

Abstract
A vast share of the population attributable risk for autism relates to inherited polygenic risk. A growing number of recent studies have indicated that inherited susceptibility may operate through a finite number of early developmental liabilities which, in various permutations and combinations, jointly predict familial recurrence of the convergent syndrome of social-communicative disability that defines the condition. Here we synthesize this body of research to derive evidence for a novel developmental sub structure for autism, which has profound implications for ongoing discovery efforts to elucidate its neurobiological causes, and to inform future clinical and biomarker studies, early intervention, and personalized approaches to therapy.
Keywords: Development, Causation, Canalization, Infancy, Endophenotype, Inheritance
Introduction
The autism spectrum disorders (ASDs) are common conditions of childhood (Maenner et al., 2020) for which the vast proportion of population risk is attributable to inheritance (Sandin et al., 2017), mediated principally by polygenic risk. The sibling recurrence rate is one order of magnitude higher than general population risk (Hansen et al., 2019), and the rate for the identical (monozygotic) co-twin of an individual with autism is 80 times higher than general population risk (Tick et al., 2016). In recent years, the association of numerous de novo mutations (germline chromosomal rearrangements or DNA sequence variants) with autism has advanced understanding of the biological underpinnings of ASD, but these are individually rare and, by definition, do not account for the formidable inherited transmission of autism in the population. Moreover, most rare monogenic syndromes associated with autism are also accompanied by substantial cognitive impairment (Myers et al., 2020), which affects no more than a third of all individuals affected by autism (Maenner et al., 2020).
In autism, the vast majority of affected children are born to unaffected parents. Given the recurrence patterns observed in families, this is presumed to occur on the basis of genetic variants being carried by unaffected parents and transmitted to their offspring. This is possible when numerous allelic variants—each individually contributing only slightly to the total risk—accumulate to the level of a clinical threshold in a given offspring while being at lower-than-threshold (or otherwise compensated) in either or both of his/her parents. Polygenic mechanisms of causation are responsible for many complex inherited neuropsychiatric conditions and pose significant obstacles to the development of therapeutic interventions; it is currently impossible to simultaneously target a plethora of genes or their protein products—in ASD numbering up to thousands believed to contribute to susceptibility in a given patient. So, in contrast to gene-based strategies for monogenic syndromes (e.g. an antisense oligonucleotide designed to disrupt overexpression of a specific gene), an alternate approach for inherited polygenic syndromes is to identify intermediate phenotypes through which groups of genetic susceptibilities exert their influence in causing a condition.
A comparable example is the approach to biological therapy for ADHD, a condition that commonly co-occurs with ASD and that is also highly heritable and largely a function of polygenic causation (. Here, pharmacologic and behavioral interventions targeted toward hyperactivity, impulsivity, and inattention have been successful in attenuating both symptomatology and the potentially severe public health consequences of the condition in adulthood (Lichtenstein et al., 2012). Another instructive example is hypertension, an elevation of the singular quantitative trait of blood pressure. The function of maintaining blood pressure can be decomposed into a finite set of underlying processes (e.g. vascular resistance, stroke volume of the heart, fluid and electrolyte balance)—each with its own genetic and mechanistic structure—that participate in an intricate system of checks and balances. Targeting disruption in cardiovascular dynamics from this granular perspective has identified numerous “levers” for the therapeutic adjustment of blood pressure during disease states. In retrospect, it would seem statistically misguided at best to try to relate genes to “blood pressure” without resolving the relationships between specific gene sets and the disparate functions which are interacting with (and at times compensating) one another dynamically in a living system.
In this article we will describe the manner in which diverse combinations of early inherited liabilities may influence the development of autism across different affected individuals, and how such causal heterogeneity may contribute to the persistent difficulty in tracing autism to a common set of inherited genetic variants; the most recent attempts to relate polygenic risk scores to autism still account for less than 10 per cent of the known heritability (Grove et al., 2019). Given the transmission of quantitative (sub clinical) autistic traits in affected families (Piven and Palmer, 1999; Constantino, 2011), and overlap of inherited liability between autism and other neuropsychiatric conditions at both the phenotypic (Jokiranta-Olkoniemi et al. 2018) and molecular genetic levels (Grove et al., 2019), we and others have considered whether specification of a developmental sub structure might serve as a more appropriate model for understanding the effects of inherited influences on autism. Under such a model, autism (like hypertension) may arise from any number of distinct disruptions (or combinations thereof) to brain or developmental processes that contribute to human social development and behavior. Such a substructure might also explain why it has been so difficult to link common genetic variation to the singular phenotype of autism, especially if different subsets of patients represent different permutations or combinations of heritable atypicalities giving rise to phenocopies of a common syndrome. Clarification of the contributing components and their timing might allow for more precise approaches to early identification or treatment of individual cases, just as has occurred for the treatment of hypertension when the physiologic underpinnings are more precisely understood.
As is the case for schizophrenia and Alzheimer’s Disease, one window in which to explore the developmental sub structure of a condition is the period of time before it develops; for autism this is from the time of conception through the first 12-18 months of life. In this article we review and synthesize new information about predictors of the condition that have been identified during this early period. Studies that address the developmental sub structure of autism have yielded surprising new insights into early risk prediction and the mechanisms by which inherited susceptibility (engendered by polygenic risk) may contribute to specific brain and behavioral processes underlying normative social development. These neurodevelopmental processes likely interact with others in complex or compensatory ways reminiscent of the checks and balances inherent in various facets of cardiovascular dynamics, yielding further clues for understanding early human social development, the discovery of reliable biomarkers, and tracing specific subsets of polygenic liability more precisely to contributory brain and behavioral processes.
We should note at the outset that, historically, attempts to “parse” the autistic syndrome into component parts have traditionally done so in a different way, i.e. by dissecting its characterizing symptoms, classically described as a “triad” of social deficits, communicative deficits, and repetitive behaviors (Wing, 1981). At face value, these symptoms appear to be distinct and potentially independent from one another. Twenty years of research, however, has indicated somewhat the opposite: that these symptom clusters, which by definition co-occur in autism, are tightly inter correlated not only in individuals with autism but throughout the general population (Frazier et al., 2015), and are extremely stable over the life course for all people (Wagner et al., 2019). Twin and family studies have demonstrated that the genetic causes of autism overlap with those of sub clinical autistic traits (Robinson et al., 2011; Robinson et al., 2016). Such traits aggregate among the unaffected family members of individuals (Virkud et al., 2009; Constantino et al., 2010), and throughout the population the estimated heritability of sub clinical autistic traits is nearly identical to that of autism itself (Ronald et al., 2006). This makes it unlikely that autism represents the coincidental overlap of symptoms with independent etiologies.
In contrast to strong inter correlations between the characterizing symptoms of autism, some of its behavioral predictors appear to arise independently from one another in the population (Pohl et al., 2019; Hawks and Constantino, 2019). This is schematically illustrated in Figure 1, which contrasts a traditional conceptualization of genetic causation with what the new data suggest. Here, panel A depicts the former, in which inherited liabilities responsible for each symptom domain are theorized to co-occur within an individual to produce autism. Panel B depicts the latter, in which combinations and permutations of disparate, causally-independent developmental liabilities engender a convergent latent liability (“autism”) which secondarily manifests in the three recognized domains of symptoms, usually early in development, although the age at which these are recognizable and distinct as the syndrome we clinically recognize as autism varies from individual-to-individual. In this article we review the recent evidence for this reconceptualization, and elaborate key implications for clinical care, early pre-emptive intervention, and future research.
Figure 1.
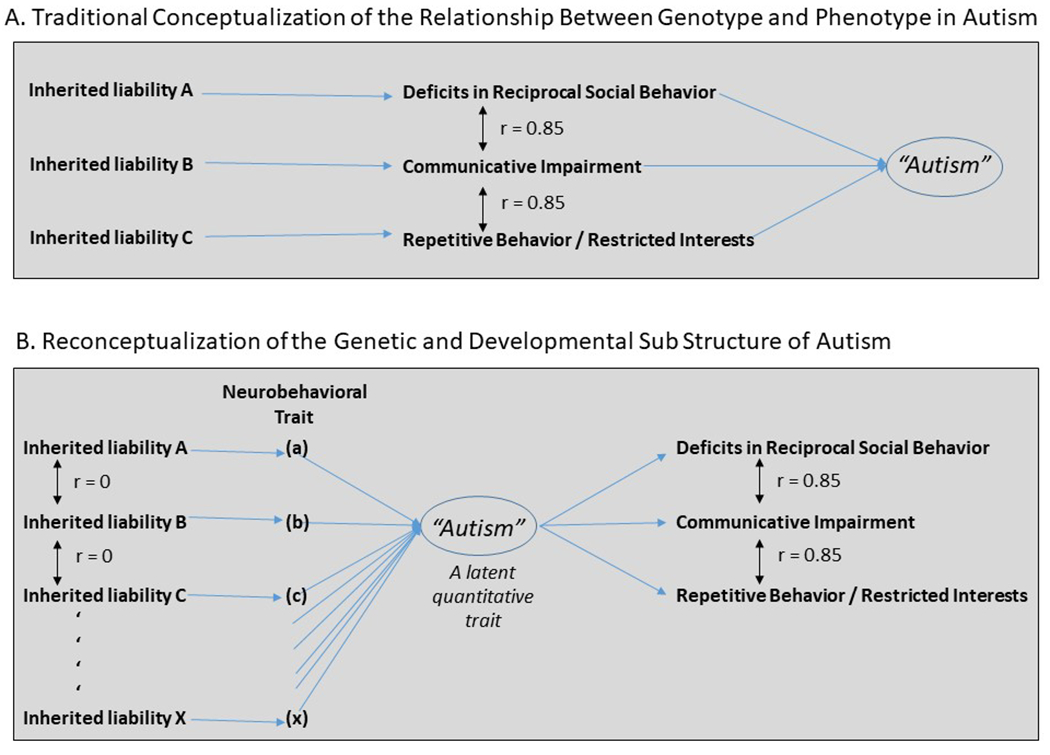
Competing models for the causal sub structure of autism. In the traditional model (Panel A), inherited liabilities corresponding to the three characterizing symptom manifestations of autism (the so-called “autism triad”) contribute to genetic susceptibility for autism (See Frazier et al., 2014) In a reconceptualization informed by recent developmental research (Panel B), permutations and combinations of independent inherited liabilities (A through X, none specific to a particular autism symptom domain nor to autism itself) contribute to allostatic load for a singular latent quantitative trait (“autism”) which secondarily gives rise to the array of characterizing traits and features of the autistic syndrome. Notable implications of reconceptualization B include: (i) a unitary factor structure for the manifest symptoms of autism (i.e. that they arise from a singular underlying biological deficit), and (ii) genotype-phenotype associations will be more pronounced for the relationship between each inherited liability (A-X) and a sub structural neurobehavioral trait (a-x) than for genotypic association with “autism”, especially if autism can arise from numerous combinatorial aggregations of a-x, the effects of which might be accentuated by de novo mutations, perinatal complications, or stochastic influences on brain development.
Combinatorial effects of inherited liabilities may underlie causation and heterogeneity in familial ASD
To complement this picture of the genetic structure of autism, it is now well established that the genetic causes of the characterizing behavioral features of ASD overlap with those of sub clinical traits of attention deficit hyperactivity disorder (ADHD), developmental coordination disorder (DCD), and Tourette Syndrome (Lichtenstein et al., 2010). At the level of clinical-range symptomatology, autism is four times more common among the siblings of individuals with ADHD than in the general population (Jokiranta-Olkoniemi et al. 2016;Miller et al., 2019)); in contrast, autism is only slightly more common among the siblings of individuals with intellectual disability than in the general population (Xie et al., 2019). In a recent sibling study it was estimated that just over half of the recurrence of autism within a family can be accounted for by the presence of ADHD, DCD, or both (Mous et al., 2016); the same proportion of variance in autistic trait scores in the general population can be predicted by variation in sub clinical traits of the same conditions, in both school-aged children (Reiersen et al., 2015); and in infants (Pohl et al., 2019). These observations are corroborated at the molecular genetic level—where substantial overlap between ASD and other common neuropsychiatric disorders has been identified (Brainstorm Consortium, 2018; Grove et al., 2019)—although the proportion of all causal variance accounted for in molecular genetic studies still remains much more limited than that captured in twin and family studies (i.e. genetic epidemiologic research designs). In general, the characterizing traits of these distinct neurodevelopmental syndromes (ADHD and DCD) are highly heritable and themselves continuously distributed in the general population. The observation that there may exist a set of inherited behavioral liabilities that contribute to multiple neuropsychiatric syndromes (ADHD and autism, for example) is consistent with the phenomenon of pleiotropy (the production of two or more apparently unrelated effects of an underlying causal influence). Pleiotropy has repeatedly been observed among patients with highly deleterious single-gene mutations—where loss of function in the same gene may result in intellectual disability in an affected individual in one family, epilepsy in another, autism in another, and so on. In some cases the cause of such pleiotropic outcomes has been traced to the interactions between disease-causing mutations and background genetic characteristics of a family which determine whether the mutation will result in clinical impairment in one or more disability domains (Finucane et al., 2016). Figure 2 illustrates a range of frameworks within which inherited liabilities that contribute both to autism and to an example disorder (ADHD) can be conceptualized.
Figure 2.
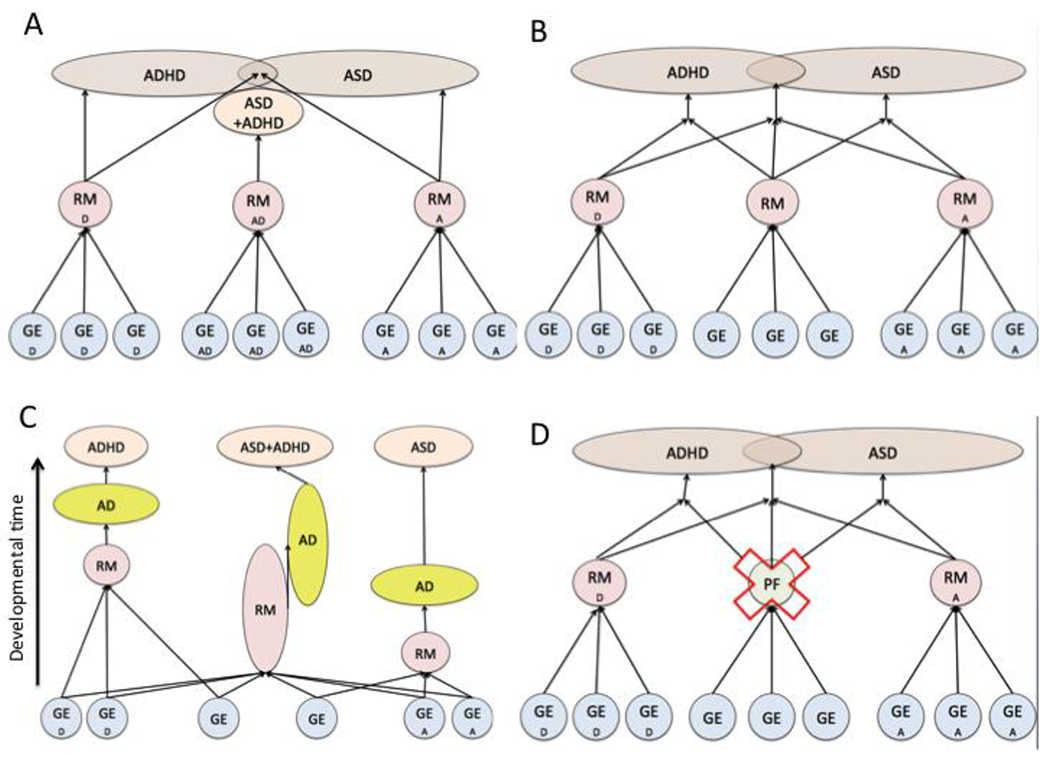
shows four possible models of the developmental emergence of behavioural symptoms of ASD and ADHD. For simplicity, bidirectional interactions between genetic and environmental risk factors, intermediate phenotypes and behavior over developmental time are not shown. A: ASD and ADHD are associated with condition-specific liabilities; in addition, there are some inherited liabilities that specifically lead to comorbid ASD and ADHD. B: Here, ASD and ADHD are caused by a combination of transdiagnostic inherited liabilities, and condition-specific liabilities. C: Here, common inherited liabilities and adaptive processes are activated at condition-specific points in development. Comorbidity is created by a longer period of activation. Condition-specific genetic and environmental factors affect the timing of expression of common inherited liabilities. D: Inherited liabilities for ASD and ADHD are condition-specific, but require the absence of condition-general protective factors to be expressed. Here, comorbidity simply results from the true statistical overlap of the presence of inherited liabilities for ASD and ADHD. Reproduced with permission from Johnson et al., 2015.
Key: RM = Risk Marker; PF = Protective Factor; A = ASD; D = ADHD; AD = Adaptive response. GE = genetic and/or environmental risk factors.
A corollary of the observation of pleiotropy—whether at the level of familial liability or specific molecular genetic susceptibility—is that any individual case of autism may arise from a distinct combination of contributing liabilities. Recently, for example, it has been demonstrated that both polygenic risk and rare monogenic variants can contribute additively to autism susceptibility within an individual (Weiner et al., 2017; McKenna et al., 2018). The search for pleiotropic developmental contributors to autism risk represents a major departure from traditional approaches to elucidating its origins, which have historically focused on “factors that are specific to autism,” an investigative bias which has unfortunately constrained the search space for causal pathways leading to the condition. In retrospect, the potential contributions of non-autism-specific liabilities to the development of autism should have been recognized long ago when it first became apparent that disparate monogenic syndromes (e.g., Fragile X Syndrome, Tuberous Sclerosis) commonly (but not always) give rise to “phenocopies” of the autistic syndrome. To this end, the common disorders that exhibit genetic overlap with autism (DCD and ADHD) have historically been regarded as “comorbidities” , a potentially misleading characterization when considering that they may actually represent contributing causes of autism (Hawks and Constantino, 2019). Thus, heritable neuropsychiatric traits of other conditions which have been now shown to predict autism (occurrence, familial recurrence, or both) represent a first set of candidates for an underlying developmental sub structure of the syndrome.
Another prototypic contributor to a developmental sub structure of ASD is atypicality in social visual engagement. This has primarily been ascertained by eye tracking infants’ viewing of dynamic social scenes in the first 18 months of life, beginning prior to the time when overt pathognomonic deficiencies in eye contact are first appreciable in most children with autism. Jones and Klin (2013) observed that among infant siblings of children with autism, decline in visual orientation to eyes over serial measurements began in the second month of life and predicted autism recurrence as well as the severity of social symptomatology among affected children. The developmental interval in which the contrast between later-born sibs with and without autism peaked was at approximately 18 months of age, at which point low eye- and mouth-looking was found to be near universal in two replication cohorts of 18 month olds with autism (Constantino et al., 2017). Using identical methodology, ascertainment of social visual engagement among epidemiologically-ascertained, 18 month-old infant twins from the general population revealed two significant qualifications of the association: i) approximately ten percent of typically-developing infants in the general population—children who did not go on to develop autism—displayed levels of visual social disengagement (low eye-looking) in the atypical range that characterized toddlers with autism, suggesting that low eye-looking may be “necessary-but-not-sufficient” to engender autism; ii) the degree of eye looking was continuously distributed in the general population and exquisitely heritable, both at the level of total proportion of visual attention allocated to eyes/mouths, and at the level of moment-to-moment pathways of visual fixation in response to dynamic social scenes (Constantino et al., 2017). Taken together these findings suggest that atypicality in social visual engagement represents a common inherited developmental liability to autism, possibly with higher positive predictive value among siblings of affected individuals (among whom other independent inherited susceptibilities to ASD may be present) than in the general population. Variation in patterns of social visual engagement constitute individual-specific differences in the manner in which children assimilate socially salient information from the visual environment. Johnson, Jones and colleagues (2015) have reported a number of convergent findings supporting a general hypothesis that such individual differences may relate to critical adaptive developmental mechanisms (such as ‘niche construction’) that are triggered by earlier relative deficiencies and which account for the emergence of autism, rather than autism arising as a direct consequence of neurodevelopmental atypicality. Rather than invoking a notion of determinism or inevitability, this view of autism as a developmental adaptation to genetic susceptibility and its consequences is aligned with observations of the genetic, developmental and symptom structure of autism described above.
A third candidate in a developmental sub structure for autism is polygenic liability specific to ASD, inter-generationally transmitted and indexed by subclinical autistic traits (Constantino et al, 2010; Lyall et al., 2013). Recently, we observed that such traits in parents are independent of other developmental predictors of autism within individuals (i.e. variation in motor coordination and attention in their offspring), and that all additively predict variation in quantitative autistic trait variation among the children (Pohl et al., 2019). Evidence for the manner in which ASD-specific risk interacts with the effects of other developmental predictors is still emerging, through studies that systematically explore the effects of joint liability, how pronounced and how early the liabilities must manifest to predict adverse clinical outcomes, and that compare prediction in the general population to within-family-recurrence among multiplex autism families.
All of the above candidate contributors to the developmental sub structure of autism lie within the realm of behavioral variation. An exciting new generation of studies, more limited in sample size because of the technologies involved, is beginning to identify neural signatures of the condition, for which time will tell whether the respective phenotypes add predictive power or overlap with the predictions made by early behavioral observations. Either way, they hold the promise of profound new insights into mechanisms by which genes and behavior are linked in the causation of ASD. Early brain overgrowth ascertained by serial brain MRI of infant siblings of children with autism has had a first demonstration of first-year-of-life prediction of ASD (Hazlett et al., 2017), a finding which is in the process of replication attempts in the U.S. and U.K. In the realm of functional brain activity, whole-brain signatures of neural complexity and communication ((Gabard-Durnam et al., 2019; Haartsen et al., 2019) and atypical electrophysiologic or haemodynamic responses to social experience ((E. J. H. Jones et al., 2016; Lloyd‐Fox et al., 2018)) are important candidate predictors that are under intensive study (see below).
A final robust predictor of autism across nearly all of its diverse genetic causes—a notable exception being rare, highly-damaging single-gene mutations engendering autism, intellectual disability and dysmorphism (Miles et al., 2012)--is male sex. There are many clues to the nature of the universally-observed 3:1 sex ratio in autism, which arises by the time the disorder first develops (second year of life, see Messinger et al., 2015), at a time when sex hormone levels are essentially identical for males and females. Importantly, the M:F recurrence ratio within families is the same as the general population sex ratio, indicating that sex differential expression of inherited risk operates within and across disparate the pathways to familial autism described herein. This observation has given rise to theories about a “female protective effect” for autism. Somewhat inconsistent with this model, however, is the repeatedly-demonstrated absence of a Carter effect for clinical autistic syndromes (Goin-Kochel et al., 2007; Gronborg et al., 2013) meaning that the family recurrence rate in relation to an affected female is generally equivalent to that in relation to an affected male, and that sisters of affected individuals do not exhibit a significant increase in risk of autism to offspring in comparison to brothers of affected individuals (Bai et al., 2020). These data favor an hypothesis of greater male sensitivity to the phenotypic effects of genetic susceptibility in relation to the mean risk population-wide (rather than a female protective effect per se), or greater variability in the effects of risk genes in males versus females; with affected individuals (male and female) exhibiting much lower-than-average likelihood of reproduction. Specifying the mechanism of sex-differential expression is a high priority for ongoing research given the potential magnitude of impact of treatments if they were capable of targeting convergent mechanisms of either resilience or sensitivity parsimoniously driven by sex.
Clinical and Translational Implications
Such understanding of the origins of autism sets the stage for a developmental approach to autism science from genetics through brain development to behavior (also see (Thapar & Riglin, 2020)). To achieve this vision, a next set of priorities involves continued work toward identification of a more complete list of contributing influences and understanding how inherited liabilities operate together to incur risk for autism; as well as mapping the interactions between inherited polygenic susceptibility, penetrant de novo variants and environmental adversity in the accumulation of risk for clinical autistic syndromes.
1. Implications for a next generation of clinical research studies on the development of autism
Although we propose several candidate inherited liabilities above, there are likely many more to be identified. The developmental “footprints” of specific inherited liabilities are most likely to be identified in the period of development prior to the emergence of diagnostic symptoms, before they become confounded by downstream effects of the experience of autism itself. Prospective longitudinal studies of infants with older siblings with ASD have convincingly shown that behavioral symptoms of ASD emerge gradually over the first three years of life (Emily J.H. Jones et al., 2014; Tiede & Walton, in press). In the first six months, symptom-relevant behaviors are near-absent in infants with later ASD; there then follows a gradual decline in social interest and communication (possibly reflecting a near-universal profile of regression in these domains, (Emily J.H. Jones et al., 2014; Ozonoff & Iosif, 2019) such that by 12-14 months, behavioral measures of autistic traits are moderately predictive of a later diagnosis (Pierce et al., 2019). Early autism diagnoses are typically maintained for life (McCauley et al., 2020); the proportion of children with ASD who receive a diagnosis increases with time and stabilizes around age 3 ((Ozonoff et al., 2015); although even in prospective studies of ASD some affected children are not clearly recognized until mid-childhood (Ozonoff et al., 2018). Variability in the timing of diagnosis even within prospective studies reflects heterogeneity in the duration of the period of apparent near-typical development before significant delays or regression become clear ( Pearson et al., 2018). Taken together, this literature suggests that autism-related variation in social development reflects a developmental process that can be variable in timing, but that unfolds and stabilizes over the first few years of life. Of note, few studies have directly examined the coupling between the emergence of social communication and restrictive/repetitive behaviors over this period ; cross-domain coupling methods such as parallel processes models will be important to test whether the early “coalescence” of characterizing traits and features of autism emerges in a uniform way across individuals and how the emergence of symptoms relate to the neurocognitive systems that shape early development.
In addition to the strong candidate inherited liabilities we introduce above, there are a range of other candidates that represent good targets for further investigation. For example, alterations in basic sensory processing, including speech perception, have been recently recognized as part of the core autism phenotype (Robertson & Baron-Cohen, 2017; Tryfon et al., 2017; Edwards et al., 2017). Recent evidence indicates that sensory atypicalities emerge in the first year of life, with a general pattern of exaggerated reactivity to simple sensory stimuli in infants with later autism. e.g. larger constriction of the pupil in response to bright light (Nyström et al., 2018), more accurate identification of a distinct visual object amongst homogenous distractors (Cheung et al., 2018), and exaggerated neural responses to repeated sounds (Kolesnik et al., 2019). Such alterations could have cascading effects on later development by affecting the critical sensory systems through which infants learn about the world. Secondly, converging evidence suggests reduced specialization of ‘social brain’ regions in young infants with later autism, with evidence of decreased responsiveness to naturalistic social interactions in key temporal brain regions (Braukmann et al., 2018; Lloyd‐Fox et al., 2018) that typically specialize for social processing within hours after birth (Farroni et al., 2013). These disruptions to social brain specialization could be a downstream consequence of the reductions in social engagement discussed above (Klin et al., 2015); alternatively, they may arise from difficulties in sensory processing of specific perceptual features like structural details of the face (Dawson et al., 2005; E. J. H. Jones et al., 2016) or syllables within language streams (Seery et al., 2013). Regardless, reduced specialization in key brain regions could compromise or reflect differences in the efficiency with which social interaction is understood. In the second year of life, another potential liability emerges in the guise of greater internally-directed attentional focus. Possible manifestations of this include slower shifting of attention to a novel stimulus when interest is already captured (Elison et al., 2013; Elsabbagh et al., 2013), and increased connectivity in the alpha range (suggestive of greater internal focus) during viewing of dynamic videos (Haartsen et al., 2019). Such focus could be related to the monotropism described in first-person accounts from autistic people and may reduce the influence of social partners in shaping children’s attentional focus.
The degree to which these candidates represent independent inherited liabilities or are actually manifestations of a common process is a critical next step for investigation. Work to combine different predictors and examine their joint or individual effects remains rare; however, there is some emerging evidence from both statistical modelling (Bedford et al., 2012) and machine-learning approaches (Tye et al., in press) that social and ‘non-social’ predictors may act additively to raise liability for later autism. Sophisticated developmental modelling will be required to move from this broad profile of individual susceptibilities towards a coherent map of autism risk that would allow for the possibility that disparate developmental competencies may include nuanced capabilities for compensation, i.e. “checks and balances” in brain and behavioral development that allow deficits in one domain to be buffered by adjustments in others. Finally, some infant predictors of later autism may be neural precursors of the behaviorally-expressed sub clinical autistic traits that run in families and were discussed above. One way to examine this is to test how putative liabilities vary with dimensional autistic traits measured among first degree relatives. In one example, E. J. H. Jones et al., (2016) showed that several measures of social attention predicted later ASD within a longitudinal cohort of infants with ASD. The same markers were associated with parental variation in heritable autistic trait measures within a population of neurotypical infants (Emily J. H. Jones, Venema, et al., 2017). As discussed above, work in general population samples may be particularly important because it allows researchers to disentangle markers that are specific to autistic trait liability from markers of pleiotropic developmental disruption.
Integration of molecular genetics remains one of the most powerful tools we have for interrogating a condition that is as strongly inherited as ASD. As in clinically-referred samples, both polygenic risk and copy number variants contribute to prediction of autism outcome within cohorts of infants with family histories of ASD (D’Abate et al., 2019); thus, it is tractable to examine the relation between genetic variation and putative inherited liabilities with infant sibling samples (e.g., (Gui, Mason, et al., 2020)). However, it may be problematic to exclusively use existing PGRS scores for this endeavor because they are calculated on the basis of categorical diagnosis. Generating PGRS for quantitative measures of autistic traits may be a more powerful approach, particularly in identifying the neurocognitive basis of the familial aggregation of behaviorally-expressed subclinical autistic traits (Constantino, 2011; Warrier et al., 2019). Further, additional genes may be associated with individual inherited liabilities but missed in PGRS generation studies because they are too weakly associated with categorical diagnosis; large-scale infant genetics consortia could also be used to identify new risk genes for particular neurodevelopmental liabilities. An alternative approach may be to use network analysis or generate polygenic scores for particular suites of genes based on their functional expression profiles at key developmental stages or within key brain regions (e.g.(Parikshak et al., 2013)), and examine their association with transdiagnostic phenotypes (Sullivan & Geschwind, 2019). Finally, integrating epigenetics may be important (Gui, Jones, et al., 2020) since measuring gene expression may add critical mechanistic insight into the development of genotype-phenotype relations (Sullivan & Geschwind, 2019); while recognizing that the ascertainment of gene expression in peripheral tissues is at best an indirect reflection of such variation in brain, which is experimentally inaccessible except in post-mortem samples.
Cross-diagnostic examination of the early predictors of later neurodevelopmental traits is also critical (Hawks & Constantino, 2020), since it is likely that there are both distinct and common inherited liabilities for different neurodevelopmental disorders detectable from infancy, as depicted in Figure 2. Indeed, researchers have begun to prospectively follow cohorts of infants with family histories of both autism and/or ADHD (Begum Ali et al., 2020), and to examine multiple neurodevelopmental outcomes within each cohort (Shephard et al., 2019). Such research has identified some predictors that appear unique to co-occurring conditions such as ADHD and anxiety within cohorts of infants with a family history of ASD (e.g.(Gui, Mason, et al., 2020; Miller et al., 2019; Shephard et al., 2019; Ersoy et al., in press). One domain that is consistently related to a broad range of developmental outcomes is effortful control/executive functioning (Moffitt et al., 2011). Effortful control refers to a set of higher-level regulatory skills that shape cognition and affect in the service of abstract goals, and that emerge gradually through during development (Hendry et al., 2016). In general, better effortful control skills appear protective against ASD, ADHD and anxiety (though not callous-unemotional traits); this may act in interaction (rather than additively) with other putative inherited liabilities. Alternatively, deficits in cerebellar function provide highly parsimonious candidates for widely distributed impairments in the developmental capacity for error-based learning or predictive modeling (von der Luhe, 2016), social motivation (Clements et al., 2018); and specific aspects of cerebellar learning that might exert joint abnormality in the domains of social and motor functioning (Valnegri et al., 2017; Nystrom et al., 2018). Notably, Limperopoulos and colleagues (2010) have described a previously under-recognized form of prematurity-related cerebellar parenchymal injury in up to 20% of extremely preterm infants, a group that is at known elevated risk for the development of ASD; moreover infants with rare, isolated cerebellar hemorrhages have highly-elevated rates of ASD outcome, and suggest that cerebellar dysfunction represents a convergent neural abnormality that may tie together disparate manifestations of social and motor disability—including oculomotor function mediated by cranial nerve nuclei under cerebellar control--in the early development of autism.
2. Advancing new understanding of a latent trait encompassing autism-related variation in human social development
The proposal of a unitary dimension underlying the spectrum of autistic traits does not necessarily imply returning to a traditional ‘single deficit’ model of autism. Single deficit theories traditionally proposed one cognitive or brain system that played a substantive role in autism (while often acknowledging that other systems may play a smaller role), and have been broadly criticized both for their empirical failures and their focus on a simplistic medical model of autism (Astle & Fletcher-Watson, 2020). Most larger studies of autism show modest or absent case/control differences on measures associated with leading ‘single deficit’ accounts (e.g. (e.g. Frazier et al., 2017; Moessnang et al., 2020; Muth et al., 2014; ) with effect sizes decreasing by up to 80% over time (Rødgaard et al., 2019). The substantial heterogeneity in the degree, direction and nature of brain and cognitive differences shown by each individual with autism has popularized approaches that involve summing individual deviations across multiple neurocognitive measures (already routinely used clinically) (e.g. (Jacob et al., 2019; Marquand et al., 2019; Zabihi et al., 2019) and/or disaggregating groups with autism into different stratified subtypes (Wolfers et al., 2019). Our conceptualization of autism related variation in social behavior as a latent trait does not necessarily imply that this trait can be localized in one area of the brain in all individuals or encompass a single domain of cognition. Rather, it may be that such liability reflects a coherent process through which characteristic behavioral traits emerge as a homeostatic response to the way in which the infant’s brain represents the world. This does not preclude fractionation (by the degree of influence of different inherited liabilities, for example), but predicts that fractionation at the level of distal causal paths may not necessarily readily map to later symptom profiles.
Such accounts build on a long history of ‘transactional’ models of developmental psychopathology in which development is shaped by complex interactions between genetic, neurobiological, cognitive and environmental factors; and views psychiatric symptoms as adaptive responses to an adverse external (or internal) environment (e.g.(Frankenhuis & Panchanathan, 2011; Mark H. Johnson et al., 2015). For the typically developing child, early social interaction provides a scaffolded environment in which social partners increase the complexity of exchanges as the child’s skills grow to maintain a moderate level of contingency on the child’s behavior (Elmlinger et al., 2019). This is akin to progress within a computer game, or the training algorithms within an online chess engine. In typical development, moderate parental contingency and informativeness prompts children to actively seek more social interaction. However, if alterations in the fidelity of early sensory processing or atypical social engagement affect the child’s opportunities to learn about social cues, their understanding of social situations will increasingly lag behind the social ‘level’ at which society expects them to be; as a result, social situations become progressively less rewarding, and the child may progressively exploit other non-social niches that better suit their processing style (Mark H. Johnson et al., 2015). This unfolding developmental process and the brain state in which it results may ultimately provide a much closer approximation to the latent trait than is afforded by traditional concepts of diagnostic symptom clusters.
Under such models, neurobiological signatures of autism might be expected to be detectable at the whole-brain level, since they are proposed to reflect a coordinated state of the brain that is predisposed by specific profiles of inherited liability. Further, neural signatures of autism-related variation in social behavior would be likely to change over early development as adaptation progresses. Measures of functional brain activity using EEG provide converging evidence of a migration of neural signatures of autism from sensory regions to anterior cortex over the first two years of life (e.g. (W. J. Bosl et al., 2018; Gabard-Durnam et al., 2019). For example, Bosl and colleagues applied a machine-learning approach to nonlinear measures of EEG complexity taken from 3 to 26 months and showed that the features that were predictive of later ASD were primarily in left temporal and right temporo-parietal regions in early infancy, with later divergence around 18 months in frontal regions. Importantly, the same machine-learning model that predicted ASD outcome also predicted dimensional variation in autism trait scores within the group as a whole. However, other studies do identify alterations in frontal regions that are predictive from the first year (e.g. in delta and gamma power from 3 to 12 months; (Gabard-Durnam et al., 2019); replication and assimilation of these analysis approaches in large cohorts will be needed to develop a coherent picture. Deeper understanding may need to rely upon methods that allow us to jointly examine changes in brain trajectories and the emergence of autistic behaviors.
The model of a developmental sub structure described herein raises the question of whether it is even possible to recover neural signatures of causation after autism develops. If not, it would explain why robust diagnostic ‘biomarkers’ have been very challenging to identify, and why biomarkers of clinical affectation may be highly distinct from early biomarkers of risk and/or causation. To take one example, the N170 is a rapid neural response that is sensitive to faces and is generally accepted to index face expertise. Alterations in the N170 are visible in infants with later ASD (E. J. H. Jones et al., 2016), and also in children and adults with a diagnosis relative to neurotypical controls (Kang et al., 2018), although the experimental results in relation to the latter are mixed and highly dependent on localization of brain regions in which the signals are recorded (Sysoeva et al., 2017). Notably, biological signatures of causal influences may exhibit stronger covariance with individual differences in behavior among unaffected individuals than among clinically-affected subjects, given data from research on identical twins demonstrating substantially weaker twin-twin correlations among the latter than among the former, suggesting that clinical-level impairment may be associated with disruption of usual/expectable relationships between genetic causation and variation-in-severity within the affected group(Castelbaum et al., 2020). This is consistent with emerging results from large studies of autism that show clear case-control differences for selected traits that do not necessarily exhibit concomitant dimensional associations within the group with autism (Del Biano et al, 2020).
3. Advancing Understanding of the Phenomenon of “Canalization” in Typical Development
These findings on the erosion of expectable associations between causal factors and variation-in-severity above the clinical threshold invoke the notion that clinical-level impairment reflects a reduction in the capacity for developmental buffering or “canalization,” in the sense that phenotypic development above the clinical threshold becomes both more exaggerated (less constrained within the window of phenotypes that allows adaptive behavior – the ‘norm of reaction’(Sarkar, 1999)) and more strongly influenced by stochastic and random environmental fluctuations (Castelbaum et al., 2020). Under this model, clinical status may be more likely to be reached when strong inherited liabilities are coupled with the removal or reduction of developmental buffering mechanisms. This is consistent with the complete overlap in range of polygenic scores between affected and non-affected groups in the Simons Simplex Collection; (D’Abate et al., 2019), which suggests that susceptibility to a clinical state of impairment may become amplified beyond the level predicted by inherited liabilities. Such decoupling of liability and outcome might only occur when buffering is removed, or when there are high levels of inherited liabilities, pushing the system toward a condition where buffering is required. Moderators that remove a buffering process might not necessarily act additively, as is possible for the role of sex in differentiating the phenotypic expression of a broad array of inherited liabilities to ASD.
What mechanisms are relevant to developmental buffering? As discussed above, development is shaped by a suite of mechanisms including canalization (which can be conceptualized as the suppression of phenotypic variation between individuals), stability (the suppression of phenotypic variation within an individual), and morphological integration (how variability is structured by the underlying developmental and genetic relation between traits; (Hallgrímsson et al., 2002)). The idea that these processes are relevant to neurodevelopmental disorders is longstanding – Down’s syndrome has been interpreted as a syndrome of developmental instability and fluctuating asymmetries (a sign of developmental instability) have been proposed as risk markers for developmental delay. One important way in which genetic or environmental extremes can reduce canalization is by producing developmental configurations that have not been selected for canalization. This is potentially why within animal models, mutant phenotypes are often more variable than wildtype (Wilkins, 2002). Stressful environments also reduce canalization, because they represent environments to which an organism has not adapted (Burla & Taylor, 1982). Indeed, stressful environments such as institutionalization have been associated with both elevated risk for autistic-like behaviors but also elevated risk for a broad range of other developmental impacts (Humphreys et al., 2017).
In addition to an altered starting state, in autism there may also be direct mutations in the processes that enable canalization itself. Transcription and translation processes have been widely implicated in the genetics of autism (de la Torre-Ubieta et al., 2016) and interactions between transcriptional regulators may play important roles in developmental canalization (Ozbudak et al., 2002; Siegal & Bergman, 2002). Gene translation is also an important source of developmental noise (Raser & O’Shea, 2005). Related to both are epigenetic alterations, which have been implicated as a source of variability between MZ twins (Wong et al., 2014) and have begun to be studied in infant populations (Gui, Jones, et al., 2020). Buffering can also be considered at the brain network level, particularly in the important role of inhibition-excitation coordination. Inhibition/excitation coordination is important in maintaining homeostatic balance in brain circuits, is intimately related to sensitive periods (Dorrn et al., 2010), and for the development of adaptive exploratory behavior (Hellyer et al., 2017). Impairments in inhibition/excitation coordination have been strongly implicated in ASD ((Lee et al., 2017), possibly linked to disruptions in relevant neurotransmitters (GABA or glutamate, e.g.(Horder et al., 2018); notably a shift in the timing of the transition of GABA to its mature function as an inhibitory neurotransmitter has been implicated in ASD and may happen later in males. An excess of excitation may explain the high prevalence of epilepsy in individuals with ASD (Frye et al., 2016). Understanding the nature of disruptions of canalization—particularly any that could be prevented or buffered—may have extremely important implications for intervention approaches aimed at amelioration of the severity of the condition among affected individuals.
4. Implications for the search for pre-emptive interventions targeting intermediate phenotypes
Identification of intermediate phenotypes on the pathways to later ASD allows us to identify potential intervention targets that relate neither to the genetic substrate level, nor directly to the clinical manifestations of ASD when fully expressed as a clinical syndrome. When correctly timed in the appropriate developmental window it may be possible, for example, to enhance aspects of social engagement and social attention via ‘environmental enrichment’ interventions such as parent mediated dyadic communication/engagement programs (Landa, 2018). Drawing on approaches developed from the broader infant ‘positive parenting’ literature, which works with parents using videofeedback to help them to understand and adapt to their infant’s individual communication style to promote social and communicative development (Bakermans-Kranenburg et al., 2003), Green and colleagues (Green et al., 2015) demonstrated that a 12-week parent mediated intervention from 7-10 months of age in infants at elevated familial likelihood of ASD led to a short-term increase in parental non-directiveness and (non-significantly) infant attentiveness to parent in a pilot randomized controlled trial (RCT). At follow-up, there was an overall reduction in autism traits and an increase in parent non-directiveness/synchrony and child attentiveness/communication initiation through to age 36 months (Green et al., 2017). However, two recent RCTs of similar parent mediated interventions with infants from community samples identified via early ASD behavioural screening measures at 12 months of age have more mixed findings; with (Whitehouse et al., 2019) reporting no differences in parenting behavior or in child dyadic communication and increases in parental responsiveness but no changes in infant early autism symptoms, adaptive functioning or language. Employing direct electrophysiological and habituation measures of social attention, Jones et al. (Emily J. H. Jones, Dawson, et al., 2017) demonstrated that following a similar 10-week parenting programme delivered from 9-11 months of age infants at enhanced familial likelihood of ASD showed changes in neurocognitive measures of social attention (greater reduction in habituation times to face versus objects, greater increase in frontal EEG theta power, and P400 response to faces versus objects) that resembled, post-intervention, those seen in typically-developing infants. Future studies that couple the effects of direct neurocognitive measures of the candidate intermediate phenotypes that lead to later ASD and direct measures of emergent autistic traits/symptoms will be important to clarify the mechanisms of effect of such approaches.
In these studies it was not the case that all infants who were recruited between 6 and 12 months of age were on the trajectory to a later ASD presentation; indeed in the elevated family likelihood cohorts it was likely only a minority were. In fact, infants who show earliest signs of autism in the first year of life are more likely to be severely impaired, many with co-occurring cognitive impairments, a subgroup who may be less responsive to early intervention than asymptomatic infants at increased familial risk. Part of the motivation for initiating such studies was the rationale that targeting the intermediate phenotype of social attention/engagement at a very early but developmentally appropriate point in time might lessen or ameliorate future autism severity. A corollary of our proposition that intermediate phenotypes early in development are both shared and influenced by other neurodevelopmental conditions, and not specific per se – at least in early development – to ASD, is that pre-emptive intervention might be targeted toward a broad array of developmental competencies, and that appropriate ascertainment of outcome should be broader than a reduction in core early ASD symptoms. Following this logic, such pre-emptive interventions are not about “preventing autism”. These interventions act on transdiagnostic, intermediate neurocognitive/neurobehavioral phenotypes and would be expected to cause shifts in neurodevelopmental, experiential and adaptive processes that will have much broader downstream neurodevelopmental effects. It is currently unknown whether these approaches or other approaches such as those that target another of our intermediate phenotypes – early executive attention – might also impact transdiagnostically on the later emergence of common cooccurring traits such as ADHD or anxiety difficulties (Talbott & Miller, 2020); although proof-of-concept pilot RCTs with infants at elevated familial likelihood of ADHD and pre-term infants are currently underway (Goodwin et al., 2016; Perra et al., 2020).
To date, these have been mostly small-scale, proof-of-concept studies but the logistic challenges in studying infants ‘at risk’ and offering universal as opposed to targeted interventions are considerable. Such pre-emptive intervention approaches also raise some novel but important ethical challenges, both from the point of view of the infants and the parents involved in the studies. These include the ethics of providing parenting interventions in relation to infants who would have gone on to be typically developing; although the programs are derived from ‘positive parenting’ approaches that have been implemented with a wide range of samples often with some indicative (infant or parent) risk for adverse outcomes (Juffer et al., 2017). The elevated familial likelihood studies, many of which have been associated with better-than-expected outcomes of infants under close surveillance within the programs (Micheletti et al., 2020), involve directly engaging in discussions about parenting behaviors and the influence of these behaviors on infants’ developmental progress and outcomes. By design, these studies include families in which there will be enhanced rates of the broader autism phenotype presentation, including in some of the parents themselves. Parents have reported finding the approach a positive experience, giving them an enriched sense of their infant child and increased enjoyment in parent child interaction, reflecting the generic quality of the positive parenting approach, designed to be adaptable across both typical and atypical development.
5. Access to early intervention and support should not be delayed due the lack of an ASD diagnosis
In many communities and clinical practice settings, a ‘watch and wait’ principle still holds sway in pediatric services. When a child is seen for a developmental assessment in the first few years of life but the presentation is not clear enough to meet criteria for ASD or another developmental disorder they are advised to come back at a later date, even when the parent has concerns about their development or behavior. Such ‘reassuring’ or passive responses by practitioners are strongly associated with delays in obtaining an ASD diagnosis, often by many years (Zuckerman et al., 2015; Constantino et al., 2020). Over-insistence on absolute certainty of diagnosis in the early years when neural plasticity potential might be at its highest ends up acting as a barrier to whatever service/intervention pathways are available. Conceptually and empirically, early diagnostic challenges are to be expected from the account that we outline – at earlier ages there are transdiagnostic perturbations to neurocognitive and behavioral development that are shared in common in infants and toddlers whose presentation will later resolve to that of a child with ASD, ADHD, language delay or general developmental delay, alone or in some combination. In other words, the traits that will later lead to ASD in infancy will not be an infant ‘version’ of the later clinical diagnosis. ‘Watch and wait’ also makes little clinical sense. Two-year-olds with emerging neurodevelopmental disorders struggle to communicate effectively, their communication and behavior restricts their opportunities to learn and develop, and this impacts on their parents who find their behavior perplexing and challenging to manage. Furthermore, many of the interventions to support children’s communication development and to help parents to anticipate, regulate and manage their young child’s behavior will be both needed and of benefit whatever the eventual diagnostic categories the child meets criteria for later in their development. Rather than being diagnosis-led, clinical services should offer a stepped approach that matches assessed needs for broader and more general interventions to increasingly specific interventions as the child’s neurodevelopmental difficulties (and diagnoses) and profiles of strengths and needs emerges over time (Monteiro et al., 2016).
6. Individualizing the approach to assessing liability and early diagnosis
The data that support the existence of a developmental sub structure for ASD have strong implications for clinical developmental assessment. In this framework, children should be viewed as an amalgam of developmental competencies (general cognition, affect regulation, impulse control, capacity for reciprocal social behavior, verbal and non-verbal communicative ability); each can be quantified, rather than constraining evaluation to a determination of whether any one competency is “abnormal” to conform to a unilaterally-defined diagnosis. Moreover, the profiles and interactions between those critical developmental competencies have yet to be subjected to standardized characterization (in the same way that clinical implication of a weight measurement is not judged in absolute terms, only in the context of height, and based on population-normed height versus weight tables). We eagerly await a next generation of developmental-epidemiological studies (some of which are currently underway) that will map the ways in which quantitative variations in cognition, capacity for reciprocal social behavior, language, attention, emotional regulation, sensorimotor function, and interpersonal experience interact with one another over the course of development from infancy to adulthood. It is possible that such studies will pave the way for a new system of characterizing syndromes of developmental delay, along measurable quantitative axes, each of which might allow more precise associations with contributing neural mechanisms (Constantino 2019). This motivates a call for personalized/individualized medicine approaches to embrace quantification of developmental risk along axes of genetic, behavioral, and environmental axes. Such information will be extremely useful in the analysis of intervention studies to better understand moderating and mediator (essentially predictors and mechanisms) of treatment outcomes ((Ng & Weisz, 2016)).
7. Practical Considerations for Biomarker Discovery
The notion that each occurrence of familial autism represents one of many possible permutations and combinations of independent developmental liabilities of varying (but specifiable) levels of severity creates new opportunity for personalized approaches to intervention as well as pragmatic guidance for nearly every aspect of biomarker discovery in autism. Relating biomarkers to inheritance and to the developmental liabilities described in this review (recognizing that the heritability of the condition diverges from the causes of its severity) may improve statistical power to identify robust biological signatures of the condition. Biomarkers have been powerful allies in advancing translational research in most branches of medicine from cancer to hypertension to Alzheimer’s Disease, and for inherited forms of autism the search for biomarkers has thus far proven most productive when focused in infancy, prior to the period when autism emerges—key examples including total brain (Hazlett et al, 2017) and CSF volume (Shen et al., 2017, 2018), early abnormalities in social visual engagement (Jones and Klin, 2013; Schultz et al., 2018), atypical electrophysiologic recordings , and very recently, newborn CSF vasopressin levels (Oztan et al., 2020). Of note, effective stratification biomarkers that identify the biological or developmental correlates of a particular inherited liability may not produce subgroups that meaningfully differ in symptom profiles at the clinical level (because the same symptom profile could result from many other liabilities), but may nonetheless enable targeted treatment that ameliorates symptoms in the subgroup for whom that liability is particularly penetrant. One area that remains to be determined is whether it is possible to identify biomarkers of the latent trait that underpins behavioral symptoms of autism (that we could think of as autism related variation in reciprocal social behavior). These would likely be observed at the whole brain level (e.g. Hazlett et al., 2017), and if a latent trait underlying autism represents a developmental process, would be more fruitfully examined by measuring change over early development than at any static point in time.
Furthermore, when studying any aspect of the relationship between genotype and phenotype, or between a putative biomarker and its association with development and behavior, an overarching implication of the developmental sub structure described herein is that many typical individuals in the population will have aggregations of one or more trait liabilities that contribute to the development of autism. If so, failure to measure those characteristics among “control” subjects in case control research designs will diminish any signal that differentiates groups (on the basis of linkage to a specific contributing trait). Therefore, taking stock of predictors of recurrence in autism, as summarized above, and including their ascertainment in the characterization of control subjects should serve as a new priority in the design of biomarker studies. Further, weak but replicable case-control differences should not be discarded; rather, they may provide valuable indications of putative liabilities that could then be entered into multivariate analyses to detect profiles that shift the probabilities of diagnosis with larger effect. A corollary of the proposed developmental sub structure for intervention research is that it is possible that even minor improvements in the severity of a contributing liability could tip the balance of an allostatic load in favor of typical development, perhaps most potently if applied before (not after) the usual time of onset of signs and symptoms of autism. And even if contributing developmental liabilities prove to be secondary to more proximate brain developmental processes, new understanding of the role of non-shared environmental influences offers additional hope for novel strategies to ameliorate the severity of the condition in affected individuals. This may include buffering the effect of stochastic influences on brain and behavioral development, which have long been implicated in psychopathology, and for which excess vulnerability may represent a signature of “clinical status” not only in autism but in other psychiatric disorders.
Conclusions
Autism is a highly heritable neurodevelopmental disorder that affects the quality of life of millions of individuals worldwide. Despite decades of effort, there remains very little understanding of the mechanisms through which genetic risk factors lead to a clinical diagnosis, and no licensed treatments for the core symptomatology. In the present article we have presented coalescing evidence to suggest that our historical focus on finding specific and necessary factors associated with a categorical diagnosis of autism, and parallel efforts to fractionate the disorder based on its clinical presentation, may have been somewhat misguided. Rather, we present a view of autism as the outcome of a multifactorial process that is prompted by a series of complex interactions between sets of both specific and general inherited liabilities over early development. Such a view sets clear priorities for empirical and clinical progress. Implications for research include increasing focus at early developmental stages, prior to canonical symptom emergence; accelerating work on early transdiagnostic predictors and their relation to a range of neurodevelopmental outcomes; the importance of more complex statistical modelling approaches that incorporate interactions between multiple predictors; and the need to largely decouple stratification biomarker development from symptom profiles. Clinically, the currently presented view suggests it is imperative to focus early intervention efforts on transdiagnostic domains of functioning that may boost outcomes for all engaged children; strongly argues against ‘watch and wait’ approaches to early support; and raises the important possibility that individuals with autism may be more susceptible to environmental risk factors, which sadly they may also more commonly experience and be more adversely affected by, in comparison to typically developing individuals. Finally, we note the critical importance of joining forces with researchers studying related neurodevelopmental conditions who may have been looking at the same “elephant” (Saxe, 1872) from another angle, and with whom we may piece together more of the holistic picture.
Key Points.
--Recent studies have highlighted combinatorial effects of independent early inherited liabilities that may account for both causation and heterogeneity in familial autism.
--This new body of research suggests the existence of a previously-unrecognized developmental sub structure for autism in which a finite number of distinct intermediate phenotypes—including, but not necessarily limited to, social visual disengagement, inattention, deficits in predictive modeling and/or impairment in motor coordination—contribute additively to a singular latent trait encompassing autism-related variation in human social development.
--Clinical-level abnormality in this latent trait is hypothesized to precede and secondarily engender the disparate symptom clusters that characterize the condition: impairment in social communication, and restricted interests and repetitive behaviors; which have been found to be tightly inter-correlated not only in autism but in the general population.
--If such a sub structure underlies inherited forms of autism (which constitute the majority of cases in the population), it has significant scientific implications for linking genes, brain and behavior in biomarker research, and clinical implications for the identification of pre-symptomatic treatment targets, which would differ from autism symptoms per se, which are the typical focus of intervention after the clinical syndrome emerges.
Future Directions.
--These data suggest new opportunities to define patterns of liability that distinguish causal origins of autism across different affected individuals and families, and therefore set the stage for personalized approaches to optimizing early treatment and ameliorating the severity of the condition.
--A next set of priorities involves continued work toward identification of a more complete list of contributing influences and understanding how inherited liabilities operate together to incur risk for autism; as well as mapping the interactions between inherited polygenic susceptibility, penetrant de novo variants and environmental adversity in the accumulation of risk for clinical autistic syndromes. Greater understanding of the mechanisms by which additive early liabilities converge on a singular latent trait will advance knowledge of the phenomenon of “canalization” that characterizes typical developmental trajectories and that is believed to be qualitatively disrupted in enduring syndromes of atypical brain and behavioral development.
--A next translational step is to determine whether ameliorating the impact of any one contributing liability in infancy might tip the balance of an allostatic load in favor of typical development, perhaps most potently if applied before (not after) the usual time of onset of signs and symptoms of autism. The data reviewed here suggest that for young children at elevated risk or showing early signs of the condition, access to early intervention and support should not be delayed due to the lack of a definitive autism diagnosis.
--Recently-identified candidates for early neural signatures of autism, detectable in the first year of life through advanced neuroimaging and/or electrophysiologic technologies, can be explored in tandem with the behaviorally-defined developmental liabilities whose detection in larger studies has principally driven the reconceptualization described in this review; to determine the extent to which they map to one another or rather highlight independent parameters of risk that may complement measurable behavioral liabilities to allow for an even higher level of risk prediction.
--Future forays in biomarker research, including neuroimaging and genotype-phenotype association studies of autism, must recognize that “control” subjects possess quantitative, sub clinical aggregations of the same causal liabilities that contribute to autism; therefore optimizing statistical power to elucidate signatures of the disorder may depend upon phenotypic ascertainment of these same liabilities in both cases and controls.
--A next set of priorities involves continued work toward identification of a more complete list of contributing influences and understanding how inherited liabilities operate together to incur risk for autism; as well as mapping the interactions between inherited polygenic susceptibility, penetrant de novo variants and environmental adversity in the accumulation of risk for autism.
Acknowledgement and Funding
This work was supported by grant P50HD103525 (the Intellectual and Developmental Disabilities Research Center at Washington University in St. Louis) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) to Dr. Constantino. Drs. Jones and Charman were supported by the UK Medical Research Council (MR/T003057/1) and the Innovative Medicines Initiative 2 (IMI2) Joint Undertaking under grant agreement No 777394. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA and AUTISM SPEAKS, Autistica, SFARI.
Footnotes
Disclosure Statement
Dr. Constantino receives royalties from Western Psychological Services for the commercial distribution of the SRS-2. Dr. Charman has served as a paid consultant to F. Hoffmann-La Roche Ltd. and Servier; and has received royalties from Sage Publications and Guilford Publications.
Contributor Information
John N. Constantino, Department of Psychiatry, Washington University School of Medicine, St. Louis, MO, USA.
Tony Charman, Department of Psychology, King’s College London Institute of Psychiatry, Psychology and Neuroscience, London, England, UK.
Emily Jones, Centre for Brain & Cognitive Development, Birkbeck, University of London, London, England, UK.
References
- Astle DE, Fletcher-Watson S. 2020. Beyond the Core-Deficit Hypothesis in Developmental Disorders. Current Directions in Psychological Science, 0963721420925518. 10.1177/0963721420925518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakermans-Kranenburg MJ, van IJzendoorn MH, Juffer F. 2003. Less is more: Meta-analyses of sensitivity and attachment interventions in early childhood. Psychological Bulletin, 129(2), 195–215. [DOI] [PubMed] [Google Scholar]
- Bedford R, Elsabbagh M, Gliga T, Pickles A, Senju A, et al. 2012. Precursors to Social and Communication Difficulties in Infants At-Risk for Autism: Gaze Following and Attentional Engagement. Journal of Autism and Developmental Disorders, 42(10), 2208–2218. [DOI] [PubMed] [Google Scholar]
- Bedford R, Gliga T, Hendry A, Jones EJH, Pasco G, et al. 2019. Infant regulatory function acts as a protective factor for later traits of autism spectrum disorder and attention deficit/hyperactivity disorder but not callous unemotional traits. Journal of Neurodevelopmental Disorders, 11(1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum Ali J, Charman T, Johnson MH, Jones EJH, BASIS/STAARS Team. 2020. Early Motor Differences in Infants at Elevated Likelihood of Autism Spectrum Disorder and/or Attention Deficit Hyperactivity Disorder. Journal of Autism and Developmental Disorders. 10.1007/s10803-020-04489-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosl WJ, Tager-Flusberg H, Nelson CA. 2018. EEG Analytics for Early Detection of Autism Spectrum Disorder: A data-driven approach. Scientific Reports, 8(1), 6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainstorm Consortium, Anttila V, Bulik-Sullivan B, Finucane HK, Walters R Bras W. 2018. Analysis of shared heritability in common disorders of the brain. Science. 360(6395):eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braukmann R, Lloyd‐Fox S, Blasi A, Johnson MH, Bekkering H, Buitelaar JK, Hunnius S. 2018. Diminished socially selective neural processing in 5‐month‐old infants at high familial risk of autism. The European Journal of Neuroscience, 47(6), 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussu G, Llera A, Jones EJH, Tye C, Johnson MH, Beckmann C, Buitelaar J. 2020. Uncovering neurodevelopmental paths to Autism Spectrum Disorder through an integrated analysis of developmental measures and neural sensitivity to faces. Journal of Psychiatry and Neuroscience. https://jpn.ca/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussu G, Jones EJH, Charman T, Johnson MH, Buitelaar JK, et al. 2019. Latent trajectories of adaptive behaviour in infants at high and low familial risk for autism spectrum disorder. Molecular Autism, 10(1), 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelbaum L, Sylvester CM, Zhang Y, Yu Q, Constantino JN. 2020. On the nature of monozygotic twin concordance and discordance for autistic trait severity: A quantitative analysis. Behav. Genet 50(4):263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charman T, 2019. Editorial: Trials and Tribulations in Early Autism Intervention Research. J Am Acad Child Adolesc Psychiatry, 58(9):846–848. [DOI] [PubMed] [Google Scholar]
- Cheung CHM, Bedford R, Johnson MH, Charman T, Gliga T, & BASIS team. (2018). Visual search performance in infants associates with later ASD diagnosis. Developmental Cognitive Neuroscience, 29, 4–10. 10.1016/j.dcn.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements CC, Zoltowski AR, Yankowitz LD, Yerys BE, Schultz RT, Herrington JD. 2018. Evaluation of the social motivation hypothesis of autism: A systematic review and meta-analysis. JAMA Psychiatry. 75(8):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN. 2011. The quantitative nature of autistic social impairment. Pediatr Res. 6955R–62R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN, Kennon-McGill S, Weichselbaum C, Marrus N, Haider A, et al. 2017. Infant viewing of social scenes is under genetic control and is atypical in autism. Nature. 547:340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN. 2019. Early behavioral indices of inherited liability to autism. Pediatr Res. 85(2):127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN, Abbacchi AM, Saulnier C, Klaiman C, Mandell DS, et al. 2020. Timing of the diagnosis of autism in African American children. Pediatrics. (published online Aug. 24, 2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Abate L, Walker S, Yuen RKC, Tammimies K, Buchanan JA, et al. 2019. Predictive impact of rare genomic copy number variations in siblings of individuals with autism spectrum disorders. Nature Communications, 10(1), 5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. 2016. Advancing the understanding of autism disease mechanisms through genetics. Nature Medicine, 22(4), 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bianco T, Mason L, Charman T, Tillman J, Loth E, et al. 2020. Temporal profiles of social attention are different across development in autistic and neurotypical people. Biological Psychiatry: Cognitive Neurosciences and Nueroimaging. [DOI] [PubMed] [Google Scholar]
- Dorrn AL, Yuan K, Barker AJ, Schreiner CE, Froemke RC. 2010. Developmental sensory experience balances cortical excitation and inhibition. Nature, 465(7300), 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LA, Wagner JB, Tager-Flusberg H, Nelson CA. 2017. Differences in neural correlates of speech perception in 3 month olds at high and low risk for autism spectrum disorder. J Autism Dev Disord. 47:3125–3138 [DOI] [PubMed] [Google Scholar]
- Elison JT, Paterson SJ, Wolff JJ, Reznick JS, Sasson NJ, et al. 2013. White Matter Microstructure and Atypical Visual Orienting in 7-Month-Olds at Risk for Autism. American Journal of Psychiatry, 170(8), 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmlinger SL, Schwade JA, Goldstein MH. 2019. The ecology of prelinguistic vocal learning: Parents simplify the structure of their speech in response to babbling. Journal of Child Language, 46(5), 998–1011. [DOI] [PubMed] [Google Scholar]
- Elsabbagh M, Fernandes J, Jane Webb S, Dawson G, Charman T, Johnson MH, British Autism Study of Infant Siblings Team. 2013. Disengagement of visual attention in infancy is associated with emerging autism in toddlerhood. Biological Psychiatry, 74(3), 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersoy M, Charman T, Pasco G, Carr E, Johnson MH, et al. In Press. Developmental paths to anxiety in an autism-enriched infant cohort: The role of temperamental reactivity and regulation. Journal of Autism & Developmental Disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farroni T, Chiarelli AM, Lloyd-Fox S, Massaccesi S, Merla A, Di Gangi V, Mattarello T, Faraguna D, Johnson MH. 2013. Infant cortex responds to other humans from shortly after birth. Scientific Reports, 3. 10.1038/srep02851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane B, Challman D, Martin CL, Ledbetter D. 2016. Shift happens: family background influences clinical variability in genetic neurodevelopmental disorders. Genet Med. 18(4):302–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenhuis WE, Panchanathan K. 2011. Balancing sampling and specialization: An adaptationist model of incremental development. Proceedings of the Royal Society B: Biological Sciences, 278(1724), 3558–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier TW, Strauss M, Klingemier EW, Zetzer EE, Hardan AY, Eng C, Youngstrom EA. 2017. A Meta-Analysis of Gaze Differences to Social and Nonsocial Information Between Individuals With and Without Autism. Journal of the American Academy of Child & Adolescent Psychiatry, 56(7), 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier TW, Ratliff KR, Gruber C, Zhang Y, Law PA, Constantino JN. 2014. Confirmatory factor analytic structure and measurement invariance of quantitative autistic traits measured by the social responsiveness scale-2. Autism. 18(1):31–44. [DOI] [PubMed] [Google Scholar]
- Gabard-Durnam LJ, Wilkinson C, Kapur K, Tager-Flusberg H, Levin AR, Nelson CA. 2019. Longitudinal EEG power in the first postnatal year differentiates autism outcomes. Nature Communications, 10(1), 4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JB, Swanson MR, Meera SS, Grzadzinski RL, Shen MD, et al. 2020. Quantitative trait variation in ASD probands and toddler sibling outcomes at 24 months J Neurodev Disord. 12(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin A, Salomone S, Bolton P, Charman T, Jones EJH., et al. 2016. Attention training for infants at familial risk of ADHD (INTERSTAARS): Study protocol for a randomised controlled trial. Trials, 17(1), 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J, Charman T, Pickles A, Wan MW, Elsabbagh M, et al. 2015. Parent-mediated intervention versus no intervention for infants at high risk of autism: A parallel, single-blind, randomised trial. The Lancet. Psychiatry, 2(2), 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J, Pickles A., Pasco G, Bedford R, Wan MW, et al. 2017. Randomised trial of a parent-mediated intervention for infants at high risk for autism: Longitudinal outcomes to age 3 years. Journal of Child Psychology and Psychiatry, and Allied Disciplines, 58(12), 1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui A, Jones EJH, Wong CCY, Meaburn E, Xia B, et al. 2020. Leveraging epigenetics to examine differences in developmental trajectories of social attention: A proof-of-principle study of DNA methylation in infants with older siblings with autism. Infant Behavior and Development, 60, 101409. [DOI] [PubMed] [Google Scholar]
- Gui A, Mason L, Gliga T, Hendry A, Begum-Ali J, et al. 2020. Look duration at the face as a developmental endophenotype: Elucidating pathways to Autism and ADHD. Development and Psychopathology https://www.cambridge.org/core/journals/development-and-psychopathology [DOI] [PubMed] [Google Scholar]
- Haartsen R, Jones EJH, Orekhova EV, Charman T, Johnson MH. 2019. Functional EEG connectivity in infants associates with later restricted and repetitive behaviours in autism; a replication study. Translational Psychiatry, 9(1), 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallgrímsson B, Willmore K, Hall BK. 2002. Canalization, Developmental Stability, and Morphological Integration in Primate Limbs. American Journal of Physical Anthropology, Suppl 35, 131–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SN, Schendel DE, Francis RW, Windham GC, Bresnahan M, et al. 2019. Recurrence Risk of Autism in Siblings and Cousins: A Multinational, Population-Based Study. Journal of the American Academy of Child and Adolescent Psychiatry, 58(9), 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawks ZW, Constantino JN. 2020. Neuropsychiatric “Comorbidity” as Causal Influence in Autism. Journal of the American Academy of Child & Adolescent Psychiatry, 59(2), 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett HC, Gu H, Munsell BC, Kim SH, Styner M, et al. 2017. Early brain development in infants at high risk for autism spectrum disorder. Nature, 542(7641), 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellyer PJ, Clopath C, Kehagia AA, Turkheimer FE, Leech R. 2017. From homeostasis to behavior: Balanced activity in an exploration of embodied dynamic environmental-neural interaction. PLOS Computational Biology, 13(8), e1005721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry A, Jones EJH, Charman T. 2016. Executive function in the first three years of life: Precursors, predictors and patterns. Developmental Review, 42, 1–33. [Google Scholar]
- Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, et al. 2018. Glutamate and GABA in autism spectrum disorder—A translational magnetic resonance spectroscopy study in man and rodent models. Translational Psychiatry, 8(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys KL, Fox NA, Nelson CA, Zeanah CH. 2017. Psychopathology Following Severe Deprivation: History, Research, and Implications of the Bucharest Early Intervention Project. In Rus AV, Parris SR, & Stativa E (Eds.), Child Maltreatment in Residential Care: History, Research, and Current Practice (pp. 129–148). Springer International Publishing. 10.1007/978-3-319-57990-0_6 [DOI] [Google Scholar]
- Jacob S, Wolff JJ, Steinbach MS, Doyle CB, Kumar V, Elison JT. 2019. Neurodevelopmental heterogeneity and computational approaches for understanding autism. Translational Psychiatry, 9(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Mark H, Jones EJH, Gliga T. 2015. Brain adaptation and alternative developmental trajectories. Development and Psychopathology, 27(2), 425–442. [DOI] [PubMed] [Google Scholar]
- Jokiranta-Olkoniemi E, Cheslack-Postava K, Sucksdorff D, Suominen A, Gyllenberg D. et al. 2016. Risk of psychiatric and neurodevelopmental disorders among siblings of probands with autism spectrum disorders. JAMA Psychiatry 73:622–9. [DOI] [PubMed] [Google Scholar]
- Jones W, Klin A. 2013. Attention to eyes is present but in decline in 2–6 month-olds later diagnosed with autism. Nature. 504:427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EJH, Venema K, Earl R, Lowy R, Barnes K et al. 2016. Reduced engagement with social stimuli in 6-month-old infants with later autism spectrum disorder: A longitudinal prospective study of infants at high familial risk. Journal of Neurodevelopmental Disorders, 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EJH, Dawson G, Kelly J, Estes A, Webb SJ. 2017. Parent-delivered early intervention in infants at risk for ASD: Effects on electrophysiological and habituation measures of social attention. Autism Research: Official Journal of the International Society for Autism Research, 10(5), 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EJH, Venema K, Earl RK, Lowy R, Webb SJ. 2017. Infant social attention: An endophenotype of ASD-related traits? Journal of Child Psychology and Psychiatry, 58(3), 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EJH, Gliga T, Bedford R, Charman T, Johnson MH. 2014. Developmental pathways to autism: A review of prospective studies of infants at risk. Neuroscience & Biobehavioral Reviews, 39, 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juffer F, Bakermans-Kranenburg MJ, van IJzendoorn MH. 2017. Pairing attachment theory and social learning theory in video-feedback intervention to promote positive parenting. Current Opinion in Psychology, 15, 189–194. [DOI] [PubMed] [Google Scholar]
- Kang E, Keifer CM, Levy EJ, Foss-Feig JH, McPartland JC, Lerner MD. 2018. Atypicality of the N170 Event-Related Potential in Autism Spectrum Disorder: A Meta-analysis. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging, 3(8), 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klin A, Shultz S, Jones W. 2015. Social visual engagement in infants and toddlers with autism: Early developmental transitions and a model of pathogenesis. Neuroscience & Biobehavioral Reviews, 50, 189–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnik A, Ali JB, Gliga T, Guiraud J, Charman T, Johnson MH, Jones EJH. 2019. Increased cortical reactivity to repeated tones at 8 months in infants with later ASD. Translational Psychiatry, 9(1), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landa RJ. 2018. Efficacy of early interventions for infants and young children with, and at risk for, autism spectrum disorders. International Review of Psychiatry (Abingdon, England), 30(1), 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Lee J, Kim E. 2017. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biological Psychiatry, 81(10), 838–847. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. 2010. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 167:1357–63. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Halldner L, Zetterqvist J, Sjölander A, Serlachius E, et al. 2012. Medication for attention deficit-hyperactivity disorder and criminality. N Engl J Med. 367(21):2006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limperopoulos C 2010. Extreme prematurity, cerebellar injury, and autism. Semin Pediatr Neruol. 17(1):25–9. [DOI] [PubMed] [Google Scholar]
- Lloyd‐Fox S, Blasi A, Pasco G, Gliga T, Jones EJH., et al. 2018. Cortical responses before 6 months of life associate with later autism. European Journal of Neuroscience, 47(6), 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall K, Constanitno JN, Weisskopf MG, Roberts AL, Ascherio A, Santagelo SL. 2014. Parental social responsiveness and risk of autism spectrum disorder in offspring. JAMA Psychiatry. 71(8):936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenner JM, Shaw KA, Baio J, EDS1, Washington A, et al. 2020. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. 69(4):1-12. Erratum. MMWR Morb Mortal Wkly Rep. 2020. 69(16):503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquand AF, Kia SM, Zabihi M, Wolfers T, Buitelaar JK., Beckmann CF. 2019. Conceptualizing mental disorders as deviations from normative functioning. Molecular Psychiatry, 24(10), 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley JB, Elias R, Lord C. 2020. Trajectories of co-occurring psychopathology symptoms in autism from late childhood to adulthood. Development and Psychopathology, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna B, Koomar T, Vervier K, Kremsreiter J, Michaelson JJ. 2018. Whole-genome sequencing in a family with twin boys with autism and intellectual disability suggests multimodal polygenic risk. Cold Spring Harb Mol Case Stud. 4(6):a003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Musser ED, Young GS, Olson B, Steiner RD, Nigg JT. 2019. Sibling Recurrence Risk and Cross-aggregation of Attention-Deficit/Hyperactivity Disorder and Autism Spectrum Disorder. JAMA Pediatrics, 173(2), 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessnang C, Baumeister S, Tillmann J, Goyard D, Charman T, et al. 2020. Social brain activation during mentalizing in a large autism cohort: The Longitudinal European Autism Project. Molecular Autism, 11(1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt TE, Arseneault L, Belsky D, Dickson N, Hancox RJ, et al. 2011. A gradient of childhood self-control predicts health, wealth, and public safety. Proceedings of the National Academy of Sciences of the United States of America, 108(7), 2693–2698. JSTOR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro SA, Dempsey J, Broton S, Berry L, Goin-Kochel RP, Voigt RG. 2016. Early Intervention Before Autism Diagnosis in Children Referred to a Regional Autism Clinic. Journal of Developmental and Behavioral Pediatrics: JDBP, 37(1), 15–19. [DOI] [PubMed] [Google Scholar]
- Mous SE, Jiang A, Agrawal A, Constantino JN. 2017. Attention and motor deficits index non-specific background liabilities that predict autism recurrence in siblings. Journal of Neurodevelopmental Disorders. 9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth A, Hönekopp J, Falter CM. 2014. Visuo-Spatial Performance in Autism: A Meta-analysis. Journal of Autism and Developmental Disorders, 12(44), 3245–3263. [DOI] [PubMed] [Google Scholar]
- Myers SM, Challman TD, Bernier R, Bourgeron T, Chung WK, et al. 2020. Insufficient evidence for “autism-specific” genes. Am J Hum Genet. 106(5):587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström P, Gliga T, Jobs EN, Gredebäck G, Charman T, et al. 2018. Enhanced pupillary light reflex in infancy is associated with autism diagnosis in toddlerhood. Nature Communications. 9(1), 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. 2002. Regulation of noise in the expression of a single gene. Nature Genetics, 31(1), 69–73. [DOI] [PubMed] [Google Scholar]
- Ozonoff S, Iosif A-M. 2019. Changing conceptualizations of regression: What prospective studies reveal about the onset of autism spectrum disorder. Neuroscience and Biobehavioral Reviews, 100, 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S, Young GS, Brian J, Charman T, Shephard E, Solish A, Zwaigenbaum L. 2018. Diagnosis of Autism Spectrum Disorder After Age 5 in Children Evaluated Longitudinally Since Infancy. Journal of the American Academy of Child and Adolescent Psychiatry, 57(11), 849–857.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonoff S, Young GS, Landa RJ, Brian J, Bryson S, et al. 2015. Diagnostic stability in young children at risk for autism spectrum disorder: A baby siblings research consortium study. Journal of Child Psychology and Psychiatry, and Allied Disciplines, 56(9), 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oztan O, Garner JP, Constantino JN, Parker KJ. 2020. Neonatal CSF vasopressin concentration predicts later medical record diagnosis of autism spectrum disorder. Proc Natl Acad Sci USA. 117(19):10609–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R., Zhang A, Won H, Lowe JK, et al. 2013. Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism. Cell, 155(5), 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson N, Charman T, Happé F, Bolton PF, McEwen FS. 2018. Regression in autism spectrum disorder: Reconciling findings from retrospective and prospective research. Autism Research, 11(12), 1602–1620. [DOI] [PubMed] [Google Scholar]
- Perra O, Wass S, McNulty A, Sweet D, Papageorgiou K, et al. 2020. Training attention control of very preterm infants: Protocol for a feasibility study of the Attention Control Training (ACT). Pilot and Feasibility Studies, 6, 17. 10.1186/s40814-020-0556-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce K, Gazestani VH, Bacon E, Barnes CC, Cha D, et al. 2019. Evaluation of the Diagnostic Stability of the Early Autism Spectrum Disorder Phenotype in the General Population Starting at 12 Months. JAMA Pediatrics. 10.1001/jamapediatrics.2019.0624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piven J, Palmer P. 1999. Psychiatric disorder and the broad autism phenotype: evidence from a family study of multiple-incidence autism families. Am J Psychiatry. 156(4):557–63 [DOI] [PubMed] [Google Scholar]
- Pohl A, Jones WR, Marrus N, Zhang Y, Klin A, Constantino JN. 2019. Behavioral predictors of autism recurrence are genetically independent and influence social reciprocity: evidence that polygenic ASD risk is mediated by separable elements of developmental liability. Transl Psychiatry. 9, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM., O’Shea EK. 2005. Noise in Gene Expression: Origins, Consequences, and Control. Science (New York, N.Y.), 309(5743), 2010–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiersen AM, Constantino JN, Todd RD. 2008. Co-occurrence of motor problems and autistic symptoms in attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 47:662–72. [DOI] [PubMed] [Google Scholar]
- Robertson CE, Baron-Cohen S. 2017. Sensory perception in autism. Nature Reviews Neuroscience, 18(11), 671–684. [DOI] [PubMed] [Google Scholar]
- Robinson EB, Koenen KC, McCormick MC, Munir K, Hallett V, Happé F, Plomin R, Ronald A. 2011. Evidence that autistic traits show the same etiology in the general population and at the quantitative extremes (5%, 2.5%, and 1%). Arch Gen Psychiatry 68:1113–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson EB, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, et al. 2016. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nature Genetics, 48(5), 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rødgaard EM, Jensen K, Vergnes JN, Soulières I., Mottron L. 2019. Temporal Changes in Effect Sizes of Studies Comparing Individuals With and Without Autism: A Meta-analysis. JAMA Psychiatry, 76(11), 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. 2017. The heritability of autism spectrum disorder. JAMA. 318, 1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S 1999. From the Reaktionsnorm to the Adaptive Norm: The Norm of Reaction, 1909–1960. Biology and Philosophy, 14(2), 235–252. [Google Scholar]
- Saxe JG. 1872. The poems of John Godfrey Saxe. Boston, MA: Houghton, Mifflin and Company. [Google Scholar]
- Seery AM, Vogel-Farley V, Tager-Flusberg H, Nelson CA. 2013. Atypical lateralization of ERP response to native and non-native speech in infants at risk for autism spectrum disorder. Developmental Cognitive Neuroscience, 5, 10–24. 10.1016/j.dcn.2012.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MD, Nordahl CW, Li DD, Lee A, Angkustsiri K, et al. 2018. Extra-axial cerebrospinal fluid in high-risk children with autism aged 2–4 years: a case-control study. Lancet Psychiatry. 5(11):895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shephard E, Bedford R, Milosavljevic B, Gliga T, Jones EJH, et al. 2019. Early developmental pathways to childhood symptoms of attention-deficit hyperactivity disorder, anxiety and autism spectrum disorder. Journal of Child Psychology and Psychiatry, 60(9), 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal ML, Bergman A. 2002. Waddington’s canalization revisited: Developmental stability and evolution. Proceedings of the National Academy of Sciences of the United States of America, 99(16), 10528–10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Geschwind DH. 2019. Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell, 177(1), 162–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbott MR, Miller MR. 2020. Future Directions for Infant Identification and Intervention for Autism Spectrum Disorder from a Transdiagnostic Perspective. Journal of Clinical Child and Adolescent Psychology: The Official Journal for the Society of Clinical Child and Adolescent Psychology, American Psychological Association, Division 53, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapar A, Riglin L. 2020. The importance of a developmental perspective in Psychiatry: What do recent genetic-epidemiological findings show? Molecular Psychiatry, 25(8), 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F. 2016. Heritability of autism spectrum disorders: A meta-analysis of twin studies. Journal of Child Psychology and Psychiatry, and Allied Disciplines, 57(5), 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiede GM, Walton KM. (in press). Social endophenotypes in autism spectrum disorder: A scoping review. Development and Psychopathology, 1–29. [DOI] [PubMed] [Google Scholar]
- Tryfon A, Foster NEV, Sharda M, Hyde KL. 2018. Speech perception in autism spectrum disorder: An activation likelihood estimation meta-analysis. Behav Brain Res 338:118–127. [DOI] [PubMed] [Google Scholar]
- Tye C, Bussu G, Gliga T, Elsabbagh M. Pasco. In Press. Understanding the nature of face processing in early autism: A prospective study. Journal of Abnormal Psychology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valnegri P, Huang J, Yamada T, Yang Y, Mejia LA, et al. 2017. RNF8/UBC13 ubiquitin signaling suppresses synapse formation in the mammalian brain. Nat Commun. 8:1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lühe T, Manera V, Barisic I, Becchio C, Vogeley K, et al. 2016. Interpersonal predictive coding, not action perception, is impaired in autism. Philos Trans R Soc Lond B Biol Sci. 5;371(1693):20150373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RE, Zhang Y, Gray T, Abbachhi A, Deporres C, et al. 2019. Autism-related variation in reciprocal social behavior: a longitudinal study. Child Dev. 90, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier V, Toro R, Won H, Leblond CS Cliquet F, et al. 2019. Social and non-social autism symptoms and trait domains are genetically dissociable. Communications Biology, 2, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner D, Wigdor EM, Ripke S, Walters RK, Kosmicki JA. 2017. Polygenic Transmission Disequilibrium Confirms That Common and Rare Variation Act Additively to Create Risk for Autism Spectrum Disorders. Nat Genet. 49(7):978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse AJO, Varcin KJ, Alvares GA, Barbaro J, Bent C, et al. 2019. Pre-emptive intervention versus treatment as usual for infants showing early behavioural risk signs of autism spectrum disorder: A single-blind, randomised controlled trial. The Lancet. Child & Adolescent Health, 3(9), 605–615. [DOI] [PubMed] [Google Scholar]
- Wing L 1981. Language, social, and cognitive impairments in autism and severe mental retardation. Journal of Autism and Developmental Disorders, 11(1), 31–44. [DOI] [PubMed] [Google Scholar]
- Wolfers T, Floris DL, Dinga R, van Rooij D, Isakoglou C, et al. 2019. From pattern classification to stratification: Towards conceptualizing the heterogeneity of Autism Spectrum Disorder. Neuroscience and Biobehavioral Reviews, 104, 240–254. [DOI] [PubMed] [Google Scholar]
- Wong CCY, Meaburn EL, Ronald A, Price TS, Jeffries AR, et al. 2014. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Molecular Psychiatry, 19(4), 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabihi M, Oldehinkel M, Wolfers T, Frouin V, Goyard D, et al. 2019. Dissecting the Heterogeneous Cortical Anatomy of Autism Spectrum Disorder Using Normative Models. Biological Psychiatry. Cognitive Neuroscience and Neuroimaging, 4(6), 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman KE, Lindly OJ, Sinche BK. 2015. Parental concerns, provider response, and timeliness of autism spectrum disorder diagnosis. The Journal of Pediatrics, 166(6), 1431–1439.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]