

Abstract
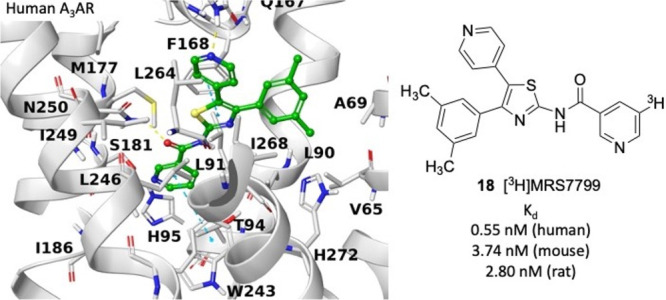
The A3 adenosine receptor (A3AR) is a target for pain, ischemia, and inflammatory disease therapy. Among the ligand tools available are selective agonists and antagonists, including radioligands, but most high-affinity non-nucleoside antagonists are limited in selectivity to primate species. We have explored the structure–activity relationship of a previously reported A3AR antagonist DPTN 9 (N-[4-(3,5-dimethylphenyl)-5-(4-pyridyl)-1,3-thiazol-2-yl]nicotinamide) for radiolabeling, including 3-halo derivatives (3-iodo, MRS7907), and characterized 9 as a high -affinity radioligand [3H]MRS7799. A3AR Kd values were (nM): 0.55 (human), 3.74 (mouse), and 2.80 (rat). An extended methyl acrylate (MRS8074, 19) maintained higher affinity (18.9 nM) than a 3-((5-chlorothiophen-2-yl)ethynyl) derivative 20. Compound 9 had an excellent brain distribution in rats (brain/plasma ratio ∼1). Receptor docking predicted its orthosteric site binding by engaging residues that were previously found to be essential for AR binding. Thus the new radioligand promises to be a useful species-general antagonist tracer for receptor characterization and drug discovery.
Keywords: Adenosine receptor, G-protein-coupled receptor, antagonist, molecular dynamics, radioligand
The Gi-coupled A3 adenosine receptor (A3AR) is a therapeutic target of interest in pain, neurodegeneration, cancer, ischemia of the heart and brain, autoimmune inflammatory diseases, and other conditions.1−8 In humans, the A3AR is expressed highly in the lung, liver, kidney, and heart. Two A3AR agonists are in advanced clinical trials for psoriasis, COVID-19, hepatocellular carcinoma (HCC), and nonalcoholic steatohepatitis (NASH).1,3,9 A3AR agonists are being explored for the treatment of chronic neuropathic pain, stroke, and other nervous system conditions.7,10,11 The moderately selective A3AR agonists already in clinical trials are IB-MECA 1 and Cl-IB-MECA 2 (Chart 1). However, we have recently expanded the A3AR agonist structure–activity relationship (SAR) to include more highly selective (N)-methanocarba (bicyclo[3.1.0]hexyl) agonists such as 4.7,12
Chart 1. Select Ligands and Radioligands Used to Study the A3ARa.
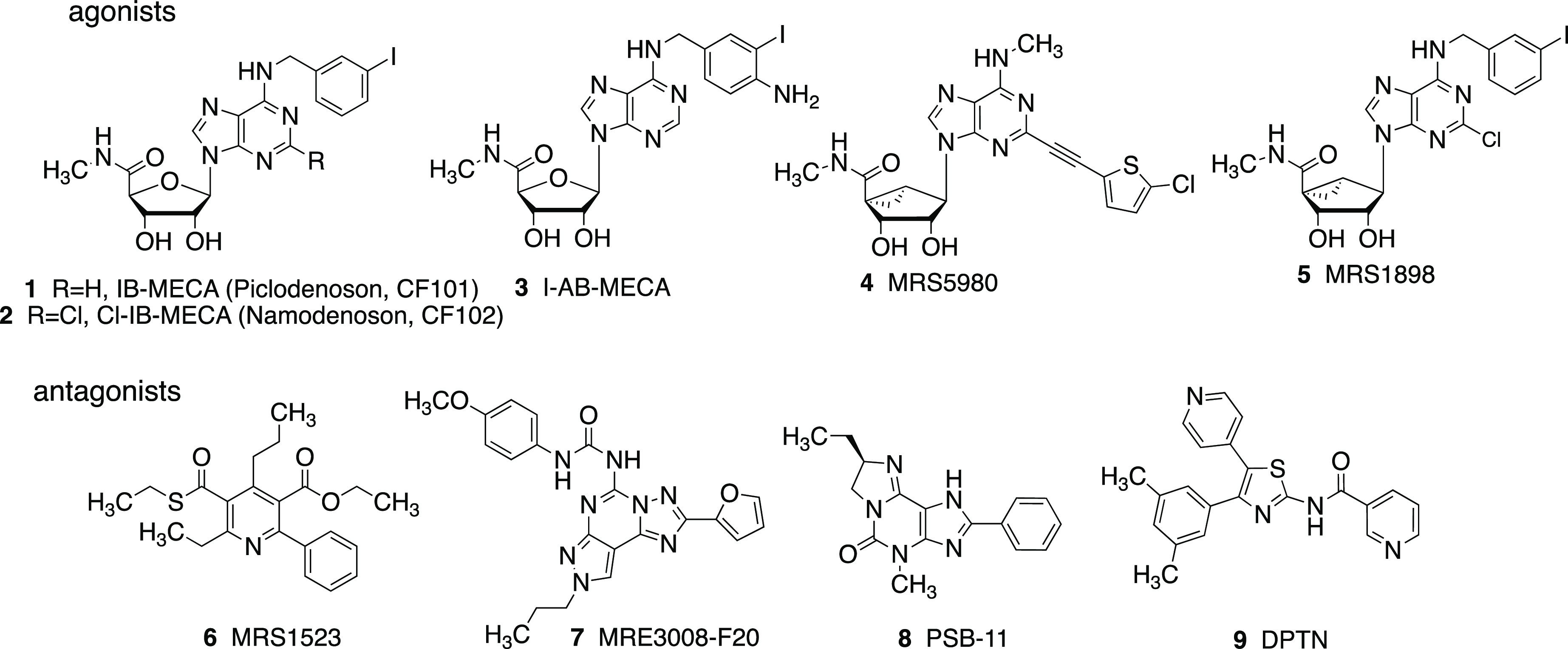
a Agonists 3 and 5 are used in radioiodinated form, and antagonists 7 and 8 have been tritiated. Binding Ki values at the hA3AR (nM) are: 1, 1.8; 2, 1.4; 3, ∼1; 4, 0.70; 5, 1.4; 6, 43.9; 7, 1.13; 8, 3.51.1,13
A3AR ligands, in particular, antagonists, are subject to interspecies differences in affinity.13−16 Thus many of the reported highly selective human (h) A3AR antagonists, such as [1,2,4]triazolo[1,5-c]quinazolin-5-amines and 1,4-dihydropyridines, are not at all or are marginally A3AR-selective in rats and mice. Furthermore, the most widely used agonist radioligand, [125I]I-AB-MECA 3, is only slightly selective for that subtype, although it has a ∼1 nM Kd value at the rat A3AR.17 Efforts to use [125I]3 for autoradiography to exclusively label the A3AR in brain tissue were problematic because it requires the addition of a selective A1AR antagonist to remove that component of binding. Other nucleoside radioligands reported include 76Br derivatives for A3AR positron emission tomography (PET) studies.18 [125I]MRS1898 5 proved to be a selective A3AR agonist radioligand in binding experiments (Kd 0.17 nM, rat A3AR).19 Antagonist radioligands [3H]MRS3008F20 7 and [3H]PSB11 8 are suitable only for the primate A3AR, as their affinities at the mouse (Ki > 10 and 6.36 μM, respectively) and rat (r) A3ARs (both: Ki > 10 μM) were much weaker.13 A pyridine derivative 6 is currently the best general purpose A3AR antagonist for use in the mouse (Ki 349 nM) or rat (Ki 216 nM).13,20 It has proven selective in various in vivo studies to define the action at the A3AR.7 However, efforts to convert this pyridine series into a radioligand, specifically a 18F PET ligand in the form of a closely related analogue, FE@SUPPY (not shown), were also not straightforward chemically or pharmacologically.21 A new chemotype (5-(4-chlorophenyl)thiophene-2-carboxamide) in moderately potent hA3AR and rA3AR antagonists was recently reported, but it does not have sufficiently high affinity for use as a rA3AR radioligand.22
We recently resynthesized an antagonist, DPTN 9 (N-[4-(3,5-dimethylphenyl)-5-(4-pyridyl)-1,3-thiazol-2-yl]nicotinamide), as reported in 2008 by Miwatashi et al.,23 and confirmed that it can serve as a selective A3AR antagonist across species (≥20-fold selectivity compared with other AR subtypes).13 Here we have explored in limited scope the SAR of DPTN for radioisotope incorporation and prepared and characterized a high-affinity tritiated radioligand.
Chemical Synthesis
The synthetic route to 9 followed the original publication23 with modification and allowed the introduction of a aromatic halogen substitution (Scheme 1). Specifically, a fluorine, bromine, or iodine atom (16b–d) was inserted in the place of a 3-methyl group present in 9. The inclusion of the Weinreb–Nahm amide 12 leading to the Weinreb ketone2413 provided a higher overall yield than that by the original route. It was possible to acylate arylamine 15 with several nicotinoyl derivatives via their acid chlorides to yield 9 and 17 (Scheme 1B). The tritiated radioligand [3H]18 ([3H]5-bromo-N-(4-(3,5-dimethylphenyl)-5-(pyridin-4-yl)thiazol-2-yl)nicotinamide, MRS7799) was synthesized by the catalytic reduction of bromo precursor 17 with tritium gas. [3H]18 was isolated by HPLC with a specific activity of 23.9 Ci/mmol (stored in an EtOH solution at a concentration of 16.24 μg/mL). The radiochemical purity was 97.5% (Supporting Information).
Scheme 1. Synthesis of Halo Analogues of 9 and Its Tritiated Form 18.
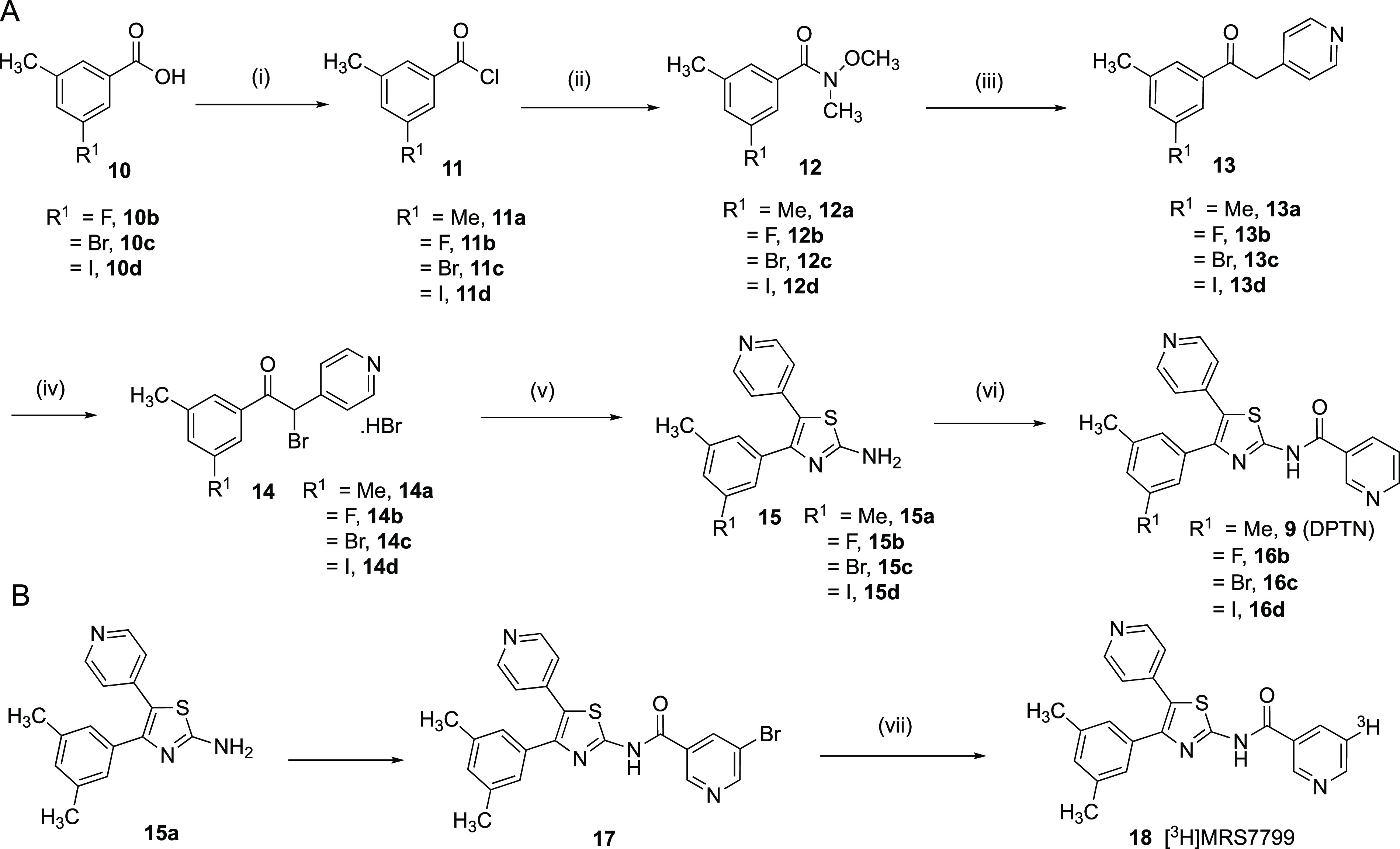
Reagents and conditions: (i) (COCl)2 (1.5 to 2.0 equiv), cat. DMF (10 μL), toluene, 0 °C to room temperature, 89–96%; (ii) N,O-dimethylhydroxylamine hydrochloride (1.2 to 1.5 equiv), K2CO3 (2.0 equiv), EtOAc–H2O (2:1), 0 °C to room temperature, 16–18 h, 86–98%; (iii) 4-picoline (1.2 to 1.5 equiv), LDA (1.0 M in THF/hexane (1.0–4.0 equiv), THF, −78 °C to room temperature, 2 to 3 h, 58–71%; (iv) Br2 (1.0 equiv), AcOH, 4 h, 80 °C, 43–90%; (v) methylthiourea (1.1 equiv), Et3N (2.1 to 3.0 equiv), ACN, reflux, 3 h, 38–87%; (vi) nicotinoyl chloride hydrochloride or 5-bromonicotinoyl chloride (1.5 equiv), DMAP (0.3 equiv) in DMA or NMP, 80 °C, 16 h; sat. NaHCO3, 39–87%; (vii) Pd/C, tritum gas.
Compounds 16d and 17 also served as intermediates for the substitution of functionalized chains at the 3-iodophenyl position (Scheme 2). Heck (19) and Sonogashira (20) reactions were utilized to install extended, rigidified substituents. These analogues were intended to probe the environment of the A3AR binding site of 9. If one or more chain-extended analogues would show considerable affinity, then it would enable the design of additional conjugates at those positions. A similar strategy of appending arylethynyl or alkenyl groups to an aromatic ring was successfully applied with 1,4-dihydropyridines as hA3AR antagonists.13
Scheme 2. Synthesis of Chain-Extended Analogues of 9.

Reagents and conditions: (i) methyl acrylate, Pd(OAc)2 (10 mol %), P(o-tol)3 (10 mol %), DMA, 90 °C, 20 h, 33%; (ii) 5-chloro-thien-2-yl acetylene, Pd(PPh3)2Cl2 (10 mol %), CuI (10 mol %), Et3N, DMF, 80 °C,16 h, 59%.
We measured the AR affinities of 9 and its analogues (Table 1) using standard radioligand binding assays with radiolabeled A1 and A2A antagonists and an A3 agonist.12,13,15,25−27 The ARs were expressed in HEK293 cell membranes, as previously reported. The inhibition constants (Ki) of 9 at the human, mouse, and rat A3ARs were 1.65, 9.61, and 8.53 nM using [125I]3 as the radioligand.13 Curiously, unlike most other reported A3AR antagonists, there was moderate affinity of 9 at the A2BAR (Ki ≈ 200 nM in the three species) and the A1AR and A2AAR.13 Other analogues 16b–16d with halo substitution of the nicotinyl ring were moderately reduced in hA3AR affinity. The rank order of potency for five-position substituents in A3AR binding was: CH3 > I, Br > F. 3-Bromo 16c and 3-iodo 16d analogues were 230- and 1400-fold selective, respectively, for hA3AR compared with hA1AR, although the A1AR binding inhibition by 16c was incomplete (Figure S1, Supporting Information). At the mARs, the 3-iodo derivative 16d was ∼200-fold selective for mA3AR compared with mA1AR. However, 3-fluoro analogue 16b is a mixed hA1 and hA3AR antagonist. The products of the Heck 19 and Sonogashira 20 reactions were similarly tested in hA3AR binding, and the results indicated the affinity of methyl acrylate derivative 19 to be four times greater than that of 5-chloro-thien-2-yl-ethynyl derivative 20 and only three times less potent than that of the parent 16d. Compound 19 was 83-fold selective for hA3AR compared with hA1AR and displayed moderate mA3AR affinity. Thus compounds 16c, 16d, 17, and 20 were weaker than the reference antagonist 9 in A1 and A2AAR binding, and 16d was more A3AR-selective than 9, although it was of lower affinity. The same arylalkyne moiety in 20 is present at the adenine C2 position of highly potent and selective A3AR agonists.12 Similarly, 4-arylalkyne moieties on a 1,4-dihydropyridine scaffold were previously found to induce high hA3AR-selectivity in antagonists.1
Table 1. Binding Affinity of Various Known and Newly Synthesized A3AR Antagonists (Affinity at the Human Receptors, Unless Noted; m, Mouse; r, Rat)a.
| compound | A1 (Ki, nM or % at 10 μM) | A2A (Ki, nM or % at 10 μM) | A2B (Ki, nM or % at 10 μM)b | A3 (Ki, nM) |
|---|---|---|---|---|
| 6c | 35.4 ± 4.2%, 27.4 ± 2.0% (m), 25.5 ± 1.9% (r) | 16.4 ± 2.9%, 25.1 ± 8.1% (m), 7.4 ± 3.2% (r) | 34.5 ± 4.6%, 20.2 ± 5.0% (m), 38.8 ± 4.7% (r) | 43.9 ± 7.6, 349 ± 72 (m), 216 ± 65 (r) |
| 9c | 162 ± 49, 411 ± 113 (m), 333 ± 58 (r) | 121 ± 42, 830 ± 92 (m), 1150 ± 80 (r) | 230 ± 40, 189 ± 61 (m), 163 ± 23 (r) | 1.65 ± 0.57, 9.61 ± 2.27 (m), 8.53 ± 1.22 (r) |
| 16b | 78.7 ± 3.3 | 3550 | ND | 69.2 ± 11.9 |
| 16c | 1450 ± 600, 252 ± 64 (m) | –7.3 ± 7.2%, 29 ± 5% (m) | ND | 6.34 ± 1.79, 22.3 ± 6.1 (m) |
| 16d | 8770 ± 800, 2640 (m) | –15 ± 19%, 7.4 ± 4.2% (m) | ND | 6.12 ± 1.92, 12.4 ± 1.6 (m) |
| 17 | 38 ± 2% | >1000 | ND | 26.6 ± 7.6 |
| 19 | 1570 ± 390 | 15 ± 17% | ND | 18.9 ± 10.9, 120 (m) |
| 20 | 21 ± 2% | –0.5 ± 8.9% | ND | 80.7 ± 5.8 |
Radioligands used (concentration): [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]DPCPX, 21, A1, 0.5 nM), [3H]4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethyl]phenol ([3H]ZM241385, 22, A2A, 1.0 nM) and [3H]21 (A2B, 5 nM), and [125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I-AB-MECA 3, A3, 0.1 nM), during 60 min incubations at 25 °C. N-Ethylcarboxamido-adenosine (NECA, 23, 100 μM) was used to define nonspecific binding.
ND, not determined.
Data from Gao et al.13
The tritiated compound 18 was evaluated as a radiotracer at human, mouse, and rat A3ARs (Figure 1). Specific binding was saturable at the three species homologues, with the highest level of specific compared with nonspecific binding occurring at the hA3AR. Kd values determined at A3ARs in the three species were (nM, n = 4): 0.55 ± 0.17 (human), 3.74 ± 0.75 (mouse), and 2.80 ± 0.29 (rat). Association and dissociation rates were determined at the human A3AR (Figure 1G,I), which provided a kinetic Kd value of 0.76 nM, in close agreement with the other affinity measures of unlabeled 9 and labeled 18. The binding inhibition by known A3AR agonists (1, 23) and selective antagonists (24, 25) is shown in Figure 1H,J, in which the use of agonist radioligand [125I]3 was compared with [3H]18. The Ki values were comparable to previously determined values, and there was consistency between the two radioligands in antagonist affinity. The incomplete inhibition by antagonists 24 and 25 (Figure 1J) has been previously observed with other hydrophobic antagonists and may be related to a long residence time.13,28 However, agonists displayed higher affinity using the agonist radioligand compared with [3H]18.
Figure 1.

Pharmacology of radioligand [3H]18 in A3AR binding saturation, inhibition, and kinetic experiments. Specific binding saturation (A,C,E) and total (●) and nonspecific (■) binding (B,D,F) at three species homologues (human, mouse, rat) of A3AR expressed in HEK293 cells using 100 μM 23 to define nonspecific binding. Data shown are representative experiments performed in duplicate; the Kd values from four independent experiments are listed in the text. Association (G, k1 = 0.297 min–1) and dissociation (I, k–1 = 0.209 min–1; t1/2 = 3.32 min) kinetics of [3H]18 (1 nM) at the hA3AR. (H,J) Binding inhibition at hA3AR and comparison of [3H]18 (1 nM) with the agonist radioligand [125I]3 (0.1 nM) as a tracer. Competing ligands were agonists (A3-selective 1, nonselective 23) and hA3-selective antagonists (N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzeneacetamide, MRS1220 24; 1,4-dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyridinedicarboxylic acid 3-ethyl-5-[(3-nitrophenyl)methyl] ester, MRS1334 25).13Ki values (nM) for the inhibition of antagonist [3H]18 binding: 24, 3.24; 25, 6.49; 23, 44.1; 1, 0.96. Ki values (nM) for the inhibition of agonist [125I]3 binding: 24, 2.97; 25, 10.6; 23, 19.6; 1, 0.37. Corresponding published Ki values for the inhibition of [125I]3 binding: 24, 0.96 ± 0.32; 25, 4.58 ± 0.89; 23, 26; 1, 1.74 ± 0.36.13,27,29
Off-target activities of selected antagonist derivatives were measured in radioligand binding assays by the NIMH Psychoactive Drug Screening Program (PDSP).30 The data are represented as either a Ki value or a % inhibition at 10 μM, if <50%. Compound 9 had no significant binding (>50% inhibition at 10 μM) at any of the 45 receptors, transporters, or channels examined.13 However, compound 16d (Table S1, Supporting Information) had several weak binding hits (Ki in μM), that is, at the σ2 receptor (3.69), TSPO (1.33 ± 0.23 μM), and D3 (0.34 ± 0.06 μM) and D4 (1.24 ± 0.02 μM) dopamine receptors. Compound 19 bound to the M5 muscarinic (3.7 μM) and 5-HT2A (0.96 ± 0.06 μM) and 5-HT2C (0.79 ± 0.31 μM) serotonin receptors.
Because one objective was to determine the suitability of this chemical series for PET ligands for A3AR in vivo brain imaging, we calculated the CNS multiparameter optimization (MPO) score, predictive of crossing the blood–brain barrier (BBB).31 The score for 9 was 3.81, which indicates possible bioavailability in the brain. Also, on the basis of the criteria of Rankovic et al. (tPSA (topological surface area) of 68 Å2 and MW 386),32 the likelihood of crossing the BBB was judged to be >50%. The Lipinski properties were also favorable for potential oral bioavailability (1 HBD, 4 HBA, LogP 4.68).33
Therefore, the in vivo pharmacokinetics (PK) of 9 was studied in male Sprague–Dawley rats. The brain distribution of 9 was determined (Figure 2, Table 2) following a bolus injection of 1 mg/kg (i.v.). A good tissue distribution (Vd) was observed, with moderate-to-high clearance from the body (CL) corresponding to 70% of liver blood flow. There was excellent brain distribution (brain/plasma ratio ∼1). However, the compound was highly bound in brain and plasma, with a low free fraction in brain and plasma (Table S2, Supporting Information). The brain/plasma ratio was 1.0 to 1.8. These observations were consistent with a lipophilic compound that readily crosses the BBB.
Figure 2.

Plasma and brain concentration–time profiles for compound 9 (i.v. bolus, in 10% DMSO/30% PEG400 in 60% PBS, pH 8.5) in male Sprague–Dawley rats.
Table 2. PK Parameters in Male Sprague Dawley Rats Following a 1.0 mg/kg Bolus Dose of 9.
| matrix | C0 (ng/mL) | Cavg (ng/mL) | CL (mL/min/kg) | Vd (L/kg) | T1/2 (h) | Fub |
|---|---|---|---|---|---|---|
| plasma | 628 | 52 | 40 | 1.8 | 0.8 | 0.03 |
| brain | 760 | 95 | 43 | 1.9 | 0.7 | 0.03 |
The estimated percent receptor occupancy of unbound 9 based on the previously published Ki value at the rat A3AR23 as a function of time following a 3 mg/kg intravenous dose was close to 100% at early time points (Table S3, Figure S2, Supporting Information). The calculation was performed with no residence time assumptions.
In the absence of an experimental structure, we and others have used hA3AR homology modeling to predict the binding modes of A3AR antagonists.8,12,22,23,26,28,29 Considering the high sequence identity of hA3AR and hA1AR (46% full sequence, 56% transmembrane (TM) helices, as reported in GPCRdb42), an antagonist-bound X-ray structure of hA1AR (PDB ID: 5UEN(43)) was used as a template for homology modeling of hA3AR. In addition, the extracellular loop (EL) 2 was modeled using a previously generated intermediate-state, agonist-bound hA3AR model as a template, to be consistent with previous works.44 The same templates were also used to generate a model of mA3AR, which has high sequence identity with hA3AR (72% full sequence, 83% TM). Both hA3AR and mA3AR models were refined by minimizing the nonconserved residues and by the induced fit docking of known antagonists: MRS1220 (hA3AR Ki ≈ 0.7 nM) and 6 (mA3AR Ki ≈ 349 nM) for hA3AR and mA3AR,13 respectively (Figures S4 and S5). The newly generated models show the typical characteristics of inactive ARs as compared with the previously reported intermediate agonist-bound hA3AR model,44 such as a translocation of TM3, lack of the bulge of TM5, outward movement of the TM7 intracellular portion and inward movement of the TM5 and TM6 intracellular portions,46 placing the “toggle switch” W243 (hA3AR) closer to TM3 (Figure S6).
The obtained models were used to generate a hypothetical binding mode of 9 at the hA3AR and mA3AR orthosteric binding sites through docking,29 and representative docking poses at the two receptors are shown for comparison (Figure 3). The conformations of the human Q167 and V169 and of the murine H168 and R170 were optimized with an induced fit docking procedure. The predicted binding modes of 9 at hA3AR and mA3AR are similar, with the thiazole scaffold at the center of the orthosteric binding pocket. The thiazole is involved in a π–π stacking interaction with F168/F169 (hA3AR/mA3AR) on EL2 and a hydrophobic contact with L246/L247 on TM6. The sulfur atom is proximal to the carbonyl group of N250/N251 (TM6), which might correspond to an uncommon, but already reported, carbonyl–S interaction, although with suboptimal geometry.45 The nicotinamide is located deep within the binding pocket, where it is surrounded by L90/L91, L91/L92, T94/T95, and H95/H96 on TM3; M177/M178 and I186/I187 on TM5; and W243/W244 and N250/N251 on TM6. The nicotinamide ring appears to be involved in a π–π T-shaped interaction with W243/W244, whereas the amide carbonyl moiety is H-bonded to N250/N251, reflecting the importance of the carbonyl group in this antagonist SAR.23 The A3AR affinity-enhancing23 4-pyridyl moiety at position five points toward the receptor’s extracellular portion in proximity to Q167 (hA3AR) or H168 and R170 (mA3AR) on EL2, which can potentially stabilize the pyridyl nitrogen. The aryl group at position four is surrounded by TMs 1, 2, 3, and 7, with a potential π–π stacking with H272/H273 on TM7. The hypothetical binding mode of compound 9 is compatible with the extended methyl acrylate moiety of 19, which maintains considerable binding affinity at both the human (Ki ≈ 18.9 nM) and mouse (Ki ≈ 120 nM) receptors. The extended methyl acrylate group of 19 is, in fact, predicted to occupy a cleft between TM2 and TM3 (Figure S7). The rigid extensions at the adenine two position of highly selective A3AR agonists also are predicted to point toward TM2 when receptor-bound.12
Figure 3.
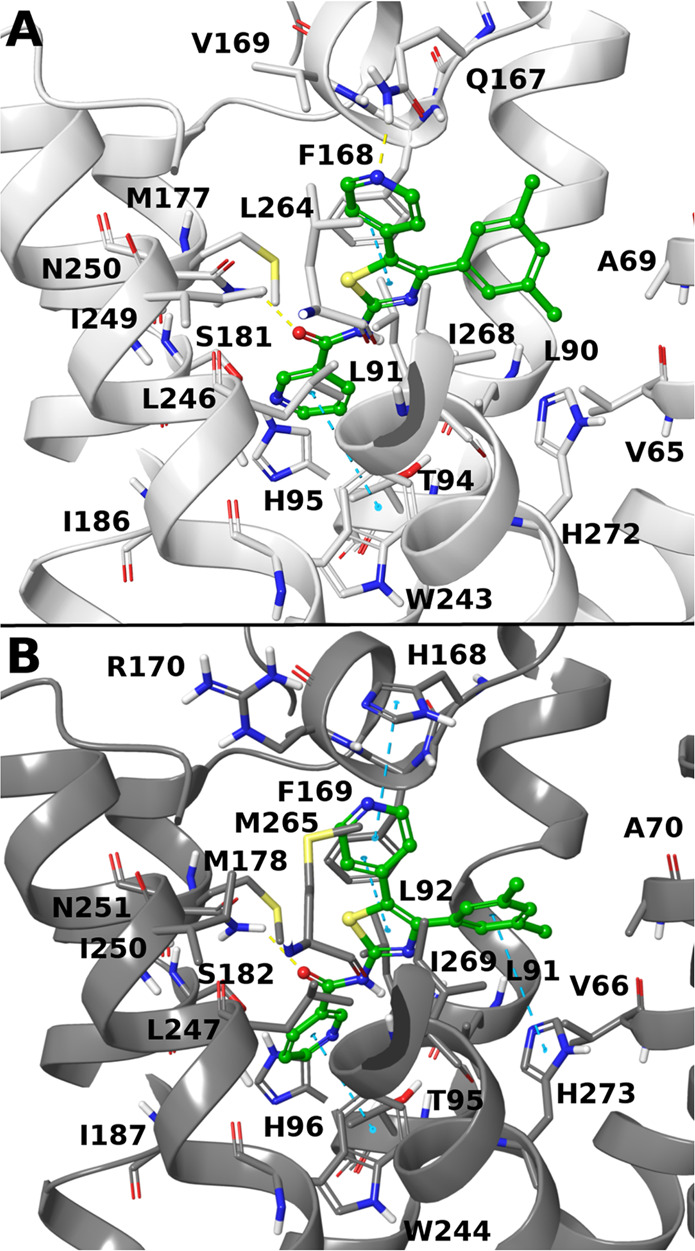
Docking pose of compounds 9 (green) at hA3AR (white, A) and mA3AR (gray, B) homology models (coordinate files in Supporting Information). Residues within 3 Å of the ligand are rendered by sticks. Yellow lines indicate H bonds, and cyan lines indicate π–π stacking interactions between the ligand and the receptor.
In conclusion, compounds 9 and 19 were predicted to bind with a similar conformation at hA3AR and mA3AR; in fact, most (∼75%) of the residues predicted to be in ligand contact (within 5 Å) are conserved between the two homologues, with the only differences being M66/I67 (TM2), T87/S88 (TM3), L89/V90 (TM3), Q167/H168 (EL2), V169/R170 (EL2), M174/L175 (TM5), I253/S254 (TM6), V259/I260 (EL3), L264/M265 (TM7), and Y265/C266 (TM7).
The Gi-coupled A3AR is a therapeutic target for inflammatory and ischemic conditions. Although diverse chemotypes have been found as A3AR-selective antagonists,18,22,23,25,29,34−37 their utility is mostly limited to higher species.13 We now report the more complete characterization of a reported antagonist 9 that is less variable across species, including its use as radioligand 18, along with a small set of analogues.
Other thiazole and thiazol-2-amine derivatives have been explored as antagonists of the A3AR and other ARs.25,27−30,34−41 Structures of the key reported thiazol-2-amine derivatives that have high hA3AR affinity in relation to compound 9 are shown in Table S4 (Supporting Information).
Compound 9 is one of the few A3AR antagonists suitable for use in rodent,13 and primate species. The only other compound in that category is pyridine derivative 6, but compound 9 is 25, 36, and 27 times more potent than 6 at rat, mouse, and human A3ARs, respectively. Interestingly, a 3-iodo derivative 16d was shown to be potent and selective for the A3AR in both humans and mice.
In conclusion, we have expanded the SAR of N-(4-aryl-5-(pyridin-4-yl)thiazol-2-yl)nicotinamides as A3AR antagonists. Radiolabeling of 9 now provides a useful and versatile radiotracer for binding studies in multiple species, and the unlabeled compound was shown to readily cross the BBB. Our data suggest that this antagonist in both labeled and unlabeled forms will be widely applicable and will fill an unmet need for A3AR characterization and drug discovery.
Acknowledgments
We acknowledge support from Astrocyte Pharmaceuticals (NIDDK CRADA 15-8056), the NIH Intramural Research Program (NIDDK, ZIADK031117), and the National Institutes of Health (NHLBI R01 grant HL133589). We thank John Lloyd (NIDDK) for mass spectral determinations. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and the National Institute of Mental Health’s Psychoactive Drug Screening Program (contract no. HHSN-271-2008-00025-C) for screening data.
Glossary
Abbreviations
- AR
adenosine receptor
- HEK293
human embryonic kidney 293
- DAT
dopamine transporter
- GPCR
G-protein-coupled receptor
- MD
molecular dynamics
- PDSP
NIMH Psychoactive Drug Screening Program
- PEG
polyethyleneglycol
- PK
pharmacokinetics
- rmsd
root-mean-square deviation
- SAR
structure–activity relationship
- TM
transmembrane helical domain
- TSPO
translocator protein.
Supporting Information Available
docking pose coordinates (pdb) of (h); molecular formula strings. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00685.
Chemical synthetic procedures and spectra, PDSP screening results, and molecular modeling methods (PDF)
Docking pose coordinates (pdb) of 9 (h) (PDB)
Docking pose coordinates (pdb) of 9 (m) (PDB)
Docking pose coordinates (pdb) of 24 (h) (PDB)
Docking pose coordinates (pdb) of receptor-bound 6 (m) (PDB)
Molecular formula strings (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Jacobson K. A.; Merighi S.; Varani K.; Borea P. A.; Baraldi S.; Aghazadeh Tabrizi M.; Romagnoli R.; Baraldi P. G.; Ciancetta A.; Tosh D. K.; Gao Z.-G.; Gessi S. A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072. 10.1002/med.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S.; Stemmer S. M.; Zozulya G.; Ochaion A.; Patoka R.; Barer F.; Bar-Yehuda S.; Rath-Wolfson L.; Jacobson K. A.; Fishman P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011, 226, 2438–2447. 10.1002/jcp.22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani K.; Vincenzi F.; Targa M.; Paradiso B.; Parrilli A.; Fini M.; Lanza G.; Borea P. A. The stimulation of A3 adenosine receptors reduces bone-residing breast cancer in a rat preclinical model. Eur. J. Cancer. 2013, 49, 482–491. 10.1016/j.ejca.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Ranjan A.; Iyer S. V.; Iwakuma T. Suppressive roles of A3AR and TMIGD3 i1 in osteosarcoma malignancy. Cell Cycle 2017, 16 (10), 903–904. 10.1080/15384101.2017.1308153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannone R.; Miele L.; Maiolino P.; Pinto A.; Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am. J. Cancer Res. 2014, 4, 172–181. [PMC free article] [PubMed] [Google Scholar]
- Fishman P.; Cohen S. The A3 adenosine receptor (A3AR): therapeutic target and predictive biological marker in rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. 10.1007/s10067-016-3202-4. [DOI] [PubMed] [Google Scholar]
- Little J.; Ford A.; Symons-Liguori A. M.; Chen Z.; Janes K.; Doyle T.; Xie J.; Luongo L.; Tosh D.; Maione S.; Bannister K.; Dickenson A.; Vanderah T. W.; Porreca F.; Jacobson K. A.; Salvemini D. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138, 28–35. 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonioli L.; Lucarini E.; Lambertucci C.; Fornai M.; Pellegrini C.; Benvenuti L.; Di Cesare Mannelli L.; Spinaci A.; Marucci G.; Blandizzi C.; Ghelardini C.; Volpini R.; Dal Ben D. The anti-inflammatory and pain-relieving effects of AR170, an adenosine A3 receptor agonist, in a rat model of colitis. Cells 2020, 9 (6), 1509. 10.3390/cells9061509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman P.; Cohen S.; Itzhak I.; Amer J.; Salhab A.; Barer F.; Safadi R. The A3 adenosine receptor agonist, namodenoson, ameliorates non-alcoholic steatohepatitis in mice. Int. J. Mol. Med. 2019, 44, 2256–2264. 10.3892/ijmm.2019.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Li X.; Deng P.; Wang D.; Bai X.; Li Y.; Luo C.; Belguise K.; Wang X.; Wei X.; Xia Z.; Yi B. Activation of adenosine A3 receptor reduces early brain injury by alleviating neuroinflammation after subarachnoid hemorrhage in elderly rats. Aging (Albany NY) 2021, 13, 694–713. 10.18632/aging.202178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston T. E.; Hinz S.; Müller C. E.; Holstein D. M.; Wendling J.; Melton R. J.; Campbell M.; Korinek W. S.; Suresh R. R.; Sethre-Hofstad D.; Gao Z. G.; Tosh D. K.; Jacobson K. A.; Lechleiter J. D. Nucleotide P2Y1 receptor agonists are In vitro and In vivo prodrugs of A1/A3 adenosine receptor agonists: Implications for roles of P2Y1 and A1/A3 receptors in health and disease. Purinergic Signalling 2020, 16, 543–559. 10.1007/s11302-020-09732-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Salmaso V.; Rao H.; Campbell R.; Bitant A.; Gao Z. G.; Auchampach J. A.; Jacobson K. A. Direct comparison of (N)-methanocarba and ribose-containing 2-arylalkynyladenosine derivatives as A3 receptor agonists. ACS Med. Chem. Lett. 2020, 11, 1935–1941. 10.1021/acsmedchemlett.9b00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Suresh R. R.; Jacobson K. A. Pharmacological characterization of DPTN and other selective A3 adenosine receptor antagonists. Purinergic Signalling 2021, 17, 737–746. 10.1007/s11302-021-09823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C. T.; Li A.; Banerjee J.; Gao Z. G.; Kambayashi T.; Jacobson K. A.; Civan M. M. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signalling 2014, 10, 465–475. 10.1007/s11302-014-9409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Blaustein J.; Gross A. S.; Melman N.; Jacobson K. A. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem. Pharmacol. 2003, 65, 1675–1684. 10.1016/S0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. N.; Wang Y.; Garcia-Roves P. M.; Bjornholm M.; Fredholm B. B. Adenosine A3 receptors regulate heart rate, motor activity and body temperature. Acta Physiol 2010, 199, 221–230. 10.1111/j.1748-1716.2010.02091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M. E.; Gallo-Rodriguez C.; Jacobson K. A.; Stiles G. L. 125I-4-Aminobenzyl-5′-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol. 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Heitman L. H.; IJzerman A. P.; van der Es D. Molecular probes for the human adenosine receptors. Purinergic Signalling 2021, 17, 85–108. 10.1007/s11302-020-09753-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Teng B.; Wu H.; Joshi B. V.; Griffiths G. L.; Jacobson K. A. Synthesis and pharmacological characterization of [125I]MRS1898, a high affinity, selective radioligand for the rat A3 adenosine receptor. Purinergic Signalling 2009, 5, 31–37. 10.1007/s11302-008-9107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnouri M. W.; Jepards S.; Casari A.; Schiedel A. C.; Hinz S.; Müller C. E. Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signalling 2015, 11 (3), 389–407. 10.1007/s11302-015-9460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balber T.; Singer J.; Berroterán-Infante N.; Dumanic M.; Fetty L.; Fazekas-Singer J.; Vraka C.; Nics L.; Bergmann M.; Pallitsch K.; Spreitzer H.; Wadsak W.; Hacker M.; Jensen-Jarolim E.; Viernstein H.; Mitterhauser M. Preclinical In Vitro and In Vivo evaluation of [18F]FE@SUPPY for cancer PET imaging: Limitations of a xenograft model for colorectal cancer. Contrast Media Molecular Imaging 2018, 2018, 1–9. 10.1155/2018/1269830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan K.; Lagarias P.; Stampelou M.; Stamatis D.; Hoare S.; Safitri D.; Klotz K.-N.; Vrontaki E.; Kolocouris A.; Ladds G. Pharmacological characterisation of novel adenosine A3 receptor antagonists. Sci. Rep. 2020, 10, 20781. 10.1038/s41598-020-74521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwatashi S.; Arikawa Y.; Matsumoto T.; Uga K.; Kanzaki N.; Imai Y. N.; Ohkawa S. Synthesis and biological activities of 4-phenyl-5-pyridyl-1,3-thiazole derivatives as selective adenosine A3 antagonists. Chem. Pharm. Bull. (Tokyo) 2008, 56 (8), 1126–1137. 10.1248/cpb.56.1126. [DOI] [PubMed] [Google Scholar]
- Nahm S.; Weinreb S. M. N-methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22 (39), 3815–3818. 10.1016/S0040-4039(01)91316-4. [DOI] [Google Scholar]
- Jung K.-Y.; Kim S.-K.; Gao Z.-G.; Gross A. S; Melman N.; Jacobson K. A; Kim Y.-C. Structure activity relationships of thiazole and thiadiazole derivatives as potent adenosine A3 receptor antagonists. Bioorg. Med. Chem. 2004, 12, 613–623. 10.1016/j.bmc.2003.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Salmaso V.; Rao H.; Bitant A.; Fisher C. L.; Lieberman D. I.; Vorbrüggen H.; Reitman M. L.; Gavrilova O.; Gao Z. G.; Auchampach J. A.; Jacobson K. A. Truncated (N)-methanocarba nucleosides as partial agonists at mouse and human A3 adenosine receptors: Affinity enhancement by N6-(2-phenylethyl) substitution. J. Med. Chem. 2020, 63, 4334–4348. 10.1021/acs.jmedchem.0c00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman A.; Gao Z. G.; Kumar D.; Wan T. C.; Gizewski E.; Auchampach J. A.; Jacobson K. A. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg. Med. Chem. Lett. 2008, 18, 2813–2819. 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L.; Burger W. A. C.; van Veldhoven J. P. D.; Kuiper B. J.; van Duijl T. T.; Lenselink E. B.; Paasman E.; Heitman L. H.; IJzerman A. P. Structure-affinity relationships and structure-kinetics relationships of pyrido[2,1-f]purine-2,4-dione derivatives as human adenosine A3 receptor antagonists. J. Med. Chem. 2017, 60 (17), 7555–7568. 10.1021/acs.jmedchem.7b00950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Tosh D. K.; Gao Z.-G.; Yu J.; Suresh R. R.; Rao H.; Romagnoli R.; Baraldi P. G.; Aghazadeh Tabrizi M. Chapter 7. Medicinal chemistry of the A3 adenosine receptor. The Adenosine Receptors, The Receptors 2018, 34, 169–198. 10.1007/978-3-319-90808-3_7. [DOI] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X.-P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R. C.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–439. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z. Retraction of “CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain”. J. Med. Chem. 2019, 62 (3), 1699. 10.1021/acs.jmedchem.8b01388. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Hosey C. M.; Ursu O.; Oprea T. I. BDDCS, the Rule of 5 and drugability. Adv. Drug Delivery Rev. 2016, 101, 89–98. 10.1016/j.addr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press N.; Keller T.; Tranter P.; Beer D.; Jones K.; Faessler A.; Heng R.; Lewis C.; Howe T.; Gedeck P. New highly potent and selective adenosine A3 receptor antagonists. Curr. Top. Med. Chem. 2004, 4, 863–870. 10.2174/1568026043451023. [DOI] [PubMed] [Google Scholar]
- Abdelrahman A.; Yerande S. G.; Namasivayam V.; Klapschinski T. A.; Alnouri M. W.; El-Tayeb A.; Müller C. E. Substituted 4-phenylthiazoles: Development of potent and selective A1, A3 and dual A1/A3 adenosine receptor antagonists. Eur. J. Med. Chem. 2020, 186, 111879. 10.1016/j.ejmech.2019.111879. [DOI] [PubMed] [Google Scholar]
- Pandya D. H.; Sharma J. A.; Jalani H. B.; Pandya A. N.; Sudarsanam V.; Kachler S.; Klotz K. N.; Vasu K. K. Novel thiazole-thiophene conjugates as adenosine receptor antagonists: synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. Lett. 2015, 25 (6), 1306–1309. 10.1016/j.bmcl.2015.01.040. [DOI] [PubMed] [Google Scholar]
- Yaziji V.; Rodríguez D.; Gutiérrez-De-Terán H.; Coelho A.; Caamaño O.; García-Mera X.; Brea J.; Loza M. I.; Cadavid M. I.; Sotelo E. Pyrimidine derivatives as potent and selective A3 adenosine receptor antagonists. J. Med. Chem. 2011, 54 (2), 457–471. 10.1021/jm100843z. [DOI] [PubMed] [Google Scholar]
- Webb T. R.; Melman N.; Lvovskiy D.; Ji X. d.; Jacobson K. A. The utilization of a unified pharmacophore query in the discovery of new antagonists of the adenosine receptor family. Bioorg. Med. Chem. Lett. 2000, 10, 31–34. 10.1016/S0960-894X(99)00583-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Muijlwijk-Koezen J. E.; Timmerman H.; Vollinga R. C.; Frijtag von Drabbe Kunzel J.; de Groote M.; Visser S.; IJzerman A. P. Thiazole and thiadiazole analogues as a novel class of adenosine receptor antagonists. J. Med. Chem. 2001, 44, 749–762. 10.1021/jm0003945. [DOI] [PubMed] [Google Scholar]
- Inamdar G. S.; Pandya A. N.; Thakar H. M.; Sudarsanam V.; Kachler S.; Sabbadin D.; Moro S.; Klotz K.-N.; Vasu K. K. New insight into adenosine receptors selectivity derived from a novel series of [5-substituted-4-phenyl-1,3-thiazol-2-yl] benzamides and furamides. Eur. J. Med. Chem. 2013, 63, 924–934. 10.1016/j.ejmech.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Trifilieff A.; Keller T. H.; Press N. J.; Howe T.; Gedeck P.; Beer D.; Walker C. CGH2466, a combined adenosine receptor antagonist, p38 mitogen-activated protein kinase and phosphodiesterase type 4 inhibitor with potent in vitro and in vivo anti-inflammatory activities. Br. J. Pharmacol. 2005, 144 (7), 1002–1010. 10.1038/sj.bjp.0706132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pándy-Szekeres G.; Munk C.; Tsonkov T. M.; Mordalski S.; Harpsøe K.; Hauser A. S.; Bojarski A. J.; Gloriam D. E. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. 10.1093/nar/gkx1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhova A.; Thal D. M.; Nguyen A. T.; Vecchio E. A.; Jörg M.; Scammells P. J.; May L. T.; Sexton P. M.; Christopoulos A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877. 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- Tosh D. K.; Salmaso V.; Rao H.; Bitant A.; Fisher C. L.; Lieberman D. I.; Vorbrüggen H.; Reitman M. L.; Gavrilova O.; Gao Z.-G.; Auchampach J. A.; Jacobson K. A. Truncated (N)-methanocarba nucleosides as partial agonists at mouse and human A3 adenosine receptors: Affinity enhancement by N6-(2-phenylethyl) substitution. J. Med. Chem. 2020, 63, 4334–4348. 10.1021/acs.jmedchem.0c00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- Carpenter B.; Lebon G. Human adenosine A2A receptor: molecular mechanism of ligand binding and activation. Front. Pharmacol. 2017, 8, 898. 10.3389/fphar.2017.00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.