

Summary
Pediatric renal cell carcinomas (RCC) differ from their adult counterparts not only in histologic subtypes but also in clinical characteristics and outcome. However, the underlying biology is still largely unclear. For this reason, we performed whole-exome and transcriptome sequencing analyses on a cohort of 25 pediatric RCC patients with various histologic subtypes, including 10 MiT family translocation (MiT) and 10 papillary RCCs. In this cohort of pediatric RCC, we find only limited genomic overlap with adult RCC, even within the same histologic subtype. Recurrent somatic mutations in genes not previously reported in RCC were detected, such as in CCDC168, PLEKHA1, VWF, and MAP3K9. Our papillary pediatric RCCs, which represent the largest cohort to date with comprehensive molecular profiling in this age group, appeared as a distinct genomic subtype differing in terms of gene mutations and gene expression patterns not only from MiT-RCC but also from their adult counterparts.
Subject areas: Genomics, Cancer
Graphical abstract
Highlights
-
•
WES and RNA-seq of 25 pediatric RCCs with various histologic subtypes
-
•
Detected only limited genomic overlap with adult RCC
-
•
Revealed recurrent somatic mutations in genes not previously reported in RCC
-
•
Discovery of a CRK-PITPNA fusion gene in a pediatric papillary RCC
Genomics; Cancer
Introduction
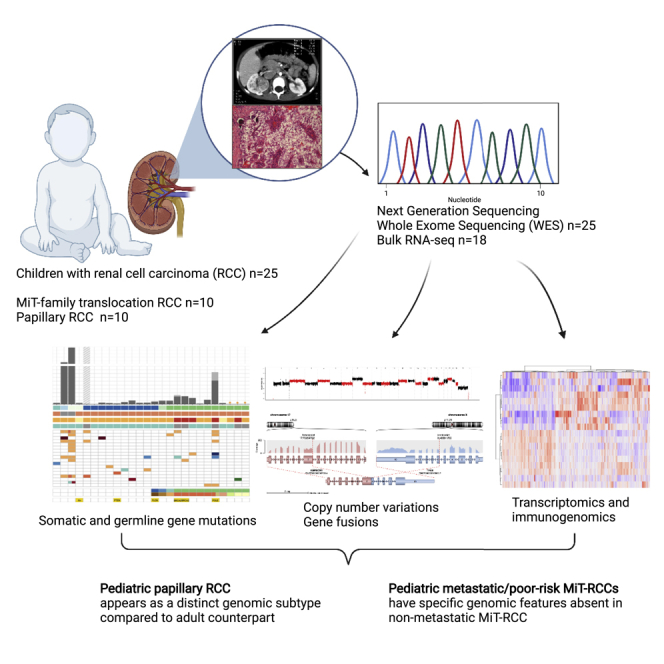
Pediatric renal cell carcinomas (pRCC) are rare tumors, which still represent the second most common renal cancer in children and adolescents after Wilms tumors, accounting for 3.5% of all renal tumors in children aged 0–14 years, rising to 50%–70% in adolescents aged 14–19 years (van der Beek et al., 2020). They differ from their adult counterparts both in histopathologic features and clinical behavior (Cajaiba et al., 2018; Geller et al., 2015; Selle et al., 2006). Approximately half of pRCCs (41%–59%) are MiT family translocation (MiT) RCCs (Cajaiba et al., 2018; Geller et al., 2020; van der Beek et al., 2021), whereas this subtype represents only 1%–4% of adult cases (Calio et al., 2019). In contrast, clear-cell RCC (ccRCC) represents the predominant subtype in adults (>75%) but rarely occurs in children. Affecting 16%–29% of pRCC cases, the papillary subtype represents the second most common pRCC histology (Cajaiba et al., 2018; van der Beek et al., 2020). Regional lymph node metastases occur more frequently associated with smaller primary tumors in pRCC than in adults (Geller et al., 2015). Further, the prognosis of RCC with regional lymph node metastases is apparently more favorable in children with an overall survival (OS) over 70%, in contrast to <30% in adults (Geller et al., 2020; Geller and Dome, 2004). The outcome of MiT-RCC also seems to be associated with age, showing a significantly better OS in younger patients in some studies (Ellis et al., 2014). The biological and molecular background of this different clinical behavior of pediatric and adult RCC is still largely unclear.
In contrast to the ever-growing body of data regarding the molecular genetics of adult RCC using next-generation sequencing (NGS) over the past few years, our understanding of the distinct genomic background of pRCC is still limited. As a result, a molecularly tailored, mechanism-of-action-based therapy in more advanced pRCC remains to be determined. The goal of this study was to analyze a representative cohort of pRCC from different histological subtypes by applying whole-exome sequencing (WES) and RNA sequencing (RNAseq), aiming to reveal more specific genomic characteristics of pRCC in comparison to their adult counterparts.
Furthermore, we focused on discovering differences between the histological and clinical subgroups of pRCC, especially aiming for potential specific genomic features in the metastatic and high-risk cases. This may impact the development of more effective treatment approaches for these patients, which is urgently needed in distant metastatic pRCC with a 5-year overall survival (OS) below 50% (Geller et al., 2020; van der Beek et al., 2021). Through the integration of these analyses, we provide an in-depth overview of the genomic landscape of pediatric RCC.
Results
Cohort characteristics
In this retrospective study, the genomic characteristics of RCC in 25 children and adolescents (15 boys, 10 girls; median age: 12.2 years [range 1.2–17]) were investigated, including 10/25 (40%) MiT-pRCC, 10/25 papillary pRCC (ppRCC), one ALK-rearranged RCC, one chromophobe RCC, one postneuroblastoma (NB)-RCC, and two histologically unclassified RCCs (Table S1). One of these initially histologically unclassified RCCs (RCC380) was recognized as harboring a germline FH mutation with bi-allelic FH inactivation after the sequencing data analysis presented here, i.e., as the FH-deficient RCC subtype. In two patients, the RCC occurred as a secondary malignancy after an earlier different malignant tumor, in one case neuroblastoma and in the other case ependymoma, treated with alkylating chemotherapy 6.8 and 10 years prior to the RCC diagnosis, respectively. One patient had a known tumor predisposition syndrome, i.e., a PTEN hamartoma tumor syndrome (RCC1430). Fifty-two percent of pRCC patients in this cohort had localized disease (11/25: T1/2, 2/25: T3N0M0), 4/25 (16%) patients had regional lymph node metastases (N1M0), and another 4/25 (16%) patients had distant metastases (N0-1M1). In four cases the regional lymph node or distant metastases status was unclear. All distant metastatic (M1) pRCC cases were MiT-RCCs. Twenty out of twenty-five patients were alive and disease free after a median follow-up of 3.75 years (range: 0.4–20.7 years), and 5/25 patients died due to RCC 1–11 years after diagnosis (3/5 M1, 1/5 T2N1M0, 1/5 T3N0M0 at diagnosis).
Somatic gene aberrations
Overall, we found 2,930 nonsynonymous somatic single-nucleotide variations (SNVs) and small insertions/deletions (indels) in 32 tumor samples from 22 patients with patient-matched controls (Table S2). The two secondary-malignancy-RCC cases RCC1230 and RCC419 harbored the highest numbers of mutations, with 786 (22.9 mutations/Mb) and 356 (10.6 mutations/Mb), respectively. Compared with all nonmetastatic and nonsecondary-malignancy cases, the three distant metastatic (M1) RCC cases (3/4; one case without matching control sample) displayed a higher tumor mutational burden (median TMB 0.66 mutations/Mb [range 0.52–2.56]; median SNV count 21 [range 16–62]; median indel count 2 [range 0–22]). In the remaining 16 nonmetastatic and nonsecondary-malignancy pRCC cases, we found very low TMB (median 0.12 mutations/Mb [range 0–0.36]; median SNV count 3.5 [range 0–11.5]; median indel count 0 [range 0–1.5]), independent of the histological subtype (Figure 1). One thousand five hundred twenty-six SNV/Indels were detected in RCC987, most likely including a substantial fraction of FFPE artefacts that could not be filtered further. RCC987 was nevertheless included into the cohort despite poor-quality FFPE tumor material, as this was the only case of an initially localized ppRCC with fatal disease course, but subsequently excluded from mutational burden analysis.
Figure 1.

Genomic aberrations in pediatric renal cell carcinomas (pRCC, n = 25)
Tumor mutational burden, number of SNV/Indels in WES, genes affected by recurrent nonsynonymous somatic aberrations# and canonical adult RCC-associated genes bearing somatic aberrations, as well as gene fusions and germline predisposition genes in different histologic and clinical subgroups of the pRCC cohort.
# Only the most important recurrent somatic aberrations are selected; in total 39 recurrent SNV/Indels were detected after exclusion of RCC987; complete list in Table S3.
∗ Cases without control blood DNA.
§ RCC380: initially histologically classified as “unclassified” subtype, but after sequencing data analysis recognized as harboring a germline FH-mutation with bi-allelic FH-inactivation, i.e., as a FH-deficient RCC subtype.
▪ Gray shaded bar: RCC987 is probably highly biased by FFPE artefacts. For this sample, all SNV/Indels shown in the somatic aberrations have been manually verified in IGV. A slightly higher SNV/Indel count is expectable, as this was the only case of a primarily localized papillary RCC with a fatal disease course following a disseminated metastatic relapse within 2 years (plus initial high LDH [3.047 U/l] and high ferritin/CrP level).
For patients with more than one tumor sample taken from different locations of the tumor piece of the same disease episode, the mean of the SNV and indel number of these tumor samples was calculated for each patient and used for calculation of tumor mutational burden (TMB) and in this figure.
RCC544_I/RCC_544_II: tumor Samples from the same patient with 10 years in between (RCC544_I from initial phase abdominal tumor material; RCC544_II from lung metastasis after 10 years).
∗ Acyl-CoA-related genes: ACSBG2 (3 SNVs) in RCC544_I, ACSM3 in RCC751, ACSM2B in RCC419, ACSL5 in RCC1230, ACBD7 in RCC987, ACOT13 and ACOX1 in RCC1230, ACADSB in RCC 419.
∗CHD: CHD1 in RCC1230, CHD4 in RCC751, CHD5 in RCC987, CHD6 in RCC419.
∗ MAPK: MAP3K9 in RCC996 und RCC1230, MAP2K5 in RCC1230, MAP2K7 in RCC419, MAP4K1 in RCC544_I, MAP4K3 in RCC751∗ SWI/SNF: SMARCA4 in RCC987, SMARCA2 in RCC419, SMARCC1 and SMARCC2 in RCC1230.
In total, 1,307 protein-coding genes harbored nonsynonymous somatic mutations (SNVs and/or indels) among the 31 samples from 21 pRCCs (without RCC987). After excluding both secondary-malignancy-RCCs, only 243 unique genes were found to be affected by a nonsynonymous somatic mutation at least once. Focusing solely on the 16 nonmetastatic and nonsecondary malignant RCCs, only 107 genes affected by somatic SNVs or indels remained. Among the nonsynonymous somatic mutations found in the pRCCs, 39 genes were affected recurrently (after excluding RCC987), mostly only twice (Table S3). Only one gene, namely CCDC168, harbored somatic nonsynonymous SNVs in the tumor samples of three different patients (Figure 1). CCDC168 is a protein-coding gene with poorly understood function and has also been found to be mutated in colorectal cancers (Wang et al., 2018a).
Among the other recurrently (i.e., in two different patients) mutated genes in our cohort, only TP53 is also frequently mutated in adult RCC. Other well-known somatic alterations in adult RCC affecting MTOR, DLEC1, ERCC2, PIK3CA, LRP2, SMARCA4, SMARCC2, PTEN, and TSC2 were only found in one case each in this pRCC cohort (5/9 of them in the same secondary-malignancy case; Figure 1). Additional mutations discovered in chromatin-remodeling genes in adult MiT-RCC (Malouf et al., 2014) were only found nonrecurrently in a few pRCC cases. Nonsynonymous SNVs in CHD4 and CHD5 were detected in one metastatic MiT-pRCC case and in the only relapsed papillary RCC case, respectively. The two secondary-malignancy (non-MiT) RCC cases harbored nonsynonymous SNVs in SMARCA2, SMARCC1, CHD1, and CHD6 and a splice-site affecting SNV in SMARCC2. In summary, only a minority (4/22 = 18%, 1/7 MiT-pRCC) of pRCCs carried a somatic mutation in the SWI/SNF gene complex or in other chromatin-remodeling genes, involving only metastatic or relapsed and fatal cases, as well as secondary-malignancy cases.
Among the remaining recurrently mutated genes in our cohort, somatic mutations affecting mitogen-activated protein kinases (MAPK) were detected. Nonframeshift deletions and a nonsynonymous SNV in the MAP3K9 gene were found in three samples from two patients. Furthermore, one frameshift deletion in MAP4K1, one nonframeshift deletion in MAP4K3, and nonsynonymous SNVs in MAP2K5 and MAP2K7 were detected. In summary, 23% (5/22) of pRCC cases carried a somatic mutation in a MAPK gene including 2/4 metastatic MiT-RCC cases (Figure 1, Table1). MAP3K9 mutations have recently been identified in lung cancer cells (Fawdar et al., 2013), MAP3K9 and MAP3K5 mutations in melanomas (Stark et al., 2011), and MAP3K1 mutations in breast cancer and ccRCC (Kandoth et al., 2013), but none or very few mutations in the other MAPK genes detected in our pRCC cohort have been observed in adult RCC (https://www.cbioportal.org/, in n = 186 adult RCCs selected). Notably, we found one splicing indel and one nonsynonymous SNV in the leucine-rich repeat kinase 2 (LRRK2) in two metastatic MiT-pRCCs. LRRK2 somatic mutations have been detected across different cancer types, and diverse alterations have been found to be associated with a worse prognosis compared with wild type (Lopez et al., 2020). Furthermore, we detected nonsynonymous SNVs in the VWF (Von Willebrand Factor) gene in two different MiT-pRCCs, including one metastatic case. VWF gene mutations have been previously reported in gliomas (Lehrer et al., 2019b) and breast cancer, and this gene has been found to influence cancer angiogenesis and apoptosis (Lehrer et al., 2019a). In addition, one frameshift deletion and one nonsynonymous SNV in PLEKHA1 occurred in two ppRCC cases. PLEKHA1 polymorphisms have been reported to be associated with chronic kidney diseases within genome-wide association studies (Chasman et al., 2012), and renal insufficiency has been demonstrated as risk factor for RCC (Tsuzuki et al., 2018), especially for papillary RCC (Woldu et al., 2014).
Table 1.
Clinical and genomic characteristics of metastatic pediatric MiT-RCC
| Case | Age at diagnosis (years) | SNV/Indels (no.) | Recurrent and/or special SNV/Indel | CNA | Chr. losses/gains | Immune-gene expression | Possible drug targets | Outcome (months) | Drug treatment/response, time to progression (TTP) |
|---|---|---|---|---|---|---|---|---|---|
| RCC544_I RCC544_II | 1 | I: 20/4, II: 21/0 |
I: MAP4K1, ACSBG2_ II: MLH3, CHEK2, ANKRD17 I/II: VWDE |
≤ 2 chr | No | PD-L1+, PD-1++, CTLA4+++, TiM3/LAG3+++ | RET, MAP4K1 | DOD (132) | IT/CT, Sunitinib, PBSCT, Vacc, Sorafenib/→PR/SD 6 years, Axitinib/Nivolumab→PD |
| RCC4039 | 3 | 16/1 | LRRK2, FIG4 | Not evaluable | Not evaluable | No RNA available | Alive NED (248) |
INFa/IL2/capecitabine/→PR →Surgery → CR | |
| RCC450 | 10 | No matched DNA | TP53 | ≥8 chr. | 1p, 3p, 4q, 9p, 11, 14, 15, 17p, 18//gain 17q | No RNA available | DOD (12) | sunitinib/→MR TTP 3 months, --> everolimus → PD |
|
| RCC751 | 16 | 62/22 | LRRK2, VWF, CCDC168, UGGT1, COL18A1, GLDN, MAP4K3, ACSM3 |
≥8 chr. | 1p, 3p, 4, 9p, q, 11, 18, 22// Gains 8, 12, 20q |
PD-L1+++, PD-1−, CTLA4−, TiM3−, LAG3(+) | MET, RET, B7-H3, MAP4K3 | DOD (19) | Sunitinib /→PR?/SD TTP 3 months, → PBSCT, Axitinib → PD |
Interestingly, somatic mutations were found in several genes coding for acyl-CoA metabolism-linked enzymes, which play a critical role in lipid metabolism like fatty acids supply in cancer cells. Two metastatic cases and two secondary malignancy pRCCs harbored mutations in acyl-CoA synthetase genes. In addition, two secondary malignancy cases and RCC987 nonsynonymous SNVs in other acyl-CoA-enzyme genes were detected (Figure 1). Acyl-CoA synthetase and thioesterase have been reported to be deregulated in different cancer diseases, and acyl-CoA-synthetases are considered potential druggable targets (Chen et al., 2016; Jung et al., 2017; Rossi Sebastiano and Konstantinidou, 2019).
Besides somatic SNVs and indels, we found chromosomal deletions on chr1p leading to CDK11A/B loss in two patients (Figure 1). CDK11 belongs to the cyclin-dependent kinases (CDK) family of serine/threonine protein kinases. CDK11 loss and/or downregulation have been reported in some tumors, such as in melanomas (Chandramouli et al., 2007), but to our knowledge not yet in RCC.
Gene pathway analysis of somatic SNVs and indels found in the pRCC cohort using the KEGG pathways revealed the FOCAL_ADHESION and the extracellular matrix (ECM)_RECEPTOR_INTERACTION pathways as significantly affected (Table S4), which are both reported to be involved in adult RCC pathogenesis (Wala et al., 2015).
Based on mutational signature analysis (single base substitution [SBS] signatures, COSMIC Signatures v3.2) (Alexandrov et al., 2020) for the secondary-malignancy-RCC samples, we found alkylating agent and platinum-treatment related signatures (SBS11/31) consistent with previous treatment and a defective DNA-repair-related signature (SBS30_Defective BER) (Figure S1). The remaining RCC cases harbored too few somatic mutations for signature analysis.
Interestingly, in the secondary-malignancy-RCC case after ependymoma (RCC1230) exome analysis revealed two inactivating somatic mutations in TSC2: a stop-gain SNV and a small frameshift deletion (Figure 1). Recently, in a morphologically distinct group of histologically unclassified adult RCCs, somatic inactivating mutations in TSC2 were reported, including cases harboring two independent mutations, which might indicate bi-allelic inactivation (Chen et al., 2019).
Germline predisposition genes
In the 22 patients with matching normal tissue, five (likely) pathogenic germline mutations were identified (Figure 1). Only in a single case a preexisting disease was known prior to the RCC manifestation, namely a clinical history of a phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome (RCC1430). Through additional whole-genome sequencing (WGS), we verified a germline deletion of exons 2-9 spanning over 150 Kb in the PTEN gene locus and a second hit in the wild-type (wt) PTEN allele in the tumor.
In RCC380, a germline structural variation in the form of a duplication of exons 2-7 spanning approximately 16 Kb within the FH gene (Figure 2C) was detected and verified with additional WGS data. In the tumor genome, the wt-FH allele was lost due to copy-neutral loss of heterozygosity (LOH). Furthermore, in this case we detected the lowest FH gene expression level across the whole pRCC cohort, implying potential functional relevance and a driver role. Germline FH mutations characterize hereditary leiomyomatosis and RCC syndrome (HLRCC) known to predispose to RCC (Ho and Jonasch, 2014; Schmidt and Linehan, 2016). However, to the best of our knowledge, the germline FH alteration described here represents a new finding not yet reported in ClinVar or other sources (Hol et al., 2020).
Figure 2.

Fusion genes found for the first time in pediatric RCC and discovery of a new germline FH mutation
(A) A complex gene fusion involving 3 different genes: TFE3, ASPSCR1, and SNX8 in RCC751.
(B) Discovery of gene fusion CRK-PITPNA in papillary pediatric RCC (RCC1176), associated with the second highest CRK expression across all pRCC.
(C) Germline structural variant within the FH gene (RCC380): duplication and intrachromosomal fusion of exon 1-7 with exon 2-10 and “second hit” copy-neutral LOH of the wild-type FH allele resulting in the lowest FH gene expression compared with all other pRCCs.
In the youngest patient (1.2 years at diagnosis) of our pRCC cohort (RCC544), a heterozygous germline frameshift insertion in BRCA2 (c.8323dupA) was found (Table S5), for which pathogenic relevance has been documented in ClinVar (rs276174904, ClinVar ID 126174). However, no additional second (somatic) hit in BRCA2 was detected in the tumor. Although germline mutations of BRCA2 have been related to an increased familial risk for breast and ovarian cancer and possibly a wider spectrum of cancers, the association with RCC remains unclear (Mersch et al., 2015). Nevertheless, heterozygous germline mutations in BRCA2 have already been reported in adult RCC cases (Carlo et al., 2018; Souza et al., 2018).
Furthermore, in one case (RCC1176) we found a heterozygous germline missense mutation in the folliculin (FLCN) gene with uncertain significance in ClinVar (NP_659434.2: p.Gln408His; https://www.ncbi.nlm.nih.gov/clinvar/variation/579468/). This patient was a 12-year-old boy without skin lesions and without a second hit in the wt-FLCN allele in the tumor sample. Mostly protein truncating but also missense mutations in FLCN are known to cause autosomal dominant Birt-Hogg-Dubé (BHD) syndrome (Ho and Jonasch, 2014; Schmidt and Linehan, 2016), which is usually clinically characterized by skin fibrofolliculomas and early onset of RCC. However, a similar RCC case in an adolescent girl with absence of skin lesions and a newly described inherited FLCN mutation has been published (Schneider et al., 2018). Interestingly, in the tumor of RCC1176 we also discovered a previously unreported CRK-PITPNA translocation (Figure 2B), as described below.
Finally, a heterozygous POLE germline mutation (c.1337G>A [p.Arg446Gln]) assessed as variant of uncertain significance (VUS) was found in a 16-year-old male suffering from a highly aggressive metastatic RCC (RCC751). The tumor sample showed a remarkably higher mutation rate (TMB 2.56 mutations/Mb, 84 SNV/indels) compared with most other pRCCs, despite no second hit. Germline POLE mutations have been described as a dominant cancer predisposition syndrome for colorectal cancers (CRC) (Valle et al., 2019), but an association with RCC is not known so far.
Gene fusions and translocations
In this pRCC cohort, 10 MiT-pRCCs were detected based on TFE3-IHC, TFE3-FISH, and/or RNA-seq data. In 8/10 cases, the exact gene fusion could be identified by bioinformatics analyses tools using WES or RNA-seq data (or by RT-PCR in one case (Macher-Goeppinger et al., 2012)) as TFE3-ASPSCR1 in 5 cases and TFE3-NONO, TFE3-SFPQ, and TFEB-CLTC in one case each (Figure 1 and S2). In the other two MiT-pRCC cases, the TFE3 fusion gene could not be discovered using WES data only. Beyond these previously known TFE3/TFEB fusion genes (Cajaiba et al., 2018; Durinck et al., 2015), we found an unreported complex TFE3-ASPSCR1-SNX8 fusion gene in one particularly aggressive metastatic case (RCC751) (Figure 2A). Furthermore, we found one ALK-rearranged pRCC harboring a TPM3-ALK gene fusion (Figure S2), as also reported by (Cajaiba et al., 2018).
Beyond that, in a pRCC, which was histologically classified as papillary RCC, we detected a CRK-PITPNA gene fusion (Figure 2B), which has so far not been described. Furthermore, in this RCC case, the second highest CRK gene expression within our whole cohort could be verified in the RNA-seq data, suggesting a biological role of this newly discovered CRK-PITPNA fusion gene.
Somatic copy number alterations
The frequency of copy number alterations (CNAs) in the pRCC cohort differed depending on the histologic RCC subtype. In contrast to the papillary pRCC (ppRCC), we found a comparatively low frequency of CNAs in most MiT-pRCC cases (Figures 3A and S3).
Figure 3.

Copy-number profiles of the pediatric RCC cohort
(A) Copy-number profiles of the papillary pediatric RCC (ppRCC, n = 10) and the MiT-family translocation pediatric RCC (MiT-pRCC, n = 9#) shown as average CNA per group. #RCC4039 was excluded due to poor DNA quality. ppRCC displayed high CNA frequency affecting predominantly gains of chr. 2, 7, 12, 16, 17, and 20. MiT-pRCC showed mostly a low CNA frequency; a high CNA count was predominantly restricted to metastatic/progredient (=poor prognosis-) RCC in older patients including losses of 1p, 3p, chr.4 or 4q, 9p, chr.11 or 11q, chr.18 and, partially, gains of 17q.
(B) Two metastatic MiT-pRCC cases selected from 3a displayed higher CNA frequency including losses of 3p, 9p, and chr.11 or 11q.
(C) Copy-number profiles of the chromophobe-RCC (RCC1348), the ALK-rearrangement-associated RCC (RCC1429), the post-neuroblastoma RCC (RCC419), and a secondary-malignancy, histologically unclassified RCC (RCC1230), each presented as a single case (copy-number profile of RCC380 see Figure 2C).
Five of nine MiT-pRCC patients in our cohort (RCC4039 was excluded because of poor DNA quality due to FFPE tumor material) showed low CNA frequency, having CNAs affecting two or less chromosomes. Among these five samples, there were 4/6 localized MiT-pRCC patients who survived long-term and one initially distant metastatic RCC patient who died due to RCC 11 years after diagnosis (RCC544). In contrast, in the high-CNA group comprising 4/9 MiT-pRCC cases, two patients had an initially metastatic disease and died within two years (RCC450 and RCC751) (Figure 3B) and one additional patient with locally advanced disease (T3) died due to relapses (RCC786) (Figure S3B), supporting the association between high CNA frequency and poor OS in MiT-RCC as reported previously (Marcon et al., 2020). Moreover, the median age in the high-CNA-group was higher compared with the low-CNA-group of MiT-pRCC patients (12.3 versus 10 years, n.s.). The most common CNA observed across the whole MiT-pRCC cohort was the loss of chromosome 1p, which has been observed in 5/9 patients and has also been reported in adult MiT-RCC (Liu et al., 2020). The high-CNA group showed CNAs affecting six or more chromosomes, including losses of 1p (4/4), 3p (2/4), 4 or 4q (2/4), 11 or 11q (3/4), 18 (3/4), and in particular exclusively in fatal cases losses of 9p (3/4), which is known as a poor-prognostic marker and preferentially found in older patients (Liu et al., 2020; Marcon et al., 2020) as well as 17q gains (2/4). Beyond that, in one of these metastatic MiT-pRCC patients (RCC450), chr17p loss was detected in addition to the TP53 mutation on the remaining 17p allele (Figure S3B).
All ppRCC in the cohort revealed uniformly high numbers of CNAs. Each sample harbored CNAs at least affecting eight chromosomes, whereas in 6/10 cases CNAs were found in ten or more chromosomes. We observed predominantly gains of whole chromosomes, most frequently found for chromosomes 2 (9/10), 7 (8/10), 12 (9/10), 16 (8/10), 17 (10/10), and 20 (8/10) and a smaller number of chromosome losses, mostly of chr21 (4/10) (Figure 3A and S3A).
In the single NB-associated RCC case in the cohort, we found a higher number of CNAs with losses of chromosomes 19, 21, and 22 and focal losses in 1p, 2q, and 6q, supporting the hypothesis of distinct underlying biological processes in the NB-associated RCC in comparison to the only other second-malignancy-RCC case in the cohort, which occurred after an anaplastic ependymoma and did not show any CNAs in the RCC tumor genome (Figure 3C). The single case of chromophobe RCC of the cohort showed only loss of chromosome 1. However, in contrast to the adult counterparts, no concurrent pathognomonic losses of further chromosomes additionally to the chr1 loss were detected (Huang and Hsieh, 2020). In the one ALK-rearrangement-associated pRCC case, we found losses in chromosomes 14 and 22 (Figure 3C).
Tumor heterogeneity and evolution
In 4/8 pRCC cases with more than one tumor sample from the same disease time point, we found a highly different SNV pattern without or with only minimal SNV overlap (Figure S1), whereas in one case only the CNA profile varied clearly between the different tumor samples (Figure S3). Interestingly, in one of these eight cases, a somatic TP53 stopgain SNV was only detected in one out of three tumor samples from the same disease time point. In this tumor, clearly different histological features between the samples were also noticed. In contrast, the TFE3/TFEB fusion genes, the well-known driver mutations, were present in all samples from the same patient.
In one patient suffering from a metastatic MiT-pRCC harboring a TFE3-ASPSCR1 fusion, we could analyze two sequential samples (RCC544_I/II; Figure 1, Table1) taken from metastatic lesions with a 10-year interval in between within the course of persisting disease. Despite many different treatment lines, the girl never achieved a complete tumor remission, but survived 11 years with metastatic lesions. Besides the TFE3 fusion, both analyzed samples shared a common somatic SNV in VWDE (von Willebrand factor D and EGF domains), which has functions still largely unclear in human biology (Leigh et al., 2020). Overall, we did not find a significant increase in the number of somatic SNV/indels in the later sample (RCC544_II) (Figure S1B), but only few shared coding SNVs between the two samples as well as different mutational signatures, obviously remarkably influenced by the treatment of the later sample (Figure S1A). For example, SNVs affecting the CHEK2 (check point kinase 2) and the MLH3 (MutL Homolog 3) gene were detected only in the tumor sample of the later time point.
Transcriptome analysis
Unsupervised hierarchical clustering of the transcriptome data (n = 18 samples with available RNAseq data) based on the top 1,000 most differentially expressed genes showed MiT-pRCC and ppRCC as clearly separated clusters, suggesting specific gene expression patterns within each subgroup (Figure 4A). The finding of a distinct MiT-RCC-associated transcriptional signature is consistent with previous results (Camparo et al., 2008; Malouf et al., 2014). The one chromophobe pRCC case was found to cluster closely with the MiT-pRCC.
Figure 4.

Gene expression analysis (RNA-seq data) of pediatric RCC (pRCC; n = 18). Among the 18/25 pRCC cases with available RNA, 6 MiT-pRCC cases (4 with ASPSCR1-TFE3 fusion, 1 with TFE3-SFPQ fusion, and 1 with TFEB-CLTC fusion), 8 papillary pRCC (6/8 histologically papillary type 1), one chromophobe, one ALK-rearranged, one post-NB, and one FH-deficient RCC were included
(A) Unsupervised hierarchical clustering of pRCC samples based on the top 1,000 most differentially expressed genes (DEGs).
(B) Volcano-plot illustration of the significantly DEGs in MiT-pRCC versus papillary pRCC.
(C) Volcano-plot illustration of the significantly DEGs in metastatic fatal MiT-pRCC versus nonmetastatic surviving MiT-pRCC.
(D) MET and RET gene expression in MiT-pRCC and papillary pRCC (ppRCC) compared with Wilms tumor relapses (INFORM), primary Wilms tumors, and normal pediatric kidney (TARGET) as well as adult clear cell RCC, adult papillary RCC, and adult normal kidney (TCGA). MET gene expression significantly higher in ppRCC versus MiT-pRCC, log2FC = 1.6, p < 0.0001, q < 0.01 (Wald test). RET gene expression significantly higher in MiT-pRCC versus ppRCC, log2FC = 1.15, p = 0.013, q = 0.056 (Wald test).
Differential gene expression analysis of MiT-pRCC versus ppRCC yielded 1,272 significantly differentially expressed protein-coding genes (DEGs) at a false discovery rate (FDR) < 0.01 and | log2FoldChange | >2. We identified NMRK2, DPP6, BRINP2, CHRM4, NROB2, MLPH, NTSR2, GDNF, HSD17B6, HHATL, SV2B, FSTL4, PTGER3, and CYP17A1 as the most significantly overexpressed genes in MiT-pRCC. In ppRCC, we detected DNAAF1, ZMYND10, DNAI1, FHL1, and SHH as being most significantly higher expressed than in MiT-pRCC (Figure 4B; Table S6).
Gene set enrichment analysis (GSEA) of these DEGs using the KEGG pathway database revealed the “Neuroactive ligand-receptor interaction” gene set as most significantly upregulated in the MiT-pRCC, which includes the above-mentioned NTSR2, CHRM4, and PTGER3 (Table S7). In addition, GDNF, BRINP2, SV2B, and DPP6 exhibit neurotrophic, neurotransmitter, or other neural-related functions and are associated with neurological disorders. Beyond that, most of them have been reported to be involved in different cancers (Chen et al., 2021; Fielder et al., 2018; Millstein et al., 2020; Zhang et al., 2020b). Among them, we found to be highest upregulated the cell-surface G-protein-coupled receptor, neurotensin receptor 2 (NTSR2) gene in MiT-pRCC (Figure 4B). NTSR2 is physiologically highly expressed in brain and participates in signal transduction of neurotensin (NT), which acts as a neurotransmitter in the central nervous system as well as a peripheral hormone in the intestine among others. In addition, dysregulation of NT and NTSR1/2 has been described with oncogenic impact in different types of cancers (Ouyang et al., 2017) which includes apoptosis regulation (Abbaci et al., 2018) as well as activation of the extracellular signal-regulated kinases 1/2 (Ayala-Sarmiento et al., 2015).
Applying the Hallmark pathway analysis to the DEGs, the XENOBIOTIC_METABOLISM pathway emerged as significantly upregulated in pMiT-RCC compared with ppRCC, which includes for example CYP17A1 (Table S7). Moreover, among the above-mentioned significantly overexpressed genes in MiT-pRCC, NMRK2, NROB2, and HSD17B6 were identified to be related to diverse metabolism functions. The NMRK2 (nicotinamide riboside kinase 2) gene was most significantly overexpressed in the MiT-pRCC samples and encodes an enzyme involved in energy metabolism and the oxidative phosphorylation system via NAD biosynthesis, which is normally highly restricted to muscle (Ratajczak et al., 2016) and could play a role in the aberrant metabolism in RCC.
The gene expression profile of the MiT-pRCC cohort corresponds well with the results reported (Camparo et al., 2008; Pflueger et al., 2013) in adult MiT-RCC cases with 25/46 and 23/48 concordantly upregulated signature genes (i.e., HHATL, CYP17A1, SV2B, NTSR2, and TRIM63).
Focusing on ppRCC, the Hallmark pathway analysis revealed the G2M_CHECKPOINT, E2F_TARGETS, and MITOTIC_SPINDLE pathways as significantly upregulated in comparison to the MiT-pRCC. Furthermore, we identified DNAAF1 (Dynein Axonemal Assembly Factor 1), DNAI1 (Dynein Axonemal Intermediate Chain 1), and ZMYND10 (Zinc finger MYND-type-containing 10) as the three most significantly higher expressed genes in ppRCC compared with MiT-pRCC. All three genes encode proteins involved in the formation and assembly of the dynein arms and are required for normal motile ciliary ultrastructure and function, which is consistent with the enriched cilium signature reported in adult papillary RCC transcriptome (Huang and Hsieh, 2020; Ricketts et al., 2018).
Furthermore, MET emerged as significantly higher expressed in ppRCC compared with MiT-pRCC (Figure 4D), which was expected based on adult papillary RCC studies (Albiges et al., 2014; Durinck et al., 2015) and is consistent with the frequent chr7 gains. Among the MiT-pRCCs, only one particularly aggressive metastatic case (RCC751) demonstrated a slightly higher MET expression than the median expression value in Wilms tumors and adult ccRCC, corresponding to the results of an IHC study in a predominantly adult MiT-RCC cohort showing a significant correlation between high MET protein expression and clinical aggressiveness (Calio et al., 2020).
Focusing on additional potential drug targets, we studied the expression of the proto-oncogene RET, a receptor tyrosine kinase recently detected to be significantly upregulated in a PRCC-TFE3-fusion-bearing mouse model (Baba et al., 2019) and subsequently verified by IHC-staining in some MiT-RCCs. However, RET was found to be only borderline significantly overexpressed (log2FC1.15, p = 0.013) in MiT-pRCCs compared with ppRCCs (Figure 4D).
In addition, we performed differential gene expression analysis between patients who died due to metastatic MiT-pRCC and nonmetastatic surviving MiT-pRCC patients (Table S8), which revealed 167 protein-coding DEGs (FDR<0.01, | log2FoldChange | >2). TFAP2A, GBP1, EFEMP1, TBX18, DSC3, GATA4, and CA3 emerged as the highest significantly overexpressed genes in the fatal cases (Figure 4C). Except for CA3, all these genes have been reported to be potentially involved in tumorigenesis and their overexpression have been frequently associated with poor prognosis (Pan et al., 2014; Tapia-Carrillo et al., 2019; Wang et al., 2018b, 2020; Zhao et al., 2019; Zhou et al., 2018).
Immune profiling
Besides tumor cell genomics, we investigated the tumor associated immune microenvironment (TME), which also affects tumor evolution in RCC (Chevrier et al., 2017). Using the quanTIseq immune cell deconvolution algorithm based on bulk RNA-seq data, we observed macrophages as the predominant immune cell population, with a mean proportion of 35% of all immune cells (IC) across pRCC samples of all histologic subtypes representing the main IC fraction in 10/18 cases, mostly consisting of M2 macrophages (26%). Macrophages dominated among the IC in a higher proportion of cases in MiT-pRCC (5/6) compared with ppRCC (4/8) (Figures 5A–5C). The highest macrophage fractions (except for one case) were found in the two metastatic MiT-pRCC with fatal course, which is consistent with studies in adult ccRCC (Mier, 2019; Stenzel et al., 2020). Beyond that, we found substantial differences in the composition of TME in different histologic subtypes of pRCC. The fraction of dendritic cells (DC) in ppRCC (mean 22.5%) was significantly higher than in MiT-pRCC (mean 4%) (Welch two sample t test, p = 0.026). A higher CD4-T cell fraction has also been observed in ppRCC (mean 14%) compared with MiT-pRCC (mean 4%) (Figures 5A–5C). A substantial fraction of CD8-T cells was found only in one exceptional metastatic MiT-pRCC. The mean fractions of CD4−, CD8−, Tregs, B cells, natural killer (NK) cells, DC, as well as neutrophils were 8%, 5%, 4%, 3%, 7%, 13%, and 22% of total IC respectively across all pRCC (Table S9).
Figure 5.

Tumor immune microenvironment (TME) deconvolution and gene expression of immune-regulatory, checkpoint or immunosuppressive activity-related genes in pediatric RCC (pRCC; n = 18)
(A) Fraction of different immune cell populations in each pRCC case.
(B) Overview of immune cell composition (median value of each cell type) in all pRCC, MiT-pRCC, and papillary pRCC (ppRCC).
(C) Fractions of selected immune cell types in all pRCC, MiT-pRCC, and ppRCC; dendritic cell fraction was significantly different between MiT-pRCC and ppRCC, p = 0.026 (Welch two sample t test).
(D) Unsupervised hierarchical clustering based on gene expression profiles of 59 immune-regulatory, checkpoint or immunosuppression-associated genes (Samstein et al., 2020) in pRCC.
(E) Anti-tumoral cytolytic activity (CYTscore) in all pRCC, MiT-pRCC, and ppRCC.
(F) Gene expression of selected immune checkpoint genes in all pRCC, MiT-pRCC, and ppRCC; ∗PD1 (p < 0.001, Wald test), ∗PD-L1 (p < 0.001, Wald test), ∗CTLA4 (p = 0.01, Wald test), and ∗TIGIT (p = 0.002, Wald test) significantly different between MiT-pRCC and ppRCC.
In addition to tumor immune microenvironment deconvolution, we analyzed the expression of immune checkpoint-related molecules and calculated the anti-tumoral cytolytic activity score (CYTscore) from the cytolytic proteins granzyme A and perforin-RNA expression (Rooney et al., 2015). The median gene expression level of PDCD1 (PD1), PD-L1 (CD274), CTLA4, and TIGIT was significantly higher in MiT-pRCC compared with ppRCC (Welch two sample t test, p<=0.01). In contrast, VTCN1 was higher expressed in ppRCC. Beyond that, no significant differential gene expression between the MiT-pRCC and ppRCC was found for HAVCR2 (TIM3), LAG3, B7-H3 (CD276), IDO1, A2AR (ADORA2A), BTLA, and the CYTscore (Figures 5E, 5F, and S5D and Table S10).
Interestingly, the two metastatic MiT-pRCC patients with contrasting clinical courses (RCC751, RCC544) (Table 1) exhibited different patterns of immune checkpoint molecule expression. The particularly aggressive case RCC751 had the second highest PD-L1 expression and the highest B7-H3 expression, which both are known as a poor prognostic marker in adult RCC (Lavacchi et al., 2020). Furthermore, the lowest CYTscore and gene expression of CTLA4, TIM3, and TIGIT, as well as the second lowest gene expression of PD1 among all pRCCs was observed in RCC751. In contrast, the highest CYTscore and highest gene expression of CTLA4, LAG3, and TIGIT as well as the second highest PD1 expression and only moderate PD-L1 expression (Tables 1 and S10) were found in RCC544, who survived for 11 years.
Finally, we applied unsupervised hierarchical clustering of gene expression values of a gene set consisting of 59 genes involved in immune-regulatory, checkpoint or immunosuppressive activities published recently (Samstein et al., 2021). Samples from ppRCC and MiT-pRCC formed distinct subtype-specific clusters (Figure 5D), suggesting specific TME patterns in each pRCC subtype.
Discussion
We present here the genomic landscape of a broad spectrum of pediatric RCCs and to the best of our knowledge, the largest RCC cohort in this age group with comprehensive NGS analyses to date.
In 23% (5/22) of the pRCC cases we found germline gene mutations in cancer predisposition genes, which is a remarkably higher frequency compared with other pediatric tumor cohorts (7.6%) (Grobner et al., 2018). So far, two of these germline gene mutations (POLE and BRCA2) have not been recognized as predisposing in RCC. In addition, in two cases, RCC appeared as a secondary malignancy after either neuroblastoma or ependymoma, yet no germline alterations were found in these patients.
Coding somatic mutations were detected at a very low frequency in most pRCC samples, which is consistent with other pediatric cancers (Grobner et al., 2018), but in contrast to adult RCC, in particular to adult papillary RCC (median 61 or 71 SNV/indels) (Ball et al., 2017; Durinck et al., 2015).
CNAs were the most pronounced and common genomic aberrations in ppRCC, which corresponds well with adult papillary RCC; however, in contrast, gains of chromosome 3 were rare, whereas gains of chromosome 2 were more frequently detected in ppRCC (Durinck et al., 2015; Liu et al., 2020). Further, only a minority of MiT-pRCC, i.e., a smaller proportion compared with adult MiT-RCC, showed an elevated CNA frequency, affecting predominantly metastatic or regionally advanced poor-prognosis cases. Only fatal pMiT-RCC exhibited a loss of chr9p, in line with adult data (Marcon et al., 2020), as well as loss of chr3p and chr11, which to the best of our knowledge has not yet been reported in MiT-RCC as a poor prognosis marker.
Overall, gene fusions were detected in 12 cases of our pRCC cohort, with TFE3 being the most frequent gene fusion partner (n = 9) as well as the most frequent recurrent somatic alteration detected in this cohort, as previously described (Cajaiba et al., 2018). However, we also found an unreported TFE3-ASPSCR1-SNX8 gene fusion in one particularly aggressive metastatic case (RCC751). The fusion partner sorting nexin 8 (SNX8), which retained its functional PX protein domain in the SNX8-ASPSCR1 fusion, belongs to the sorting nexin protein family, whose members are involved in endocytosis and endosomal sorting and signaling (Dyve et al., 2009). Although the underlying mechanisms remain unknown, the complex triple fusion may have contributed to the particularly fatal clinical course of this pRCC patient.
Furthermore, we discovered a CRK-PITPNA translocation in one sample classified histologically as papillary RCC, which so far, to the best of our knowledge, has not yet been reported in cancer. The CRK gene encodes an adaptor protein that acts as a molecular bridge between tyrosine kinases and their substrates. The CRK protein has been identified as an oncoprotein involved in the regulation of a wide spectrum of biological processes including cell motility, invasion, survival, and apoptosis (Kumar et al., 2014). We detected the second highest CRK gene expression level across the whole pRCC cohort in this case, suggesting a potential biological role of the aberrant CRK-PITPNA fusion gene. Interestingly, CRK is higher expressed in all papillary type 1 pRCCs compared with other subtypes, yet no further CRK fusion has been detected in this ppRCC cohort. The phosphatidylinositol transfer protein alpha (PITPNA) gene has also been shown to be involved in oncogenesis as a fusion gene partner of MLL (KMT2A) in pediatric leukemia (Ney Garcia et al., 2017).
We found only limited overlap between somatic mutations in pediatric and adult RCC, even within the same histological subtype. Canonical genes mutated in adult papillary RCC including MET, CDKN2A/B, SETD2, NF2, NFE2L2, SLC5A3, KDM6A, SMARCB1, PBRM1, BAP1, FAT1, PNKD, MLL3, STAG2, CHD3, CPQ, CRTC1, and SLC9A3R1 (Cancer Genome Atlas Research et al., 2016; Chen et al., 2016; Durinck et al., 2015; Hsieh et al., 2018; Liu et al., 2020) were not found to be mutated in pediatric papillary RCC or in other pediatric RCC subtypes. In addition, TERT promotor or ATM mutations, which were previously described in adult MiT-RCC (Marcon et al., 2020), were not found in pediatric MiT-RCC.
Overall, only a limited number of recurrent somatic mutations were detected in our pediatric RCC samples, most notably CCDC168, LRRK2, PLEKHA1, VWF, and MAP3K9, with the majority of them being reported in RCC in general for the first time. Interestingly, LRRK2, VWF, and various MAPK genes (i.e., MAP4K3, MAP4K1) mutated in metastatic MiT-pRCC cases have been reported as potential drug targets, mostly in pre-clinical models (Goh et al., 2021; Johnson et al., 2019; Mancini et al., 2020). Furthermore, analysis of differentially expressed genes revealed a limited number of potential drug targets unreported so far for MiT-pRCC such as NTSR2, CYP17A1, CHRM4, CA3, and PTGER3, besides VEGFA/HIF1A and RET with a lower degree of overexpression. For example, CYP17A1 inhibition has been shown to result in significant tumor suppression in a ccRCC-mouse xenograft model (Lee et al., 2017). Moreover, in the immune checkpoint gene expression analysis, the two metastatic MiT-pRCC exhibited either an exceptionally high PD-L1 expression or a high PD1/LAG3/TiM3+ expression together with the highest CD8-T cell fraction, suggesting possible benefit of a PD1/PD-L1-directed therapy in these patients (Simonaggio et al., 2021). In addition, the TIGIT, TIM3, and B7-H3 expression in MiT-pRCC makes them possible drug targets to consider for future clinical trials (Andrews et al., 2019; Li et al., 2020; Scribner et al., 2020). Further studies addressing the genomic characteristics of pRCCs are necessary to verify our findings in larger cohorts and broaden the spectrum of suitable drug targets, particularly focusing on MiT-pRCC patients, who urgently need substantially improved adjuvant treatment options in metastatic cases.
In conclusion, pediatric RCC represents a particular type of tumor with limited genomic overlap with adult RCC and other pediatric malignancies, rarely harboring somatic mutations typically known for adult RCC or other pediatric solid tumors (Ma et al., 2018). The ppRCC emerged as a distinct subtype, with somatic mutations and gene expression profiles being different not only from MiT-pRCC but also from adult papillary RCC (Durinck et al., 2015; Linehan and Ricketts, 2013). We discovered a CRK-PITPNA gene fusion for the first time and observed a complex TFE3-ASPSCR1-SNX8 triple gene fusion. Potentially tumor-predisposing mutations in POLE and BRCA2 were observed in RCC for the first time. Furthermore, several recurrent somatic mutations were found, which were not reported in RCC to date. This study sheds light onto the genomics of pediatric RCC through comprehensive computational analyses and lays the foundation for future studies to better understand the biology of this rare tumor type.
Limitations of the study
Our pediatric RCC study is restricted by some limitations, first of all by the relatively small sample size and the retrospective characteristic of the analysis. Furthermore, due to the rarity of pRCC and limited availability of tumor material, heterogeneous tumor samples were examined from different sites and time points of tumor evolution. Apart from that, DNA quality has been limited due to FFPE specimens from cases for which fresh frozen tumor material was not available. The results of the differential gene expression analyses may be limited, partly because of the restricted comparisons between MiT-pRCC and ppRCC only, lacking healthy control tissue from pediatric kidney. Finally, deconvolution of the TME based on the bulk RNA-seq data lack wet-lab validation.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Natalie Jäger (n.jaeger@kitz-heidelberg.de).
Materials availability
This study did not generate new unique reagents.
Experimental models and subject details
Tumor samples of 25 children and adolescents <18 years (15 boys and 10 girls) with RCC diagnosed between 1999 and 2018 in Germany were analyzed. In 20/25 cases, fresh frozen tumor material was available, collected sequentially, unselected from patients within the German SIOP93-01/GPOH and SIOP2001/GPOH studies. For 19 of these 20 cases, we also obtained patient-matched normal tissue (blood). For 8 patients more than one tumor sample from the same disease time-point, but a spatially different tumor location, was sequenced (Table S1). In addition, for one of these 20 patients, tumor samples from two sequential/different time-points in the disease course were available and included in the analysis (RCC544). Tumor samples of five additionally selected patients with particularly interesting features such as distant metastatic RCC (n = 3) or fatal disease course in primary localized RCC (n = 2) were included. From four of these five patients we obtained formalin-fixed and paraffin-embedded (FFPE) tumor samples from the Kiel Paediatric Tumor Registry (KTR), in the other case from the regional pathology (Heidelberg), and in 3 of these 5 cases matched normal tissue (2/3 blood, 1/3 lymph node) was available. Clinical data were taken from medical reports and registry forms of the SIOP/GPOH-nephroblastoma trial head office in Homburg/Germany. For the histologic diagnosis the tumor specimens were reviewed in KTR by I. Leuschner or CV, including TFE3-immunohistochemistry (IHC)/FISH in 23/25 cases. Tumors were classified according to the 2016 WHO classification, and in consideration of the International Society of Urological Pathology (ISUP) consensus conference on molecular pathology of genitourinary tumors in 2019 as well as the Genitourinary Pathology Society (GUPS), as clear-cell, papillary, chromophobe, MiT family translocation-associated, FH-deficient, unclassified RCC and the emerging, provisional entities “RCCs associated with ALK rearrangement” and “RCCs in neuroblastoma survivors” (Moch et al., 2016; Trpkov et al., 2021; Williamson et al., 2020). The tumor stages were classified according to the TNM-staging system proposed by the World Health Organization (WHO) in accordance with the 2010 UICC/AJCC standard TNM staging for RCC (Edge and Compton, 2010). All subjects or their guardians provided informed consent for clinical data registry and tumor analysis. The SIOP-93-01/GPOH and SIOP 2001/GPOH studies were approved by the ethics committee of the medical association from Saarland/Germany. The clinical and genetic characteristics of these patients are described in Table S1 in the Supplementary Appendix.
Method details
Whole-exome sequencing (WES) and RNA sequencing data from all 25 patients (including the two INFORM cases INF_KO_00D and RCC544_II) were sequenced and analysed as described below.
Whole exome sequencing (WES) and RNA sequencing
Libraries for whole-exome sequencing were prepared using the Agilent SureSelect Human All Exon kit v5 (without UTRs) and sequenced on Illumina HiSeq 4000 HiSeq 3000_4000 in paired-end mode. Strand-specific, polyA+ RNA sequencing libraries were prepared using the Illumina TruSeq protocol and sequenced on the Illumina HiSeq 4000 HiSeq 3000_4000 in paired-end mode. All samples were submitted to the Genomics and Proteomics Core Facility (GPCF) of the German Cancer Research Center (DKFZ) for WES and RNA sequencing and were only included for library preparation after passing all standard quality controls.
Whole exome sequencing (WES) data processing
Alignments were performed according to the standards defined for ICGC PanCancer (Stein et al., 2015). All reads were aligned against the phase II reference of the 1000 Genomes Project including decoy sequences d5 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/ phase2_reference_assembly_sequence/hs37d5.fa.gz) using BWA MEM (v0.7.15 using standard values except for invoking -T 0). The raw BAM files were sorted and duplicates were marked using sambamba (SAMBAMBA MARKDUP_VERSION = 0.6.5) (Tarasov et al., 2015).
Transcriptome data processing
The STAR aligner was used for alignment (version 2.5.3a) (Dobin et al., 2013). Reads were aligned to a STAR index generated from the 1000 genomes assembly, gencode 19 gene models and for asjbdOverhang of 200. The tool featureCounts (Liao et al., 2014) was used to perform gene specific read counting over exon features based on the gencode 19 gene models. Both reads of a paired fragment were used for counting and the quality threshold was set to 255 (which indicates that STAR found a unique alignment). Strand unspecific counting was used. A custom script was used to calculate RPKM and TPM expression values. For total library abundance calculations, all genes on chromosomes X, Y, MT and rRNA and tRNA genes were omitted as they are likely to introduce library size estimation biases.
Mutation calling of somatic and germline SNVs and indels
Detection of somatic and germline SNVs and insertions or deletions (indels) in the WES data was performed using the DKFZ in-house pipeline “Roddy”. The pipeline is based on SAMtools (v.0.1.19) (Li et al., 2009) mpileup and bcftools using parameter adjustments allowing for SNV calling even with low allele frequency in the tumor, as previously described in (Jones et al., 2012). In short, variants were first called in the tumour sample and then queried in the matching control sample. The raw calls were subsequently annotated using multiple publicly available tracks such as 1000 Genome variants, single nucleotide polymorphism database (dbSNP), repeats and other elements. The functional effect of the mutations was annotated using Annovar (Wang et al., 2010) and the variants were assessed for their confidence (based in read depth, sequence context, and many more parameters) and split into somatic and germline calls. The workflow is available at https://github.com/DKFZ-ODCF/SNVCallingWorkflow, SNVCallingWorkflow:1.2.166-1 was used in this project. The small insertion/deletion (indel) detection workflow is based on Platypus (Rimmer et al., 2014) with extensive quality control additions for the DKFZ developed workflow management system “Roddy” and is available at https://github.com/DKFZ-ODCF/IndelCallingWorkflow, IndelCallingWorkflow:1.2.177 was used in this project.
The analysis of samples without available germline controls was performed using an in-house developed ‘No-control workflow’ pipeline. Aligned tumor.bam files are softlinked along with the creation of fake-control.bam files. Using the Roddy version 3.5.8 framework (https://github.com/TheRoddyWMS/Roddy), standard DKFZ pipeline SNVCallingWorkflow 1.4.2 (https://github.com/DKFZ-ODCF/SNVCallingWorkflow) was used for analysing single-nucleotide variants(SNVs), while the indels were called using a Platypus (Rimmer et al., 2014) based workflow (https://github.com/DKFZ-ODCF/IndelCallingWorkflow). The output .vcf files were further analysed for somatic and germline variants.
For patients with more than one tumor sample taken from different locations of the tumor piece of the same disease episode, the mean of the SNV and indel number of these tumor samples was calculated for each patient and used in the downstream analyses and figures.
Tumor mutational burden (TMB) calculation
TMB was defined as the total number of somatic, non-synonymous mutations (including indels) normalized to the exonic coverage of the respective whole-exome kit in megabases (mutations per megabase). Only on-target mutations were counted.
Gene fusion identification
Gene fusions were identified using the Arriba algorithm (version 1.0.1) (Uhrig et al., 2021). The results were manually curated according to the output value “SUPPORTING READ COUNT”, only gene fusions with the top five highest numbers of supporting reads were considered for further analysis.
Structural variants identification
The structural variants (SVs) were called using the tool SOPHIA (https://github.com/DKFZ-ODCF/SophiaWorkflow). Particular interesting structural variants were further confirmed and characterized using IGV (Robinson et al., 2011).
Copy number variation identification
The copy number variations (CNVs) were called using the tool CNVkit v0.9.3 (Talevich et al., 2016). The average CNV plots for the papillary and MiT family translocation subgroups (Figure 3A) were generated from Log2 copynumber ratio obtained from the cnr output files from CNVkit. These files were converted to BED files, concatenated to a single file, sorted and merged using standard Linux commands and BEDTools v2.27.1 (Quinlan and Hall, 2010). BEDTools merge options were set so that bookended fragments were not merged. Using a custom python script, the resulting merged BED file was tiled into bins spanning 10000 positions each with the log2ratio of each bin being calculated as weighted average from the fragments overlapping the bin. The results were plotted using a custom R script. The single samples were processed the same.
Pathway analysis
Pathway analysis of somatic SNVs and indels (Table S4) as well as pathway analysis of differentially expressed protein-coding genes between the papillary and MiT family translocation subgroups (Table S7) was done using the Bioconductor package clusterProfiler v3.16.1 (Wu et al., 2021). Information about HALLMARK and KEGG pathways was downloaded from MSigDB (Liberzon et al., 2011, 2015; Subramanian et al., 2005).
Unsupervised hierarchical clustering based on gene expression profiles
Unsupervised hierarchical clustering was performed on the pediatric RCC samples based on expression profiles. log2(TPMs + 1) transformation of the Transcript per Million (TPM) values was performed and the top 1000 most differentially expressed genes (DEGs) were selected. A “row-centered” expression value of each of these DE-Gs were then calculated by subtracting the row median value (median expression value of each gene in the pediatric RCC cohort). These “row-centered” expression values were then used for unsupervised clustering with the R package ComplexHeatmap (Gu et al., 2016) with the parameters: clustering_distance_rows = “pearson”, clustering_method_rows = “ward.D”, clustering_distance_columns = “pearson”, clustering_method_columns = “ward.D”. Heatmap (Figure 4A) was generated with the package ComplexHeatmap.
Differential gene expression analysis
Differential gene expression analysis between the MiT family translocation subgroup and papillary subgroup as well as dead of disease (DOD)/metastasized versus alive/non-metastasized cases within the MiT family translocation subgroup were performed using the R package Deseq2 v1.18.1 (Love et al., 2014). Protein-coding genes were selected for further analysis using a custom python script. The R packages ggplot2 v3.3.3, ggrepel v0.9.1 and dbplyr v2.1.1 were used to generate the volcano plots (Figures 4B and 4C). Genes with padjust (q-value) < 0.01 and | log2(FoldChange) | > 2 were defined as significantly differentially expressed (Tables S6 and S8).
Batch effect correction
Batch effect correction was performed across the pediatric RCC cohort, the INFORM pediatric relapse Wilms tumor German cases, the TARGET pediatric Wilms tumor and normal pediatric renal tissue, the TCGA adult clear cell RCC (KIRC project) and adult papillary RCC (KIRP project) and adult normal renal tissue samples (included in KIRC and KIRP projects). The function “ComBat_seq()” built in the R package sva v3.36.0 (Zhang et al., 2020a) has been applied for batch effect correction. Raw counts of the pediatric RCC, INFORM, TARGET and TCGA cohorts has been assigned to four batches with the “batch” parameter, no specific groups were assigned (“group = NULL”). A custom python script has been used to transform the batch effect corrected raw counts into TPMs. Comparison of gene expression values across cohorts are based on the batch effect corrected TPM values (Figures 4D, S4A, S5B, and S5D).
Prediction of tumor-infiltrating immune cell fraction
QuanTIseq (Finotello et al., 2019) was applied on bulk RNAseq data according to developers instructions (https://icbi.i-med.ac.at/software/quantiseq/doc/index.html) with the following command line options “--pipelinestart = decon --tumour = TRUE --method = lsei” to perform tumor immune microenvironment (TME) deconvolution for all pediatric RCC samples (Figures 5A and 5C). The median value of cell fractions in each cell type was calculated for all pediatric RCC samples, the papillary subgroup and the MiT family translocation subgroup and illustrated in Figure 5B accordingly. No batch effect correction was applied on the pediatric RCC cohort, the INFORM pediatric relapse Wilms tumor German cases, the TARGET pediatric Wilms tumor and normal pediatric renal tissue, the TCGA adult clear cell RCC (KIRC project) and adult papillary RCC (KIRP project) and adult normal renal tissue samples (included in KIRC and KIRP projects) upstream the TME deconvolution pipeline (Figures S5A and S5C), since batch effect correction led to error-prone deconvolution results.
Gene expression of immune-regulatory, checkpoint or immunosuppressive activity-related genes
Expression of an immunoregulatory gene set consisting of 59 immune-regulatory, checkpoint or immunosuppressive activity-related genes published in (Samstein et al., 2021) has been studied across the pediatric RCC cohort. Unsupervised hierarchical clustering was performed as described in the section “Unsupervised hierarchical clustering based on gene expression profiles” (Figure 5D).
Mutational signature analysis
Mutational signature analysis was performed with the software YAPSA v1.16.0 (Hubschmann et al., 2021) based on the COSMIC signatures v3.2. The “PCAWG_valid_norm” signature results were used for the final figure (Figure S1).
Quantification and statistical analysis
We performed all statistical tests and analyses with R version R3.6.3. Differential gene expression analysis of the genes MET (p < 0.0001, Wald test), RET (p = 0.013, Wald test) and selected immune checkpoint genes including PD1 (p < 0.001, Wald test), PD-L1 (p < 0.001, Wald test), CTLA4 (p = 0.01, Wald test) and TIGIT (p = 0.002, Wald test) between pediatric MiT family translocation RCC (MiT-pRCC) and pediatric papillary RCC (ppRCC) were performed with DESEq2 v1.18.1. Significant difference between the fractions of dendritic cells between MiT-pRCC and ppRCC was tested with the Welch Two Sample t-test (p = 0.026). Visualization has been performed with the R package ‘ggplot2’ with R version R3.6.3.
Acknowledgments
We thank Andrea Wittmann (DKFZ, B062) for sample handling and sequencing submissions. We acknowledge the DKFZ's ODCF and GPCF core facilities for supporting the genomic sequencing and data processing. PB is funded by the DKFZ International PhD Program (Annemarie Poustka fellowship). The following sources are acknowledged for their support to the INFORM (INdividualized therapy FOr Relapsed Malignancies in childhood) registry: German Cancer Aid (111234), the German Childhood Oncology Foundation (DKS 2014.12), the German Federal Ministry of Education and Research (01KX2025), German Federal Ministry of Health (ZMVI1-2520IGW004), German Cancer Consortium (DKTK, Heidelberg, Germany), Bild e.V. “Ein Herz für Kinder” (PÄ-24151), and the Scheu Family donation.
Author contributions
P.B., B.S., S.M.P., and N.J. conceptualized the study. B.S., N.J., and S.M.P. provided supervision. P.B., N.J., L.M., and A.G. performed data analyses. Data visualization was conducted by P.B., L.M., and N.J. Data were collected and curated by B.S., D.T.W.J., C.V., A.N., I.B.B., M.E., J.W., N.G., and M.G. The original draft was written by B.S., P.B., and N.J., and all co-authors reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104167.
Supplemental information
Data and code availability
-
•
The published article contains all data sets generated or analyzed during this study. The WES and bulk RNA sequencing data generated in this study have been deposited in the European Genome-phenome archive (EGA) under the accession number EGAS00001006057.
-
•
This study did not generate original code.
-
•
Any additional information required to reanalyze the data included in this study will be provided by the lead contact upon request.
References
- Abbaci A., Talbot H., Saada S., Gachard N., Abraham J., Jaccard A., Bordessoule D., Fauchais A.L., Naves T., Jauberteau M.O. Neurotensin receptor type 2 protects B-cell chronic lymphocytic leukemia cells from apoptosis. Oncogene. 2018;37:756–767. doi: 10.1038/onc.2017.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albiges L., Guegan J., Le Formal A., Verkarre V., Rioux-Leclercq N., Sibony M., Bernhard J.C., Camparo P., Merabet Z., Molinie V., et al. MET is a potential target across all papillary renal cell carcinomas: result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin. Cancer Res. 2014;20:3411–3421. doi: 10.1158/1078-0432.CCR-13-2173. [DOI] [PubMed] [Google Scholar]
- Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews L.P., Yano H., Vignali D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 2019;20:1425–1434. doi: 10.1038/s41590-019-0512-0. [DOI] [PubMed] [Google Scholar]
- Ayala-Sarmiento A.E., Martinez-Fong D., Segovia J. The internalization of neurotensin by the low-affinity neurotensin receptors (NTSR2 and vNTSR2) activates ERK 1/2 in glioma cells and allows neurotensin-polyplex transfection of tGAS1. Cell Mol. Neurobiol. 2015;35:785–795. doi: 10.1007/s10571-015-0172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M., Furuya M., Motoshima T., Lang M., Funasaki S., Ma W., Sun H.W., Hasumi H., Huang Y., Kato I., et al. TFE3 Xp11.2 translocation renal cell carcinoma mouse model reveals novel therapeutic targets and identifies GPNMB as a diagnostic marker for human disease. Mol. Cancer Res. 2019;17:1613–1626. doi: 10.1158/1541-7786.MCR-18-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M.W., Gorin M.A., Drake C.G., Hammers H.J., Allaf M.E. The landscape of whole-genome alterations and pathologic features in genitourinary malignancies: an analysis of the cancer genome Atlas. Eur. Urol. Focus. 2017;3:584–589. doi: 10.1016/j.euf.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajaiba M.M., Dyer L.M., Geller J.I., Jennings L.J., George D., Kirschmann D., Rohan S.M., Cost N.G., Khanna G., Mullen E.A., et al. The classification of pediatric and young adult renal cell carcinomas registered on the children's oncology group (COG) protocol AREN03B2 after focused genetic testing. Cancer. 2018;124:3381–3389. doi: 10.1002/cncr.31578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calio A., Brunelli M., Segala D., Pedron S., Remo A., Ammendola S., Munari E., Pierconti F., Mosca A., Bollito E., et al. Comprehensive analysis of 34 MiT family translocation renal cell carcinomas and review of the literature: investigating prognostic markers and therapy targets. Pathology. 2020;52:297–309. doi: 10.1016/j.pathol.2019.11.006. [DOI] [PubMed] [Google Scholar]
- Calio A., Segala D., Munari E., Brunelli M., Martignoni G. MiT family translocation renal cell carcinoma: from the early descriptions to the current knowledge. Cancers (Basel) 2019;11:1110. doi: 10.3390/cancers11081110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camparo P., Vasiliu V., Molinie V., Couturier J., Dykema K.J., Petillo D., Furge K.A., Comperat E.M., Lae M., Bouvier R., et al. Renal translocation carcinomas: clinicopathologic, immunohistochemical, and gene expression profiling analysis of 31 cases with a review of the literature. Am. J. Surg. Pathol. 2008;32:656–670. doi: 10.1097/PAS.0b013e3181609914. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N., Linehan W.M., Spellman P.T., Ricketts C.J., Creighton C.J., Fei S.S., Davis C., Wheeler D.A., Murray B.A., Schmidt L., et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016;374:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlo M.I., Mukherjee S., Mandelker D., Vijai J., Kemel Y., Zhang L., Knezevic A., Patil S., Ceyhan-Birsoy O., Huang K.C., et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. 2018;4:1228–1235. doi: 10.1001/jamaoncol.2018.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouli A., Shi J., Feng Y., Holubec H., Shanas R.M., Bhattacharyya A.K., Zheng W., Nelson M.A. Haploinsufficiency of the cdc2l gene contributes to skin cancer development in mice. Carcinogenesis. 2007;28:2028–2035. doi: 10.1093/carcin/bgm066. [DOI] [PubMed] [Google Scholar]
- Chasman D.I., Fuchsberger C., Pattaro C., Teumer A., Boger C.A., Endlich K., Olden M., Chen M.H., Tin A., Taliun D., et al. Integration of genome-wide association studies with biological knowledge identifies six novel genes related to kidney function. Hum. Mol. Genet. 2012;21:5329–5343. doi: 10.1093/hmg/dds369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Zhang Y., Senbabaoglu Y., Ciriello G., Yang L., Reznik E., Shuch B., Micevic G., De Velasco G., Shinbrot E., et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep. 2016;14:2476–2489. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.Y., Wen Y.C., Lin S.R., Yeh H.L., Jiang K.C., Chen W.H., Lin Y.S., Zhang Q., Liew P.L., Hsiao M., et al. Nerve growth factor interacts with CHRM4 and promotes neuroendocrine differentiation of prostate cancer and castration resistance. Commun. Biol. 2021;4:22. doi: 10.1038/s42003-020-01549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.B., Mirsadraei L., Jayakumaran G., Al-Ahmadie H.A., Fine S.W., Gopalan A., Sirintrapun S.J., Tickoo S.K., Reuter V.E. Somatic mutations of TSC2 or MTOR characterize a morphologically distinct subset of sporadic renal cell carcinoma with eosinophilic and vacuolated cytoplasm. Am. J. Surg. Pathol. 2019;43:121–131. doi: 10.1097/PAS.0000000000001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S., Levine J.H., Zanotelli V.R.T., Silina K., Schulz D., Bacac M., Ries C.H., Ailles L., Jewett M.A.S., Moch H., et al. An immune Atlas of clear cell renal cell carcinoma. Cell. 2017;169:736–749.e718. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S., Stawiski E.W., Pavia-Jimenez A., Modrusan Z., Kapur P., Jaiswal B.S., Zhang N., Toffessi-Tcheuyap V., Nguyen T.T., Pahuja K.B., et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 2015;47:13–21. doi: 10.1038/ng.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyve A.B., Bergan J., Utskarpen A., Sandvig K. Sorting nexin 8 regulates endosome-to-Golgi transport. Biochem. Biophys. Res. Commun. 2009;390:109–114. doi: 10.1016/j.bbrc.2009.09.076. [DOI] [PubMed] [Google Scholar]
- Edge S.B., Compton C.C. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010;17:1471–1474. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- Ellis C.L., Eble J.N., Subhawong A.P., Martignoni G., Zhong M., Ladanyi M., Epstein J.I., Netto G.J., Argani P. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: impact of fusion subtype, age, and stage. Mod. Pathol. 2014;27:875–886. doi: 10.1038/modpathol.2013.208. [DOI] [PubMed] [Google Scholar]
- Fawdar S., Trotter E.W., Li Y., Stephenson N.L., Hanke F., Marusiak A.A., Edwards Z.C., Ientile S., Waszkowycz B., Miller C.J., Brognard J. Targeted genetic dependency screen facilitates identification of actionable mutations in FGFR4, MAP3K9, and PAK5 in lung cancer. Proc. Natl. Acad. Sci. U S A. 2013;110:12426–12431. doi: 10.1073/pnas.1305207110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder G.C., Yang T.W., Razdan M., Li Y., Lu J., Perry J.K., Lobie P.E., Liu D.X. The GDNF family: a role in cancer? Neoplasia. 2018;20:99–117. doi: 10.1016/j.neo.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotello F., Mayer C., Plattner C., Laschober G., Rieder D., Hackl H., Krogsdam A., Loncova Z., Posch W., Wilflingseder D., et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34. doi: 10.1186/s13073-019-0638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller J.I., Cost N.G., Chi Y.Y., Tornwall B., Cajaiba M., Perlman E.J., Kim Y., Mullen E.A., Glick R.D., Khanna G., et al. A prospective study of pediatric and adolescent renal cell carcinoma: a report from the Children's Oncology Group AREN0321 study. Cancer. 2020;126:5156–5164. doi: 10.1002/cncr.33173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller J.I., Dome J.S. Local lymph node involvement does not predict poor outcome in pediatric renal cell carcinoma. Cancer. 2004;101:1575–1583. doi: 10.1002/cncr.20548. [DOI] [PubMed] [Google Scholar]
- Geller J.I., Ehrlich P.F., Cost N.G., Khanna G., Mullen E.A., Gratias E.J., Naranjo A., Dome J.S., Perlman E.J. Characterization of adolescent and pediatric renal cell carcinoma: a report from the Children's Oncology Group study AREN03B2. Cancer. 2015;121:2457–2464. doi: 10.1002/cncr.29368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh C.Y., Patmore S., Smolenski A., Howard J., Evans S., O'Sullivan J., McCann A. The role of von Willebrand factor in breast cancer metastasis. Transl Oncol. 2021;14:101033. doi: 10.1016/j.tranon.2021.101033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobner S.N., Worst B.C., Weischenfeldt J., Buchhalter I., Kleinheinz K., Rudneva V.A., Johann P.D., Balasubramanian G.P., Segura-Wang M., Brabetz S., et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Ho T.H., Jonasch E. Genetic kidney cancer syndromes. J. Natl. Compr. Canc Netw. 2014;12:1347–1355. doi: 10.6004/jnccn.2014.0129. [DOI] [PubMed] [Google Scholar]
- Hol J.A., Jongmans M.C.J., Littooij A.S., de Krijger R.R., Kuiper R.P., van Harssel J.J.T., Mensenkamp A., Simons M., Tytgat G.A.M., van den Heuvel-Eibrink M.M., van Grotel M. Renal cell carcinoma in young FH mutation carriers: case series and review of the literature. Fam. Cancer. 2020;19:55–63. doi: 10.1007/s10689-019-00155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J.J., Le V., Cao D., Cheng E.H., Creighton C.J. Genomic classifications of renal cell carcinoma: a critical step towards the future application of personalized kidney cancer care with pan-omics precision. J. Pathol. 2018;244:525–537. doi: 10.1002/path.5022. [DOI] [PubMed] [Google Scholar]
- Huang J.J., Hsieh J.J. The pan-omics landscape of renal cell carcinoma and its implication on future clinical practice. Kidney Cancer. 2020;4:121–129. doi: 10.3233/KCA-200085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubschmann D., Jopp-Saile L., Andresen C., Kramer S., Gu Z., Heilig C.E., Kreutzfeldt S., Teleanu V., Frohling S., Eils R., Schlesner M. Analysis of mutational signatures with yet another package for signature analysis. Genes Chromosomes Cancer. 2021;60:314–331. doi: 10.1002/gcc.22918. [DOI] [PubMed] [Google Scholar]
- Johnson E., McTigue M., Gallego R.A., Johnson T.W., Timofeevski S., Maestre M., Fisher T.S., Kania R., Sawasdikosol S., Burakoff S., Cronin C.N. Multiple conformational states of the HPK1 kinase domain in complex with sunitinib reveal the structural changes accompanying HPK1 trans-regulation. J. Biol. Chem. 2019;294:9029–9036. doi: 10.1074/jbc.AC119.007466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T., Jager N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y.J., Pugh T.J., Hovestadt V., Stutz A.M., et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S.H., Lee H.C., Hwang H.J., Park H.A., Moon Y.A., Kim B.C., Lee H.M., Kim K.P., Kim Y.N., Lee B.L., et al. Acyl-CoA thioesterase 7 is involved in cell cycle progression via regulation of PKCzeta-p53-p21 signaling pathway. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2017.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Fajardo J.E., Birge R.B., Sriram G. Crk at the quarter century mark: perspectives in signaling and cancer. J. Cell Biochem. 2014;115:819–825. doi: 10.1002/jcb.24749. [DOI] [PubMed] [Google Scholar]
- Lavacchi D., Pellegrini E., Palmieri V.E., Doni L., Mela M.M., Di Maida F., Amedei A., Pillozzi S., Carini M., Antonuzzo L. Immune checkpoint inhibitors in the treatment of renal cancer: current state and future perspective. Int. J. Mol. Sci. 2020;21:4691. doi: 10.3390/ijms21134691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.T., Han C.S., Kwon Y.S., Patel R., Modi P.K., Kwon S.J., Faiena I., Patel N., Singer E.A., Ahn H.J., et al. Intracrine androgen biosynthesis in renal cell carcinoma. Br. J. Cancer. 2017;116:937–943. doi: 10.1038/bjc.2017.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer S., Green S., Dembitzer F.R., Rheinstein P.H., Rosenzweig K.E. Increased RNA expression of von Willebrand factor gene is associated with infiltrating lobular breast cancer and normal PAM50 subtype. Cancer Genomics Proteomics. 2019;16:147–153. doi: 10.21873/cgp.20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer S., Rheinstein P.H., Green S., Rosenzweig K.E. von Willebrand factor gene expression in primary lower grade glioma: mutually Co-occurring mutations in von Willebrand factor, ATRX, and TP53. Brain Tumor Res. Treat. 2019;7:33–38. doi: 10.14791/btrt.2019.7.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh N.D., Sessa S., Dragalzew A.C., Payzin-Dogru D., Sousa J.F., Aggouras A.N., Johnson K., Dunlap G.S., Haas B.J., Levin M., et al. von Willebrand factor D and EGF domains is an evolutionarily conserved and required feature of blastemas capable of multitissue appendage regeneration. Evol. Dev. 2020;22:297–311. doi: 10.1111/ede.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv.1303.3997v2.
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Huang C., Zhang Z., Feng Y., Wang Z., Tang X., Zhong K., Hu Y., Guo G., Zhou L., et al. MEK inhibitor augments antitumor activity of B7-H3-redirected bispecific antibody. Front Oncol. 2020;10:1527. doi: 10.3389/fonc.2020.01527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Subramanian A., Pinchback R., Thorvaldsdottir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan W.M., Ricketts C.J. The metabolic basis of kidney cancer. Semin. Cancer Biol. 2013;23:46–55. doi: 10.1016/j.semcancer.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.J., Houldsworth J., Emmadi R., Dyer L., Wolff D.J. Assessing genomic copy number alterations as best practice for renal cell neoplasia: an evidence-based review from the cancer genomics Consortium workgroup. Cancer Genet. 2020;244:40–54. doi: 10.1016/j.cancergen.2020.04.004. [DOI] [PubMed] [Google Scholar]
- Lopez G., Lazzeri G., Rappa A., Isimbaldi G., Cribiu F.M., Guerini-Rocco E., Ferrero S., Vaira V., Di Fonzo A. Comprehensive genomic analysis reveals the prognostic role of LRRK2 copy-number variations in human malignancies. Genes (Basel) 2020;11:846. doi: 10.3390/genes11080846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Liu Y., Liu Y., Alexandrov L.B., Edmonson M.N., Gawad C., Zhou X., Li Y., Rusch M.C., Easton J., et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macher-Goeppinger S., Roth W., Wagener N., Hohenfellner M., Penzel R., Haferkamp A., Schirmacher P., Aulmann S. Molecular heterogeneity of TFE3 activation in renal cell carcinomas. Mod. Pathol. 2012;25:308–315. doi: 10.1038/modpathol.2011.169. [DOI] [PubMed] [Google Scholar]
- Malouf G.G., Su X., Yao H., Gao J., Xiong L., He Q., Comperat E., Couturier J., Molinie V., Escudier B., et al. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clin. Cancer Res. 2014;20:4129–4140. doi: 10.1158/1078-0432.CCR-13-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini A., Mazzocchetti P., Sciaccaluga M., Megaro A., Bellingacci L., Beccano-Kelly D.A., Di Filippo M., Tozzi A., Calabresi P. From synaptic dysfunction to neuroprotective strategies in genetic Parkinson's disease: lessons from LRRK2. Front Cell Neurosci. 2020;14:158. doi: 10.3389/fncel.2020.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon J., DiNatale R.G., Sanchez A., Kotecha R.R., Gupta S., Kuo F., Makarov V., Sandhu A., Mano R., Silagy A.W., et al. Comprehensive genomic analysis of translocation renal cell carcinoma reveals copy-number variations as drivers of disease progression. Clin. Cancer Res. 2020;26:3629–3640. doi: 10.1158/1078-0432.CCR-19-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersch J., Jackson M.A., Park M., Nebgen D., Peterson S.K., Singletary C., Arun B.K., Litton J.K. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269–275. doi: 10.1002/cncr.29041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mier J.W. The tumor microenvironment in renal cell cancer. Curr. Opin. Oncol. 2019;31:194–199. doi: 10.1097/CCO.0000000000000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millstein J., Budden T., Goode E.L., Anglesio M.S., Talhouk A., Intermaggio M.P., Leong H.S., Chen S., Elatre W., Gilks B., et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann. Oncol. 2020;31:1240–1250. doi: 10.1016/j.annonc.2020.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moch H., Cubilla A.L., Humphrey P.A., Reuter V.E., Ulbright T.M. The 2016 WHO classification of tumours of the urinary system and male genital organs-Part A: renal, penile, and testicular tumours. Eur. Urol. 2016;70:93–105. doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- Ney Garcia D.R., de Souza M.T., de Figueiredo A.F., Othman M.A.K., Rittscher K., Abdelhay E., Capela de Matos R.R., Meyer C., Marschalek R., Land M.G.P., et al. Molecular characterization of KMT2A fusion partner genes in 13 cases of pediatric leukemia with complex or cryptic karyotypes. Hematol. Oncol. 2017;35:760–768. doi: 10.1002/hon.2299. [DOI] [PubMed] [Google Scholar]
- Ouyang Q., Zhou J., Yang W., Cui H., Xu M., Yi L. Oncogenic role of neurotensin and neurotensin receptors in various cancers. Clin. Exp. Pharmacol. Physiol. 2017;44:841–846. doi: 10.1111/1440-1681.12787. [DOI] [PubMed] [Google Scholar]
- Pan J., Chen Y., Mo C., Wang D., Chen J., Mao X., Guo S., Zhuang J., Qiu S. Association of DSC3 mRNA down-regulation in prostate cancer with promoter hypermethylation and poor prognosis. PLoS One. 2014;9 doi: 10.1371/journal.pone.0092815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflueger D., Sboner A., Storz M., Roth J., Comperat E., Bruder E., Rubin M.A., Schraml P., Moch H. Identification of molecular tumor markers in renal cell carcinomas with TFE3 protein expression by RNA sequencing. Neoplasia. 2013;15:1231–1240. doi: 10.1593/neo.131544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak J., Joffraud M., Trammell S.A.J., Ras R., Canela N., Boutant M., Kulkarni S.S., Rodrigues M., Redpath P., Migaud M.E., et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016;7:13103. doi: 10.1038/ncomms13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts C.J., De Cubas A.A., Fan H., Smith C.C., Lang M., Reznik E., Bowlby R., Gibb E.A., Akbani R., Beroukhim R., et al. The cancer genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:313–326.e315. doi: 10.1016/j.celrep.2018.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmer A., Phan H., Mathieson I., Iqbal Z., Twigg S.R.F., Consortium W.G.S., Wilkie A.O.M., McVean G., Lunter G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014;46:912–918. doi: 10.1038/ng.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi Sebastiano M., Konstantinidou G. Targeting long Chain acyl-CoA synthetases for cancer therapy. Int. J. Mol. Sci. 2019;20:3624. doi: 10.3390/ijms20153624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein R.M., Krishna C., Ma X., Pei X., Lee K.W., Makarov V., Kuo F., Chung J., Srivastava R.M., Purohit T.A., et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer. 2021;1:1188–1203. doi: 10.1038/s43018-020-00139-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt L.S., Linehan W.M. Genetic predisposition to kidney cancer. Semin. Oncol. 2016;43:566–574. doi: 10.1053/j.seminoncol.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M., Dinkelborg K., Xiao X., Chan-Smutko G., Hruska K., Huang D., Sagar P., Harisinghani M., Iliopoulos O. Early onset renal cell carcinoma in an adolescent girl with germline FLCN exon 5 deletion. Fam. Cancer. 2018;17:135–139. doi: 10.1007/s10689-017-0008-8. [DOI] [PubMed] [Google Scholar]
- Scribner J.A., Brown J.G., Son T., Chiechi M., Li P., Sharma S., Li H., De Costa A., Li Y., Chen Y., et al. Preclinical development of MGC018, a duocarmycin-based antibody-drug conjugate targeting B7-H3 for solid cancer. Mol. Cancer Ther. 2020;19:2235–2244. doi: 10.1158/1535-7163.MCT-20-0116. [DOI] [PubMed] [Google Scholar]
- Selle B., Furtwangler R., Graf N., Kaatsch P., Bruder E., Leuschner I. Population-based study of renal cell carcinoma in children in Germany, 1980-2005: more frequently localized tumors and underlying disorders compared with adult counterparts. Cancer. 2006;107:2906–2914. doi: 10.1002/cncr.22346. [DOI] [PubMed] [Google Scholar]
- Simonaggio A., Epaillard N., Pobel C., Moreira M., Oudard S., Vano Y.A. Tumor microenvironment features as predictive biomarkers of response to immune checkpoint inhibitors (ICI) in metastatic clear cell renal cell carcinoma (mccRCC) Cancers (Basel) 2021;13:231. doi: 10.3390/cancers13020231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza V.C., de Freitas Vinhas C., Miguel D., Athanazio D.A., Trpkov K. Renal cell carcinoma morphologically similar to fumarate hydratase-deficient RCC in a patient with BRCA2 germline mutation. Pathol. Int. 2018;68:541–542. doi: 10.1111/pin.12688. [DOI] [PubMed] [Google Scholar]
- Stark M.S., Woods S.L., Gartside M.G., Bonazzi V.F., Dutton-Regester K., Aoude L.G., Chow D., Sereduk C., Niemi N.M., Tang N., et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat. Genet. 2011;44:165–169. doi: 10.1038/ng.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L.D., Knoppers B.M., Campbell P., Getz G., Korbel J.O. Data analysis: create a cloud commons. Nature. 2015;523:149–151. doi: 10.1038/523149a. [DOI] [PubMed] [Google Scholar]
- Stenzel P.J., Schindeldecker M., Tagscherer K.E., Foersch S., Herpel E., Hohenfellner M., Hatiboglu G., Alt J., Thomas C., Haferkamp A., et al. Prognostic and predictive value of tumor-infiltrating leukocytes and of immune checkpoint molecules PD1 and PDL1 in clear cell renal cell carcinoma. Transl Oncol. 2020;13:336–345. doi: 10.1016/j.tranon.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016;12 doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Carrillo D., Tovar H., Velazquez-Caldelas T.E., Hernandez-Lemus E. Master regulators of signaling pathways: an application to the analysis of gene regulation in breast cancer. Front Genet. 2019;10:1180. doi: 10.3389/fgene.2019.01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trpkov K., Hes O., Williamson S.R., Adeniran A.J., Agaimy A., Alaghehbandan R., Amin M.B., Argani P., Chen Y.B., Cheng L., et al. New developments in existing WHO entities and evolving molecular concepts: the Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 2021;34:1392–1424. doi: 10.1038/s41379-021-00779-w. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T., Iwata H., Murase Y., Takahara T., Ohashi A. Renal tumors in end-stage renal disease: a comprehensive review. Int. J. Urol. 2018;25:780–786. doi: 10.1111/iju.13759. [DOI] [PubMed] [Google Scholar]
- Uhrig S., Ellermann J., Walther T., Burkhardt P., Frohlich M., Hutter B., Toprak U.H., Neumann O., Stenzinger A., Scholl C., et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021;31:448–460. doi: 10.1101/gr.257246.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle L., de Voer R.M., Goldberg Y., Sjursen W., Forsti A., Ruiz-Ponte C., Caldes T., Garre P., Olsen M.F., Nordling M., et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Aspects Med. 2019;69:10–26. doi: 10.1016/j.mam.2019.03.001. [DOI] [PubMed] [Google Scholar]
- van der Beek J.N., Geller J.I., de Krijger R.R., Graf N., Pritchard-Jones K., Drost J., Verschuur A.C., Murphy D., Ray S., Spreafico F., et al. Characteristics and outcome of children with renal cell carcinoma: a narrative review. Cancers (Basel) 2020;12:1776. doi: 10.3390/cancers12071776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Beek J.N., Hol J.A., Coulomb-l'Hermine A., Graf N., van Tinteren H., Pritchard-Jones K., Houwing M.E., de Krijger R.R., Vujanic G.M., Dzhuma K., et al. Characteristics and outcome of pediatric renal cell carcinoma patients registered in the International Society of Pediatric Oncology (SIOP) 93-01, 2001 and UK-IMPORT database: a report of the SIOP-Renal Tumor Study Group. Int. J. Cancer. 2021;148:2724–2735. doi: 10.1002/ijc.33476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala S.J., Karamchandani J.R., Saleeb R., Evans A., Ding Q., Ibrahim R., Jewett M., Pasic M., Finelli A., Pace K., et al. An integrated genomic analysis of papillary renal cell carcinoma type 1 uncovers the role of focal adhesion and extracellular matrix pathways. Mol. Oncol. 2015;9:1667–1677. doi: 10.1016/j.molonc.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang V.G., Kim H., Chuang J.H. Whole-exome sequencing capture kit biases yield false negative mutation calls in TCGA cohorts. PLoS One. 2018;13 doi: 10.1371/journal.pone.0204912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wang Y., Liu F. A 44-gene set constructed for predicting the prognosis of clear cell renal cell carcinoma. Int. J. Mol. Med. 2018;42:3105–3114. doi: 10.3892/ijmm.2018.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Kang J., Lian J., Huang L., Xie W., Zhao D., Ma H., Lin Z. EFEMP1 as a potential biomarker for diagnosis and prognosis of osteosarcoma. Biomed. Res. Int. 2020;2020:5264265. doi: 10.1155/2020/5264265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson S.R., Gill A.J., Argani P., Chen Y.B., Egevad L., Kristiansen G., Grignon D.J., Hes O. Report from the international society of urological pathology (ISUP) consultation conference on molecular pathology of urogenital cancers: III: molecular pathology of kidney cancer. Am. J. Surg. Pathol. 2020;44:e47–e65. doi: 10.1097/PAS.0000000000001476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldu S.L., Weinberg A.C., RoyChoudhury A., Chase H., Kalloo S.D., McKiernan J.M., DeCastro G.J. Renal insufficiency is associated with an increased risk of papillary renal cell carcinoma histology. Int. Urol. Nephrol. 2014;46:2127–2132. doi: 10.1007/s11255-014-0780-4. [DOI] [PubMed] [Google Scholar]
- Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. The Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Lin E., Zhuang H., Xie L., Feng X., Liu J., Yu Y. Construction of a novel gene-based model for prognosis prediction of clear cell renal cell carcinoma. Cancer Cell Int. 2020;20:27. doi: 10.1186/s12935-020-1113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Li X., Liu L., Cao J., Goscinski M.A., Fan H., Li H., Suo Z. Oncogenic role of guanylate binding protein 1 in human prostate cancer. Front Oncol. 2019;9:1494. doi: 10.3389/fonc.2019.01494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Chang H., Yang B. GATA4 is upregulated in nasopharyngeal cancer and facilitates epithelial-mesenchymal transition and metastasis through regulation of SLUG. Exp. Ther. Med. 2018;16:5318–5326. doi: 10.3892/etm.2018.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The published article contains all data sets generated or analyzed during this study. The WES and bulk RNA sequencing data generated in this study have been deposited in the European Genome-phenome archive (EGA) under the accession number EGAS00001006057.
-
•
This study did not generate original code.
-
•
Any additional information required to reanalyze the data included in this study will be provided by the lead contact upon request.