

Abstract

RNA polymerase I (Pol I) transcribes ribosomal DNA (rDNA) into the 47S ribosomal RNA (rRNA) precursor. Further processing produces the 28S, 5.8S, and 18S rRNAs that are assembled into mature ribosomes. Many cancers exhibit higher Pol I transcriptional activity, reflecting a need for increased ribosome biogenesis and protein synthesis and making the inhibition of this process an attractive therapeutic strategy. Lead molecule BMH-21 (1) has been established as a Pol I inhibitor by affecting the destruction of RPA194, the Pol I large catalytic subunit. A previous structure–activity relationship (SAR) study uncovered key pharmacophores, but activity was constrained within a tight chemical space. This work details further SAR efforts that have yielded new scaffolds and improved off-target activity while retaining the desired RPA194 degradation potency. Pharmacokinetic profiling was obtained and provides a starting point for further optimization. New compounds present additional opportunities for the development of Pol I inhibitory cancer therapies.
Keywords: RNA polymerase I, cancer, inhibitor, RPA194
RNA polymerase I (Pol I) is a DNA-dependent RNA polymerase that is responsible for transcription of the 47S ribosomal RNA (rRNA) precursor in the nucleolus. This 47S pre-rRNA is further processed into the mature 28S, 5.8S, and 18S rRNAs that are assembled into ribosomes. Pol I transcriptional activity is frequently deregulated in cancers, reflecting a need for increased ribosome biogenesis and protein synthesis. Increased Pol I activity is generally not attributed to gain-of-function mutations or amplification in the core Pol I transcription apparatus. Rather, increased rDNA transcription can be accomplished by the activation of oncogenes and upstream signaling pathways that promote preinitiation complex assembly or loss-of-function mutations in tumor suppressors that repress Pol I transcription. Although there is currently no evidence that suggests that increased Pol I transcription is a causative factor of cancer formation, it is certainly possible that cancer cells can become reliant on the process and subsequently become selectively vulnerable to therapeutics that inhibit Pol I.1 Instead of targeting specific features of certain cancers, inhibiting a process that is critical for a wide range of cancers can provide a therapeutic benefit.2
The morphology of the nucleolus in tumors has been appreciated by pathologists for over a century, and prognostic markers staining the nucleolar components have been developed.3 As our understanding of the roles of Pol I and the nucleolus in malignancy has evolved,4 interest in developing compounds that specifically target Pol I is increasing.5 During the normal function of Pol I transcription, the tumor suppressor, p53, is sequestered by the E3 ubiquitin ligase, Mdm2.6 This interaction keeps p53 levels low, as it is constantly ubiquitinated and degraded. Induction of nucleolar stress activates the ribosomal surveillance pathway, resulting in the accumulation of p537 and potentially leading to outcomes such as apoptosis and cell-cycle arrest in cancer cells. Furthermore, as demonstrated by our previous work8 and others,9 normal cells are able to recover from this treatment. Thus the inhibition of Pol I transcription in the nucleolus and the induction of nucleolar stress on target cancer cells is an attractive therapeutic strategy. Whereas several chemotherapeutics such as Actinomycin D10 and CX-546111 have Pol I inhibition as part of their multimodal mode of action, no specific Pol I inhibitor is in clinical use.2
A proof of principle has been established for lead compound BMH-21 (1), shown in Figure 1.
Figure 1.
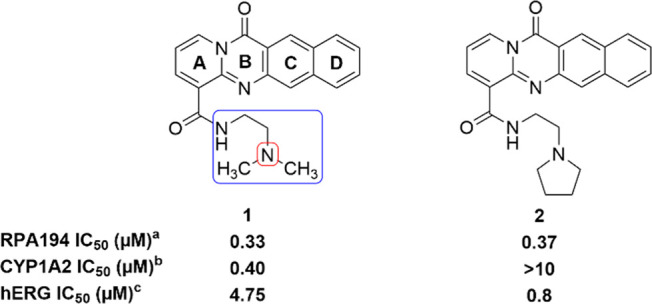
Comparison of key data for previously reported compounds 1 and 2. aRPA194 degradation measured in A375 cells. IC50 represents the mean of duplicate independent biological experiments performed in triplicate. bCYP1A2 inhibition analysis performed using human hepatic CYP450s (baculovirus–insect–cell expression system) expressing the isoform 1A2. chERG inhibition analysis performed using HEK293 cells stably transfected with hERG cDNA and measured by QPatch. IC50 value represents the mean of n = 3.
Compound 1 was found to inhibit Pol I transcription in a p53-independent manner by inducing the proteasome-mediated degradation of RPA194, the large catalytic subunit of Pol I, as a result of intercalating into GC-rich rDNA without eliciting a DNA damage response.8 Additionally, the destruction of RPA194 has been correlated with cancer cell killing.8 This offers a novel mechanism of action of inhibition of Pol I transcription and the basis for determining the on-target potency of compounds. The key components of this mechanism include the absence of eliciting a DNA damage response and independence of p53 activity. Compound 1 induces nucleolar stress12 but is still efficacious in the absence of p53. This highlights the importance that RPA194 has in rDNA transcription, and a quantitative assay measuring the extent of RPA194 degradation after compound treatment in A375 (human malignant melanoma) cells has been developed and is used to determine the potency of compounds.8 A375 cells are treated with compounds in an eight-point titration for 3 h. After fixing, permeabilizing, and blocking, cells are immunostained for RPA194 and observed by fluorescence microscopy. Using this assay, previous findings indicated that a four-ring tetracycle intercalator, secondary amide, and two-carbon linker between amide and terminal basic amine were optimal for potency (Figure 1).12 Whereas the presence of a basic amine is important for both potency and solubility, it engenders some hERG inhibitory activity (4.75 μM), an undesirable off-target. The human ether-a-go-go related gene (hERG) protein is involved in cardiac repolarization. The inhibition of hERG can cause QT prolongation and result in torsades de pointes and cardiac arrhythmia.13 Furthermore, CYP1A2 inhibition has been seen as a consistent feature of 1 and similar compounds. CYP1A2 is part of the cytochrome P450 enzyme superfamily and is responsible for the metabolism of numerous commonly used drugs and endogenous compounds.14 The inhibition of CYP1A2 could cause drug–drug interactions, especially in an oncology setting where combination therapy is common. Herein we describe efforts to improve this class of RNA Pol I inhibitor, especially in regards to unwanted hERG and CYP1A2 activity.
Initial structure–activity relationships (SARs) suggested that although the dimethylamino side chain present in compound 1 was optimal, similar RPA194 potency could be achieved when the methyl groups were wrapped into rings, as demonstrated by compounds 2 and 3 (Table 1). The pyrrolidine side chain in 2 was found to greatly reduce CYP1A2 inhibition while maintaining adequate potency (Figure 1). As shown in Table 1, additional conservative changes in these structures were explored to modulate physical properties such as basicity and lipophilicity that may eventually help address potential hERG13 and CYP1A2 inhibition liabilities.
Table 1. Compound 1 Side-Chain Modifications.
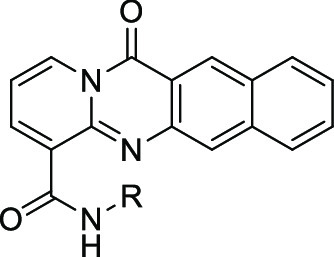

RPA194 degradation measured in A375 cells. IC50 represents the mean of duplicate independent biological experiments performed in triplicate.
Offsetting the ring by incorporating one carbon of the linker into the ring allowed compounds 4, 5, and 6 to retain some potency (IC50 of 0.78, 0.59, and 0.92 μM, respectively). Conversion of the piperidine to an offset capped morpholine 7 resulted in reduced potency. Installation of hydroxymethyl 8 was moderately tolerated, allowing a potential avenue for the reduction of lipophilicity, but no constraint resulted in improved potency. Furthermore, capping the secondary amine generated from these constraints led to decreases in activity (compounds 9, 10, and 11 IC50 of 3.94, 0.99, and 10 μM, respectively). Attempting to modulate the basicity of the amine by replacing a hydrogen atom with a fluorine led to a substantial loss of potency or inactivity (compounds 12–16). Interestingly, the four-position monofluoro analog 17 was moderately tolerated in addition to other four-position-substituted analogs (compounds 18 and 19 IC50 of 0.60 and 0.74 μM, respectively). However, replacing the piperidine ring with thiomorpholine 20 or the corresponding sulphone 21 led to a loss of activity. With the SAR tolerability of the side chain being seemingly more sensitive than originally predicted, another effort was made to modify the central tetracyclic core.
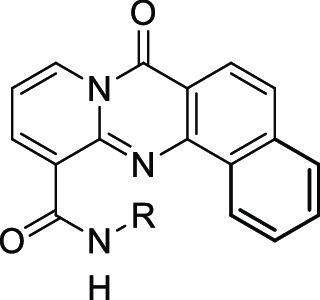
A prior SAR suggested that the four-fused-ring tetracycle of 1 was optimal.12 Importantly, this ring structure is somewhat basic, which imparts improved solubility of the compounds at slightly acidic pH. Truncating the core to a tricycle has previously been shown to decrease RPA194 potency,12 so new heterocycles, depicted in Table 2, were designed to further probe the sensitivity of RPA194 potency to changes in the core structure. Replacing the “D” ring (Figure 1) with various aliphatic rings in the case of compounds 22, 23, 24, and 25 resulted in lower potency (IC50 of 1.04, 1.39, 0.54, and 0.42 μM, respectively). Similar to previous observations, substituted tricyclic cores 26 and 27 showed decreased potency (IC50 of 1.16 and 6.86 μM, respectively). An attempt was made to introduce a conservative change in the tetracycle core to affect the electronics of the ring system, but this was also not tolerated (compound 28, IC50 of 5.5 μM). Removing the carbonyl of the tetracycle was also not tolerated and possibly disrupted an essential hydrogen-bonding interaction with rDNA (exemplified by compounds 29 and 30; IC50 of 1.7 and 6 μM, respectively). Introducing a turn in the tetracycle between the “C” and “D” rings resulted in similar potency to that of reference compound 1 (Compound 31, IC50 of 0.21 μM). Remarkably, introducing a turn in the opposite direction was not tolerated, as exemplified by compounds 32 and 33 (IC50 of >10 and 4.2 μM). With the discovery of compound 31, a further SAR was conducted to determine optimal side chains to pair with the new core, as summarized in Table 3.
Table 2. Modification of the Tetracyclic Core.

RPA194 degradation measured in A375 cells. IC50 represents the mean of duplicate independent biological experiments performed in triplicate.
Table 3. Compound 31 Analogs.

RPA194 degradation measured in A375 cells. IC50 represents the mean of duplicate independent biological experiments performed in triplicate.
CYP1A2 inhibition analysis performed using human hepatic CYP450s (baculovirus–insect–cell expression system) expressing the isoform 1A2.
hERG inhibition analysis performed using HEK293 cells stably transfected with hERG cDNA and measured by QPatch. IC50 value represents the mean of n = 3.
As previously seen, a basic amine was required for activity. Replacing the amine with a hydroxyl essentially resulted in inactivity (compound 34, IC50 of 9.5 μM). The preference for the amine to exist as a secondary or tertiary amine for activity is also worth noting (compounds 35, 36, and 37; IC50 of >10, 0.47, and 0.60 μM, respectively). Tying back the methyl groups into rings such as pyrrolidines and piperidines was tolerated, but this was not the case with piperazine (compounds 31, 39, and 40; IC50 of 0.21, 0.64, and >10 μM, respectively). Compound 31 was also observed to notably decrease the CYP1A2 inhibition, a desirable quality for reducing potential drug–drug interactions. Further derivatization of the pyrrolidine side chain showed that some potency could be retained while introducing another vector for an additional SAR, and that this activity was dependent on the stereochemistry of the substituted pyrrolidine (compounds 41 and 42, IC50 of >10 and 1.9 μM, respectively). Finally, the two-carbon linker was again observed to be optimal (compounds 31 and 37 vs 43 and 44; IC50 of 0.21 and 0.16 μM vs >10 and >10 μM, respectively). However, incorporating the linker into a ring was not tolerated (compounds 45 and 46, IC50 of >10 and >10 μM, respectively). With these results, compound 31 was identified as the optimal analog and exhibited similar RPA194 potency to that of 1 but a more than 100-fold improvement in CYP1A2 inhibition. In addition, compound 31 did not invoke a DNA damage response, as measured by the immunofluorescence of γ-H2AX, a commonly used biomarker for DNA damage.15 Compound 31 was also a potent inhibitor of A375 cell viability, with an IC50 of 38 nM (Figure 2).
Figure 2.

Cell viability analysis was determined in A375 cells treated with increasing concentrations of 31 for 3 days using the CellTiter-Glo luminescent cell viability assay. Data shown represent the mean of four experiments.
As seen in Table 3, changes to the side chain that are consistent with potency in the RPA194 assay did not substantially change the hERG activity, suggesting an overlap of the SAR between the two assays in this region of the molecule. More remarkable was the difference in hERG potency between 31 (3.6 μM) and 32 (0.07 μM) just by changing the direction of the turn introduced to the tetracycle core.
To determine selectivity for Pol I inhibition versus Pol II and Pol III, we performed quantitative polymerase chain reaction (qPCR), as shown in Figure 3. A375 cells were treated with compounds (1 μM) for 6 h. RNA was isolated using the Qiagen RNeasy kit, reverse-transcribed, and used to perform qPCR with SYBR GREEN master mix.16 Both 1 and 31 showed the selective, robust transcription inhibition of Pol I rRNA transcript 5′ETS851, whereas Pol II gene DHFR and Pol III gene tRNA-valine were unaffected.
Figure 3.

qPCR analysis of A375 cells treated with 1 μM of compounds 1 and 31 for 6 h normalized to GAPDH. Data shown represent the mean of three independent biological experiments performed in triplicate. P values were calculated using ordinary one-way ANOVA in GraphPad Prism 9.3.1.
A pharmacokinetic (PK) profile in CD-1 mice for compound 31 was obtained by administering a single intravenous (IV) dose of 1 mg/kg, an oral (PO) dose of 30 mg/kg, or an intraperitoneal (IP) dose of 30 mg/kg. Relevant PK parameters are summarized in Table 4.
Table 4. Intravenous (IV), Oral (PO), and Intraperitoneal (IP) Pharmacokinetic Parameters for 31 in CD-1 Micea.
| route | Cmax (ng/mL) | AUC(0–t) (h·ng/mL) | t1/2 (h) | Clobs (mL/min/kg) | Vss_obs (L/kg) | F (%) |
|---|---|---|---|---|---|---|
| IV 1 mg/kg | 30.8 ± 2.0 | 1.22 ± 0.12 | 508 ± 27 | 33.2 ± 4.1 | ||
| PO 30 mg/kg | 13.9 ± 6.2 | 122 ± 40 | 13.0 ± 4.2 | |||
| IP 30 mg/kg | 83.0 ± 5.9 | 518 ± 96 | 7.52 ± 1.72 |
Pharmacokinetic parameters were determined after a single dose administered intravenously (1 mg/kg, n = 3), orally (30 mg/kg, n = 3), or intraperitoneally (30 mg/kg, n = 3). IV compound was formulated in 30% dimethylacetamide (DMA) + 10% polyethylene glycol 200 (PEG200) + 5% Kolliphor ELP in Milli-Q water. PO and IP compounds were formulated in 0.2 M phosphate buffer, pH 6.8.
After IV administration, the calculated maximum plasma concentration (Cmax) and area under the curve (AUC) values were 45.6 ng/mL and 30.8 h·ng/mL, respectively. A short half life (t1/2) of 1.22 h was observed along with an extremely high clearance (Clobs) value of 508 mL/min/kg, possibly influenced by the high volume of distribution. Following PO administration, the bioavailability was low; however, IP administration saw improved exposure, which suggests a route for examining this class of compounds for in vivo efficacy. The metabolic stability for compound 31 was also measured in mouse liver microsomes and rat liver hepatocytes. Compound 31 exhibited intrinsic clearance values of 132 μL/min/mg and 1057.8 mL/min/kg, respectively. Taken together, these values serve as a starting point for further PK property optimization.
Compound 1 was synthesized as previously reported12 by the acid-promoted cyclocondensation of 2-chloronicotinic acid and 3-amino-2-napthoic acid followed by TBTU-mediated amide coupling. Commercially available diamines were coupled in this fashion to furnish amide analogs (compounds 2–11). Diamines that were not commercially available were prepared, as represented in Scheme 1. Boc-protected diamine was produced by the nucleophilic substitution of 2-(Boc-amino)ethyl bromide (47) with various secondary amines (48a–j). Following Boc deprotection, TBTU amide coupling with 50 allowed the production of the corresponding amide analogs (compounds 12–21). Several alternate cores were synthesized in a similar fashion as the 1 tetracycle. Summarized in Scheme 2, an acid-promoted cyclocondensation of 2-chloronicotinic acid (51) and various anthranilic acids (52a–e) followed by hydrolysis of the ester and TBTU-mediated amide coupling produced the corresponding amides with modifications to the tetracycle (compounds 22–24, 26, and 27).
Scheme 1. General Scheme for the Synthesis to Provide Compounds 1–21.

Reagents and conditions: (a) DIPEA, MeCN, rt. (b) 4 M HCl in 1,4-dioxane, 1,4-dioxane, rt or TFA, DCM, rt. (c) amine, TBTU, DIPEA, DMF, rt.
Scheme 2. General Scheme for the Synthesis to Provide Compounds 22–24, 26, and 27.

Reagents and conditions: (a) HCl, EtOH, reflux. (b) HCl, 100 °C. (c) 2-pyrrolidin-1-ylethanamine, TBTU, DIPEA, DMF/THF, rt.
However, the cyclocondensation chemistry was not amenable to all substrates. An alternative route based on a cyclocarbonylation report by Xu and Alper17 was envisioned to produce tetracyclic cores that could not be furnished by the acid-promoted cyclocondensation method. Scheme S1 summarizes the synthesis of 25. Following the initial palladium-catalyzed cross-coupling and selective bromination to produce the key bromoanilinopyridine intermediate, subsequent amide coupling and palladium-catalyzed cyclocarbonylation were performed to give 25. As summarized in Scheme S2, 28 was synthesized in a similar fashion.
The synthesis of 31 and subsequent analogs is summarized in Scheme 3. Buchwald–Hartwig cross-coupling produced the desired key intermediate 56. Successful palladium-catalyzed cyclocarbonylation allowed closing of the ring and insertion of the carbonyl. Hydrolysis of the ester followed by amide coupling with the corresponding amine led to the formation of compounds 31 and 34–46.
Scheme 3. Synthesis of Compounds 31 and 34–46.

Reagents and conditions: (a) Pd(OAc)2 (4 mol %), rac-BINAP (6 mol %), Cs2CO3, toluene, 130 °C. (b) Pd(OAc)2 (10 mol %), Xantphos (15 mol %), Xantphos Pd G3 (5 mol %), K3PO4, Mo(CO)6, toluene, 100 °C. (c) 2 M NaOH, MeOH, rt. (d) amine, TBTU, DIPEA, DMF, rt.
In the case of 29 and 30, unexpected results led to additional unique structures. Scheme 4 shows the overall transformation to produce 29. During the initial palladium-catalyzed cross-coupling, a second cross-coupling event between the pyridyl nitrogen and the aryl bromide resulted in a ring closing producing the 6–5–6–6 ring system and notably lacking the carbonyl seen in many of the other tetracycles. A similar result occurred with 30. After the initial cross-coupling, a bromination attempt to give the desired key intermediate instead resulted in the SNAR product, giving another 6–5–6–6 ring system lacking the carbonyl.
Scheme 4. Synthesis of Compounds 29 and 30.

Reagents and conditions: (a) Pd(OAc)2 (5 mol %), Xantphos (10 mol %), Cs2CO3, toluene, 130 °C. (b) 1 M NaOH, MeOH, rt. (c) 2-pyrrolidin-1-ylethanamine, TBTU, DIPEA, DMF, rt. (d) NBS, DCM, rt.
Scheme S3 summarizes the production of 32. After preparation of the two cross-coupling partners and subsequent coupling, palladium-catalyzed cyclocarbonylation successfully closed the ring and inserted the carbonyl, as desired. Ester hydrolysis followed by amide coupling with the corresponding amine furnished compound 32.
Finally, compound 33 was synthesized, as summarized in Scheme S4. Following the initial cross-coupling, a selective bromination was attempted. Bromination was achieved but at a different position than originally predicted. This intermediate was carried forward and successfully underwent palladium-catalyzed cyclocarbonylation to ultimately afford 33 after ester hydrolysis and amide coupling.
In summary, the optimization of 1 to reduce the hERG and CYP activity while maintaining the potency demonstrated that only modest changes to the basic side chain were tolerated. The requirement for a basic amine for RPA194 potency has thus far prevented the complete elimination of hERG activity. Undesired CYP1A2 inhibition was successfully removed, while potency was maintained by utilizing cyclic amines. Whereas a tetracyclic aromatic core structure is required for robust activity, subtle changes such as changing the orientation of the terminal ring (compound 32) can have a remarkable effect on the potency. From this work, compound 31 emerged as an important lead compound, retaining substantial potency while reducing CYP1A2 inhibition by over 100-fold as compared with compound 1 without a substantial increase in the hERG activity. PK profiling provided a starting point for further optimization. Future experiments will focus on improving the pharmacokinetics, namely, improving the clearance and obtaining a structural understanding of how the compounds interact with the DNA–Pol I complex assembly.
Acknowledgments
We thank WuXi AppTec and IntelliSyn for chemistry support.
Glossary
Abbreviations
- AUC
area under the curve
- CYP1A2
cytochrome P450 isoform 1A2
- hERG
human ether-a-go-go potassium channel
- γ-H2AX
serine 139 phosphorylation of histone protein family member 2AX
- IP
intraperitoneal
- IV
intravenous
- PK
pharmacokinetic
- PO
per os, oral
- Pol I
RNA polymerase I
- r
ribosomal
- SAR
structure–activity relationship
- TBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00660.
Additional chemistry schemes, synthesis and characterization of compounds, and biochemical and pharmacokinetic assay protocols (PDF)
Author Present Address
▽ Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702, United States
This work was supported in part by NIGMS R01GM121404, NCI P30 CA006973, the Lieber Institute for Brain Development, and Bluefield Innovations LLC.
The authors declare the following competing financial interest(s): T.E.D., P.d.L., A.B., H.L., N.V.R., M.L., and J.C.B. hold patents or patent applications to RNA Pol I inhibitors.
Supplementary Material
References
- Hein N.; Hannan K. M.; George A. J.; Sanij E.; Hannan R. D. The Nucleolus: An Emerging Target for Cancer Therapy. Trends Mol. Med. 2013, 19 (11), 643–654. 10.1016/j.molmed.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Ferreira R.; Schneekloth J. S.; Panov K. I.; Hannan K. M.; Hannan R. D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9 (2), 266. 10.3390/cells9020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derenzini M.; Montanaro L.; Treré D. What the Nucleolus Says to a Tumour Pathologist. Histopathology 2009, 54 (6), 753–762. 10.1111/j.1365-2559.2008.03168.x. [DOI] [PubMed] [Google Scholar]
- Bywater M. J.; Pearson R. B.; McArthur G. A.; Hannan R. D. Dysregulation of the Basal RNA Polymerase Transcription Apparatus in Cancer. Nat. Rev. Cancer 2013, 13 (5), 299–314. 10.1038/nrc3496. [DOI] [PubMed] [Google Scholar]
- Drygin D.; Rice W. G.; Grummt I. The RNA Polymerase I Transcription Machinery: An Emerging Target for the Treatment of Cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50 (1), 131–156. 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- Deisenroth C.; Zhang Y. Ribosome Biogenesis Surveillance: Probing the Ribosomal Protein-Mdm2-P53 Pathway. Oncogene 2010, 29 (30), 4253–4260. 10.1038/onc.2010.189. [DOI] [PubMed] [Google Scholar]
- Bursac S.; Brdovcak M. C.; Donati G.; Volarevic S. Activation of the Tumor Suppressor P53 upon Impairment of Ribosome Biogenesis. Biochim. Biophys. Acta - Mol. Basis Dis. 2014, 1842 (6), 817–830. 10.1016/j.bbadis.2013.08.014. [DOI] [PubMed] [Google Scholar]
- Peltonen K.; Colis L.; Liu H.; Trivedi R.; Moubarek M. S.; Moore H. M.; Bai B.; Rudek M. A.; Bieberich C. J.; Laiho M. A Targeting Modality for Destruction of RNA Polymerase I That Possesses Anticancer Activity. Cancer Cell 2014, 25 (1), 77–90. 10.1016/j.ccr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater M. J.; Poortinga G.; Sanij E.; Hein N.; Peck A.; Cullinane C.; Wall M.; Cluse L.; Drygin D.; Anderes K.; Huser N.; Proffitt C.; Bliesath J.; Haddach M.; Schwaebe M. K.; Ryckman D. M.; Rice W. G.; Schmitt C.; Lowe S. W.; Johnstone R. W.; Pearson R. B.; McArthur G. A.; Hannan R. D. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of P53. Cancer Cell 2012, 22 (1), 51–65. 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K.; Mühl B.; Harasim T.; Rohrmoser M.; Malamoussi A.; Orban M.; Kellner M.; Gruber-Eber A.; Kremmer E.; Hölzel M.; Eick D. Chemotherapeutic Drugs Inhibit Ribosome Biogenesis at Various Levels. J. Biol. Chem. 2010, 285 (16), 12416–12425. 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno P. M.; Lu M.; Dennis K. A.; Inam H.; Moore C. J.; Sheehe J.; Elledge S. J.; Hemann M. T.; Pritchard J. R. The Primary Mechanism of Cytotoxicity of the Chemotherapeutic Agent CX-5461 Is Topoisomerase II Poisoning. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (8), 4053–4060. 10.1073/pnas.1921649117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colis L.; Ernst G.; Sanders S.; Liu H.; Sirajuddin P.; Peltonen K.; Depasquale M.; Barrow J. C.; Laiho M. Design, Synthesis, and Structure-Activity Relationships of Pyridoquinazolinecarboxamides as RNA Polymerase i Inhibitors. J. Med. Chem. 2014, 57 (11), 4950–4961. 10.1021/jm5004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal Chemistry of HERG Optimizations: Highlights and Hang-Ups. J. Med. Chem. 2006, 49 (17), 5029–5046. 10.1021/jm060379l. [DOI] [PubMed] [Google Scholar]
- Gunes A.; Dahl M. L. Variation in CYP1A2 Activity and Its Clinical Implications: Influence of Environmental Factors and Genetic Polymorphisms. Pharmacogenomics 2008, 9 (5), 625–637. 10.2217/14622416.9.5.625. [DOI] [PubMed] [Google Scholar]
- Ivashkevich A.; Redon C. E.; Nakamura A. J.; Martin R. F.; Martin O. A. Use of the γ-H2AX Assay to Monitor DNA Damage and Repair in Translational Cancer Research. Cancer Lett. 2012, 327 (1–2), 123–133. 10.1016/j.canlet.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen K.; Colis L.; Liu H.; Jäämaa S.; Moore H. M.; Enbäck J.; Laakkonen P.; Vaahtokari A.; Jones R. J.; Af Hällström T. M.; Laiho M. Identification of Novel P53 Pathway Activating Small-Molecule Compounds Reveals Unexpected Similarities with Known Therapeutic Agents. PLoS One 2010, 5 (9), e12996 10.1371/journal.pone.0012996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T.; Alper H. Synthesis of Pyrido[2,1- b ]Quinazolin-11-Ones and Dipyrido[1,2-a:2′,3′- d ]Pyrimidin-5-Ones by Pd/DIBPP-Catalyzed Dearomatizing Carbonylation. Org. Lett. 2015, 17 (6), 1569–1572. 10.1021/acs.orglett.5b00452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.