

Abstract

The 6-trifluoro substituted 8-nitrobenzothiazinones (BTZs) represent a novel type of antitubercular agents, and their high antimycobacterial activity is related to the inhibition of decaprenylphosphoryl-β-d-ribose 2′-oxidase (DprE1), an enzyme essential for the biosynthesis of mycobacterial cell wall. While extraordinary whole-cell activity was reported for the clinically advanced compound PBTZ169, its poor aqueous solubility signals the potential low bioavailability. To ameliorate the BTZ physiochemical property, a series of 6-methanesulfonyl substituted compounds were designed and prepared, and their antitubercular activity and DprE1 inhibition ability were evaluated. Among these compounds, MsPBTZ169 and compounds 2 and 8 exhibited minimum inhibitory concentrations (MICs) of less than 40 nM; moreover, these compounds displayed increased aqueous solubility and acceptable metabolic stability. Taken together, this study suggested that the 6-methanesulfonyl substituted 8-nitrobenzothiazinone derivatives, in combination with side chain modification, might provide BTZ type antitubercular agents with improved drug-like properties.
Keywords: Antitubercular agents, benzothiazinone derivatives, DprE1 inhibitors
Tuberculosis is considered a global health problem that used to claim more lives than any other infectious diseases, and its persistent prevalence results in approximately two million deaths every year.1 The scientific community is challenged by the emergence of multidrug resistant (MDR) and extensive drug resistant (XDR) strains,2 which emphasizes the growing need for antitubercular agents with novel scaffolds and mechanisms of action.
Discovered though phenotype screening, compounds with 8-nitrobenzothiaziones (BTZs) scaffolds are reported to be highly effective against Mycobacterium tuberculosis.3 Further study revealed that the mechanism of action of BTZs relies on the covalent inhibition of decaprenylphosphoryl-β-d-ribose 2′ epimerase (DprE1),4 an essential enzyme responsible for the biosynthesis of the cell wall component arabinose. Among the BTZ compounds, PBTZ169 was elected and advanced to clinic trail.5 However, despite PBTZ169 demonstrating the highest potency, good pharmacodynamic property, and low toxicity, it still shows some liabilities due to its poor solubility (less than 0.01 μg/mL in PBS buffer at pH7.4). Consequently, research aimed to improve the physicochemical properties of BTZ compounds has been a continuous effort, and different modification strategies focused on the side chain by introducing a polar moiety,6 disruption of the molecular symmetry,7 or other approaches likewise8,9 have been reported.
In this regard, we first set out to replace the electron withdrawing 6-trifluoromethyl with a more hydrophilic sulfonyl group (Figure 1); this bioisosteric replacement has been successfully applied for the 4-hydroxy-phenylpyruvate dioxygenase inhibitors Sulcotrione and NTBC.10 The methanesulfonyl substituted compound MsPBTZ169 proved to be less potent than PBTZ169, displaying a minimum inhibitory concentration (MIC) of 0.01 μM11 (vs MIC 0.5 nM of PBTZ169); nevertheless, this value is lower than that of the first line drugs isoniazid and rifampicin (MIC 0.26 μM and MIC 0.02 μM, respectively).12
Figure 1.

Structures of PBTZ169 and MsPBTZ169.
To further improve the compound activity, we continued our effort with the modifications at the side chain position. It was shown that the cyclohexyl ring is metabolically liable and oxidative hydroxylation occurred at different positions.13 We assumed that the incorporation of a heteroatom into the cyclohexyl might block or interfere with the metabolic pattern while increasing water solubility and/or metabolic stability of the compounds14 (Figure 2, compounds 2–5). Replacement of the cyclohexyl with an aromatic ring (Figure 2, compounds 6 and 7) was an alternative strategy. We also conceived other commonly implemented designs like introduction of a spirocyclic ring (Figure 2, compounds 8 and 9) or of the amide group (compound 10), or of the nonsymmetrical 7-member ring that might benefit the compound flexibility (compounds 11–16). Finally, analogs with side chain R attachment to the piperazinyl/1,4-diazepanyl through the amide or sulfonamide bond (Figure 2, compounds 17–28) were also proposed to extensively explore the side chain portion.
Figure 2.

Proposed analogues in this study.
The synthesis of compounds 2–10 is described in Scheme 1. Starting from the commercial 2-chloro-5-(methylthio) benzoic acid, the sulfide group was oxidized to sulfone 29 by oxone and then further treated with potassium nitrate in sulfuric acid at 90 °C to give the nitro substituted compound 30. Next, the carboxyl group was converted to acyl chloride and then to isothiocyanate 31 in one pot; the reaction of 31 with the corresponding amine gave the final cyclization products 2–10. The amines used for compounds 2–10 were prepared and shown in Scheme 1. The corresponding amines 33a–33e were prepared starting from the commercial aldehydes, reductive amination reaction of the amine with 1-Boc-piperazine gave intermediates 32a–32e, and removing the Boc group afforded the key intermediates 33a–33e. The amines 33f–33i were furnished according to the reported methods.14
Scheme 1. Synthetic Route for Compounds 2–10.
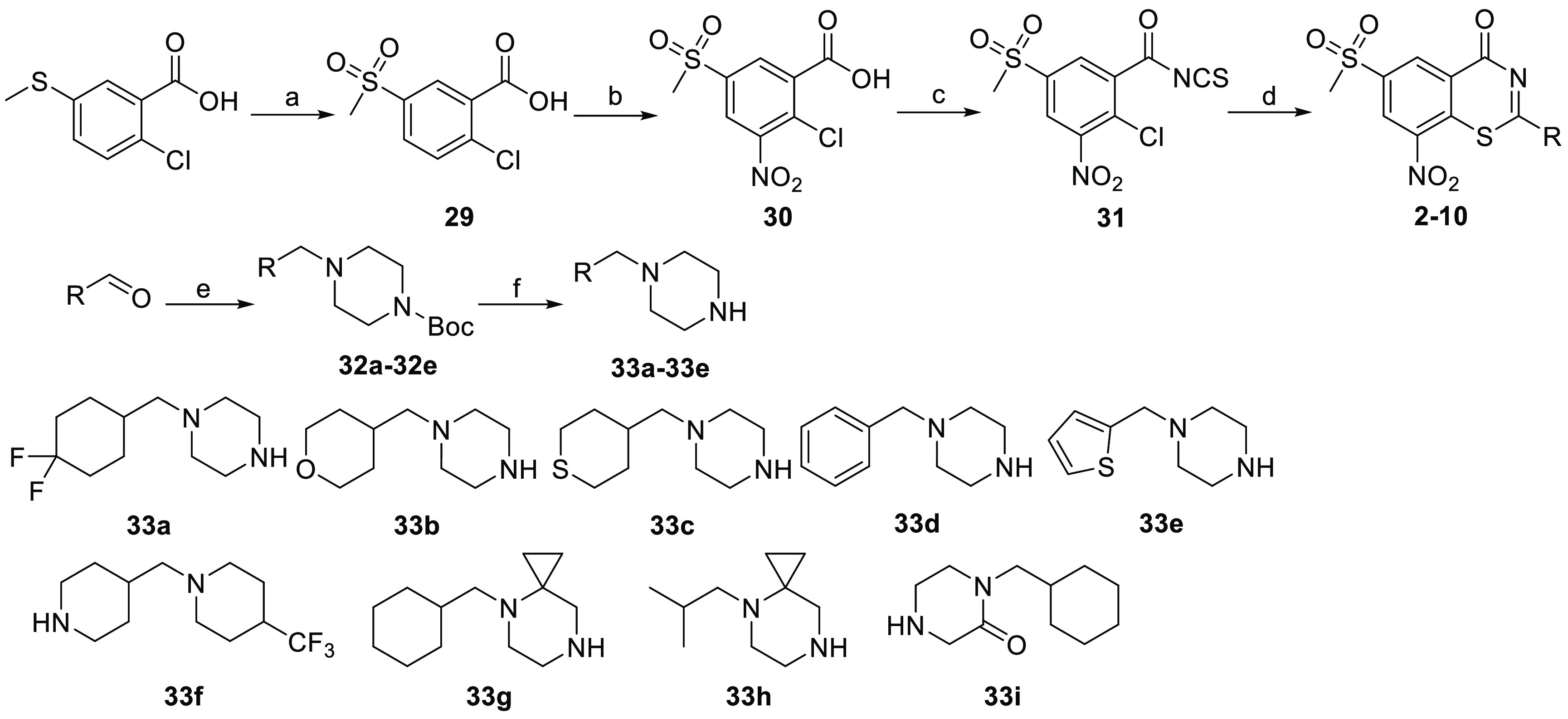
Reagents and conditions: (a) Oxone, MeOH, rt, 4 h, 86%; (b) KNO3, conc. H2SO4, 90 °C, 3 h, 80%; (c) oxyl chloride, DCM, then KNCS; (d) intermediates 33a–33e, yield 32%–55%; (e) 1-Boc-piperazine, NaBH3CN, rt, DCM, 88–93%; (f) CF3COOH, DCM, rt.
The synthetic route for compounds 11–16 is shown in Scheme 2. Reductive amination reaction between mono-Boc protected 1,4-diazepane and the corresponding aldehyde provided intermediates 34a–34f, and removing the Boc group gave the key intermediates 35a–35f, which were reacted with 31 to furnish the synthesis of the final compounds 11–16.
Scheme 2. Synthetic Route for Compounds 11–16.

Reagents and conditions: (a) Corresponding amine 35a–35f, DCM, rt, 44%–61%; (b) aldehyde then NaBH3CN, rt, DCM, 88%–92%; (c) CF3COOH, DCM, rt.
The synthetic route for compounds 17–28 is shown in Scheme 3. Briefly, the common reaction intermediate 31 was reacted with mono-Boc protected piperazine or 1,4-diazepane to give 36a/36b. After removing the Boc protective group using trifluoroacetic acid, the obtained intermediates 37a/37b were further reacted with the corresponding acyl chloride to furnish compounds 17 and 18 or reacted with the corresponding sulfonyl chloride in the presence of triethylamine affording compounds 19–28.
Scheme 3. Synthetic Route for Compounds 17–28.

Reagents and conditions: (a) 1-Boc-piperizine or mono-Boc-1,4-diazepane, DCM, 2 h, 44%–63%; (b) CF3COOH, DCM, rt, 3 h, 92–94%; (c) acyl chloride, Et3N, DCM, 88–90%; (d) corresponding sulfonyl chloride, Et3N, DCM, 86–91%.
All compounds were evaluated for their whole-cell activity against the H37Rv M. tuberculosis strain, MICs are show in Table 1. Among them, compound 2 with the para gem-fluoro-substitution exhibited a comparable MIC to that of compound 1 (MsPBTZ169). By contrast, the oxygen and sulfur incorporated compounds 3–5 displayed a pronounced loss of activity; specifically, the MIC of compound 5 increased over 100-fold. Compound 6 with benzyl or compound 7 with thiophenyl substitution displayed 6–20-fold increased MICs, indicating that aromatic rings are less favorable. Compound 8 with a cyclopropyl fused to the piperazine ring exhibited good antibacterial activity with a low MIC 0.04 μM, while compound 9 with a substitution of cyclohexyl by isobutyl displayed decreased activity. Compound 10 with the introduction of a ketone displayed a MIC of 0.07 μM. Overall, the 7-membered 1,4-diazepanyl linker was tolerated but not superior to cyclohexyl. Compounds 11 and 13 showed MICs of 0.06 and 0.07 μM, being more potent than compounds 12, 14, and 15, suggesting that both the piperazine/1,4-diazepanyl and the attached R groups could influence the compound inhibitory activity. For this set, compounds 2 and 8 exhibited the highest activity, with MICs lower than 0.04 μM.
Table 1. Compounds 1–16 and Their MICs against M. tuberculosis H37Rv.
| compd | MIC, μM |
|---|---|
| 1 (MsPBTZ169) | 0.01 |
| 2 | 0.03 |
| 3 | 0.51 |
| 4 | 0.10 |
| 5 | 1.87 |
| 6 | 0.21 |
| 7 | 0.25 |
| 8 | 0.04 |
| 9 | 0.17 |
| 10 | 0.07 |
| 11 | 0.07 |
| 12 | 0. 15 |
| 13 | 0.07 |
| 14 | 0.49 |
| 15 | 0.49 |
| 16 | 0.48 |
It has been reported that some 2-sulfonylpiperazin sPBTZ derivatives displayed high antitubercular activity and, more importantly, better water solubility and enhanced stability in microsomal assay.15 The same strategy with a side chain incorporation of sulfonamide/amide was exploited in compounds 17–28, with the activity changing dramatically as shown in Table 2. The two 2-acylpiperazin compounds 17 and 18 exhibited significant activity loss. For the rest sulfonamide incorporated compounds in this group, compound 21 with a flexible n-butyl chain exhibited the highest activity (MIC 0.09 μM) while all other compounds displayed modest to weak activity with MICs higher than 0.2 μM.
Table 2. Compounds 17–28 and Their MICs against M. tuberculosis H37Rv.
| compd | MIC, μM |
|---|---|
| 17 | 0.96 |
| 18 | >64 |
| 19 | 108 |
| 20 | 0.30 |
| 21 | 0.09 |
| 22 | 0.26 |
| 23 | 0.33 |
| 24 | >3.5 |
| 25 | >3.5 |
| 26 | 0.94 |
| 27 | >2.0 |
| 28 | >2.0 |
To confirm the on target inhibitory activity, selected active compounds with different side chain features were assayed against DprE1 enzyme activity.16 The determined IC50 values are shown in Table 3. The most potent MsPBTZ169 exhibited the highest enzyme inhibition activity (IC50 0.05 μM); the difluoro substituted compound 2 displayed a 4-fold increased IC50, a value in line with its MIC. The oxygen incorporated compound 3 had IC50 1.32 μM, indicating that the oxygen atom is not well tolerated in the enzyme binding pocket. The 1,4-diazepane linker compounds 11–13 and sulfonamide incorporated compounds 20–22 exhibited IC50 values of 0.36–0.54 μM; compound 26 exhibited the weakest enzymatic activity, consistent with the observed antibacterial activity. Overall, the determined enzymatic inhibition IC50 values well correlated with the antimycobacterial MICs, indicating the compounds on target inhibitory activity.
Table 3. DprE1 Inhibitory Activity for Selected Compounds.
| compd | IC50, μM |
|---|---|
| MsPBTZ169 | 0.05 ± 0.03 |
| 2 | 1.1 ± 0.1 |
| 3 | 10.3 ± 0.2 |
| 8 | 3.9 ± 0.2 |
| 11 | 1.1 ± 0.1 |
| 12 | 4.4 ± 0.3 |
| 13 | 1.3 ± 0.2 |
| 20 | 7.1 ± 0.4 |
| 21 | 1.4 ± 0.2 |
| 22 | 6.7 ± 0.3 |
| 26 | 16.7 ± 0.4 |
| PBTZ169 | 0.01 ± 0.03 |
Our objective was to improve the compound solubility; therefore, selected compounds MsPBTZ169, 2, and 8 along with PBTZ169 were examined for aqueous solubility in phosphate buffer pH 7.417 (Table 4). Compared to PBTZ169, all three 6-methanesulfonyl substituted compounds exhibited improved water solubility, although still low, with compound 2 displaying the highest solubility (1.6 μM). The plasma protein binding (PPB)7 property was also determined: the free fraction of MsPBTZ169 increased to 2.5% versus less than 0.1% PBTZ169, suggesting the increased possibility for improvement of the compound drug-like properties.
Table 4. Thermodynamic Solubility and Plasma Protein Binding of Compounds MsPBTZ169, 2, 8, and PBTZ169.
| compd | solubility (μg/mL) | PPB, % |
|---|---|---|
| MsPBTZ169 | 0.64 | 97.5 |
| 2 | 0.81 | 97.9 |
| 8 | 0.70 | 98.4 |
| PBTZ169 | <0.01 | >99.9% |
Based on the above results, we further examined the liver microsomal metabolic stability of the compounds, in both human (HLM) and mouse (MLM) microsomes; the results are shown in Table 5. Among the three tested compounds, incorporation of difluoro (2) exhibited the highest metabolic stability in human liver microsomes, displaying t1/2 38.6 min and a lower clearance rate Clint 45.1 mL/min/kg; compound MsPBTZ169 is less metabolically stable, as indicated by the shorter t1/2 and higher Clint; the cyclopropyl ring fused compound 8 displayed a faster clearance rate in both human and mouse liver microsomes. Interestingly, it was noticed the 6-methanesulfonyl substituted derivatives exhibited a metabolic pattern different from that of the 6-trifluoromethyl PBTZ169, with a lower clearance rate in MLM than in HLM, while PBTZ169 exhibited the opposite pattern. Since all 6-methanesulfonyl substituted compounds displayed a higher clearance rate than the control compound, our results indicate that both the side chain and the 6-position group play important roles for the metabolic stability of the compounds.
Table 5. Liver Microsomal Stability of Compounds MsPBTZ169, 2, 8, and PBTZ169.
| compd | species | t1/2 (min) | Clint, (mL/min/kg) |
|---|---|---|---|
| MsPBTZ169 | HLM | 28.8 | 60.5 |
| MLM | 42.1 | 129.2 | |
| 2 | HLM | 38.6 | 45.1 |
| MLM | 59.0 | 92.4 | |
| 8 | HLM | 22.6 | 77.0 |
| MLM | 33.5 | 163.0 | |
| PBTZ169 | HLM | 62.1 | 28.1 |
| MLM | 31.5 | 173.6 |
Finally, the pharmacokinetic property of compound 2 was examined in BALB/C mice after intravenous (i.v.) (2 mg/kg) and oral (p.o.) (10 mg/kg) administration. Compound 2 displayed an acceptable pharmacokinetics (PK) profile with only dosing i.v. (Supporting Information, Table S1). Unfortunately, no compound signal was detected in the case of oral dosing, suggesting either poor membrane permeability or rapid clearance rate in vivo.
In conclusion, to improve the physiochemical property of BTZ type compounds, we designed and prepared a number of BTZ derivatives with 6-methanesulfonyl substitution along with side chain modification. The implemented strategy led to the discovery of compounds 2 and 8, with MICs of less than 40 nM. The DprE1 enzyme inhibitory assay confirmed the compounds’ on target inhibitory activity. Finally, we demonstrated that the compounds had enhanced water solubility, improved plasma protein binding, and acceptable liver microsomal stability. However, further development of the compounds as antimycobacterial agent was suspended due to the compound in vivo PK profiles. Nevertheless, the study provided complementary insights for the BTZ compound properties, and further optimization of the 6-methanesulfonyl benzothiazinone core in combination with alternative side chain modification is undergoing and will be reported in due course.
Acknowledgments
The support from the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD) is appreciated.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00652.
Synthetic procedures for all compounds, and compound 1H NMR, 13C NMR, and HRMS data; MIC determination procedure; compound IC50 determination; liver microsomal metabolic stability test; cytotoxicity examination for compound 2 and 8; in vivo PK profile for compound 2 (PDF)
1H NMR and 13C NMR original spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Global Tuberculosis Report 2021; World Health Organization.
- Shetye G. S.; Franzblau S. G.; Cho S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. 10.1016/j.trsl.2020.03.007. [DOI] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikušová K.; Mollmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Beláňová M.; Bobovská A.; Dianišková P.; Korduláková J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer C.; Škovierová H.; Buroni S.; Bobovská A.; Nenci S.; Molteni E.; Pojer F.; Pasca M. R.; Makarov V.; Cole S. T.; Riccardi G.; Mikušová K.; Johnsson K. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-D-ribofuranose 2’-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- https://www.newtbdrugs.org/pipeline/clinical.
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Howe M.; Aldrich C. C. Spirocyclic and bicyclic 8-nitrobenzothiazinones for Tuberculosis with improved physicochemical and pharmacokinetic properties. ACS Med. Chem. Lett. 2019, 10, 348–351. 10.1021/acsmedchemlett.8b00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv K.; You X.; Wang B.; Wei Z.; Chai Y.; Wang B.; Wang A.; Huang G.; Liu M.; Lu Y. Identification of better pharmacokinetic benzothiazinone derivatives as new antitubercular agents. ACS Med. Chem. Lett. 2017, 8, 636–641. 10.1021/acsmedchemlett.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Lv K.; Wang B.; Li L.; Wang B.; Liu M.; Guo H.; Wang A.; Lu Y. Design, synthesis and antitubercular evaluation of benzothiazinones containing an oximido or amino nitrogen heterocycle moiety. RSC Adv. 2017, 7, 1480–1483. 10.1039/C6RA25712G. [DOI] [Google Scholar]
- Lin H.; Yang J.; Wang D.; Hao G.; Dong J.; Wang Y.; Yang W.; Wu J.; Zhan C.; Yang G. Molecular insights into the mechanism of 4-hydroxyphenylpyruvate dioxygenase inhibition: enzyme kinetics, X-ray crystallography and computational simulations. FEBS J. 2019, 286, 975. 10.1111/febs.14747. [DOI] [PubMed] [Google Scholar]
- Fan D.; Wang B.; Stelitano G.; Savková K.; Shi R.; Huszár S.; Han Q.; Mikusová K.; Chiarelli L. R.; Lu Y.; Qiao C. Structural and activity relationships of 6-sulfonyl-8- nitrobenzothiazinones as antitubercular agents. J. Med. Chem. 2021, 64, 14526–14539. 10.1021/acs.jmedchem.1c01049. [DOI] [PubMed] [Google Scholar]
- Gumbo T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob. Agents Chemother. 2010, 54, 1484–1491. 10.1128/AAC.01474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaggiari D.; Desfontaine V.; Cruchon S.; Guinchard S.; Vocat A.; Blattes E.; Pitteloud J.; CiulliniID L.; Bardinet C.; Ivanyuk A.; Makarov V.; Ryabova O.; Buclin T.; Cole S. T.; Decosterd L. A. Development and validation of a multiplex UHPLC-MS/MS method for the determination of the investigational antibiotic against multi-resistant tuberculosis macozinone (PBTZ169) and five active metabolites in human plasma. PLoS One 2019, 14, e0217139. 10.1371/journal.pone.0217139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhou J.; Ji M.; Zhu Z.; Cao R.; Chen X.; Xu B. Discovery of 2-substituted 1H-benzo[d]imidazole-4-carboxamide derivatives as novel poly(ADP-ribose)polymerase-1 inhibitors with in vivo anti-tumor activity. Eur. J. Med. Chem. 2017, 132, 26–41. 10.1016/j.ejmech.2017.03.013. [DOI] [PubMed] [Google Scholar]; b Qiao C., Fan D.. Benzothiazinone derivatives as anti-tuberculosis drugs and their preparation, pharmaceutical compositions and use in the treatment of mycobacterium tuberculosis. WO 2021203812, Oct 14, 2021.
- Piton J.; Vocat A.; Lupien A.; Foo C. S.; Riabova O.; Makarov V.; Cole S. T. Structure-based drug design and characterization of sulfonyl-piperazine benzothiazinone inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62 (10), e00681-18. 10.1128/AAC.00681-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Kong C.; Fumagalli M.; Savkova K.; Xu Y.; Huszar S.; Sammartino J. C.; Fan D.; Chiarelli L. R.; Mikusova K.; Sun Z.; Qiao C. Design, synthesis and evaluation of covalent inhibitors of DprE1 as antitubercular agents. Eur. J. Med. Chem. 2020, 208, 112773. 10.1016/j.ejmech.2020.112773. [DOI] [PubMed] [Google Scholar]
- Bergstrom C. A. S.; Wassvik C. M.; Johansson K.; Hubatsch I. Poorly soluble marketed drugs display solvation limited solubility. J. Med. Chem. 2007, 50, 5858–5862. 10.1021/jm0706416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.