

Abstract
In cystic fibrosis (CF), the deletion of phenylalanine 508 (F508del) in the CF transmembrane conductance regulator (CFTR) leads to misfolding and premature degradation of the mutant protein. These defects can be targeted with pharmacological agents named potentiators and correctors. During the past years, several efforts have been devoted to develop and approve new effective molecules. However, their clinical use remains limited, as they fail to fully restore F508del-CFTR biological function. Indeed, the search for CFTR correctors with different and additive mechanisms has recently increased. Among them, drugs that modulate the CFTR proteostasis environment are particularly attractive to enhance therapy effectiveness further. This Perspective focuses on reviewing the recent progress in discovering CFTR proteostasis regulators, mainly describing the design, chemical structure, and structure–activity relationships. The opportunities, challenges, and future directions in this emerging and promising field of research are discussed, as well.
1. Introduction
Cystic fibrosis (CF) is a lethal genetic disease caused by defects in the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-dependent chloride and bicarbonate ion channel that is widely expressed at the plasma membrane (PM) of several epithelial cells.1 CFTR is composed by two membrane spanning domains (MSDs) that form an anion-selective pore, two nucleotide binding domains (NBD1 and NBD2), which contain ATP binding sites and a regulatory region (R).2,3 Currently, over 2000 mutations have been identified,4 but to date, the pathogenicity has been confirmed only for 382 mutations.5 CF-causing mutations are commonly classified into six classes: (1) class I mutations result in the absence of CFTR mRNA and/or protein; (2) class II mutations cause protein misfolding and its premature degradation; (3) class III mutations impair the gating of the channel; (4) class IV mutations lead to decreased channel conductance; (5) class V mutations reduce the amount of CFTR channels at the PM; and (6) class VI mutations reduce the stability of the CFTR protein at the PM.6,7 The most prevalent CFTR mutation (around 80%) is the deletion of phenylalanine 508 (F508del), a class II mutation that is associated with misfolding and defective gating of the mutant protein.8,9 The misfolding defect results in reduced stability of F508del-CFTR, retention of the mutant channel at the endoplasmic reticulum (ER), and premature degradation by the ubiquitin–proteasome system (UPS), which causes a reduced expression of F508del-CFTR at the PM.10−12 The gating defect results in reduced activity of the mutant channel due to its abnormal persistence in the closed state.13 As a consequence, the epithelial fluid transport in the airway is dysregulated and leads to the production of a thickened mucus that favors chronic bacterial colonization, inflammation, and ultimately leads to lethal respiratory failure.7 Depending on the specific basic defect stemming from the CFTR mutation, distinct drugs, namely, CFTR modulators, with different mechanism of actions are necessary.14 CFTR modulators include correctors, potentiators, stabilizers, and amplifiers. Correctors increase the number of mutant CFTR channels at the PM acting as pharmacological chaperones or as proteostasis regulators. Pharmacological chaperones are thought to act directly on the mutant CFTR, stabilizing or improving specific domains’ interaction. Instead, proteostasis regulators target components of the CFTR regulome, such as chaperones, cochaperones, kinases, or ubiquitin ligases that affect the synthesis, folding, stability, and trafficking to the plasma membrane of the mutant channel.15,16 Potentiators improve channel gating of CFTR proteins expressed at the PM, directly binding to the mutant channel.17,18 Amplifiers increase CFTR mRNA translation and stimulate protein expression.19 Stabilizers increase the quantity of CFTR channels at the PM by anchoring the defective protein20 or stabilizing its interaction with other membrane components.21
During the past 10 years, several efforts have been made to develop and approve new effective therapeutic strategies to restore CFTR biological function in a large cohort of CF patients. For example, ivacaftor or VX-770 (1, Figure 1)22 is a potentiator that first received marketing authorization in 2012 (with the commercial name of Kalydeco) to treat CF patients who have at least one copy of the G551D mutation, subsequently expanded to a selection of class III and IV mutations. Instead, VX-809, also known as lumacaftor (2, Figure 1),23 was the first corrector to be approved for therapeutic use in CF patients carrying the F508del mutation and, along with the potentiator 1, constitutes the combination drug Orkambi, approved by both FDA and EMA in 2015. Many clinical studies highlighted positive results on lung function, increased body mass index (BMI), reduction of sweat Cl– concentration, and lung clearance index (LCI).24,25 However, frequent drug intolerance and respiratory adverse effects were observed in patients treated with Orkambi.26,27 VX-661, also known as tezacaftor (3, Figure 1), is an analogue of 2 with improved pharmacokinetics and less side effects, and the 3/1 co-therapy (trade name Symdeko) received marketing authorization in 2018.28 Different clinical trials showed that the 3/1 combination displays effects similar to those of Orkambi in F508del homozygous patients.29 Notably, patients heterozygous F508del with G551D or with residual function mutations are more responsive to 3/1 combination than F508del homozigous.30,312 and 3 are considered first-generation correctors and act as pharmacological chaperones that stabilize the CFTR structure by improving the interdomain interactions and CFTR folding.12,32,33 Instead, the next-generation corrector VX-445, also known as elexacaftor (4, Figure 1), likely acts on different binding sites than the first-generation correctors.34 Indeed, VX-445 exhibits additive or synergistic effects in combination with a first-generation corrector (and with the potentiator 1), leading to markedly increased PM expression of F508del-CFTR.34,35 Recent phase 3 studies by Vertex confirmed safety and benefits of 4 in CF patients who are homozygous for the F508del mutation or heterozygous for the F508del and a minimal function (MF) mutation.35 In the F508del cohort who was receiving standard 1/3 treatment, the addition of 4 resulted in 11.0 point rise in ppFEV1 (percent predicted forced expiratory volume in 1 s).35 In the MF group, treatment with 1/3/4 resulted in an increase in ppFEV1 of 13.8 percentage points.36 Strikingly, 4 is now included in a triple drug combination (trade name Trikafta in the U.S. and Kaftrio in Europe) together with 1 and 3, for CF patients 12 years and older who have at least one F508del mutation, or another mutation known to be responsive to the drug (for the complete list of mutations, see Trikafta.com).36,37
Figure 1.
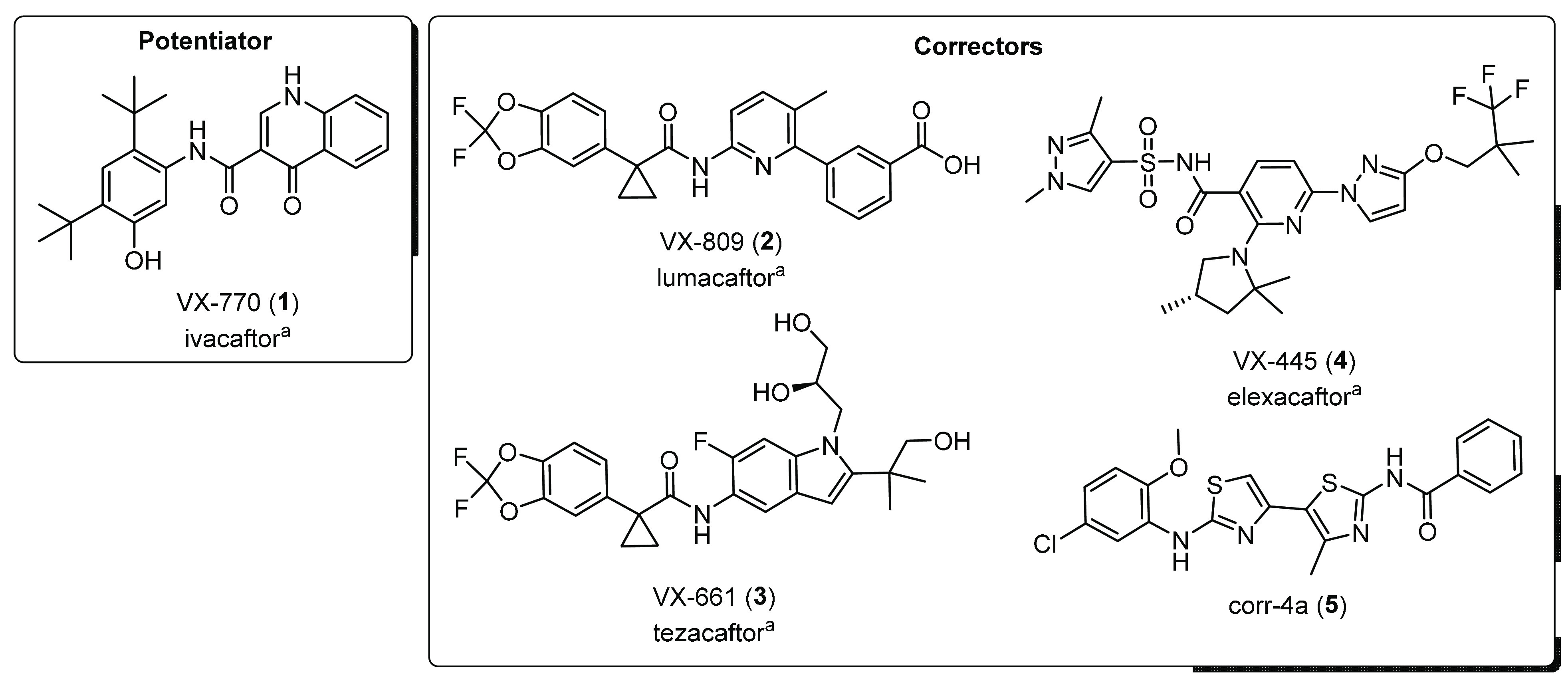
Structures of the potentiator VX-770 (1) and of the correctors VX-809 (2), VX-661 (3), VX-445 (4), and corr-4a (5). aApproved drugs.
Orkambi and Symdeko still represent a standard care for many CF patients, albeit their clinical effects remain moderate.38 Although Trikafta has undoubtedly proved to be a breakthrough in CF treatment, by significantly slowing down CF progress with substantiated clinical benefits,39 it fails to fully restore mutant CFTR function.34,40 Indeed, it has been demonstrated that the 3/4 combination can rescue F508del-CFTR activity only up to 65% of the wild-type (WT) CFTR activity level.34,40 Hence, development of new pharmacological chaperones acting with different mechanisms or with ameliorated characteristics is currently a thriving research field. Two successful examples are the recent search for optimized analogues of the bithiazole corr-4a (5, Figure 1)41,42 and the development of multitarget compounds able to simultaneously act as antiviral agents and F508del-CFTR correctors.43,44 For more detailed information about the promising pharmacological chaperone correctors under clinical trials or currently in study, there are several recent reviews available.45−47 On the other hand, no CFTR proteostasis regulator has entered the market to date. Nevertheless, the research in this field is flourishing, and many proteostasis targets have been recently uncovered due to siRNA-mediated silencing techniques, proteomic and interactomic studies,15,48−51 while others remain elusive yet. Indeed, the development of new compounds targeting biological components of the CFTR physiological pathway may be useful to optimize combination therapies for those patients with mutations (in particular, affecting protein maturation, trafficking, and stability at the PM), poorly responsive to current treatments.
This review summarizes the recent progress in the discovery of CFTR proteostasis regulators. In particular, we will review and discuss studies that led to discovering active compounds affecting different CFTR-related targets. Understanding their mechanism of action would facilitate structure–activity relationships (SAR) and could inspire the medicinal chemistry community to develop novel promising molecules with clinical potentiality. Likewise, the new compounds might represent effective chemical probes useful to dissect biological processes involved in CFTR dysfunctions that lead to CF.
2. Targeting RNA Binding Proteins
As CFTR is a monomeric polytopic membrane protein, its biosynthesis occurs at the ER and CFTR assembly and domains folding involve both co- and post-translational translocation events.52 CFTR polypeptide is synthesized by ribosomes present in the rough ER and, as the nascent chain emerges from the ribosome, is co-translationally translocated to the ER lumen by the cytosolic signal recognition particle (SRP).53 Subsequent assembly of MSDs and NBDs requires cytosolic and lumenal chaperones including Hsp70, Hsp40, Hsp90, and others.54−56 Once CFTR transmembrane domains are folded properly, the polypeptide associates with a complex set of cellular proteins that facilitate translocation across the ER membrane and integration into the lipid bilayer.52 However, the SRP-dependent co-translational translocation is reported to direct correct topology for less than half of nascent CFTR chains.52 Furthermore, mutations that reduce CFTR mRNA levels or impair CFTR translation, as well as those that lead to misfolded, unstable, or defective proteins, exacerbate inefficiencies of CFTR biosynthesis, folding, and trafficking.57,58
Several pharmaceutical companies gained interest into associations of molecules with different mechanisms. In order to identify novel classes of molecules exhibiting synergy with potentiator 1 and corrector 2, Proteostasis Therapeutics Inc. recently performed a phenotypic high-throughput strategy (HTS) of approximately 54000 small molecules selected for novelty and drug-like properties.59 With this strategy, the novel class of CFTR modulators called amplifiers was identified. Indeed, the phenylisoxazole PTI-CH (6, Figure 2), which was selected as a representative compound of this novel class of small molecules, nearly doubled the activity of 1 and 2 when coadministered in primary human bronchial epithelial (HBE) cells. These results suggested that 6 might possess a distinct mechanism relative to known modulators. Further in vitro experiments highlighted that 6 increased CFTR protein expression across different mutations, including F508del, by increasing CFTR mRNA levels by ∼1.5 to ∼2-fold in HBE cells. This enhancement was specific for CFTR transcript and did not lead to induction of cytosolic or ER-associated cellular stress responses.59 The same year, Bear and co-workers provided the first evidence that the amplifier 6 could enhance Orkambi efficacy in nasal cultures from patients bearing the rare mutation ΔI1234_R1239.19 This effect was further corroborated using a CRISPR/Cas9-edited HBE cell line harboring this rare mutation. In this cell model, treatment with compound 6 increased ΔI1234_R1239-CFTR mRNA, and when combined with Orkambi, it significantly enhanced CFTR channel activity compared to that in Orkambi treatment alone.19 Afterward, Miller and co-workers investigated the mechanism through which amplifiers stabilize CFTR mRNA and showed that they might enhance translational efficacy by increasing CFTR mRNA association with polysomes.60 Indeed, using chemical proteomics, the authors showed that the phenylisoxazole PTI-CV (7, Figure 2), an analogue of 6 with better pharmacokinetic and drug-like properties, could bind to the poly-r(C) binding protein 1 (PCBP1). PCBP1 is a RNA binding protein that was reported to regulate CFTR mRNA levels in mouse oocytes.61 Notably, amplifier 7 showed an affinity for RNA-bound PCBP1 higher than that of free PCBP1,60 suggesting that amplifiers might increase CFTR expression through promoting translation, an innovative mechanism that is independent of the CF-causing mutation and genotype. Thanks to these outstanding results, Proteostasis Therapeutics Inc. advanced to early phase clinical trials the phenylisoxazole amplifier PTI-428, also known as nesolicaftor (8, Figure 2), chosen as the most promising drug candidate of the class.
Figure 2.
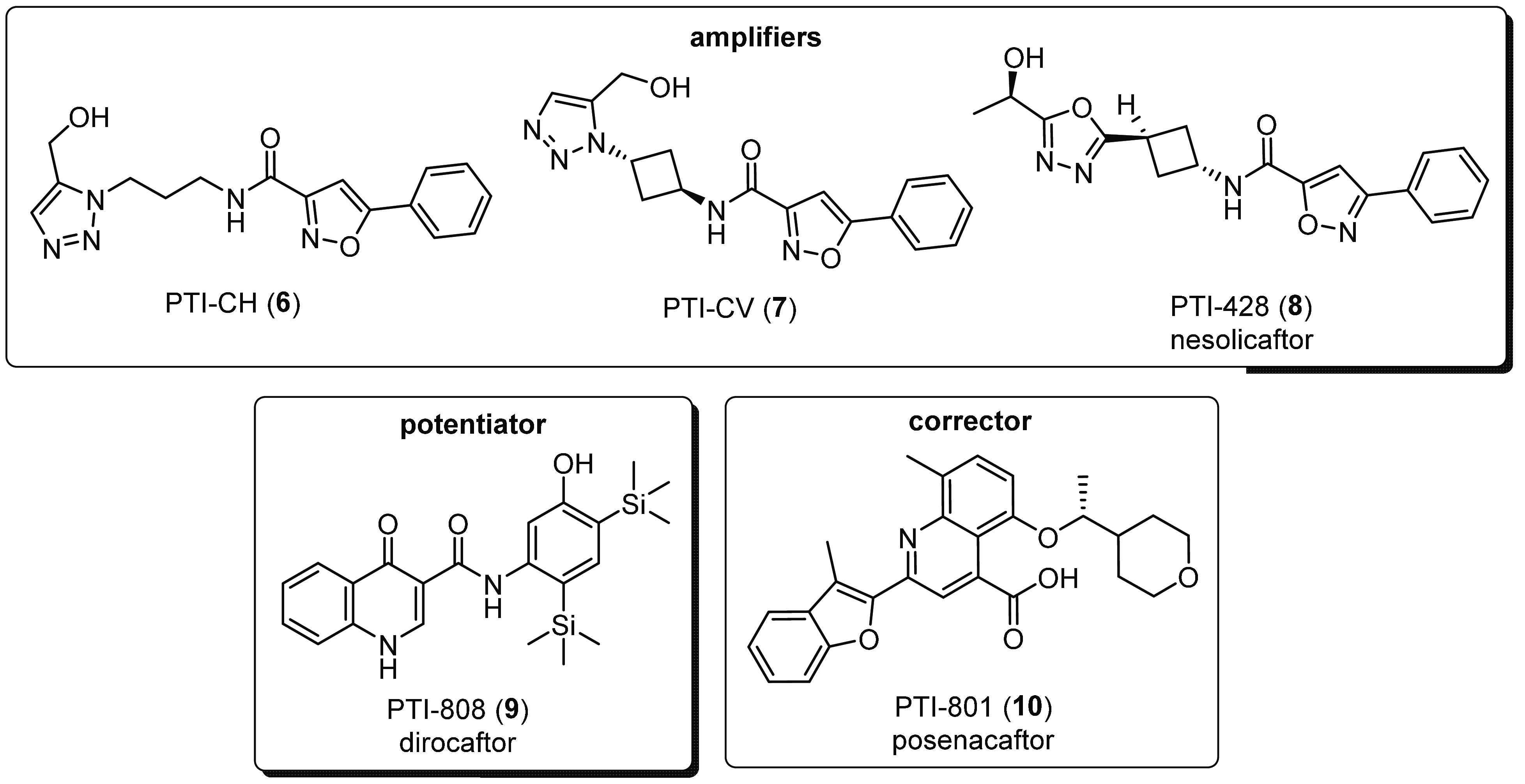
Structures of the amplifiers PTI-CH (6), PTI-CV (7), and PTI-428 (8) of the potentiator PTI-808 (9) and of the corrector PTI-801 (10) developed by Proteostasis Therapeutics Inc.
Notably, the company is also developing other molecules for combination therapies: the 4-oxo-1,4-dihydroquinoline potentiator PTI-808, also known as dirocaftor (9, Figure 2), and the quinoline-4-carboxylic acid corrector PTI-801, also known as posenacaftor (10, Figure 2). The triple combination 8/9/10 increased the CFTR-dependent chloride secretion to almost normal levels in F508del-expressing cells. Furthermore, in a phase 1/2 study (NCT03500263), this triple combination regimen has an acceptable safety and tolerability profile and led to a statistically significant reduction in sweat chloride concentration and improvement in lung function (8% in ppFEV1) compared to placebo in F508del homozygous patients.62 Currently, a phase 1/2 clinical trial is assessing the triple combination treatments’ effectiveness in 180 patients either homozygous for the F508del or heterozygous for the F508del CFTR genotype, treated for a longer period (28 days, NCT03251092). These modulator drugs were also included in a HIT-CF project in February 2019 with the purpose of testing 8, 9, and 10 in intestinal organoids of patients carrying rare CF genotypes.63 No results have been published yet. Other early stage clinical trials are currently ongoing to assess the safety and efficacy of 8 in CF patients on stable treatment with Kalydeco (NCT03258424), Orkambi (NCT02718495), and Symdeko (NCT03591094). Just early results from the phase 2 clinical trial on 24 CF patients (ages ≥18 years) homozygous for F508del and receiving background Orkambi therapy were released. Treatment for 28 days with daily doses of 50 mg of 8 or placebo, followed by a 7 day follow-up period, caused an increase by 5.2% points over days 14–28 in the ppFEV1 of the treated ones, and the therapy was well-tolerated.64 Additional data are expected shortly. However, recent in vitro investigations by Galietta and co-workers showed that 8, as expected, was effective in improving the rescue of F508del-CFTR but failed to increase the rescue of other CFTR mutants, such as G542X-CFTR or W1282X-CFTR, in combination with read-through agents and/or nonsense-mediated mRNA decay (NMD) inhibitors.65 Further experiments on HBE cells showed that 8 could also significantly enhance ENaC and TMEM16A channels’ activities. Such results suggest that CFTR amplifiers may alter the expression and/or function of other proteins involved in transepithelial ion transport,65 calling for the need for further investigations.
3. Targeting Heat Shock Proteins and Cochaperones
Two main chaperone systems are involved in the biosynthesis of the CFTR protein, mainly assisting the folding and assembly of the cytosolic domains of CFTR: the Hsp70/Hsc70 chaperone system is involved in early steps of CFTR folding,66,67 preferentially recognizing unfolded proteins, while the Hsp90 system is involved in later steps, binding to partially folded intermediates.68 These two complexes help CFTR to fold properly, protect the channel from aggregation, and trigger the degradation of non-native conformers.
3.1. Hsp70/Hsc70 Chaperone System
In the human cytosol, the major Hsp70 chaperones are the constitutively expressed heat shock cognate protein Hsc70 and its stress-inducible homologue Hsp70, which are closely related. Structure prediction using AlphaFold69 revealed that Hsp70 is formed by an amino-terminal ATP-binding domain (NBD), a C-terminal substrate-binding domain (SBD), and an α-helical subdomain that forms a flexible lid (Figure 3). Both Hsp70 and Hsc70 share an ATP-dependent mechanism that is regulated by two classes of cochaperones: DNAJs stimulate ATP hydrolysis and substrate binding to Hsp70/Hsc70 SBD, whereas NEFs promote the release of ADP and Hsp70/Hsc70 dissociation from the substrate.70 Proper folding of NBD1 of CFTR at the ER is highly dependent on Hsc70 and its cochaperone DNAJA1.71 Using a proteomic approach, it was found that more Hsc70 was associated with misfolded F508del-CFTR than with WT, consistent with the engagement of chaperones in trying to refold mutant CFTR.72,73 Direct evidence was obtained with DNAJA1 knockdown, which decreased CFTR folding and trafficking and increased the degradation of both WT- and F508del-CFTR.74 Instead, up-regulation of the inducible Hsp70 and its cochaperone Hsp40 was seen to lead to a modest but significant improvement in trafficking, stabilization, and activity of F508del-CFTR at the PM.75,76 However, the Hsc70/Hsp70 chaperone complex is also involved in CFTR degradation and in cell-surface quality control (QC). In the cytosol, Hsc70 in complex with the soluble E3 ubiquitin ligase CHIP binds more tightly to misfolded F508del-CFTR than to WT-CFTR, and thus the mutant protein is incapable of exiting the ER and is degraded by the proteasome.73,77 In parallel at the ER level, Hsc70 promotes the degradation of F508del-CFTR dependent on the membrane-anchored E3 ligases gp78 and RMA1.74 The Hsc70-CHIP complex also functions in the clearance system at the PM by promoting internalization by endocytosis and lysosomal degradation of misfolded CFTR.77,78 How these opposite roles of Hsc70 are balanced remains unclear.
Figure 3.
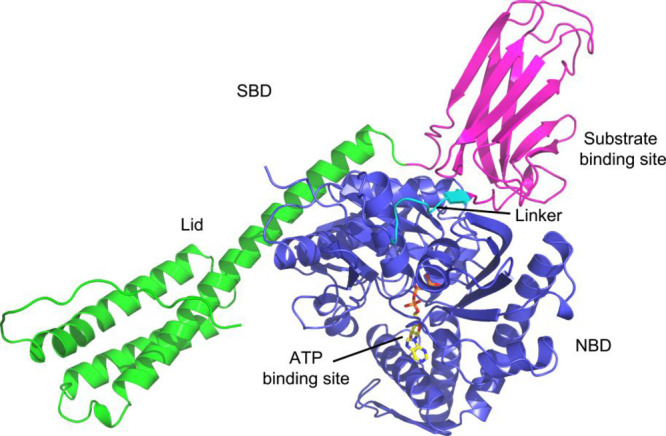
Predicted structure of Heat shock 70 kDa protein 1A by AlphaFold.69 The N-terminal nucleotide-binding domain (NBD) (also known as the ATPase domain) is represented in blue. The C-terminal substrate-binding domain (SBD) (magenta) contains a substrate-binding pocket that interacts with client/substrate proteins. An α-helical subdomain from the C-terminal side of SBD forms a flexible lid (green).
Even if evidence suggests that targeting the Hsc/Hsp70 complex might have pleiotropic effects, it is believed that both profolding and antidegradation strategies could be therapeutically interestesting. For that, small molecules that target components of the Hsc/Hsp70 system can either block mutant CFTR degradation or promote its folding and therefore have potential applications as CF therapeutics.
In recent years, small molecule inhibitors of Hsc70 have become available, and many are being studied for their antitumor properties. The growing evidence that Hsc70 inhibition can rescue defective cellular processing of mutant CFTR prompted the evaluation of these inhibitors’ activity on membrane trafficking of F508del-CFTR. For example, the rhodacyanine derivative MKT-077 (11, Figure 4) is an allosteric inhibitor with high affinity for the ADP-bound state of Hsp70.79,80 Young and co-workers proved that 11 could enhance levels of both mature protein and F508del-CFTR, by slowing turnover and allowing delayed maturation, respectively.81 Thus, 11 appears to increase the stability of F508del-CFTR against ER-associated degradation (ERAD), allowing the accumulation of further rescued protein. Furthermore, when combined with the corrector 2, 11 was able to efficiently correct the trafficking defect of F508del-CFTR and boost the mutant channel activity.81 However, compound 11 was withdrawn from phase 1 clinical study due to its poor metabolic stability.82 Since then, many researchers have been looking for analogues of 11 with a similar effect on Hsc70 and improved safety and pharmacokinetic profiles.83,84 Despite that, an analysis of the efficacy of those new Hsc70 inhibitors on CFTR rescue is still missing, and we believe that it could be of great interest for the CF community.
Figure 4.

Structure of the allosteric Hsc70 inhibitor MKT-077 (11).
Sulfogalactolipids (SGL), such as sulfogalactosylceramide (SGC), were found binding to a putative sulfatide binding site on a variety of Hsp70s.85−88 This sulfatide SGL binding site lies within the N-terminal ATPase domain of Hsp70,85 and SGL binding is known to decrease ATPase activity.89 On the basis of this knowledge, Whetstone and Lingwood synthesized a water-soluble analogue of SGC that could mimic the structural and functional features of the natural glycolipid.90 For that, commercially available 3′-sulfogalactosyl ceramide (3′-SGC, 12, Figure 5) was subjected to deacylation, and the generated amine coupled to the carboxyl group of α-adamantaneacetic acid, yielding compound adaSGC (13, Figure 5) with an α-adamantane rigid frame.90 Single-turnover assays indicated that the resulting conjugate 13 inhibited the Hsp40-stimulated ATPase activity of the Hsp70 chaperone, with a Ki of ∼10 μM. Interestingly, 13 was seen to be associated with the N-terminal domain of Hsp70, directly hindering the Hsp40 binding site and reducing the C-terminal peptide binding. In transfected baby hamster kidney (BHK) cells, 13 increased the levels of immature F508del-CFTR, suggesting that inhibition of Hsp70s ATPase activity by 13 might suppress the ERAD pathway. Increased maturation and iodide influx, however, were observed only after low-temperature glycerol rescue of F508del-CFTR in 13-treated cells.91 Furthermore, the binding site of these glycolipids is only loosely defined, and their selectivity has yet to be firmly established. Despite these insights, the authors did not investigate further the role of 13 on CFTR rescue but instead moved forward and proposed 13 as a new tool to manipulate mammalian glycosphingolipid metabolism in lysosomal storage disease.92
Figure 5.
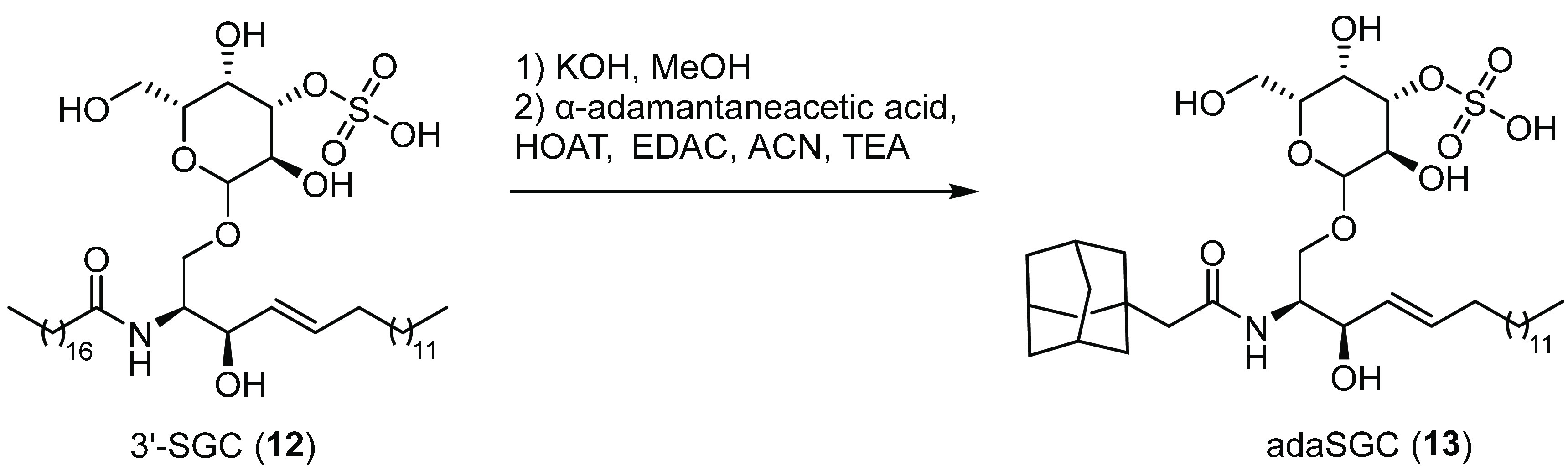
Chemical modification of 3′-SGC (12) to obtain the water-soluble analogue adaSGC (13), a Hsp40–Hsp70 chaperone complex inhibitor.91
Looking for molecules with apoptosis-inducing activity as potential cancer therapeutics, Shin and co-workers used a cell-based assay to screen a previously synthesized imidazole library of 216 derivatives.93,94 One of them, Apoptozole (Az, 14, Figure 6A), was seen to regulate apoptosis by inhibiting the function of Hsp70 and/or Hsc70.94 Further studies showed that 14 blocked the ATPase activity of Hsp70 by 55% at 200 μM, based on the malachite green assay, by binding to its ATPase domain, as demonstrated by affinity chromatography. To explore the utility of the Hsc70 inhibitor 14 in rescuing defective F508del-CFTR cellular processing, the same authors initially determined the detailed binding mode of 14 to Hsc70.95 For that, they employed a ligand-directed protein labeling method with a 14-conjugated probe, obtained by connecting 14 to diethylaminocoumarin via a sulfonate reactive group (Figure 6B). Briefly, when the purified Hsc70 was incubated with the 14 probe, the binding of the 14 moiety promoted a SN2-type reaction of the sulfonate group with a nucleophilic amino acid residue located near the binding site of the protein (Figure 6C). This chemical event promoted protein labeling because the 14-containing moiety was released from the probe. Then the labeled amino acids were identified by mass spectrometry (MS) after trypsin digestion. Next, molecular modeling studies of the complex between the 14 probe and Hsc70 were performed, suggesting that 14 might inhibit Hsc70 activity by interacting with the ATP binding pocket. Treatment of F508del-CFTR cells with nanomolar concentrations of 14 induced cAMP-stimulated CFTR chloride channel activity by increasing the expression of the mutant channel at the PM. Further measurements of the half-life of rescued F508del-CFTR suggested that 14 could also increase the stability of the cell-surface mutant CFTR. In addition, the study suggested that 14-induced membrane trafficking of F508del-CFTR might be caused by the disruption of mutant CFTR association with Hsc70 and CHIP, thus suppressing its ubiquitination and causing escape from the ERQC.95 However, the authors did not investigate the effect of 14 on other CFTR processing events, and after this initial interest for 14 in CF, the group moved to study this small molecule for the development of new anticancer therapies.96
Figure 6.
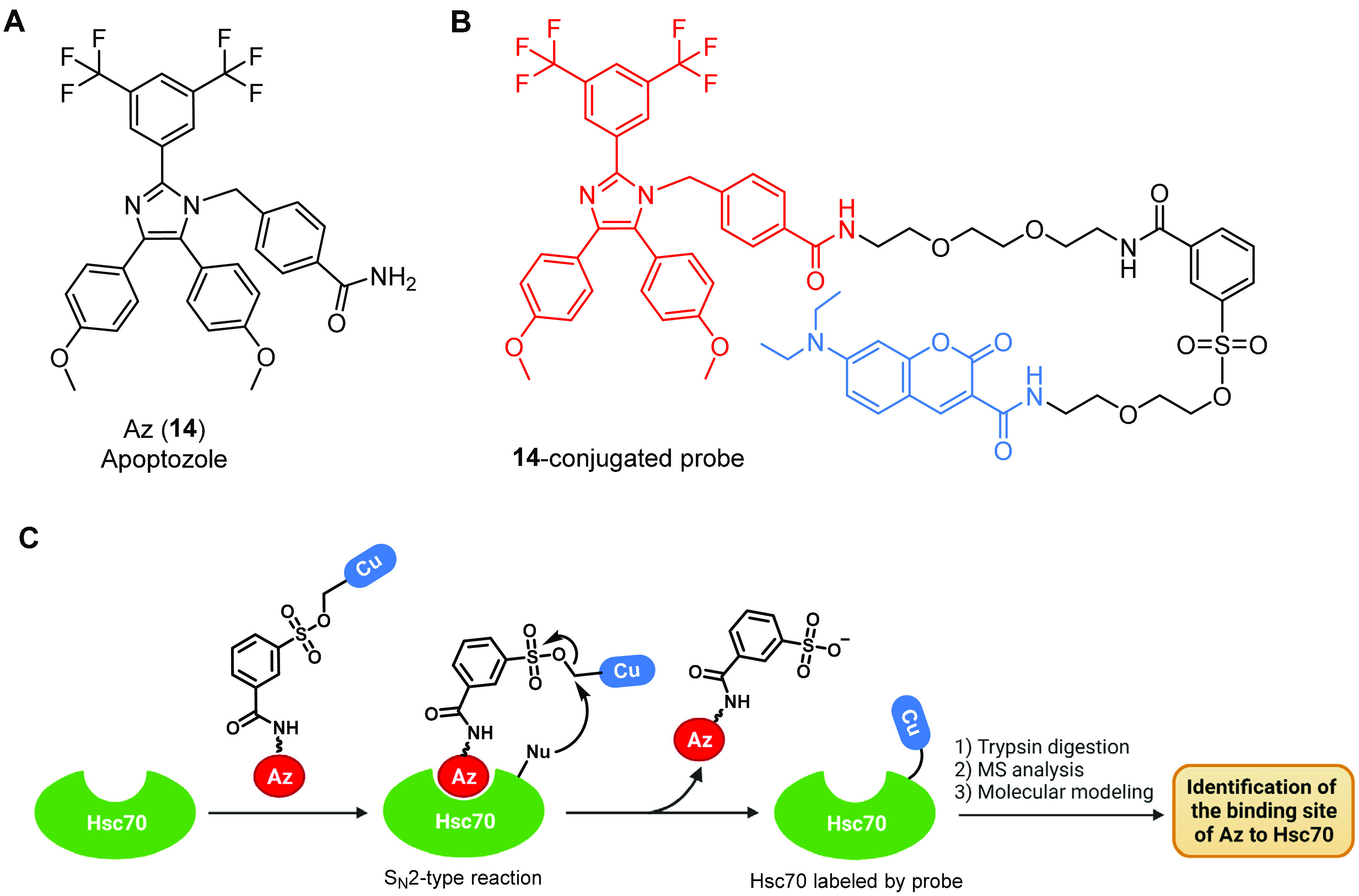
Structure of the Hsc70 ATPase activity inhibitor Az (14) in the study by Shin and co-workers and ligand-directed protein labeling technique used to determine the detailed binding mode of 14 to Hsc70. (A) Chemical structure of 14. (B) Chemical structure of the probe employed in the ligand-directed protein labeling assay, where 14 (in red) is conjugated with diethylaminocoumarin (in blue) via a sulfonate group. (C) Schematic illustration of the strategy used to identify the binding site of 14 on Hsc70 using the 14-conjugated probe. Cu = diethylaminocoumarin; Nu = a nucleophilic amino acid residue.95 Created with BioRender.com.
In contrast to the inhibition of Hsc70, which has proven to be more effective in preventing the growth of a variety of cancer cells, researchers have been prompted to look for small molecules that could cause an increase of the activity of Hsp70 or other chaperones that might prevent protein aggregate accumulation. Indeed, enhanced Hsp70 activity by compounds that act as Hsp70 agonists could improve CFTR folding and prove beneficial to promote F508del-CFTR rescue. With this idea, Brodsky and co-workers monitored the stability of F508del-CFTR in the presence of MAL1-271 (15, Figure 7), a previously reported dihydropyrimidinone activator of Hsp70. In vitro studies showed that 15 accelerated the ATPase and protein-folding activity of Hsp70 in the presence of Hsp40 by binding to the cochaperone binding site of Hsp70 and thus regulating Hsp70–Hsp40 complex assembly.97 Using an immunoblot assay, they examined the effect of 15 on the ER glycosylated and Golgi glycosylated forms of F508del-CFTR. They found that 30 μM of 15 increased the amount of the ER glycosylated form (∼2.1-fold) to a greater extent than the corrector 2 without increasing the level of the Golgi glycosylated form of CFTR. Perhaps this reflects the fact that a portion of the immature protein had folded into a proteasome-resistant state. Based on these results, they assayed 12 derivatives of 15, which were synthesized through modification of the free acid at the end of the flexible hydrocarbon chain. Interestingly, compound DWN-723-23 (16, Figure 7), generated by alkylation of the cesium salt of 15 to obtain a derivative containing a nitrile instead of the free acid, was almost equally active as 15 in the F508del-CFTR maturation assay. Therefore, although further modifications are in order to increase the activity of this class of derivatives, the authors speculated that the cyanomethyl ester moiety of 16 could convey biological absorption potential superior to that of the carboxylate in 15. Meanwhile, this function should maintain similar binding affinity due to the polar nitrile group that might act as hydrogen bond acceptor. Finally, none of the derivatives exhibited cellular toxicity nor induced cellular stress response pathways, in contrast to what was observed with Hsp70 inhibitors, which are mostly cytotoxic. Taken together, these results serve as a gateway for the development of new Hsp70 agonists and for further optimization of the pharmacokinetic properties of this pyrimidinone–peptoid class of compounds.98
Figure 7.

Chemical structure of the Hsp70 agonist MAL1-271 (15) and synthesis of DWN-723-23 (16), the most promising new derivative.98
3.2. Hsp90–Aha1 Chaperone Complex
The Hsp90 cytosolic chaperone system has been broadly implicated in the folding process of more than 300 specific client proteins,99 including nascent CFTRs.100,101 Initially, the proof-of-concept of the role of Hsp90 in CFTR post-translational folding was demonstrated with Hsp90 inhibitors such as geldanamycin, a 1,4-benzoquinone ansamycin antibiotic.102 The immature CFTR molecule was detected in association with Hsp90, and this interaction was found to have a major impact on the fate of nascent CFTR.100 Therefore, geldanamycin, binding to the ADP/ATP-binding pocket of Hsp90, was able to nearly completely abrogate the maturation of nascent WT-CFTR and enhance its degradation. These results provided the evidence of the role of Hsp90 in the maturation of newly synthesized, incompletely folded, or assembled cytoplasmic CFTRs.100
Hsp90 is a flexible homodimer consisting of three domains per monomer: the N-terminal domain (NTD) containing the ATP-binding site, the middle domain (MD) where the unfolded client proteins are assembled, and the C-terminal domain (CTD) (Figure 8).68 The function of Hsp90 is regulated by ATP-induced large conformational changes, which represent the rate-limiting step of the formation of the Hsp90 catalytically active state.103 Among the variety of cochaperones that participate to the ATPase cycle of Hsp90,104,105 the activator of the Hsp90 ATPase (Aha1) had a major role.106 By MS approaches, Balch and co-workers demonstrated that the C-terminal domain of Aha1 binds to the NTD of Hsp90.101 This interaction accelerates the formation of an N-terminally closed state of Hsp90 that triggers the assembling of the unfolded substrates on Hsp90 MD (Figure 8).101,107 Subsequently, the hydrolysis of ATP leads to the opening of the NTD of Hsp90 dimer and to the release of the mature client protein.108
Figure 8.
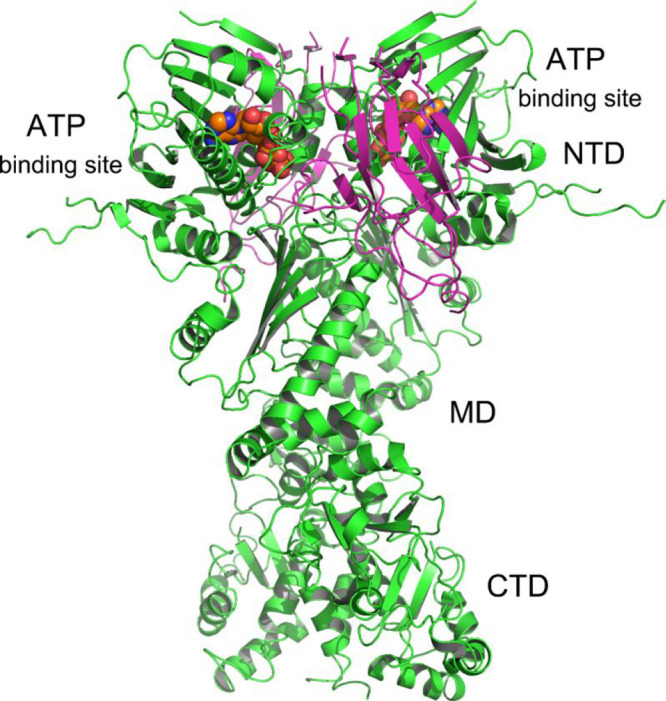
Crystal structure of full-length yeast Hsp90 (green) in complex with an ATP analogue (sphere, orange) and the cochaperone p23/Sba1 (magenta) (PDB: 2CG9).109
When F508del-CFTR is processed by the Hsp90–Aha1 complex, the mutation impairs folding and kinetically restricts F508del-CFTR to a folding intermediate, which is prematurely degraded by the ERAD pathway (Figure 9, panel 1).56,101 Strikingly, the knockdown of Aha1 was seen rescuing the trafficking of F508del-CFTR to the cell surface and restoring channel function.56 This suggests that the Hsp90–Aha1 machinery governing the folding of F508del-CFTR can be manipulated pharmacologically to promote folding and transport of the mutant channel to the PM. Accordingly, disrupting the interaction of Hsp90 with Aha1 may slow down the ATP-driven conformational cycle of Hsp90, increasing the folding efficacy of F508del-CFTR and protecting the mutant fold from degradation.101
Figure 9.

Hsp90 ATPase chaperone cycle and potential CF therapeutic strategies. (1) The client protein, such as F508del-CFTR, is loaded onto the middle domain (MD) of Hsp90. Aha1 increases Hsp90 ATPase activity, thus contributing to the closing of the N-terminal domain (NTD). Finally, ATP hydrolysis causes the release of the misfolded CFTR channel. The open conformation of Hsp90 is restored for the next chaperone cycle. F508del-CFTR is targeted by the QC system and degraded. (2) A12 (17) and A16 (18) might inhibit Hsp90–Aha1 chaperone complex formation, slowing down the conformational changes of Hsp90 and enhancing its folding efficacy. (3) HAM-1 (19) might interfere with the catalytic function of the cochaperone Aha1 C-terminal domain (C), without preventing Aha1 N-domain (N) binding to Hsp90 MD. This inhibition might increase the dwell time with F508del-CFTR and therefore enhance its folding efficacy. Created with BioRender.com.
The Hsp90–Aha1 complex formation can be, in principle, inhibited with protein–protein interaction (PPI) inhibitors, as shown by Obermann and co-workers (Figure 9, panel 2).110 For that, they adapted the Hsp90–Aha1 complex to serve as target for inhibitor screening using amplified luminescence proximity homogeneous assay (Alpha Technology). Using this system, 14,400 drug-like molecules of the Maybridge HitFinder collection were tested, and among them, eight candidates inhibited Hsp90–Aha1 interaction. Next, the drug-like molecules were assayed for their potency to preserve the CFTR residual channel activity in BHK cells stably expressing F508del-CFTR, using the iodide efflux assay. The 1,2,4-triazolic compound A12 (17, Figure 10) and the naphthalenone A16 (18, Figure 10) increased the iodide efflux ∼2.5-fold compared to that of the untreated control. Further biological evaluations showed that 17 and 18 were most effective in combination with the corrector 2, increasing the iodide transport ∼25-fold or ∼15-fold, respectively, compared to untreated cells. Thus, the authors suggested that the two molecules, by acting as Hsp90–Aha1 chaperone complex formation inhibitors, could be beneficial to further enhance F508del-CFTR channel activity as they showed potentiated synergistic effects in combination with 2.110 However, the regulation of the Hsp90–Aha1 interaction through PPI inhibitors is still going through an embryonic stage and requires further research. Furthermore, 17 inhibited multiple targets, including (1,6)-β-glucan synthesis111 and Notum carboxylesterase activity, which recently led to its application in the treatment of central nervous system disorders and in cancer and metabolic disorder therapy.112 For these reasons, 17 is unlikely to represent a CF drug scaffold.
Figure 10.

Chemical structures of the Hsp90–Aha1 complex inhibitors A12 (17), A16 (18), and of the Aha1-stimulated Hsp90 ATPase activity inhibitors HAM-1 (19) and SEW84 (20).
In a different proof-of-principle study, Buchner and co-workers showed that a drug-like small molecule could specifically interfere with the catalytic function of the cochaperone Aha1, without affecting complex formation with Hsp90 (Figure 9, panel 3).113 For that, they screened a library of ∼15,000 chemical compounds from ChemDiv by a FRET-based assay that monitored the interaction between Hsp90 and Aha1 and identified nearly 40 compounds as possible modulators. Among them, the dihydropyranopyrazole HAM-1 (19, Figure 10) was the strongest inhibitor, able to almost completely suppress the stimulatory effect of Aha1 on the Hsp90 ATPase without preventing binding of Aha1 to Hsp90, as determined by SPR measurements. NMR studies revealed that 19 could bind to the Hsp90 NTD, thus sterically blocking its interaction with Aha1that is required to accelerate the formation of the Hsp90 N-terminally closed state. Instead, the Hsp90 MD appeared to be unaffected in its interaction with Aha1 N-domain by 19, and therefore, this inhibitory compound could not dissociate the Aha1–Hsp90 complex. Further in vivo studies demonstrated that 19 affected the activation and processing of Hsp90–Aha1-dependent client proteins. Then, because Hsp90–Aha1 machinery targets F508del-CFTR for degradation, the effect of 19 on this misfolded protein stability was evaluated. As expected, in the presence of 19, higher amounts and a prolonged lifetime of F508del-CFTR were observed.113
In 2020, Singh et al. identified a novel inhibitor of the Aha1-stimulated Hsp90 ATPase activity, the isothiosemicarbazide SEW84 (20, Figure 10), using a modified quinaldine red-based HTS.11420 emerged to bind to the C-terminal domain of Aha1, causing the weakening of its binding to Hsp90 without affecting the basal ATPase activity of Hsp90. Therefore, similarly to 19, 20 could maintain the basal activity of Hsp90, potentially avoiding the toxic effects reported with the use of common Hsp90 inhibitors.114−116 However, the authors only evaluated the effects of 20 on tau phosphorylation and on the trophic activity of androgen receptor variants but did not investigate its impact on CFTR protein folding.114 Nevertheless, these results highlight the potential benefits of small molecule inhibitors of the Aha1-stimulated Hsp90 ATPase activity in managing proteostatic diseases such as CF, and hopefully, this will be further substantiated with in vivo experiments.
4. Targeting Ubiquitination/Deubiquitination Enzymes
The network of the ERQC factors, known as ERAD, is responsible for disposing of newly synthesized F508del-CFTRs that fail to reach their proper conformations.117 Misfolded F508del-CFTR accumulates in a kinetically trapped conformation, which is retained in the ER and ubiquitinylated by the sequential action of a ubiquitin activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). Next, the poly-ubiquitinylated F508del-CFTR is prematurely degraded by the UPS, leading to reduced PM expression of the mutant channel.118 The ubiquitination process is regulated by deubiquitinating enzymes (DUBs) that catalyze the deconjugation of ubiquitin chains from substrates. At the peripheral level, ubiquitination and deubiquitination processes regulate endocytosis, endocytic recycling, and lysosomal degradation of CFTRs.119,120 Therefore, the plasma abundance of this membrane protein is largely dependent on the balance between ubiquitin ligation by E3 ligases and deubiquitination by DUBs.
Efforts to manipulate the UPS to promote the functional expression of the CFTR channel have focused on different strategies, such as (i) inhibiting the proteasome, (ii) inhibiting E1 or E3 ligases, and (iii) activating endogenous DUBs. However, all three approaches have several limitations, due to their widespread role in proteostasis that may lead to off-target effects. Early studies investigated whether proteasome inhibitors could rescue F508del-CFTR, even though it is a relatively nonspecific way to target ubiquitin-dependent protein degradation.121 However, it was shown that proteasome inhibition leads to accumulation of insoluble, multi-ubiquitinylated F508del-CFTR proteins, with no detectable increase in the level of folded CFTRs.122 Based on these results, targeting earlier and more specific steps in the ERAD cascade, such as enzymes involved in the ubiquitination or deubiquitination process, might be more reasonable to achieve an increase of the pool of correctable F508del-CFTR.
4.1. Ubiquitin Ligase RNF5/RMA1
In 2006, Younger and co-workers identified an ER-associated ubiquitin ligase complex, containing the membrane-anchored E3 ubiquitin ligase RNF5 (also known as RMA1), the E2 Ubc6e, and the transmembrane QC factor Derlin-1.123 This complex cooperates with the cytosolic Hsc/Hsp70-CHIP E3 ligase complex to monitor the conformation and to triage nascent WT-CFTR and F508del-CFTR.124 However, while CHIP QC checkpoint inspects the folding status of CFTR’s cytosolic domains,71,77,124 the RMA1 QC checkpoint senses the assembly status of amino terminal regions of CFTR.123 Moreover, the CHIP E3 complex performs its function after CFTR’s NBD2 synthesis (post-translational role), while RNF5 E3 complex acts prior to NBD2 synthesis (co-translational role). Although these two sequential QC checkpoints are both responsible for F508del-CFTR degradation, RNF5 was seen to be particularly relevant, as its loss by RNA interference strongly increases the folding of F508del-CFTR and synergizes with the bithiazole corrector 5 (Figure 1)125 to rescue F508del-CFTR folding.126 Given our group’s interest in the identification of novel targets and chemical compounds for the development of innovative therapeutic approaches for CF, we screened a siRNA library targeting known CFTR interactors.127 Our analysis showed that silencing RNF5 elicited a 70–80% increase in F508del-CFTR function in the microfluorimetric assay based on the halide-sensitive yellow fluorescent protein (HS-YFP) and displayed an additive effect with the known corrector 2. As validation of in vivo efficacy of RNF5 modulation, RNF5 knockdown in F508del-CFTR transgenic mice exhibited improved intestinal absorption and increased CFTR activity in intestinal epithelial cells, relative to animals expressing WT-RNF5.127 These findings validated RNF5 as a drug target for CF and provided the basis for the development of RNF5-targeting molecules that could inhibit its activity.
With this aim, using a computational approach, we generated a homology model of RNF5 RING domain (Figure 11A,B) and performed high-throughput docking selecting a first set of 1623 ligands.128 A second diversity set of 1000 ligands, based on molecular fingerprinting chemical diversity, was extracted from the LifeChemicals database. In total, 2623 molecules were purchased and tested as F508del-CFTR correctors, using the HS-YFP functional assay in a bronchial epithelial cell line (CFBE41o-). This primary biological screening identified two potential hit compounds having a clear dose-dependent effect, the thiadiazolylidene derivative inh-2 (21, Figure 11C) and the benzooxazolylthiolic compound inh-5 (22, Figure 11C). To evaluate the best hit as a F508del-CFTR corrector, we used electrophysiological techniques on human primary bronchial epithelia. As a result, 21 increased the F508del-CFTR-mediated current, whereas 22 had no activity as a corrector. The lack of consistency between results obtained with 22 in immortalized and primary bronchial cells could be explained by assuming a possible effect of 22 on other targets than RNF5 that, in primary cells, hinder mutant CFTR rescue. On the other hand, ubiquitination experiments confirmed that 21 decreased ubiquitination of mutant CFTR, thus stabilizing its mature form. To further investigate the mechanism of action of the putative RNF5 inhibitor 21, we exploited known RNF5 downstream targets, such as the regulator of basal autophagy ATG4B and the actin cytoskeleton factor Paxillin. Functional and biochemical experiments confirmed that 21 decreases the ubiquitinylated form of ATG4B and increases the basal level of autophagy, while scratch/wound healing assays confirmed that 21 increases cell motility, consistent with what has been described for RNF5 knockdown. Taken together, all of these data demonstrated that 21 could act as RNF5 inhibitor able to rescue the F508del-CFTR trafficking defect and pioneered 21 optimization through a medicinal chemistry campaign, which is currently ongoing.
Figure 11.

Representative conformational state of the RNF5 ring domain extracted from the molecular dynamics trajectory conducted on the model built by homology modeling that was used to select RNF5 ligands.128 (A) Cartoon representation of RNF5 ring domain; zinc ions are represented as spheres. (B) Surface representation of RNF5 ring domain. (C) Chemical structures of hits inh-2 (21) and inh-5 (22) identified from the primary screening.
4.2. Ubiquitin-Activating Enzyme E1
Another way to achieve F508del-CFTR stabilization through suppression of the ubiquitin-dependent degradation pathway may be the inhibition of the ubiquitin-activating enzyme E1. While E3s determine the substrate specificity of ubiquitination and, therefore, some hundreds of different mammalian E3s are involved in different cellular processes, there is just one major E1 in human.129 Whereas the proteasome represents the final destination for many ubiquitinylated proteins, E1 is the common first step in ubiquitination, thus activating and transferring ubiquitin (Ub) to tens of different E2s involved in different downstream pathways (Figure 12).130 Indeed, compounds developed to inhibit E1 enzyme could enable F508del-CFTR to pass through both sequential cytosolic and ER-associated QC checkpoints, thus efficiently suppressing F508del-CFTR degradation.
Figure 12.
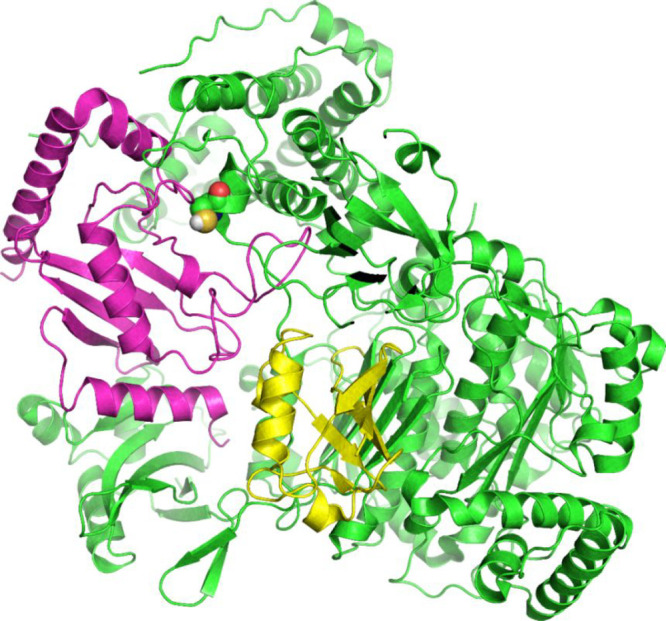
Complex of E1 (green), E2 (magenta), and ubiquitin (yellow) of S. pombe (PDB: 5KNL).131 Catalytic Cys593 of E1 is shown as spheres.
Therefore, using the pyrazone PYR-41 (23, Figure 13), a previously reported E1 inhibitor used in cancer therapy,132 Brodsky and co-workers showed that it is possible to significantly increase F508del-CFTR stability, trafficking to, and activity at the PM when a E1 inhibitor is combined with different types of correctors.133 However, the use of 23 is limited due to its toxicity, perhaps linked to its 5-nitrofuroyl moiety. To identify small molecule analogues with lower toxicity and increased potency, the authors performed a SAR exploration around 23 by purchasing 22 different compounds from Sigma-Aldrich.134 All of the analogues left the pyrazolidinedione core unaltered while bearing different combinations of substituents in the 4-position of the central ring and on the phenyl ring in the 2-position. In particular, they focused on the removal of either nitro or furan moieties, while in some cases, the furan ring was replaced with different phenyl ring systems. In addition, some compounds had unique substituents at the 4-position of the pyrazolidinedione core, such as an indolinone, a pyrimidinetrione, or an isopropyl moiety. Other modifications included the methylation of the central pyrazolidine or the exploration of different substituents on the phenyl ring in the 2-position. These modifications are summarized in Figure 13. Ubiquitination experiments of both WT- and F508del-CFTR showed that the analogue 24 (Figure 13), bearing an electron-rich nitrobenzodioxolyl substituent in the 4-position and a 3-chloro-4-methylphenyl ring in the 2-position, markedly inhibited ubiquitination, meanwhile lacking the toxicity of the parent compound 23. Further in vitro experiments showed that when corrector 2 was combined with 24, a significant increase in maturation, expression, and activity of F508del-CFTR at the PM was observed in comparison to the combination of 23 and 2. Finally, in silico modeling studies confirmed the proposed mechanism of action of 23 and 24, with the molecules binding through extensive hydrogen bonds to a pocket near the active Cys593 of E1 (Figure 12). Overall, these results suggested that the suppression of F508del-CFTR ubiquitination by an E1 inhibitor could synergize with other known correctors, thus opening the way for further optimization of 24.134 However, we should consider that 23 and 23-related compounds were first proposed as therapeutics in cancer for their capacity to kill transformed cells.132 During the characterization of 23, Yang and co-workers suggested that 23 could potentially function by covalently modifying E1, perhaps through heteroconjugate addition of the E1 cysteine residue to the α,β-unsaturated pyrazolidinedione.132 Subsequent studies on chemical reactivity of 23 provided some insight into the covalent protein cross-linking potential and partial target selectivity of 23.135 Indeed, along with the inhibition of E1, 23 emerged to have equal or greater inhibitory activity against several DUBs.135 Furthermore, 23 mediated cross-linking of specific protein kinases such as Bcr-Abl and Jak2, causing the inhibition of their signaling activity.135 Therefore, further studies on 24’s mechanism of action and selectivity are needed in order to address possible side effects and to refine the hit optimization strategy for this emerging class of E1 inhibitors.
Figure 13.

Depiction of the SAR strategy around E1 inhibitor PYR-41 (23) and structure of the optimized analogue 24.134
4.3. Deubiquitinating Enzymes DUBs
The human genome encodes approximately 100 DUBs, among which some are responsible for the deubiquitination of WT-CFTR, although their role in the PM turnover of conformationally defective F508del-CFTR mostly remains unclear. Very recently, Colecraft and co-workers successfully demonstrated that selective ubiquitin chain removal could rescue trafficking-deficient CFTR via the development of engineered DUBs (enDUBs).136 For this proof-of-concept study, they prepared an enDUB comprising the catalytic component of ubiquitin-specific protease USP21 (enDUB-U21) and engineered CFTR protein to probe the impact of six distinct CF-causing mutations of class II and class IV that all impair channel surface density. Applying corrector 2 in combination with enDUB-U21 afforded a synergistic increase of four out of six of the CFTR mutants’ surface density from flow cytometry experiments. Strikingly, two mutations (N1303K and 4279insA) displayed expression levels equivalent to those of WT following the same treatment. Moreover, human embryonic kidney (HEK293) cells coexpressing enDUB-U21 and either N1303K CFTR or 4326ΔT CFTR yielded substantially increased 1-potentiated currents. In order to enable enDUB-mediated functional rescue of endogenous CFTR channels, the authors adapted a selective nanobody (E3h) against CFTR to the enDUB-U21 system (enDUB-U21CEE3h). Remarkably, in Fischer rat thyroid (FRT) epithelial cells expressing the pharmacotherapy-resistant N1303K mutation, enDUB-U21CEE3h in combination with the potentiator 1 and the corrector 2 rescued N1303K CFTR currents to ∼40% of WT levels. Furthermore, in the same cell system, enDUB-U21CEE3h synergized with Trikafta to increase N1303K CFTR currents up to 80% of WT levels. Concerning the F508del mutation, the authors exploited the previously reported Cerulean-nanobody T2a137 in order to stabilize the F508del-CFTR channel upon binding of enDUB-U21CET2a. In FRT cells expressing F508del-CFTR, the enDUB-U21CET2a/1/2 combination substantially rescued F508del-CFTR to ∼45% of WT levels. Notably, enDUB-U21CET2a in combination with Trikafta increased F508del-CFTR currents to beyond WT levels.136 Although the application of enDUBs finds its place in gene therapy, the authors hypothesized that this therapeutic modality could be of inspiration for bivalent small molecules able to induce endogenous DUBs to target specific substrates, too.
In both academic and industrial settings, targeted protein degradation (TPD) and targeted protein stabilization (TPS) have recently emerged as powerful drug discovery approaches.138−140 Proteolysis targeting chimeras (PROTACs) are the earliest example of TPD effectors and consist of heterobifunctional molecules formed by a protein-targeting ligand linked to an E3 ligase recruiter. Their purpose is to achieve the proteasomal degradation of a specific protein through induced proximity of the substrate with the E3 ubiquitination enzyme.138−140 Recently, Henning and co-workers extended induced proximity paradigm to develop a heterobifunctional stabilizer, such as a deubiquitinase-targeting chimera (DUBTAC) for CFTR stabilization.141 Using chemoproteomic approaches, the authors identified OTUB1 as a candidate DUB for covalent ligand screening and the acrylamide EN523 (25, Figure 14A) as a OTUB1 recruiter. Further evaluations showed that the cysteine-reactive acrylamide of 25 was able to selectively target the noncatalytic cysteine C23 of OTUB1, without interfering with its deubiquitination activity. Then the authors synthesized two different DUBTACs by linking 25 to the corrector 2 through C3 or C5 alkyl linkers. Between them, treatment of CFBE41o- cells expressing F508del-CFTR with the C5-DUBTAC NJH-2-057 (26, Figure 14B) showed a dose–responsive increase of CFTR stabilization, based on Western blotting data. Proteomic analysis of treated cells confirmed that CFTR was among the most stabilized proteins and showed that CFTR stabilization occurred through ∼60% of OUTB1 occupancy. These data suggested that F508del-CFTR stabilization by a fully synthetic DUBTAC might be possible, and the relatively minimal OTUB1 occupancy of the covalent 26 might still allow endogenous DUB function.141 However, no demonstration of increased CFTR mutants’ surface density and activity by 26 was obtained from this proof-of-concept study. Furthermore, the alteration of deubiquitination process is likely to lead to dysregulation of other biological networks DUB-associated. Indeed, OTUB1 has recently emerged as an essential regulator of a variety of physiological processes, such as immune signaling and DNA damage response, although its functions remain largely unclear.142 For that, elucidation of the mechanism of deubiquitination of CFTR and better understanding the consequences on protein homeostasis are pivotal prior to consider DUBTACs or enDUBs as CF therapeutic strategies.
Figure 14.
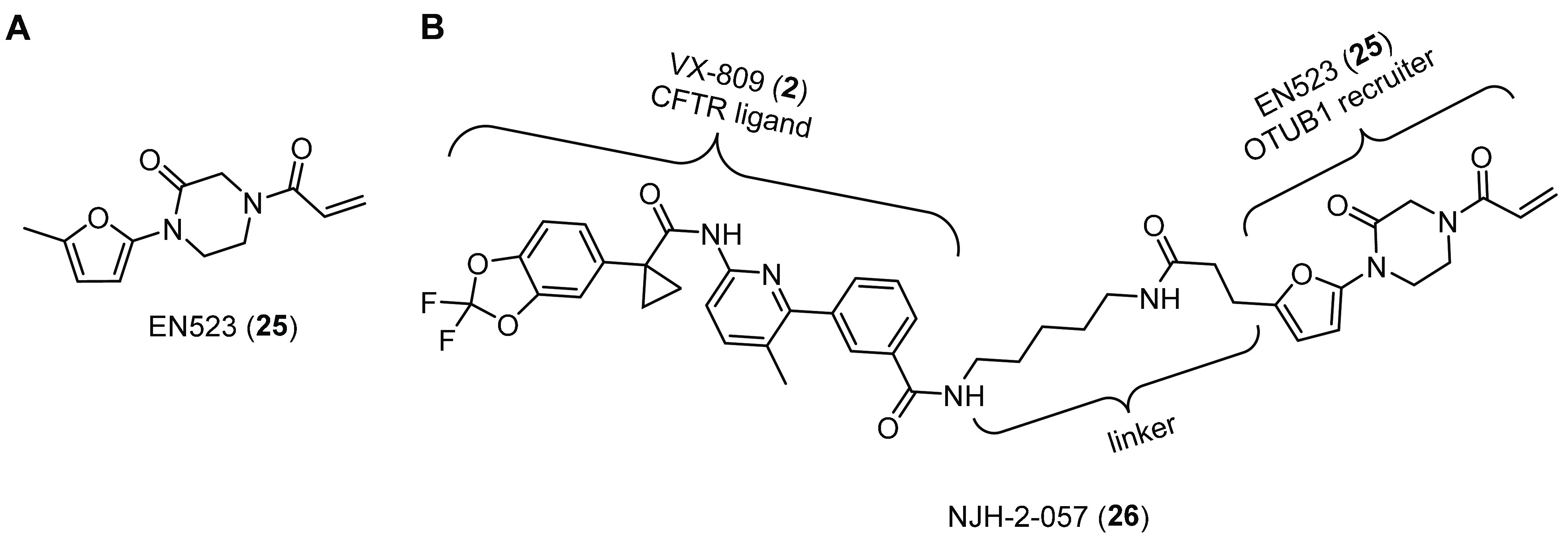
Deubiquitinase-targeting chimera (DUBTAC) for CFTR stabilization in study by Henning and co-workers.141 (A) Chemical structures of OTUB1 recruiter EN523 (25). (B) DUBTAC NJH-2-057 (26) synthesized by linking the CFTR ligand VX-809 (2) to the OTUB1 recruiter 25 through a C5 linker.
5. Targeting Poly-ADP Ribose Polymerases
The family of nuclear enzymes poly(ADP ribose) polymerases (PARPs) is a group of 17 enzymes that catalyze the attachment of polymers of ADP ribose to different target proteins, a modification known as poly-ADP (ribosyl)ation (PARylation).143,144 The PARP family has been involved in the regulation of multiple basal cellular processes, including DNA repair, cell division, protein homeostasis, oxidative stress, and viral infection. PARP-1–6 have shown to transfer polymeric chains of ADP ribose to substrates inside the cell, whereas PARP-7–17 are either presumed or proven to attach one ADP ribose unit per time.145−147 PARP-1 is the most abundant and best-characterized isoform of the PARP family for its involvement in DNA damage repair and genome maintenance.146 Consequently, PARP-1 has been primarily considered to be an attractive drug target, and several inhibitors are currently under investigation for cancer therapy.148 In CF, the presence of defective CFTR appears to produce a redox imbalance in epithelial cells and extracellular fluids and to cause an abnormal generation of reactive oxygen species (ROS).149 Thus, CF patients display increased susceptibility to oxidative-induced DNA damage, although this appears to be independent of clinical status.150 In view of the central role of PARP-1 in cellular stress response,143 Thomas and co-workers decided to investigate its role in CF and demonstrated that PARP-1 activity is 2.9-fold higher in HBE cells from patients homozygous for F508del and 2.5-fold higher in CFBE41o- cells than in non-CF cells.151 Therefore, they tested various well-known PARP-1 inhibitors for their effect on CFTR function and expression. Among them, the benzimidazole inhibitor ABT888 (Veliparib 27, Figure 15)152 partially restored F508del-CFTR activity and trafficking in CFBE41o- cells at low concentrations (maximal inhibition observed at 1 nM). Similarly, treatment ex vivo of ileum tissues from CF mice with 27 partially rescued F508del-CFTR activity to 7% of WT levels and in vivo to 7.8% by measuring salivary secretion. Moreover, when PARP-1 activity was inhibited pharmacologically or by siRNA-mediated silencing, F508del-CFTR maturation was altered, with an increase in the fraction of the mature CFTR glycoform. In addition, the effect of PARP-1 inhibition was seen to be F508del-CFTR specific, as no improvement in the maturation of WT-CFTR was observed. Therefore, the same authors speculated that attenuation of PARP-1 activity could lower oxidative stress that is particularly high in CF and increase expression and folding of F508del-CFTR, at least partially, by altering PARylation of key members of CFTR folding interactome.151 However, further studies are required to uncover 27 mechanism of action in the context of CFTR proteostasis regulation.
Figure 15.

Chemical structures of the PARP-1 inhibitor ABT888 (27),152 the alkaloids latonduines (28–30), and MCG315 (31), a more potent derivative of 28.153
In the same year, to diversify the collection of CFTR correctors previously discovered, the same research group screened a marine extract collection derived from South Pacific sponges using a bioassay-guided fractionation.154 With this method, they identified the class of latonduine heterocyclic compounds 28–30 (Figure 15),153 which corrected F508del-CFTR trafficking up to 45% of WT-CFTR surface expression. Among these alkaloids, latonduine A (28) strongly rescued F508del-CFTR misfolding in both BHK and CFBE41o- cells, and this result was confirmed in both ex vivo and in vivo studies in F508del-CFTR mice. Using pull-down experiments and MS studies, the authors identified PARP-1–5 as target proteins of 28. Finally, combined treatment of 28 with other F508del-CFTR correctors, such as 2, gave increased levels of correction, thus suggesting that 28 might define a novel class of potential CF therapeutics acting through PARP inhibition.154
In order to identify analogues of 28 with improved CFTR corrector potency and to understand the mechanism by which PARP inhibition improves F508del-CFTR trafficking, Thomas and co-workers synthesized a small set of analogues of 28.155 The removal of the aminopyrimidine moiety and the replacement of the pyrrole with a phenyl ring led to the tetrahydrobenzoazepinone analogue MCG315 (31, Figure 15), a 10-fold more potent corrector than the parent compound, as evinced by short-circuit current measurements on CFBE41o- monolayers in Ussing Chamber assays. Moreover, enzyme inhibition assays showed that 28 and 31 in vitro behave as strong inhibitors of both PARP isozymes 3 and 16, perhaps through binding to the nicotinamide binding pocket, as evinced from molecular modeling studies. Further analysis showed that siRNA inhibition of both PARP-3 and PARP-16 resulted in a decrease in the concentration of 31 necessary for maximal F508del-CFTR rescue.155 Intriguingly, PARP-16 is an ER membrane-associated protein, which can ADP ribosylate the stress sensor IRE-1, ultimately triggering the activation of the unfolded protein response (UPR), whose role is to eliminate aberrant proteins.156 Therefore, in line with other works that showed that modulation of IRE-1 and UPR pathway can rescue F508del-CFTR trafficking,15728 and its analogue 31 might trigger F508del-CFTR correction by inhibiting PARP-16-mediated UPR activation and by simultaneously inhibiting PARP-3.155
In order to confirm this proposed dual-target F508del-CFTR corrector mechanism of action of 28 and 31, the same authors recently performed a chemical campaign around the tetrahydrobenzoazepinone scaffold to obtain compounds selective against PARP-3 or PARP-16 enzymes.158 Therefore, they reported the discovery of the two selective inhibitors 32 and 33 (Figure 16). Photochemical reaction of phthalimides 34 and 35 with alkenes 36 and 37 yielded the benzoazepinediones 38 and 39, respectively, which were, in turn, reduced by NaBH4 to afford the corresponding alcohols 32 and 33 as racemic mixtures. Purification of the desired isomers followed by single-crystal X-ray diffraction and NMR analysis confirmed the cis configuration of (+/−) 32 and (+/−) 33.
Figure 16.
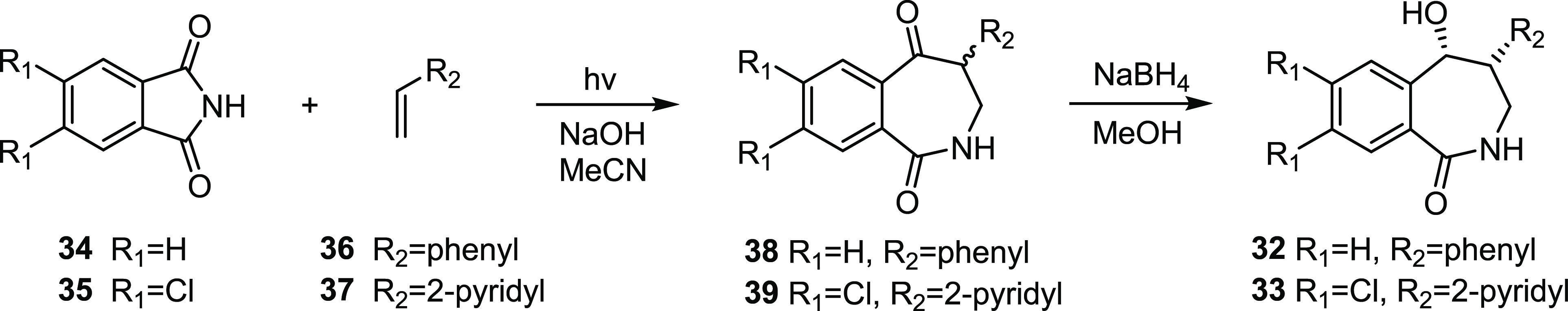
Synthesis of the two PARP-3 and PARP-16 selective inhibitors 32 and 33.158
In vitro evaluation of compounds ability to inhibit PARPs showed that 32 was a modestly potent PARP-3 selective inhibitor (IC50 = 3.1 μM) relative to PARP-16 (IC50 = 296.3 μM), whereas compound 33 displayed strong inhibition of PARP-16 (IC50 = 0.362 μM) with no significant effect on PARP-3 (IC50 = 74.1 μM). Interestingly, HTS and FMP cell-based assays showed that neither 32 nor 33 alone at either 1 or 10 μM elicited F508del-CFTR corrector activity, while the combination of these two selective inhibitors at both 1 or 10 μM each produced the same functional correction generated with an equal amount of 28 (Figure 17). These data strongly confirmed the authors’ hypothesis that the F508del-CFTR rescue exhibited by 28 and 31 could be caused by the dual-target simultaneous inhibition of PARP-3 and PARP-16.158 However, the mechanism of CFTR rescue enhanced by PARPi needs to be explained in more detail in order to anticipate possible side effects.
Figure 17.
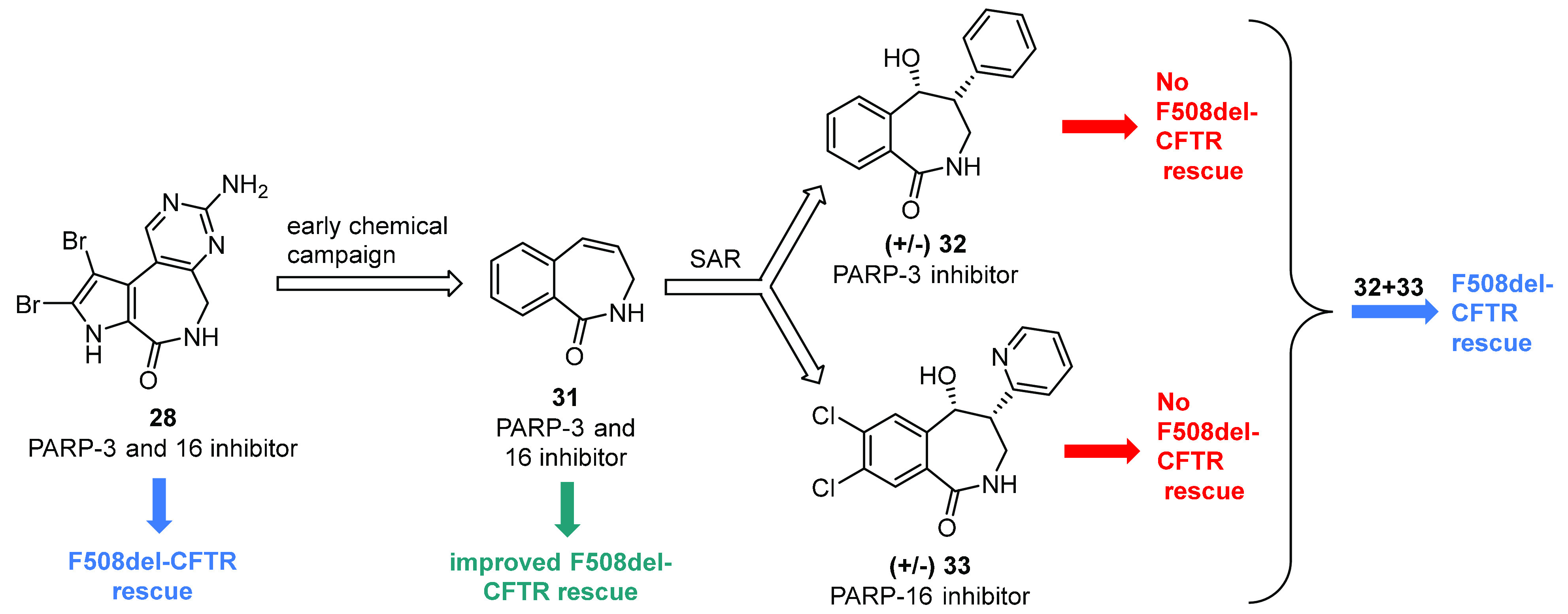
Latonduine A (28) optimization and strategy to prove that the PARP-mediated F508del-CFTR trafficking correction could be achieved by administrating either a single dual-target inhibitor (28 or 31) or a combination of two selective single-target inhibitors (32 and 33).158
6. Targeting CFTR-Associated PDZ Domain Protein
The long-lasting PM expression of WT-CFTR depends on the endocytic trafficking events that occur at the cell surface, such as CFTR internalization by clathrin-mediated endocytosis (CME) and efficient recycling back from the endosomes to the PM.159,160 On the contrary, rescued F508del-CFTR shows a short PM half-life, due to increased endocytosis,161 selective ubiquitination by peripheral protein QC machineries, and rapid lysosome degradation of the mutant protein.162 Several protein partners regulate CFTR stability on the PM, among which PDZ domain-containing proteins (PDZ proteins) are most relevant.163,164 The C-terminus of CFTR binds two types of PDZ proteins: Na+/H+ exchanger regulatory factors 1 and 2 (NHERF-1 and -2) that work as scaffold proteins and stabilize CFTR on the PM by coupling it to the actin cytoskeleton165−167 and CFTR-associated ligand (CAL) that negatively regulates CFTR abundance by promoting its lysosomal degradation.168 Intriguingly, RNA interference-targeting CAL specifically increased cell-surface expression of F508del-CFTR by 4.4-fold and reduced CAL-mediated degradation,169 suggesting that selective inhibitors of the CAL-CFTR interaction could provide a novel generation of CFTR proteostasis regulators. The structure of CAL PDZ domain (CALP) bound to CFTR was solved by resolution NMR and showed interactions between the four C-terminal residues of CFTR peptide (residues Asp-Thr-Arg-Leu) and CALP.170 Madden and co-workers initially validated the possibility of selective PPIs disruption with the discovery of a peptidyl inhibitor of the CFTR-CAL interaction able to bind to CALP with high affinity and therefore hypothetically displace the natural binding partner CFTR.171 Their approach involved the synthesis of up to 6000 different cellulose-bond peptides through SPOT technology, with free C-terminal domains. Peptide screening and iterative optimization using substitutional analysis finally resulted in the identification of a decameric peptide inhibitor iCAL-36 (40, Table 1) with exhibited affinity of 22.6 ± 8.0 μM for CALP172 and no interaction with the NHERF PDZ domains, as determined by fluorescence polarization (FP) measurements and pull-down experiments.171,173 Remarkably, a control decameric C-terminal CFTR sequence (CFTR10, Table 1) exhibited interactions with the CAL PDZ domain (Ki = 390 μM) weaker than those exhibited by 40.
Table 1. Sequences and Affinity Constants of Peptides Binding to the PDZ Domain of CAL.
| peptide | sequencea | Ki (μM) | ref |
|---|---|---|---|
| CFTR10 | Thr-Glu-Glu-Glu-Val-Gln-Asp-Thr-Arg-Leu-OH | 390 ± 20 | (171) |
| iCAL36 (40) | Ala-Asn-Ser-Arg-Trp-Pro-Thr-Ser-Ile-Ile-OH | 22.6 ± 8.0 | (172) |
| kCAL01 (41) | Ac-Trp-Gln-Val-Thr-Arg-Val-OH | 2.3 ± 0.2 | (174) |
Ac = acetyl.
To visualize this new inhibitor functional effect on CFTR activity, a N-terminally fluorosceinated analogue of 40 (F*-40) was synthesized for Ussing Chamber assays. The treatment of CFBE bronchial epithelial cells from CF patients expressing F508del-CFTR with F*-40 increased the half-life and the amount of apical F508del-CFTR channels, and this CFTR rescue effect was magnified when 40 was combined with the first-generation corrector 5.173 New structural insights were obtained later with the determination of a high-resolution structure of CALP in complex with 40 (PDB ID: 4E34)172 and by chemically modifying side chains at different positions along the CALP binding cleft.175 This studies revealed that 40 could bind through canonical class 1 PDZ binding interactions, allowing the ligand C-terminal residue (P0) to form a critical interaction with the carboxylate-binding loop,172 while side chain interactions of residues P-1, P-3, P-4, and P-5 might be responsible for CALP affinity and specificity (Figure 18).175
Figure 18.
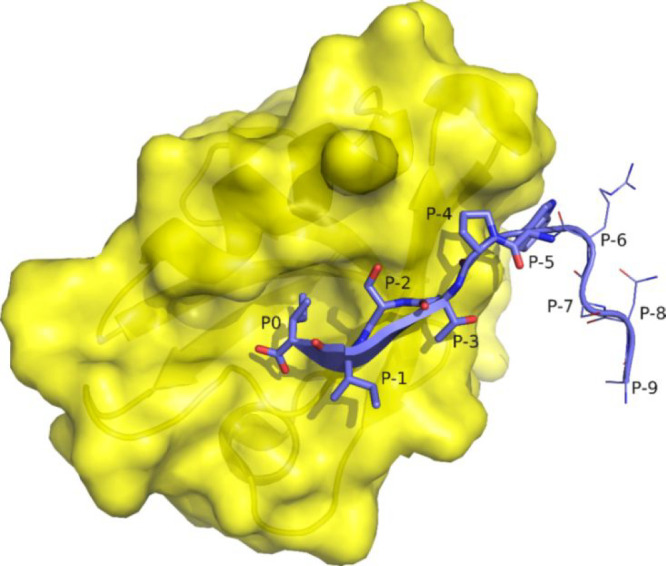
Interaction between PDZ peptide-binding domain (surface, yellow) and the decameric peptide iCAL36 (40) (stick, cyan) (PDB: 4E34).172
To further expand their work on CAL/CFTR PPI disruption, Madden and co-workers used a new computational protein design algorithm (K*) to rationally develop a better binding-efficient competitive peptide CAL inhibitor.174 Using K* to calculate accurate predictions of peptide–CALP binding affinities, they screened up to 8000 hexameric C-terminal peptides from the HumLib library. The top-ranked 11 peptides predicted with the K* CAL-CFTR design were purchased from NEO BioScience, and their Ki values were determined using FP. All examined sequences showed high CAL affinity in the μM range, with kCAL01 (41) representing the tightest hexameric binder (Table 1; Ki = 2.3 ± 0.2 μM). Despite its smaller size, 41 yielded a higher affinity than the decamer 40 and a 170-fold stronger binding than natural CFTR C-terminus. Furthermore, 41 restored F508del-CFTR-mediated chloride efflux in CFBE cells in Ussing Chamber experiments, similarly to the previously available inhibitor F*-40 or to the corrector 5.174 Structure and energy landscape analysis of the crystal structure of 41:CALP (PDB ID: 6OV7) showed that the tighter binding efficiency of 41 could stem from entropic effect at P0 and favorable substitutions at P-1 and P-4 with long polar and charged residues (from Ile and Pro of 40 to Arg and Gln of 41, respectively).176
It is well-known that peptides present inherent limitations in metabolic stability and cell permeability that prevent their use as pharmacological treatment. To overcome these limitations, Pei and co-workers designed a disulfide-cyclized analogue of 41 by incorporating a short amphipathic Cys-Arg-Arg-Arg-Arg-Phe sequence (cell-permeating peptide, CPP) to its N-terminus and replacing Val at position P-3 with Cys to allow intramolecular disulfide bond formation (peptide 42, Figure 19A).177 The obtained peptide was labeled with fluorescein isothiocyanate (FITC), and flow cytometry was used to demonstrate that 42 was readily cell-permeable and had a superior serum stability. Furthermore, fluorescence anisotropy (FA) analysis showed that only the reduced, linear form of peptide 42 could bind the CAL-PDZ domain with Kd of 490 ± 130 nM, whereas the cyclic form could not. Treatment of CFBE41o- cells with a combination of 42 and the corrector 2 increased the activity of F508del-CFTR by 77%. Therefore, the authors hypothesized that peptide 42 can exist as a disulfide cyclized form with improved proteolytic stability when outside the cell and, due to the CPP motif, can show high cell permeability, too. Upon entering the cell, intracellular thiols convert 42 into its linear form, which can expose the CAL binding sequence for efficient displacement of the CFTR-CAL interaction. This effect may be responsible for the increase of F508del-CFTR stability, through hypothetical reduction of lysosome-mediated degradation of the mutant protein.177
Figure 19.

Development of disulfide-cyclized peptidyl inhibitors of the CAL-CFTR interaction from Dougherty et al.177,178 (A) Depiction of the SAR optimization of the peptide 42 to obtain PGD97 (43). The CPP sequence (in black) allows efficient cell permeation of the disulfide-cyclized conjugates, and the CAL binding sequence (in red) allows binding to CALP. (B) Strategy for cyclized peptide 43 cellular entry and conversion to the linear form 44. When outside the cell, the CPP conjugate 43 is proteolytically stable and cell-permeable. Once inside the cytosol, 43 is reduced into its linear, biologically active form 44 by intracellular thiols, such as glutathione (GSH).
To obtain great improvement of peptide 42 potency, selectivity, and pharmacokinetic properties, the same authors recently performed a modeling-guided medicinal chemistry campaign through in silico binding evaluation of a library of peptide analogues, followed by the synthesis and FP-based competition assay of the sequences containing the best residues.178 First, they focused on enhancing CAL binding efficacy. With this strategy, they selected tert-butyl-l-alanine (Tle) as the P0 residue, which increased the binding affinity by 2.7-fold and proteolytic stability due to its bulky tert-butyl side chain. At the P-3 position, l-penicillamine (Pen) was incorporated, yielding a more conformationally defined disulfide bond, whereas at P-6 Phe was replaced with a larger hydrophobic 3-(2-naphthyl)-l-alanine (2-Nal), resulting in a 5-fold increase in CAL affinity. Concerning the CPP sequence, in order to optimize cell permeability and proteolytic stability, the number of arginine residues was reduced from four to three, and at the P-9 position a d-arginine was incorporated, yielding higher cytosolic entry efficacy as determined by flow cytometry. To further enhance peptide 42 CAL selectivity over NHERF, Gln at P-4 and Trp at P-5 were replaced by pipecolic acid (Pip) and 3-(3-benzothienyl)-l-alanine (Bta), respectively. All of these modifications ultimately produced the disulfide-cyclized peptide PGD97 (43, Figure 19A), which showed great cellular entry efficacy and high stability in human serum compared to the parent peptide 42. Furthermore, the linear form of 43, peptide 44 (Figure 19B), was highly potent and selective, with Kd = 6 nM and ≥130-fold selectivity for CALP vs NHERF. To gain insight about the structural basis of the exceptional binding affinity of peptide 44, the authors analyzed its predicted binding mode with CALP (Figure 20). In particular, the C-terminal carboxylate of Tle (P0) could form key hydrogen bonds with the backbone amides of Leu299, Gly300, and Ile301, while the tert-butyl side chain of the same residue could interact with an adjacent hydrophobic area. Instead, Pip at P-4 might facilitate the peptide to assume an optimal conformation that might position the benzothienyl ring of Bta at P-5 for a critical π–π interaction with His309. Biological evaluation of 43 indicated that it strongly increased the surface expression, stability, and function of F508del-CFTR in CFBE41o- cells. Furthermore, in CF-patient-derived HBE cells, 43 increased F508del-CFTR ion channel activity by ∼3-fold (EC50 ∼ 10 nM) and further potentiated the therapeutic effect of the known corrector 3 by ∼2-fold.178 All of these data demonstrated that the authors successfully developed a drug-like cyclized peptidyl molecule as a potent, selective, and with high proteolytic stability inhibitor of CAL-CFTR PPI. This creates interest in further optimizing 43 for clinical trial evaluations and in developing other peptidyl inhibitors to rescue F508del-CFTR PM stability.
Figure 20.

Diagram showing the key interactions between peptide 44 and the CFTR associated ligand (CAL) PDZ domain. Pink arrows indicate hydrogen bonds, and a green line indicates π–π stacking interaction. PDB from Dougherty et al.178
Noteworthy, there have also been efforts to develop small molecule inhibitors of this PPI. Madden and co-workers performed a comparative HTS using peptide 40 as control and either FRET or AS proximity assays as primary screen.179 Of the 3161 tested chemical compounds of the St. Jude bioactive collection, 12 hits were identified with both approaches, and among them, HSQC footprints of the CALP identified two compounds giving residue-specific chemical-shift perturbations. One of them, the methyl-3,4-dephostatin MD (45, Figure 21) did not exhibit cytotoxic and cytostatic effects when applied to F508del-CFBE monolayers, but unfortunately, it failed to increase F508del-CFTR chloride current in Ussing Chamber experiments when tested in the same cell model. Crystallographic and NMR studies showed that 45 could interact in a distinct site than the canonical peptide-binding domain of CALP. Further investigations revealed that the catechol 45 and its close analogue ethyl-3,4-dephostatin ED (46, Figure 21) might function by covalently binding to CAL by forming a cysteine adduct. Therefore, despite 45 and 46 representing the first example of small molecules able to regulate PDZ-CFTR interaction, their utility as drug scaffolds remains limited because of their ability to covalently modify proteins. Moreover, 45 and 46 are likely to be pan-interference compounds (PAINS)180 and exhibited involvement in several regulation pathways, which might lead to undesired off-target effects.179,181−183
Figure 21.
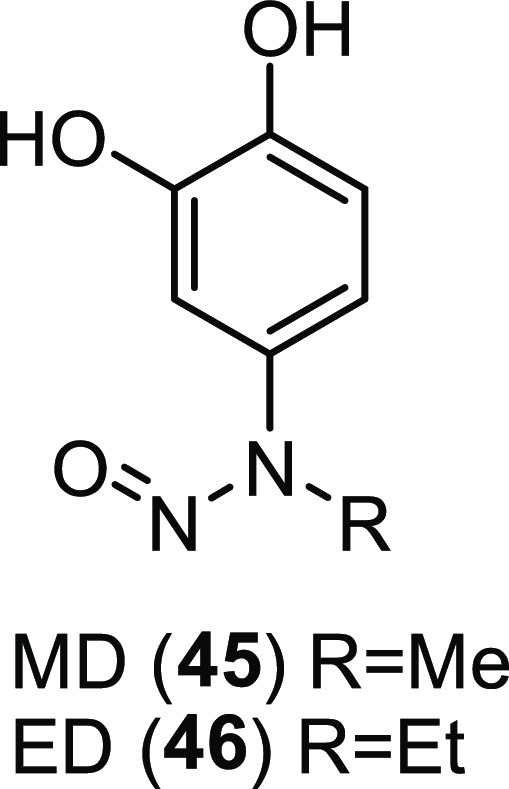
Chemical structures of the catecholic compounds MD (45) and ED (46).
7. Additional Pharmacological Strategies under Investigation
7.1. Restoring Defective Autophagy
There is an emerging interest for autophagy modulating compounds in controlling pathogenesis of CF disease,184 although this field of research remains controversial for many reasons. Raia and co-workers have been some of the major exponents of the research aimed at finding autophagy inducers as CF therapeutics. They initially demonstrated that human CF airways are autophagy-deficient, a condition that leads to decreased clearance of aggresomes (misfolded proteins aggregates).185,186 Autophagy is a key process in cellular clearance of protein aggregates and removal of ROS sources.187 Dysfunctional F508del-CFTR is believed to induce the generation of ROS that lead to an increase of the activity of profibrotic tissue transglutaminase 2 (TG2).186 The increase in TG2 activity, in turn, drives the sequestration of beclin-1 and the corresponding accumulation of p62, two key proteins in autophagosome formation.188 These events trap misfolded CFTR at the ER level, thus leading to rapid F508del-CFTR degradation and decreased trafficking to the PM. To investigate whether the restoration of autophagy could revert these sequential events and allow rescue of F508del-CFTR trafficking, the same authors showed that, by overexpressing beclin-1 or knocking down p62, the level of F508del-CFTR at the cell surface increased. In addition, the TG2 inhibitor cysteamine (47, Figure 22A), already approved for the treatment of orphan disease cystinosis, had similar results, thus partially restoring in vivo expression of beclin-1 and slightly increasing PM expression of F508del-CFTR at a concentration of 250 μM in nasal epithelial cells from CF patients.186,189 Although the effect of 47 was modest and a high concentration was required to activate autophagy, the authors moved forward by performing a phase 2 pilot clinical study with 10 F508del-CFTR homozygous patients. Their results showed that the combination of 47 and the natural epigallocatechin 3-gallate (EGCG, 48, Figure 22A)190 activated autophagy and improved mutant CFTR function. They speculated that the addition of 48 could modulate a different related pathway, such as the protein kinase CK2 involved in proteolytic degradation of mutant CFTR. Therefore, by inhibiting CK2, 48 might increase F508del-CFTR stability at the PM after 47-mediated rescue.189,191,192 However, to our knowledge, the effect of 47 alone or in combination with 48 on F508del-CFTR rescue has not been confirmed by other researchers to date. Indeed, three different research groups could not detect F508del-CFTR functional rescue using the same concentration of 47 in well-differentiated HBE cells, and among them, our group even reported deleterious effects on CFTR expression and activity after treatment with 47/48 combination.193−195 Similarly, despite promising initial data in cell and animal models, 48 failed in clinical trials for different proteinopathies.196 One of the reasons for its negative outcomes could be the lack of a clear understanding of the mechanism of action for 48 and its critical molecular targets.196 These results raise some concern about the activity of the autophagy modulator 47 and its combination with the antioxidant 48 and call for other confirmational evidence. Furthermore, 47 administration comes with many problems, such as the low potency, unpleasant thiol smell and taste, and short half-life. The chronic treatment with high doses of 47 is therefore not feasible and can lead to undesirable off-target and side effects.
Figure 22.

Autophagy activators in study as F508del-CFTR proteostasis regulators. (A) Chemical structures of cysteamine (47) and EGCG (48). (B) Strategy for 47 potency and stability optimization to obtain CAT-5571 (51).193
In order to find a more potent, yet safe and effective autophagy activator for further proof-of-concept studies, Liu and co-workers used their expertise in fatty acid conjugates to synthesize a covalent conjugate of 47 and docosahexaenoic acid (DHA, 49, Figure 22B),193 a ω-3 fatty acid that previously demonstrated to induce autophagy.197,198 With this strategy, the authors believed that the conjugate 50 (Figure 22B) could allow delivery of equimolar concentrations of 47 and 49 inside the cells, thus enabling synergism of the two bioactive components in terms of autophagy activation. Further biological experiments showed that 50 could increase beclin-1 levels and activate autophagy at concentrations (3 μM) lower than those of 47 in primary homozygous F508del-CFTR HBE cells. Interestingly, the individual components (47 and 49) either alone or in combination were not able to replicate the same level of activation, even at concentration of 250 μM. When compound 50 was used in triple combination with the potentiator 1 and the corrector 2, an additive effect was obtained, as evinced from immunoblot and F508del-CFTR chloride conductance assays. However, the authors reported solubility issues of 50 at concentrations greater than 3 μM, and a wide range of responses were observed depending on the primary CF-HBE cells used. Furthermore, conjugate 50 showed intrinsic instability, perhaps due to the sulfhydryl moiety. To enhance the stability of 50, the authors synthesized a small set of analogues, by converting the sulfhydryl group to an amide or a disulfide bound carrying different functionalities. Among them, conjugate CAT-5571 (51, Figure 22B) with a sterically hindered methylpropylnicotinamide moiety adjacent to the disulfide bound showed improved stability in rat, mouse, dog, and human plasma and was orally available due to a self-emulsifying dispersion formulation.193 Moreover, the authors reasoned that the new potent and bioavailable autophagy activator 51 could have a significant impact on intracellular clearance of bacteria, too, which is highly recommendable in CF patients that are subjected to chronic lung bacterial infections. Therefore, 51 has the characteristics for representing a potential new proteostasis regulator with multiple therapeutic effects and for that was included in preclinical studies at Catabasis Pharmaceuticals. Treatment with 51 in vitro and in vivo efficiently restored autophagy and caused a significant reduction in the intracellular bacterial load of Pseudomona aeruginosa and Burkholderia cenocepacia.199 In addition, 51 was able to enhance cell-surface trafficking and function of F508del-CFTR in combination with 1 and 2.199 At present, Catabasis is conducting a preclinical study in collaboration with the Bill & Melinda Gates Medical Research Institute to evaluate 51 as a potential oral therapy to promote autophagy and clear persistent lung infections in patients with both drug-sensitive and drug-resistant tuberculosis (TB).200
Recently, Romani et al. investigated the effect on F508del-CFTR rescue of thymosin alpha 1 (Tα1, 52), a well-known polypeptide in immunotherapy with sequence Ser-Asp-Ala-Ala-Val-Asp-Thr-Ser-Ser-Glu-Ile-Thr-Thr-Lys-Asp-Leu-Lys-Glu-Lys-Lys-Glu-Val-Val-Glu-Glu-Ala-Glu-Asn.20152 is commercialized under the trade name of Zadaxin for the treatment of viral infections, immunodeficiencies, malignancies, and HIV/AIDS.202 Its mechanism of action involves the induction of the immunoregulatory enzyme indoleamine 2,3-dioxygenase 1 (IDO1) in the bronchial epithelium, thus potentiating immune tolerance in the lung, reducing inflammation and activating autophagy.203 In accordance with their previous works on 47, the authors believed that, through promoting autophagy, 52 could affect trafficking and expression of F508del-CFTR. Therefore, they performed in vitro and in vivo experiments reporting that 52 could suppress inflammation and at the same time rescue F508del-CFTR maturation, stability, and activity in CFBE41o- cells, primary HBE cells, and in a CF murine model.201 Furthermore, they reported that 52 also increased the chloride current of the calcium-activated chloride channel (CaCC) in three of five patients examined, thus promoting a compensatory chloride secretion. For all of these reasons, the authors proposed 52 as a single-molecule-based therapy for CF patients with F508del.201,204 Since then, our group along with five other independent CF research groups have been trying to reproduce these results but failed to obtain any correction of F508del-CFTR or activation of CaCC in several bronchial cell cultures and using different measurement protocols.195,205,206 All of these results do not exclude the beneficial effects of 52 on the immune system and inflammation but call into question its use as a CFTR proteostasis regulator.
A further noteworthy study in this field is represented by the work of Coppinger and co-workers. Using protein interaction profiling and global bioinformatics analysis, these authors identified PI3K/Akt/mTOR as an additional signaling pathway involved in autophagy that could be targeted to obtain F508del-CFTR rescue.207 This axis has a central role in cell growth and stress response,208,209 with the serine/threonine kinase rapamycin (mTOR) tightly regulating autophagosome formation and Akt directly phosphorylating beclin-1.210,211 The authors’ analysis showed that mTOR activity was up-regulated in F508del CFBE41o- cells and therefore hypothesized that small molecule inhibitors of the PI3K/Akt/mTOR complex could be useful to promote F508del-CFTR stability and function. For that, a small set of six known inhibitors targeting different components of the pathway were purchased from commercial sources and tested for their effect on F508del-CFTR and on defective autophagy restoration in F508del CFBE41o- cells. Among them, the mTOR/Akt inhibitor MK-2206 (53, Figure 23) displayed the highest increase in mutant CFTR maturation, stability, and expression and strong induction of autophagy. Furthermore, PI3K/Akt/mTOR inhibition also decreased the levels of BAG3, a cochaperone of the Hsp70/Hsc70 complex involved in the autophagic degradation of misfolded protein aggregates. Thus, the authors speculated that the mechanism of action of 53 might involve the activation of autophagy through inhibition of both PI3K/Akt/mTOR and BAG3 axes, which, in turn, could decrease the sequestration of mutant CFTR into aggregates and lead to channel rescue to the PM. However, PI3K/Akt/mTOR inhibitors are associated with several off-targets and side effects, such as severe hepatotoxicity and pneumonitis, which have restricted their application and clinical significance.212,213 This limits possible use of 53 in CF therapy and suggests that further studies are needed to address its utility as a proteostasis regulator scaffold.
Figure 23.

Chemical structure of the mTOR/Akt inhibitor MK-2206 (53).
7.2. Targeting Kinases
Protein kinases are involved in several cellular pathways and processes, including CFTR degradation, where they presumably affect specific chaperones that normally control the maturation of the protein (e.g., Hsp70/Hsc70, Hsp90 chaperone complexes). Rotin and co-workers have been working for many years on the identification of the kinases that could play a role in CFTR processing and if their inhibition could lead to rescue of F508del-CFTR function. First, using a high-content screening protocol, the authors performed a kinase inhibitor screen with a library of 231 compounds, including FDA-approved drugs or compounds in clinical trials for the treatment of different diseases, mainly cancer and inflammation.214 The kinase inhibitors were purchased from different vendors or synthesized when not commercially available and were chosen to target as many kinases as possible. In vitro treatment with several compounds resulted in an increase of the F508del-CFTR activity. The 41 inhibitors showing the higher F508del-CFTR rescue were further validated by biochemical and electrophysiological techniques in different cell types, including primary HBE cells from CF patients. The results showed that several compounds increased F508del-CFTR maturation, expression, and activity at nanomolar concentrations. In particular, inhibitors of the receptor Tyr kinases (FGFRs, VEGFR, and PDGFR), such as the indolinone SU5402 (54, Figure 24) and its analogue SU6668 (55, Figure 24), already in clinical trials mainly for cancer therapy, showed a robust rescue of the mutant CFTR channel.214 However, how the identified kinase inhibitors could possibly rescue F508del-CFTR needed further investigations.
Figure 24.

Chemical structures of some of the hit kinase inhibitors identified by Rotin et al. and Bruchez et al. that showed an enhancement of F508del-CFTR rescue.214,216
In order to complement the small molecule screen, in 2015, the same authors performed an additional kinome screen with a library of endoribonuclease-prepared short interfering RNAs (esiRNA)-targeting 759 different kinases.157 With this strategy, several genes were identified eliciting significant F508del-CFTR rescue when knocked down, including those of the previously reported FGFR pathway. Indeed, silencing of FGFRs and their downstream proteins further validated the involvement of this signaling cascade in F508del-CFTR trafficking and maturation. Treatment of F508del-CFTR mice or intestinal organoids generated from the same animals with the FGFR1 inhibitor 54 led to a promising rescue of the mutant channel. In addition, in vitro combination of 54 with the corrector 2 resulted in an additive effect, suggesting that the two bioactive molecules might have different mechanisms of action. Indeed, the authors claimed that FGFR inhibition by 54 might regulate different chaperones involved in F508del-CFTR maturation, as evinced from chaperone array analysis.157 However, 54 exhibits significant off-target activities, such as the inhibition of several tyrosine kinases different from FGFR1.215 Thus, 54 is far from having a possible therapeutic value for the treatment of CF, and the development of more specific and potent analogues is pivotal for further investigations.
More recently, Bruchez and co-workers used an innovative HTS methodology to identify druggable kinases able to enhance F508del-CFTR trafficking and stability at the cell surface.216 Indeed, they previously developed and validated a fluorogen-activating protein (FAP)-based platform for quick and selective detection of F508del-CFTR surface expression and overall protein content onto the PM.217,218 Using this technology, the authors performed the screening of the siRNA kinase library (Dharmacon ON-TARGETplus SMARTpool) targeting 715 different kinases, with or without corrector 2 co-treatment.216 In brief, to determine the ratio of exposed F508del-CFTR channels over the total protein expressed, the authors used different types of fluorogens to label first the surface and then the intracellular CFTRs present in HEK293 cells expressing FAP-F508del-CFTR. In particular, the malachite green sulfonated analogue MG-B-Tau was used for CFTR surface labeling, while for intracellular CFTRs, a novel type of cell-permeable dye was synthesized, the n-butylamine MGnBu. When the FAP protein fused to F508del-CFTR binds to the fluorogens, the dye becomes fluorescent and can be revealed. Interestingly, with this technology, several target kinases were identified, whose suppression showed an increase in F508del-CFTR surface density in combination with treatment with 2. Among them, kinases CAMKK1 and RAF1 were the most promising targets. Further validation with selective RAF1 and CAMKK1 inhibitors, GW5074 (56, Figure 24) and STO609 (57, Figure 24), respectively, showed significant F508del-CFTR rescue compared to 2 treatment alone, as measured by flow cytometry. These results confirmed the sensitivity and robustness of the high-throughput methodology proposed by the authors, paving the way for its application to different cellular targets in future. Although the identification of druggable targets maintains a role of primary importance, fewer efforts have been done to date to discover new drug-like molecules acting as kinase inhibitors able to rescue mutant CFTR defects. Once this gap will be filled with new proof-of-concept studies, we believe that a better understanding of the role and consequences of these kinases inhibition will be available and may guide new CF research projects.
7.3. Targeting Histone Deacetylases HDACs
Histone acetyl transferases (HATs) and deacetylases (HDACs) have been extensively studied for their role in modulating transcriptional events implicated in several human diseases.219 These enzymes catalyze the post-translational addition or removal of acetyl functional groups to histones, transcription factors, and other cytosolic components (e.g., the chaperon Hsp90), thus regulating their activity.219,220 Histone deacetylase inhibitors (HDACi) serve as transcriptional activators, and they have been proven to be beneficial in many pathological conditions, as reported in several recent reviews.221−223 Mammalian genome encodes 11 HDACs categorized into four different classes, but the precise role of each isoform in cellular function is not yet completely understood.224 HDACi have represented useful tools to help clarify these issues, as they inhibit different HDACs mostly by interacting with their catalytic sites, leading to decreased deacetylation of histones and proteins. Moreover, HDACi could be potentially useful to influence misfolding protein maturation and function, such as F508del-CFTR, through either activating transcription of CFTR-related genes or modulating the activity of proteins involved in CFTR processing.
Balch and co-workers showed that the suberoylanilide hydroxamic acid (SAHA, trade name Vorinostat, 58, Figure 25A), the first FDA-approved HDACi for T-cell lymphoma treatment,225 yielded a notable correction of F508del-CFTR trafficking defect in the human lung CFBE41o- cell line.226 To identify the specific target of 58, siRNA screening of several types of HDACs was performed, revealing that HDAC7 could be the key enzyme involved in 58-mediated F508del-CFTR trafficking and activity rescue.226 Thus, the authors claimed that HDAC7 could be a viable pharmacological target for mutant CFTR correction in CF, even though 58 showed a very modest increase of F508del-CFTR activity in primary HBE cells. Hence, our group227 along with Bergougnoux et al.228 independently reported that 58 was not able to increase F508del-CFTR maturation and chloride secretion in primary airway epithelial cells from CF patients227 or in an ex vivo model of differentiated nasal cells obtained by scraping from CF patients.228 These different observed effects on the F508del-CFTR chloride channel could be explained by alterations in the baseline chromatin state between primary bronchial epithelial cells and immortalized cell lines. Therefore, these results suggest that the activity of 58 on F508del-CFTR rescue might have been overestimated from Balch and co-workers and call into question its efficacy and potential use for CF treatment.
Figure 25.
HDAC inhibitors in study as F508del-CFTR correctors. (A) Chemical structures of the known inhibitors SAHA (58), LBH-589 (61), and FK-228 (62). (B) Strategy for apicidin (59) F508del-CFTR corrector efficacy optimization to obtain the cyclic tetrapeptide 60.229
Balch and co-workers further expanded their research on CFTR proteostasis regulators with an epigenetic mechanism by focusing on synthetic conformationally biased cyclic tetrapeptides related to the natural compound apicidin (59, Figure 25B), a known HDAC inhibitor.229 Cyclic tetrapeptide inhibitors are characterized by the presence of functionally critical Zn2+-coordinating chains containing terminal α,β-epoxyketone, ethylketone, amide, or carboxylic acid. These unnatural amino acids can be considered isosteres of acetylated Lys and therefore may interact with the HDAC zinc-binding domain by mimicking an acetylated Lys residue of a substrate protein.230 For this property, cyclic tetrapeptides and pseudotetrapeptides have been extensively studied for their strong HDAC inhibition profile.230−232 Indeed, the authors screened for their ability to rescue F508del-CFTR trafficking a library of cyclic tetrapeptide HDAC inhibitors designed to cover a broad range of pharmacophore configurations. The compounds of interest were synthesized by varying each one of the four amino acids, through amide alkylation, addition of side chain functionalities or zinc coordinating moieties, and introduction of β-amino acids or triazoles in the backbone. Among them, the 13-membered ring tetrapeptide 60 (Figure 25B) showed efficacy as the F508del-CFTR corrector higher than that of the parent apicidin 59 and compared to the previously evaluated inhibitor 58, with levels of mutant CFTR rescue of nearly 40%. However, full HDAC inhibition profiling showed that 58 was a more potent and broad HDACs inhibitor than compound 60, although 60 was a better F508del-CFTR corrector compared to 58.229 From these results, the relationship between HDAC inhibition and F508del-CFTR rescue to the cell surface appears questionable, although cyclic tetrapeptide 60 may represent a novel type of CFTR corrector and further structure–activity optimizations may be explored in the future.
More recently, Balch and co-workers examined the impact of three additional FDA-approved HDACi on F508del-CFTR trafficking and function rescue in F508del-CFBE cells and in primary HBE cells from F508del homozygous patients.233 Two of the tested compounds, the indole LBH-589 (trade name Panobinostat, 61, Figure 25A) and the cyclic peptide derivative FK-228 (trade name Romidepsin, 62, Figure 25A) restored the functionality and PM expression of the mutant channel, and their effects were synergized with the corrector 2. Interestingly, the authors further evaluated the effects of 61 and 62 on the rescue of additional types of CFTR mutants carrying less common class II and class III variants, which are responsible for differential onset and progression of CF disease. Although the two molecules gave different responses with respect to the CFTR variants taken into consideration, they could provide functional correction of the mutant proteins, both alone and in combination with 2. Thus, the authors speculated that, by globally modulating the expression or activation of multiple cellular pathways, HDACi might facilitate the folding of different CFTR variants.233 Taken all together, HDAC inhibition remains a promising and questionable field of interest in CF research and ought to be further investigated by other independent researchers.
8. Challenges and Opportunities in Developing New Proteostasis Regulators
To date, numerous potential proteostasis regulator drug targets have been identified through genetic screening, proteomic and interactomic studies, and combinational approaches.15,48−51 However, this has yet to correlate with improved medicines in the clinic, with only the amplifier 8 reaching early clinical trials and the autophagy activator 51 being under preclinical evaluation.
From this overview, it appears that proteostasis regulator effects are additive with other correctors and therefore are expected to have a higher therapeutic ceiling and expand pharmacological treatment applicability to CF patients bearing mutations poorly responsive to already developed modulators. In particular, the most promising proteostasis regulators are those showing additive effects over the triple combination drug (Trikafta), which is by far the most effective treatment available—at the moment—for CF patients bearing eligible genotypes. However, most proteostasis regulators have been discovered in the pre-4 era, thus their efficacy in combination with approved CFTR modulators has been evaluated mainly toward 2 only. Investigating the efficacy of proteostasis modulators in combination with 4/3 as well as the identification of those more effective on mutations poorly responsive to approved drugs could be helpful to prioritize them for further development.
Few research groups undertook the challenging path of evaluating novel proteostasis regulators on CF-causing mutations other than F508del. Perhaps this is due to differences in the CFTR interacting networks among the various CFTR genotypes, which may be responsible for different responses to proteostasis regulators treatment. Nevertheless, a possible use of such approaches to rescue less common CFTR mutants has much potential for CF, meeting a need that the CFTR pharmacotherapy from the previous 20 years has struggled to fill.
One of the largest barriers to the development of more drugs acting as CFTR proteostasis regulators is the complexity of CFTR regulome. For example, some target proteins may have pleiotropic effects on CFTR processing, such as the Hsc/Hsp70 and Hsp90 complexes that are implicated in both degradation and folding of mutant CFTR.66 Therefore, targeting these pathways may fall into the risk of unbalancing the delicate processes of CFTR biogenesis and rescue. Furthermore, both Hsc70 and Hsp90 systems have a crucial role in cell signaling and protein homeostasis, and therefore, their pharmacological manipulation hides many challenges at first glance, with many opportunities for toxicity. Many research groups and companies have studied Hsp90 and Hsc70 inhibitors with successful results over the past few decades.99,234 Taking as example Hsp90 inhibitors, most of the clinical trials that were initiated have been terminated due to lack of efficacy and severe off-target effects such as hepatotoxicity and ocular toxicity. Consequently, none has been approved by FDA until now.235,236 Additionally, Hsp90 and Hsc70 chaperone systems function together as a multiprotein dynamic complex.237 For that, the intervention with PPI inhibitors is complicated due to the highly dynamic interactions between Hsp90/Hsc70 and their cochaperones.
Other strategies under investigation, such as targeting E1 or DUBs, are a relatively nonspecific way to achieve mutant CFTR rescue and, for this reason, may not be amenable to therapeutic development. Indeed, inhibiting E1 might block the degradation of all proteins targeted by the UPS, leading to undesirable off-target effects. Additionally, the alteration of the ubiquitination/deubiquitination process might lead to the dysregulation of those biological networks in which ubiquitination plays an important regulatory role. Conversely, given the selectivity of ubiquitination provided by E3s, targeting RNF5 may overcome the specificity issues encountered with E1 inhibitors and DUB activators. Thus, inhibiting RNF5 ligase is expected to affect only a limited set of substrates, leading to fewer side effects. However, RNF5 regulates the degradation of proteins involved in different cellular process, such as autophagy,238 inflammation,239 cell migration,240 and innate antiviral response,241 with possible pathophysiological implications. For that, the concept of inhibiting this E3 ligase to treat CF remains controversial, and further investigations are required to consider it as potential therapeutic.
Concerning CFTR stabilization on the PM, difficulties may arise due to the generally low binding selectivity of CFTR-associated PDZ domain proteins, such as CAL.242 PDZ proteins play a crucial role in several molecular and physiological pathways, such as signal transduction, cell polarity, cell adhesion, and protein trafficking, which have made PDZ proteins very attractive targets for drug discovery through the years.243 However, under this light, selectivity in targeting CAL appears to be a difficult but crucial task for a successful CF therapy, in order to decrease cross-reactivity and improve efficacy.
Some candidate proteostasis regulators, such as PARPi or HDACi, still have an unknown or under investigation mechanism of F508del-CFTR correction, which make the hit optimization campaign challenging. Further evidence is therefore required in order to gain in-depth knowledge about the therapeutic relevance of these new approaches and to anticipate possible side effects. Even those proteins acting on CFTR trafficking whose structures or binding sites are unknown represent CF drug discovery issues, as they make novel small molecules difficult to design.
While preparing this paper, we also noted that the suitability of the molecular target chosen is an important point of discussion in proteostasis regulator research. Many studies focused on targets found in cell models that were different from human airway cells, and for this reason, sometimes they failed to rescue mutant CFTR when tested in primary bronchial epithelial cell models. This typically happens because there might be differences in the QC mechanisms between cell lines and primary cell models. Thus, validation of novel corrector compounds in well-differentiated airway cells appears mandatory in order to have meaningful data of efficacy that could guide the drug discovery process and, more importantly, to prioritize those whose mechanism of action is more effective on disease-relevant cell models.
As shown throughout this review, a variety of mechanisms have been proposed to further enhance CFTR folding, trafficking, and function in a variety of systems and cell lines. However, the number of druggable targets belonging to the CFTR interactome and regulome that remain uninvestigated is high. To develop more effective drugs, a more complete understanding of them is necessary. For example, the interactions between CFTR and the PM it resides in has not been extensively explored. Lipids are known to have a large impact on other ABC transporters’ stability and function.244 Therefore, it is very likely that lipids modulate the function and stability of CFTR, too. It is well-documented that CF epithelial cells have an imbalance in the levels of polyunsaturated fatty acids (PUFAs), with reduced levels of DHA and increased levels of arachidonic acid (AA), which could lead to a proinflammatory state.245−247 CF cells also show increased levels of ceramide and glucosylceramide,248−251 which may contribute to chronic inflammation and increased susceptibility to P. aeruginosa seen in CF patients.252,253 Furthermore, CF cells expressing mutated CFTR channels are characterized by a decrease in glycosphingolipid GM1 content, which results in a diminished capacity for cell wound repair after injury.254,255 Mancini et al. recently demonstrated that the recovery of GM1 PM levels by its exogenous administration could stabilize the rescued F508del-CFTR protein and its PM interactome, leading to a significant improvement in the chloride transport of the mutant channel in association with 1 and 2.255 From these data, it appears that the CFTR protein needs a proper organization of the PM environment to exert its function, and that GM1 could play a key role therein. Restoring the PM proper composition is fundamental for CFTR stability and function. Indeed, Garić et al. demonstrated that the proinflammatory imbalance in fatty acids could be causally linked to the lack of functional CFTR at the PM, as the correction of CFTR protein deficiency normalized the imbalance among ceramide species.249 The authors also demonstrated that fenretinide (63, Figure 26), a synthetic analogue of retinoic acid in study for cancer therapy, could rebalance ceramide levels by down-regulating ceramide synthase 5 (Cers5) enzyme activity.249 In CFTR knockout mice, 63 decreased AA levels and simultaneously increased DHA levels, thus efficiently correcting the proinflammatory lipid imbalance.247 Additionally, 63 treatment significantly decreased bacterial burden upon infection of CF mice with P. aeruginosa.248 Due to its pleiotropic effects, the underlying mechanism of 63 is still unclear. Likewise, the mechanistic connection between lipid modulation by 63 and CFTR expression and function remains unexplored. Nevertheless, this evidence suggests that only a fine coordination of PM interactome creates the proper PM environment for innate immunity, host defense, CFTR stability, and activity, thus opening a new scenario toward developing new alternative treatments for CF.
Figure 26.

Chemical structure of fenretinide (63).
9. Future Prospects and Final Thoughts
CF is the most frequent among the autosomal recessive, lethal rare genetic diseases. Types and severity of symptoms can differ widely from person to person. While the CFTR genotype correlates with pancreatic status, the correlations between the CFTR genotype and lung and gastrointestinal phenotypes are debated, with CF patients bearing the same genotype displaying heterogeneous phenotypes.256 Over the last 30 years, advances in specialized CF care and in the CF therapeutic landscape have increased the expectancy and quality of life of people with CF. Industry and academic institutions have done extensive work to develop efficient CFTR potentiator and corrector molecules, as approximately 80% of CF patients can now benefit from one or more approved drugs. However, these therapies can only afford a partial rescue of the mutant CFTR channel,40 and their cost remains very high, leading to accessibility disparities worldwide. While for the F508del mutation, the efficacy of current corrector compounds, although partial, may be sufficient to achieve a marked clinical benefit,35,257 other mutations, displaying defective processing and trafficking to the PM, still lack effective therapies. In this way, proteostasis regulators shall have a wide applicability, as they target specific steps in CFTR processing that may create bottlenecks in its rescue. In addition, by addressing different cellular pathways, proteostasis regulators are likely to be useful tools to target protein trafficking diseases other than CF. For that, this innovative strategy holds great promise, but its application still has a long way to go. The way in which proteostasis regulators are implemented will be key to their success. First, it must be accepted that these compounds have effectiveness only in combination with other CFTR rescuing modulators. Indeed, cellular QC mechanisms are functionally redundant, and the modulation of one of those elements may not have a significant effect on the global biological outcome due to adaptive responses. In this way, proteostasis regulators alone will probably never reach levels of therapeutic efficacy equal to currently approved corrector/potentiator combinations. If a proteostasis regulator is to be developed, one must consider the greater potential for side effects of its underlying mechanism, as all the biological pathways involved in CFTR processing are interconnected. Alternatively, if proteostasis regulator compounds are implemented to specifically target those pathways that create the main bottlenecks in CFTR rescue, it is possible that through their combination with current therapies the dosage and therefore side effects of these treatments could be lowered. Additionally, many proteostasis regulators have shown positive secondary effects along with CFTR rescue activity, such as anti-inflammatory or innate host defense stimulatory effects against invading bacteria.199,258 Those pleiotropic effects, if well-calibrated, could have a great impact on CF patients suffering from chronic lung infections and inflammation. With these considerations in place, the future of proteostasis regulators could be bright, new research projects involved in drug-like molecules discovery will grow, and hopefully, this will be reflected into the clinic.
Future research should employ new cellular models that best represent CF disease and that can best mimic a given patient response to different treatments. This includes two-dimensional primary cultures from CF patients’ nasal brushing and three-dimensional organoids from airway or intestinal cells.259−261 Concerning the high costs and difficulties associated with drug development for this orphan disease, drug-repositioning technology may represent an auxiliary option to reduce both costs and likelihood of adverse effects. However, this field has to be considered complementary to up-to-date CF drug discovery pipelines. In this regard, the use of novel bioinformatics virtual screening protocols, such as high-performance computing (HPC), will allow the handling of large libraries of chemical compounds for new CFTR-rescuing hit identification, as recently seen for other diseases.262 Additionally, machine learning algorithm and artificial intelligence shall be exploited to speed up the extraction of results from omics data libraries and to power chemical synthetic processes.263
Given the need to develop novel CF treatment options, cross-disciplinary communication appears vital. CF foundations, companies, and academic laboratories have to find new ways to exchange knowledge and collaborate in order to encourage the exploration of previously uninvestigated targets. This will be fundamental for the expansion of the pioneering approach of CFTR proteostasis regulators in the future, which confidently will lead to new effective therapies for CF.
Acknowledgments
This work was supported by a grant from the Fondazione per la Ricerca sulla Fibrosi Cistica (FFC#09/2019), by the Istituto Italiano di Tecnologia (IIT) and the University of Bologna - Alma Mater Studiorum.
Glossary
Abbreviations
- 2-Nal
3-(2-naphthyl)-l-alanine
- AA
arachidonic acid
- Aha1
activator of the Hsp90 ATPase
- AS
AlphaScreen
- Az
Apoptozole
- BAG3
Bcl2-associated athanogene 3
- BHK
baby hamster kidney cells
- Bta
3-(3-benzothienyl)-l-alanine
- CaCC
calcium activated chloride channel
- CAL
CFTR-associated ligand
- CALP
CAL PDZ domain
- Cers5
ceramide synthase 5
- CF
cystic fibrosis
- CFBE
bronchial epithelial cells
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHIP
C-terminus of Hsc70-interacting protein
- CK2
casein kinase 2
- CME
clathrin-mediated endocytosis
- CPP
cell permeating peptide
- DHA
docosahexaenoic acid
- DUBs
deubiquitinating enzymes
- DUBTAC
deubiquitinase-targeting chimera
- EGCG
epigallocatechin-3-gallate
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum associated degradation
- ERQC
endoplasmic reticulum quality control
- esiRNA
endoribonuclease-prepared short interfering RNA
- FA
fluorescence anisotropy
- FAP
fluorogen-activating-protein
- FICT
fluorescein isothiocyanate
- FMP
FLIPR membrane potential assay
- FP
fluorescence polarization
- FRT
Fischer rat thyroid cells
- GSH
glutathione
- HATs
histone acetyl transferases
- HBE
primary human bronchial epithelial cells
- HDACs
histone deacetylases
- HDACi
histone deacetylase inhibitors
- Hsc
heat shock cognate
- Hsp
heat shock protein
- HS-YFP
halide-sensitive yellow fluorescent protein
- IDO1
indoleamine 2,3-dioxygenase 1
- NEF
nucleotide exchange factors
- NHERF
Na+/H+ exchanger regulatory factors
- NMD
nonsense-mediated mRNA decay
- PAINS
pan-interference compounds
- PARP
poly(ADPribose) polymerase
- PARPi
PARP inhibitor
- PCBP
poly-r(C) binding protein
- PDB
protein data bank
- Pen
l-penicillamine
- Pip
pipecolic acid
- PM
plasma membrane
- ppFEV1
percent predicted forced expiratory volume in 1 s
- PPI
protein protein interaction
- PROTACs
proteolitically targeting chimeras
- PUFA
polyunsaturated fatty acid
- RNF5
ring finger protein 5
- SAHA
suberoylanilid hydroxamic acid
- SGC
sulfagalactosylceramide
- SGL
sulfagalactolipid
- siRNA
small interfering RNA
- SRP
signal recognition particle
- TG2
transglutaminase 2
- Tle
tert-butyl-l-alanine
- TPD
targeted protein degradation
- TPS
targeted protein stabilization
- Tα1
thymosin alpha 1
- Ub
ubiquitin
- UPR
unfolded protein response
- UPS
ubiquitin-proteasome system
- F508del
deletion of phenylalanine 508
Biographies
Irene Brusa got her MSc degree in Chemistry and Pharmaceutical Technologies at University of Bologna in 2017. During her Master thesis, she spent a research period at the Ruhr-Universität Bochum (Germany) working on the synthesis and biological investigation of opioid peptide conjugates. In 2018, under the supervision of Prof. Andrea Cavalli (Italian Institute of Technology & UniBo) and Prof. Marinella Roberti (UniBo), she started her PhD in Medicinal Chemistry, focused on the design and synthesis of novel inhibitors of the ubiquitin-ligase RNF5 for cystic fibrosis disease.
Elvira Sondo joined the Laboratory of Medical Genetics of the Giannina Gaslini Institute in Genoa (Italy) in 2005. She obtained her PhD degree in Genetics in 2005 at the University Genoa. She has been enrolled in several projects funded by the Italian Cystic Fibrosis Foundation aimed at the identification of novel therapeutic strategies for cystic fibrosis. In particular, she focused her attention on CFTR pharmacology with the identification of CFTR correctors and potentiators by high-throughput methods and their evaluation in native epithelial cells. In 2019, she received the Gerd Doring Award for her contribution to cystic fibrosis research.
Federico Falchi obtained his PhD degree in Medicinal Chemistry in 2010 at “La Sapienza” University of Rome (Italy). He is a Senior Assistant Professor at the Department of Pharmacy and Biotechnology of the University of Bologna (Italy). His main areas of interest are virtual screening, pharmacophore modeling, 3D-QSAR, molecular docking, molecular dynamics, and drug design.
Nicoletta Pedemonte is a Principal Investigator at the Laboratory of Medical Genetics of the Giannina Gaslini Institute in Genoa (Italy), where she leads a group of seven researchers. She has a strong expertise in drug discovery strategies and functional genomics. Her research activity is focused on transepithelial ion transport mechanisms in airway epithelial cells, mechanisms associated with mutant protein maturation and degradation, and identification of novel small molecules to rescue mutant CFTR protein in cystic fibrosis. She has authored more than 80 publications in peer-reviewed journals, and she is an inventor in several international patents.
Marinella Roberti is Professor of Medicinal Chemistry at the University of Bologna. Her research field regards the design and the synthesis of biologically active small molecules, which can be either innovative lead candidates for drug discovery or valuable chemical tools to study molecular pathways. The therapeutic field of main interest is cancer and recently she has been involved in a project aimed to obtain new RNF5 inhibitors as correctors of the mutant CFTR chloride channel in cystic fibrosis. The scientific research activity is documented by more than 100 publications in peer-reviewed journals.
Andrea Cavalli is Professor of Medicinal Chemistry at the University of Bologna and Director of Computational and Chemical Biology at the Italian Institute of Technology, Genova, where he is also Deputy Director for Computational Sciences. Prof. Cavalli’s research has combined computational chemistry with drug discovery, focusing on neurodegenerative diseases, cancer, and neglected tropical diseases. In particular, he has been a pioneer in the use of molecular dynamics approaches to drug discovery. These new methods have led to the identification and characterization of lead candidates within the framework of multitarget drug discovery and polypharmacology. He is an author of more than 230 scientific papers and inventor in several international patents. He has delivered more than 120 invited lectures and seminars.
Author Present Address
⊥ Department of Pharmacy and Biotechnology, University of Bologna, 40126, Bologna, Italy
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Gadsby D. C.; Vergani P.; Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006, 440, 477–483. 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford B. CFTR structure: lassoing cystic fibrosis. Nat. Struct. Mol. Biol. 2017, 24, 13–14. 10.1038/nsmb.3353. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Liu F.; Chen J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 12757–12762. 10.1073/pnas.1815287115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cystic Fibrosis Mutation Database (CFTR1); http://www.genet.sickkids.on.ca/ (accessed 2022-02-06).
- CFTR2 Database; https://cftr2.org/ (accessed 2022-02-06).
- Bareil C.; Bergougnoux A. CFTR gene variants, epidemiology and molecular pathology. Arch. Pediatr. 2020, 27, eS8–eS12. 10.1016/S0929-693X(20)30044-0. [DOI] [PubMed] [Google Scholar]
- O’Sullivan B. P.; Freedman S. D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- Thibodeau P. H.; Richardson J. M. 3rd; Wang W.; Millen L.; Watson J.; Mendoza J. L.; Du K.; Fischman S.; Senderowitz H.; Lukacs G. L.; Kirk K.; Thomas P. J. The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2010, 285, 35825–35835. 10.1074/jbc.M110.131623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasevich I.; Yang Z. R.; Wang C.; Atwell S.; Zhao X.; Emtage S.; Wetmore D.; Hunt J. F.; Brouillette C. G. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010, 19, 1917–1931. 10.1002/pro.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. H.; Gregory R. J.; Marshall J.; Paul S.; Souza D. W.; White G. A.; O’Riordan C. R.; Smith A. E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Welch W. J. Role of quality control pathways in human diseases involving protein misfolding. Semin. Cell Dev. Biol. 2004, 15, 31–38. 10.1016/j.semcdb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T.; Veit G.; Dekkers J. F.; Bagdany M.; Soya N.; Xu H.; Roldan A.; Verkman A. S.; Kurth M.; Simon A.; Hegedus T.; Beekman J. M.; Lukacs G. L. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggino W. B.; Stanton B. A. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006, 7, 426–436. 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- Laselva O.; Guerra L.; Castellani S.; Favia M.; Di Gioia S.; Conese M. Small-molecule drugs for cystic fibrosis: Where are we now?. Pulm. Pharmacol. Ther. 2022, 72, 102098. 10.1016/j.pupt.2021.102098. [DOI] [PubMed] [Google Scholar]
- Sondo E.; Pesce E.; Tomati V.; Marini M.; Pedemonte N. RNF5, DAB2 and Friends: Novel Drug Targets for Cystic Fibrosis. Curr. Pharm. Des. 2017, 23, 176–186. 10.2174/1381612822666161006161033. [DOI] [PubMed] [Google Scholar]
- Li H.; Pesce E.; Sheppard D. N.; Singh A. K.; Pedemonte N. Therapeutic approaches to CFTR dysfunction: From discovery to drug development. J. Cyst. Fibros. 2018, 17, S14–S21. 10.1016/j.jcf.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Liu F.; Zhang Z.; Levit A.; Levring J.; Touhara K. K.; Shoichet B. K.; Chen J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. 10.1126/science.aaw7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh H.-I.; Qiu L.; Sohma Y.; Conrath K.; Zou X.; Hwang T.-C. Identifying the molecular target sites for CFTR potentiators GLPG1837 and VX-770. J. Gen. Physiol. 2019, 151, 912–928. 10.1085/jgp.201912360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinski S. V.; Ahmadi S.; Ip W.; Ouyang H.; Villella A.; Miller J. P.; Lee P. S.; Kulleperuma K.; Du K.; Di Paola M.; Eckford P. D. W.; Laselva O.; Huan L. J.; Wellhauser L.; Li E.; Ray P. N.; Pomès R.; Moraes T. J.; Gonska T.; Ratjen F.; Bear C. E. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. 10.15252/emmm.201607137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz S.; Sousa M.; Moraes B. J.; Mendes A. I.; Palma M.; Barreto C.; Fragata J. I.; Amaral M. D.; Matos P. HGF Stimulation of Rac1 Signaling Enhances Pharmacological Correction of the Most Prevalent Cystic Fibrosis Mutant F508del-CFTR. ACS Chem. Biol. 2013, 8, 432–442. 10.1021/cb300484r. [DOI] [PubMed] [Google Scholar]
- Alshafie W.; Chappe F. G.; Li M.; Anini Y.; Chappe V. M. VIP regulates CFTR membrane expression and function in Calu-3 cells by increasing its interaction with NHERF1 and P-ERM in a VPAC1- and PKCε-dependent manner. Am. J. Physiol. Cell Physiol. 2014, 307, C107–C119. 10.1152/ajpcell.00296.2013. [DOI] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Cao D.; Neuberger T.; Turnbull A.; Singh A.; Joubran J.; Hazlewood A.; Zhou J.; McCartney J.; Arumugam V.; Decker C.; Yang J.; Young C.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18825–18830. 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Stack J. H.; Straley K. S.; Decker C. J.; Miller M.; McCartney J.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. A. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18843–18848. 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratjen F.; Hug C.; Marigowda G.; Tian S.; Huang X.; Stanojevic S.; Milla C. E.; Robinson P. D.; Waltz D.; Davies J. C.; Rosenfeld M.; Starner T.; Retsch-Bogart G.; Chmiel J.; Orenstein D.; Milla C.; Rubenstein R.; Walker S.; Cornell A.; Asfour F.; Black P.; Colombo J.; Froh D.; McColley S.; Ruiz F.; Quintero D.; Casey A.; Mueller G.; Flume P.; Livingston F.; Rock M.; O’Sullivan B.; Schmidt H.; Lahiri T.; McNamara J.; Chidekel A.; Sass L.; Keens T.; Schaeffer D.; Solomon M.; Chilvers M.; Lands L.; Junge S.; Griese M.; Staab D.; Pressler T.; van Koningsburggen-Rietschel S.; Naehrlich L.; Reid A.; Balfour-Lynn I.; Urquhart D.; Lee T.; Munck A.; Gaudelus I. S.; De Boeck C.; Reix P.; Malfroot A.; Bui S.; Selvadurai H.; Robinson P.; Wainwright C.; Clements B.; Hilton J.; Hjelte L. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. 10.1016/S2213-2600(17)30215-1. [DOI] [PubMed] [Google Scholar]
- Milla C. E.; Ratjen F.; Marigowda G.; Liu F.; Waltz D.; Rosenfeld M. Lumacaftor/Ivacaftor in Patients Aged 6–11 Years with Cystic Fibrosis and Homozygous forF508del-CFTR. Am. J. Respir. Crit. Care Med. 2017, 195, 912–920. 10.1164/rccm.201608-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert D.; Chiron R.; Camara B.; Grenet D.; Prévotat A.; Bassinet L.; Dominique S.; Rault G.; Macey J.; Honoré I.; Kanaan R.; Leroy S.; Desmazes Dufeu N.; Burgel P.-R. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J. Cyst. Fibros. 2017, 16, 388–391. 10.1016/j.jcf.2017.03.003. [DOI] [PubMed] [Google Scholar]
- Jennings M. T.; Dezube R.; Paranjape S.; West N. E.; Hong G.; Braun A.; Grant J.; Merlo C. A.; Lechtzin N. An Observational Study of Outcomes and Tolerances in Patients with Cystic Fibrosis Initiated on Lumacaftor/Ivacaftor. Ann. Am. Thorac. Soc. 2017, 14, 1662–1666. 10.1513/AnnalsATS.201701-058OC. [DOI] [PubMed] [Google Scholar]
- Guerra L.; Favia M.; Di Gioia S.; Laselva O.; Bisogno A.; Casavola V.; Colombo C.; Conese M. The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis. Expert Opin. Drug Discovery 2020, 15, 873–891. 10.1080/17460441.2020.1750592. [DOI] [PubMed] [Google Scholar]
- Boyle M. P.; Bell S. C.; Konstan M. W.; McColley S. A.; Rowe S. M.; Rietschel E.; Huang X.; Waltz D.; Patel N. R.; Rodman D. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir. Med. 2014, 2, 527–538. 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- Donaldson S. H.; Pilewski J. M.; Griese M.; Cooke J.; Viswanathan L.; Tullis E.; Davies J. C.; Lekstrom-Himes J. A.; Wang L. T. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. 10.1164/rccm.201704-0717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. M.; Daines C.; Ringshausen F. C.; Kerem E.; Wilson J.; Tullis E.; Nair N.; Simard C.; Han L.; Ingenito E. P.; McKee C.; Lekstrom-Himes J.; Davies J. C. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha C. M.; King-Underwood J.; Sousa M.; Correia A. R.; Henriques B. J.; Roxo-Rosa M.; Da Paula A. C.; Williams J.; Hirst S.; Gomes C. M.; Amaral M. D. Revertants, Low Temperature, and Correctors Reveal the Mechanism of F508del-CFTR Rescue by VX-809 and Suggest Multiple Agents for Full Correction. Chem. Biol. 2013, 20, 943–955. 10.1016/j.chembiol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Veit G.; Xu H.; Dreano E.; Avramescu R. G.; Bagdany M.; Beitel L. K.; Roldan A.; Hancock M. A.; Lay C.; Li W.; Morin K.; Gao S.; Mak P. A.; Ainscow E.; Orth A. P.; McNamara P.; Edelman A.; Frenkiel S.; Matouk E.; Sermet-Gaudelus I.; Barnes W. G.; Lukacs G. L. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. 10.1038/s41591-018-0200-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G.; Roldan A.; Hancock M. A.; Da Fonte D. F.; Xu H.; Hussein M.; Frenkiel S.; Matouk E.; Velkov T.; Lukacs G. L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. 10.1172/jci.insight.139983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating D.; Marigowda G.; Burr L.; Daines C.; Mall M. A.; McKone E. F.; Ramsey B. W.; Rowe S. M.; Sass L. A.; Tullis E.; McKee C. M.; Moskowitz S. M.; Robertson S.; Savage J.; Simard C.; Van Goor F.; Waltz D.; Xuan F.; Young T.; Taylor-Cousar J. L. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy S. M. Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs 2019, 79, 2001–2007. 10.1007/s40265-019-01233-7. [DOI] [PubMed] [Google Scholar]
- Vertex Pharmaceuticals Incorporated . Positive Phase 3 Study Results for TRIKAFTA® (elexacaftor/tezacaftor/ivacaftor and ivacaftor) in People Ages 12 and Older With Cystic Fibrosis Who Have One Copy of the F508del Mutation and One Gating or Residual Function Mutation. Press release Jul 20, 2020; https://investors.vrtx.com/node/27601/pdf (accessed 2022-02-06).
- Wainwright C. E.; Elborn J. S.; Ramsey B. W.; Marigowda G.; Huang X.; Cipolli M.; Colombo C.; Davies J. C.; De Boeck K.; Flume P. A.; Konstan M. W.; McColley S. A.; McCoy K.; McKone E. F.; Munck A.; Ratjen F.; Rowe S. M.; Waltz D.; Boyle M. P. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijerman H. G. M.; McKone E. F.; Downey D. G.; Van Braeckel E.; Rowe S. M.; Tullis E.; Mall M. A.; Welter J. J.; Ramsey B. W.; McKee C. M.; Marigowda G.; Moskowitz S. M.; Waltz D.; Sosnay P. R.; Simard C.; Ahluwalia N.; Xuan F.; Zhang Y.; Taylor-Cousar J. L.; McCoy K. S.; McCoy K.; Donaldson S.; Walker S.; Chmiel J.; Rubenstein R.; Froh D. K.; Neuringer I.; Jain M.; Moffett K.; Taylor-Cousar J. L.; Barnett B.; Mueller G.; Flume P.; Livingston F.; Mehdi N.; Teneback C.; Welter J.; Jain R.; Kissner D.; Patel K.; Calimano F. J.; Johannes J.; Daines C.; Keens T.; Scher H.; Chittivelu S.; Reddivalam S.; Klingsberg R. C.; Johnson L. G.; Verhulst S.; Macedo P.; Downey D.; Connett G.; Nash E.; Withers N.; Lee T.; Bakker M.; Heijerman H.; Vermeulen F.; Van Braeckel E.; Knoop C.; De Wachter E.; van der Meer R.; Merkus P.; Majoor C. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capurro V.; Tomati V.; Sondo E.; Renda M.; Borrelli A.; Pastorino C.; Guidone D.; Venturini A.; Giraudo A.; Mandrup Bertozzi S.; Musante I.; Bertozzi F.; Bandiera T.; Zara F.; Galietta L. J. V.; Pedemonte N. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. 10.3390/ijms22105262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N. Small-molecule correctors of defective F508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanò V.; Barreca M.; Cilibrasi V.; Genovese M.; Renda M.; Montalbano A.; Galietta L. J. V.; Barraja P. Evaluation of Fused Pyrrolothiazole Systems as Correctors of Mutant CFTR Protein. Molecules 2021, 26, 1275. 10.3390/molecules26051275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassini S.; Sun L.; Lanko K.; Crespan E.; Langron E.; Falchi F.; Kissova M.; Armijos-Rivera J. I.; Delang L.; Mirabelli C.; Neyts J.; Pieroni M.; Cavalli A.; Costantino G.; Maga G.; Vergani P.; Leyssen P.; Radi M. Discovery of Multitarget Agents Active as Broad-Spectrum Antivirals and Correctors of Cystic Fibrosis Transmembrane Conductance Regulator for Associated Pulmonary Diseases. J. Med. Chem. 2017, 60, 1400–1416. 10.1021/acs.jmedchem.6b01521. [DOI] [PubMed] [Google Scholar]
- Tassini S.; Langron E.; Delang L.; Mirabelli C.; Lanko K.; Crespan E.; Kissova M.; Tagliavini G.; Fontò G.; Bertoni S.; Palese S.; Giorgio C.; Ravanetti F.; Ragionieri L.; Zamperini C.; Mancini A.; Dreassi E.; Maga G.; Vergani P.; Neyts J.; Radi M. Multitarget CFTR Modulators Endowed with Multiple Beneficial Side Effects for Cystic Fibrosis Patients: Toward a Simplified Therapeutic Approach. J. Med. Chem. 2019, 62, 10833–10847. 10.1021/acs.jmedchem.9b01416. [DOI] [PubMed] [Google Scholar]
- Bardin E.; Pastor A.; Semeraro M.; Golec A.; Hayes K.; Chevalier B.; Berhal F.; Prestat G.; Hinzpeter A.; Gravier-Pelletier C.; Pranke I.; Sermet-Gaudelus I. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. 10.1016/j.ejmech.2021.113195. [DOI] [PubMed] [Google Scholar]
- Ghelani D. P.; Schneider-Futschik E. K. Emerging Cystic Fibrosis Transmembrane Conductance Regulator Modulators as New Drugs for Cystic Fibrosis: A Portrait of in Vitro Pharmacology and Clinical Translation. ACS Pharmacol. Transl. Sci. 2020, 3, 4–10. 10.1021/acsptsci.9b00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanò V.; Montalbano A.; Carbone A.; Scudieri P.; Galietta L. J. V.; Barraja P. An overview on chemical structures as ΔF508-CFTR correctors. Eur. J. Med. Chem. 2019, 180, 430–448. 10.1016/j.ejmech.2019.07.037. [DOI] [PubMed] [Google Scholar]
- Liessi N.; Pedemonte N.; Armirotti A.; Braccia C. Proteomics and Metabolomics for Cystic Fibrosis Research. Int. J. Mol. Sci. 2020, 21, 5439. 10.3390/ijms21155439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub M. D.; McCray P. B. Jr. Transcriptomic and Proteostasis Networks of CFTR and the Development of Small Molecule Modulators for the Treatment of Cystic Fibrosis Lung Disease. Genes 2020, 11, 546. 10.3390/genes11050546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M. D.; Hutt D. M.; Tomati V.; Botelho H. M.; Pedemonte N. CFTR processing, trafficking and interactions. J. Cyst. Fibros. 2020, 19 (Suppl 1), S33–S36. 10.1016/j.jcf.2019.10.017. [DOI] [PubMed] [Google Scholar]
- Tomati V.; Pesce E.; Caci E.; Sondo E.; Scudieri P.; Marini M.; Amato F.; Castaldo G.; Ravazzolo R.; Galietta L. J. V.; Pedemonte N. High-throughput screening identifies FAU protein as a regulator of mutant cystic fibrosis transmembrane conductance regulator channel. J. Biol. Chem. 2018, 293, 1203–1217. 10.1074/jbc.M117.816595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Xiong X.; Helm A.; Kimani K.; Bragin A.; Skach W. R. Co- and Posttranslational Translocation Mechanisms Direct Cystic Fibrosis Transmembrane Conductance Regulator N Terminus Transmembrane Assembly. J. Biol. Chem. 1998, 273, 568–576. 10.1074/jbc.273.1.568. [DOI] [PubMed] [Google Scholar]
- Walter P.; Blobel G. Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981, 91, 551–556. 10.1083/jcb.91.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M. D. CFTR and Chaperones: Processing and Degradation. J. Mol. Neurosci. 2004, 23, 041–048. 10.1385/JMN:23:1-2:041. [DOI] [PubMed] [Google Scholar]
- Skach W. R. CFTR: New Members Join the Fold. Cell 2006, 127, 673–675. 10.1016/j.cell.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Wang X.; Venable J.; LaPointe P.; Hutt D. M.; Koulov A. V.; Coppinger J.; Gurkan C.; Kellner W.; Matteson J.; Plutner H.; Riordan J. R.; Kelly J. W.; Yates J. R.; Balch W. E. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Egan M. E. Genetics of Cystic Fibrosis. Clin. Chest Med. 2016, 37, 9–16. 10.1016/j.ccm.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Ong T.; Ramsey B. W. New Therapeutic Approaches to Modulate and Correct Cystic Fibrosis Transmembrane Conductance Regulator. Pediatr. Clin. North Am. 2016, 63, 751–764. 10.1016/j.pcl.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano K. A.; Wachi S.; Drew L.; Dukovski D.; Green O.; Bastos C.; Cullen M. D.; Hauck S.; Tait B. D.; Munoz B.; Lee P.-S.; Miller J. P. Use of a High-Throughput Phenotypic Screening Strategy to Identify Amplifiers, a Novel Pharmacological Class of Small Molecules That Exhibit Functional Synergy with Potentiators and Correctors. SLAS Discovery 2018, 23, 111–121. 10.1177/2472555217729790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukovski D.; Villella A.; Bastos C.; King R.; Finley D.; Kelly J. W.; Morimoto R. I.; Hartl F. U.; Munoz B.; Lee P.-S.; Zecevic M.; Miller J. P. Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. J. Cyst. Fibros. 2020, 19, 733–741. 10.1016/j.jcf.2020.02.006. [DOI] [PubMed] [Google Scholar]
- Xia M.; He H.; Wang Y.; Liu M.; Zhou T.; Lin M.; Zhou Z.; Huo R.; Zhou Q.; Sha J. PCBP1 is required for maintenance of the transcriptionally silent state in fully grown mouse oocytes. Cell Cycle 2012, 11, 2833–2842. 10.4161/cc.21169. [DOI] [PubMed] [Google Scholar]
- Silva I. A. L.; Ramalho A. S.; Vonk A. M.; Bierlaagh M.; Suen S. W. F.; Pott J.; Amaral M. D.; Vermeulen F.; De Boeck C.; Beekman J. M.; van der Ent K.. First results of the HIT-CF ex vivo organoid study show rescue of CFTR with ultra-rare mutations by a novel triple combination of CFTR modulators. In Abstracts of the 44th European Cystic Fibrosis Conference (ECFS 2021); Elsevier Inc., 2021; Vol. 20, p S17. [Google Scholar]
- Lopes-Pacheco M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. 10.3389/fphar.2019.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flume P.; Sawicki G.; Pressler T.; Schwarz C.; Fajac I.; Layish D.; Bialek P.; Wilson S.; Kang L.; Mclaughlin B.; Scafidi S.; Lee P.-S.; Gilmartin G.. Phase 2 initial results evaluating PTI-428, a novel CFTR amplifier, in patients with cystic fibrosis. In Abstracts of the 41st European Cystic Fibrosis Conference (ECFS 2018); Elsevier B.V.: Belgrade, Serbia, 2018; Vol. 17, pp S1–S2. [Google Scholar]
- Venturini A.; Borrelli A.; Musante I.; Scudieri P.; Capurro V.; Renda M.; Pedemonte N.; Galietta L. J. V. Comprehensive Analysis of Combinatorial Pharmacological Treatments to Correct Nonsense Mutations in the CFTR Gene. Int. J. Mol. Sci. 2021, 22, 11972. 10.3390/ijms222111972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J. C. The role of the cytosolic HSP70 chaperone system in diseases caused by misfolding and aberrant trafficking of ion channels. Dis. Model. Mech. 2014, 7, 319–329. 10.1242/dmm.014001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. E.; Hipp M. S.; Bracher A.; Hayer-Hartl M.; Ulrich Hartl F. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Pearl L. H.; Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006, 75, 271–294. 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M. P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. 10.1016/j.tibs.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Meacham G. C.; Lu Z.; King S.; Sorscher E.; Tousson A.; Cyr D. M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppinger J. A.; Hutt D. M.; Razvi A.; Koulov A. V.; Pankow S.; Yates J. R.; Balch W. E. A chaperone trap contributes to the onset of cystic fibrosis. PLoS One 2012, 7, e37682. 10.1371/journal.pone.0037682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Ward T. S.; Amaral M. D. Deletion of Phe508 in the first nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator increases its affinity for the heat shock cognate 70 chaperone. FEBS J. 2009, 276, 7097–7109. 10.1111/j.1742-4658.2009.07421.x. [DOI] [PubMed] [Google Scholar]
- Grove D. E.; Fan C. Y.; Ren H. Y.; Cyr D. M. The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol. Biol. Cell 2011, 22, 301–314. 10.1091/mbc.e10-09-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo-Kang L. R.; Zeitlin P. L. Induction of HSP70 promotes DeltaF508 CFTR trafficking. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L58–68. 10.1152/ajplung.2001.281.1.L58. [DOI] [PubMed] [Google Scholar]
- Bagdany M.; Veit G.; Fukuda R.; Avramescu R. G.; Okiyoneda T.; Baaklini I.; Singh J.; Sovak G.; Xu H.; Apaja P. M.; Sattin S.; Beitel L. K.; Roldan A.; Colombo G.; Balch W.; Young J. C.; Lukacs G. L. Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat. Commun. 2017, 8, 398. 10.1038/s41467-017-00444-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham G. C.; Patterson C.; Zhang W.; Younger J. M.; Cyr D. M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T.; Barrière H.; Bagdány M.; Rabeh W. M.; Du K.; Höhfeld J.; Young J. C.; Lukacs G. L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousaki A.; Miyata Y.; Jinwal U. K.; Dickey C. A.; Gestwicki J. E.; Zuiderweg E. R. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J. Mol. Biol. 2011, 411, 614–632. 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya K.; Li Y.; Wang H.; Ukai T.; Tatsuta N.; Kawakami M.; Shishido; Chen L. B. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996, 56, 538–543. [PubMed] [Google Scholar]
- Kim Chiaw P.; Hantouche C.; Wong M. J. H.; Matthes E.; Robert R.; Hanrahan J. W.; Shrier A.; Young J. C. Hsp70 and DNAJA2 limit CFTR levels through degradation. PLoS One 2019, 14, e0220984. 10.1371/journal.pone.0220984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Propper D. J.; Braybrooke J. P.; Taylor D. J.; Lodi R.; Styles P.; Cramer J. A.; Collins W. C. J.; Levitt N. C.; Talbot D. C.; Ganesan T. S.; Harris A. L. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo-resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. 10.1023/A:1008336904585. [DOI] [PubMed] [Google Scholar]
- Li X.; Srinivasan S. R.; Connarn J.; Ahmad A.; Young Z. T.; Kabza A. M.; Zuiderweg E. R. P.; Sun D.; Gestwicki J. E. Analogues of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, As Anti-Cancer Agents. ACS Med. Chem. Lett. 2013, 4, 1042–1047. 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H.; Li X.; Moses M. A.; Gilbert L. A.; Kalyanaraman C.; Young Z. T.; Chernova M.; Journey S. N.; Weissman J. S.; Hann B.; Jacobson M. P.; Neckers L.; Gestwicki J. E. Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein–Protein Interactions with Heat Shock Protein 70 (Hsp70). J. Med. Chem. 2018, 61, 6163–6177. 10.1021/acs.jmedchem.8b00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamelak D.; Lingwood C. The ATPase domain of hsp70 possesses a unique binding specificity for 3′-sulfogalactolipids. J. Biol. Chem. 2001, 276, 449–456. 10.1074/jbc.M006732200. [DOI] [PubMed] [Google Scholar]
- Boulanger J.; Faulds D.; Eddy E. M.; Lingwood C. A. Members of the 70 kDa heat shock protein family specifically recognize sulfoglycolipids: Role in gamete recognition and mycoplasma-related infertility. J. Cell. Physiol. 1995, 165, 7–17. 10.1002/jcp.1041650103. [DOI] [PubMed] [Google Scholar]
- Mamelak D.; Lingwood C. Glycoconj. J. 1997, 14, 715–722. 10.1023/A:1018569417218. [DOI] [PubMed] [Google Scholar]
- Mamelak D.; Mylvaganam M.; Whetstone H.; Hartmann E.; Lennarz W.; Wyrick P. B.; Raulston J.; Han H.; Hoffman P.; Lingwood C. A. Hsp70s Contain a Specific Sulfogalactolipid Binding Site. Differential Aglycone Influence on Sulfogalactosyl Ceramide Binding by Recombinant Prokaryotic and Eukaryotic Hsp70 Family Members. Biochemistry 2001, 40, 3572–3582. 10.1021/bi001643u. [DOI] [PubMed] [Google Scholar]
- Bukau B.; Horwich A. L. The Hsp70 and Hsp60 Chaperone Machines. Cell 1998, 92, 351–366. 10.1016/S0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Whetstone H.; Lingwood C. 3′Sulfogalactolipid binding specifically inhibits Hsp70 ATPase activity in vitro. Biochemistry 2003, 42, 1611–1617. 10.1021/bi026735t. [DOI] [PubMed] [Google Scholar]
- Park H. J.; Mylvaganum M.; McPherson A.; Fewell S. W.; Brodsky J. L.; Lingwood C. A. A soluble sulfogalactosyl ceramide mimic promotes Delta F508 CFTR escape from endoplasmic reticulum associated degradation. Chem. Biol. 2009, 16, 461–470. 10.1016/j.chembiol.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamani M.; Mylvaganam M.; Tian R.; Rigat B.; Binnington B.; Lingwood C. Adamantyl Glycosphingolipids Provide a New Approach to the Selective Regulation of Cellular Glycosphingolipid Metabolism. J. Biol. Chem. 2011, 286, 21413–21426. 10.1074/jbc.M110.207670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. R.; Lee M.-R.; Song Y.-A.; Ko S.-K.; Kim G.-H.; Shin I. Synthetic Small Molecules that Induce Neurogenesis in Skeletal Muscle. J. Am. Chem. Soc. 2007, 129, 9258–9259. 10.1021/ja072817z. [DOI] [PubMed] [Google Scholar]
- Williams D. R.; Ko S. K.; Park S.; Lee M. R.; Shin I. An apoptosis-inducing small molecule that binds to heat shock protein 70. Angew. Chem., Int. Ed. 2008, 47, 7466–7469. 10.1002/anie.200802801. [DOI] [PubMed] [Google Scholar]
- Cho H. J.; Gee H. Y.; Baek K. H.; Ko S. K.; Park J. M.; Lee H.; Kim N. D.; Lee M. G.; Shin I. A small molecule that binds to an ATPase domain of Hsc70 promotes membrane trafficking of mutant cystic fibrosis transmembrane conductance regulator. J. Am. Chem. Soc. 2011, 133, 20267–20276. 10.1021/ja206762p. [DOI] [PubMed] [Google Scholar]
- Ko S. K.; Kim J.; Na D. C.; Park S.; Park S. H.; Hyun J. Y.; Baek K. H.; Kim N. D.; Kim N. K.; Park Y. N.; Song K.; Shin I. A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem. Biol. 2015, 22, 391–403. 10.1016/j.chembiol.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Wisén S.; Bertelsen E. B.; Thompson A. D.; Patury S.; Ung P.; Chang L.; Evans C. G.; Walter G. M.; Wipf P.; Carlson H. A.; Brodsky J. L.; Zuiderweg E. R.; Gestwicki J. E. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of hsp70-hsp40. ACS Chem. Biol. 2010, 5, 611–622. 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang A. N.; Liang M.; Dominguez-Meijide A.; Masaracchia C.; Goeckeler-Fried J. L.; Mazzone C. S.; Newhouse D. W.; Kendsersky N. M.; Yates M. E.; Manos-Turvey A.; Needham P. G.; Outeiro T. F.; Wipf P.; Brodsky J. L. Synthesis and evaluation of esterified Hsp70 agonists in cellular models of protein aggregation and folding. Bioorg. Med. Chem. 2019, 27, 79–91. 10.1016/j.bmc.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Wang L.; You Q.-D.; Xu X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. 10.1021/acs.jmedchem.9b00940. [DOI] [PubMed] [Google Scholar]
- Loo M. A.; Jensen T. J.; Cui L.; Hou Y.; Chang X. B.; Riordan J. R. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998, 17, 6879–6887. 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulov A. V.; LaPointe P.; Lu B.; Razvi A.; Coppinger J.; Dong M. Q.; Matteson J.; Laister R.; Arrowsmith C.; Yates J. R.; Balch W. E. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. 10.1091/mbc.e09-12-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T. W.; Akinaga S.; Soga S.; Sullivan W.; Stensgard B.; Toft D.; Neckers L. M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100–108. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C. The ’active life’ of Hsp90 complexes. Biochim. Biophys. Acta 2012, 1823, 614–623. 10.1016/j.bbamcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Soroka J.; Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Siligardi G.; Hu B.; Panaretou B.; Piper P. W.; Pearl L. H.; Prodromou C. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 2004, 279, 51989–51998. 10.1074/jbc.M410562200. [DOI] [PubMed] [Google Scholar]
- Panaretou B.; Siligardi G.; Meyer P.; Maloney A.; Sullivan J. K.; Singh S.; Millson S. H.; Clarke P. A.; Naaby-Hansen S.; Stein R.; Cramer R.; Mollapour M.; Workman P.; Piper P. W.; Pearl L. H.; Prodromou C. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 2002, 10, 1307–1318. 10.1016/S1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- Li J.; Richter K.; Reinstein J.; Buchner J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol. 2013, 20, 326–331. 10.1038/nsmb.2502. [DOI] [PubMed] [Google Scholar]
- Shiau A. K.; Harris S. F.; Southworth D. R.; Agard D. A. Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Ali M. M. U.; Roe S. M.; Vaughan C. K.; Meyer P.; Panaretou B.; Piper P. W.; Prodromou C.; Pearl L. H. Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrig V.; Obermann W. M. J. Identifying Inhibitors of the Hsp90-Aha1 Protein Complex, a Potential Target to Drug Cystic Fibrosis, by Alpha Technology. SLAS Discovery 2017, 22, 923–928. 10.1177/2472555216688312. [DOI] [PubMed] [Google Scholar]
- Katsuhiro K.; Kazuo K.; Tetsunori F.; Chikanori M.; Takashi S.. Preparation of heterocyclic compounds as fungicides. Patent Appl. WO2005077948, 2005.
- Fish P. V.; Mahy W.; Willis N. J.; Woodward H.; Atkinson B. N.; Bayle E. D.; Sipthorp J.; Jones E. Y.; Zhao Y.; Vecchia L.; Ruza R. R.. Inhibitors of secreted carboxy esterase, Notum, and their use in increasing Wnt protein signalling and treatment of central nervous system and other disorders. Patent Appl. WO2020043866, 2020.
- Stiegler S. C.; Rübbelke M.; Korotkov V. S.; Weiwad M.; John C.; Fischer G.; Sieber S. A.; Sattler M.; Buchner J. A chemical compound inhibiting the Aha1-Hsp90 chaperone complex. J. Biol. Chem. 2017, 292, 17073–17083. 10.1074/jbc.M117.797829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J. K.; Hutt D. M.; Tait B.; Guy N. C.; Sivils J. C.; Ortiz N. R.; Payan A. N.; Komaragiri S. K.; Owens J. J.; Culbertson D.; Blair L. J.; Dickey C.; Kuo S. Y.; Finley D.; Dyson H. J.; Cox M. B.; Chaudhary J.; Gestwicki J. E.; Balch W. E. Management of Hsp90-Dependent Protein Folding by Small Molecules Targeting the Aha1 Co-Chaperone. Cell Chem. Biol. 2020, 27, 292–305. 10.1016/j.chembiol.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaze E. R.; Lambert A. L.; Smith A. C.; Page J. G.; Johnson W. D.; McCormick D. L.; Brown A. P.; Levine B. S.; Covey J. M.; Egorin M. J.; Eiseman J. L.; Holleran J. L.; Sausville E. A.; Tomaszewski J. E. Preclinical toxicity of a geldanamycin analog, 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG), in rats and dogs: potential clinical relevance. Cancer Chemother. Pharmacol. 2005, 56, 637–647. 10.1007/s00280-005-1000-9. [DOI] [PubMed] [Google Scholar]
- Iyer G.; Morris M. J.; Rathkopf D.; Slovin S. F.; Steers M.; Larson S. M.; Schwartz L. H.; Curley T.; DeLaCruz A.; Ye Q.; Heller G.; Egorin M. J.; Ivy S. P.; Rosen N.; Scher H. I.; Solit D. B. A phase I trial of docetaxel and pulse-dose 17-allylamino-17-demethoxygeldanamycin in adult patients with solid tumors. Cancer Chemother. Pharmacol. 2012, 69, 1089–1097. 10.1007/s00280-011-1789-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Kartner N.; Lukacs G. L. Limited proteolysis as a probe for arrested conformational maturation of delta F508 CFTR. Nat. Struct. Biol. 1998, 5, 180–183. 10.1038/nsb0398-180. [DOI] [PubMed] [Google Scholar]
- Kreda S. M.; Mall M.; Mengos A.; Rochelle L.; Yankaskas J.; Riordan J. R.; Boucher R. C. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell 2005, 16, 2154–2167. 10.1091/mbc.e04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R.; Okiyoneda T. Peripheral Protein Quality Control as a Novel Drug Target for CFTR Stabilizer. Front. Pharmacol. 2018, 9, 1100. 10.3389/fphar.2018.01100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L.; Dunn R. Regulation of Membrane Protein Transport by Ubiquitin and Ubiquitin-Binding Proteins. Annu. Rev. Cell Dev. Biol. 2003, 19, 141–172. 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Gelman M. S.; Kannegaard E. S.; Kopito R. R. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2002, 277, 11709–11714. 10.1074/jbc.M111958200. [DOI] [PubMed] [Google Scholar]
- Gelman M. S.; Kopito R. R. Rescuing protein conformation: prospects for pharmacological therapy in cystic fibrosis. J. Clin. Invest. 2002, 110, 1591–1597. 10.1172/JCI0216786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J. M.; Chen L.; Ren H. Y.; Rosser M. F.; Turnbull E. L.; Fan C. Y.; Patterson C.; Cyr D. M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Younger J. M.; Ren H. Y.; Chen L.; Fan C. Y.; Fields A.; Patterson C.; Cyr D. M. A foldable CFTR{Delta}F508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J. Cell Biol. 2004, 167, 1075–1085. 10.1083/jcb.200410065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo C. L.; Yu G. J.; Yang B.; Robins L. I.; Verkman A. S.; Kurth M. J. 4′-Methyl-4,5′-bithiazole-based correctors of defective delta F508-CFTR cellular processing. Bioorg. Med. Chem. Lett. 2008, 18, 2610–2614. 10.1016/j.bmcl.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove D. E.; Rosser M. F.; Ren H. Y.; Naren A. P.; Cyr D. M. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol. Biol. Cell 2009, 20, 4059–4069. 10.1091/mbc.e08-09-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomati V.; Sondo E.; Armirotti A.; Caci E.; Pesce E.; Marini M.; Gianotti A.; Ju Jeon Y.; Cilli M.; Pistorio A.; Mastracci L.; Ravazzolo R.; Scholte B.; Ronai Z.; Galietta L. J. V.; Pedemonte N. Genetic Inhibition Of The Ubiquitin Ligase Rnf5 Attenuates Phenotypes Associated To F508del Cystic Fibrosis Mutation. Sci. Rep. 2015, 5, 12138. 10.1038/srep12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondo E.; Falchi F.; Caci E.; Ferrera L.; Giacomini E.; Pesce E.; Tomati V.; Mandrup Bertozzi S.; Goldoni L.; Armirotti A.; Ravazzolo R.; Cavalli A.; Pedemonte N. Pharmacological Inhibition of the Ubiquitin Ligase RNF5 Rescues F508del-CFTR in Cystic Fibrosis Airway Epithelia. Cell Chem. Biol. 2018, 25, 891–905. 10.1016/j.chembiol.2018.04.010. [DOI] [PubMed] [Google Scholar]
- Pickart C. M. Back to the Future with Ubiquitin. Cell 2004, 116, 181–190. 10.1016/S0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- Schulman B. A.; Wade Harper J. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z.; Rickman K. A.; Yuan L.; Williams K.; Selvam S. P.; Woosley A. N.; Howe P. H.; Ogretmen B.; Smogorzewska A.; Olsen S. K. S. pombe Uba1-Ubc15 Structure Reveals a Novel Regulatory Mechanism of Ubiquitin E2 Activity. Mol. Cell 2017, 65, 699–714. 10.1016/j.molcel.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Kitagaki J.; Dai R.-M.; Tsai Y. C.; Lorick K. L.; Ludwig R. L.; Pierre S. A.; Jensen J. P.; Davydov I. V.; Oberoi P.; Li C.-C. H.; Kenten J. H.; Beutler J. A.; Vousden K. H.; Weissman A. M. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res. 2007, 67, 9472–9481. 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- Chung W. J.; Goeckeler-Fried J. L.; Havasi V.; Chiang A.; Rowe S. M.; Plyler Z. E.; Hong J. S.; Mazur M.; Piazza G. A.; Keeton A. B.; White E. L.; Rasmussen L.; Weissman A. M.; Denny R. A.; Brodsky J. L.; Sorscher E. J. Increasing the Endoplasmic Reticulum Pool of the F508del Allele of the Cystic Fibrosis Transmembrane Conductance Regulator Leads to Greater Folding Correction by Small Molecule Therapeutics. PLoS One 2016, 11, e0163615. 10.1371/journal.pone.0163615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeler-Fried J. L.; Aldrin Denny R.; Joshi D.; Hill C.; Larsen M. B.; Chiang A. N.; Frizzell R. A.; Wipf P.; Sorscher E. J.; Brodsky J. L. Improved correction of F508del-CFTR biogenesis with a folding facilitator and an inhibitor of protein ubiquitination. Bioorg. Med. Chem. Lett. 2021, 48, 128243. 10.1016/j.bmcl.2021.128243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapuria V.; Peterson L. F.; Showalter H. D. H.; Kirchhoff P. D.; Talpaz M.; Donato N. J. Protein cross-linking as a novel mechanism of action of a ubiquitin-activating enzyme inhibitor with anti-tumor activity. Biochem. Pharmacol. 2011, 82, 341–349. 10.1016/j.bcp.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Kanner S. A.; Shuja Z.; Choudhury P.; Jain A.; Colecraft H. M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 2020, 17, 1245–1253. 10.1038/s41592-020-00992-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigoillot M.; Overtus M.; Grodecka M.; Scholl D.; Garcia-Pino A.; Laeremans T.; He L.; Pardon E.; Hildebrandt E.; Urbatsch I.; Steyaert J.; Riordan J. R.; Govaerts C. Domain-interface dynamics of CFTR revealed by stabilizing nanobodies. Nat. Commun. 2019, 10, 2636. 10.1038/s41467-019-10714-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Crews C. M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalawansha D. A.; Crews C. M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher B. P.; Ward C. C.; Nomura D. K. Ligandability of E3 Ligases for Targeted Protein Degradation Applications. Biochemistry 2021, 10.1021/acs.biochem.1c00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning N. J.; Boike L.; Spradlin J. N.; Ward C. C.; Belcher B.; Brittain S. M.; Hesse M.; Dovala D.; McGregor L. M.; McKenna J. M.; Tallarico J. A.; Schirle M.; Nomura D. K.. Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization. BioRxiv 2021, 10.1101/2021.1104.1130.441959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Fu Y.; Li L.; Liu C. H.; Zhang L. The functions and regulation of Otubains in protein homeostasis and diseases. Ageing Res. Rev. 2021, 67, 101303. 10.1016/j.arr.2021.101303. [DOI] [PubMed] [Google Scholar]
- Luo X.; Kraus W. L. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amé J. C.; Spenlehauer C.; de Murcia G. The PARP superfamily. Bioessays 2004, 26, 882–893. 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- Petermann E.; Keil C.; Oei S. L. Importance of poly(ADP-ribose) polymerases in the regulation of DNA-dependent processes. Cell. Mol. Life Sci. 2005, 62, 731–738. 10.1007/s00018-004-4504-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. J.; Ball S. S.; Bowater R. P.; Wormstone I. M. PARP-1 inhibition influences the oxidative stress response of the human lens. Redox Biol. 2016, 8, 354–362. 10.1016/j.redox.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuny C. V.; Sullivan C. S. Virus-Host Interactions and the ARTD/PARP Family of Enzymes. PLoS Pathog. 2016, 12, e1005453. 10.1371/journal.ppat.1005453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau M.; Patel A.; Hendzel M. J.; Kaufmann S. H.; Poirier G. G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli F.; Battistoni A.; Gambari R.; Pompella A.; Bragonzi A.; Pilolli F.; Iuliano L.; Piroddi M.; Dechecchi M. C.; Cabrini G. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta 2012, 1822, 690–713. 10.1016/j.bbadis.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Brown R. K.; McBurney A.; Lunec J.; Kelly F. J. Oxidative damage to DNA in patients with cystic fibrosis. Free Radic. Biol. Med. 1995, 18, 801–806. 10.1016/0891-5849(94)00172-G. [DOI] [PubMed] [Google Scholar]
- Anjos S. M.; Robert R.; Waller D.; Zhang D. L.; Balghi H.; Sampson H. M.; Ciciriello F.; Lesimple P.; Carlile G. W.; Goepp J.; Liao J.; Ferraro P.; Phillipe R.; Dantzer F.; Hanrahan J. W.; Thomas D. Y. Decreasing Poly(ADP-Ribose) Polymerase Activity Restores ΔF508 CFTR Trafficking. Front. Pharmacol. 2012, 3, 165. 10.3389/fphar.2012.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donawho C. K.; Luo Y.; Luo Y.; Penning T. D.; Bauch J. L.; Bouska J. J.; Bontcheva-Diaz V. D.; Cox B. F.; DeWeese T. L.; Dillehay L. E.; Ferguson D. C.; Ghoreishi-Haack N. S.; Grimm D. R.; Guan R.; Han E. K.; Holley-Shanks R. R.; Hristov B.; Idler K. B.; Jarvis K.; Johnson E. F.; Kleinberg L. R.; Klinghofer V.; Lasko L. M.; Liu X.; Marsh K. C.; McGonigal T. P.; Meulbroek J. A.; Olson A. M.; Palma J. P.; Rodriguez L. E.; Shi Y.; Stavropoulos J. A.; Tsurutani A. C.; Zhu G.-D.; Rosenberg S. H.; Giranda V. L.; Frost D. J. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- Linington R. G.; Williams D. E.; Tahir A.; van Soest R.; Andersen R. J. Latonduines A and B, new alkaloids isolated from the marine sponge Stylissa carteri: structure elucidation, synthesis, and biogenetic implications. Org. Lett. 2003, 5, 2735–2738. 10.1021/ol034950b. [DOI] [PubMed] [Google Scholar]
- Carlile G. W.; Keyzers R. A.; Teske K. A.; Robert R.; Williams D. E.; Linington R. G.; Gray C. A.; Centko R. M.; Yan L.; Anjos S. M.; Sampson H. M.; Zhang D.; Liao J.; Hanrahan J. W.; Andersen R. J.; Thomas D. Y. Correction of F508del-CFTR trafficking by the sponge alkaloid latonduine is modulated by interaction with PARP. Chem. Biol. 2012, 19, 1288–1299. 10.1016/j.chembiol.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Carlile G. W.; Robert R.; Matthes E.; Yang Q.; Solari R.; Hatley R.; Edge C. M.; Hanrahan J. W.; Andersen R.; Thomas D. Y.; Birault V. Latonduine Analogs Restore F508del-Cystic Fibrosis Transmembrane Conductance Regulator Trafficking through the Modulation of Poly-ADP Ribose Polymerase 3 and Poly-ADP Ribose Polymerase 16 Activity. Mol. Pharmacol. 2016, 90, 65–79. 10.1124/mol.115.102418. [DOI] [PubMed] [Google Scholar]
- Jwa M.; Chang P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1α-mediated unfolded protein response. Nat. Cell Biol. 2012, 14, 1223–1230. 10.1038/ncb2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzcińska-Daneluti A. M.; Chen A.; Nguyen L.; Murchie R.; Jiang C.; Moffat J.; Pelletier L.; Rotin D. RNA Interference Screen to Identify Kinases That Suppress Rescue of ΔF508-CFTR. Mol. Cell. Proteom. 2015, 14, 1569–1583. 10.1074/mcp.M114.046375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centko R. M.; Carlile G. W.; Barne I.; Patrick B. O.; Blagojevic P.; Thomas D. Y.; Andersen R. J. Combination of Selective PARP3 and PARP16 Inhibitory Analogues of Latonduine A Corrects F508del-CFTR Trafficking. ACS Omega 2020, 5, 25593–25604. 10.1021/acsomega.0c02467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L.; Segal G.; Kartner N.; Grinstein S.; Zhang F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem. J. 1997, 328, 353–361. 10.1042/bj3280353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M.; Chang X. B.; Cui L.; Wu Y.; Ozols V. V.; Choudhury A.; Pagano R. E.; Riordan J. R. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 2684–2696. 10.1091/mbc.e04-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatecka-Urban A.; Brown A.; Moreau-Marquis S.; Renuka J.; Coutermarsh B.; Barnaby R.; Karlson K. H.; Flotte T. R.; Fukuda M.; Langford G. M.; Stanton B. A. The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 2005, 280, 36762–36772. 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Pampinella F.; Nemes C.; Benharouga M.; So J.; Du K.; Bache K. G.; Papsin B.; Zerangue N.; Stenmark H.; Lukacs G. L. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris B. Z.; Lim W. A. Mechanism and role of PDZ domains in signaling complex assembly. J. Cell Sci. 2001, 114, 3219–3231. 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Farinha C. M.; Canato S. From the endoplasmic reticulum to the plasma membrane: mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. 10.1007/s00018-016-2387-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favia M.; Guerra L.; Fanelli T.; Cardone R. A.; Monterisi S.; Di Sole F.; Castellani S.; Chen M.; Seidler U.; Reshkin S. J.; Conese M.; Casavola V. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol. Biol. Cell 2010, 21, 73–86. 10.1091/mbc.e09-03-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short D. B.; Trotter K. W.; Reczek D.; Kreda S. M.; Bretscher A.; Boucher R. C.; Stutts M. J.; Milgram S. L. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J. Biol. Chem. 1998, 273, 19797–19801. 10.1074/jbc.273.31.19797. [DOI] [PubMed] [Google Scholar]
- Wang S.; Raab R. W.; Schatz P. J.; Guggino W. B.; Li M. Peptide binding consensus of the NHE-RF-PDZ1 domain matches the C-terminal sequence of cystic fibrosis transmembrane conductance regulator (CFTR). FEBS Lett. 1998, 427, 103–108. 10.1016/S0014-5793(98)00402-5. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Wang H.; Guggino W. B. Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J. Biol. Chem. 2004, 279, 1892–1898. 10.1074/jbc.M308640200. [DOI] [PubMed] [Google Scholar]
- Wolde M.; Fellows A.; Cheng J.; Kivenson A.; Coutermarsh B.; Talebian L.; Karlson K.; Piserchio A.; Mierke D. F.; Stanton B. A.; Guggino W. B.; Madden D. R. Targeting CAL as a negative regulator of DeltaF508-CFTR cell-surface expression: an RNA interference and structure-based mutagenetic approach. J. Biol. Chem. 2007, 282, 8099–8109. 10.1074/jbc.M611049200. [DOI] [PubMed] [Google Scholar]
- Piserchio A.; Fellows A.; Madden D. R.; Mierke D. F. Association of the cystic fibrosis transmembrane regulator with CAL: structural features and molecular dynamics. Biochemistry 2005, 44, 16158–16166. 10.1021/bi0516475. [DOI] [PubMed] [Google Scholar]
- Vouilleme L.; Cushing P. R.; Volkmer R.; Madden D. R.; Boisguerin P. Engineering peptide inhibitors to overcome PDZ binding promiscuity. Angew. Chem., Int. Ed. 2010, 49, 9912–9916. 10.1002/anie.201005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amacher J. F.; Cushing P. R.; Bahl C. D.; Beck T.; Madden D. R. Stereochemical determinants of C-terminal specificity in PDZ peptide-binding domains: a novel contribution of the carboxylate-binding loop. J. Biol. Chem. 2013, 288, 5114–5126. 10.1074/jbc.M112.401588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing P. R.; Vouilleme L.; Pellegrini M.; Boisguerin P.; Madden D. R. A stabilizing influence: CAL PDZ inhibition extends the half-life of ΔF508-CFTR. Angew. Chem., Int. Ed. 2010, 49, 9907–9911. 10.1002/anie.201005585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. E.; Cushing P. R.; Boisguerin P.; Madden D. R.; Donald B. R. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput. Biol. 2012, 8, e1002477. 10.1371/journal.pcbi.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amacher J. F.; Zhao R.; Spaller M. R.; Madden D. R. Chemically modified peptide scaffolds target the CFTR-associated ligand PDZ domain. PLoS One 2014, 9, e103650. 10.1371/journal.pone.0103650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt G. T.; Jou J. D.; Gill N. P.; Lowegard A. U.; Martin J. W.; Madden D. R.; Donald B. R. Computational Analysis of Energy Landscapes Reveals Dynamic Features That Contribute to Binding of Inhibitors to CFTR-Associated Ligand. J. Phys. Chem. B 2019, 123, 10441–10455. 10.1021/acs.jpcb.9b07278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Xu X.; Amacher J. F.; Madden D. R.; Cormet-Boyaka E.; Pei D. Intracellular Delivery of Peptidyl Ligands by Reversible Cyclization: Discovery of a PDZ Domain Inhibitor that Rescues CFTR Activity. Angew. Chem., Int. Ed. 2015, 54, 5874–5878. 10.1002/anie.201411594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty P. G.; Wellmerling J. H.; Koley A.; Lukowski J. K.; Hummon A. B.; Cormet-Boyaka E.; Pei D. Cyclic Peptidyl Inhibitors against CAL/CFTR Interaction for Treatment of Cystic Fibrosis. J. Med. Chem. 2020, 63, 15773–15784. 10.1021/acs.jmedchem.0c01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Cushing P. R.; Smithson D. C.; Pellegrini M.; Pletnev A. A.; Al-Ayyoubi S.; Grassetti A. V.; Gerber S. A.; Guy R. K.; Madden D. R. Cysteine modifiers suggest an allosteric inhibitory site on the CAL PDZ domain. Biosci. Rep. 2018, 38, BSR20180231. 10.1042/BSR20180231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Li M.; Shandilya S. M.; Carpenter M. A.; Rathore A.; Brown W. L.; Perkins A. L.; Harki D. A.; Solberg J.; Hook D. J.; Pandey K. K.; Parniak M. A.; Johnson J. R.; Krogan N. J.; Somasundaran M.; Ali A.; Schiffer C. A.; Harris R. S. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M.; Zhao Z. Positive effects of SH2 domain-containing tyrosine phosphatase SHP-1 on epidermal growth factor- and interferon-gamma-stimulated activation of STAT transcription factors in HeLa cells. J. Biol. Chem. 1997, 272, 23376–23381. 10.1074/jbc.272.37.23376. [DOI] [PubMed] [Google Scholar]
- Culmsee C.; Gerling N.; Landshamer S.; Rickerts B.; Duchstein H. J.; Umezawa K.; Klumpp S.; Krieglstein J. Nitric oxide donors induce neurotrophin-like survival signaling and protect neurons against apoptosis. Mol. Pharmacol. 2005, 68, 1006–1017. 10.1124/mol.105.013086. [DOI] [PubMed] [Google Scholar]
- Bodas M.; Vij N. Adapting Proteostasis and Autophagy for Controlling the Pathogenesis of Cystic Fibrosis Lung Disease. Front. Pharmacol. 2019, 10, 20. 10.3389/fphar.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani A.; Villella V. R.; Esposito S.; Brunetti-Pierri N.; Medina D. L.; Settembre C.; Gavina M.; Raia V.; Ballabio A.; Maiuri L. Cystic fibrosis: A disorder with defective autophagy. Autophagy 2011, 7, 104–106. 10.4161/auto.7.1.13987. [DOI] [PubMed] [Google Scholar]
- Luciani A.; Villella V. R.; Esposito S.; Brunetti-Pierri N.; Medina D.; Settembre C.; Gavina M.; Pulze L.; Giardino I.; Pettoello-Mantovani M.; D’Apolito M.; Guido S.; Masliah E.; Spencer B.; Quaratino S.; Raia V.; Ballabio A.; Maiuri L. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- Levine B.; Kroemer G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani A.; Villella V. R.; Esposito S.; Gavina M.; Russo I.; Silano M.; Guido S.; Pettoello-Mantovani M.; Carnuccio R.; Scholte B.; De Matteis A.; Maiuri M. C.; Raia V.; Luini A.; Kroemer G.; Maiuri L. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012, 8, 1657–1672. 10.4161/auto.21483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefano D. D.; Villella V. R.; Esposito S.; Tosco A.; Sepe A.; Gregorio F. D.; Salvadori L.; Grassia R.; Leone C. A.; Rosa G. D.; Maiuri M. C.; Pettoello-Mantovani M.; Guido S.; Bossi A.; Zolin A.; Venerando A.; Pinna L. A.; Mehta A.; Bona G.; Kroemer G.; Maiuri L.; Raia V. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. 10.4161/15548627.2014.973737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle D. G.; Ferreira D.; Zhou Y.-D. Epigallocatechin-3-gallate (EGCG): Chemical and biomedical perspectives. Phytochemistry 2006, 67, 1849–1855. 10.1016/j.phytochem.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-S.; Quon M. J.; Kim J.-a. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195. 10.1016/j.redox.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosco A.; De Gregorio F.; Esposito S.; De Stefano D.; Sana I.; Ferrari E.; Sepe A.; Salvadori L.; Buonpensiero P.; Di Pasqua A.; Grassia R.; Leone C. A.; Guido S.; De Rosa G.; Lusa S.; Bona G.; Stoll G.; Maiuri M. C.; Mehta A.; Kroemer G.; Maiuri L.; Raia V. A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016, 23, 1380–1393. 10.1038/cdd.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu C. B.; Bridges R. J.; Pena-Rasgado C.; Lacerda A. E.; Bordwell C.; Sewell A.; Nichols A. J.; Chandran S.; Lonkar P.; Picarella D.; Ting A.; Wensley A.; Yeager M.; Liu F. Fatty Acid Cysteamine Conjugates as Novel and Potent Autophagy Activators That Enhance the Correction of Misfolded F508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). J. Med. Chem. 2017, 60, 458–473. 10.1021/acs.jmedchem.6b01539. [DOI] [PubMed] [Google Scholar]
- Hanrahan J. W.; Sato Y.; Carlile G. W.; Jansen G.; Young J. C.; Thomas D. Y. Cystic Fibrosis: Proteostatic correctors of CFTR trafficking and alternative therapeutic targets. Expert Opin. Ther. Targets 2019, 23, 711–724. 10.1080/14728222.2019.1628948. [DOI] [PubMed] [Google Scholar]
- Armirotti A.; Tomati V.; Matthes E.; Veit G.; Cholon D. M.; Phuan P.-W.; Braccia C.; Guidone D.; Gentzsch M.; Lukacs G. L.; Verkman A. S.; Galietta L. J. V.; Hanrahan J. W.; Pedemonte N. Bioactive Thymosin Alpha-1 Does Not Influence F508del-CFTR Maturation and Activity. Sci. Rep. 2019, 9, 10310. 10.1038/s41598-019-46639-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G. The recent failure of the PROMESA clinical trial for multiple system atrophy raises the question-are polyphenols a viable therapeutic option against proteinopathies?. Ann. Transl. Med. 2020, 8, 719. 10.21037/atm.2020.01.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing K.; Song K.-S.; Shin S.; Kim N.; Jeong S.; Oh H.-R.; Park J.-H.; Seo K.-S.; Heo J.-Y.; Han J.; Park J.-I.; Han C.; Wu T.; Kweon G.-R.; Park S.-K.; Yoon W.-H.; Hwang B.-D.; Lim K. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. 10.4161/auto.7.11.16658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Bey Y.; Boularan C.; Vural A.; Huang N.-N.; Hwang I.-Y.; Shan-Shi C.; Kehrl J. H. Omega-3 Free Fatty Acids Suppress Macrophage Inflammasome Activation by Inhibiting NF-κB Activation and Enhancing Autophagy. PLoS One 2014, 9, e97957. 10.1371/journal.pone.0097957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Amer A.; Krause K.; Bonfield T. L.; Fletcher D.; Richards C.; Reilly J. F.; Nichols A. J.; Vu C. B.. CAT-5571: an autophagy activator that enhances the clearance of intracellular bacteria. In Abstracts of the 40th European Cystic Fibrosis Conference, Seville, Spain, 2017; Vol. 16, p S13.
- Catabasis Pharmaceuticals Inc. Catabasis Pharmaceuticals and Bill & Melinda Gates Medical Research Institute to Study CAT-5571 in Drug-Sensitive and Drug-Resistant Tuberculosis. Press release Aug 04, 2020; https://ir.catabasis.com/node/9091/pdf (accessed 2021-11-11).
- Romani L.; Oikonomou V.; Moretti S.; Iannitti R. G.; D’Adamo M. C.; Villella V. R.; Pariano M.; Sforna L.; Borghi M.; Bellet M. M.; Fallarino F.; Pallotta M. T.; Servillo G.; Ferrari E.; Puccetti P.; Kroemer G.; Pessia M.; Maiuri L.; Goldstein A. L.; Garaci E. Thymosin α1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat. Med. 2017, 23, 590–600. 10.1038/nm.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R.; Tuthill C.. Immune Modulation with Thymosin Alpha 1 Treatment. In Thymosins; Litwack G., Ed.; Academic Press, 2016; Vol. 102, pp 151–178. [DOI] [PubMed] [Google Scholar]
- Chaudhary K.; Shinde R.; Liu H.; Gnana-Prakasam J. P.; Veeranan-Karmegam R.; Huang L.; Ravishankar B.; Bradley J.; Kvirkvelia N.; McMenamin M.; Xiao W.; Kleven D.; Mellor A. L.; Madaio M. P.; McGaha T. L. Amino Acid Metabolism Inhibits Antibody-Driven Kidney Injury by Inducing Autophagy. J. Immunol. 2015, 194, 5713–5724. 10.4049/jimmunol.1500277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincardini C.; Renga G.; Villella V.; Pariano M.; Oikonomou V.; Borghi M.; Bellet M. M.; Sforna L.; Costantini C.; Goldstein A. L.; Garaci E.; Romani L. Cellular proteostasis: a new twist in the action of thymosin alpha1. Expert Opin. Biol. Ther. 2018, 18, 43–48. 10.1080/14712598.2018.1484103. [DOI] [PubMed] [Google Scholar]
- Matthes E.; Hanrahan J. W.; Cantin A. M. F508del-CFTR is not corrected by thymosin α1. Nat. Med. 2018, 24, 890–891. 10.1038/s41591-018-0079-6. [DOI] [PubMed] [Google Scholar]
- Tomati V.; Caci E.; Ferrera L.; Pesce E.; Sondo E.; Cholon D. M.; Quinney N. L.; Boyles S. E.; Armirotti A.; Ravazzolo R.; Galietta L. J. V.; Gentzsch M.; Pedemonte N. Thymosin α-1 does not correct F508del-CFTR in cystic fibrosis airway epithelia. JCI Insight 2018, 3, e98699. 10.1172/jci.insight.98699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly R.; Mroz M. S.; Dempsey E.; Wynne K.; Keely S. J.; McKone E. F.; Hiebel C.; Behl C.; Coppinger J. A. Targeting the PI3K/Akt/mTOR signalling pathway in Cystic Fibrosis. Sci. Rep. 2017, 7, 7642. 10.1038/s41598-017-06588-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S.; Loewith R.; Hall M. N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Conn C. S.; Qian S.-B. mTOR signaling in protein homeostasis. Cell Cycle 2011, 10, 1940–1947. 10.4161/cc.10.12.15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. C.; Yuan H. X.; Guan K. L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. C.; Wei Y.; An Z.; Zou Z.; Xiao G.; Bhagat G.; White M.; Reichelt J.; Levine B. Akt-Mediated Regulation of Autophagy and Tumorigenesis Through Beclin 1 Phosphorylation. Science 2012, 338, 956–959. 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S.; Ng Y.; James D. E. Next-generation Akt inhibitors provide greater specificity: effects on glucose metabolism in adipocytes. Biochem. J. 2011, 435, 539–544. 10.1042/BJ20110040. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yan H.; Xu Z.; Yang B.; Luo P.; He Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. 10.1080/17425255.2019.1663169. [DOI] [PubMed] [Google Scholar]
- Trzcińska-Daneluti A. M.; Nguyen L.; Jiang C.; Fladd C.; Uehling D.; Prakesch M.; Al-awar R.; Rotin D. Use of Kinase Inhibitors to Correct ΔF508-CFTR Function. Mol. Cell. Proteomics 2012, 11, 745–757. 10.1074/mcp.M111.016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudernova I.; Vesela I.; Balek L.; Buchtova M.; Dosedelova H.; Kunova M.; Pivnicka J.; Jelinkova I.; Roubalova L.; Kozubik A.; Krejci P. Multikinase activity of fibroblast growth factor receptor (FGFR) inhibitors SU5402, PD173074, AZD1480, AZD4547 and BGJ398 compromises the use of small chemicals targeting FGFR catalytic activity for therapy of short-stature syndromes. Hum. Mol. Genet. 2016, 25, 9–23. 10.1093/hmg/ddv441. [DOI] [PubMed] [Google Scholar]
- Perkins L. A.; Fisher G. W.; Naganbabu M.; Schmidt B. F.; Mun F.; Bruchez M. P. High-Content Surface and Total Expression siRNA Kinase Library Screen with VX-809 Treatment Reveals Kinase Targets that Enhance F508del-CFTR Rescue. Mol. Pharmaceutics 2018, 15, 759–767. 10.1021/acs.molpharmaceut.7b00928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran J. P.; Glover M. L.; Peters K. W.; Bertrand C. A.; Watkins S. C.; Jarvik J. W.; Frizzell R. A. Pharmacological Rescue of the Mutant Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Detected by Use of a Novel Fluorescence Platform. Mol. Med. 2012, 18, 685–696. 10.2119/molmed.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. B.; Hu J.; Frizzell R. A.; Watkins S. C. Simple image-based no-wash method for quantitative detection of surface expressed CFTR. Methods 2016, 96, 40–45. 10.1016/j.ymeth.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M.; Montgomery R. L.; Olson E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggins B. T.; Robzyk K.; Wang D.; Marcu M. G.; Tsutsumi S.; Beebe K.; Cotter R. J.; Felts S.; Toft D.; Karnitz L.; Rosen N.; Neckers L. An Acetylation Site in the Middle Domain of Hsp90 Regulates Chaperone Function. Mol. Cell 2007, 25, 151–159. 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Zhang X.-H.; Qin M.; Wu H.-P.; Khamis M. Y.; Li Y.-H.; Ma L.-Y.; Liu H.-M. A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem. 2021, 64, 1362–1391. 10.1021/acs.jmedchem.0c01782. [DOI] [PubMed] [Google Scholar]
- Adhikari N.; Jha T.; Ghosh B. Dissecting Histone Deacetylase 3 in Multiple Disease Conditions: Selective Inhibition as a Promising Therapeutic Strategy. J. Med. Chem. 2021, 64, 8827–8869. 10.1021/acs.jmedchem.0c01676. [DOI] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Marks P. A. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA). Cell Cycle 2004, 3, 532–533. 10.4161/cc.3.5.824. [DOI] [PubMed] [Google Scholar]
- Hutt D. M.; Herman D.; Rodrigues A. P.; Noel S.; Pilewski J. M.; Matteson J.; Hoch B.; Kellner W.; Kelly J. W.; Schmidt A.; Thomas P. J.; Matsumura Y.; Skach W. R.; Gentzsch M.; Riordan J. R.; Sorscher E. J.; Okiyoneda T.; Yates J. R. 3rd; Lukacs G. L.; Frizzell R. A.; Manning G.; Gottesfeld J. M.; Balch W. E. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010, 6, 25–33. 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondo E.; Tomati V.; Caci E.; Esposito A. I.; Pfeffer U.; Pedemonte N.; Galietta L. J. V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Cell Physiol. 2011, 301, C872–C885. 10.1152/ajpcell.00507.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergougnoux A.; Petit A.; Knabe L.; Bribes E.; Chiron R.; De Sario A.; Claustres M.; Molinari N.; Vachier I.; Taulan-Cadars M.; Bourdin A. The HDAC inhibitor SAHA does not rescue CFTR membrane expression in Cystic Fibrosis. Int. J. Biochem. Cell Biol. 2017, 88, 124–132. 10.1016/j.biocel.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Hutt D. M.; Olsen C. A.; Vickers C. J.; Herman D.; Chalfant M. A.; Montero A.; Leman L. J.; Burkle R.; Maryanoff B. E.; Balch W. E.; Ghadiri M. R. Potential Agents for Treating Cystic Fibrosis: Cyclic Tetrapeptides That Restore Trafficking and Activity of ΔF508-CFTR. ACS Med. Chem. Lett. 2011, 2, 703–707. 10.1021/ml200136e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero A.; Beierle J. M.; Olsen C. A.; Ghadiri M. R. Design, Synthesis, Biological Evaluation, and Structural Characterization of Potent Histone Deacetylase Inhibitors Based on Cyclic α/β-Tetrapeptide Architectures. J. Am. Chem. Soc. 2009, 131, 3033–3041. 10.1021/ja809508f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C. A.; Ghadiri M. R. Discovery of Potent and Selective Histone Deacetylase Inhibitors via Focused Combinatorial Libraries of Cyclic α3β-Tetrapeptides. J. Med. Chem. 2009, 52, 7836–7846. 10.1021/jm900850t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne W. S.; Olsen C. A.; Beierle J. M.; Montero A.; Ghadiri M. R. Probing the Bioactive Conformation of an Archetypal Natural Product HDAC Inhibitor with Conformationally Homogeneous Triazole-Modified Cyclic Tetrapeptides. Angew. Chem., Int. Ed. 2009, 48, 4718–4724. 10.1002/anie.200805900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglès F.; Hutt D. M.; Balch W. E. HDAC inhibitors rescue multiple disease-causing CFTR variants. Hum. Mol. Genet. 2019, 28, 1982–2000. 10.1093/hmg/ddz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C. G.; Chang L.; Gestwicki J. E. Heat Shock Protein 70 (Hsp70) as an Emerging Drug Target. J. Med. Chem. 2010, 53, 4585–4602. 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri K.; Taldone T.; Modi S.; Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Liu Y. Recent Advances in the Discovery of Novel HSP90 Inhibitors: An Update from 2014. Curr. Drug Targets 2020, 21, 302–317. 10.2174/1389450120666190829162544. [DOI] [PubMed] [Google Scholar]
- Pratt W. B.; Toft D. O. Regulation of Signaling Protein Function and Trafficking by the hsp90/hsp70-Based Chaperone Machinery. Exp. Biol. Med. (Maywood) 2003, 228, 111–133. 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- Kuang E.; Qi J.; Ronai Z. e. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem. Sci. 2013, 38, 453–460. 10.1016/j.tibs.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y.; Khateb A.; Li Y.; Tinoco R.; Zhang T.; Bar-Yoseph H.; Tam M. A.; Chowers Y.; Sabo E.; Gerassy-Vainberg S.; Starosvetsky E.; James B.; Brown K.; Shen-Orr S. S.; Bradley L. M.; Tessier P. A.; Ronai Z. e. A. Regulation of S100A8 Stability by RNF5 in Intestinal Epithelial Cells Determines Intestinal Inflammation and Severity of Colitis. Cell Rep. 2018, 24, 3296–3311. 10.1016/j.celrep.2018.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. Roles of E3 ubiquitin ligases in cell adhesion and migration. Cell Adh Migr 2010, 4, 10–18. 10.4161/cam.4.1.9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B.; Zhang Y.; Tan B.; Liu T.-T.; Wang Y.-Y.; Shu H.-B. The E3 Ubiquitin Ligase RNF5 Targets Virus-Induced Signaling Adaptor for Ubiquitination and Degradation. J. Immunol. 2010, 184, 6249–6255. 10.4049/jimmunol.0903748. [DOI] [PubMed] [Google Scholar]
- Munz M.; Hein J.; Biggin P. C. The Role of Flexibility and Conformational Selection in the Binding Promiscuity of PDZ Domains. PLoS Comput. Biol. 2012, 8, e1002749. 10.1371/journal.pcbi.1002749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella C.; Visconti L.; Malagrinò F.; Pagano L.; Bufano M.; Nalli M.; Coluccia A.; La Regina G.; Silvestri R.; Gianni S.; Toto A. Targeting PDZ domains as potential treatment for viral infections, neurodegeneration and cancer. Biol. Direct 2021, 16, 15. 10.1186/s13062-021-00303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom F. J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman S. D.; Blanco P. G.; Zaman M. M.; Shea J. C.; Ollero M.; Hopper I. K.; Weed D. A.; Gelrud A.; Regan M. M.; Laposata M.; Alvarez J. G.; O’Sullivan B. P. Association of Cystic Fibrosis with Abnormalities in Fatty Acid Metabolism. N. Engl. J. Med. 2004, 350, 560–569. 10.1056/NEJMoa021218. [DOI] [PubMed] [Google Scholar]
- Freedman S. D.; Katz M. H.; Parker E. M.; Laposata M.; Urman M. Y.; Alvarez J. G. A membrane lipid imbalance plays a role in the phenotypic expression of cystic fibrosis in cftr–/– mice. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 13995–14000. 10.1073/pnas.96.24.13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbault C.; Wojewodka G.; Saeed Z.; Hajduch M.; Matouk E.; De Sanctis J. B.; Radzioch D. Cystic Fibrosis Fatty Acid Imbalance Is Linked to Ceramide Deficiency and Corrected by Fenretinide. Am. J. Respir. Cell Mol. Biol. 2009, 41, 100–106. 10.1165/rcmb.2008-0279OC. [DOI] [PubMed] [Google Scholar]
- Guilbault C.; De Sanctis J. B.; Wojewodka G.; Saeed Z.; Lachance C.; Skinner T. A. A.; Vilela R. M.; Kubow S.; Lands L. C.; Hajduch M.; Matouk E.; Radzioch D. Fenretinide Corrects Newly Found Ceramide Deficiency in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 38, 47–56. 10.1165/rcmb.2007-0036OC. [DOI] [PubMed] [Google Scholar]
- Garić D.; De Sanctis J. B.; Wojewodka G.; Houle D.; Cupri S.; Abu-Arish A.; Hanrahan J. W.; Hajduch M.; Matouk E.; Radzioch D. Fenretinide differentially modulates the levels of long- and very long-chain ceramides by downregulating Cers5 enzyme: evidence from bench to bedside. J. Mol. Med. 2017, 95, 1053–1064. 10.1007/s00109-017-1564-y. [DOI] [PubMed] [Google Scholar]
- Duchesneau P.; Besla R.; Derouet M. F.; Guo L.; Karoubi G.; Silberberg A.; Wong A. P.; Waddell T. K. Partial Restoration of CFTR Function in cftr-Null Mice following Targeted Cell Replacement Therapy. Mol. Ther. 2017, 25, 654–665. 10.1016/j.ymthe.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loberto N.; Mancini G.; Bassi R.; Carsana E. V.; Tamanini A.; Pedemonte N.; Dechecchi M. C.; Sonnino S.; Aureli M. Sphingolipids and plasma membrane hydrolases in human primary bronchial cells during differentiation and their altered patterns in cystic fibrosis. Glycoconj. J. 2020, 37, 623–633. 10.1007/s10719-020-09935-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R.; Caretti A.; Signorelli P. Role of Sphingolipids in the Pathobiology of Lung Inflammation. Mediators Inflamm. 2015, 2015, 1–19. 10.1155/2015/487508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichgräber V.; Ulrich M.; Endlich N.; Riethmüller J.; Wilker B.; De Oliveira-Munding C. C.; van Heeckeren A. M.; Barr M. L.; von Kürthy G.; Schmid K. W.; Weller M.; Tümmler B.; Lang F.; Grassme H.; Döring G.; Gulbins E. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008, 14, 382–391. 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- Itokazu Y.; Pagano R. E.; Schroeder A. S.; O’Grady S. M.; Limper A. H.; Marks D. L. Reduced GM1 ganglioside in CFTR-deficient human airway cells results in decreased β1-integrin signaling and delayed wound repair. Am. J. Physiol. Cell Physiol. 2014, 306, C819–C830. 10.1152/ajpcell.00168.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini G.; Loberto N.; Olioso D.; Dechecchi M. C.; Cabrini G.; Mauri L.; Bassi R.; Schiumarini D.; Chiricozzi E.; Lippi G.; Pesce E.; Sonnino S.; Pedemonte N.; Tamanini A.; Aureli M. GM1 as Adjuvant of Innovative Therapies for Cystic Fibrosis Disease. Int. J. Mol. Sci. 2020, 21, 4486. 10.3390/ijms21124486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore F.; Scudiero O.; Castaldo G. Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am. J. Med. Genet. 2002, 111, 88–95. 10.1002/ajmg.10461. [DOI] [PubMed] [Google Scholar]
- Middleton P. G.; Mall M. A.; Drevinek P.; Lands L. C.; McKone E. F.; Polineni D.; Ramsey B. W.; Taylor-Cousar J. L.; Tullis E.; Vermeulen F.; Marigowda G.; McKee C. M.; Moskowitz S. M.; Nair N.; Savage J.; Simard C.; Tian S.; Waltz D.; Xuan F.; Rowe S. M.; Jain R. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang E.; Okumura C. Y. M.; Sheffy-Levin S.; Varsano T.; Shu V. C.-W.; Qi J.; Niesman I. R.; Yang H.-J.; Lopez-Otin C.; Yang W. Y.; Reed J. C.; Broday L.; Nizet V.; Ronai Z. A. Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection. PLoS Genet. 2012, 8, e1003007. 10.1371/journal.pgen.1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkers G.; van Mourik P.; Vonk A. M.; Kruisselbrink E.; Dekkers J. F.; de Winter-de Groot K. M.; Arets H. G. M.; Marck-van der Wilt R. E. P.; Dijkema J. S.; Vanderschuren M. M.; Houwen R. H. J.; Heijerman H. G. M.; van de Graaf E. A.; Elias S. G.; Majoor C. J.; Koppelman G. H.; Roukema J.; Bakker M.; Janssens H. M.; van der Meer R.; Vries R. G. J.; Clevers H. C.; de Jonge H. R.; Beekman J. M.; van der Ent C. K. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708. 10.1016/j.celrep.2019.01.068. [DOI] [PubMed] [Google Scholar]
- Li F.; Zhang P.; Wu S.; Yuan L.; Liu Z. Advance in Human Epithelial-Derived Organoids Research. Mol. Pharmaceutics 2021, 18, 3931–3950. 10.1021/acs.molpharmaceut.1c00452. [DOI] [PubMed] [Google Scholar]
- Keegan D.; Brewington J. Nasal Epithelial Cell-Based Models for Individualized Study in Cystic Fibrosis. Int. J. Mol. Sci. 2021, 22, 4448. 10.3390/ijms22094448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar K. B.; Bhat A. H.; Amin S.; Hamid R.; Anees S.; Anjum S.; Reshi B. A.; Zargar M. A.; Masood A.; Ganie S. A. Modern Computational Strategies for Designing Drugs to Curb Human Diseases: A Prospect. Curr. Top. Med. Chem. 2019, 18, 2702–2719. 10.2174/1568026619666190119150741. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang Y.; Byrne R.; Schneider G.; Yang S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. 10.1021/acs.chemrev.8b00728. [DOI] [PubMed] [Google Scholar]