

Abstract

The NLRP3 inflammasome has now emerged as one of the most appealing drug targets for many inflammation-related diseases. Velutone F, a natural NLPR3 inhibitor, identified in our previous study has been limited in application by its low in planta abundance, weak activity, and complicated synthetic routes. To address these needs, structural optimization of velutone F led to a series of novel NLRP3 inhibitors. Among them, compound 14c exerted remarkable inhibitory activity with an IC50 value in the nanomolar range (251.1 nM) and was approximately 5-fold more potent than velutone F. Moreover, the synthesis method of 14c was simple, easy to handle, and scalable. Compound 14c could suppress NLRP3 inflammasome activation by attenuating ASC speck formation. Most importantly, compound 14c reduced peritoneal neutrophil influx in mice and IL-1β in the spleen in the MSU-induced peritonitis in LPS-primed mouse model. Taken together, compound 14c is a prospective lead compound in the discovery of NLRP3 inflammasome inhibitors.
Keywords: Chalcone analogues, NLRP3 inflammasome, SAR, Anti-inflammation, Selectivity
NLRP3 inflammasome, a key regulator of the inflammatory responses, is formed by an intracellular sensor NLRP3, adaptor (ASC, apoptosis-associated speck-like protein containing a CARD), and effector pro-caspase-1.1 The pyrin domain of NLRP3 could interact with ASC to initiate the inflammasome assembly, which then oligomerizes to recruit pro-caspase-1 through CARD–CARD interactions and enables pro-caspase-1 self-cleavage. Activated caspase-1 continuously cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18 and subsequently triggers a series of inflammatory responses and pyroptosis.2,3 NLRP3 inflammasome that is abnormally activated might lead to many inflammatory and metabolic diseases, including rheumatoid arthritis, nonalcoholic steatohepatitis (NASH), atherosclerosis, inflammatory bowel disease (IBD), gout, and type 2 diabetes.4−7 Meanwhile, NLRP3 inflammasome was also reported to have a close relationship with neuroinflammation in recent years.8,9 Hence, it is considered as a prospective target for the therapy of many inflammation-associated diseases.10
Chalcones are the products of hydroxaldehyde condensation of aromatic aldehydes and ketones,11 and their α,β-unsaturated ketone system could covalently modify different proteins, causing a broad spectrum of biological activities, such as oxygen free radical scavenging, antitumor, anti-inflammatory, and antibacterial activities.12−16 Some chalcone-based compounds have been approved for marketing or have been clinically tested (Figure 1). For example, sofalcone has been used as an antiulcer and mucosal protection drug17 and metochalcone is approved as a choleretic drug.18 Applications for clinical trials of hesperidin methylchalcone have been submitted for the treatment of chronic venous lymphatic insufficiency.19 Licochalcone A was evaluated in numerous clinical research studies to examine the effect of chalcones in inflammatory skin conditions.20,21 Elafibranor is an oral selective PPAR modulator in phase III clinical development for the treatment of patients with primary biliary cholangitis (PBC).22 Ro-09-0410 and its sodium phosphate salt Ro-09-0415 were clinically used to treat rhinovirus infection.23 More recently, several chalcone structures have been reported to inhibit the inflammatory response by affecting the NLRP3 inflammasome,24−27 suggesting the potential anti-inflammation medical use of chalcones. However, the low potency of the reported compounds (IC50 values at the micromolar level), lack of selectivity research with NLRP3, and pharmacokinetics evaluation limited further preclinical study. Therefore, it is of great interest to seek a novel chalcone of high activity, selectivity, and favorable drug-like properties against NLRP3 inflammasome activation.
Figure 1.

Several structures of marketed or clinical chalcones.
In our previous research, we reported a furanochalcone velutone F isolated from Millettia velutina that exhibited an inhibitory effect against IL-1β production in THP-1 through inhibiting the activation of NLRP3 inflammasome.28 However, its application for further study with NLRP3 inflammasome was compromised by its low IC50 (1.3 μM) on IL-1β release inhibition and lack of selectivity, indicating potential immunosuppressive effects to increase the risk of infection. Herein, we report the structural modification, characterization, and structure–activity relationship (SAR) of velutone F derived chalcones. After continuous optimization efforts, compound 14c was discovered as a potent NLRP3 inhibitor.
Velutone F was first obtained through total synthesis with the route shown in Scheme 1. Khellin was reacted with KOH in H2O at 120 °C to obtain intermediate 1. Subsequently, intermediate 1 was treated with triflic anhydride at 0 °C and stirred at room temperature to give intermediate 2. Intermediate 2, Et3N, and HCO2H were successively added into DMF at 60 °C to give intermediate 3. The key intermediate 6 was synthesized through two oxidation reactions and one reduction reaction from compound 3, which then underwent an aldol reaction with acetophenone to obtain natural chalcone 7 (velutone F). The SAR study is further discussed, and the strategies for structural optimizations of velutone F are summarized in Figure 2. The specific synthesis methods are shown in the Supporting Information (synthetic methods, procedures, and NMR data).
Scheme 1. Synthesis of Velutone F (Compound 7).

Reagents and conditions: (a) KOH, H2O, 120 °C, 9 h; (b) Tf2O, DMAP, DCM, 0 °C → rt, 5 h; (c) Et3N, HCOOH, PdCl2(dppf), DPPP, DMF, 60 °C, 3 h; (d) NaClO, H2O, 1,4-dioxane, 120 °C, 2 h; (e) LiAlH4, N2, THF, 30 °C, 4 h; (f) MnO2, toluene, 100 °C, 8 h; (g) Acetophenone, 3M NaOH, MeOH, 35–60 °C, 4 h.
Figure 2.

Summarized strategy for structural optimization of velutone F.
The initial SAR study focused on the substitution of R1 (ring A) (Table 1). THP-1 cells were seeded and treated with phorbol 12-myristate 13-acetate (PMA) for 24 h to differentiate human monocytic THP-1 cells into macrophage-like cells. The following day, differentiated cells were first stimulated with LPS (1 μg/mL) for 3 h and then pretreated with compounds (2 μM) for 40 min before induced with nigericin (10 μM) for 40 min. Finally, ELISA assay was used to determine IL-1β in the supernatant to measure the inhibition of the target compounds. Compared with compound 7, replacement of ring A by a six-membered nitrogen-containing heterocycle (8a) was well tolerated. But the removal of ring A (8e) or replacement of ring A by a five-membered heterocycle or three-membered ring (8b–8d) resulted in a loss of activity. Five-membered rings, such as pyrrole (8b) and furan (8c), reduced the activity, indicating that substitution of R1 by a hydrophobic group with a certain steric hindrance is required for the inhibitory effect of IL-1β release. Thus, compounds 8f–8x with different substituted phenyl rings or pyridines were then synthesized and evaluated for their activities. Substituents, including trifluoromethyl (8g and 8h), meta-hydroxyl (8l), meta-amino (8m), fluoro (8i–8k), para-bromo (8p), and meta-methyl (8r), could increase the activity of compound 7 to varying extents or retain the activity. In particular, the 4-fluoro analogue (8k) had almost a fold increase in activity over compound 7. Other substituents, including cyano (8q) ortho-amino (8n), para-amino (8o) and methoxy (8f), were detrimental for the inhibitory effect of IL-1β release. The above results showed that when ring A is the phenyl ring, in most cases, the electronic withdrawing group at the para position or the electronic donating group at the meta position was the most favorable for the activity. Considering a slightly improved inhibition ratio when the phenyl ring was replaced with 2-bromopyridine (8s), thiophene, benzene, and trifluoromethyl were then introduced to replace Br in 8s by the Suzuki coupling reaction to obtain compounds 8u, 8w, and 8x, respectively. The results showed a significantly decreased inhibition ratio compared to that of compound 7, revealing that this position could not tolerate bulky groups. Some other substituents on pyridine (8t, 8v) also displayed weaker activities.
Table 1. Structure and Inhibition of Compounds 7 (Velutone F) and 8a–8x.


N.A. = no activity. Data are the average of experiments repeated three times. The % inhibition values are shown as mean ± SD.
THP-1 cells (2 × 105 cells/mL) were seeded in 48-well plates and then induced with PMA (100 ng/mL) for 24 h to differentiate into macrophages. The medium, instead of Opti-MEM, was used the next day, and differentiated cells were first stimulated with LPS (1 μg/mL) for 3 h and then pretreated with compounds for 40 min before induced with nigericin (10 μM) for 40 min.
We then chose four promising substitutions as shown in Table 1 (phenyl-, 4-fluorophenyl-, 3-(trifluoromethyl)phenyl-, 1-(6-bromo-2-pyridinyl)-) as R1 and used key intermediate 6 as the starting material to synthesize compounds with different linkers to study the necessity of the α,β-unsaturated ketone moiety (9a–9d). The SAR study showed that the α,β-unsaturated carbonyl group of the chalcone providing conjugation between aromatic groups on both ends was the pharmacophore of this class of compounds (Table 2). The corresponding saturated ketone led to the complete loss of activity (9a–9d). And exchanging the positions of ring A and ring B could also significantly reduce potency (10). It has been reported that an acrylamide linker can be used in anti-inflammatory compounds,29,30 and structures with an acrylamide linker have also been reported in the design of NLRP3 inhibitors.31,32 Thus, amide linkage was introduced into the structures 11a–11d. We next changed the α,β-unsaturated carbonyl group to an appendix position to explore if it could improve the activity (13a–13d). Unfortunately, these new backbone structures were unfavorable for the activities.
Table 2. Structures and Inhibitions of Compounds 9a–13d.

N.A. = no activity. Data are the average of experiments repeated three times. The % inhibition values are shown as mean ± SD.
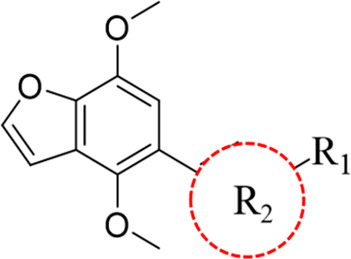
Having identified the necessity of the α,β-unsaturated ketone moiety, we chose compound 8k as a lead compound to further explore the influence of the furan ring on ring B (Table 3). First, the furan ring of 8k was removed to obtain compound 14a. Unexpectedly, this modification resulted in an apparent advance for the activity. Then furan was replaced with methyl (14b), which slightly reduced the inhibitory activity compared with the unsubstituted compound 14a. To evaluate the influence of substituents, we subsequently explored various substituents at the C-4 position of ring B. Among them, compound 14c showed the best activity with an almost 6-fold increase in the inhibitory effect of IL-1β release to positive control compound 7. Replacement of the bromine atom with a substituted benzene ring, heterocycle, or propenyl afforded compounds 15a–15j. Notably, comparing with all of these compounds, only the incorporation of a sulfone group could benefit the activities (15c and 15d). This positive result led us to further evaluate the influence of sulfone. Compound 14c was reacted with different sulfonylamines to provide compounds 16a and 16b. Unfortunately, these compounds showed no activity.
Table 3. Structure and Inhibition of Compounds 14a–16b.


N.A. = no activity. Data are the average of experiments repeated three times. The % inhibition values are shown as mean ± SD.
On the basis of their inhibitory effects on IL-1β release, we determined the intrinsic cytotoxicity of 8k, 14c, 15c, and 15d against THP-1 cells by performing the CCK8 assay. As shown in Figure 3A, all compounds showed no cytotoxicity at 0.05– 6.4 μM. Then their IC50 values of IL-1β release inhibition were further determined by ELISA assay. As shown in Figure 3B and C, 14c displayed the strongest inhibitory activity with an IC50 value of 0.251 μM, approximately 5-fold higher than that of velutone F (IC50: 1.3 μM),28 while 8k, 15c, and 15d exhibited IC50 values of 0.656, 0.325, and 0.407 μM, respectively. Therefore, compound 14c was chosen for the subsequent experiments. Western blot results revealed that caspase-1 p20 and IL-1β processing was dose-dependently reduced in supernatants in THP-1 cells treated with compound 14c (Figure 3D) and 14c also inhibited activation of NLRP3 inflammasome induced by LPS/MSU (Figure S118). Meanwhile, compound 14c treatment did not reduce pro-IL-1β or pro-caspase-1 in cell lysates (Figure 3D). A propidium iodide staining assay was performed to exclude the eventuality that cell death leads to decreased IL-1β release. Figure 3E and F shows that compound 14c is not toxic to THP-1 cells and could avoid nigericin-induced pyroptosis.
Figure 3.

Compound 14c suppresses caspase-1 activation and IL-1β secretion. (A) Cytotoxic evaluation of compounds 8k, 14c, 15c, and 15d. THP-1 cells were preincubated with the tested compounds for 1.5 h. Cell viability was determined by the CCK8 assay. (B) After induction with LPS, THP-1 cells were treated with variable doses of compounds 8k, 14c, 15c, and 15d for 40 min and then stimulated with nigericin for 40 min. Supernatants (SN) were used to detect the levels of cleaved IL-1β. (C) LPS-primed THP-1 cells were treated with various doses of compound 14c for 40 min and then stimulated with nigericin for 40 min. ELISA was used to evaluate the IL-1β levels. (D) Supernatants (SN) were used to detect the levels of IL-1β and caspase-1, and cell extracts (Input) were analyzed for NLRP3, pro-IL-1β, and so on by immunoblotting. (E) THP-1 cells were primed with LPS for 3 h or pretreated with LPS (3 h) in the presence or absence of 14c before nigericin treatment for 40 min. After activation, the cells were incubated with propidium iodide for 10 min at room temperature. Images were captured on a NIKON Eclipse Ts2R/FL inverted microscope. (F) Pyroptosis of cells represented as histograms. Statistical differences were analyzed using one-way ANOVA: # means successful modeling, **p < 0.01, ***p < 0.001, ns, not significant.
To determine whether 14c was a specific inhibitor of NLRP3-dependent IL-1β production, we tested its effects against other well-characterized inflammasomes in differentiated THP-1. Results showed that 14c would affect neither NLRC4 nor AIM2 inflammasomes at concentrations below 1 μM (Figure S117). When the concentration increased, it could dose-dependently inhibit those two inflammasomes (Figure S117), suggesting that 14c was not a selective inhibitor of NLRP3 inflammasome.
Activation of NLRP3 inflammasome is regarded as a two-step process, including priming and activation. The priming phase of NLRP3 inflammasome activation regulates NLRP3 and IL-1β genes and also NLRP3 post-translation.33,34 We then next investigated whether compound 14c inhibited the two-step process. When THP-1 cells were treated with 14c before LPS treatment, compound 14c with a concentration of less than 1 μM had no effects on LPS-induced NLRP3, pro-IL-1β, and TNF-α expression (Figure 4A and B), suggesting that 14c did not affect LPS-induced TLR signaling or the priming phase of NLRP3 inflammasome activation at low concentrations. However, it did inhibit the priming phase when 14c was used at higher concentrations (greater than 1 μM). When THP-1 cells were treated with 14c after LPS treatment, no effects on LPS-induced NLRP3, pro-IL-1β, and TNF-α expression were observed (Figure 4C and D). ASC oligomerization has been considered as an indispensable event for the activation of NLRP3.1 Immunofluorescent (IF) microscopy for endogenous ASC in THP-1 cells showed that ASC oligomerizes into a large protein speck upon activation with nigericin. Treatment of 14c obviously reduced the formation of ASC specks (Figure 4E and F). And NLRP3 deficiency in THP-1 cells ablated nigericin-induced ASC speck formation (Figure 4E and F). Similar conclusions were obtained by immunoblotting. 14c prevented ASC oligomerization in THP-1 cells, while this was absent in NLRP3 KO cells (Figure 4G). As a Michael acceptor, the α,β-unsaturated ketone moiety can covalently bind with cysteine sulfhydryl group in proteins, so it is regarded as an important active group. First, we tested this by using the drug affinity responsive target stability (DARTS) approach35 and CETSA,36 using THP-1 cells or NLRP3ΔLRR protein (Figure S119), which was obtained following the protocol described in the Supporting Information. Increasing doses of 14c (0.01–1 mM) protected NLRP3 in THP-1 cells and NLRP3ΔLRR protein from pronase-mediated degradation (Figure 4H and I). The results showed that 14c improved the thermal stability of NLRP3 in THP-1 cells compared to DMSO blank (Figure 4J). Meanwhile, 14c dose-dependently enhanced the level of NLRP3ΔLRR protein at 57 °C, suggesting the rising stability of NLRP3ΔLRR protein with 14c (Figure 4K). These results showed that compound 14c directly bind to NLRP3.
Figure 4.

Compound 14c could interrupt ASC oligomerization and suppress LPS-induced NLRP3 priming through directly interacting with NLRP3. (A, B) THP-1 cells were treated with 14c for 40 min and then primed with LPS for 3 h. Medium supernatants were analyzed for TNF-α and cell extracts (Input) were analyzed for pro-IL-1β and NLRP3 by immunoblotting. (C, D) THP-1 cells were primed with LPS for 3 h and then treated with 14c for 40 min. (E) Immunofluorescent microscopy was conducted to observe ASC specks. (F) Proportion of cells with ASC specks represented as histograms. (G) Immunoblot analysis of ASC oligomerization in lysates of THP-1 cells treated with 14c. (H) DARTS was used to analyze the relationship between 14C and NLRP3 in THP-1 lysates. (I) DARTS was used to analyze the relationship between 14C and NLRP3△LRR. (J) CETSA assay was performed to detect the stabilization of NLRP3. (K) CETSA performed on purified NLRP3ΔLRR. Statistical differences were analyzed using one-way ANOVA: # means successful modeling, *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
The pharmacokinetic parameters of compound 14c were then investigated. Compound 14c was administrated intravenously (i.v., 10 mg/kg) and orally (p.o., 10, 30, and 90 mg/kg), respectively. LC/MS/MS was performed to quantify plasma levels at multiple time points. As summarized in Table 4, compound 14c exhibited extremely low exposure (14.6–23.53 μg·h/L), poor bioavailability (2.47–13.79%), and high plasma clearance (2201.58–5551.12 L/h/kg) after different doses for oral administration. Following intravenous administration, compound 14c demonstrated relatively lower plasma clearance (133.75 L/h/kg) and higher exposure (105.88 μg·h/L) compared with oral administration. We finally chose intravenous administration for further in vivo characterization.
Table 4. Pharmacokinetic Properties for Compound 14ca,b.
| mode of administration | dosage (mg/kg) | AUC0–t(μg·h/L) | CL (L/h/kg) | Cmax(μg/L) | T1/2 (h) | Tmax (h) | %F |
|---|---|---|---|---|---|---|---|
| intravenous administration | 10 | 105.88 | 133.75 | 81.97 | 3.13 | 0.11 | |
| oral administration | 10 | 14.60 | 2201.58 | 3.35 | 7.43 | 2.11 | 13.79% |
| 30 | 15.84 | 2583.27 | 16.42 | 7.92 | 1.26 | 4.99% | |
| 90 | 23.53 | 5551.12 | 13.59 | 6.08 | 4.21 | 2.47% |
Data are mean value.
Abbreviations: AUC, area under the concentration–time curve; CL, clearance; Cmax, highest plasma concentration of a drug after administration; T1/2, half-life period; Tmax, time to reach Cmax; %F, bioavailability, based on AUC.
The efficacy of compound 14c was further examined in vivo in a PK/PD experiment in MSU-induced peritonitis in a LPS-primed mouse model, which is shown to be a canonical NLRP3 inflammasome dependent model37,38 to understand the relationship between IL-1β changes with compound 14c exposure in the spleen. As shown in Figure 5A, 7 week old male C57BL/6J mice were pretreated with LPS (1 mg/kg, diluted in sterile PBS) at t = −3 h for priming, followed by an intraperitoneal (i.p.) injection of MSU suspension (100 mg/kg) and intravenous injection (i.v.) of compound 14c (10 mg/kg) or velutone F (10 mg/kg) into each mouse at t = 0 h. At the different time points demonstrated in Figure 5A, the mice were sacrificed and spleen samples were collected to analyze the concentrations of IL-1β after the treatment of compound 14c. As further demonstrated in Figure 5B, the level of IL-1β in spleens of LPS/MSU treated mice gradually decreased with the decreasing concentration of 14c. Indeed, as shown in Figure 5C, after 6 h, IL-1β release in the spleen of mice treated with 14c was significantly reduced comparing with those of the model animals (1519 ± 229 vs 805 ± 141) and substantially better than reference compound (velutone F) (805 ± 141 vs 1181 ± 125), indicating that compound 14c could effectively relieve peritonitis in mice. The protection effects of compound 14c was also evaluated by the ratio of neutrophils influx with the same peritonitis model (Figure 5D). Mice that were primed with LPS and then injected with MSU displayed significant peritoneal neutrophil influx (CD11b+ Ly6G+), while administration of 14c (10 mg/kg) significantly reduced the increase of peritoneal neutrophil influx compared with the control group and demonstrated better activity than velutone F (Figure 5E and F).
Figure 5.

PK/PD experiment of compound 14c in MSU-induced peritonitis in LPS-primed mice model. (A) PK/PD experiment timeline scheme. (B) The IL-1β level in spleen was detected by ELISA at 11 time points (0–24 h), and spleen concentrations of compound 14c were analyzed with LC/MS/MS at four time points (30 min, 2 h, 6 h, and 10 h), n = 6/group. (C) Efficiency of compound 14c at 6 h. (D) Timeline scheme of peritonitis influx analysis experiment. (E) Flow cytometry analysis of peritoneal cells isolated from C57BL/6J mice pretreated with LPS (1 mg/kg) or vehicle control stained with Ly6G and CD11b after i.p. administration of MSU (100 mg/kg) and intravenous injection of compound 14c (10 mg/kg) and velutone F (10 mg/kg), n = 6/group. (F) Incidence of CD11b+ Ly6G+ cells in the total population represented as histograms. Statistical differences were analyzed using one-way ANOVA: # means successful modeling, *p < 0.05, ***p < 0.001, ns, not significant.
In summary, 53 novel compounds based on velutone F were designed and synthesized as NLRP3 inflammasome inhibitors. The bioassay results showed that some chalcone derivatives exhibited favorable inhibitory effects of IL-1β release in THP-1 cells. The multiple optimizations guided by SAR analysis for velutone F (IC50: 1.3 μM) helped us discover compound 14c (IC50: 0.251 μM) as the most active compound among all the tested compounds. 14c inhibited the LPS-induced NLRP3 priming process and NLRP3-dependent ASC oligomerization. 14c could also inhibit the activation of AIM2 and NLRC4 inflammasomes. The results of DARTS and CETSA showed that 14c bind sdirectly to NLRP3. In vivo, 14c showed a potent therapeutic effect in a MSU-induced peritonitis model by reducing the expression of IL-1β in the spleen and decreasing neutrophils. More significantly, the synthesis of 14c was simple, with inexpensive starting materials, which satisfied the economic principle of drug design and optimization. Therefore, compound 14c has the potential to be further developed as an effective inhibitor for treating NLRP3 related diseases.
Acknowledgments
The authors are thankful for the financial support from the National Natural Science Foundation of China (81874297, 82174077, and 81973542), the Science and Technology Planning Project of Sichuan Province (2020YJ0056 and 2021YFS0230), and the Foundation of Sichuan University (2018SCUH0067).
Glossary
Abbreviations
- NLRP3
NOD-like receptor family, pyrin domain-containing protein 3
- IL-1β
interleukin-1β
- NASH
nonalcoholic steatohepatitis
- ASC
apoptosis-associated speck-like protein containing a CARD
- IBD
inflammatory bowel disease
- IF
immunofluorescent
- SAR
structure–activity relationship
- LPS
lipopolysaccharide
- ATP
adenosine triphosphate
- THP-1
human myeloid leukemia mononuclear cells
- AIM2
interferon-inducible protein
- ELISA
enzyme-linked immunosorbent assay
- NLRC4
NLR family CARD domain-containing protein 4
- MSU
monosodium urate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00597.
Synthetic methods, procedures, NMR data; pharmacological methods and procedures; characterization of all new compounds; pharmacology (PDF)
Author Contributions
# R.Z., F.H., and M.Z. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Swanson K. V.; Deng M.; Ting J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A.; Masters S. L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. 10.1038/nri.2016.151. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zhang S.; Xiao Y.; Zhang W.; Wu S.; Qin T.; Yue Y.; Qian W.; Li L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longevity 2020, 2020, 4063562. 10.1155/2020/4063562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoseini Z.; Sepahvand F.; Rashidi B.; Sahebkar A.; Masoudifar A.; Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. 10.1002/jcp.25930. [DOI] [PubMed] [Google Scholar]
- Zhen Y.; Zhang H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. 10.3389/fimmu.2019.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhang J. J.; Lin J. H.; Yen G. C. Beneficial Properties of Phytochemicals on NLRP3 Inflammasome-Mediated Gout and Complication. J. Agr. Food Chem. 2018, 66, 765–772. 10.1021/acs.jafc.7b05113. [DOI] [PubMed] [Google Scholar]
- Cocco M.; Pellegrini C.; Martínez-Banaclocha H.; Giorgis M.; Marini E.; Costale A.; Miglio G.; Fornai M.; Antonioli L.; López-Castejón G.; Tapia-Abellán A.; Angosto D.; Hafner-Bratkovič I.; Regazzoni L.; Blandizzi C.; Pelegrín P.; Bertinaria M. Development of an Acrylate Derivative Targeting the NLRP3 Inflammasome for the Treatment of Inflammatory Bowel Disease. J. Med. Chem. 2017, 60, 3656–3671. 10.1021/acs.jmedchem.6b01624. [DOI] [PubMed] [Google Scholar]
- Wang S.; Yuan Y. H.; Chen N. H.; Wang H. B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. 10.1016/j.intimp.2018.12.019. [DOI] [PubMed] [Google Scholar]
- Ising C.; Venegas C.; Zhang S.; Scheiblich H.; Schmidt S. V.; Vieira-Saecker A.; Schwartz S.; Albasset S.; McManus R. M.; Tejera D.; Griep A.; Santarelli F.; Brosseron F.; Opitz S.; Stunden J.; Merten M.; Kayed R.; Golenbock D. T.; Blum D.; Latz E.; Buée L.; Heneka M. T. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. H.; Yang Y. X.; Meng X.; Luo X. Y.; Li X. M.; Shuai Z. W.; Ye D. Q.; Pan H. F. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun. Rev. 2018, 17, 694–702. 10.1016/j.autrev.2018.01.020. [DOI] [PubMed] [Google Scholar]
- Zhuang C.; Zhang W.; Sheng C.; Zhang W.; Xing C.; Miao Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z. Y.; Jin Q. H.; Qu Y. L.; Guan L. P. Chalcone derivatives bearing chromen or benzo[f]chromen moieties: Design, synthesis, and evaluations of anti-inflammatory, analgesic, selective COX-2 inhibitory activities. Bioorg. Med. Chem. Lett. 2019, 29, 1909–1912. 10.1016/j.bmcl.2019.05.051. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xue S.; Li R.; Zheng Z.; Yi H.; Li Z. Synthesis and biological evaluation of novel synthetic chalcone derivatives as anti-tumor agents targeting Cat L and Cat K. Bioorgan. Med. Chem. 2018, 26, 8–16. 10.1016/j.bmc.2017.09.019. [DOI] [PubMed] [Google Scholar]
- Xu M.; Wu P.; Shen F.; Ji J.; Rakesh K. P. Chalcone derivatives and their antibacterial activities: Current development. Bioorg. Chem. 2019, 91, 103133. 10.1016/j.bioorg.2019.103133. [DOI] [PubMed] [Google Scholar]
- Mahapatra D. K.; Bharti S. K.; Asati V. Chalcone Derivatives: Anti-inflammatory Potential and Molecular Targets Perspectives. Curr. Top. Med. Chem. 2017, 17, 3146–3169. 10.2174/1568026617666170914160446. [DOI] [PubMed] [Google Scholar]
- Jung J. C.; Lee Y.; Min D.; Jung M.; Oh S. Practical Synthesis of Chalcone Derivatives and Their Biological Activities. Molecules 2017, 22, 1872. 10.3390/molecules22111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi K.; Watanabe T.; Tanigawa T.; Tominaga K.; Fujiwara Y.; Arakawa T. Sofalcone, a gastroprotective drug, promotes gastric ulcer healing following eradication therapy for Helicobacter pylori: a randomized controlled comparative trial with cimetidine, an H2-receptor antagonist. J. Gastroen. Hepatol. 2010, 25 (Suppl 1), S155–160. 10.1111/j.1440-1746.2010.06232.x. [DOI] [PubMed] [Google Scholar]
- Sahu N. K.; Balbhadra S. S.; Choudhary J.; Kohli D. V. Exploring pharmacological significance of chalcone scaffold: a review. Curr. Med. Chem. 2012, 19, 209–225. 10.2174/092986712803414132. [DOI] [PubMed] [Google Scholar]
- Boyle P.; Diehm C.; Robertson C. Meta-analysis of clinical trials of Cyclo 3 Fort in the treatment of chronic venous insufficiency. Int. Angiol. 2003, 22, 250–262. [PubMed] [Google Scholar]
- Boonchai W.; Varothai S.; Winayanuwattikun W.; Phaitoonvatanakij S.; Chaweekulrat P.; Kasemsarn P. Randomized investigator-blinded comparative study of moisturizer containing 4-t-butylcyclohexanol and licochalcone A versus 0.02% triamcinolone acetonide cream in facial dermatitis. J. Cosme. Dermatol. -US 2018, 17, 1130–1135. 10.1111/jocd.12499. [DOI] [PubMed] [Google Scholar]
- Sulzberger M.; Worthmann A. C.; Holtzmann U.; Buck B.; Jung K. A.; Schoelermann A. M.; Rippke F.; Stäb F.; Wenck H.; Neufang G.; Grönniger E. Effective treatment for sensitive skin: 4-t-butylcyclohexanol and licochalcone A. J. Eur. Acad. Dermato: 2016, 30 (Suppl 1), 9–17. 10.1111/jdv.13529. [DOI] [PubMed] [Google Scholar]
- Schattenberg J. M.; Pares A.; Kowdley K. V.; Heneghan M. A.; Caldwell S.; Pratt D.; Bonder A.; Hirschfield G. M.; Levy C.; Vierling J.; Jones D.; Tailleux A.; Staels B.; Megnien S.; Hanf R.; Magrez D.; Birman P.; Luketic V. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J. Hepatolo. 2021, 74, 1344–1354. 10.1016/j.jhep.2021.01.013. [DOI] [PubMed] [Google Scholar]
- Phillpotts R. J.; Higgins P. G.; Willman J. S.; Tyrrell D. A.; Lenox-Smith I. Evaluation of the antirhinovirus chalcone Ro 09-0415 given orally to volunteers. J. Antimicrob. Chemoth. 1984, 14, 403–409. 10.1093/jac/14.4.403. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Yue H.; Sun P.; Hua L.; Liang S.; Ou Y.; Wu D.; Wu X.; Chen H.; Hao Y.; Hu W.; Yang Z. Discovery of chalcone analogues as novel NLRP3 inflammasome inhibitors with potent anti-inflammation activities. Eur. J. Med. Chem. 2021, 219, 113417. 10.1016/j.ejmech.2021.113417. [DOI] [PubMed] [Google Scholar]
- Choi H. R.; Lim H.; Lee J. H.; Park H.; Kim H. P. Interruption of Helicobacter pylori-Induced NLRP3 Inflammasome Activation by Chalcone Derivatives. Biomol. Ther. 2021, 29, 410–418. 10.4062/biomolther.2020.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S.; Watanabe T.; Tanigawa T.; Shimada S.; Nadatani Y.; Miyazaki T.; Iimuro M.; Fujiwara Y. Isoliquiritigenin Ameliorates Indomethacin-Induced Small Intestinal Damage by Inhibiting NOD-Like Receptor Family, Pyrin Domain-Containing 3 Inflammasome Activation. Pharmacology 2018, 101, 236–245. 10.1159/000486599. [DOI] [PubMed] [Google Scholar]
- Zeng J.; Chen Y.; Ding R.; Feng L.; Fu Z.; Yang S.; Deng X.; Xie Z.; Zheng S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 2017, 14, 119. 10.1186/s12974-017-0895-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X.; Zhao M.; Tang M. H.; Xue L. L.; Zhang R. J.; Liu L.; Ni H. F.; Cai X. Y.; Kuang S.; Hong F.; Wang L.; Chen K.; Tang H.; Li Y.; Peng A. H.; Yang J. H.; Pei H. Y.; Ye H. Y.; Chen L. J. Flavonoids with Inhibitory Effects on NLRP3 Inflammasome Activation from Millettia velutina. J. Nat. Prod. 2020, 83, 2950–2959. 10.1021/acs.jnatprod.0c00478. [DOI] [PubMed] [Google Scholar]
- Zeng F.; Li S.; Yang G.; Luo Y.; Qi T.; Liang Y.; Yang T.; Zhang L.; Wang R.; Zhu L.; Li H.; Xu X. Design, synthesis, molecular modeling, and biological evaluation of acrylamide derivatives as potent inhibitors of human dihydroorotate dehydrogenase for the treatment of rheumatoid arthritis. Acta. Pharm. Sin. B 2021, 11, 795–809. 10.1016/j.apsb.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; Samanta S.; Xue D.; Petrunak E. M.; Stuckey J. A.; Han Y.; Sun D.; Wu Y.; Neamati N. Structure-Based Design of N-(5-Phenylthiazol-2-yl)acrylamides as Novel and Potent Glutathione S-Transferase Omega 1 Inhibitors. J. Med. Chem. 2019, 62, 3068–3087. 10.1021/acs.jmedchem.8b01960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco M.; Miglio G.; Giorgis M.; Garella D.; Marini E.; Costale A.; Regazzoni L.; Vistoli G.; Orioli M.; Massulaha-Ahmed R.; Détraz-Durieux I.; Groslambert M.; Py B. F.; Bertinaria M. Design, Synthesis, and Evaluation of Acrylamide Derivatives as Direct NLRP3 Inflammasome Inhibitors. ChemMedChem. 2016, 11, 1790–1803. 10.1002/cmdc.201600055. [DOI] [PubMed] [Google Scholar]
- Cocco M.; Garella D.; Di Stilo A.; Borretto E.; Stevanato L.; Giorgis M.; Marini E.; Fantozzi R.; Miglio G.; Bertinaria M. Electrophilic warhead-based design of compounds preventing NLRP3 inflammasome-dependent pyroptosis. J. Med. Chem. 2014, 57, 10366–10382. 10.1021/jm501072b. [DOI] [PubMed] [Google Scholar]
- Mangan M. S. J.; Olhava E. J.; Roush W. R.; Seidel H. M.; Glick G. D.; Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discovery 2018, 17, 588–606. 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- Afonina I. S.; Zhong Z.; Karin M.; Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. 10.1038/ni.3772. [DOI] [PubMed] [Google Scholar]
- Lomenick B.; Hao R.; Jonai N.; Chin R. M.; Aghajan M.; Warburton S.; Wang J.; Wu R. P.; Gomez F.; Loo J. A.; Wohlschlegel J. A.; Vondriska T. M.; Pelletier J.; Herschman H. R.; Clardy J.; Clarke C. F.; Huang J. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21984–21989. 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Q.; Xing Y.; Liu J.; Dong D.; Liu Y.; Qiao H.; Zhang Y.; Hu L. Lonicerin targets EZH2 to alleviate ulcerative colitis by autophagy-mediated NLRP3 inflammasome inactivation. Acta. Pharm. Sin. B 2021, 11, 2880–2899. 10.1016/j.apsb.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Ting J. P. Inflammasome Assays In Vitro and in Mouse Models. Curr. Protoc Immuno. 2020, 131, e107. 10.1002/cpim.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M.; Guo H.; Tam J. W.; Johnson B. M.; Brickey W. J.; New J. S.; Lenox A.; Shi H.; Golenbock D. T.; Koller B. H.; McKinnon K. P.; Beutler B.; Ting J. P. Platelet-activating factor (PAF) mediates NLRP3-NEK7 inflammasome induction independently of PAFR. J. Exp. Med. 2019, 216, 2838–2853. 10.1084/jem.20190111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.