

Abstract
N ε-lysine acetylation within the lumen of the endoplasmic reticulum is a recently characterized protein quality control system that positively selects properly folded glycoproteins in the early secretory pathway. Overexpression of the endoplasmic reticulum acetyl-CoA transporter AT-1 in mouse forebrain neurons results in increased dendritic branching, spine formation and an autistic-like phenotype that is attributed to altered glycoprotein flux through the secretory pathway. AT-1 overexpressing neurons maintain the cytosolic pool of acetyl-CoA by upregulation of SLC25A1, the mitochondrial citrate/malate antiporter and ATP citrate lyase, which converts cytosolic citrate into acetyl-CoA. All three genes have been associated with autism spectrum disorder, suggesting that aberrant cytosolic-to-endoplasmic reticulum flux of acetyl-CoA can be a mechanistic driver for the development of autism spectrum disorder.
We therefore generated a SLC25A1 neuron transgenic mouse with overexpression specifically in the forebrain neurons.
The mice displayed autistic-like behaviours with a jumping stereotypy. They exhibited increased steady-state levels of citrate and acetyl-CoA, disrupted white matter integrity with activated microglia and altered synaptic plasticity and morphology. Finally, quantitative proteomic and acetyl-proteomic analyses revealed differential adaptations in the hippocampus and cortex.
Overall, our study reinforces the connection between aberrant cytosolic-to-endoplasmic reticulum acetyl-CoA flux and the development of an autistic-like phenotype.
Keywords: lysine acetylation, autism spectrum disorder, acetyl-CoA, secretory pathway, white matter
Rigby et al. report that mice with neuronal overexpression of SLC25A1 exhibit aberrant citrate/acetyl-CoA homeostasis and an ASD-like phenotype with disrupted white matter integrity, altered synaptic plasticity and morphology, and widespread proteomic alterations.
Introduction
Neurons require efficient protein quality control measures in order to deliver properly folded proteins for synaptic formation and maintenance. Nε-lysine acetylation within the lumen of the endoplasmic reticulum (ER) is a recently characterized protein quality control system that performs two important cellular functions: (i) positive selection of properly folded glycoproteins in the early secretory pathway; and (ii) regulation of reticulophagy (ER-specific autophagy) to dispose of protein aggregates within the ER.1-21 Aberrant ER-based Nε-lysine acetylation can dramatically impact glycoprotein flux through the secretory pathway and alter intracellular crosstalk between the ER, mitochondria and nucleus,12,17–20 which can result in a variety of disease states including neurodegeneration and autistic-like features.10,12,22,23 For example, patients with gene duplication events in the 3q25.31 locus containing SLC33A1/AT-1, the ER acetyl-CoA antiporter that brings acetyl-CoA into the ER in exchange for CoA, display intellectual disability, dysmorphism and autism spectrum disorder (ASD).24 On the contrary, mutations in SLC33A1/AT-1 are associated with hereditary spastic paraplegia, developmental disability and premature death.22,23 As such, balanced ER-based Nε-lysine acetylation, in part by properly regulated acetyl-CoA import into the ER, is vitally important for neuron physiology.
Several well-known monogenic forms of ASD have common mechanisms involving disrupted synaptic development, plasticity or signalling.25 We recently reported that forebrain neuron overexpression of SLC33A1/AT-1 results in a dramatic increase in dendritic branching, spine formation and an autistic-like phenotype in the mouse.12 The phenotype of this AT-1 neuron transgenic (nTg) mouse was attributed to excessive cytosolic-to-ER import of acetyl-CoA impacting glycoprotein flux through the secretory pathway and altering the biochemical composition of the synapse. In dissecting the cellular adaptations that occurred as a result of SLC33A1/AT-1 overexpression, it became obvious that the neuron made a concerted effort to maintain the cytosolic pool of acetyl-CoA. This was accomplished by upregulating the expression of SLC25A1/CIC, the mitochondrial antiporter that pumps citrate into the cytosol in exchange for malate, as well as ATP citrate lyase (ACLY), which converts cytosolic citrate into acetyl-CoA.12 Importantly, in addition to SLC33A1/AT-1, gene duplication events of SLC25A1 (contained in 22q11.21), ACLY (contained in 17q21.2) and the plasma membrane sodium/citrate synporter SLC13A5/NaCT/INDY (contained in 17p13.1) are all associated with ASD.24 Thus, there appears to be a mechanistic link between the intracellular flux of citrate/acetyl-CoA and the development of ASD, which is at least in part related to changes in ER-based lysine acetylation. Therefore, we hypothesized that altering intracellular citrate/acetyl-CoA flux through the upregulation of SLC25A1 would recapitulate the AT-1 nTg autistic-like phenotype through changes in synapse formation and plasticity.
In this study, we report the generation and phenotypic characterization of the SLC25A1 nTg mouse that demonstrates autistic-like behaviour with jumping stereotypy. We show that both white matter integrity as well as synaptic structure and function are altered in our transgenic mice. Finally, we elucidate both the proteomic and acetyl-proteomic changes within the hippocampus and cortex that highlight the complex adaptations occurring across the cell as a result of SLC25A1 overexpression.
Materials and methods
Transgenic mouse generation
Camk2a-tTA;TRE-SLC25A1 (referred to as SLC25A1 nTg) mice were generated as previously described.12 Briefly, human cDNA was isolated by polymerase chain reaction from SLC25A1-pCMV6 plasmid (Origene; RC200657) and subcloned into pTRE-Tight vector (Takara Bio, Inc.) using HindIII and EcoRI restriction sites. Xho1-linearized pTRE-Tight vector containing the SLC25A1 cDNA was injected into C57BL/6J mice (The Jackson Laboratory; Stock No. 000664) and monogenic offspring were crossed with B6.Cg-Tg(Camk2atTA)1Mmay/DboJ (Camk2a-tTA) mice (The Jackson Laboratory; Stock No. 007004) to generate SLC25A1 nTg mice. Genotyping from tail DNA was performed using the following primers: SLC25A1 forward (5′-CCATCCGCTTCTTCGTCAT-3′), SLC25A1 reverse (5′-CCTGCATCCGGGTCTTAATC-3′), Camk2a-tTA forward (5′-CGCTGTGGGGCATTTTACTTTAG-3′) and Camk2a-tTA reverse (5′-CATGTCCAGATCGAAATCGTC-3′).
Animals
Mice were housed in standard cages provided by the University Laboratory Animal Resources and grouped with littermates, one to five mice per cage; animals were supplied standard chow and water ad libitum. Animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison (protocol #M005120). Non-transgenic, wild-type C57BL/6J mice (The Jackson Laboratory; Stock No. 000664) were used as controls throughout the study. The specific age and sex of animals at time of experimentation are specified in the figure legends.
Behaviour testing
All behavioural assays were conducted within the Waisman Center Behavioral Testing Service (Madison, WI, USA). The experimenter was blind to the genotype of the mice during testing. All mice received a minimum of 30 min acclimation time to the testing room prior to each behaviour assay. The following behavioural assays have been previously described12: novel object recognition, marble burying assay, fear conditioning paradigm and social interaction.
Open field exploration
Open field exploration sessions lasted 30 min and each mouse received 1 session. Each mouse was removed from its home cage and placed in the centre of the arena. The Omnitech Fusion system used photobeams to continuously monitor and record the animal’s placement during the session. Testing variables included total distance travelled (in centimetres), vertical activity episode count and time spent in the centre portion of the open field versus the periphery (in seconds). Data were recorded using the Omnitech Fusion system with a centre ratio zone map.
Light/dark exploration
Each mouse was placed into a split arena for a 10-min assay. Time spent and number of entries into the light and dark portions of the arena were recorded.
Jumping activity
During the dark cycle, videos were taken with mice in their home cages using a red light for visualization. The number of jumps per mouse was quantified over a 50-s time period.
Primary neuron culturing
Primary neurons were generated, cultured and stained as previously described12 on microdissected hippocampus and cortex, and cultures were maintained for a maximum of 28 days. For immunocytofluorescence analysis of synapse formation, the following primary antibodies were used: Syn-1 (Cell Signaling Technology; 5927S; 1:200), Homer1 (Synaptic Systems; 160-011; 1:100), NF-200 (Thermo Fisher; PA3-16753; 1:1000), Psd-95 (Thermo Fisher; MAI-045; 1:500) and Vglut2 (Synaptic Systems; 135–043; 1:500). Images were taken on a Nikon A1 inverted confocal microscope using NIS-Elements AR version 5.11.01 software with 405 nm (blue channel), 488 nm (green channel), 561 nm (red channel) and 640 nm (far red) laser wavelengths using the Galvano scan head. Z-stack images (1024 × 1024 pixels with 25 z-steps every 0.15 µm) were acquired using a 60× oil objective [numerical aperture (NA) = 1.4; 0.21 µm/pixel] at a pinhole size of 39.59 µm. Neurons and neurites were identified by positive NeuN and NF-200 staining, respectively. The .nd2 images were imported into Imaris (Bitplane; Version 9.5) and converted to native .ims format. For neuron morphology analysis, the Filament Tracer module with the Autopath method was used to detect dendrites starting from the soma with the thinnest diameter of 1.5 µm; dendrite seed points were removed within 30 µm of the soma. Dendritic spines were subsequently detected with a seed point diameter of 0.7 µm and maximum length of 7 µm. A Sholl analysis was conducted using 1-µm spaced spheres. For synapse formation analysis, signal from the nucleus was first removed by creating masks of the pre- and postsynaptic marker channels using a surface reconstruction of the 4′,6-diamidino-2-phenylindole (DAPI) signal (setting voxels within the surface to zero). Spots of 2 µm diameter were fit to the pre- and postsynaptic marker signals and spots were co-localized if within 1 µm of each other. The spot counts were normalized to neurite volume, which was estimated from surface reconstruction of the NF-200 signal less the surface reconstruction of the DAPI signal.
For multi-electrode array (MEA), 48-well MEA plates (Axion Biosystems; M768-tMEA-48B-5) were coated in filter-sterilized 0.1% polyethyleneimine (Sigma-Aldrich; 181978-5G) diluted in borate buffer (Thermo Fisher; 28341) for 1 h. The wells were rinsed four times with sterile water then dried overnight. Primary neuron cell suspensions were supplemented with mouse laminin (Thermo Fisher; 23017015; 1 µg/ml) and plated in the centre of the well using 50 000 cells/well in a volume between 5 and 10 µl. After a 1-h incubation at 37°C and 5% CO2, 200 µl neuron culture media was added per well. The cells were maintained at 37°C and 5% CO2 with a half media change every 3–4 days (avoided on the day of recording) and spontaneous activity recordings (extracellular field potentials) were taken every 7 days in vitro (DIV) using the Maestro Pro Multiwell Multielectrode Array and Impedance System (Axion Biosystems). Before recording, the plate was allowed to equilibrate in the MEA recording chamber for 5 min at 37°C and 5% CO2. Data were collected for 10 min using the Axis Navigator software (Axion Biosystems; Version 2.0.2.5) with the Neural Real Time configuration for continuous spontaneous activity. A band-pass filter of 3000 Hz (low-pass) to 200 Hz (high-pass) was applied with a variable threshold spike detector at ±6 standard deviations of the root mean squared of the background noise. A minimum spike rate of 5 spikes/min was used to determine an active electrode, and a mature network was considered to be a well with ≥8 of 16 active electrodes. Bursts were detected in mature networks using the inter-spike interval threshold with a maximum inter-spike interval of 100 ms and a minimum number of five spikes. Network bursts were detected in mature networks with a maximum inter-spike interval of 100 ms, a minimum number of 50 spikes and a minimum of 35% participating electrodes. A synchronicity window of 20 ms was applied. Mean firing rate (Hz) and burst rate (Hz) represent neural excitability while network burst rate (Hz) and network synchronicity (index value between 0 and 1) represent network synchronization.
Neuron isolation
Postnatal Day 7 (P7) pups were euthanized by decapitation and brain processed into a single cell suspension using the neural tissue dissociation kit for postnatal neurons (Miltenyi Biotec; 130–094-802) with the gentleMACS dissociator (Miltenyi Biotec; 130–093-235). Up to 107 cells were processed using the neuron isolation kit (Miltenyi Biotec; 130–115-389) to separate neurons from non-neurons using the QuadroMACS separator (Miltenyi Biotec; 130–090-976).
Diffusion-weighted imaging
On P120, mice were anaesthetized with isoflurane then transcardially perfused with phosphate-buffered solution (PBS) followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. Brains were extracted and post-fixed in PFA. Imaged brains were placed in a custom-built holder immersed in Fluorinert (FC-3283, 3M) and imaged with a 4.7-T Agilent MRI system with a 3.5-cm diameter quadrature volume radiofrequency (RF) coil. Multi-slice, diffusion-weighted, spin echo images were used to acquire 10 non-diffusion-weighted images (b = 0 s/mm2) and 75 diffusion-weighted images (DWIs; 25: b = 800 s/mm2, 50: b = 2000 s/mm2), using non-colinear diffusion-weighting directions. Other imaging parameters include the following: echo time/repetition time = 24.17/2000-ms, field of view = 30 × 30 mm2, matrix = 192 × 192 reconstructed to 256 × 256 for an isotropic voxel size of 0.25-mm over two signal averages.
Raw data files were converted to NIfTI format, and FSL was used to correct for eddy current artefacts. A diffusion tensor imaging (DTI)-based mouse brain atlas26 was used as a template and to define regions of interest, including the left and right hippocampus, amygdala and corpus callosum. Multishell diffusion data were fit with the Microstructure Diffusion Toolbox27 to models of Neurite Orientation Dispersion and Density Imaging (NODDI) and ActiveAx. An additional compartment of isotropic restriction was included to account for potential fixative effects as recommended.28 Whole-brain voxel-wise differences between wild-type and SLC25A1 nTg animals for all MC-DWI indices were determined as previously described with tract-based spatial statistics (TBSS).29 A fractional anisotropy threshold of 0.2 was applied for the creation of the skeleton and permutation test results for multiple comparisons and threshold-free cluster enhancement was implemented with FSL’s Randomize to compare the experimental SLC25A1 nTg group to the wild-type control group, with P < 0.05 as threshold for significance. Region-based differences between wild-type and SLC25A1 nTg animals were determined by calculating the indices of DWI in each diffusion model [fractional anisotropy, mean diffusivity, neurite dispersion index (NDI), orientation dispersion index (ODI), intracellular, extracellular, and stat] for each region of interest and comparing the values between wild-type and SLC25A1 nTg animals via a Student’s t-test (significance P < 0.05). Statistically significant differences were then determined after controlling for multiple comparisons with the Benjamini–Hochberg procedure with the false discovery rate (FDR) set at 0.05.
Electrophysiology
Extracellular recordings of field excitatory postsynaptic potentials (fEPSPs) and long-term potentiation (LTP) were conducted as previously described12 with the following modifications. Slice preparation solution contained 124 mM NaCl, 1.25 mM NaH2PO4, 3 mM KCl, 25 mM NaHCO3, 10 mM glucose, 1 mM sodium ascorbate, 3 mM kynurenic acid, 3.6 mM MgSO4 and 0.8 mM CaCl2. Recording artificial CSF contained 124 mM NaCl, 1.25 mM NaH2PO4, 3 mM KCl, 25 mM NaHCO3, 15 mM glucose, 0.8 mM sodium ascorbate, 1.3 mM MgSO4 and 2.5 mM CaCl2. All solutions were buffered to pH 7.3 when saturated with carbogen and had an osmolality between 294 and 297 mOsm. Recordings were taken from coronal slices using fire-polished borosilicate glass recording pipettes filled with 1 M NaCl (3–5 MΩ) and Pt/Ir concentric bipolar stimulating electrodes. LTP was induced using three theta burst trains, each of which consisted of 40 stimuli grouped into 10 bursts of four stimuli each, with an intraburst frequency of 100 Hz and burst delivered at a rate of 5 Hz. Potentiation was defined as the mean fEPSP slope during the last 10 min of the recording compared to the average of the 10 min immediately preceding the theta burst stimulus.
Synaptosome preparation
Crude synaptosomes were prepared from fresh or snap-frozen hippocampus and cortex using Syn-PER synaptic protein extraction reagent (Thermo Fisher; 87793) supplemented with protease inhibitor cocktail (Millipore; 11836170001) following the manufacturer’s instructions.
Western blotting
Western blotting was conducted as previously described.1,3,10 The following primary antibodies were used in this study: Slc25a1 (Thermo Fisher; 15235–1-AP; 1:1000), β-actin (Cell Signaling Technology; 3700 or 4967; 1:1000 to 1:5000), Neurexin 1 (Thermo Fisher; 18730; 1:1000), Neuroligin 3 (Thermo Fisher; PA5-18849; 1:1000), Rap2a (Thermo Fisher; 23298; 1:1000); Ampa2/3/4 (Cell Signaling Technologies; 2460; 1:1000), mGluR5 (Millipore; AB5675; 1:2000), Synaptogyrin 1 (Abcam; ab113886; 1:1000), Rab12 (Thermo Fisher; PA5-48179; 1:1000), Psd-95 (Cell Signaling Technologies or Thermo Fisher; 3409 or MAI-045; 1:2000 or 1:1000), Syn-1 (Cell Signal Technologies; 5927S; 1:1000), Homer1 (Synaptic Systems; 160–011; 1:1000) and Vglut2 (Synaptic Systems; 135–043; 1:1000).
Donkey anti-rabbit, goat anti-mouse and donkey anti-goat IRDye 800CW, 680RD and 680LT-conjugated secondary antibodies (LI-COR Biosciences; 925–32213, 925–32210, 926–68073, 926–68070, 926–68024) were used for infrared imaging (LICOR Odyssey Infrared Imaging System; LI-COR Biosciences). The original uncropped western blot images included in the manuscript can be found in Supplementary Figs 8 and 9.
Histology and immunostaining
Histology and immunostaining techniques were performed as described previously.10,12,13,30 Klüver–Barrera staining was performed according to kit instructions (Electron Microscopy Sciences; 26681). Golgi staining was performed as previously described.12 The following primary antibodies were used: Gfap (Thermo Fisher; PIMA512023; 1:1000), Iba1 (Abcam; ab178847; 1:1000), Iba1 (Millipore; MABN92; 1:300), NeuN (Millipore; ABN91MI; 1:1000), myelin basic protein (MBP; Abcam; ab40390; 1:200), APC/CC-1 (Millipore; OP80; 1:50), Olig2 (Millipore; AB9610; 1:300) and Slc25a1 (Thermo Fisher; 15235–1-AP; 1:100).
Bright-field images were acquired using a Leica DM4000 B microscope with a 10× or 20× air objective using Image-Pro version 6.3. For high-magnification imaging of Golgi-stained sections, bright-field images were acquired using the Zeiss Axioplan2 upright microscope with a 100× oil objective using Köhler illumination and Q-imaging Retiga 2000r (1.92 MP 12 bit) monochrome camera (7.4 µm × 7.4 µm pixel resolution); z-stacks were collected every 1 µm for a total thickness of 40 µm using StereoInvesigator version 2021 software. All fluorescently labelled slides were imaged on a Nikon A1 inverted confocal microscope using NIS-Elements AR version 5.11.01 software with 405 nm (blue channel), 488 nm (green channel), 561 nm (red channel) and 640 nm (far red) laser wavelengths using the Galvano scan head. For Gfap/Iba1/NeuN and Slc25a1/NeuN-stained slides, single z-slice images (1024 × 1024 pixels) were acquired using a 10× air objective (NA = 0.3; 1.24 µm/pixel) and 60× oil objective (NA = 1.4; 0.21 µm/pixel) at a pinhole size of 255.43 µm (Gfap/Iba1/NeuN) or 220.95 µm (Slc25a1/NeuN). For MBP/NeuN-stained slides, single z-slice images (1024 × 1024 pixels; 1.24 µm/pixel) were acquired using a 10× air objective (NA = 0.3) at a pinhole size of 166.03 µm. For CC-1/Olig2/NeuN-stained slides, single z-slice images (1024 × 1024 pixels; 0.63 µm/pixel) were acquired using a 20× air objective (NA = 0.75) at a pinhole size of 166.03 µm. For Iba1/SLC25A1/NeuN-stained slides, single z-slice images (1024 × 1024 pixels) were acquired using a 60× oil objective (NA = 1.4; 0.21 µm/pixel) at a pinhole size of 132.82 µm.
Microglia (Iba1-positive cells) were quantified using ImageJ (Version 1.52) by making binary images via an intensity threshold and counting objects using the Particle Analyzer tool. Golgi staining images were preprocessed in ImageJ (Version 1.52) by first inverting the images then using the Background Subtract tool three consecutive times with a pixel size of 100, 50 and 25. Images were saved as .tiff, imported into Imaris (Bitplane; Version 9.5) and converted into native .ims format. Secondary dendritic branches were manually traced using the Autopath method of the Filament Tracer module with a diameter of 0.25 µm followed by automatic dendrite volume detection. For spine detection, a mask was first created to eliminate signal distant from the reconstructed dendrites to reduce computation time. Spines were identified with a minimum diameter of 0.25 µm, a maximum length of 5 µm and allowing for branch spines. Dendrite spine density (in spines per 10 µm dendrite length) and spine volume (µm3) were extracted for analysis.
Citrate and acetyl-CoA assays
Hemi-cortex was weighed and fractionated into total ER and cytosol (supernatant after the final centrifugation) using the ER enrichment kit (Novus; NPB2-29482) according to kit instructions. The total ER pellet was resuspended in 100 µl ice-cold acetyl-CoA assay buffer (Abcam; ab87546). Cytoplasmic fractions were extracted from hemi-hippocampus using a nuclear extraction kit (Abcam; ab113474) adjusting the amount of pre-extraction buffer added depending on tissue mass.
All samples were deproteinized (BioVision; #K808) according to kit instructions. Acetyl-CoA concentrations were determined by an acetyl-CoA assay kit (Abcam; ab87546) according to kit instructions, measuring fluorescence on a GloMax plate reader (Promega) using a green fluorescence filter (excitation = 525 nm; emission = 580–640 nm). Citrate concentrations were determined by a citrate assay kit (Abcam; ab83396) according to colorimetric kit instructions, measuring absorbance at 570 nm on a VersaMax plate reader. Cortical ER and cytosolic values were normalized to initial tissue mass.
Proteomics
Cortical and hippocampal tissues were lysed in 8 M urea buffer containing protease and phosphatase inhibitors (Thermo Fisher) followed by 1 min of sonication. The lysate was reduced with 10 mM dithiothreitol at 56°C for 30 min, then alkylated with 20 mM iodoacetamide at room temperature in the dark for an additional 15 min. Approximately 100 μg of protein was then digested with trypsin at a trypsin/protein ratio of 1:100 overnight at 37°C. Peptides were extracted and dried (SpeedVac; Eppendorf), then concentrations of the peptide mixture were measured. Samples were lyophilized and stored at −80°C.
Dimethylated leucine (DiLeu) tags were synthesized as previously described.31 DiLeu tags were activated in anhydrous dimethylformamide (DMF) combined with 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM) and N-methylmorpholine (NMM) at 0.7× molar ratio and vortexed at room temperature for 45 min. After centrifugation, the supernatant was used immediately for peptide labelling. DiLeu labelling was performed by the addition of labelling solution at a 20:1 tag to digested peptide ratio by weight and vortexed at room temperature for 2 h. The labelling reaction was quenched by the addition of hydroxylamine to a concentration of 0.25% and the labelled peptide samples were dried in vacuo. The samples were combined and cleaned with SCX SpinTips (Protea Biosciences) and desalted with Omix C18 pipet tips (Agilent).
The liquid chromatography–tandem mass spectrometry (LC-MS/MS) detection system consisted of a nanoflow high-performance liquid chromatograph instrument (Dionex UltiMate 3000 UPLC system; Thermo Fisher) coupled to an Q Exactive HF orbitrap mass spectrometer (Thermo Fisher) with a nanoelectrospray ion source (Thermo Fisher). In brief, 0.5 μg of peptide mixture dissolved in buffer A (0.1% formic acid) was loaded onto a 75 μm × 15 cm fabricated column filled with 1.7 μm Bridged Ethylene Hybrid packing materials (130 Å; Waters) over a 126-min linear gradient of 3–45% Mobile Phase B (buffer A, 0.1% formic acid in water; buffer B, 0.1% formic acid in acetonitrile) at a flow rate of 300 nl/min. The v analysis was performed in a data-dependent manner using an Orbitrap mass analyser. For a full MS survey scan, the target value was 105, and the scan ranged from 300 to 1500 m/z at a resolution of 60 000, with a maximum injection time of 100 ms. For the MS2 scan, up to 15 of the most intense precursor ions from a survey scan were selected for MS/MS and detected by the Orbitrap at a mass resolution of 15 000 at m/z 400. Only precursor ions with charge states of 2–6 were selected for fragmentation by higher-energy collision dissociation with a normalized collision energy of 30%. The automatic gain control for MS/MS was set to 8e3, with maximum ion injection times of 100 ms. Dynamic exclusion time was 45 s, and the window for isolating the precursors was 1.4 m/z.
Protein and peptide identification and quantification were conducted through MaxQuant version 1.5.3.8.32 Raw files were searched against the UniProt mouse reviewed database (August, 2019) using the integrated Andromeda search engine with FDR < 1% at the peptide and protein level. Trypsin was selected as the enzyme with at most two missed cleavages. A reverse database for the decoy search was generated automatically in MaxQuant. Enzyme specificity was set to ‘Trypsin’, and a minimum number of seven amino acids were required for peptide identification. Static modifications included carbamidomethylation of cysteine residues (+57.02146 Da) and DiLeu labelling on N-terminus and lysine residues (+145.12801 Da). Dynamic modifications included oxidation of methionine residues (+15.99492 Da) and deamidation of asparagine and glutamine residues (+0.98402 Da). The first search mass tolerance was 20 ppm and the main search peptide tolerance was 4.5 ppm. The FDRs of the peptide–spectrum matches and proteins were set to less than 1%. Quantitation was performed using Perseus software.33 Briefly, the raw reporter ion intensity in each DiLeu channel was corrected for isotope impurities31 and normalized for mixing differences by equalizing the total signal in each channel. In cases where no signal was detected in a channel, the missing value was assigned with the noise level of the original spectrum (noise-band capping of missing channels) and the resultant intensity was not corrected for impurities or normalized for uneven mixing.
Stoichiometry of protein acetylation
Cortical and hippocampal tissue was prepared and analysed as previously described.17,34 Acetyl stoichiometry values are reported as a range from 0 to 1 representing 0% to 100% of a detected lysine site being endogenously acetylated, respectively. Changes in acetyl stoichiometry values (Δ acetyl stoichiometry) are consistently reported as the nTg value minus the wild-type value. Pathway analysis and network plot construction was conducted using the R package enrichplot implementing an overrepresentation analysis with the Mus musculus organism database.35 Relevant parameters included a minimum and a maximum number of genes for a category of 5 and 2000, respectively, with the Benjamini–Hochberg method for multiple test adjustment to a FDR of 0.05.
Statistical analysis
Data analysis was performed using GraphPad Prism version 9.0.1. Data are expressed as mean ± standard deviation (SD) unless otherwise specified. Comparison of the means was performed using Student’s t-test for two groups and ordinary one-way or two-way ANOVA for three or more groups followed by either Tukey–Kramer (comparison between all groups) or Dunnett’s (comparison to one control group) multiple comparisons test. If no sex differences were found, data were combined for ease of visualization. Statistical test details are described in the figure legends. Grubb’s test was used to remove outliers determined at P < 0.05. Differences in the mean were declared statistically significant if P < 0.05 and the following statistical significance indicators are used: *P < 0.05; **P < 0.005; ***P < 0.0005.
Data availability
The proteomics data that support the findings of this study have been deposited to the ProteomeXchange Consortium (ID number PXD025415). The acetyl-proteomics data that support the findings of this study have been deposited to the ProteomeXchange Consortium (ID number PXD025424) and the MassIVE partner repository (ID number MSV000087209). The authors declare that all other data supporting the findings of this study are available within the paper, the Supplementary Tables and Supplementary material.
Results
SLC25A1 neuron transgenic mice display autistic-like behaviour
To investigate the role of SLC25A1 in the development of ASD, we created transgenic C57BL/6J mice that specifically overexpress human SLC25A1 in forebrain neurons, termed SLC25A1 nTg mice, as previously described.12 To ensure the SLC25A1 transgene was being properly expressed, we first evaluated SLC25A1 expression in our SLC25A1 nTg mice compared to wild-type controls. As expected, we observed a robust expression of SLC25A1 protein in isolated neurons (Fig. 1A). Additionally, the SLC25A1 protein could be seen specifically expressed within neurons as assessed by co-localization with the neuronal marker NeuN (Fig. 1B). Because SLC25A1 transports citrate into the cytoplasm in exchange for malate, we expected an increase in the steady-state levels of citrate within the neuron cytoplasm in our transgenic mice. To test this, we performed subcellular fractionation on cortical tissue to obtain both enriched ER and cytosol; as expected, the cytosolic fractions had a higher level of citrate in the SLC25A1 nTg samples compared to wild-type controls (Fig. 1C). We performed a similar experiment on hippocampus, but due to the small amount of starting material, we performed a simple subcellular fractionation to obtain nuclear and cytoplasmic fractions; again, we observed an increase in citrate in the SLC25A1 nTg cytoplasmic fractions compared to wild-type (Fig. 1D). Because cytosolic citrate can be converted into acetyl-CoA and enter the ER via the acetyl-CoA transporter AT-1, we assessed whether steady-state levels of acetyl-CoA in the cytosol and ER changed in our SLC25A1 nTg mice. Using the same samples prepared in Fig. 1C, we observed an increase in acetyl-CoA levels in both the enriched ER as well as the cytosolic fraction (Fig. 1E). These data show successful generation of a transgenic mouse line that overexpresses SLC25A1 within forebrain neurons resulting in increased steady-state levels of citrate and acetyl-CoA in the cytosol and acetyl-CoA in the ER.
Figure 1.

SLC25A1 nTg mice overexpress SLC25A1 in neurons and have elevated citrate and acetyl-CoA levels. (A) Protein expression of Slc25a1 in neurons isolated from P7 mice. (B) Paraffin-embedded brain slices immunostained for SLC25A1 and NeuN (neuronal marker). Mice are 3-month-old males. (C) Levels of cytosolic citrate in cortical tissue. Absorbance values (A570) were normalized to cortex mass and expressed as a fold change over wild-type. Data are mean ± SD, n = 6 mice per genotype (three males, three females). **P < 0.005 via two-way ANOVA (Genotype × Sex). (D) Levels of cytoplasmic citrate in hippocampal tissue. Data are expressed as fold change over wild-type and shown as mean ± SD, n = 6 mice per genotype (three males, three females). **P < 0.005 via two-way ANOVA (Genotype × Sex). (E) Levels of acetyl-CoA in cytosolic and ER fractions from cortical tissue. Arbitrary fluorescence units (AFU) were normalized to cortex mass and expressed as a fold change over wild-type. Data are mean ± SD, n = 6 mice per genotype (three males, three females). **P < 0.005 via two-way ANOVA (Genotype × Sex). Mice in panels C–E are 10–13 months of age.
We next assessed the phenotypic consequence of forebrain neuron overexpression of SLC25A1. Shortly after generating the transgenic mouse line, it was obvious that the SLC25A1 nTg mice exhibit a repetitive jumping behaviour in their home cage (Supplementary Video 1). Observed in both the light and dark cycle, the SLC25A1 nTg mice jumped repeatedly for prolonged periods of time while the wild-type littermates did not. Formal quantification of this stereotyped behaviour revealed a faster jumping rate in female versus male transgenic mice (Fig. 2A). We also performed formalized behavioural assessments on wild-type versus SLC25A1 nTg mice aimed at evaluating hippocampal function and autistic-like behaviours like assessed in the AT-1 nTg model.12 A fear conditioning paradigm revealed a significant lack of freezing behaviour in the SLC25A1 nTg mice in both the training and challenge phases of the paradigm (Fig. 2B), and in a novel object recognition paradigm, the SLC25A1 nTg mice spent significantly more time investigating both the novel and familiar objects without a change in the percentage of time investigating the novel object (Fig. 2C). Additionally, the SLC25A1 nTg mice preferred the light side of a light–dark box (Fig. 2D) and exhibited reduced marble burying behaviour (Fig. 2E). Our SLC25A1 nTg mice did not display noteworthy differences from wild-type controls in open field or social interaction assays (Supplementary Fig. 1). Overall, our SLC25A1 nTg mice displayed jumping stereotypy while in their home cage with evidence of aberrant behaviour in the fear conditioning, novel object recognition, light–dark exploration and marble burying paradigms. Several of these behavioural abnormalities have been observed in other rodent models of ASD including the AT-1 nTg,12,36–39 and as such we conclude our SLC25A1 nTg mice exhibit an autistic-like behavioural phenotype.
Figure 2.

SLC25A1 nTg mice exhibit repetitive jumping and aberrant behaviours. (A) Number of jumps in a 50-s period. Wild-type (WT), n = 6 females; nTg female, n = 7; nTg male, n = 5. ***P < 0.0005 via one-way ANOVA with Tukey’s multiple comparison test. (B) Fear conditioning (FC) paradigm. Wild-type, n = 10; Tg, n = 10; equal males/females. *P < 0.05, **P < 0.005 via two-way ANOVA (Genotype × Sex). (C) Novel object recognition (NOR). Wild-type, n = 10; Tg, n = 10; equal males/females. *P < 0.05 via two-way ANOVA (Genotype × Sex). (D) Light–dark (LD) exploration. Wild-type, n = 10; Tg, n = 10; equal males/females. *P < 0.05 via two-way ANOVA (Genotype × Sex). (E) Marble burying (MB) assay. Wild-type, n = 10; Tg, n = 10; equal males/females. **P < 0.005 via two-way ANOVA (Genotype × Sex). All data are displayed as mean ± SD and are from mice at 4–7 months of age.
SLC25A1 neuron transgenic mice have altered white matter integrity
We next sought to explore the cellular and molecular changes that underlie the behavioural abnormalities seen in our SLC25A1 nTg mice. We began with ex vivo DWI as an unbiased way to assess white matter integrity in our transgenic mice, as studies commonly show disrupted white matter integrity in individuals with ASD.40,41 We used both region of interest and TBSS analyses with three diffusion models: DTI, NODDI and ActiveAx. We selected the regions of interest right/left hippocampus and right/left amygdala based upon the behavioural abnormalities observed, and we used the right/left corpus callosum regions of interest as a comparison that we did not expect to change. In the DTI analysis, there was a significant reduction in fractional anisotropy in the right/left hippocampus and right/left amygdala as well as multiple regions of decreased fractional anisotropy in the TBSS analysis (Fig. 3A and B); we did not observe changes in mean diffusivity (Supplementary Fig. 2). NODDI analysis revealed an increase in the ODI in the same regions of interest as well as in the TBSS analysis (Fig. 3A and B) without changes in the NDI (Supplementary Fig. 2). Finally, ActiveAx revealed a decrease in the ‘intra-neuronal’ intracellular parameter in the right hippocampus and right/left amygdala as well as increased ‘glial’ stat parameter values in multiple regions in the TBSS analysis (Fig. 3A and B). No changes in the ‘extra-neuronal’ extracellular parameter were observed (Supplementary Fig. 2). Overall, these data suggest disrupted white matter integrity, as supported by decreased DTI fractional anisotropy, with either an increase in neurite dispersion or interfering non-neuronal processes within the white matter tracts supported by the NODDI and ActiveAx analyses.
Figure 3.

SLC25A1 nTg mice have altered white matter integrity and synaptic plasticity. (A) Ex vivo DWI of male wild-type and SLC25A1 nTg mice at 4 months old, n = 6 mice per genotype. Fractional anisotropy (FA), orientation dispersion index (ODI), and intraneuronal compartment (IC)/glial stat compartment parameters were calculated from a region of interest analysis using DTI, NODDI and ActiveAx models, respectively. Data are mean ± SD. *P < 0.05, **P < 0.005, ***P < 0.0005 via Student’s t-test with Benjamini–Hochberg procedure with FDR = 0.05. (B) Whole-brain voxel-wise TBSS analysis of DWI in panel A. Coloured voxels are statistically different between wild-type and SLC25A1 nTg (P < 0.05). (C) Paraffin-embedded brain slices immunostained for Iba1 (microglia), Gfap (astrocytes) and NeuN (neuronal marker). Quantification of Iba1-positive cells is shown as mean ± SD, n = 6 mice per genotype (three males, three females) at 9–12 months of age. (D) Theta-burst (3×) LTP in hippocampal brain slices. Data are mean ± SEM. **P < 0.005 via Student’s t-test of the average potentiation value in the last 10 min, n = 8 slices per genotype (from two male and three female wild-type mice and three male and three female SLC25A1 nTg mice). Mice are 6–8 months old. (E) Golgi staining of CA1 pyramidal neuron apical dendrites (secondary branches). Representative images along with unbiased dendrite and spine reconstructions are shown. Dendrite spine density: data are shown as mean ± SD with each data-point representing one animal. Spine volume: data are a violin plot of each reconstructed spine’s volume with the solid line representing the median and dashed lines representing the 25th and 75th quartiles. *P < 0.05, ***P < 0.0005 via Student’s t-test, n = 6 mice per genotype (three males, three females) at 3 months of age.
To investigate whether changes in the glial populations could explain the imaging findings,42 we performed immunofluorescence staining for microglia (Iba1) and astrocytes (Gfap). In the SLC25A1 nTg hippocampus, we saw a robust increase in the number of microglia that appeared to exhibit a more ramified morphology compared to wild-type controls; we did not see significant changes in astrocyte number or morphology (Fig. 3C). Importantly, we did not observe overexpression of SLC25A1 within the microglia themselves (Supplementary Fig. 3), suggesting the changes are not due to SLC25A1 transgene expression in cells other than forebrain neurons. We also performed histological assessment of myelin and immunofluorescence staining for oligodendrocytes, but did not observe significant changes from wild-type (Supplementary Fig. 4). Therefore, we conclude the SLC25A1 nTg mice have disrupted white matter integrity that may be in part explained by an expansion of the microglial population in response to neuronal alterations.
SLC25A1 neuron transgenic mice have altered synaptic structure and function
In addition to white matter integrity, we also assessed synaptic structure and function in our transgenic mice, as dramatic changes in synaptic density, biochemical composition, and plasticity occurred in the AT-1 nTg model.12 To begin, we performed hippocampal brain slice electrophysiology to assess synaptic plasticity of the Schaffer collateral pathway. Using a theta burst stimulation paradigm to induce LTP, the SLC25A1 nTg mice exhibited increased potentiation compared to wild-type controls (Fig. 3D). Because of the observed changes in LTP as well as the behavioural changes described above, we performed several experiments to examine neuron morphology and function. To begin, we performed Golgi method staining to visualize the neurons in vivo. Unlike the AT-1 nTg model,12 there were no obvious differences in neuron morphology, namely abundance of dendritic branches, at low magnification in our SLC25A1 nTg mice (Supplementary Fig. 5); however, high-magnification imaging of CA1 pyramidal neuron apical dendrites revealed an increase in dendrite spine density with a concomitant decrease in spine volume in the SLC25A1 nTg compared to wild-type controls (Fig. 3E). Additionally, we isolated synaptosomes from our SLC25A1 nTg mice to evaluate the expression of proteins involved in neuronal outgrowth and synaptic plasticity, specifically focusing on those that were increased in the AT-1 nTg autistic-like model.12 We found minimal differences in protein expression in the SLC25A1 nTg synaptosomes from the cortex and hippocampus compared to wild-type controls (Supplementary Fig. 6). Therefore, in vivo, our SLC25A1 nTg have abnormal synaptic plasticity as demonstrated by enhanced LTP in the Schaffer collateral to CA1 synapse as well as increased dendritic spine density in CA1 pyramidal neurons. Moreover, we found several unexpected differences in the SLC25A1 nTg model compared to the AT-1 nTg model.
We also cultured primary neurons from our transgenic mice in order to assess both neuron morphology as well as synapse formation and function. We cultured embryonic cortical and hippocampal primary neurons and analysed them in the same manner. At 15 DIV, the SLC25A1 nTg cortical neurons exhibited increased dendritic branching compared to wild-type littermates as demonstrated by a Sholl analysis; these neurons did not have a difference in dendritic spine density but did have a robust increase in dendritic spine volume (Fig. 4A). To assess spontaneous synapse formation in vitro, we performed immunostaining for pre- and postsynaptic markers and assessed for co-localization at 15 DIV, implying the formation of a synapse between adjacent neurons.43 When normalized to neurite volume, we observed an increase in puncta formation of the presynaptic markers Syn-1 and Vglut2 as well as the postsynaptic marker Psd-95 in our transgenic cortical neurons compared to wild-type littermates; importantly, we also observed an increased amount of co-localized Syn-1/Homer1 and Vglut2/Psd-95 puncta, suggesting a higher density of synapses in the SLC25A1 nTg cortical neurons (Fig. 4B). To assess the function of these synapses, we plated our cortical neurons on MEAs and monitored their spontaneous electrical activity every 7 days up to 28 DIV. The spontaneous activity of the networks gradually increased over time as evident by monitoring the number of active electrodes at each time point. Interestingly, the SLC25A1 nTg cortical neurons had a higher percentage of active electrodes at 14 and 21 DIV, suggesting the networks matured more rapidly compared to wild-type littermates (Fig. 4C). Additionally, the transgenic cortical neurons exhibited a higher mean firing rate compared to wild-type littermates at 14 DIV (Fig. 4C); there was no difference in the burst frequency, network burst frequency and network synchrony at any time point between the transgenic and wild-type cortical neurons (Supplementary Fig. 7). In the SLC25A1 nTg hippocampal primary neurons, we observed similar changes in neuron morphology compared to wild-type controls with an increase in dendritic branching and spine volume (Fig. 5A). We also observed an increase in Vglut2 and Psd-95 puncta as well as co-localized puncta relative to wild-type littermates (Fig. 5B). We did not see changes in Syn-1 and Homer1 puncta or co-localized puncta in the hippocampal neurons like we did in the cortical neurons (Fig. 5B). Finally, the SLC25A1 nTg hippocampal neurons had a higher percentage of active electrodes at 7 DIV with an increase in mean firing rate at all time points except 28 DIV compared to wild-type littermates (Fig. 5C). We again did not see differences in burst frequency, network burst frequency or network synchrony at any time point between the transgenic and wild-type hippocampal neurons (Supplementary Fig. 7). Overall, these data demonstrate the SLC25A1 nTg primary neurons in vitro exhibit increased dendritic branching, dendritic spine volume and synaptic density with a concomitant increase in neural excitability.
Figure 4.
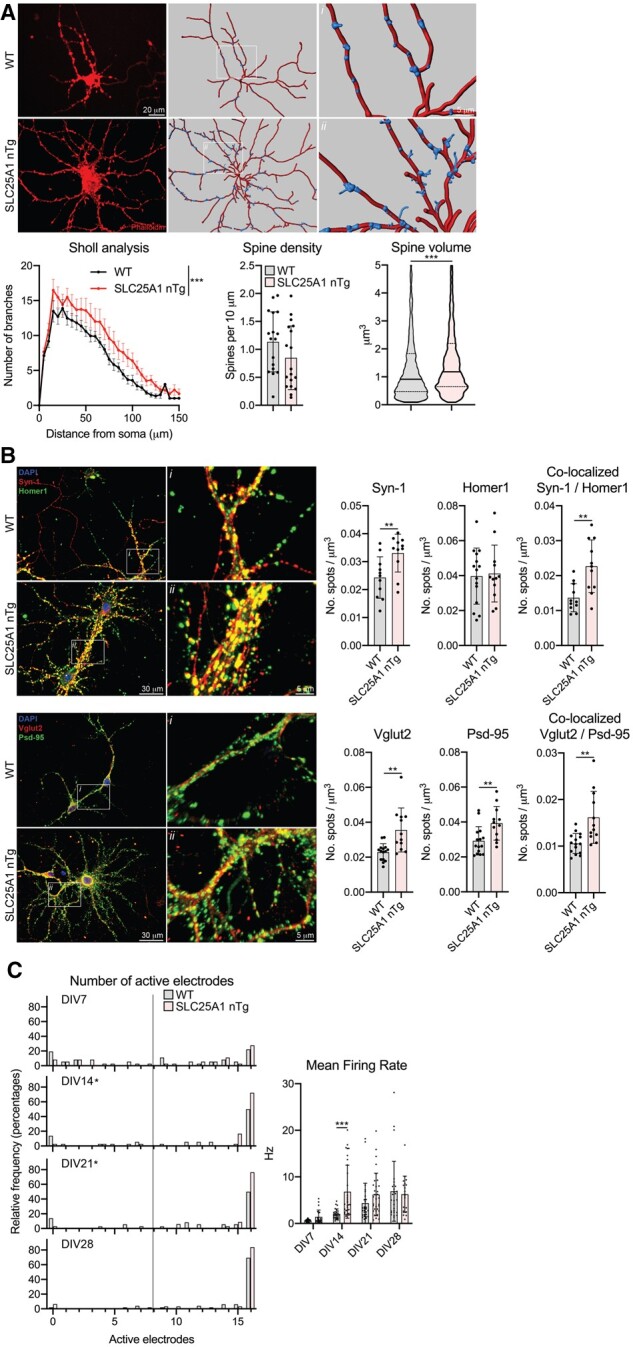
Primary cortical neurons from SLC25A1 nTg mice have increased dendritic branching, altered spine morphology and increased spontaneous activity. (A) Morphological assessment of cultured neurons at 15 DIV. Phalloidin staining (left) and unbiased computer-driven reconstruction (right) are shown along with quantification from n = 3 embryos per genotype. Sholl analysis: data are mean ± SEM. ***P < 0.0005 via two-way ANOVA. Spine density: data are mean ± SD with each data-point representing one neuron. Spine volume: data are displayed as a violin plot of each reconstructed spine’s volume with the solid line representing the median and dashed lines representing the 25th and 75th quartiles. *P < 0.0005 via Student’s t-test (spine volume). (B) Immunostaining of cultured neurons for pre-/postsynaptic marker pairs Syn-1/Homer1 and Vglut2/Psd-95 at 15 DIV. Puncta were fit with 2 µm spots and normalized to neuron volume and spots co-localized if they were within 1 µm of each other. Data are mean ± SD with each data-point representing one neuron, n = 4 wild-type (WT) and n = 3 SLC25A1 nTg embryos. **P < 0.005 via Student’s t-test. (C) MEA spontaneous activity. Left: Histogram showing the number of active electrodes per network expressed as the relative frequency in per cent. The vertical line demarks eight active electrodes, which is the minimum value required to be considered a mature network. *P < 0.05 via Mann–Whitney test comparing frequency distributions. Right: Spontaneous activity measured by mean firing rate. Each data-point is an independent network of cultured neurons and exhibits at least 8 of 16 active electrodes. ***P < 0.0005 via mixed effects analysis with Sidak’s multiple comparison test. Data are from six embryos per genotype.
Figure 5.
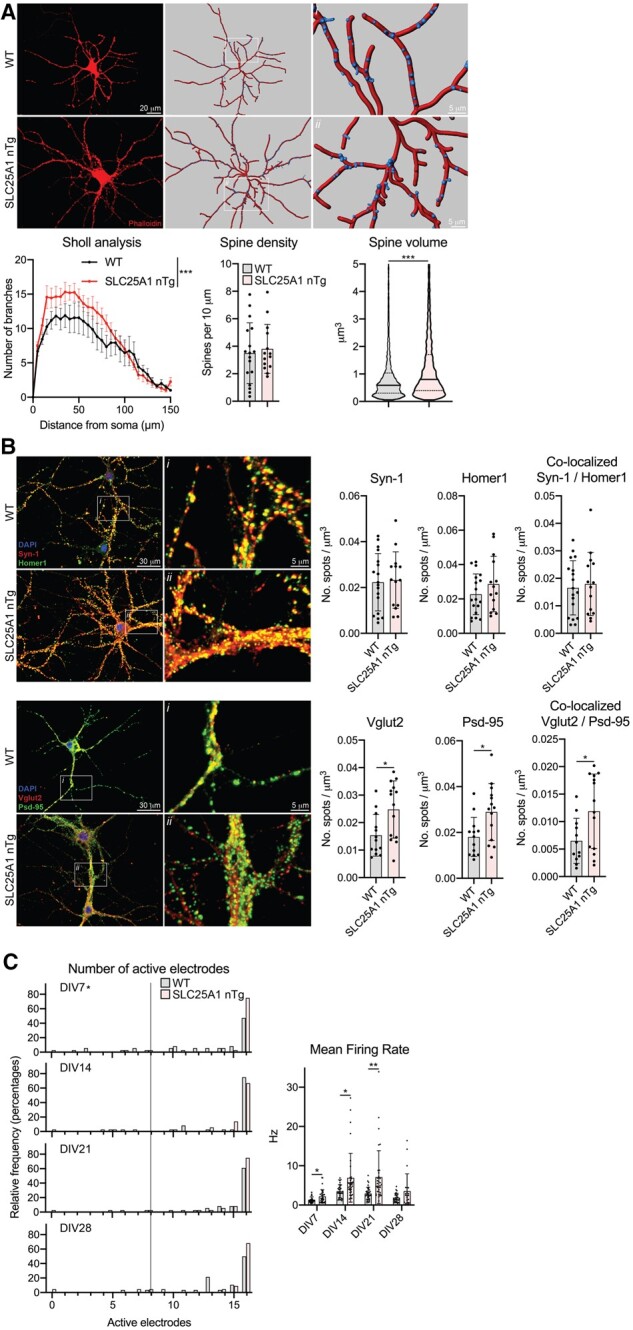
Primary hippocampal neurons from SLC25A1 nTg mice have increased dendritic branching, altered spine morphology and increased spontaneous activity. (A) Morphological assessment of cultured neurons at 15 DIV. Phalloidin staining (left) and unbiased computer-driven reconstruction (right) are shown along with quantification from n = 3 embryos per genotype. Sholl analysis: data are mean ± SEM. ***P < 0.0005 via two-way ANOVA. Spine density: data are mean ± SD with each data-point representing one neuron. Spine volume: data are displayed as a violin plot of each reconstructed spine’s volume with the solid line representing the median and dashed lines representing the 25th and 75th quartiles. ***P < 0.0005 via Student’s t-test (spine volume). (B) Immunostaining of cultured neurons for pre-/postsynaptic marker pairs Syn-1/Homer1 and Vglut2/Psd-95 at 15 DIV. Puncta were fit with 2 µm spots and normalized to neuron volume and spots co-localized if they were within 1 µm of each other. Data are mean ± SD with each data-point representing one neuron, n = 3 wild-type (WT) and n = 4 SLC25A1 nTg embryos. *P < 0.05 via Student’s t-test. (C) MEA spontaneous activity. Left: Histogram showing the number of active electrodes per network expressed as the relative frequency in percent. The vertical line demarks eight active electrodes, which is the minimum value required to be considered a mature network. *P < 0.05 via Mann–Whitney test comparing frequency distributions. Right: Spontaneous activity measured by mean firing rate. Each data-point is an independent network of cultured neurons and exhibits at least 8 of 16 active electrodes. *P < 0.05, **P < 0.005 via mixed effects analysis with Sidak’s multiple comparison test. Data are from six embryos per genotype.
SLC25A1 neuron transgenic mice display differential changes in the proteome and acetyl-proteome
To resolve the potential molecular mechanisms of the SLC25A1 nTg phenotype, we performed quantitative proteomics on cortical and hippocampal tissue from our SLC25A1 nTg mice compared to wild-type controls. We detected a total of 1887 and 1623 proteins in the hippocampus and cortex, respectively; of these proteins, 99 and 227 in the hippocampus and cortex, respectively, had a statistically significant change in expression compared to wild-type controls (P < 0.05; Fig. 6A). Interestingly, the proteomic adaptations in the SLC25A1 nTg hippocampus and cortex differed significantly. First, we observed a difference in the overall distribution of protein expression changes from wild-type detected in the two tissue types (Fig. 6B). Additionally, only 13 significantly changed proteins were commonly shared between hippocampus and cortex, and all but 3 exhibited a fold change in the opposite direction (Fig. 6C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the significantly changed proteins revealed distinct categories between the hippocampus and cortex, but despite these differences, multiple categories were pertinent to the SLC25A1 nTg phenotype including glutamatergic synapse, dopaminergic synapse and LTP (Fig. 6D). Moreover, there were several categories related to protein synthesis and secretion, including salivary secretion, insulin secretion and ribosome, which may be related to changes occurring in the secretory pathway (discussed later). Finally, we constructed gene-network plots with the significantly changed proteins using the gene ontology (GO) cellular component function database (Fig. 6E). Several categories relevant to the phenotype were enriched including neuron to neuron synapse, postsynaptic density and synaptic vesicle. Therefore, despite differential proteomic changes between the hippocampus and cortex, the cellular functions affected were quite similar and closely related to the phenotype of the SLC25A1 nTg mouse.
Figure 6.

SLC25A1 nTg mice display global proteomic changes in the hippocampus and cortex. (A) Volcano plots displaying all quantified proteins in SLC25A1 nTg hippocampus (left) and cortex (right) compared with wild-type (WT) controls. Statistically significant proteins are shown in green (99 in hippocampus and 227 in cortex; P < 0.05 via Fisher’s method) with all other proteins in grey. n = 4 male mice per genotype at 2–5 months of age. (B) Histogram and overlaid Gaussian distribution showing the distribution of all proteins’ log2 fold change from wild-type in hippocampus and cortex. A box-and-whisker plot is shown above with the box representing 25th/75th percentiles, middle line representing mean and whiskers representing 1st/99th percentiles. ***P < 0.0005 via the Kolmogorov–Smirnov test. (C) Significantly changed proteins with overlap between the hippocampus and cortex. All proteins represented in green shown in panel A are included. The heat map shows the expression profile of the 13 overlapping proteins between hippocampus and cortex. (D) The fold enrichment of KEGG pathways determined from significantly changed proteins in the hippocampus and cortex compared with wild-type controls. The top 10 categories sorted by enrichment score are shown with a filtered FDR score of 0.05. (E) Gene-network plots of significantly changed proteins in the SLC25A1 nTg hippocampus and cortex. Plots constructed using an overrepresentation analysis using the GO cellular component function database. The dot size of each network category is scaled by the number of overlapping proteins within the category. The top 15 results are shown with a filtered FDR score of 0.05.
To assess how altered acetyl-CoA metabolism (Fig. 1E) via SLC25A1 overexpression affects protein acetylation, we performed quantitative acetyl-proteomics. Protein acetylation is a mechanism of post-translational regulation that can impact protein interactions and function and can be independent of protein expression changes.44 We again used hippocampal and cortical tissue from our SLC25A1 nTg compared to wild-type control mice. We performed a subcellular fractionation (cytoplasmic, nuclear/membrane and chromatin-associated) before measuring the stoichiometry of acetylation in order to increase detection resolution.34 In the three fractions combined, we detected 2506 and 3886 protein acetylation sites (henceforth called acetylpeptides) in the hippocampus and cortex, respectively. Of these acetylpeptides, the following number had a stoichiometry value that was statistically different from wild-type: cytoplasmic, 25 in the hippocampus and 51 in the cortex; nuclear/membrane, 25 in the hippocampus and 64 in the cortex; chromatin-associated, 37 in the hippocampus and 60 in the cortex (Fig. 7A and B). In the cortex, several acetylpeptides that exhibited a statistically significant stoichiometry change from wild-type were detected in multiple fractions and this did not occur in the hippocampus (Fig. 7B). When the significant acetylpeptides were combined from all three fractions, the hippocampus and cortex had a total of 87 and 166 significant acetylpeptides, respectively; nine of these acetylpeptides were the same between the two tissue types with four exhibiting a stoichiometry change in the same direction and five in the opposite direction (Fig. 7C). KEGG pathway analysis of the proteins harbouring the significantly changed acetylation sites revealed a strong predominance of oxidative phosphorylation-related categories including oxidative phosphorylation, carbon metabolism, citrate cycle (tricarboxylic acid cycle) and pyruvate metabolism (Fig. 7D). Gene-network plots with these same proteins using the GO cellular component function database revealed categories related to the SLC25A1 nTg phenotype including neuron to neuron synapse, postsynaptic density and axon part (Fig. 7E). Overall, similar to the proteomic analysis, the hippocampus and cortex acetyl-proteomic adaptations involved different proteins and/or sites, but impacted similar cellular functions that were relevant to the SLC25A1 nTg phenotype.
Figure 7.

SLC25A1 nTg mice display changes in the stoichiometry of acetylation in the hippocampus and cortex. (A) Volcano plots displaying all quantified acetylpeptides in the SLC25A1 nTg versus wild-type hippocampus (top) and cortex (bottom) across the three subcellular fractions (cytosolic, nuclear/membrane and chromatin-associated). Change in acetylation stoichiometry (Δ acetyl stoichiometry) is calculated using the SLC25A1 nTg stoichiometry value minus the wild-type stoichiometry value. Statistically significant acetylpeptides (P < 0.05) are shown in green with all other acetylpeptides in grey, n = 4 male mice per genotype at 2 months of age. (B) Overlap of the statistically significant acetylpeptides across the three subcellular fractions in both the hippocampus and cortex. (C) Overlap between the hippocampus and cortex of all the statistically significant acetylpeptides (combined from the three subcellular fractions). A heat map of the nine overlapping acetylpeptides between the two tissue types is shown. The change in acetylation stoichiometry for the following acetylpeptides is displayed (detected in multiple cortex subcellular fractions): Atp5b K451, nuclear/membrane; Dctn1 K993, cytoplasmic. (D) The fold enrichment of KEGG pathways determined from proteins harbouring the acetylation sites that were significantly changed from wild-type in the hippocampus and cortex. The top 10 categories sorted by enrichment score are shown with a filtered FDR score of 0.05. (E) Gene-network plots of proteins harbouring the acetylation sites that were significantly changed from wild-type in the hippocampus and cortex. Plots constructed via an overrepresentation analysis using the GO cellular component function database. The dot size of each network category is scaled by the number of overlapping proteins within the category. The top 10 categories sorted by enrichment score are shown with a filtered FDR score of 0.05. Because proteins could be found in multiple fractions and potentially acetylated on multiple lysine sites, the Δ acetyl stoichiometry value displayed in the network plot is as follows: (i) if the acetylpeptide was detected in multiple subcellular fractions, the fraction selected for illustration was prioritized as follows: cytoplasmic, nuclear/membrane, then chromatin-associated; and (ii) if multiple sites per protein exhibited significantly changed acetyl stoichiometry, the lowest number lysine residue was selected for illustration.
Because we were able to characterize both the proteome and acetyl-proteome of our SLC25A1 nTg mice, we compared the list of significantly affected proteins from both analyses and found a small overlap in both the hippocampus and cortex (Fig. 8A). To further illustrate this, we manually constructed a cluster plot using the proteins within the four overlapping GO cellular component categories in both the proteome and acetyl-proteome (see gene-network plots in Figs 6E and 7E): postsynaptic density, asymmetric synapse, postsynaptic specialization and neuron-to-neuron synapse (Fig. 8B). This plot revealed most proteins were regulated at either the proteome level or acetyl-proteome level, but rarely both. Additionally, some proteins exhibited complex modes of regulation such as multiple acetylation changes in different directions. In summary, the SLC25A1 nTg mouse exhibited differential changes in both the proteome and acetyl-proteome in the hippocampus and cortex, but despite these differences, the affected cellular functions were strikingly similar and related to the observed mouse phenotype.
Figure 8.

SLC25A1 nTg mice display minimal overlap in their proteome and acetyl-proteome. (A) Overlap between the proteins with significantly different expression from wild-type (proteome) and proteins harbouring the acetylation sites that were significantly changed from wild-type (acetyl-proteome) in both the hippocampus and cortex. (B) Proteins from the overlapping GO cellular component categories shown in Figs 6E and 7E are displayed (postsynaptic density, asymmetric synapse, postsynaptic specialization and neuron-to-neuron synapse) with denoted changes at the level of the proteome or acetyl-proteome. All proteins displayed are found within the 4 GO cellular component categories with these proteins unique in the following categories: postsynaptic specialization: Nlgn2, Nlgn3, Gabrb3; neuron-to-neuron synapse: Calb1, Syt1, Sh3gl2.
Discussion
In this study, we reported that mice with overexpression of human SLC25A1 in forebrain neurons display increased steady-state levels of citrate and acetyl-CoA into the cytosol, increased flux of acetyl-CoA into the ER lumen and autistic-like behaviours. These mice had evident changes in white matter integrity with an unexpected expansion of the microglial population. Additionally, synaptic structure and function were affected at multiple levels, which were in part due to both proteomic and acetyl-proteomic adaptations.
We have previously reported that aberrant intracellular flux of acetyl-CoA from the cytosol to the ER appears to be a mechanistic driver of ASD, which is in part due to the cellular crosstalk occurring between the ER, mitochondria and nucleus.12,17,19,20 This is substantiated by the fact that gene duplication events that can impact intracellular acetyl-CoA flux, including AT-1/SLC33A1, SLC25A1, SLC13A5 and ACLY, are all associated with ASD.24 Therefore, we were not surprised to find that overexpression of the mitochondrial citrate/malate antiporter SLC25A1 resulted in increased levels of citrate and acetyl-CoA in the cytosol, increased levels of acetyl-CoA in the ER and autistic-like behaviours in the mouse. It is worth mentioning that although there are some overlapping findings, the specific behavioural aberrations we observed in the SLC25A1 nTg mice do not directly match with our previously reported AT-1 nTg autistic-like model.12 Additionally, the SLC25A1 nTg mice also have a jumping stereotypy not observed in the AT-1 nTg but observed in other models of ASD, such as the Jakmip1 knockout and C58/J inbred mouse strain.12,36,38 Indeed, the differences observed in the fear conditioning and novel object recognition assays as well as the normal behaviour in the social interaction paradigm (specifically the recognition trial) do not show clear evidence of cognitive dysfunction in the SLC25A1 nTg mice but rather reveal erratic behaviour, such as the dramatic lack of freezing behaviour in the fear conditioning paradigm, that significantly deviates from wild-type controls. There is continued disagreement in the exact behavioural aberrations that define ASD in rodents, and there is likely considerable overlap with other neurodevelopmental diseases such as attention deficit hyperactivity disorder (ADHD).37,39,45 Therefore, although we conclude the SLC25A1 nTg phenotype is indeed autistic-like, we have not excluded the possibility of modelling other forms of neurodevelopmental diseases. Additionally, we have not specifically tested our SLC25A1 nTg mice for behavioural hallmarks of ADHD such as motor impulsiveness,46 which may warrant further investigation.
Our DWI analysis on the SLC25A1 nTg mice revealed disrupted white matter integrity as evident by reduced DTI fractional anisotropy, which is a common observation in children with ASD.40,41 Additionally, advanced diffusion modelling (NODDI and ActiveAx) revealed changes consistent with neuroinflammation,42 which was corroborated by the increased microglial density and ramification observed in the SLC25A1 nTg hippocampus. To our surprise, we did not find proteomic or acetyl-proteomic changes consistent with activated neuroinflammatory pathways, nor did we see other characteristic hallmarks of neuroinflammation such as reactive astrocytosis. Therefore, we presume there is some type of neuron-to-microglia signalling resulting in microglial activation that deviates from the typical neuroinflammatory cascade, which could include secreted factors or extracellular vesicles from the SLC25A1 overexpressing neurons.47,48 Elucidating the molecular underpinnings of the microglial activation in our SLC25A1 nTg mice could reveal a new mode of neuron-to-microglia communication during neurodevelopment.
N ε-lysine acetylation within the lumen of the ER is a recently characterized post-translational modification that serves as an important quality control mechanism within the secretory pathway. In the AT-1 nTg, excess acetyl-CoA import into the ER resulted in a dramatic increase in glycoprotein flux through the secretory pathway, which caused increased dendritic branch and spine formation, significant upregulation of multiple proteins at the synapse and altered synaptic plasticity.12 We observed some of the same molecular and cellular events in the SLC25A1 nTg, such as enhanced hippocampal LTP and increased dendritic branching in vitro. Additionally, our KEGG pathway analysis of the SLC25A1 nTg proteome revealed widespread alterations in protein secretion, which could implicate ER-based Nε-lysine acetylation. However, key features of the AT-1 nTg phenotype were not observed in the SLC25A1 nTg mouse; most striking was the lack of protein expression changes in the synapse in vivo. It is possible that our crude synaptosome preparations were not pure enough to detect subtle changes in protein expression at the synapse. In the AT-1 nTg study, purified postsynaptic densities were used rather than crude synaptosomes.12 While we predict altered Nε-lysine acetylation to be—at least in part—an underlying mechanism for the SLC25A1 nTg phenotype, especially due to the increased ER acetyl-CoA steady-state levels, mechanisms outside of ER acetylation are likely important. Reduced gene dosage of SLC25A1 occurs in DiGeorge syndrome (22q11.2 hemizygous microdeletion), where patients routinely demonstrate features of ASD, ADHD and other neuropsychiatric symptoms; recent work has demonstrated the SLC25A1-SLC25A4 interactome modulates the risk of neurodevelopmental disorders and can impact synaptic morphology and function.49,50 Additionally, loss-of-function mutations in SLC25A1 result in a severe neurodevelopmental phenotype and combined d-2- and l-2-hydroxyglutaric aciduria, while overexpression results in fatty acid accumulation, highlighting the critical importance of SLC25A1 in cellular metabolism.51,52 Modulation of various components of the citrate/acetyl-CoA intracellular flux pathway, such as through inhibition of ACLY53 or the ER-based acetyltransferases ATase1 and ATase2,4,11,13,16 could aid in dissecting the contributing components to our SLC25A1 nTg phenotype.
The synaptic changes occurring in our SLC25A1 nTg mice were more complicated than what was observed in the AT-1 nTg12 and warrant discussion. While our Golgi staining analysis showed increased dendritic spine density and decreased spine volume in vivo, the primary neuron analysis in vitro showed no change in dendritic spine density but increased spine volume. However, there was increased co-localization of synaptic proteins in the primary neurons, suggesting a higher density of synapses, but not dendritic spines, formed in vitro. The underlying significance of this distinction is unclear, but the differences in vivo versus in vitro may have to do with the presence or absence of microglia; co-cultures of neurons and microglia could evaluate this hypothesis.54 It is also worth mentioning that the MEA data showed increased neural excitability as evident by mean firing rate and number of active electrodes, but the transgenic networks did not have changes in synchronicity as evident by unaltered network burst frequency and synchronicity index. This could be interpreted as the SLC25A1 nTg primary neurons forming more synapses in vitro, but these synapses are not enhancing neuron-to-neuron communication. Rather, SLC25A1 overexpression increases the basal neuron firing rate that does not affect the activity of adjacent neurons. Additional electrophysiologic experiments would need to be conducted to narrow down the potential rationale for this observed phenomenon.
In our proteomic and acetyl-proteomic data, we observed distinct changes in proteins and acetylpeptides between the hippocampus and cortex. However, to our surprise, very similar pathways were affected as revealed by the GO cellular component and KEGG pathway analyses, and the primary neurons derived from hippocampus or cortex exhibited a similar phenotype. Therefore, while different mechanisms are likely resulting in changes in protein abundance and acetylation as a result of SLC25A1 overexpression, they are working in concert to regulate similar cellular functions. Whether this is a consequence of different expression levels of SLC25A1 in the hippocampus and cortex or more complex tissue-specific gene or protein regulatory events is an open question. Additionally, the KEGG pathway analyses of the proteome revealed many categories related to secretion while the acetyl-proteome contained many related to oxidative phosphorylation; it is plausible that different cellular functions in the SLC25A1 nTg mice, such as glycoprotein synthesis or glucose metabolism, are being regulated at different levels (protein expression or lysine acetylation).
In conclusion, we have demonstrated that overexpression of SLC25A1 in mouse forebrain neurons results in an autistic-like phenotype with jumping stereotypy, disrupted white matter integrity and altered synaptic function. These data strongly support that aberrant cytosolic-to-ER acetyl-CoA flux can be a mechanistic driver of an autistic-like phenotype.
Supplementary Material
Acknowledgements
We would like to thank Kathy Krentz at the Genome Editing & Animal Models Core of UW-Madison for the generation of the SLC25A1 transgenic mice; Heather Mitchell at the UW-Madison Waisman Behavior Testing Service for conducting mouse behaviour testing; and B. Sheehan and Y. Peng for technical assistance.
Funding
This research was supported by National Institutes of Health (R01 NS094154, R01 GM065386, F30 AG066329, RF1 AG052324, U01 CA231081, R01 DK071801, P41 GM108538) and a core grant to the Waisman Center from NICHD-U54 HD090256. M.J.R. was supported in part by the Wisconsin Distinguished Graduate Fellowship. I.A.D. was supported by T32 AG000213. J.J.Y. was supported by the Clinical and Translational Science Award (CTSA) programme, through the National Institutes of Health National Center for Advancing Translational Sciences (NCATS), grant UL1TR002373. Additional imaging support was provided by the University of Wisconsin Carbone Cancer Center Support Grant P30CA014520 and Waisman Core Grant P30 HD003352-45 and U54 AI117924-03. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
The authors declare the following competing interests: J.M.D. is a co-founder of Galilei BioScience Inc. and a consultant for Evrys Bio. The remaining authors have no competing interests to disclose.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- ASD
autism spectrum disorder
- DIV
days in vitro
- DTI
diffusion tensor imaging
- DWI
diffusion weighted imaging
- ER
endoplasmic reticulum
- GO
gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LTP
long-term potentiation
- MEA
multi-electrode array
- MS
mass spectrometry
- NA
numerical aperture
- NODDI
neurite orientation dispersion and density imaging
- nTg
neuron transgenic
- TBSS
tract-based spatial statistics
References
- 1. Costantini C, Ko MH, Jonas MC, Puglielli L.. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407(3):383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ko MH, Puglielli L.. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 2009;284(4):2482–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jonas MC, Pehar M, Puglielli L.. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J Cell Sci. 2010;123(Pt 19):3378–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ding Y, Ko MH, Pehar M, et al. Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces beta-secretase (BACE1) levels and Abeta generation. J Biol Chem. 2012;287(11):8424–8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pehar M, Jonas MC, Hare TM, Puglielli L.. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J Biol Chem. 2012;287(35):29921–29930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pehar M, Lehnus M, Karst A, Puglielli L.. Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by N(epsilon)-lysine acetylation in the lumen of the organelle and predicts broad biological impact. J Biol Chem. 2012;287(27):22436–22440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pehar M, Puglielli L.. Lysine acetylation in the lumen of the ER: A novel and essential function under the control of the UPR. Biochim Biophys Acta. 2013;1833(3):686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding Y, Dellisanti CD, Ko MH, Czajkowski C, Puglielli L.. The endoplasmic reticulum-based acetyltransferases, ATase1 and ATase2, associate with the oligosaccharyltransferase to acetylate correctly folded polypeptides. J Biol Chem. 2014;289(46):32044–32055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mak AB, Pehar M, Nixon AM, et al. Post-translational regulation of CD133 by ATase1/ATase2-mediated lysine acetylation. J Mol Biol. 2014;426(11):2175–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peng Y, Li M, Clarkson BD, et al. Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J Neurosci. 2014;34(20):6772–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duran-Aniotz C, Cornejo VH, Hetz C.. Targeting endoplasmic reticulum acetylation to restore proteostasis in Alzheimer's disease. Brain. 2016;139(Pt 3):650–652. [DOI] [PubMed] [Google Scholar]
- 12. Hullinger R, Li M, Wang J, et al. Increased expression of AT-1/SLC33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J Exp Med. 2016;213(7):1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peng Y, Kim MJ, Hullinger R, et al. Improved proteostasis in the secretory pathway rescues Alzheimer's disease in the mouse. Brain. 2016;139(Pt 3):937–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peng Y, Puglielli L.. N-lysine acetylation in the lumen of the endoplasmic reticulum: A way to regulate autophagy and maintain protein homeostasis in the secretory pathway. Autophagy. 2016;12(6):1051–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farrugia MA, Puglielli L.. Nε-lysine acetylation in the endoplasmic reticulum—A novel cellular mechanism that regulates proteostasis and autophagy. J Cell Sci. 2018;131(22):jcs221747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peng Y, Shapiro SL, Banduseela VC, et al. Increased transport of acetyl-CoA into the endoplasmic reticulum causes a progeria-like phenotype. Aging Cell. 2018;17(5):e12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dieterich IA, Lawton AJ, Peng Y, et al. Acetyl-CoA flux regulates the proteome and acetyl-proteome to maintain intracellular metabolic crosstalk. Nat Commun. 2019;10(1):3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rigby MJ, Ding Y, Farrugia MA, et al. The endoplasmic reticulum acetyltransferases ATase1/NAT8B and ATase2/NAT8 are differentially regulated to adjust engagement of the secretory pathway. J Neurochem. 2020;154(4):404–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dieterich IA, Cui Y, Braun MM, et al. Acetyl-CoA flux from the cytosol to the ER regulates engagement and quality of the secretory pathway. Sci Rep. 2021;11(1):2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rigby MJ, Lawton AJ, Kaur G, et al. Endoplasmic reticulum acetyltransferases Atase1 and Atase2 differentially regulate reticulophagy, macroautophagy and cellular acetyl-CoA metabolism. Commun Biol. 2021;4:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheehan BK, Orefice NS, Peng Y, Shapiro SL, Puglielli L.. ATG9A regulates proteostasis through reticulophagy receptors FAM134B and SEC62 and folding chaperones CALR and HSPB1. ISCIENCE. 2021;24(4):102315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huppke P, Brendel C, Kalscheuer V, et al. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am J Hum Genet. 2012;90(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin PF, Li JW, Liu QJ, et al. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am J Hum Genet. 2008;83(6):752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sanders SJ, Ercan-Sencicek AG, Hus V, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70(5):863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ebrahimi-Fakhari D, Sahin M.. Autism and the synapse: Emerging mechanisms and mechanism-based therapies. Curr Opin Neurol. 2015;28(2):91–102. [DOI] [PubMed] [Google Scholar]
- 26. Jiang Y, Johnson GA.. Microscopic diffusion tensor atlas of the mouse brain. Neuroimage. 2011;56(3):1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harms RL, Fritz FJ, Tobisch A, Goebel R, Roebroeck A.. Robust and fast nonlinear optimization of diffusion MRI microstructure models. Neuroimage. 2017;155:82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alexander DC, Hubbard PL, Hall MG, et al. Orientationally invariant indices of axon diameter and density from diffusion MRI. Neuroimage. 2010;52(4):1374–1389. [DOI] [PubMed] [Google Scholar]
- 29. Barnett BR, Torres-Velazquez M, Yi SY, et al. Sex-specific deficits in neurite density and white matter integrity are associated with targeted disruption of exon 2 of the Disc1 gene in the rat. Transl Psychiatry. 2019;9(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pehar M, O'Riordan KJ, Burns-Cusato M, et al. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9(2):174–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frost DC, Greer T, Li L.. High-resolution enabled 12-plex DiLeu isobaric tags for quantitative proteomics. Anal Chem. 2015;87(3):1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tyanova S, Temu T, Cox J.. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11(12):2301–2319. [DOI] [PubMed] [Google Scholar]
- 33. Tyanova S, Temu T, Sinitcyn P, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731–740. [DOI] [PubMed] [Google Scholar]
- 34. Baeza J, Lawton AJ, Fan J, et al. Revealing dynamic protein acetylation across subcellular compartments. J Proteome Res. 2020;19(6):2404–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu G. enrichplot : Visualization of Functional Enrichment Result [computer program]. Version R package version 1.8.1. Accessed 16 December 2021. https://github.com/GuangchuangYu/enrichplot
- 36. Berg JM, Lee C, Chen L, et al. JAKMIP1, a novel regulator of neuronal translation, modulates synaptic function and autistic-like behaviors in mouse. Neuron. 2015;88(6):1173–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lewis MH, Tanimura Y, Lee LW, Bodfish JW.. Animal models of restricted repetitive behavior in autism. Behav Brain Res. 2007;176(1):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryan BC, Young NB, Crawley JN, Bodfish JW, Moy SS.. Social deficits, stereotypy and early emergence of repetitive behavior in the C58/J inbred mouse strain. Behav Brain Res. 2010;208(1):178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silverman JL, Yang M, Lord C, Crawley JN.. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11(7):490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hiremath CS, Sagar KJV, Yamini BK, et al. Emerging behavioral and neuroimaging biomarkers for early and accurate characterization of autism spectrum disorders: A systematic review. Transl Psychiatry. 2021;11(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li D, Karnath HO, Xu X.. Candidate biomarkers in children with autism spectrum disorder: A review of MRI studies. Neurosci Bull. 2017;33(2):219–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yi SY, Barnett BR, Torres-Velazquez M, et al. Detecting microglial density with quantitative multi-compartment diffusion MRI. Front Neurosci. 2019;13(81). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deshpande A, Yadav S, Dao DQ, et al. Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep. 2017;21(10):2678–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Verdin E, Ott M.. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16(4):258–264. [DOI] [PubMed] [Google Scholar]
- 45. Leo D, Gainetdinov RR.. Transgenic mouse models for ADHD. Cell Tissue Res. 2013;354(1):259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sagvolden T, Russell VA, Aase H, Johansen EB, Farshbaf M.. Rodent models of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(11):1239–1247. [DOI] [PubMed] [Google Scholar]
- 47. Eyo UB, Wu LJ.. Bidirectional microglia–neuron communication in the healthy brain. Neural Plast. 2013;2013:456857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T.. Bidirectional microglia–neuron communication in health and disease. Front Cell Neurosci. 2018;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gokhale A, Hartwig C, Freeman AAH, et al. Systems analysis of the 22q11.2 microdeletion syndrome converges on a mitochondrial interactome necessary for synapse function and behavior. J Neurosci. 2019;39(18):3561–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rojas-Charry L, Nardi L, Methner A, Schmeisser MJ.. Abnormalities of synaptic mitochondria in autism spectrum disorder and related neurodevelopmental disorders. J Mol Med (Berl). 2021;99(2):161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang JH, Li SQ, Khan MAK, Garre V, Vongsangnak W, Song YD.. Increased lipid accumulation in mucor circinelloides by overexpression of mitochondria! Citrate transporter genes. Ind Eng Chem Res. 2019;58(6):2125–2134. [Google Scholar]
- 52. Palmieri F, Scarcia P, Monne M.. Diseases caused by mutations in mitochondrial carrier genes SLC25: A review. Biomolecules. 2020;10(4):655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. 2018;157:1276–1291. [DOI] [PubMed] [Google Scholar]
- 54. Park LC, Zhang H, Gibson GE.. Co-culture with astrocytes or microglia protects metabolically impaired neurons. Mech Ageing Dev. 2001;123(1):21–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics data that support the findings of this study have been deposited to the ProteomeXchange Consortium (ID number PXD025415). The acetyl-proteomics data that support the findings of this study have been deposited to the ProteomeXchange Consortium (ID number PXD025424) and the MassIVE partner repository (ID number MSV000087209). The authors declare that all other data supporting the findings of this study are available within the paper, the Supplementary Tables and Supplementary material.