

Abstract
Phospholipase D (PLD) is a phospholipase enzyme responsible for hydrolyzing phosphatidylcholine into the lipid signaling molecule, phosphatidic acid, and choline. From a therapeutic perspective, PLD has been implicated in human cancer progression as well as a target for neurodegenerative diseases, including Alzheimer’s. Moreover, knockdown of PLD rescues the ALS phenotype in multiple Drosophila models of ALS (amyotrophic lateral sclerosis) and displays modest motor benefits in an SOD1 ALS mouse model. To further validate whether inhibiting PLD is beneficial for the treatment of ALS, a brain penetrant small molecule inhibitor with suitable PK properties to test in an ALS animal model is needed. Using a combination of ligand-based drug discovery and structure-based design, a dual PLD1/PLD2 inhibitor was discovered that is single digit nanomolar in the Calu-1 cell assay and has suitable PK properties for in vivo studies. To capture the in vivo measurement of PLD inhibition, a transphosphatidylation pharmacodynamic LC-MS assay was developed, in which a dual PLD1/PLD2 inhibitor was found to reduce PLD activity by 15–20-fold.
Keywords: Phospholipase D, PLD inhibitor, ALS, structure-based drug design, transphosphatidylation
Amyotrophic lateral sclerosis (ALS) is a progressive and devastating neurodegenerative disease that affects motor neurons in the brain and spinal cord. ALS patients have gradual reduction in motor function, and most will advance to losing the ability to use their arms, walk, speak and breathe, succumbing to respiratory failure 2–5 years after disease onset.1 In 2014, the Centers for Disease Control and Prevention approximated that ∼16 000 people in the United States had ALS (1 in 20 000 persons).2 There are no approved disease-modifying treatments for ALS. The quick progression of ALS coupled with no effective treatments underscores the high unmet need for the development of novel therapies.
ALS pathology can be classified into sporadic (sALS, no family history, 95% ALS patients) and familial (fALS, patients with a positive family history, 5% of ALS patients).3 Over 25 different genes have been identified to contribute to the etiology of ALS; the SOD1 mutation and C9orf72 repeat expansion mutation are the most common genetic causes. Although recent discoveries have vastly improved our understanding of the different disease pathways in ALS, a deeper understanding of the complex genetic architecture is necessary to effectively identify novel druggable targets for disease modifying therapies.
To discover and evaluate new targets for the treatment of ALS, a genome-wide screen was conducted to discover potential modifiers of Drosophila models of degenerative eye phenotypes associated with the expression of human FUS transgenes carrying fALS mutations.4 After cross-referencing the findings from the Drosophila screen with postmortem gene expression in motor neurons from sALS patients,5 several genes were implicated, including three that are components of the phospholipase D pathway (ARFGAP3, RALB, and PLD).4 Phospholipase D (PLD) is a phospholipase enzyme responsible for hydrolyzing phosphatidylcholine into the lipid signaling molecule, phosphatidic acid (PA), and choline.6 PA is a critical lipid constituent in cell membranes; c-Raf and mTOR directly bind PA to mediate translocation or activation, respectively.7 PA also has been associated with a variety of signaling cascades involving cell growth, proliferation and survival.8
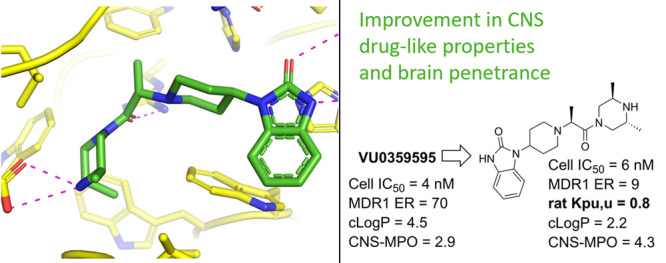
Historical interest in PLD as a target for therapeutic intervention for oncology had resulted in the identification of several published PLD inhibitors.9 One of the first reported PLD inhibitors was derived from halopemide (1, Figure 1), a D2 antagonist and moderate PLD1/PLD2 dual inhibitor which inspired the discovery by Novartis that potency for PLD2 inhibition could be increased by replacing the fluorophenyl amide with bicyclic heterocycles such as indole (not pictured).10 Capitalizing on this discovery, Lindsley and co-workers further advanced the halopemide scaffold to deliver isoform selective inhibitors VU0359595 (2), and the triaza-spirocyclic scaffold ML299 (3), that could be modulated to afford PLD2 or dual PLD1/PLD2 inhibition (Figure 1).11 These molecules exhibited reasonable in vitro potency but unfortunately were substrates for efflux transporter P-glycoprotein (Pgp) with efflux ratios (ER) in the MDCK-MDR1 assay > 40 and a correspondingly low ratio of unbound brain/unbound plasma concentration (Kpu,u).12 These absorption, distribution, metabolism, and elimination (ADME) properties rendered these tool inhibitors unsuitable for use in evaluating the effect of PLD inhibition in mouse models of ALS, due to their inability to achieve sufficient free brain concentrations.
Figure 1.

Known PLD inhibitors.
In the absence of either the PLD1 or PLD2 human crystal structures to guide structure-based drug design,13 a ligand-based drug design approach was pursued, starting from the reported PLD1-selective14 inhibitors, while in parallel conducting an HTS campaign. The strategic decision to focus on the PLD1-selective inhibitors was predicated on the original Drosophila findings, which identified a number of PLD1 specific pathway proteins as genetic suppressors of ALS-related phenotypes.4 It is important to note that Drosophila contains a single PLD gene and thus it was unclear from the outset whether inhibition of both isoforms in humans would be desired.4 To further our understanding of the structure–activity relationship (SAR) for driving the potency against PLD1 and PLD2, all new analogues synthesized were screened against both isoforms. The enzymatic activity of inhibitors was assessed by immunoprecipitation of PLD1 or PLD2 from HEK-293 cell lysates (referred to as IPoP) and cellular inhibition was measured as previously described in Calu-1 cells.15,16
With the goal to design inhibitors that exhibited good blood-brain barrier (BBB) permeability, the team focused on synthesizing molecules with a minimal number of hydrogen–bond (H-bond) donors as a means to reduce recognition by Pgp (also known as MDR1, Table 1).17 Building from leads disclosed by the Lindsley group, the benzimidazolone compound 4, which demonstrated good biochemical (PLD1 IC50 = 33 nM) and cellular (Calu IC50 = 16 nM) potency and human liver microsomal stability (HLM Clint <4 mL/min/kg), was deemed a suitable scaffold for optimization with the objective of improving the central nervous system (CNS) drug-like properties (Table 1, entry 1). Analogue 4 suffered from high in vivo clearance (rat IV CL = 69 mL/min/kg) and significant MDR1-mediated efflux (ER = 82), which correlated with low unbound brain exposure (Kpu,u = 0.02) when dosed in a rat infusion model. Several analogues were prepared with the aim of replacing the H-bond donor on the benzimidazolone ring. Methylation of the benzimidazolone nitrogen resulted in a complete loss of PLD potency and still maintained an unacceptable MDR1 efflux ratio of 19. Replacement of the benzimidazolone with the benzoxazolone moiety led to 5, which was a weak Pgp substrate (ER = 2.0); however, both benzoxazolone 5 and the indolinone 6 analogues resulted in a significant loss in PLD1 biochemical potency. Removal of the aryl motif while maintaining the cyclic urea in 7 demonstrated no inhibition at the highest concentration. Notably, this was one of the first modifications which demonstrated an improvement in the in vitro rat liver microsome assay (RLM CLint = 16 mL/min/kg). To probe the role of the carbonyl functionality, the benzimidazolidinone was replaced with the indazole in 8, but this also resulted in a decrease in potency (Table 1). These findings indicated that the carbonyl, NH bond donor, and aromatic ring within the benzimidazolone moiety were essential components of the pharmacophore required for PLD affinity.
Table 1. Modification of the Benzimidazolinone.
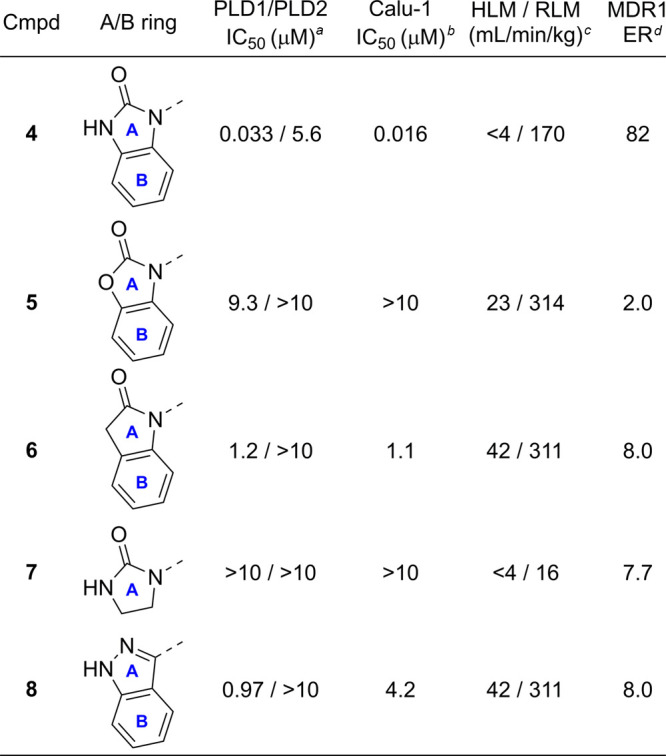
Biochemical inhibition of PLD1/2 was determined by immunoprecipitated on plate (IPoP) assay of PLD1/2 from HEK cells.
Cellular assay performed in Calu-1 cells.
Intrinsic Cl (Clint) in rat or human liver microsomes.
Efflux ratio = Papp(B – A)/Papp(A – B) MDCK-MDR1.
A cocrystal structure of compound 4 bound to hPLD218 was obtained at 2.9 Å resolution (Figure 2). The structure revealed the benzimidazolone moiety formed interactions with residues Gln642 and Asn773 within the HKD pocket. The piperidine linker orients the benzylamide moiety into a region of the active site where an electrostatic interaction occurs between the carbonyl of the benzylamide moiety and Arg486.
Figure 2.
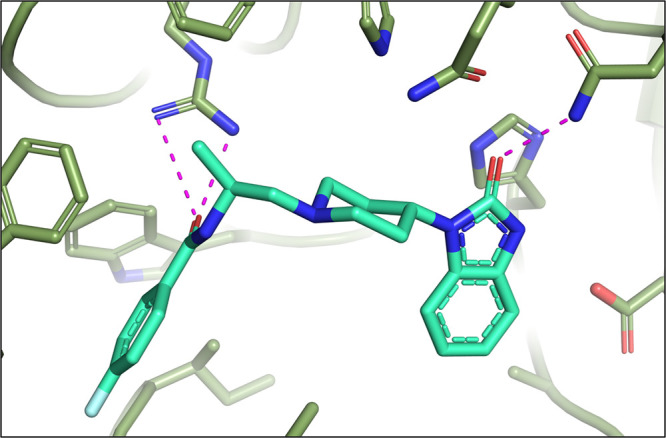
X-ray cocrystal of 4 with PLD2 (PDB# 6OHQ)
Having confirmed the carbonyl, NH bond and aromatic moieties were necessary to achieve inhibition of PLD1, a series of analogues were designed composed of novel spirocyclic cores, which maintained the putative pharmacophore while facilitating modulation of the physicochemical properties via increased Fsp3 character.19 This effort initiated from the dual PLD1/PLD2 inhibitor ML299 (3, Table 2, entry 1), which also demonstrated high Pgp ER and ADME properties that made it unsuitable as an in vivo CNS tool. Exploration of fused spirocyclic replacements for the imidazolidin-4-one moiety present in ML299 (3) provided several inhibitors with improved ER and microsomal stability as outlined in Table 2. The 3,4-dihydroquinolin-2-one 9 resulted in an improvement in the in vivo CL (rat IV CL = 8.6 mL/min/kg), although compound 9 was a Pgp substrate (MDCK-MDR1 ER = 11) with a low Kpu,u (0.02 in rat). Similarly, spirocyclic ureas 10 and 11 did not improve activity against PLD1 and, as expected, increased ER. Interestingly, the spirocyclic carbonate 12 restored the activity against PLD1 and demonstrated moderate in vivo clearance that warranted further investigation of the SAR around the fused aromatic ring. Introducing a fluorine substituent on the aryl motif led to 13, which exhibited similar PLD biochemical and Calu-1 cellular potency but was less stable in liver microsomes compared to the des-fluoro analogue 12. However, replacing the phenyl moiety with the pyridooxazinones (14 and 15) maintained inhibition of PLD1 and improved microsomal stability. Unfortunately, this improved in vitro stability was not reflected in vivo. One exciting discovery culminating from this effort was the design of 16, which was based on in silico solvent mapping of the PLD2 active site that revealed a novel pocket adjacent to the HKD binding domain into which it was hypothesized that substituents attached at C5 would project (Figure 3A). This new chemical series demonstrated drastically improved PLD1/2 potency and, more importantly, enabled the first X-ray cocrystal structure of PLD1 (Figure 3C).18 Despite 16 achieving a 5-fold increase in cellular potency compared to ML299 (3), 16 and related analogues continued to demonstrate inadequate ADME and CNS properties, leading to the deprioritization of this scaffold. The cocrystal structure of 16 bound to PLD1b revealed subtle differences in the binding mode within the active site (Figure 2 vs Figure 3C). Compound 16 maintained the key binding interactions with conserved residues Gln782 and Asn913 (in the HKD pocket, PLD1b numbering), which oriented the C5 fluorophenyl ring to bind in the cavity newly identified from the PLD2 solvent mapping, and Arg464/486 (PLD2/PLD1b) with the amide carbonyl. Comparison of the D-ring phenyl amide binding pocket and the space occupied by the alpha-methyl alkyl linkers led to observations that rationalized the isoform selectivity differences observed for reported PLD inhibitors, which are described in detail in ref (18).
Table 2. Exploration of Novel Spirocyclic Cores.

Biochemical inhibition of PLD1/2 was determined by immunoprecipitation on a plate (IPoP) of PLD1/2 from HEK cells.
Cellular assay performed with Calu-1 cell line.
Intrinsic CL (Clint) in rat or human liver microsomes.
Efflux ratio = Papp(B – A)/Papp(A – B) in MDR1-MDCK.
Kinetic solubility pH = 6.8.
Clearance determined by cassette dosing; Clu = unbound in vivo clearance (Cl/fu).
Figure 3.

(A) Solvent mapping of the PLD2 active site; (B) docking of inhibitor 16 into X-ray cocrystal of PLD2 with inhibitor 4 showing C5 substituent filling pocket adjacent to benzimidazolinone ring; (C) X-ray cocrystal structure of 16 with PLD1b (PDB# 6OHR).
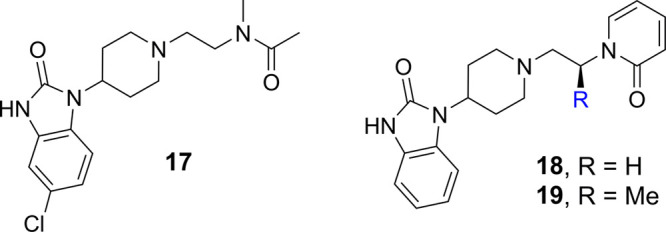
Utilizing insights gained from the hPLD1 cocrystal structure, a series of compounds were designed to further interrogate the pharmacophore required for binding in the phenyl amide (D-ring) region of the structure. Since the spirocyclic carbonate series had exhibited poor ADME properties, all further optimization was performed using the original benzimidazolone series. To reduce the MW and lipophilicity, more truncated analogues were prepared to establish the minimum pharmacophore. The N-methylacetamide analogue 17, which was designed to maintain the electrostatic interaction with Arg486 but eliminate the amide H-bond and lipophilic phenyl moiety, maintained good PLD1 and Calu-1 cellular inhibition while retaining selectivity over PLD2 (Table 3). Despite the high CNS-MPO score for 17 and superior drug-like properties that provided improved stability in HLM, these changes did not ultimately result in a reduction of Pgp-mediated efflux. Having demonstrated that the H-bond donor was not required for activity, pyridone 18 was synthesized, which was designed to maintain interaction with Arg486, but also to determine if potency could be improved by reintroduction of an aromatic ring. Interestingly, the biochemical potency was not impacted, although inhibition of Calu-1 was diminished. It was observed that introducing conformational constraint on the alkyl linker via the (S)-methyl substitution (19) improved the potency and reduced the ER value, although this improvement did not translate to an improved Kpu,u (0.02 in rat). Further exploration of scaffold 19 by introduction of electron-withdrawing groups on the pyridone (data not shown) was not effective in improving PLD inhibition or further reducing the ER.
Table 3. Removal of the Amide H-Bond.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| compd | PLD1a | PLD2a | Calu-1b | MDR1 ERc | cLogP/MPOd |
| 17 | 0.13 | >10 | 0.18 | 24 | 2.3/5.8 |
| 18 | 0.12 | >10 | 0.98 | 17 | 1.6/5.8 |
| 19 | 0.043 | >10 | 0.058 | 8.6 | 1.8/5.8 |
Biochemical inhibition of PLD1/2 was determined by immunoprecipitation on a plate (IPoP) of PLD1/2 from HEK cells.
Cellular assay performed with Calu-1 cell line.
Rat or human liver microsomes.
Efflux ratio determined by measuring Papp(B-A)/Papp(A-B) in MDR1-MDCK.
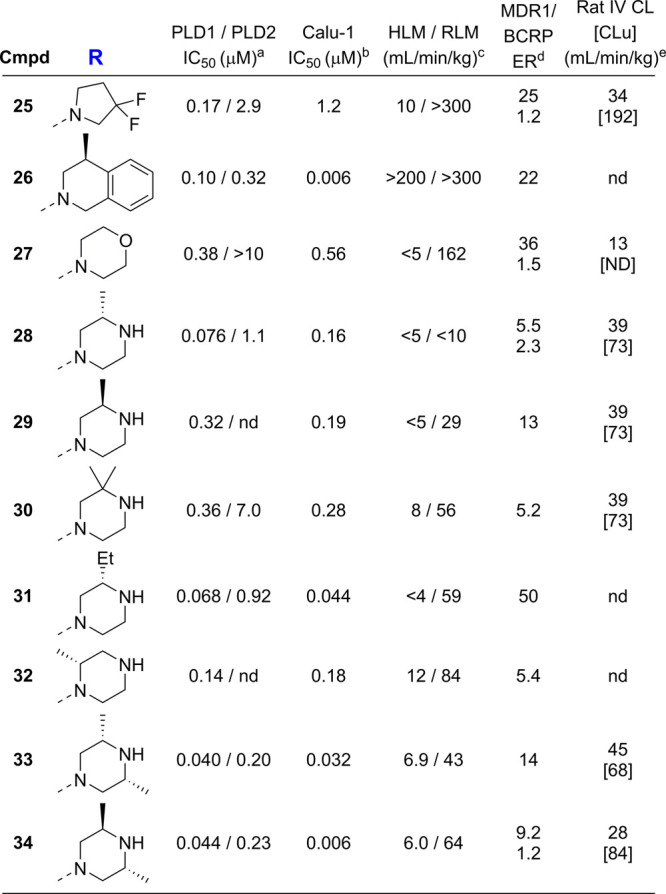
Inspired by the observation that both the phenyl (D ring) and the N–H amide moieties were not required for PLD1 activity, exploration of the linkage between the piperidine C ring and the amide carbonyl was pursued. Comparison of X-ray structures of spirocylic inhibitor 3 and benzimidazolinone 4, which exhibit differences between the two carbonyls forming key interactions with the protein (HKD and Arg464), led us to speculate that the Arg464 side chain was flexible and could reorient to maintain the critical electrostatic interaction with the amide carbonyl moiety.18 To test this hypothesis, a series of structurally distinct analogues were designed containing an alanine linker, which reversed the amide linkage compared to previously prepared analogues and positioned the carbonyl closer to the piperidine ring (Table 4). Based on the docking studies, it was perceived that introducing the S-Me alpha substituent would rigidify the molecule, directing the carbonyl moiety toward the side chain of Arg464. Since this alanine linkage was anticipated to yield a novel pharmacophore that was amenable for parallel medicinal chemistry (PMC), a library of analogues was enumerated utilizing in silico docking studies to identify amine fragments predicted to bind in this region of the protein and would improve the drug-like properties with respect to reducing the molecular weight, Fsp2 character and lipophilicity. Utilizing the commercially available 1-(piperidin-4-yl)-1,3-dihydro-2H-benzoimidazol-2-one starting material 20, the piperidine amine was alkylated, affording the ester 22, which was separated by SFC to obtain enantioenriched 23, which was saponified to provide 24 in 98% yield. The acid intermediate 24 underwent HATU-mediated amide coupling to deliver a focused library of novel alanine linker analogues (Scheme 1). Several surprising hits emerged from the library. SAR studies conducted on the amide moiety using the linker contained in compounds 1–19 showed a clear preference for the presence of an aromatic ring to fill the hydrophobic pocket defined by residues W364 and W365, L407, I409, and W519.18 As postulated by comparison of the PLD1 and PLD2 Xray structures, the presence of an aromatic ring that can engage in a π-stacking interaction with W519 imparts greater potency against PLD2, while truncated Sp3 rings demonstrated improved inhibition of PLD1 (compare 26 and 28). The observed potency and lower ER obtained by analogues containing a basic amine moiety in place of a more lipophilic aromatic substituent was surprising. The increased potency on PLD2 without the aromatic ring for π-stacking supports the hypothesis described in ref (18), that the distance between the benzimidazolinone and the chiral methyl group present on the linker is key in obtaining PLD1 selectivity, and that compounds with a smaller distance between these two moieties can be accommodated by both PLD1 and PLD2 isoforms. Comparison of morpholine 27 with piperazine 28 shows an increase in potency with the inclusion of the NH, and piperazine analogues containing substitution on that nitrogen were not tolerated, pointing to the possibility of a new favorable interaction with the protein. Piperazine 28 showed modestly improved Kpu,u = 0.14 compared to previous analogues tested. Exploration of substitution around the piperazine ring revealed a preference for 3,5-dimethyl substitution, with the 3S,5S-stereochemistry (34) demonstrating superior potency in the Calu-1 cellular assay. A cocrystal structure of 34 was obtained with PLD2 and as anticipated, a new interaction with the protein was identified, with the piperazine amine forming an H-bonding interaction with Asp518 (Figure 4A). The axial piperazine methyl group is accommodated in a hydrophobic pocket bordered by Ile411 and Trps364/365. Also present in the cocrystal structure with 34 is a water molecule that forms a bridging interaction between the benzimidazolone NH and Glu786 and Asp784, which provides rationale for the required H-bond donor at that position. Also of note is the movement of Leu514, where the alanine methyl present in 34 has a different trajectory than the methyl present on the linker in PLD1-preferring inhibitor 4 (Figure 4B) an observation in line with our previously reported hypothesis on the structural basis of isoform selectivity.18 A more intriguing discovery was the improvement in the CNS penetration observed for inhibitor 34, which demonstrated a Kpu,u = 0.8 in a rat infusion model. Further profiling of compound 34 demonstrated no BCRP efflux, hERG liability, or inhibition of CYP enzymes.20 The ADME properties and potency of analogue 34 prompted the team to move forward with this compound to explore the measurement of PLD inhibition in vivo.
Table 4. SAR of Truncated, Reversed Amides.
Biochemical inhibition of PLD1/2 was determined by immunoprecipitation on a plate (IPoP) of PLD1/2 from HEK cells.
Cellular assay performed with Calu-1 cell line.
Rat or human liver microsomes.
Efflux ratio determined by measuring Papp(B-A)/Papp(A-B).
cassette dosing used; [Clu] = Unbound clearance (Cl/fu).
Scheme 1. Synthesis of Truncated, Reversed-Amide Containing Benzimidazolinones 25–34.

Figure 4.

(A) X-ray cocrystal of 34 with hPLD2 (PDB# 7SVP); (B) overlay of X-ray cocrystals of 4 (slate) and 34 (green) in hPLD2.
With the discovery of compound 34, which had the potency and ADME properties suitable for use as an in vivo tool, the team set about developing a PLD dependent pharmacodynamic assay for determinng a PK/PD relationship for 34. Unfortunately, utilizing phosphatidic acid (PA) as a direct readout of PLD activity was deemed impractical, owing to its short in vivo half-life and existence of multiple pathways, including PLD, which contribute to the formation of phosphatidic acid.21 Therefore, an alternative approach was adopted, wherein PLD activity was correlated with its ability to catalyze a transphosphatidylation reaction of phosphatidyl choline (PC) with short-chain primary alcohols, such as ethanol or 1-butanol to yield phosphatidylethanol (PtdEtOH) or phosphatidylbutanol (PtdButOH). The transphosphatidylation reaction is exclusive to phospholipase D1/D2 and has been employed as a specific assay for assessing the enzyme activity in cells such as the Calu-1 assay and in vivo.22,23 Using LC-MS detection, PtdButOH(34:1) was confirmed to exhibit significantly higher intensity than PtdEtOH(34:1) (data not shown), therefore 1-butanol was selected for further experiments and, based on the enhanced signal sensitivity, deuterated 1-butanol-d10 was employed to improve assay specificity.24
An extensive in vivo optimization effort was necessary for establishing a time-course and concentration effect for the transphosphatidylation (PtdBut-d9) readout with respect to t-butanol-d10 to ensure maximum signal/noise ratio. These experiments determined that the optimal detection period following injection of t-butanol-d10 was 15 min, as (PtdBut-d9) levels in liver and brain declined significantly at 30 min. Using an unbiased LC-FTICR-MS assay, 18 unique phosphatidylbutanol-d9 (PtdBut-d9) species were detected from the liver and seven from the brain. The most abundant PtdBut-d9 species from the brain and the liver were different, with PtdBut-d9 (34:1) proving to be most abundant in the brain and PtdBut-d9 (34:2) in the liver (Figure 5).
Figure 5.

Expanded profile of PLD inhibitor 34 and measurement of in vivo PLD inhibition in mice treated with 34. (A) Reduction of various PtdBuOH-d9 species by 34 in liver. (B) Selectivity and rodent PK of 34. aAverage of A-B/B-A Papp in MDCK cells with low MDR1 expression. bCYPs evaluated: 1A2, 2B6, 2D6, 2C9, 2C19, 3A4. cAverage of two experiments with n = 3 rats each. dFormulation: HPMC/Tween. (C) Liver/brain concentrations of 34 at measured time points. (D) PtdBuOH-d9 species in brains of mice treated with 34.
Although 34 exhibited moderate oral bioavailability (%F = 21) when dosed orally in rat, the lengthy oral absorption profile (Tmax = 3 h) was considered impractical for the PD assay. Subcutaneous (SC) dosing of 34 in mice resulted in a shorter Tmax, 0.4 h, indicating that measurement of transphosphatidylation within 1 h of dosing compound would be possible. To understand the time course of PLD inhibition in vivo with 34, mice were first dosed subcutaneously with 34 (10 mg/kg) at 15, 30, and 60 min prior to injection with 1-butanol-d10 and tissues were collected after 15 min. Comparison of compound-treated and control mice revealed that 16 out of 18 PtdBut-d9 species in the liver showed statistically significant decreases, with only two low-abundance lipids not showing a meaningful signal (p < 0.05, Figure 5A). In contrast to the excellent Kpu,u observed in the rat infusion experiment, 34 did not show expected brain concentrations in the mice at the selected time-points and thus minimal changes were observed in the production of PtdBut-d9 species in the brain (Figure 5D). The Kpu,u of 34 in mouse at all three time points was 0.02, providing a maximum of 9× coverage of the Calu-1 IC50. In the liver, there was robust inhibition, but minimal difference between the 15, 30, or 60 min time points, presumably due to the large multiple over IC50 observed in the liver at all time points (Figure 5C). While not statistically significant, the later time points in the brain samples, which correlated with higher exposure, did show a trend toward lowering of PtdBut-d9 (34:1).
Starting from reported peripherally restricted PLD inhibitors, we identified brain penetrant PLD inhibitor 34 with improved drug-like attributes and ADME properties. Through this effort, the first cocrystal structures of human PLD1 and PLD2 isoforms were characterized and the SAR rationale for isozyme selectivity was uncovered. While 34 showed excellent brain penetration in rats, the initial PD experiment in mice with PLD1/2 inhibitor 34 failed to show a significant reduction in PLD-mediated transphophatidylation in the brain tissue, likely due to the reduced brain exposure of 34 in mice vs rats at the measured time point. The observed difference in the Kpu,u values between the mouse PD and the rat IV infusion Kpu,u measurement may be attributed to the SC dosing in mouse and measurement at non-steady-state conditions. Additional experiments are required to determine an in vivo PK/PD correlation and establish which of the in vitro potency readouts are predictive for the observed in vivo pharmacodynamic effect necessary for guiding any future medicinal chemistry effort. While the neuroPK observed in mice was disappointing, compound 34 does exhibit sufficient potency, ADME properties, and BBB permeability in rat to serve as a tool CNS-penetrant compound for evaluating PLD signaling in vivo. More importantly, discovery of the H-bond interaction between 34 and ASP518 (PLD2) should facilitate the discovery and optimization of future PLD inhibitors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00682.
Compound synthesis preparation, characterization data, and X-ray crystallography data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- van Es M. A.; Hardiman O.; Chio A.; Al-Chalabi A.; Pasterkamp R. J.; Veldink J. H.; van den Berg L. H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. 10.1016/S0140-6736(17)31287-4. [DOI] [PubMed] [Google Scholar]
- Mehta P.; Kaye W.; Raymond J.; Wu R.; Larson T.; Punjani R.; Heller D.; Cohen J.; Peters T.; Muravov O.; Horton K. Prevalence of Amyotrophic Lateral Sclerosis. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 216–218. 10.15585/mmwr.mm6707a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. P.; Van Broeckhoven C.; van der Zee J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet 2018, 34, 404–423. 10.1016/j.tig.2018.03.001. [DOI] [PubMed] [Google Scholar]
- Kankel M. W; Sen A.; Lu L.; Theodorou M.; Dimlich D. N; McCampbell A.; Henderson C. E; Shneider N. A; Artavanis-Tsakonas S. Amyotrophic Lateral Sclerosis modifiers in Drosophila reveal the Phospholipase D pathway as a potential therapeutic target. Genetics 2020, 215, 747–766. 10.1534/genetics.119.302985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kaplan A.; Spiller K. J.; Towne C.; Kanning K. C.; Choe G. T.; Geber A.; Akay T.; Aebischer P.; Henderson C. E. Transplantation of Neural Precursors Derived from Induced Pluripotent Cells Preserve Perineuronal Nets and Stimulate Neural Plasticity in ALS Rats. Neuron 2014, 81, 333–348. 10.1016/j.neuron.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rabin S. J.; Kim J. M. H.; Baughn M.; Libby R. T.; Kim Y. J.; Fan Y.; Libby R. T.; La Spada A.; Stone B.; Ravits J. Sporadic ALS has compartment-specific aberrant exon splicing and perturbation of cell-matrix adhesion biology. Hum. Mol. Genet. 2010, 19, 313–328. 10.1093/hmg/ddp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Su W.; Frohman M. A.. Phospholipase D. In Handbook of Cell Signaling, 2nd ed.; Bradshaw R. A., Dennis E. A., Eds.; Elsevier: New York, 2010; Vol. 1, pp 1167–1176. [Google Scholar]; b Selvy P. E.; Lavieri R. R.; Lindsley C. W.; Brown H. A. Phospholipase D - enzymology, functionality, and chemical modulation. Chem. Rev. 2011, 111, 6064–6119. 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ghosh S.; Strum J. C.; Sciorra V. A.; Daniel L.; Bell R. M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 1996, 271, 8472–8480. 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]; b Rizzo M. A.; Shome K.; Watkins S. C.; Romero G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem. 2000, 275, 23911–23918. 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]; c Fang Y.; Vilella-Bach M.; Bachmann R.; Flanigan A.; Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- Tanguy E.; Wang Q.; Moine H.; Vitale N. Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front. Cell Neurosci. 2019, 13, 2. 10.3389/fncel.2019.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H. A.; Thomas P. G.; Lindsley C. W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Disc. 2017, 16, 351–367. 10.1038/nrd.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cuyper H.; van Praag H. M.; Verstraeten D. The Clinical Significance of Halopemide, a Dopamine-Blocker Related to the Butyrophenones. Neuropsychobiology 1984, 12, 211–216. 10.1159/000118141. [DOI] [PubMed] [Google Scholar]; b Monovich L.; Mugrage B.; Quadros E.; Toscano K.; Tommasi R.; LaVoie S.; Liu E.; Du Z.; LaSala D.; Boyar W.; Steed P. Optimization of halopemide for phospholipase D2 inhibition. Bioorg. Med. Chem. Lett. 2007, 17, 2310–2311. 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- a Waterson A. G.; Scott S. A.; Kett N. R.; Blobaum A. L.; Brown A. B.; Lindsley C. W. Isoform selective PLD inhibition by novel, chiral 2,8-diazaspiro[4.5]decan-1-one derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 3670–3673. 10.1016/j.bmcl.2018.10.033. [DOI] [PubMed] [Google Scholar]; b Lewis J. A.; Scott S. A.; Lavieri R.; Buck J. R.; Selvy P. E.; Stoops S. L.; Armstrong M. D.; Brown H. A.; Lindsley C. W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 2009, 19, 1916–1920. 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Scott S. A.; Selvy P. E.; Buck J. R.; Cho H. P.; Criswell T. L.; Thomas A. L.; Armstrong M. D.; Arteaga C. L.; Lindsley C. W.; Brown H. A. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 2009, 5, 108–117. 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lavieri R.; Scott S. A.; Lewis J. A.; Selvy P. E.; Armstrong M. D.; Brown H. A.; Lindsley C. W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II. Identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Biorg. Med. Chem. Lett. 2009, 19, 2240–2243. 10.1016/j.bmcl.2009.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]; e O’Reilly M. C.; Scott S. A.; Brown K. A.; Oguin T. H.; Thomas P. G.; Daniels J. S.; Morrison R.; Brown H. A.; Lindsley C. W. Development of Dual PLD1/2 and PLD2 Selective Inhibitors from a Common 1,3,8-Triazaspiro[4.5]decane Core: Discovery of ML298 and ML299 That Decrease Invasive Migration in U87-MG Glioblastoma Cells. J. Med. Chem. 2013, 56, 2695–2600. 10.1021/jm301782e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kpu,u is the
ratio of free compound concentration in the brain over the free compound
concentration in the plasma at steady state, equation:
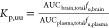 The Kpu,u values
for 1 and 2 in mouse are 0.04 and 0.09, respectively, and the approximate Kpu,u of ML299 (3) was determined using the reported
total brain/plasma ratio = 0.44 in ref (11e), using mouse plasma protein binding (%Fu =
8%) and brain binding (%Fu = 0.9%) values obtained at Biogen. This
does not represent a steady-state Kpu,u.
The Kpu,u values
for 1 and 2 in mouse are 0.04 and 0.09, respectively, and the approximate Kpu,u of ML299 (3) was determined using the reported
total brain/plasma ratio = 0.44 in ref (11e), using mouse plasma protein binding (%Fu =
8%) and brain binding (%Fu = 0.9%) values obtained at Biogen. This
does not represent a steady-state Kpu,u. - The bacterial PLD structure has been reported; however, the human and bacterial proteins have low sequence conservation.Leiros I.; Secundo F.; Zambonelli C.; Servi S.; Hough E. The first crystal structure of a phospholipase D. Structure 2000, 8, 655–667. 10.1016/S0969-2126(00)00150-7. [DOI] [PubMed] [Google Scholar]
- PLD1 activity was prioritized over its isoform PLD2, since PLD1 was found to be upregulated in human ALS patients (ref (5)). PLD2 activity was monitored but not optimized during the investigation.
- As full length PLD1/2 was not stable to isolation, the enzymatic activity of PLD1 and PLD2 were measured by immunoprecipitation of PLD1 from HEK-293 cell lysates. PLD was captured with a GTP antibody in an ELISA plate and 08.0 phospatidyl choline was used as substrate; amplex red detection of choline was used as the readout. For a previously disclosure measurement of enzymatic activity of PLD; see ref (11c).
- The cellular assay was measured in Calu-1 cells that express only PLD1 and not PLD2; the assay was a mass-spectrometry-based detection of d9-phosphobutanol arising from PLD1-catalyzed transphosphatidylation with d9-BuOH.11
- Ghose A. K.; Herbertz T.; Hudkins R. L.; Dorsey B. D.; Mallamo J. P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012, 3, 50–68. 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metrick C. M.; Peterson E. A.; Santoro J. C.; Enyedy I. J.; Murugan P.; Chen T.; Michelsen K.; Cullivan M.; Spilker K. A.; Kumar P. R.; May-Dracka T. L.; Chodaparambil J. V. Human PLD structures enable drug design and characterization of isoenzyme selectivity. Nat. Chem. Biol. 2020, 16, 391–399. 10.1038/s41589-019-0458-4. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- See the SI for the full data table.
- Bond P. Phosphatidic acid: biosynthesis, pharmacokinetics, mechanisms of action and effect on strength and body composition in resistance-trained individuals. Nutr. Metab. (Lond) 2017, 14, 12. 10.1186/s12986-017-0166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Morris A. J.; Frohman M. A.; Engebrecht J. Measurement of phospholipase D activity. Anal. Biochem. 1997, 252, 1–9. 10.1006/abio.1997.2299. [DOI] [PubMed] [Google Scholar]; b Brown H. A.; Henage L. G.; Preininger A. M.; Xiang Y.; Exton J. H. Biochemical analysis of phospholipase D. Methods Enzymol 2007, 434, 49–87. 10.1016/S0076-6879(07)34004-4. [DOI] [PubMed] [Google Scholar]
- a Scott S. A.; Xiang Y.; Mathews T. P.; Cho H. P.; Myers D. S.; Armstrong M. D.; Tallman K. A.; O'Reilly M. C.; Lindsley C. W.; Brown H. A. Regulation of Phospholipase D Activity and Phosphatidic Acid Production after Purinergic (P2Y6) Receptor Stimulation. J. Biol. Chem. 2013, 288, 20477–20487. 10.1074/jbc.M113.451708. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Philip F.; Ha E. E.; Seeliger M. A.; Frohman M. A. Measuring Phospholipase D Enzymatic Activity Through Biochemical and Imaging Methods. Methods Enzymol 2017, 583, 309–325. 10.1016/bs.mie.2016.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Oliveira T. G.; Chan R. B.; Tian H.; Laredo M.; Shui G.; Staniszewski A.; Zhang H.; Wang L.; Kim T.-W.; Duff K. E.; Wenk M. R.; Arancio O.; Di Paolo G. Phospholipase D2 Ablation Ameliorates Alzheimer’s Disease-Linked Synaptic Dysfunction and Cognitive Deficits. J. Neurosci. 2010, 30, 16419–16428. 10.1523/JNEUROSCI.3317-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untargeted high-resolution LC-FT ICR-MS assay profiled the lipid pools and distinguished PtdBut species from the interference of other lipids in tissue. Chromatograms of PtdBut species were optimized to achieve characteristic LC elution profile. The targeted LC-MRM assay was also used to quantify each PtdBut species observed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.