group, whereas the black lines indicate the survival of patients in the lower expression group. The p-Values and hazard ratio (HR) scores were computed using the log-rank (Mantel-Cox) test. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
BAF chromatin remodelling complex subunit (BCL11A), POU class 2 homeobox associating factor 1 (POU2AF1), and S100 calcium binding protein B (S100B) exhibited a difference in DMFS, higher survival with higher expression. Cancer/Testis antigen 83 (CT83), is a borderline prognostic biomarker with a p value of 0.052. We picked BCL11A, S100B, CT83 and POU2AF1, which have been identified as potential TNBC prognostic hallmark genes, based on their significant association with distant metastasis-free survival in TNBC patients.
3.5. Interaction analysis of survival analysis nominated genes
The targeted genes (S100B, POU2AF1, BCL11A and CT83) were mapped to the human protein–protein interaction network to investigate the physical and functional interactions (Fig. 8). A total of 1222, 441, 928, 172 nodes and 361838, 49426, 162840, 5964 edges are found for S100B, POU2AF1, BCL11A and CT83, respectively. Further, the top modules selected by clustering analysis resulted in the highest number of interactions in BCL11A, with 265 nodes and 49,714 edges, followed by POU2AF1, with 165 nodes and 23,102 edges (Fig. 8). Furthermore, S100B has 113 nodes with 3274 edges; and for CT83, 48 nodes with 1640 edges (Fig. 8).
Fig. 8.

Protein-protein subnetwork and top MCODE clusters of putative prognostic genes. Protein-protein interaction sub-network of putative prognostic genes (POU2AF1, S100B, BCL11A, and CT83) extracted by mapping on the human (center) protein–protein interactome, with 2428 interactions. Top MCODE clusters of all the selected prognostic genes; POU2AF1 with 165 interactions (top left), 2) S100B with 113 interactions (top right), BCL11A with 265 interactions (bottom left), and CT83 with 48 interactions (bottom right). Created with BioRender.com.
The interacting partners of selected modules were subjected to a pathway enrichment analysis using the KEGG database to understand the underlying regulatory pathways. All the enriched pathways are shown in Supplementary Fig. 1. This analysis revealed that BCL11A, S100B, and POU2AF1 have a pivotal role in cross-talking in cancer metastasis and could provide novel insights into its role in the cancer signaling pathways.
4. Discussion
TNBC is a pre-eminent classical BrCa subtype, based on characteristic markers such as ER, PR, and HER2, although its genetic properties are more complex than anticipated. The application of gene expression data in identifying TNBC biomarkers is reported by various other studies [52], [53]. However, the major drawback is the non-availability of large sample size in publicly available datasets. Thus, in this study, we attempt to address this issue by selecting multiple datasets from a publicly available database to increase the number of samples that were merged for meta-analysis. The gene expression data has a vast amount of information in intricate patterns among various genes. ML algorithms are emerging as a better approach to unravel these intricate patterns in the gene expression datasets to identify potential novel biomarkers [54], [55], [56]. However, a large number of genes (20,756) in comparison to a smaller number of samples is a hurdle in achieving a better performance of ML classifiers. To circumvent this limitation, we employed a feature selection algorithm (RFE-RF). Feature selection algorithms help in identifying the core representative and functional genes that can potentially differentiate two subtypes of a cancer. The features obtained using RFE-RF comprised genes reported to be involved in TNBC, further validating our unconventional ML-based pipeline (Fig. 2). The pipeline also enabled the identification of novel transcripts influencing the distant metastasis-free survival of TNBC patients.
As discussed above in the results section, XGBoost performs the best with the highest accuracy and AUC for the complete dataset trained with the top 25 features (CX-25), including TTYH1, ATP7B, PPP4R4, PTGFR, BCHE, TFF1 and SERPINA6 (Fig. 4). Further, for the driver dataset, XGBoost outperformed the other ML-based algorithms (Fig. 5). Notably, DX-20 included WIF1, CDKN2A, ZNF521, COL2A1, WNK4, MUC16, and S100A7.
Out of 45 genes listed in CX-25 and DX-20, several genes have established role in breast cancer, validating the employed pipeline. For instance, BBOX1 [57], AR [58], ZIC1 [59], CAPN6 [60], PCDH8 [61], and others were in agreement with our findings.
The DGE (Differential gene expression) analysis was performed on the complete dataset. A total of seventeen genes in CX-25 and seventeen genes in DX-20 were found to be differentially expressed (Fig. 6). The identification of AR, PGR and ERBB4 is of prime importance, downregulated in TNBC, further validating the efficacy of the DGE analysis and the ML pipeline. In a study based on a robust rank aggregation of gene expression profiles, Zhong et al. observed that HORMAD1, BCL11A, and CT83 are upregulated, whereas FOXA1 is downregulated [62]. D. Dill and colleagues [63] employed a network-based approach to discover TNBC drivers that regulate survival time of patients and identified that HORMAD1 is upregulated, while FOXA1, ESR1, SLC44A4 and ERBB4 are downregulated. The results of DGE analysis in the complete dataset are consistent with those of Zhong et al. and D. Dill and associates.
Survival analysis predicted four putative prognostic genes, i.e., BCL11A, CT83, POU2AF1, and S100B for DMFS (Fig. 7; Table 1). Intriguingly, the two genes (POU2AF1 and S100B) are novel potential prognostic factors for TNBC metastasis-associated survival. Further validation of the genes was performed by referring to the literature. Out of all the four predicted prognostic genes, BCL11A has a role in breast cancer-specific metastasis. Studies have shown, high expression of BCL11A in TNBC, employing qRT-PCR and immunohistochemistry techniques and its paramount role in stem and progenitor cells, thereby causing tumour development in TNBC. However, knock-down studies performed in mouse model have reported a dramatic reduction in tumourigenicity and the tumour size [64]. Furthermore, the mRNA expression of BCL11A positively correlates with ST8SIA1, which regulates metastasis by activating the FAK-AKT-mTOR signaling pathways [65]. BCL11A can also upregulate the expression of BCL2, BCL2-xL, and MDM2, which suppress p53 activities [66]. These molecular variations can occur in solid tumours, such as lung cancer. Therefore, BCL11A may have a role in carcinogenesis by affecting apoptosis, the cell cycle, and DNA damage repair, but the exact mechanisms are not known. However, Jiang and others reported high expression of BCL11A in clinical non-small cell lung cancer (NSCLC) tissue samples at transcriptional and translational levels. They also observed better survival outcomes for patients with high expression of BCL11A [67]. Seachrist et al. have reported the role of BCL11A in suppressing a splicing regulator, muscleblind-like splicing regulator 1 (MBNL1), thereby promoting cell invasion and metastasis in TNBC [68]. However, we observed that BCL11A was upregulated in TNBC and survival analysis indicates that patients with high expression of BCL11A had better survival outcomes, similar results to previous study on NSCLC. Therefore, one might wonder if BCL11A functions as a tumour suppressor and suppresses cancer metastasis or promotes metastasis. Liu et al. reported that BCL11A may be a T-cell tumor suppressor gene as they observed T-cell leukaemia in recipient mice as a result of BCL11A-knockout in murine fetal liver cells [69]. In addition to the above-mentioned putative mechanism, P21 and CHEK1, two cell cycle checkpoint members, such as G1/S and G2/M checkpoints might also be responsible for this function [70], [71]. These evidences suggest that BCL11A may target P21 and CHEK1, implying an interesting dual role for BCL11A in TNBC progression which can be explored further.
Currently, the precise role of CT83 in TNBC is poorly understood due to the limited relevant data availability. CT83 is the most specific TNBC gene that is considerably increased in TNBC but downregulated in other malignancies. It may promote carcinogenesis by causing the activation of cell cycle-related signaling pathways [72].
POU2AF1 is a protein-coding gene also known as BOB1, OBF1, OCAB, and OBF-1, found on chromosome 11q23.1, which encodes a protein of 256 amino acids (http://www.ncbi.nlm.nih.gov/geo). POU2AF1 was previously thought to be expressed only in lymphocytes, where it acts as a co-activator of OCT1 and OCT2 (octamer-binding transcription factors) to regulate immunoglobin expression and other host defence genes [73], [74], [75]. POU2AF1 itself has no intrinsic DNA binding domain, it recognizes the POU domain of OCT1 and OCT2 and thus plays an essential role in B-cell responses to antigens and is also essential for the formation of germinal centres [76], [77], [78]. However, sporadic literature suggests that POU2AF1 might play a prominent role in other cells as well. For instance, POU2AF1 has an average level of expression in the human airway epithelium when compared to all other transcription factors [79]. Similarly, in the murine intestinal follicle-associated epithelium, high expression of POU2AF1 was observed as compared to villous epithelium [80], [81]. The above-mentioned studies along with the known functions, shows that POU2AF1 plays a regulatory role in immune system. Further, the role of POU2AF1 in breast cancer, primarily focusing on TNBC, is still unknown. This study, on the other hand, is the first to mention the significance of POU2AF1 in the prognosis of distant metastasis in TNBC patients. Patients with high POU2AF1 expression had a longer distant metastasis free survival time than the patients with low POU2AF1 expression, according to the Kaplan-Meier survival analyses in this study (Fig. 7). These observations are on par with the above-mentioned studies and indicates the immunological role of POU2AF1 in limiting distant metastasis in TNBC which might be further explored.
Similarly, S100B, also known as NEF, S100, S100B, and S100 beta, is located on the chromosome 21q22.3 and encodes a 92 amino acidic, Ca2 + binding protein involved in diverse biological processes including inflammation (http://www.ncbi.nlm.nih.gov/geo). Inflammatory molecules alter metastasis-related pathways such as EMT [82]. In ER-negative breast cancer cell lines, S100B treatment greatly reduced cell movement and promoted the epithelial phenotype by activating anti-metastatic signaling pathways [83]. Our results are in concordance with the activation of anti-metastatic pathways. S100B gene expression is upregulated in TNBC, also the patients with high S100B expression have a longer distant metastasis free survival, further supporting their role in the suppressed cell migration. Further, analysis was performed to decipher the role of BCL11A, CT83, POU2AF1, and S100B in metastasis of TNBC using pathway enrichment.
The pathway enrichment analysis of these genes and their modules affirmed their association with signaling pathways involved in cancer metastasis. Metastasis cascade includes epithelial-mesenchymal transition (EMT), tumor neoangiogenesis, and the spread of malignancy to a new site. The spread of malignancy is caused by the transport of malignant cells through blood vessels to target tissues and organs, which are then invaded by infiltrating malignant cells, resulting in secondary tumors. The aberrant constitutive activation of PI3-Akt, MAPK, and focal adhesion signaling provides resistance to programmed cell death and anticancer therapy to cancer cells [84], [85]. Furthermore, Notch, Wnt, Hedgehog, TGF-β, and Nf-Kappa B signaling pathways play a prominent role in EMT, cell proliferation, migration, and motility of these cells [86], [87], [88]. The transcription factor Nf- kappa B is anti-apoptotic and pro-proliferative in tumor cells and plays a role in the synthesis of angiogenic factors such as vascular endothelial growth factors [89].
Transforming growth factor-beta (TGF-β) is known to stimulate and/or sustain tumor cell motility and metastasis by causing the loss of the epithelial marker E-cadherin. E-cadherin is correlated to aberrant EMT, tumor cell motility and invasion, and anoikis resistance [14] (Fig. 9). This study has demonstrated the potential role of POU2AF1, and S100B as novel prognostic biomarkers in metastasis and therapeutic targets for TNBC; yet, whether these gene signatures can be used for diagnosis or drug development is still a challenge since their detailed molecular mechanisms remains to be understood comprehensively.
Fig. 9.

Schematic illustration of signal transduction pathways involved in metastasis cascade. If normal epithelial cells (orange) detach from the membrane, it undergoes apoptosis, known as anoikis (brown), while the tumor cells (red) resist apoptosis through the activation of anti-apoptotic pathways such as PI3-AKT, MAPK (anti-apoptotic pathways). With the loss of E-cadherin receptors (promoted by TGF-β pathway), the tumor cells undergo a transition from non-mobile (epithelial) to mobile (mesenchymal) form (red tumor cell with a tail). These mobile or transformed tumor cells intravasate into blood vessels and transported to a new site, where they extravasate and localize (brain shown in blue). At the new site, tumor cells proliferate through the activation of Wnt, Notch, Hedgehog, and other related pathways to develop a new tumor (metastasis). New blood vessels are formed (angiogenesis) by synthesizing angiogenic factors by Nf- kappa B and other signaling pathways. The new blood vessels are employed for the tumor cells' nutrition and transport (intravasation and extravasation). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
One of the limitations of this study is the small sample size used to predict the survival analysis of these genes. The limitation is also aggravated due to an insufficient number of TNBC samples and their demographic information in publicly available datasets. As an additional limitation, cross-platform validation is also needed, as we used only the GPL570 platform in our study. Nonetheless, we established our study on the most extensively used platform, which outshines the study limitations and enables valuable insights into prognostic indicators for TNBC.
Our findings demonstrated the viability of using RFE-RF and ML to uncover biomarkers that distinguish TNBC from non-TNBC. Although more data on a larger scale is needed to corroborate our findings, we have meticulously evaluated and analyzed our results at every step, consistent with the past related research.
5. Conclusion
Summarily, the present study demonstrates an unconventional and important workflow that deduced the novel potential prognostic factors associated with TNBC. We believe that understanding the role of these prognostic genes in metastasis may further provide potential targets for future intervention and therapy. Furthermore, the application of the pipeline developed also shows the potential to explore the prognostic factors associated with other life-threatening ailments. Finally, the study demonstrates the potential of Recursive feature selection-Random Forest as a feature selection algorithm for gene expression profiles or other similar data having a large number of features as compared to the number of samples.
Data Availability Statement: The associated GEO datasets, as well as metadata of the samples, are available at NCBI GEO (https://www.ncbi.nlm.nih.gov/gds). The list of the GEO datasets used is provided in Supplementary file Table 1 and Table 2.
Funding
This work was financially supported by the Department of Biotechnology (DBT), Government of India, grant BT/PR40151/BTIS/137/5/2021. A.T. and S.R. acknowledge fellowship awarded by the Council of Scientific and Industrial Research (CSIR), Government of India, file no. 09/512(0227)/2017-EMR-I and 09/512(0212)/2016-EMR-I, respectively. H.K.J acknowledges the fellowship awarded by GlaxoSmithKline (GSK, India), registration no. RCB/PhD-BI/2020/1016. We acknowledge the use of the study datasets from the NCBI GEO. Some of the images in the publication were created using BioRender.com.
CRediT authorship contribution statement
Anamika Thalor: Conceptualization, Methodology, Data curation, Formal analysis, Validation, Investigation, Visualization, Writing – original draft, Writing – review & editing. Hemant Kumar Joon: Formal Analysis, Validation, Investigation, Visualization, Writing- review and editing. Gagandeep Singh: Visualization, Formal analysis, Validation, Writing - original draft. Shikha Roy: Data curation. Dinesh Gupta: Conceptualization, Methodology, Funding acquisition, Supervision, Project administration, Resources, Writing – original draft, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.03.019.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sung, H., et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 2021. 71(3): p. 209-249. [DOI] [PubMed]
- 2.Provenzano E., Ulaner G.A., Chin S.F. Molecular Classification of Breast Cancer. PET Clin. 2018;13(3):325–338. doi: 10.1016/j.cpet.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Raman D., et al. Breast Cancer: A Molecular and Redox Snapshot. Antioxid Redox Signal. 2016;25(6):337–370. doi: 10.1089/ars.2015.6546. [DOI] [PubMed] [Google Scholar]
- 4.Al-Thoubaity F.K. Molecular classification of breast cancer: A retrospective cohort study. Ann Med Surg (Lond) 2020;49:44–48. doi: 10.1016/j.amsu.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldhirsch A., et al. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011;22(8):1736–1747. doi: 10.1093/annonc/mdr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldhirsch A., et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013;24(9):2206–2223. doi: 10.1093/annonc/mdt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheang M.C., et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14(5):1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 8.Wahba H.A., El-Hadaad H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12(2):106–116. doi: 10.7497/j.issn.2095-3941.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X., et al. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res Treat. 2017;161(2):279–287. doi: 10.1007/s10549-016-4059-6. [DOI] [PubMed] [Google Scholar]
- 10.Denkert C., et al. Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. The Lancet. 2017;389(10087):2430–2442. doi: 10.1016/S0140-6736(16)32454-0. [DOI] [PubMed] [Google Scholar]
- 11.Prat A., et al. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br J Cancer. 2014;111(8):1532–1541. doi: 10.1038/bjc.2014.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Mahmood S., et al. Metastatic and triple-negative breast cancer: challenges and treatment options. Drug Deliv Transl Res. 2018;8(5):1483–1507. doi: 10.1007/s13346-018-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valastyan S., Weinberg R.A. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu W., Yang Z., Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 2015;9(4):317–324. doi: 10.1080/19336918.2015.1016686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroeder M.C., et al. Early and Locally Advanced Metaplastic Breast Cancer: Presentation and Survival by Receptor Status in Surveillance, Epidemiology, and End Results (SEER) 2010–2014. Oncologist. 2018;23(4):481–488. doi: 10.1634/theoncologist.2017-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin N.U., et al. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer. 2012;118(22):5463–5472. doi: 10.1002/cncr.27581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pal S.K., Childs B.H., Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat. 2011;125(3):627–636. doi: 10.1007/s10549-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z.H., et al. Artificial intelligence for assisting cancer diagnosis and treatment in the era of precision medicine. Cancer Commun (Lond) 2021 doi: 10.1002/cac2.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villemin J.P., et al. A cell-to-patient machine learning transfer approach uncovers novel basal-like breast cancer prognostic markers amongst alternative splice variants. BMC Biol. 2021;19(1):70. doi: 10.1186/s12915-021-01002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kothari C., et al. Machine learning analysis identifies genes differentiating triple negative breast cancers. Sci Rep. 2020;10(1):10464. doi: 10.1038/s41598-020-67525-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCall M.N., Bolstad B.M., Irizarry R.A. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11(2):242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautier L., et al. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 24.Durinck S., et al. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller J.A., et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinf. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.B, B., preprocessCore: A collection of pre-processing functions. R package version 1.54.0. https://github.com/bmbolstad/preprocessCore.; 2021.
- 27.Leek JT, J.W., Parker HS, Fertig EJ, Jaffe AE, Zhang Y, Storey JD, Torres LC, sva: Surrogate Variable Analysis. R package version 3.40.0. 2021.
- 28.Zhang Y., et al. Alternative empirical Bayes models for adjusting for batch effects in genomic studies. BMC Bioinf. 2018;19(1):262. doi: 10.1186/s12859-018-2263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai J., et al. Feature selection in machine learning: A new perspective. Neurocomputing. 2018;300:70–79. [Google Scholar]
- 30.Chen T.G.C. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016. XGBoost: A Scalable Tree Boosting System; p. 9. [Google Scholar]
- 31.Breiman L. Machine Learning. 2001;45(1):5–32. [Google Scholar]
- 32.Breiman L. Bagging predictors. Machine Learning. 1996;24(2):123–140. [Google Scholar]
- 33.Boser, B.E.G., I.M.; Vapnik, V.N. , A training algorithm for optimal margin classifiers. Proceedings of the fifth annual workshop on Computational learning theory – COLT . CiteSeerX; 1992.
- 34.Altman N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am Statistic. 1992;46(3):175–185. [Google Scholar]
- 35.Breiman, L.F., J.H. Olshen, R.A. Stone C.J. Classification and regression trees. Monterey, CA: Wadsworth & Brooks/Cole Advanced Books & Software; 1984.
- 36.Ghosh Sourish D.A., Aleena S. A Study on Support Vector Machine based Linear and Non-Linear Pattern Classification. IEEE. 2019:4. [Google Scholar]
- 37.Xing W., Bei Y. Medical Health Big Data Classification Based on KNN Classification Algorithm. IEEE Access. 2020;8:28808–28819. [Google Scholar]
- 38.Fürnkranz J. Encyclopedia of Machine Learning. Springer; Boston, MA: 2010. Decision Tree. [Google Scholar]
- 39.John G Tate, S.B., Harry C Jubb, Zbyslaw Sondka, David M Beare, Nidhi Bindal, Harry Boutselakis, Charlotte G Cole, Celestino Creatore, Elisabeth Dawson, Peter Fish, Bhavana Harsha, Charlie Hathaway, Steve C Jupe, Chai Yin Kok, Kate Noble, Laura Ponting, Christopher C Ramshaw, Claire E Rye, Helen E Speedy, Ray Stefancsik, Sam L Thompson, Shicai Wang, Sari Ward, Peter J Campbell, Simon A Forbes, COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res; 2019. 47: p. 7. [DOI] [PMC free article] [PubMed]
- 40.Gonzalez-Perez A., et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10(11):1081–1082. doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bailey M.H., et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell. 2018;173(2):371–385 e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gentleman R, C.V., Huber W, Hahne F, genefilter: genefilter: methods for filtering genes from high-throughput experiments. R package version 1.74.0. 2021.
- 43.Ritchie M.E., et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7) doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gyorffy B., et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 45.Szklarczyk D., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bader G.D., Hogue C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanehisa M., et al. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–D551. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim S.B., et al. An extracellular matrix-related prognostic and predictive indicator for early-stage non-small cell lung cancer. Nat Commun. 2017;8(1):1734. doi: 10.1038/s41467-017-01430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Győrffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101–4109. doi: 10.1016/j.csbj.2021.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai Q., et al. Identification of differentially expressed genes between triple and non-triple-negative breast cancer using bioinformatics analysis. Breast Cancer. 2019;26(6):784–791. doi: 10.1007/s12282-019-00988-x. [DOI] [PubMed] [Google Scholar]
- 53.Yang R., et al. Comprehensive Analysis of Differentially Expressed Profiles of lncRNAs/mRNAs and miRNAs with Associated ceRNA Networks in Triple-Negative Breast Cancer. Cell Physiol Biochem. 2018;50(2):473–488. doi: 10.1159/000494162. [DOI] [PubMed] [Google Scholar]
- 54.Li M.X., et al. Using a machine learning approach to identify key prognostic molecules for esophageal squamous cell carcinoma. BMC Cancer. 2021;21(1):906. doi: 10.1186/s12885-021-08647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan F., Lu L., Zou Q. Analysis of gene expression profiles of lung cancer subtypes with machine learning algorithms. Biochim Biophys Acta Mol Basis Dis. 2020;1866(8) doi: 10.1016/j.bbadis.2020.165822. [DOI] [PubMed] [Google Scholar]
- 56.Sinkala M., Mulder N., Martin D. Machine Learning and Network Analyses Reveal Disease Subtypes of Pancreatic Cancer and their Molecular Characteristics. Sci Rep. 2020;10(1):1212. doi: 10.1038/s41598-020-58290-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao C., et al. Identification of BBOX1 as a Therapeutic Target in Triple-Negative Breast Cancer. Cancer Discov. 2020;10(11):1706–1721. doi: 10.1158/2159-8290.CD-20-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kensler K.H., et al. Prognostic and predictive value of androgen receptor expression in postmenopausal women with estrogen receptor-positive breast cancer: results from the Breast International Group Trial 1–98. Breast Cancer Res. 2019;21(1):30. doi: 10.1186/s13058-019-1118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han W., et al. ZIC1 acts a tumor suppressor in breast cancer by targeting survivin. Int J Oncol. 2018;53(3):937–948. doi: 10.3892/ijo.2018.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Storr S.J., et al. Calpain in Breast Cancer: Role in Disease Progression and Treatment Response. Pathobiology. 2015;82(3–4):133–141. doi: 10.1159/000430464. [DOI] [PubMed] [Google Scholar]
- 61.Yu J.S., et al. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene. 2008;27(34):4657–4665. doi: 10.1038/onc.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhong G., et al. Identification of key genes as potential biomarkers for triplenegative breast cancer using integrating genomics analysis. Mol Med Rep. 2020;21(2):557–566. doi: 10.3892/mmr.2019.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dill C.D., et al. A network approach reveals driver genes associated with survival of patients with triple-negative breast cancer. iScience. 2021;24(5) doi: 10.1016/j.isci.2021.102451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khaled W.T., et al. BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat Commun. 2015;6:5987. doi: 10.1038/ncomms6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen K., et al. ST8SIA1 Regulates Tumor Growth and Metastasis in TNBC by Activating the FAK-AKT-mTOR Signaling Pathway. Mol Cancer Ther. 2018;17(12):2689–2701. doi: 10.1158/1535-7163.MCT-18-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu Y., et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med. 2012;209(13):2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang B.Y., et al. BCL11A overexpression predicts survival and relapse in non-small cell lung cancer and is modulated by microRNA-30a and gene amplification. Mol Cancer. 2013;12:61. doi: 10.1186/1476-4598-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seachrist D.D., et al. The transcriptional repressor BCL11A promotes breast cancer metastasis. J Biol Chem. 2020;295(33):11707–11719. doi: 10.1074/jbc.RA120.014018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu P., et al. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4(6):525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 70.Insinga A., et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci U S A. 2013;110(10):3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dai Y., Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen C., et al. Multiomics analysis reveals CT83 is the most specific gene for triple negative breast cancer and its hypomethylation is oncogenic in breast cancer. Sci Rep. 2021;11(1):12172. doi: 10.1038/s41598-021-91290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brunner C., Wirth T. BOB.1/OBF.1 - A Critical Regulator of B Cell Function. Curr Immunol Rev. 2006;2(1):3–12. [Google Scholar]
- 74.Teitell M.A. OCA-B regulation of B-cell development and function. Trends Immunol. 2003;24(10):546–553. doi: 10.1016/j.it.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 75.Luo Y., Roeder R.G. B-cell-specific coactivator OCA-B: biochemical aspects, role in B-cell development and beyond. Cold Spring Harb Symp Quant Biol. 1999;64:119–131. doi: 10.1101/sqb.1999.64.119. [DOI] [PubMed] [Google Scholar]
- 76.Kim U., et al. The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. 1996;383(6600):542–547. doi: 10.1038/383542a0. [DOI] [PubMed] [Google Scholar]
- 77.Nielsen P.J., et al. B lymphocytes are impaired in mice lacking the transcriptional co-activator Bob1/OCA-B/OBF1. Eur J Immunol. 1996;26(12):3214–3218. doi: 10.1002/eji.1830261255. [DOI] [PubMed] [Google Scholar]
- 78.Schubart D.B., et al. B-cell-specific coactivator OBF-1/OCA-B/Bob1 required for immune response and germinal centre formation. Nature. 1996;383(6600):538–542. doi: 10.1038/383538a0. [DOI] [PubMed] [Google Scholar]
- 79.Zhou H., et al. POU2AF1 Functions in the Human Airway Epithelium To Regulate Expression of Host Defense Genes. J Immunol. 2016;196(7):3159–3167. doi: 10.4049/jimmunol.1502400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nakato G., et al. New approach for m-cell-specific molecules screening by comprehensive transcriptome analysis. DNA Res. 2009;16(4):227–235. doi: 10.1093/dnares/dsp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mach J., et al. Development of intestinal M cells. Immunol Rev. 2005;206:177–189. doi: 10.1111/j.0105-2896.2005.00281.x. [DOI] [PubMed] [Google Scholar]
- 82.Dominguez C., David J.M., Palena C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin Cancer Biol. 2017;47:177–184. doi: 10.1016/j.semcancer.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yen M.C., et al. S100B expression in breast cancer as a predictive marker for cancer metastasis. Int J Oncol. 2018;52(2):433–440. doi: 10.3892/ijo.2017.4226. [DOI] [PubMed] [Google Scholar]
- 84.Chiarugi P., Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76(11):1352–1364. doi: 10.1016/j.bcp.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 85.Brabletz T., et al. β-Catenin Regulates the Expression of the Matrix Metalloproteinase-7 in Human Colorectal Cancer. The American Journal of Pathology. 1999;155(4):1033–1038. doi: 10.1016/s0002-9440(10)65204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kwon O.J., et al. Increased Notch signalling inhibits anoikis and stimulates proliferation of prostate luminal epithelial cells. Nat Commun. 2014;5:4416. doi: 10.1038/ncomms5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rathinam R., Berrier A., Alahari S.K. Role of Rho GTPases and their regulators in cancer progression. Front Biosci (Landmark Ed) 2011;16:2561–2571. doi: 10.2741/3872. [DOI] [PubMed] [Google Scholar]
- 88.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 89.Tabruyn S.P., et al. NF-kappaB activation in endothelial cells is critical for the activity of angiostatic agents. Mol Cancer Ther. 2009;8(9):2645–2654. doi: 10.1158/1535-7163.MCT-09-0383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.